

Abstract
Background:
Patients resuscitated from cardiac arrest are routinely sedated during targeted temperature management, while the effects of sedation on cerebral physiology and outcomes after cardiac arrest remain to be determined. We hypothesized that sedation would improve survival and neurological outcomes in mice after cardiac arrest.
Methods:
Adult C57BL/6J mice of both sexes were subjected to potassium chloride-induced cardiac arrest and cardiopulmonary resuscitation. Starting at return of spontaneous circulation or at 60 minutes after return of spontaneous circulation, mice received intravenous infusion of propofol at 40 mg η kg−1 η h−1, dexmedetomidine at 1 μg η kg−1 η h−1, or normal saline for 2 hours. Body temperature was lowered and maintained at 33°C during sedation. Cerebral blood flow was measured for 4 hours post-resuscitation. Telemetric electroencephalogram (EEG) was recorded in freely moving mice from 3 days before up to 7 days after cardiac arrest.
Results:
Sedation with propofol or dexmedetomidine starting at return of spontaneous circulation improved survival in hypothermia-treated mice (propofol [13/16, 81%] vs. no sedation [4/16, 25%], P = 0.008; dexmedetomidine [14/16, 88%] vs. no sedation [4/16, 25%], P = 0.002). Mice receiving no sedation exhibited cerebral hyperemia immediately after resuscitation and EEG power remained less than 30% of the baseline in the first 6 hours post-resuscitation. Administration of propofol or dexmedetomidine starting at return of spontaneous circulation attenuated cerebral hyperemia and increased EEG slow oscillation power during and early after sedation (40 to 80% of the baseline). In contrast, delayed sedation failed to improve outcomes, without attenuating cerebral hyperemia and inducing slow-wave activity.
Conclusions:
Early administration of sedation with propofol or dexmedetomidine improved survival and neurological outcomes in mice resuscitated from cardiac arrest and treated with hypothermia. The beneficial effects of sedation were accompanied by attenuation of the cerebral hyperemic response and enhancement of electroencephalographic slow-wave activity.
Introduction
Cardiac arrest is a major public health challenge worldwide.1 Despite advances in resuscitation methods, cardiac arrest is still associated with high mortality and morbidity.2 As part of post-resuscitation care, targeted temperature management has been used in patients who achieved return of spontaneous circulation with the aim of minimizing hypoxic-ischemic brain damage after cardiac arrest.3 Nevertheless, the burden of post-anoxic brain injury remains unacceptably high, with the majority of cardiac arrest survivors presenting in coma or with an altered level of consciousness.2
Patients undergoing targeted temperature management are routinely sedated with drugs that promote cardiorespiratory stabilization, facilitate mechanical ventilation, and control agitation, pain, anxiety, delirium, and shivering.4–6 Preclinical studies suggest that sedatives exert neuroprotective effects in animals subjected to focal or global brain ischemia and reperfusion.7,8 In cardiac arrest survivors with impaired cerebral autoregulation, sedation may protect the brain against secondary brain injury by modulating cerebral blood flow and cerebral metabolic rate of oxygen.6,9,10 On the other hand, randomized clinical trials conducted in the general intensive care unit showed that minimizing or avoiding sedation provides better outcomes, including shorter duration of mechanical ventilation and hospital length of stay.11,12 Based on these observations, the benefits of using sedation in unresponsive cardiac arrest patients have been questioned.13 However, patients with severe acute brain injury (e.g., comatose patients after cardiac arrest, ischemic or traumatic brain injury) have been excluded from previous studies.11,12 The role of pharmacological sedation in comatose cardiac arrest patients remains to be determined.
The electroencephalogram (EEG) reflects oscillatory extracellular electrical currents and potentials arising from neuronal activity in the cortical and subcortical brain structures. In healthy individuals, sedative-hypnotic agents give rise to low-frequency, high-amplitude activity that becomes slower with deepening levels of sedation/anesthesia,14 while altering EEG power within a specific frequency range (e.g., propofol-induced alpha oscillations and dexmedetomidine-induced spindle oscillations).15 Although there has been a growing interest in the use of EEG analysis for outcome prediction after cardiac arrest, knowledge about sedation-induced EEG changes in the post-cardiac arrest population is limited.16,17 Because sedation during targeted temperature management affects consciousness and potentially interferes with interpretation of EEG, optimal sedation is recommended in post-resuscitation care to allow early awakening and limit confounding in accurate prognostication.9,18 On the other hand, the ability to generate slow waves in response to propofol infusion at 4 mg η kg−1 η h−1 has been proposed as an early predictor of neurological recovery in comatose cardiac arrest patients.19,20 The slow-wave activity is recognized as a neurophysiological signature of normal brain function observed during sleep and deep sedation/anesthesia,14,21 which is hypothesized to provide rest for individual neurons and prevent long-term neuronal damage.22 Nevertheless, little is known about the significance of sedation-induced EEG changes in neurological recovery after cardiac arrest.
The objective of the current study was to elucidate the impact of pharmacological sedation on neurological outcomes in mice resuscitated from cardiac arrest managed with therapeutic hypothermia. We hypothesized that sedation would improve survival and neurological outcomes after cardiac arrest in hypothermia-treated mice. To test this hypothesis, we sought to characterize the effects of two commonly used sedatives: propofol (2,6-di-isopropylphenol), a GABA receptor agonist, and dexmedetomidine, a highly selective α2-adrenoceptor agonist, in mice resuscitated from experimental cardiac arrest with continuous EEG monitoring and cerebral blood flow measurement.
Materials and Methods
Animal Preparation
Laboratory animal housing, handling, and procedures were performed in compliance with the protocols approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital. We studied adult male (10 to 12 weeks old, 25 to 30 g) and female (15 to 20 weeks old, 21 to 26 g) C57BL/6J wild-type mice. In response to peer review, additional data from female mice were obtained. All mice were housed in a 24-hour light/dark cycle (lights on at 7:00 AM and off at 7:00 PM), temperature-controlled room (20–23°C) with free access to food and water. All mice were within a healthy body weight range at baseline. The individual animal was considered the experimental unit. The number of mice used for each experiment (survival study, brain histology, cerebral blood flow measurement, and EEG recording) is summarized in Supplementary Table 1 (n refers to the number of animals).
Mouse Model of Cardiac Arrest
Prior to cardiac arrest and cardiopulmonary resuscitation (CPR), mice were anesthetized with 5% isoflurane in 100% oxygen and intubated with a 20-gauge catheter (Angiocath; Becton Dickinson, Franklin Lakes, NJ). Mice were then mechanically ventilated with a respiratory rate of 110 breaths/minute and a tidal volume of 10 μl/g (mini-vent; Harvard Apparatus, Holliston, MA). Under isoflurane anesthesia at 1.5%, mice were instrumented with microcatheters (PE-10; Becton Dickinson, Franklin Lakes, NJ) that were placed in the femoral artery for monitoring mean arterial pressure and in the femoral vein for administrating drugs. The experimental procedures were conducted during the light phase in the previously described manner with some modifications.23 Briefly, male mice were subjected to potassium chloride-induced cardiac arrest for 8 minutes followed by chest compression in combination with resumption of mechanical ventilation with 100% oxygen and intravenous epinephrine administration. Female mice were subjected to cardiac arrest for 8.5 minutes because they are less sensitive brain ischemia than male mice.24 Isoflurane anesthesia was discontinued when cardiac arrest was induced in all mice. Core body temperature was measured throughout the procedure using an esophageal temperature probe. Bupivacaine (2 mg/kg) was injected in the surgical wounds pre-operatively in conjunction with administration of buprenorphine (0.1 mg/kg) for post-operative analgesia. All animals were treated in the same manner before, during, and after cardiac arrest and CPR in terms of pre-cardiac arrest anesthesia, mechanical ventilation, temperature control, monitoring, and post-operative care including analgesia, whether or not they received post-cardiac arrest sedation.
Post-cardiac Arrest Sedation Starting at Return of Spontaneous Circulation
Starting at return of spontaneous circulation, 30 male and 18 female mice were randomly assigned to receive continuous intravenous infusion of propofol at a rate of 40 mg η kg−1 η h−1 or dexmedetomidine at a rate of 1 μg η kg−1 η h−1 or normal saline (vehicle) for 2 hours (10 male mice and 6 female mice per group; Supplementary Table 1). The infusion rate of the drugs was determined based on our pilot experiments (Supplementary Figure 1). In addition, a group of male mice undergoing EEG recording received administration of propofol at 10 mg η kg−1 η h−1 starting at return of spontaneous circulation (Supplementary Table 1) to investigate the dose-dependent effects of propofol. All mice were weaned from mechanical ventilation at 20 minutes after return of spontaneous circulation when the spontaneous respiratory rate exceeded 100 breaths/minute, and oxygen was administered continuously through a T-piece circuit up to 40 minutes after return of spontaneous circulation. Mean arterial pressure, heart rate, and respiratory rate were recorded up to 120 minutes after return of spontaneous circulation. Body temperature was maintained at 37°C until 20 minutes after return of spontaneous circulation, and was subsequently lowered and maintained at 33°C until administration of propofol or dexmedetomidine or vehicle was discontinued. After removal of the catheters, all mice were returned to their cages where they are allowed to equilibrate at the ambient temperature. The detailed experimental timeline is shown in Supplementary Figure 2A. Mice were followed up for 10 days after cardiac arrest, and survival rates were assessed by an investigator blinded to the experimental groups. The animals were euthanized with an overdose of isoflurane at the end of the study follow-up period.
Post-cardiac Arrest Sedation Starting at 60 Minutes after Return of Spontaneous Circulation
Starting at 60 minutes after return of spontaneous circulation, 30 male mice in another group were randomly assigned to receive administration of propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 or normal saline (vehicle) for 2 hours (10 male mice per group; Supplementary Table 1). In these experiments, body temperature was maintained at 33°C up to 180 minutes after return of spontaneous circulation (Supplementary Figure 2B). Mice were followed up for 10 days after cardiac arrest, and survival rates were assessed by an investigator blinded to the experimental groups. The animals were euthanized with an overdose of isoflurane at the end of the study follow-up period.
Neurological Outcome Assessment
Neurological function was assessed at 24, 48, 72, 96, and 120 hours after cardiac arrest by an investigator blinded to the experimental groups. Animals in the different experimental groups were assessed in sequential order. The previously reported neurological function scoring system was used with minor modifications (Scale 1).23 For mice that underwent continuous EEG recording, we employed an alternative scoring system (Scale 2) to avoid applying a stimulus that could affect EEG recording, which was based partly on a scale evaluating post-ischemic neurological function in a different murine model of global cerebral ischemia.25 In both scales, higher scores indicate better neurological outcomes. Dead mice were scored at 0 points and were included in the statistical analysis. Details about Scale 1 and 2 are shown in Supplementary Table 2.
Histological Assessment
Histological examination was carried out in a separate group of male mice (n = 12) that were randomly assigned to receive no sedation (vehicle administration) or sedation with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation, or no sedation (Supplementary Table 1). Brains were harvested from mice euthanized at 24 hours after cardiac arrest in the previously described manner with minor modifications.26,27 Briefly, Fluoro-Jade B (FJB) was used to stain dying neurons in combination with counterstaining with 4′,6-Diamidino-2-phenylindole (DAPI) that allows nuclear staining. To assess the degree of neuronal degeneration in the cerebral cortex, 3 images were randomly selected from 2 different brain sections per mouse. FJB- and DAPI-positive neurons were counted by an investigator blinded to the identity of samples, using ImageJ 1.53g (National Institutes of Health, Bethesda, MD), and the percentage of FJB-positive neurons to DAPI-positive neurons was reported.
Cerebral Blood Flow Monitoring
In a separate group of male mice (n = 33) that were randomly assigned to receive sedation with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation or at 60 minutes after return of spontaneous circulation, or no sedation (vehicle administration starting at return of spontaneous circulation or at 60 minutes after return of spontaneous circulation) (Supplementary Table 1), we measured cerebral blood flow using a laser Doppler flowmeter (moorVMD-LDF1; Moor Instruments Inc., Wilmington, DE) affixed to the skull over the middle cerebral artery region. Before induction of cardiac arrest, baseline cerebral blood flow was measured for 2 minutes under isoflurane anesthesia. The laser Doppler flowmeter was calibrated to set the baseline value as 100% in each mouse. Cerebral blood flow was recorded for 4 hours after return of spontaneous circulation, and the relative value of cerebral blood flow was calculated as percent of the baseline value.
Electroencephalogram Transmitter Implantation
A telemetric EEG transmitter (ETA-F10; Data Sciences International, St. Paul, MN) was used in the current study. Under isoflurane anesthesia, the EEG transmitter was implanted subcutaneously so that the bipotential leads were positioned parallel to the long axis of the body. Two electrodes were placed at the following coordinates: the negative lead at 1.0 mm anterior and 1.0 mm lateral to Bregma, and the positive lead at 3.0 mm posterior and 3.0 mm lateral to Bregma on the contralateral side.28 After electrical contact with the dura membrane was established, dental acrylic was applied to the lead entry holes to ensure that the electrodes remain affixed to the skull. Bupivacaine (2 mg/kg) was injected in the surgical wounds pre-operatively in conjunction with administration of buprenorphine (0.1 mg/kg) for post-operative analgesia. Ten to 14 days after EEG transmitter implantation, mice were subjected to cardiac arrest and CPR, and thereafter randomly assigned to receive administration of propofol or dexmedetomidine or normal saline (vehicle) to examine the effects of sedation on EEG.
Electroencephalogram Acquisition and Processing
EEG was continuously recorded in 30 male and 5 female mice from 3 days before and up to 7 days after cardiac arrest (Supplementary Table 1). Telemetric data from the implantable EEG transmitters were digitally collected from freely moving mice at a sampling rate of 500 Hz in the Dataquest Advanced Research Technology system (Data Sciences International, St. Paul, MN) and analyzed with the use of NeuroScore 3.3.1 (Data Sciences International, St. Paul, MN). The derived EEG signals were band-pass filtered in the frequency ranges as follows: delta (0.5–4 Hz), theta (4–8 Hz), alpha (8–12 Hz), sigma (12–16 Hz), beta (16–24 Hz), and gamma (30–90 Hz) oscillations. For each frequency band, total EEG power during the light phase of the day before cardiac arrest (12 hours) was calculated as the pre-cardiac arrest baseline where both sleep and awake states were included. To describe time-dependent EEG changes after resuscitation, post-cardiac arrest EEG power was calculated for each frequency band at consecutive 1-hour time blocks up to 48 hours after return of spontaneous circulation, or at consecutive 10-minute time blocks up to 6 hours after return of spontaneous circulation. The post-cardiac arrest EEG power at each time block was expressed as percent of the pre-cardiac arrest baseline EEG power that was adjusted to match the length of the post-cardiac arrest time block (i.e., 1 hour or 10 minutes). For example, to calculate the percent post-cardiac arrest EEG power of a 1-hour time block, the post-cardiac arrest EEG power of the 1-hour time block was divided by the 1-hour pre-cardiac arrest baseline EEG power, which was obtained by dividing the total EEG power during the 12-hour light phase before cardiac arrest by 12.
MATLAB R2020a (MathWorks, Natick, MA) was used for spectral analysis. The spectrogram and power spectrum were computed using the multitaper method implemented in Chronux,29 an open-source software package including a MATLAB toolbox for signal processing of neurobiological time series data. The multitaper approach was employed to create EEG spectrograms for the purpose of providing clear and accurate high-resolution spectral estimates. Time-frequency spectrograms before cardiac arrest and in the first 24 hours after return of spontaneous circulation were generated using the Chronux function “mtspectrumc” with a time-bandwidth product TW = 3, number of tapers K = 5, and window length T = 3 seconds. Power spectral density plots were generated for 60-second windows at 6 time points during and after sedation (30, 60, 90, 120, 240, and 360 minutes after return of spontaneous circulation) to quantify the difference in the frequency distribution of EEG power between mice without sedation and those sedated with propofol or dexmedetomidine. The median power values were plotted by frequency with 95% confidence intervals derived from 1000-fold bootstrapping (calculated using the MATLAB bootstrap function “bootci”).30
Statistical Analysis
Variables were tested for normality with the Shapiro-Wilk test and Q-Q plots. Data were presented as means and standard deviations (SDs) for normally distributed variables, and median and interquartile ranges (IQRs) otherwise. For comparisons between mice without sedation and those sedated with propofol or dexmedetomidine, parametric data were analyzed using a one-way analysis of variance (ANOVA) followed by Sidak’s multiple comparisons test. The Kruskal-Wallis test followed by Dunn’s multiple comparisons test was used when data were not normally distributed. The Dunnett’s multiple comparisons test was performed as the post hoc test following a two-way repeated measures ANOVA to determine the propofol- or dexmedetomidine-induced effects on mean arterial pressure, heart rate, respiratory rate, cerebral blood flow, and quantitative EEG. Survival data was visualized using a Kaplan-Meier survival plot, and the log-rank test was used for comparing survival curves. The number of animals required for the survival study was estimated by a power analysis as 10 per group based on the assumption that the median survival times in the control and experimental treatment groups are 2 days and 9 days, respectively, over a follow-up period of 10 days (α = 0.05, β = 0.2, [Power = 80%], two-sided; PS: Power and Sample Size Calculation version 3.1.6). A priori sample size calculation was not performed in mice undergoing cerebral blood flow measurement and EEG recording because the effects of sedation on cerebral blood flow and quantitative EEG changes were unknown. The Spearman correlation coefficient was used to measure the degree of association between the relative EEG power early after resuscitation and neurological function at 24 hours after cardiac arrest. Mortality, neurological function, and physiological variables were examined with the individual animal as the unit of analysis. All statistical tests were two-tailed with significance set at P < 0.05. GraphPad Prism 9.2.0 (GraphPad Software Inc., La Jolla, CA) was used for statistical analyses.
Results
Sedation with Propofol or Dexmedetomidine Starting at Return of Spontaneous Circulation Improved Survival and Neurological Outcomes after Cardiac Arrest
Continuous intravenous infusion of propofol at 40 mg η kg−1 η h−1 modestly decreased mean arterial pressure (~10%) compared to no sedation in the first 2 hours after return of spontaneous circulation (Supplementary Figure 3A). Sedation with propofol or dexmedetomidine was associated with lower heart rate (~15%) than no sedation between 80 to 120 minutes after return of spontaneous circulation (Supplementary Figure 3B). Compared to mice that received no sedation, those sedated with propofol or dexmedetomidine exhibited improved survival at 10 days after cardiac arrest (Figure 1A; log-rank, propofol [13/16, 81%] vs. no sedation [4/16, 25%], P = 0.008; dexmedetomidine [14/16, 88%] vs. no sedation [4/16, 25%], P = 0.002; Supplementary Figure 4). The neurological function score was significantly higher in propofol- or dexmedetomidine-treated mice than in those without sedation at 5 days after cardiac arrest (Figure 1B; Kruskal-Wallis test, propofol 11.5 [9.0 – 12.0] vs. no sedation 1.5 [0.0 – 7.0], P = 0.0005; dexmedetomidine 10.0 [9.3 – 11.8] vs. no sedation 1.5 [0.0 – 7.0], P = 0.002). The range of times required for CPR is reported in Supplementary Table 3. Mice sedated with propofol or dexmedetomidine exhibited reduced neuronal death after cardiac arrest on histology, compared to those receiving no sedation (Figure 1C and 1D). These results suggest that administration of propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation prevented brain injury after cardiac arrest in hypothermia-treated mice.
Figure 1.
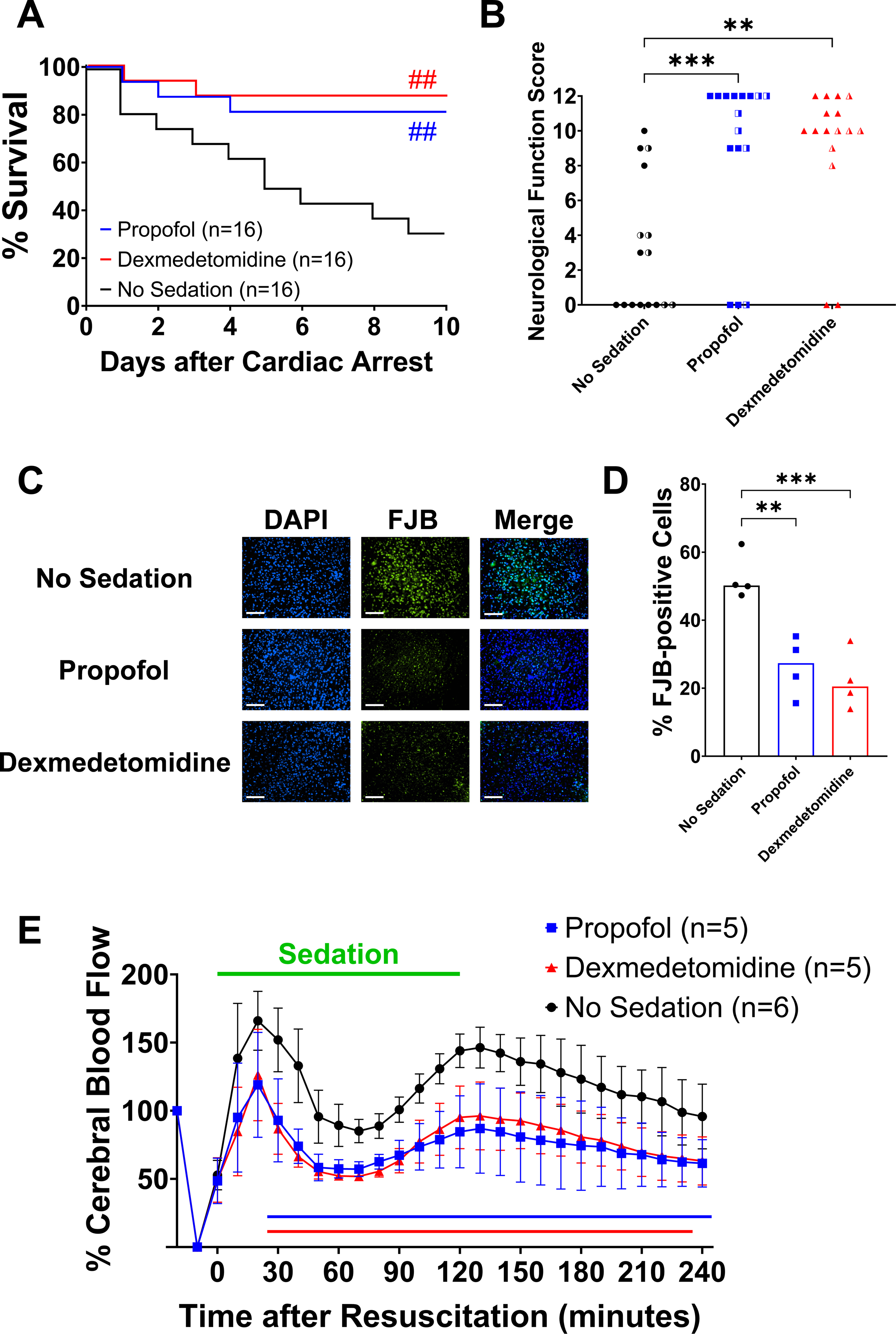
Effects of sedation with propofol or dexmedetomidine starting at return of spontaneous circulation. (A) Percent survival in the first 10 days after cardiac arrest. Ten male mice and 6 female mice per group. ##P < 0.01; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation. (B) Neurological function scores at 5 days after cardiac arrest that were assessed using the Scale 1. Ten male mice and 6 female mice per group. The half-colored symbols represent female animals. Note that dead mice (scored at 0 points) were included in the statistical analysis. **P < 0.01, ***P < 0.001; vs. no sedation. (C) Representative images of the cerebral cortex at 24 hours post-cardiac arrest from male mice without sedation and those sedated with propofol or dexmedetomidine. Neuronal degeneration was visualized by double staining with Fluoro-Jade B (FJB) and 4′,6-Diamidino-2-phenylindole (DAPI). Scale bar = 100 μm. (D) The percentage of FJB-positive cells to DAPI-positive cells in the cerebral cortex was significantly lower in male mice sedated with propofol or dexmedetomidine starting at return of spontaneous circulation (one-way ANOVA, propofol 26.3 ± 8.7 vs. no sedation 52.5 ± 6.7, P = 0.002; dexmedetomidine 22.2 ± 8.5 vs. no sedation 52.5 ± 6.7, P = 0.0009). **P < 0.01, ***P < 0.001; vs. no sedation. (E) Changes in the relative cerebral blood flow in the first 240 minutes after return of spontaneous circulation in male mice without sedation and those sedated with propofol or dexmedetomidine starting at return of spontaneous circulation. Data are presented as mean ± SD. A two-way repeated measures ANOVA with the post hoc Dunnett’s test was used (two-way repeated measures ANOVA, propofol vs. no sedation, interaction effect [group × time]: P < 0.0001; dexmedetomidine vs. no sedation, interaction effect [group × time]: P < 0.0001). The colored lines below indicate statistically significant differences from the no sedation group at specific time points after resuscitation (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test).
Sedation with Propofol or Dexmedetomidine Attenuated the Cerebral Hyperemic Response after Return of Spontaneous Circulation
To determine the effects of sedation on cerebral perfusion in mice after cardiac arrest, we continuously measured cerebral blood flow for 240 minutes after return of spontaneous circulation. In mice that received no sedation after return of spontaneous circulation, cerebral blood flow surged to ~160% of the baseline at 20 minutes after return of spontaneous circulation, returned to baseline levels temporarily, and then increased again to ~150% of the baseline around 120 minutes after return of spontaneous circulation (Figure 1E). In contrast, sedation with propofol or dexmedetomidine markedly attenuated cerebral hyperemia immediately after return of spontaneous circulation C. Even after reaching its nadir, cerebral blood flow remained lower in sedated mice than in those without sedation up to 240 minutes after return of spontaneous circulation (Figure 1E). These observations suggest that the effects of sedation with propofol or dexmedetomidine starting at return of spontaneous circulation were characterized by attenuation of the cerebral hyperemic response immediately after resuscitation.
Delayed Sedation with Propofol or Dexmedetomidine Starting at 60 Minutes after Return of Spontaneous Circulation did not Improve Outcomes after Cardiac Arrest
To further characterize the relationship between early cerebral hyperemia and beneficial effects of sedation in post-cardiac arrest mice, we delayed the start of sedation until 60 minutes after return of spontaneous circulation so that the initial peak increase of cerebral blood flow after return of spontaneous circulation would not be affected by administration of propofol or dexmedetomidine. In comparison with vehicle administration starting at 60 minutes after return of spontaneous circulation, administration of propofol starting at 60 minutes after return of spontaneous circulation decreased mean arterial pressure (~20%) (Supplementary Figure 3D). In contrast to sedation starting immediately after resuscitation, delayed sedation with propofol or dexmedetomidine failed to improve survival at 10 days after cardiac arrest (Figure 2A; log-rank, propofol [3/10, 33%] vs. no sedation [3/10, 33%], P = 0.872; dexmedetomidine [3/10, 33%] vs. no sedation [3/10, 33%], P = 0.705). No difference was found in the neurological function score among groups at 5 days after cardiac arrest (Figure 2B; Kruskal-Wallis test, propofol 0.0 [0.0 – 9.3] vs. no sedation 0.0 [0.0 – 8.0], P > 0.999; dexmedetomidine 0.0 [0.0 – 10.3] vs. no sedation 0.0 [0.0 – 8.0], P = 0.953). The cerebral blood flow changed similarly in the first 60 minutes after return of spontaneous circulation in mice that received vehicle administration and those sedated with propofol or dexmedetomidine starting at 60 minutes after return of spontaneous circulation. Delayed administration of propofol or dexmedetomidine did not decrease cerebral blood flow from 60 until 180 minutes after return of spontaneous circulation except that there was a significant reduction in cerebral blood flow at 90 and 100 minutes after return of spontaneous circulation during sedation with dexmedetomidine (Figure 2C). These results suggest that sedation with propofol or dexmedetomidine failed to improve outcomes when initiated at 60 minutes after return of spontaneous circulation.
Figure 2.
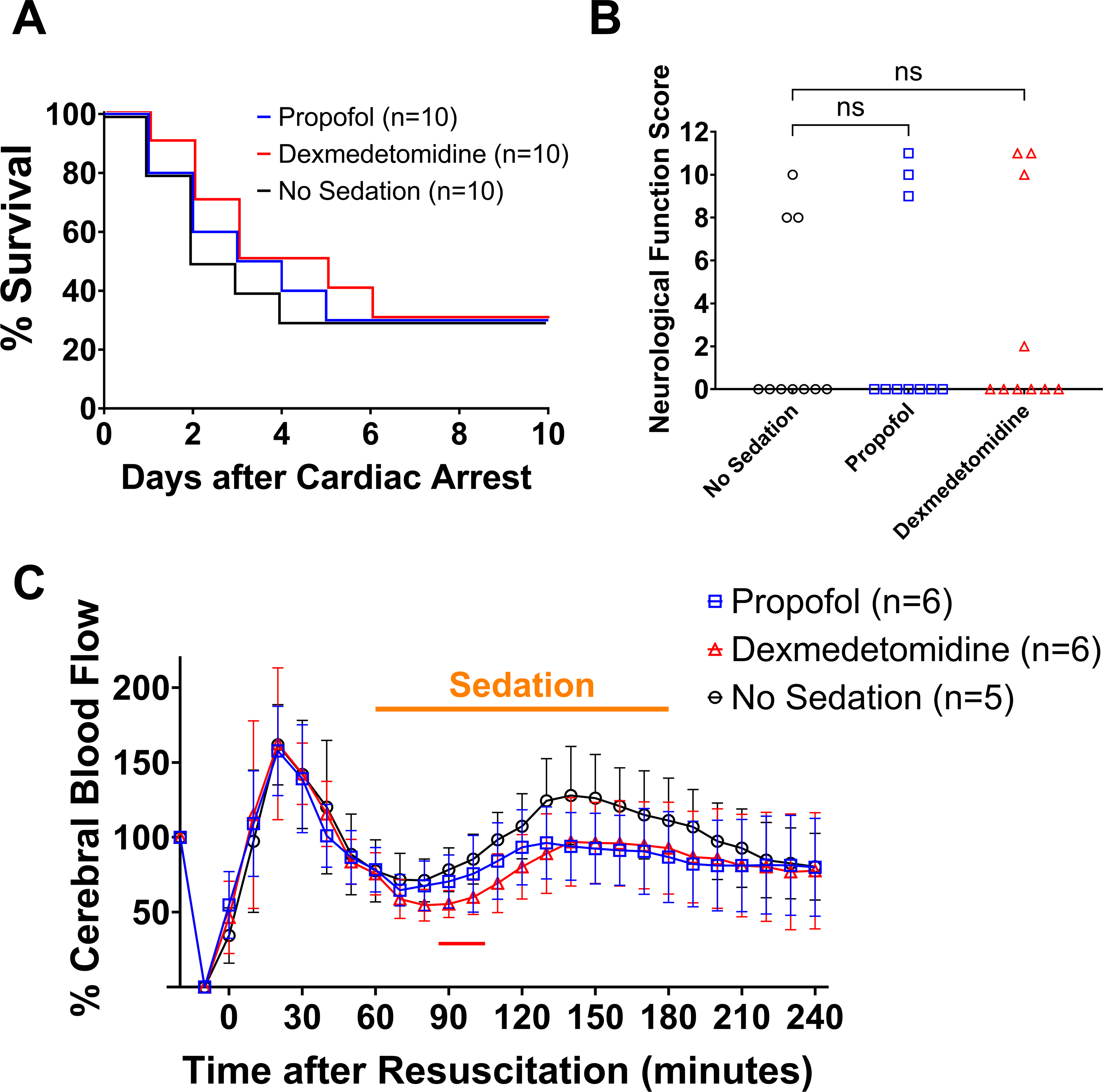
Effects of delayed sedation with propofol or dexmedetomidine starting at 60 minutes after return of spontaneous circulation. (A) Percent survival in the first 10 days after cardiac arrest. Ten male mice per group. (B) Neurological function scores at 5 days after cardiac arrest that were assessed using the Scale 1. Ten male mice per group. Note that dead mice (scored at 0 points) were included in the statistical analysis. (C) Changes in the relative cerebral blood flow in the first 240 minutes after return of spontaneous circulation in male mice without sedation and those sedated with propofol or dexmedetomidine starting at 60 minutes after return of spontaneous circulation. Data are presented as mean ± SD. A two-way repeated measures ANOVA with the post hoc Dunnett’s test was performed during and after the period of sedation (two-way repeated measures ANOVA, propofol vs. no sedation, interaction effect [group × time]: P = 0.011; dexmedetomidine vs. no sedation, interaction effect [group × time]: P = 0.074, main effect: P = 0.202). The colored line below indicates statistically significant differences from the no sedation group at specific time points after resuscitation (P < 0.05; red: dexmedetomidine vs. no sedation; Dunnett’s test).
Sedation with Propofol or Dexmedetomidine Accelerated EEG Recovery after Cardiac Arrest (0–48 Hours after Return of Spontaneous Circulation)
Because brain electrical activity is tightly coupled with cerebral metabolism that changes in tandem with cerebral blood flow,31 we hypothesized that attenuation of cerebral hyperemia with pharmacological sedation is associated with suppression of neuronal activity in the early phase of recovery after hypoxic-ischemic brain injury. To address this hypothesis, EEG was continuously recorded in freely moving mice before and after experimental cardiac arrest. Survival rates in mice that underwent EEG recording are summarized in Supplementary Figure 5 (mice that were not resuscitated from cardiac arrest were excluded).
Representative EEG spectrograms and unprocessed waveforms are displayed in Figure 3 from pre-cardiac arrest awake mice (A), post-cardiac arrest mice that received no sedation (B), sedation with propofol (C), and sedation with dexmedetomidine (D). Spectrograms for the individual animals are provided in Supplementary Figure 6. In mice that received no sedation, the EEG was isoelectric early after resuscitation, and EEG power remained lower in the first 24 hours after return of spontaneous circulation compared to the pre-cardiac arrest baseline. In contrast, mice sedated with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation exhibited pronounced EEG activity within frequencies up to 10 Hz in the first 6 hours after return of spontaneous circulation, which was followed by gradually increasing EEG power over 12 to 24 hours after resuscitation. Quantitative analysis showed that in contrast to mice without sedation, those sedated with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation exhibited electrophysiological recovery at earlier points in time after resuscitation across all frequency bands (Figure 4A – 4F). Although the body temperature recorded by an EEG transmitter showed a temporary decrease below 33°C after the experimental procedure followed by a gradual increase over time, no statistically significant differences were found in body temperature among groups up to 24 hours after return of spontaneous circulation, except at 3 points in time (4, 5, and 24 hours) (Supplementary Figure 7). These results indicate that sedation with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation accelerated electrophysiological recovery in mice after cardiac arrest.
Figure 3.
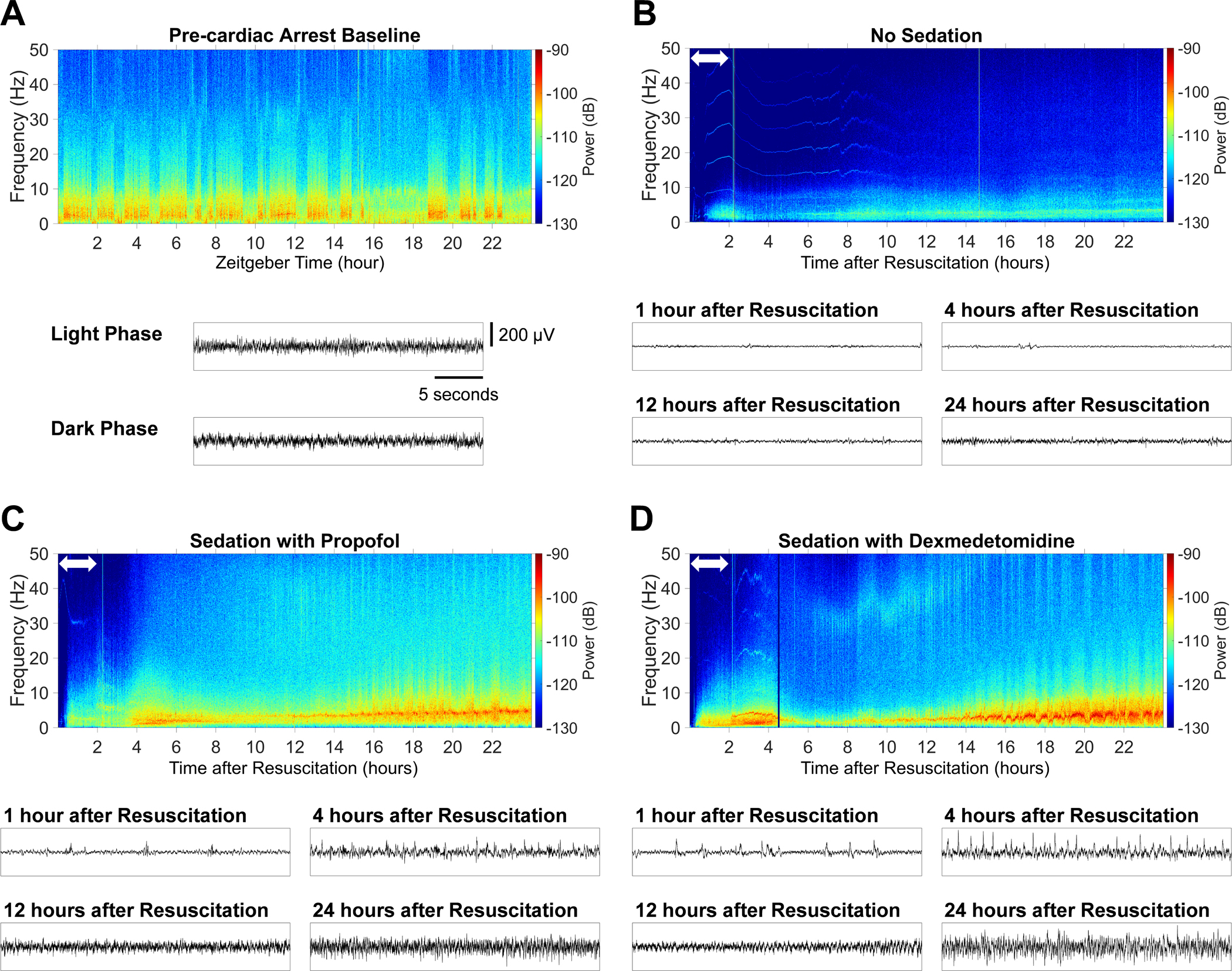
Representative EEG spectrograms before cardiac arrest and in the first 24 hours after return of spontaneous circulation. (A) Baseline spectrogram (the day before cardiac arrest). Mice were housed individually, exposed to a 24-hour light/dark cycle (light: 7:00 AM – 7:00 PM, dark: 7:00 PM – 7:00 AM). Normal cycling between sleep and wake states was observed before cardiac arrest. Unprocessed EEG waveforms during the light and dark phase are also provided. Examples of spectrograms in the first 24 hours after resuscitation from (B) male mice that received no sedation, (C) those sedated with propofol at 40 mg η kg−1 η h−1 starting at return of spontaneous circulation, and (D) those sedated with dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation. The white double-headed arrow in the spectrogram indicates the period of (B) vehicle administration, (C) sedation with propofol, or (D) sedation with dexmedetomidine. Unprocessed EEG waveforms at 1 hour, 4 hours, 12 hours, and 24 hours after resuscitation are also provided.
Figure 4.
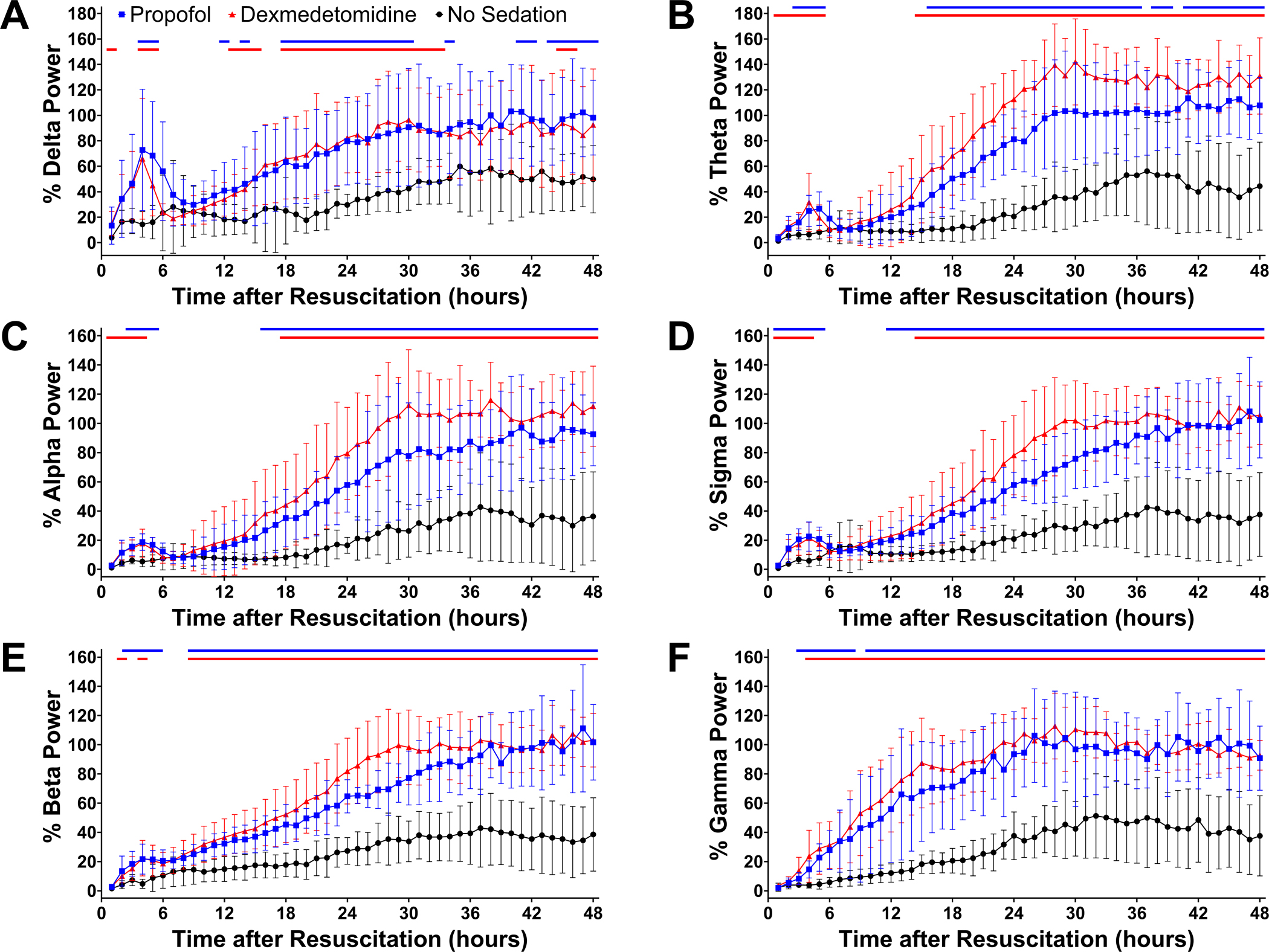
Quantitative changes in EEG power after cardiac arrest. Changes in (A) delta, (B) theta, (C) alpha, (D) sigma, (E) beta, and (F) gamma power up to 48 hours after resuscitation are provided in male and female mice sedated with propofol at 40 mg η kg−1 η h−1 starting at return of spontaneous circulation, those sedated with dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation, and those receiving no sedation. EEG power was calculated for each frequency band at consecutive 1-hour time blocks up to 48 hours after resuscitation, which was expressed as percent of the baseline EEG power before cardiac arrest. Data are presented as mean ± SD. The P-values for the interaction effect [group × time] are summarized in Supplementary Table 4. The colored lines on top indicate statistically significant differences from the no sedation group at specific time points after resuscitation (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test).
Sedation with Propofol or Dexmedetomidine Induced Slow-wave Activity in Post-cardiac Arrest Mice (0–2 Hours after Return of Spontaneous Circulation)
To further analyze propofol- or dexmedetomidine-induced EEG changes over time during sedation in comatose post-cardiac arrest mice, we generated power spectral density plots at 3 time points during sedation. Compared to mice sedated with propofol or those without sedation, mice sedated with dexmedetomidine at 1 μg η kg−1 η h−1 exhibited greater EEG power at 30 minutes after return of spontaneous circulation, while sedation with propofol at 40 mg η kg−1 η h−1 was associated with increased EEG power across frequencies below 25 Hz compared to no sedation (Figure 5A). At 60 and 90 minutes after return of spontaneous circulation, EEG power was greater across a frequency range up to approximately 35 Hz in mice sedated with propofol or dexmedetomidine than in those receiving no sedation (Figure 5B and 5C).
Figure 5.
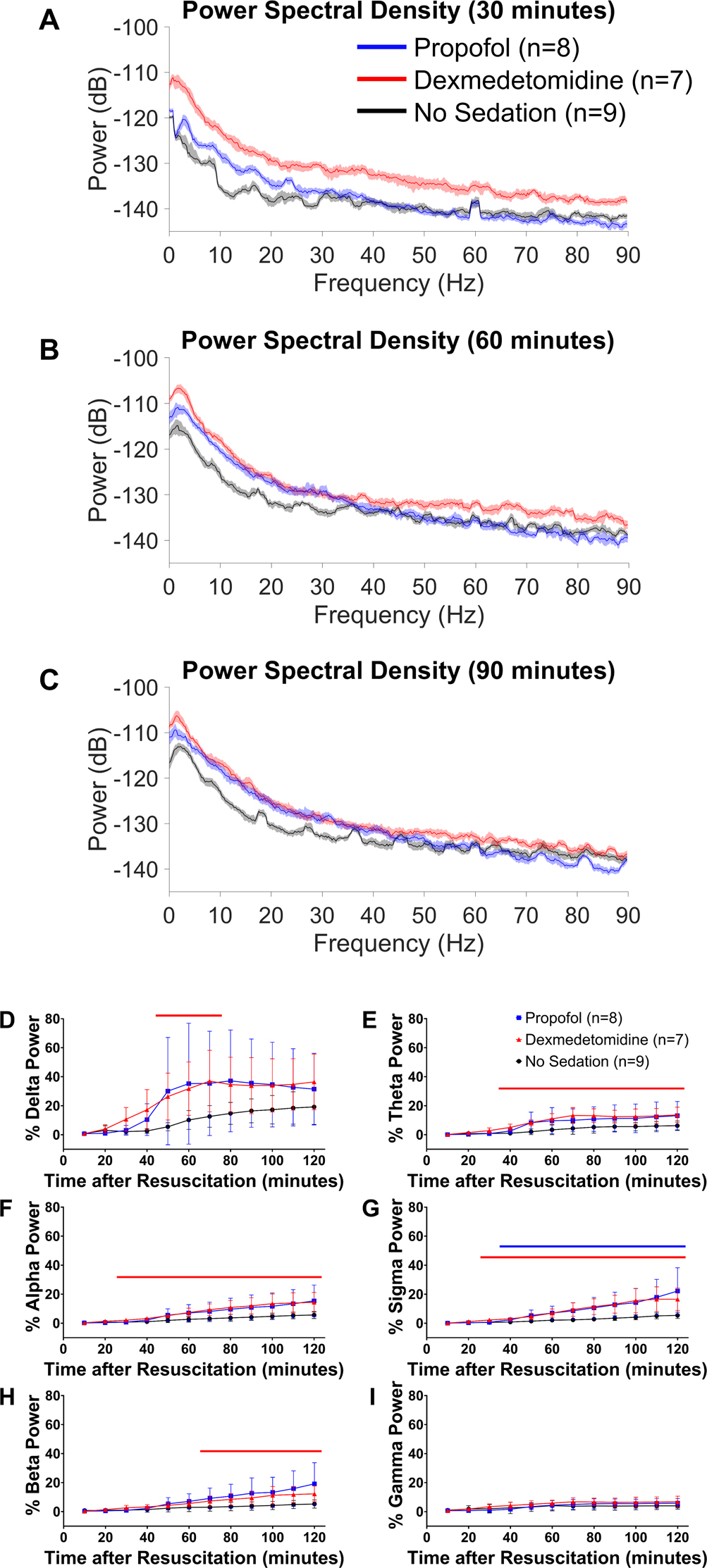
Propofol- or dexmedetomidine-induced EEG changes during post-cardiac arrest sedation (0–2 hours after return of spontaneous circulation). Power spectral density at (A) 30, (B) 60, and (C) 90 minutes after return of spontaneous circulation in male and female mice without sedation and those sedated with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation. The median and 95% confidence intervals of spectral density are displayed. Changes in (D) delta, (E) theta, (F) alpha, (G) sigma, (H) beta, and (I) gamma power in the first 120 minutes after resuscitation. EEG power was calculated for each frequency band at consecutive 10-minute time blocks, which was expressed as percent of the baseline EEG power before cardiac arrest. Data are presented as mean ± SD. The P-values for the interaction effect [group × time] are summarized in Supplementary Table 4. The colored lines in each graph indicate statistically significant differences from the no sedation group at specific time points after resuscitation (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test).
To visualize quantitative EEG changes, the relative EEG power was calculated for all EEG frequency bands in consecutive 10-minute time blocks up to 120 minutes after return of spontaneous circulation. In mice receiving no sedation, EEG power remained lower than ~20% of the baseline across all frequency bands in the first 120 minutes after return of spontaneous circulation (Figure 5D – 5I, Supplementary Figure 8). As compared with mice without sedation, there was a noticeable increase in delta power early during sedation in mice sedated with propofol or dexmedetomidine (Figure 5D, Supplementary Figure 8). Though less prominent than changes in slow oscillation power, sedation with propofol or dexmedetomidine was associated with increased EEG power in theta, alpha, sigma, and beta frequency ranges (Figure 5E – 5H). Unlike frequencies below 30 Hz, no increase in gamma power was observed during sedation (Figure 5I).
Sedation with Propofol or Dexmedetomidine Enhanced Recovery of EEG Power in the Early Hours after Sedation (2–6 Hours after Return of Spontaneous Circulation)
To quantify the time-dependent changes in EEG frequency distribution after sedation, we generated power spectral density plots at 3 time points in the first 4 hours after sedation was discontinued. EEG power was greater up to approximately 45 Hz at 120 minutes after return of spontaneous circulation (at the end of sedation) in mice sedated with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation than in those receiving no sedation (Figure 6A). Spectral analysis revealed an increase in EEG power across a broad frequency range up to 90 Hz at 240 minutes after return of spontaneous circulation (2 hours after the end of sedation) in mice sedated with propofol or dexmedetomidine (Figure 6B). At 360 minutes after return of spontaneous circulation (4 hours after the end of sedation), mice that received post-cardiac arrest sedation were associated with increased EEG power across frequencies below 5 Hz and between 10 and 90 Hz (Figure 6C).
Figure 6.
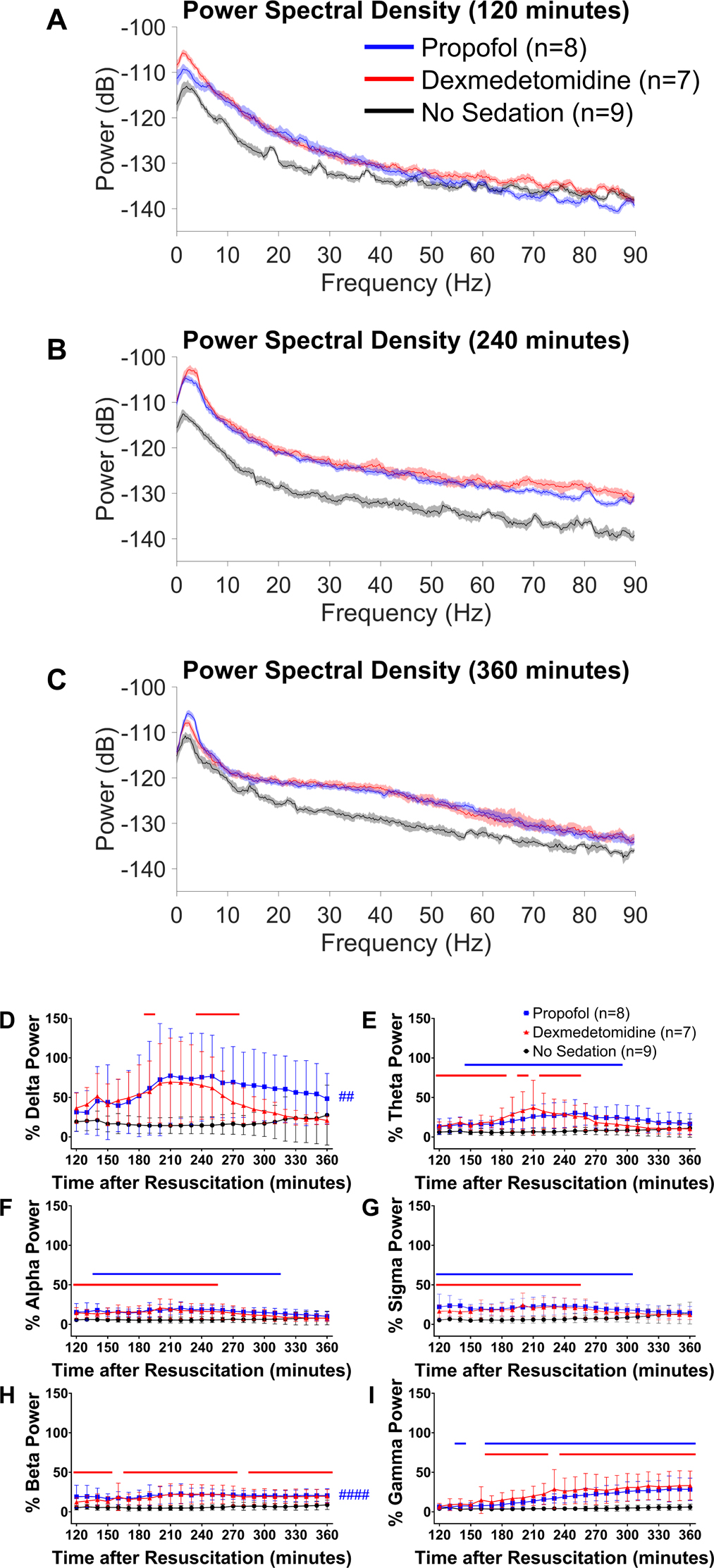
Propofol- or dexmedetomidine-induced EEG changes early after sedation (2–6 hours after return of spontaneous circulation). Power spectral density at (A) 120, (B) 240, and (C) 360 minutes after return of spontaneous circulation in male and female mice without sedation and those sedated with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation. The median and 95% confidence intervals of spectral density are displayed. Changes in (D) delta, (E) theta, (F) alpha, (G) sigma, (H) beta, and (I) gamma power from 120 minutes up to 360 minutes after resuscitation. EEG power was calculated for each frequency band at consecutive 10-minute time blocks, which was expressed as percent of the baseline EEG power before cardiac arrest. Data are presented as mean ± SD. The P-values for the interaction effect [group × time] are summarized in Supplementary Table 4. ##P < 0.01, ####P < 0.0001; blue: propofol vs. no sedation (main effect). The colored lines in each graph indicate statistically significant differences from the no sedation group at specific time points after resuscitation (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test).
Quantitative EEG changes were visualized by calculating the relative EEG power in consecutive 10-minute time blocks from 120 minutes up to 360 minutes after return of spontaneous circulation. EEG power remained lower than ~30% of the baseline levels in mice receiving no sedation, while mice sedated with propofol or dexmedetomidine exhibited a notable increase in delta power over 210 to 240 minutes after return of spontaneous circulation (Figure 6D). These sedated mice also showed increased EEG power within theta, alpha, sigma, and beta frequencies with a time course similar to that of delta power (Figure 6E – 6H). In contrast to the other frequency bands, gamma power was continuously increased after the end of sedation with propofol or dexmedetomidine (Figure 6I).
Sedation with Low-dose Propofol and Delayed Sedation with Propofol Starting at 60 Minutes after Return of Spontaneous Circulation did not Accelerate Electrophysiological Recovery in Hypothermia-treated Mice
Unlike sedation with propofol at 40 mg η kg−1 η h−1 starting at return of spontaneous circulation, the time-dependent increases in EEG were not observed in mice sedated with propofol at 10 mg η kg−1 η h−1 starting at return of spontaneous circulation or propofol at 40 mg η kg−1 η h−1 starting at 60 minutes after return of spontaneous circulation (Supplementary Figure 9 and 10). In fact, EEG power changed over time similarly in mice without sedation and those sedated with propofol starting at 60 minutes after return of spontaneous circulation. There was no apparent tendency for delta activity to increase during and after sedation with propofol at 40 mg η kg−1 η h−1 starting at 60 minutes after return of spontaneous circulation.
Early Recovery of Electrophysiological Activities Predicted Post-cardiac Arrest Outcomes in Hypothermia-treated Mice
To explore whether early recovery of electrophysiological function would predict outcomes after cardiac arrest, we investigated the association between the relative EEG power in the early post-resuscitation phase and neurological outcomes in a total of 35 mice that underwent EEG recording. There was a positive correlation between the neurological function score at 24 hours after cardiac arrest and the relative EEG power across all frequency bands in the first 6 hours after return of spontaneous circulation (Figure 7).
Figure 7.
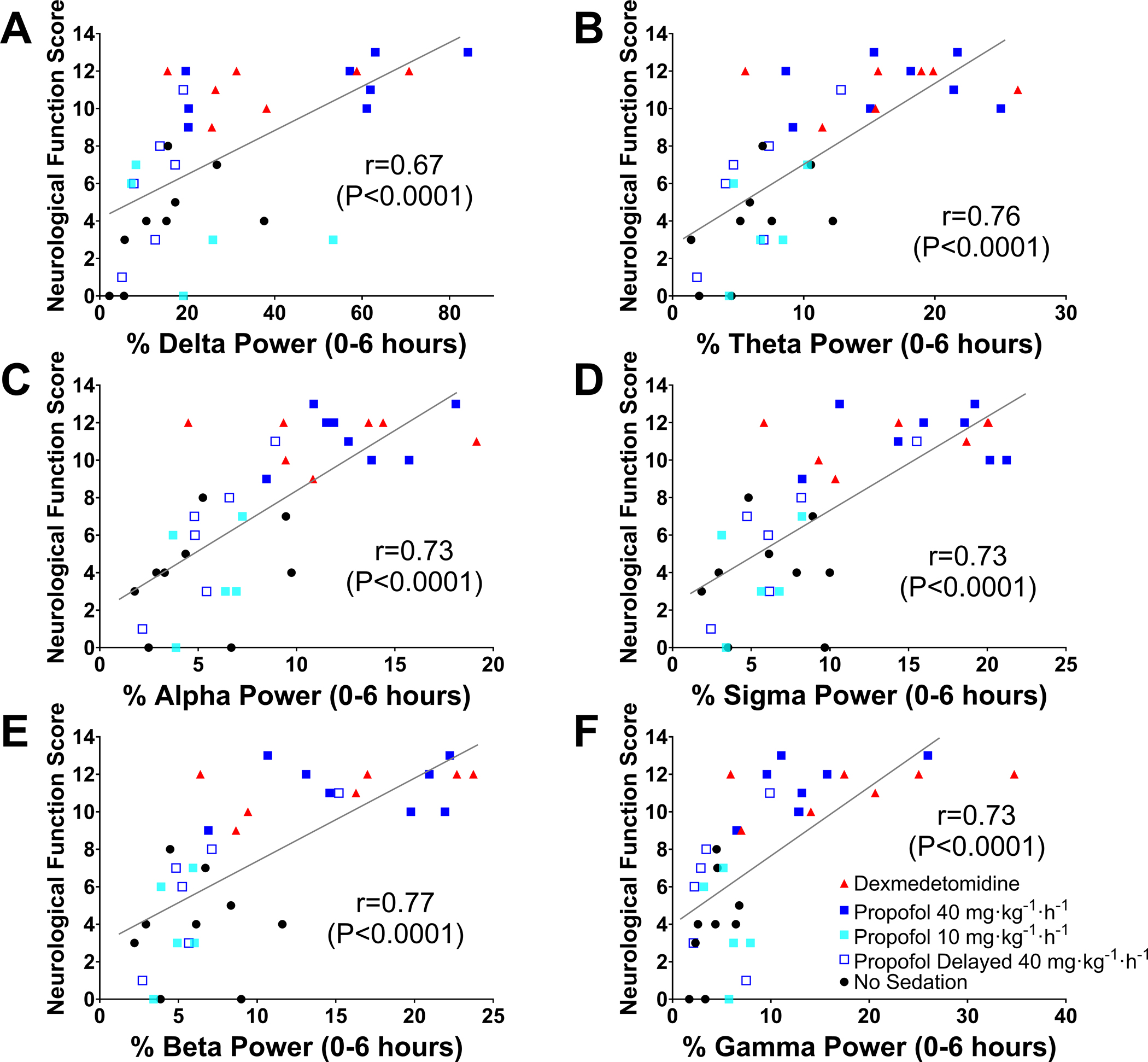
Relationship between EEG power early after resuscitation and neurological function at 24 hours post-cardiac arrest in male and female mice. The horizontal axis represents the relative EEG power in (A) delta, (B) theta, (C) alpha, (D) sigma, (E) beta, and (F) gamma frequencies in the first 6 hours after return of spontaneous circulation, and the vertical axis represents the neurological function scores at 24 hours post-cardiac arrest that were assessed using the Scale 2. The Spearman correlation coefficient is reported. Note that dead mice were included in the analysis.
Discussion
Our study uncovered the effects of post-cardiac arrest sedation in comatose hypothermia-treated mice. As compared with no sedation, mice that were sedated with propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 starting at return of spontaneous circulation had improved survival and neurological outcomes. Early administration of sedation ameliorated histological brain injury and was accompanied by attenuation of early cerebral hyperemia and enhancement of EEG slow-wave activity during and early after sedation. Our findings suggest a possible neuroprotective effect of post-cardiac arrest sedation, which is associated with modulation of slow-wave activity and prevention of cerebral hyperemia immediately after resuscitation.
In animal models of global brain ischemia, the temporal course of cerebrovascular changes after reperfusion is characterized by early cerebral hyperemia followed by a hypoperfusion phase lasting several hours.32,33,34 Preclinical studies have suggested a possible benefit of blunting post-resuscitation cerebral hyperemia.35,36 Our findings that sedation with propofol or dexmedetomidine starting at resuscitation, but not at 60 minutes after resuscitation, improved neurological outcomes would be in consistent with early evidence that barbiturates ameliorated ischemic brain damage when administered early, but not late post-ischemia.8 Propofol produces cerebral vasoconstriction indirectly as a result of reduced cerebral metabolism in the healthy human brain.37 Similarly, dexmedetomidine reduces cerebral blood flow possibly via α2-adrenoreceptor-mediated cerebral vasoconstriction.38 It is therefore conceivable that these agents altered the cerebral vasculature in post-cardiac arrest mice, thereby attenuating super-normal cerebral perfusion early after resuscitation. Sedatives may also limit cerebral oxygen supply/demand mismatch by exerting a coupled reduction of cerebral blood flow and cerebral metabolic rate of oxygen in conditions of impaired autoregulation.9 Our observations highlight the potential of pharmacological sedation as a protective intervention targeted to stabilize cerebrovascular function in the immediate post-resuscitation phase.
Because the primary intended effect of sedation is to modulate neuronal activity, we hypothesized that administration of sedative-hypnotic agents to unconscious post-cardiac arrest mice would produce a deeper state of unconsciousness, or at least sustain the comatose state during the period of pharmacological sedation. To our surprise, early administration of propofol at 40 mg η kg−1 η h−1 or dexmedetomidine at 1 μg η kg−1 η h−1 altered quantitative EEG profiles in comatose mice by inducing slow-wave activity. In contrast, the majority of mice without sedation remained in a state of prolonged unconsciousness with slow oscillation power being less than 30% of the baseline up to 24 hours after cardiac arrest. These results are in alignment with the recent clinical observation that early recovery of EEG slow-wave activity during propofol sedation is associated with favorable outcomes in comatose cardiac arrest survivors.20 Slow oscillations have been considered as a shared EEG feature of general anesthesia and sleep in healthy brains.14,21 Sedative-hypnotic agents cause neocortical neurons to oscillate at ~1 Hz between a depolarizing state of intense firing and a hyperpolarizing state of silence.39,40 The cortically generated rhythmic pattern of the electrical activity synchronizes into travelling waves over the cortical surface and gives rise to spatiotemporal slow (0.5–4 Hz) oscillations.41 Sensory deafferentation during anesthesia or sleep and rhythmic input from intrinsically oscillating thalamocortical neurons also contribute to the full expression of slow oscillations.42 Because the synchronized interaction of large neuronal populations between the cortical and subcortical areas is responsible for the formation of slow waves, it has been hypothesized that this electrophysiological phenomenon can be disrupted by severe acute brain injury.19 In the current study, the failure of delayed sedation with propofol to induce slow-wave activity and improve post-cardiac arrest outcomes may reflect ongoing or severe disruption of the delicate neuronal networks required to generate slow oscillations.
Recent evidence shows that slow oscillations during non-rapid eye movement sleep play an essential role in sleep homeostasis and higher cognitive function.43,44 There is an emerging concept that views the globally synchronized neuronal off periods as a cellular maintenance process in which minor cellular injury can be ameliorated in order to prevent progression to irreversible injury.22 In line with this conceptual framework, it is tempting to speculate that pharmacological sedation produced states of neuronal silence in the early phase of recovery after hypoxic-ischemic brain injury, thereby allowing damaged neurons to shut down before primary cellular dysfunction is exacerbated to the point of causing permanent damage. Modulation of slow-wave activity induced by post-cardiac arrest sedation with propofol or dexmedetomidine may be an electroencephalographic signature of anesthetic-induced metabolic suppression that enables brain cells to rest and recuperate from deleterious ischemic insults. Whether induction of slow-wave activity early after resuscitation serves not only as a marker of the electroencephalographic reactivity to anesthetics but also as a potential strategy of neuroprotection warrants further investigation.
Although propofol administration starting at return of spontaneous circulation dose-dependently exerted neuroprotective effects, EEG burst suppression was absent during sedation with propofol at 40 or 10 mg η kg−1 η h-1. These observations are in agreement with the findings of Warner DS et al. that barbiturates ameliorated brain damage after focal brain ischemia at low doses causing modest depression of electrical activity, with no additional benefit observed at higher doses sufficient to cause EEG burst suppression.45 Our findings imply that a possible dose-response relationship for the protective effect of propofol may be present at doses less than those required for EEG quiescence. Additional studies are required to examine detailed dose-response effects of post-cardiac arrest sedation.
There are limitations in our study. First, all mice were anesthetized with isoflurane before inducing cardiac arrest. Although isoflurane was discontinued at onset of cardiac arrest, it is possible that some isoflurane that remained in mouse tissues provided some sedative effects after resuscitation. Second, a longer arrest time was used for female mice because they are more resistant to brain ischemia than male mice.24 Although survival and neurological outcomes after cardiac arrest appear to be similar between male and female animals, the uneven distribution of female mice allocated to each treatment group could be a potential source of bias. Third, body temperature of mice decreased below 33°C after they were returned to home cages and active warming was stopped. While the body temperature was similar among groups up to 24 hours after return of spontaneous circulation, the possibility was not ruled out that this unintended hypothermia influenced the results. Given the recent results of the Targeted Temperature Management 2 trial and updated clinical guidelines that do not recommend hypothermia in post-cardiac arrest patients, the effects of sedation need to be studied in normothermia in future studies.46,47 Fourth, the decision to discontinue mechanical ventilation was not based on arterial blood gas analysis. In all mice, weaning from mechanical ventilation and cessation of oxygen administration was uniformly carried out at 20 and 40 minutes, respectively, after return of spontaneous circulation on resumption of spontaneous ventilation (> 100 breaths/minute). While there was no difference in the respiratory rate with or without sedation, the presence of hypoxemia or hypercapnia was not ascertained. Fifth, the infusion rate of propofol used in this study (40 mg η kg−1 η h−1) was selected based on our pilot experiments in mice and is considerably higher than the recommended dose for patients. Finally, a mouse model of potassium chloride-induced cardiac arrest was used in the current study. Therefore, the applicability of the current results to clinical cardiac arrest is unknown. Further clinical studies are needed to elucidate the role of post-cardiac arrest sedation on neurological outcomes.
In conclusion, post-cardiac arrest sedation with propofol or dexmedetomidine starting immediately after resuscitation improved survival and neurological outcomes in hypothermia-treated mice. The beneficial effects of pharmacological sedation early after resuscitation were associated with attenuation of cerebral hyperemia and early recovery of EEG power during and after sedation. In particular, our observations shed light on the ability of sedation to induce EEG slow-wave activity in comatose mice and improve neurological outcomes after cardiac arrest. These results should stimulate future studies to gain further insight into the effects of sedation on neurological recovery in cardiac arrest patients.
Supplementary Material
Supplementary Figure 1. Percent survival in mice treated with differing doses of propofol or dexmedetomidine starting at return of spontaneous circulation in the pilot study. (A) Percent survival in the first 10 days post-cardiac arrest in male mice sedated with propofol at 40 mg · kg−1 · h−1 or 10 mg · kg−1 · h−1. (B) Percent survival in the first 10 days post-cardiac arrest in male mice sedated with dexmedetomidine (DEX) at 1, 2, or 3 μg · kg−1 · h−1.
Supplementary Figure 2. Schematic timeline illustrating the procedure for experimental cardiac arrest and post-cardiac arrest sedation. (A) Starting at return of spontaneous circulation (ROSC) or (B) at 60 minutes after ROSC, mice received no sedation or post-cardiac arrest sedation with propofol at 40 mg · kg−1 · h−1 or dexmedetomidine (DEX) at 1 μg · kg−1 · h−1. KCl = potassium chloride. CPR = cardiopulmonary resuscitation. FiO2 = fraction of inspired oxygen.
Supplementary Figure 3. Changes in physiological parameters in mice without post-cardiac arrest sedation and those sedated with propofol or dexmedetomidine. Changes in (A) mean arterial pressure, (B) heart rate, and (C) respiratory rate before and after cardiac arrest in male and female mice without sedation and those sedated with propofol at 40 mg · kg−1 · h−1 or dexmedetomidine (DEX) at 1 μg · kg−1 · h−1 starting at return of spontaneous circulation (ROSC). Changes in (D) mean arterial pressure, (E) heart rate, and (F) respiratory rate before and after cardiac arrest in male mice without sedation and those sedated with propofol or dexmedetomidine (DEX) starting at 60 minutes after ROSC. Data are presented as mean α SD. The P-values for the interaction effect [group × time] are summarized in Supplementary Table 4. The colored lines in each graph indicate statistically significant differences from the no sedation group at specific time points after ROSC (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test).
Supplementary Figure 5. Percent survival in the first 7 days post-cardiac arrest in male and female mice that underwent EEG recording.
Supplementary Figure 4. Effects of sedation with propofol or dexmedetomidine starting at return of spontaneous circulation on neurological outcomes and survival. (A) Percent survival in the first 10 days post-cardiac arrest in male mice (log-rank, propofol [8/10, 80%] vs. no sedation [3/10, 30%], P = 0.039; dexmedetomidine (DEX) [8/10, 80%] vs. no sedation [3/10, 30%], P = 0.035). #P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation. (B) Neurological function scores at 5 days post-cardiac arrest in male mice that were assessed using the Scale 1 (Kruskal-Wallis test, propofol 12.0 [6.8 – 12.0] vs. no sedation 0.0 [0.0 – 8.25], P = 0.009; dexmedetomidine 10.5 [7.5 – 12.0] vs. no sedation 0.0 [0.0 – 8.25], P = 0.025). *P < 0.05, **P < 0.01; vs. no sedation. (C) Percent survival in the first 15 days post-cardiac arrest in female mice (log-rank, propofol [5/6, 83%] vs. no sedation [1/6, 17%], P = 0.042; dexmedetomidine [6/6, 100%] vs. no sedation [1/6, 17%], P = 0.004). #P < 0.05, ##P < 0.01; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation. (D) Neurological function scores at 5 days post-cardiac arrest in female mice that were assessed using the Scale 1 (Kruskal-Wallis test, propofol 10.5 [6.8 – 12.0] vs. no sedation 3.5 [0.0 – 5.25], P = 0.030; dexmedetomidine 10.0 [8.8 – 10.5] vs. no sedation 3.5 [0.0 – 5.25], P = 0.040). *P < 0.05; vs. no sedation.
Supplementary Figure 7. Changes in body temperature recorded by an implantable EEG transmitter in the first 24 hours after return of spontaneous circulation (ROSC) in male and female mice. Data are presented as mean α SD. A two-way repeated measures ANOVA with the post hoc Dunnett’s test was used (two-way repeated measures ANOVA, propofol vs. no sedation, interaction effect [group × time]: P < 0.0001; dexmedetomidine vs. no sedation, interaction effect [group × time]: P < 0.0001). The colored lines indicate statistically significant differences from the no sedation group at specific time points after ROSC (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test). DEX = dexmedetomidine.
Supplementary Figure 9. Quantitative changes in EEG power after cardiac arrest. Changes in (A) delta, (B) theta, (C) alpha, (D) sigma, (E) beta, and (F) gamma power up to up to 360 minutes after return of spontaneous circulation (ROSC) are provided in male mice sedated with propofol at 10 mg · kg−1 · h−1 starting at ROSC, and those sedated with propofol at 40 mg · kg−1 · h−1 starting at 60 minutes after ROSC. EEG power was calculated for each frequency band at consecutive 10-minute time blocks, which was expressed as percent of the baseline EEG power before cardiac arrest. Data are presented as mean α SD.
Supplementary Figure 8. Representative EEG spectrograms in the first 120 minutes after return of spontaneous circulation (ROSC) from (A) male mice that received no sedation, (B) those sedated with propofol at 40 mg · kg−1 · h−1 starting at ROSC, and (C) those sedated with dexmedetomidine at 1 μg · kg−1 · h−1 starting at ROSC.
Supplementary Figure 10. Representative EEG spectrograms in the first 24 hours after return of spontaneous circulation (ROSC) from (A) male mice sedated with propofol at 10 mg · kg−1 · h−1 starting at ROSC, and (B) those sedated with propofol at 40 mg · kg−1 · h−1 starting at 60 minutes after ROSC. The white double-headed arrow in the spectrogram indicates the period of sedation.
Supplementary Figure 6. EEG spectrograms in the first 24 hours after return of spontaneous circulation (ROSC) for the individual animals that received no sedation, sedation with propofol at 40 mg · kg−1 · h−1 starting at ROSC, or sedation with dexmedetomidine at 1 μg · kg−1 · h−1 starting at ROSC.
Funding Statement:
This work was supported by a grant from the ZOLL Foundation to Dr. Ikeda, and funds from the Department of Anesthesia, Critical Care and Pain Medicine, Massachusetts General Hospital to Dr. Ichinose.
Footnotes
Conflicts of Interest:
Dr. Amorim was supported during this research by the National Institute of Health (1K23NS119794), Hellman Fellows Fund, Regents of the University of California (Resource Allocation Program), CURE Epilepsy Foundation (Taking Flight Award), Weil-Society of Critical Care Medicine Research Grant, ZOLL Foundation Grant, American Heart Association (20CDA35310297).
Dr. Malhotra was supported by the National Heart, Lung, and Blood Institute (R01HL142809), the American Heart Association (18TPA34230025), and the Wild Family Foundation.
Dr. Solt was a consultant to Takeda Pharmaceuticals.
Dr. Ichinose was supported by the National Institute of Health (R01NS112373 and R21NS116671) and was a consultant to Nihon Kohden Innovation Center and received sponsored research agreement from Kyowa Hakko Bio and Cyclerion Therapautics.
Prior Presentations:
Part of the work has been presented at the following conferences:
American Society of Anesthesiologists, October 3, 2020, Virtual (The Best of Abstracts: Basic Science session)
Resuscitation Science Symposium, American Heart Association, November 14–16, 2020, Virtual
References
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee: Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019; 139:e56–e528 [DOI] [PubMed] [Google Scholar]
- 2.Geocadin RG, Callaway CW, Fink EL, Golan E, Greer DM, Ko NU, Lang E, Licht DJ, Marino BS, McNair ND, Peberdy MA, Perman SM, Sims DB, Soar J, Sandroni C; American Heart Association Emergency Cardiovascular Care Committee: Standards for Studies of Neurological Prognostication in Comatose Survivors of Cardiac Arrest: A Scientific Statement From the American Heart Association. Circulation 2019; 140:e517–e542 [DOI] [PubMed] [Google Scholar]
- 3.Madden LK, Hill M, May TL, Human T, Guanci MM, Jacobi J, Moreda MV, Badjatia N: The Implementation of Targeted Temperature Management: An Evidence-Based Guideline from the Neurocritical Care Society. Neurocrit Care 2017;27:468–87. [DOI] [PubMed] [Google Scholar]
- 4.Peberdy MA, Callaway CW, Neumar RW, Geocadin RG, Zimmerman JL, Donnino M, Gabrielli A, Silvers SM, Zaritsky AL, Merchant R, Vanden Hoek TL, Kronick SL; American Heart Association: Part 9: post-cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation 2010; 122:S768–86 [DOI] [PubMed] [Google Scholar]
- 5.Citerio G, Cormio M: Sedation in neurointensive care: advances in understanding and practice. Curr Opin Crit Care 2003; 9:120–6 [DOI] [PubMed] [Google Scholar]
- 6.Dell’Anna AM, Taccone FS, Halenarova K, Citerio G: Sedation after cardiac arrest and during therapeutic hypothermia. Minerva Anestesiol 2014; 80:954–62 [PubMed] [Google Scholar]
- 7.Young Y, Menon DK, Tisavipat N, Matta BF, Jones JG: Propofol neuroprotection in a rat model of ischaemia reperfusion injury. Eur J Anaesthesiol 1997; 14:320–6 [DOI] [PubMed] [Google Scholar]
- 8.Bleyaert AL, Nemoto EM, Safar P, Stezoski SM, Mickell JJ, Moossy J, Rao GR: Thiopental amelioration of brain damage after global ischemia in monkeys. Anesthesiology 1978; 49:390–8 [DOI] [PubMed] [Google Scholar]
- 9.Oddo M, Crippa IA, Mehta S, Menon D, Payen JF, Taccone FS, Citerio G: Optimizing sedation in patients with acute brain injury. Crit Care 2016; 20:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keegan MT: Sedation in the neurologic intensive care unit. Curr Treat Options Neurol 2008; 10:111–25 [DOI] [PubMed] [Google Scholar]
- 11.Strøm T, Martinussen T, Toft P: A protocol of no sedation for critically ill patients receiving mechanical ventilation: a randomised trial. Lancet 2010; 375:475–80 [DOI] [PubMed] [Google Scholar]
- 12.Kress JP, Pohlman AS, O’Connor MF, Hall JB: Daily interruption of sedative infusions in critically ill patients undergoing mechanical ventilation. N Engl J Med 2000; 342:1471–7 [DOI] [PubMed] [Google Scholar]
- 13.Kalbhenn J, Knörlein J, Posch MJ: “Do no further harm” - Why shall we sedate unresponsive patients? Intensive Care Med 2021; 47:807–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brown EN, Lydic R, Schiff ND: General anesthesia, sleep, and coma. N Engl J Med 2010; 363:2638–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Akeju O, Pavone KJ, Westover MB, Vazquez R, Prerau MJ, Harrell PG, Hartnack KE, Rhee J, Sampson AL, Habeeb K, Gao L, Pierce ET, Walsh JL, Brown EN, Purdon PL: A comparison of propofol- and dexmedetomidine-induced electroencephalogram dynamics using spectral and coherence analysis. Anesthesiology 2014; 121:978–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drohan CM, Cardi AI, Rittenberger JC, Popescu A, Callaway CW, Baldwin ME, Elmer J: Effect of sedation on quantitative electroencephalography after cardiac arrest. Resuscitation 2018; 124:132–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Amorim E, Rittenberger JC, Zheng JJ, Westover MB, Baldwin ME, Callaway CW, Popescu A; Post Cardiac Arrest Service: Continuous EEG monitoring enhances multimodal outcome prediction in hypoxic-ischemic brain injury. Resuscitation 2016; 109:121–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samaniego EA, Mlynash M, Caulfield AF, Eyngorn I, Wijman CA: Sedation confounds outcome prediction in cardiac arrest survivors treated with hypothermia. Neurocrit Care 2011; 15:113–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kortelainen J, Väyrynen E, Huuskonen U, Laurila J, Koskenkari J, Backman JT, Alahuhta S, Seppänen T, Ala-Kokko T: Pilot Study of Propofol-induced Slow Waves as a Pharmacologic Test for Brain Dysfunction after Brain Injury. Anesthesiology 2017; 126:94–103 [DOI] [PubMed] [Google Scholar]
- 20.Kortelainen J, Ala-Kokko T, Tiainen M, Strbian D, Rantanen K, Laurila J, Koskenkari J, Kallio M, Toppila J, Väyrynen E, Skrifvars MB, Hästbacka J: Early recovery of frontal EEG slow wave activity during propofol sedation predicts outcome after cardiac arrest. Resuscitation 2021; 165:170–6 [DOI] [PubMed] [Google Scholar]
- 21.Purdon PL, Pierce ET, Mukamel EA, Prerau MJ, Walsh JL, Wong KF, Salazar-Gomez AF, Harrell PG, Sampson AL, Cimenser A, Ching S, Kopell NJ, Tavares-Stoeckel C, Habeeb K, Merhar R, Brown EN: Electroencephalogram signatures of loss and recovery of consciousness from propofol. Proc Natl Acad Sci U S A 2013; 110:E1142–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vyazovskiy VV, Harris KD: Sleep and the single neuron: the role of global slow oscillations in individual cell rest. Nat Rev Neurosci 2013; 14:443–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Minamishima S, Bougaki M, Sips PY, Yu JD, Minamishima YA, Elrod JW, Lefer DJ, Bloch KD, Ichinose F: Hydrogen sulfide improves survival after cardiac arrest and cardiopulmonary resuscitation via a nitric oxide synthase 3-dependent mechanism in mice. Circulation 2009; 120:888–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marutani E, Morita M, Hirai S, Kai S, Grange RMH, Miyazaki Y, Nagashima F, Traeger L, Magliocca A, Ida T, Matsunaga T, Flicker DR, Corman B, Mori N, Yamazaki Y, Batten A, Li R, Tanaka T, Ikeda T, Nakagawa A, Atochin DN, Ihara H, Olenchock BA, Shen X, Nishida M, Hanaoka K, Kevil CG, Xian M, Bloch DB, Akaike T, Hindle AG, Motohashi H, Ichinose F: Sulfide catabolism ameliorates hypoxic brain injury. Nat Commun 2021; 12:3108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serge C Thal, Simone E Thal, Nikolaus Plesnila: Characterization of a 3-vessel Occlusion Model for the Induction of Complete Global Cerebral Ischemia in Mice. J Neurosci Methods 2010; 192:219–27 [DOI] [PubMed] [Google Scholar]
- 26.Hayashida K, Bagchi A, Miyazaki Y, Hirai S, Seth D, Silverman MG, Rezoagli E, Marutani E, Mori N, Magliocca A, Liu X, Berra L, Hindle AG, Donnino MW, Malhotra R, Bradley MO, Stamler JS, Ichinose F: Improvement in Outcomes After Cardiac Arrest and Resuscitation by Inhibition of S-Nitrosoglutathione Reductase. Circulation 2019; 139:815–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miyazaki Y, Marutani E, Ikeda T, Ni X, Hanaoka K, Xian M, Ichinose F: A Sulfonyl Azide-Based Sulfide Scavenger Rescues Mice from Lethal Hydrogen Sulfide Intoxication. Toxicol Sci 2021; 183:393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang X, Sanford LD: Telemetric recording of sleep and home cage activity in mice. Sleep 2002; 25:691–9 [PubMed] [Google Scholar]
- 29.Mitra P, Bokil H: Observed Brain Dynamics. Oxford; New York, Oxford University Press, 2008 [Google Scholar]
- 30.Moody OA, Zhang ER, Arora V, Kato R, Cotten JF, Solt K: D-Amphetamine Accelerates Recovery of Consciousness and Respiratory Drive After High-Dose Fentanyl in Rats. Front Pharmacol 2020; 11:585356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du F, Zhu XH, Zhang Y, Friedman M, Zhang N, Ugurbil K, Chen W: Tightly coupled brain activity and cerebral ATP metabolic rate. Proc Natl Acad Sci U S A 2008; 105:6409–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemoto EM, Snyder JV, Carroll RG, Morita H: Global ischemia in dogs: cerebrovascular CO2 reactivity and autoregulation. Stroke 1975; 6:425–31 [DOI] [PubMed] [Google Scholar]
- 33.Kofke WA, Nemoto EM, Hossmann KA, Taylor F, Kessler PD, Stezoski SW: Brain blood flow and metabolism after global ischemia and post-insult thiopental therapy in monkeys. Stroke 1979; 10:554–60 [DOI] [PubMed] [Google Scholar]
- 34.Kågström E, Smith ML, Siesjö BK: Local cerebral blood flow in the recovery period following complete cerebral ischemia in the rat. J Cereb Blood Flow Metab 1983; 3:170–82 [DOI] [PubMed] [Google Scholar]
- 35.Manole MD, Kochanek PM, Foley LM, Hitchens TK, Bayır H, Alexander H, Garman R, Ma L, Hsia CJ, Ho C, Clark RS: Polynitroxyl albumin and albumin therapy after pediatric asphyxial cardiac arrest: effects on cerebral blood flow and neurologic outcome. J Cereb Blood Flow Metab 2012; 32:560–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerchiari EL, Hoel TM, Safar P, Sclabassi RJ: Protective effects of combined superoxide dismutase and deferoxamine on recovery of cerebral blood flow and function after cardiac arrest in dogs. Stroke 1987; 18:869–78 [DOI] [PubMed] [Google Scholar]
- 37.Matta BF, Mayberg TS, Lam AM: Direct cerebrovasodilatory effects of halothane, isoflurane, and desflurane during propofol-induced isoelectric electroencephalogram in humans. Anesthesiology 1995; 83:980–5 [DOI] [PubMed] [Google Scholar]
- 38.Slupe AM, Kirsch JR: Effects of anesthesia on cerebral blood flow, metabolism, and neuroprotection. J Cereb Blood Flow Metab 2018; 38:2192–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ní Mhuircheartaigh R, Warnaby C, Rogers R, Jbabdi S, Tracey I: Slow-wave activity saturation and thalamocortical isolation during propofol anesthesia in humans. Sci Transl Med 2013; 5:208ra148. [DOI] [PubMed] [Google Scholar]
- 40.Lőrincz ML, Gunner D, Bao Y, Connelly WM, Isaac JT, Hughes SW, Crunelli V: A distinct class of slow (~0.2–2 Hz) intrinsically bursting layer 5 pyramidal neurons determines UP/DOWN state dynamics in the neocortex. J Neurosci 2015; 35:5442–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Massimini M, Huber R, Ferrarelli F, Hill S, Tononi G: The sleep slow oscillation as a traveling wave. J Neurosci 2004; 24:6862–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.David F, Schmiedt JT, Taylor HL, Orban G, Di Giovanni G, Uebele VN, Renger JJ, Lambert RC, Leresche N, Crunelli V: Essential thalamic contribution to slow waves of natural sleep. J Neurosci 2013; 33:19599–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tononi G, Cirelli C: Sleep and synaptic homeostasis: a hypothesis. Brain Res Bull 2003; 62:143–50 [DOI] [PubMed] [Google Scholar]
- 44.Marshall L, Helgadóttir H, Mölle M, Born J: Boosting slow oscillations during sleep potentiates memory. Nature 2006; 444:610–3 [DOI] [PubMed] [Google Scholar]
- 45.Warner DS, Takaoka S, Wu B, Ludwig PS, Pearlstein RD, Brinkhous AD, Dexter F: Electroencephalographic burst suppression is not required to elicit maximal neuroprotection from pentobarbital in a rat model of focal cerebral ischemia. Anesthesiology 1996; 84: 1475–84 [DOI] [PubMed] [Google Scholar]
- 46.Dankiewicz J, Cronberg T, Lilja G, Jakobsen JC, Levin H, Ullén S, Rylander C, Wise MP, Oddo M, Cariou A, Bělohlávek J, Hovdenes J, Saxena M, Kirkegaard H, Young PJ, Pelosi P, Storm C, Taccone FS, Joannidis M, Callaway C, Eastwood GM, Morgan MPG, Nordberg P, Erlinge D, Nichol AD, Chew MS, Hollenberg J, Thomas M, Bewley J, Sweet K, Grejs AM, Christensen S, Haenggi M, Levis A, Lundin A, Düring J, Schmidbauer S, Keeble TR, Karamasis GV, Schrag C, Faessler E, Smid O, Otáhal M, Maggiorini M, Wendel Garcia PD, Jaubert P, Cole JM, Solar M, Borgquist O, Leithner C, Abed-Maillard S, Navarra L, Annborn M, Undén J, Brunetti I, Awad A, McGuigan P, Bjørkholt Olsen R, Cassina T, Vignon P, Langeland H, Lange T, Friberg H, Nielsen N; TTM2 Trial Investigators: Hypothermia versus Normothermia after Out-of-Hospital Cardiac Arrest. N Engl J Med 2021; 17;384: 2283–94 [DOI] [PubMed] [Google Scholar]
- 47.Sandroni C, Nolan JP, Andersen LW, Böttiger BW, Cariou A, Cronberg T, Friberg H, Genbrugge C, Lilja G, Morley PT, Nikolaou N, Olasveengen TM, Skrifvars MB, Taccone FS, Soar J. ERC-ESICM guidelines on temperature control after cardiac arrest in adults. Intensive Care Med 2022; 48: 261–9 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Percent survival in mice treated with differing doses of propofol or dexmedetomidine starting at return of spontaneous circulation in the pilot study. (A) Percent survival in the first 10 days post-cardiac arrest in male mice sedated with propofol at 40 mg · kg−1 · h−1 or 10 mg · kg−1 · h−1. (B) Percent survival in the first 10 days post-cardiac arrest in male mice sedated with dexmedetomidine (DEX) at 1, 2, or 3 μg · kg−1 · h−1.
Supplementary Figure 2. Schematic timeline illustrating the procedure for experimental cardiac arrest and post-cardiac arrest sedation. (A) Starting at return of spontaneous circulation (ROSC) or (B) at 60 minutes after ROSC, mice received no sedation or post-cardiac arrest sedation with propofol at 40 mg · kg−1 · h−1 or dexmedetomidine (DEX) at 1 μg · kg−1 · h−1. KCl = potassium chloride. CPR = cardiopulmonary resuscitation. FiO2 = fraction of inspired oxygen.
Supplementary Figure 3. Changes in physiological parameters in mice without post-cardiac arrest sedation and those sedated with propofol or dexmedetomidine. Changes in (A) mean arterial pressure, (B) heart rate, and (C) respiratory rate before and after cardiac arrest in male and female mice without sedation and those sedated with propofol at 40 mg · kg−1 · h−1 or dexmedetomidine (DEX) at 1 μg · kg−1 · h−1 starting at return of spontaneous circulation (ROSC). Changes in (D) mean arterial pressure, (E) heart rate, and (F) respiratory rate before and after cardiac arrest in male mice without sedation and those sedated with propofol or dexmedetomidine (DEX) starting at 60 minutes after ROSC. Data are presented as mean α SD. The P-values for the interaction effect [group × time] are summarized in Supplementary Table 4. The colored lines in each graph indicate statistically significant differences from the no sedation group at specific time points after ROSC (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test).
Supplementary Figure 5. Percent survival in the first 7 days post-cardiac arrest in male and female mice that underwent EEG recording.
Supplementary Figure 4. Effects of sedation with propofol or dexmedetomidine starting at return of spontaneous circulation on neurological outcomes and survival. (A) Percent survival in the first 10 days post-cardiac arrest in male mice (log-rank, propofol [8/10, 80%] vs. no sedation [3/10, 30%], P = 0.039; dexmedetomidine (DEX) [8/10, 80%] vs. no sedation [3/10, 30%], P = 0.035). #P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation. (B) Neurological function scores at 5 days post-cardiac arrest in male mice that were assessed using the Scale 1 (Kruskal-Wallis test, propofol 12.0 [6.8 – 12.0] vs. no sedation 0.0 [0.0 – 8.25], P = 0.009; dexmedetomidine 10.5 [7.5 – 12.0] vs. no sedation 0.0 [0.0 – 8.25], P = 0.025). *P < 0.05, **P < 0.01; vs. no sedation. (C) Percent survival in the first 15 days post-cardiac arrest in female mice (log-rank, propofol [5/6, 83%] vs. no sedation [1/6, 17%], P = 0.042; dexmedetomidine [6/6, 100%] vs. no sedation [1/6, 17%], P = 0.004). #P < 0.05, ##P < 0.01; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation. (D) Neurological function scores at 5 days post-cardiac arrest in female mice that were assessed using the Scale 1 (Kruskal-Wallis test, propofol 10.5 [6.8 – 12.0] vs. no sedation 3.5 [0.0 – 5.25], P = 0.030; dexmedetomidine 10.0 [8.8 – 10.5] vs. no sedation 3.5 [0.0 – 5.25], P = 0.040). *P < 0.05; vs. no sedation.
Supplementary Figure 7. Changes in body temperature recorded by an implantable EEG transmitter in the first 24 hours after return of spontaneous circulation (ROSC) in male and female mice. Data are presented as mean α SD. A two-way repeated measures ANOVA with the post hoc Dunnett’s test was used (two-way repeated measures ANOVA, propofol vs. no sedation, interaction effect [group × time]: P < 0.0001; dexmedetomidine vs. no sedation, interaction effect [group × time]: P < 0.0001). The colored lines indicate statistically significant differences from the no sedation group at specific time points after ROSC (P < 0.05; blue: propofol vs. no sedation, red: dexmedetomidine vs. no sedation; Dunnett’s test). DEX = dexmedetomidine.
Supplementary Figure 9. Quantitative changes in EEG power after cardiac arrest. Changes in (A) delta, (B) theta, (C) alpha, (D) sigma, (E) beta, and (F) gamma power up to up to 360 minutes after return of spontaneous circulation (ROSC) are provided in male mice sedated with propofol at 10 mg · kg−1 · h−1 starting at ROSC, and those sedated with propofol at 40 mg · kg−1 · h−1 starting at 60 minutes after ROSC. EEG power was calculated for each frequency band at consecutive 10-minute time blocks, which was expressed as percent of the baseline EEG power before cardiac arrest. Data are presented as mean α SD.
Supplementary Figure 8. Representative EEG spectrograms in the first 120 minutes after return of spontaneous circulation (ROSC) from (A) male mice that received no sedation, (B) those sedated with propofol at 40 mg · kg−1 · h−1 starting at ROSC, and (C) those sedated with dexmedetomidine at 1 μg · kg−1 · h−1 starting at ROSC.
Supplementary Figure 10. Representative EEG spectrograms in the first 24 hours after return of spontaneous circulation (ROSC) from (A) male mice sedated with propofol at 10 mg · kg−1 · h−1 starting at ROSC, and (B) those sedated with propofol at 40 mg · kg−1 · h−1 starting at 60 minutes after ROSC. The white double-headed arrow in the spectrogram indicates the period of sedation.
Supplementary Figure 6. EEG spectrograms in the first 24 hours after return of spontaneous circulation (ROSC) for the individual animals that received no sedation, sedation with propofol at 40 mg · kg−1 · h−1 starting at ROSC, or sedation with dexmedetomidine at 1 μg · kg−1 · h−1 starting at ROSC.