

Summary
Of the many research challenges posed by human cytomegalovirus latency, perhaps the most notable is the requirement for primary hematopoietic cell culture. Culturing hematopoietic subpopulations while maintaining physiological relevance must be given utmost consideration. We describe a long-standing primary CD34+ hematopoietic progenitor cell (HPCs) system as an in vitro model to study human cytomegalovirus (HCMV) latent infection. Key aspects of our model include infection of primary human CD34+ HPCs prior to ex vivo expansion, maintenance of undifferentiated cells in a long-term culture with a stromal cell support, and an assay to quantitate infectious centers produced prior to and following a reactivation stimulus. Our method offers a unique way to quantitatively assess HCMV latency and reactivation to study the contribution of viral and host genes in latency and reactivation.
Keywords: Human Cytomegalovirus, CD34+ hematopoietic cells, Latency, Reactivation, Extreme limiting dilution analysis
1. Introduction
Human Cytomegalovirus, HCMV, like all herpesviruses, possesses an extraordinary ability to persist in the immunocompetent host by way of a latent infection. HCMV infects a variety of cell types in the host, including hematopoietic progenitor cells, stromal cells, monocytes, monocyte derived macrophages, endothelial cells, epithelial cells, fibroblasts, neuronal cells and smooth muscle cells (1–4). Despite this broad cell tropism, the latent infection is thought to be restricted to hematopoietic cells. HCMV latency has been associated with myeloid lineage cells ranging from CD34+ (5–8) and CD33+ (9–11) progenitor cells to CD14+ monocytes (12–15) both in vivo and in vitro. Studying HCMV latency in any of these cell types has been impeded by the difficulty of physiologically-relevant culturing primary hematopoietic cell populations and the lack of quantitative assays for understanding the molecular mechanisms of latency in these cells.
Here we describe methods to efficiently infect and culture highly purified CD34+ HPCs in a system designed to support hematopoietic progenitor populations. The methods described are designed for pure populations of infected CD34+ HPCs cells, which are achieved by isolating cells infected with recombinant virus expressing a fluorescent marker for infection (i.e., GFP, mCherry) by fluorescent activated cell sorting. The infected HPCs are cultured above a mixed support comprised of two immortalized stromal cell lines, M2–10B4 and S1/S1. These murine stromal cell clones have been engineered to express key human cytokines and have been shown to support human hematopoietic progenitor cell proliferation, differentiation and stem cell self-renewal (16). For these reasons, stromal cell co-culture represents the most physiologically relevant means to maintaining hematopoietic progenitor cells in culture. During the 10–14 day culture period, CD34+ cells maintain viral genomes, but the genome becomes quiescent, expressing few, if any, genes (17). At a frequency of 1 in approximately 10,000 cells, virus replication is reactivated by transferring infected CD34+ cells into co-culture with primary permissive fibroblasts in a variety of cytokine mixtures. Latency and reactivation from latency is determined by calculating the infectious centers produced prior to and following treatment with a reactivation stimulus. Latency is defined by a 5- to 10-fold increase in the frequency of infectious centers formation following a reactivation stimulus. The quantitative nature of this assay is able to discern a number of possible phenotypes (Figure 1). This robust method has allowed for the identification of subpopulations of CD34+ cells supporting a latent infection (5) and viral determinants of latency (18–20).
Figure 1. The potential latency phenotypes in CD34+ hematopoietic progenitor cell model.
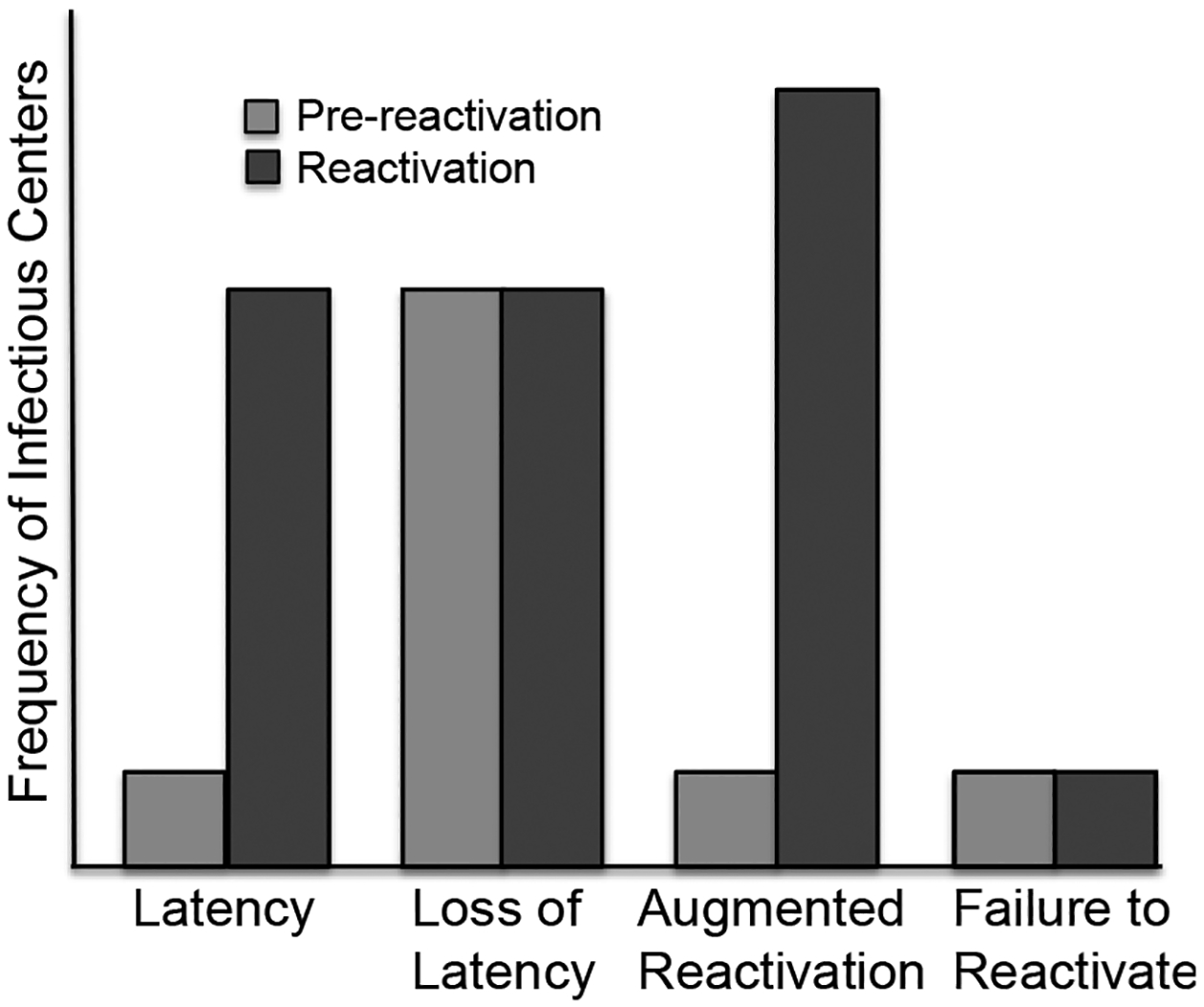
Pre-reactivation is the virus produced prior to the reactivation as determined by measuring infectious centers in the lysate controls (light grey bars). The frequency of reactivation (dark grey bars) is determined by infectious centers obtained by the co-culture of viable, latently infected CD34+ cells with fibroblast monolayers.
2. Materials
All solutions should be prepared using ultrapure water and cell-culture grade reagents. Fetal bovine serum for the culture of hematopoietic progenitors cells must be tested for its ability to support hematopoietic progenitors self-renewal and differentiation.
2.1. Human hematopoietic long term culture components
M2–10B4 murine (MG3) and S1/S1 stromal cells: Thaw two vials of each cell type using standard thawing techniques (see Note 1). Seed the cells into separate 10 cm dishes with appropriate growth media. Once confluent, pass the cells weekly at 1:50 or as appropriate for experimental needs. Do not allow cells to become overgrown. Every 2nd – 3rd passage, seed MG3 and S1/S1 into media containing Hygromycin and G418 to select for stably transfected cells (see Note 2).
MG3 growth medium: RPMI-1640 containing 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 60 μg/ml Hygromycin and 400 μg/ml geneticin (G418).
Purified CD34+ HPCs: Cells may be isolated fresh from cord blood (see Note 3) or bone marrow or purchased (Lonza, Wlakersville, MD; National Disease Research Interchange, NDRI, Philadelphia, PA). For isolation of CD34+ cells, use the CD34 MicroBead kit (catalogue # 130-046-703) and macs separation columns (catalogue # 130-042-401) from Miltenyi Biotec according to manufacturer’s instructions. Cells should be used or cryopreserved for storage in liquid nitrogen immediately upon isolation (see Note 4). Purity should be >75% CD34+ cells as determined by flow cytometry using a fluorescently-conjugated CD34 antibody (BD Biosciences or BD Pharmingen).
S1/S1 growth medium: Iscove’s modified Dulbecco’s medium (IMDM) containing 10% fetal bovine serum, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 μg/ml streptomycin, 125 μg/ml Hygromycin and 800 μg/ml Geneticin (G418).
CD34+ hematopoietic cells infection medium: IMDM supplemented with 10% BIT9500 serum substitute, 2 mM L-Glutamine, 20 ng/ml low density lipoproteins and 50 μM 2-mercaptoethanol.
Human CD34+ long-term culture medium (hLTCM): MyeloCult H5100 (Catalogue # 05150, Stem Cell Technologies, Vancouver, British Columbia) supplemented with 1 μM hydrocortisone (HC), 100 U/ml penicillin and 100 μg/ml streptomycin for complete hLTCM media.
BIT 9500 Serum substitute (Catalogue # 09500, Stem Cell Technologies, Vancouver, British Columbia).
High grade, cell culture-tested dimethyl sulfoxide (DMSO; Catalogue # D2650-Hybrimax, Sigma-Aldrich, St. Louis, MO).
DNase I (Catalogue # D-4513, cell culture-tested, Sigma-Aldrich, St. Louis, MO) reconstituted to 1mg/ml in water.
Gamma irradiator (135Cs source preferable).
Collagen (Catalogue # 04902, Stem Cell Technologies, Vancouver, British Columbia).
FACS Buffer: PBS+0.5% FBS.
SlickSeal Microfuge tubes (Catalogue # CN170S-GTS, National Scientific Supply Co, Inc., Claramont, CA).
FACSAria, BD Biosciences Immunocytometry Systems or similar cell sorting capability.
Transwell-COL (Catalog # 3491, 24mm diameter, 0.4 μm pore size, Costar, Corning, NY).
2.2. Reactivation components
Human fibroblast growth medium: Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 10mM HEPES, 1 mM sodium pyruvate, 2 mM L-Glutamine, 0.1 mM Non-essential amino acids, 100 U/ml penicillin and 100 μg/ml streptomycin.
Human primary fibroblasts are isolated from human foreskins or human embryonic lungs. The human embryonic lung fibroblast cell line, MRC5, purchased from ATCC (CCL-171™, Manassas, VA) can also be used.
Reactivation medium: RPMI supplemented with 20% fetal bovine serum (FBS), 10mM HEPES, 1 mM sodium pyruvate, 2mM L-Glutamine, 0.1 mM non-essential amino acids, 100U/ml penicillin and 100 μg/ml streptomycin. Optional: Reactivation media can be supplemented with cytokines to promote differentiation and reactivation, including 15 ng/ml IL6, 15 ng/ml G-CSF, 15 ng/ml GM-CSF, 15 ng/ml IL3 (all cytokines purchased from R&D Systems, Minneapolis, MN).
Infected CD34+ cells from long-term culture.
96-well deep well dishes for dilutions (catalogue # 40002–014, VWR, Radnor, PA).
96-well cell culture dishes.
Multichannel pipetman (12-channel).
3. Methods
3.1. Purification of CD34+ cells from cord blood or bone marrow
Use fresh cord blood or bone marrow for the isolation of CD34+ Cells (see Note 3). Dilute cord blood or bone marrow 1:1 with 1X PBS.
Layer 35 ml of blood or bone marrow on to 15ml lymphocyte separation medium (i.e. Ficoll; Catalogue # 25–072-CV, Mediatech, Inc., Manassas, VA) in 50 ml conical tubes. Centrifuge for 45 min at 450xg at 18°C with NO BRAKE.
Carefully aspirate the top layer (plasma) to within 5 ml of the interface
Using a 5 ml pipette (and pipette-aid on slow speed), carefully collect mononuclear cells from the interface in to a fresh 50 ml conical tube, being careful to leave as much of the Ficoll layer behind (Figure 2).
Fill conical tube with PBS + 0.5% tested-lot FBS and pellet cells at 450xg for 12 mins at 4°C.
Resuspend cells in cold PBS + 0.5% lot-tested FBS. Filter cells through a 70 μm strainer and pool cells into a single 50 ml conical tube.
Count cells using hemocytometer and pellet cells as in step 5.
Purify CD34+ cells using the CD34 MicroBead kit from Miltenyi Biotec as per the manufacturer’s protocol. Infect cells immediately or cryopreserve for storage in liquid nitrogen (see Note 4).
Figure 2. Purification of CD34+ cells from cord blood.
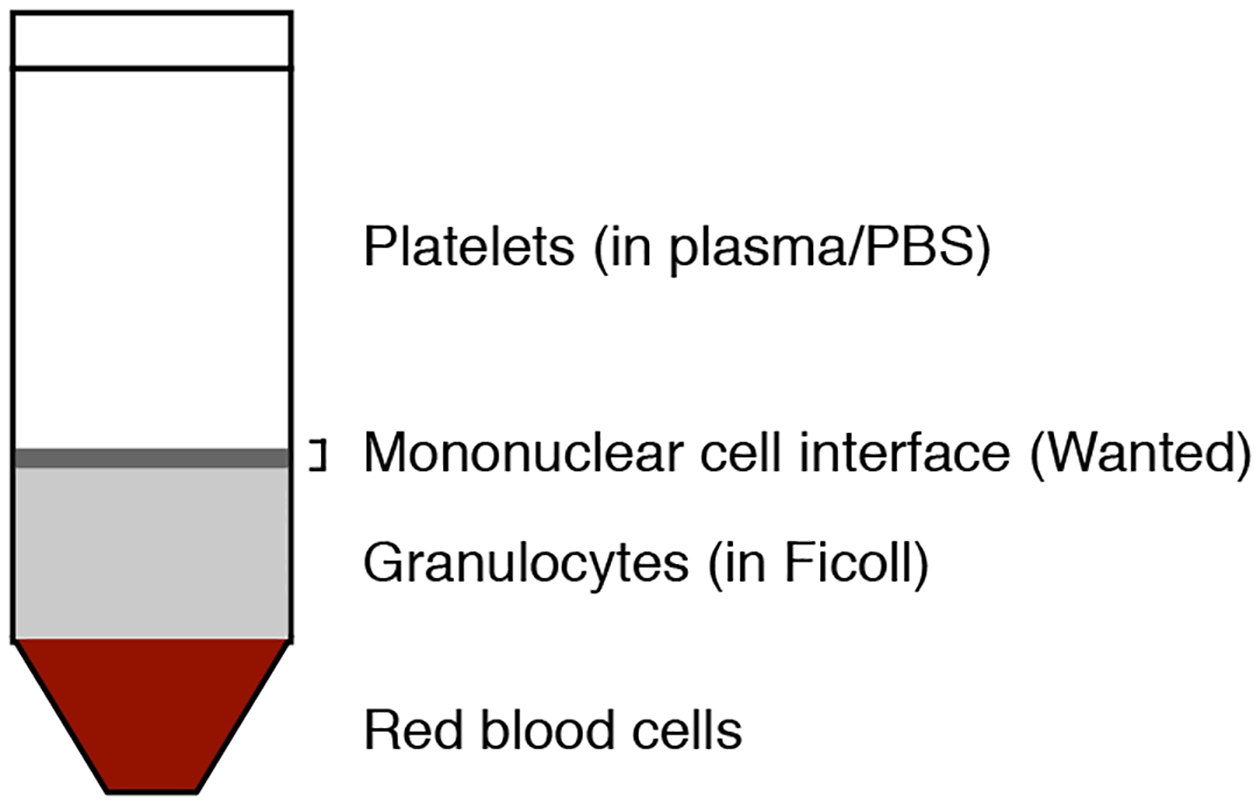
The mononuclear cells from any blood or bone marrow source are separated by Ficoll gradients and CD34+ cells are subsequently purified from the mononuclear fraction using the CD34 MicroBead kit.
3.2. Thaw CD34+ cells for infection
Prepare to thaw at least 2×107 cells per virus infection assuming 40% loss of cells in thawing process (see Note 5).
Remove vial(s) from liquid nitrogen storage and immediately transfer vial to 37°C water bath. Agitate continuously until completely thawed.
Submerge vial in 70% ethanol to disinfect and gently transfer cells from cryovial to a conical tube.
Add 100 μl of DNase I (1 mg/ml stock) for each cryovial thawed. Incubate 90 seconds (see Note 5).
Add an equal volume of IMDM + 2% FBS to the total cell suspension dropwise with gentle agitation and rest cells 3 minutes to allow equilibration.
Again double the volume by adding IMDM + 2% FBS to the cell suspension. Add slowly with gentle agitation to mix and rest cells 3 minutes.
Repeat step 5 until the total volume has reached 10–20X that of the original cell volume.
Pellet cells gently at 450Xg for 10 min. Aspirate most of the media without disturbing cell pellet.
Resuspend cell pellet in 2/3 the amount of DNase I used in step 3 by gently flicking the tube.
Add 5 ml IMDM + 2%FBS for every vial originally thawed and pellet cells at 450Xg for 10 min.
Resuspend cell pellet in 1/3 the amount of DNase I used in step 3 by gently flicking the tube.
Add 5 ml IMDM + 2%FBS for every vial originally thawed and pellet cells at 450Xg for 10 min.
Resuspend cell pellet in 5 ml of PBS for every vial originally thawed to wash away the serum.
Count cells using a hemocytometer and trypan blue. Pellet cells and aspirate media.
Resuspend cells in CD34+ hematopoietic cells infection medium and Incubate cells overnight in the 37°C + 5%CO2 incubator. (see Note 5)
3.3. Infection of CD34+ cells
To sufficiently infect CD34+ cells and analyze latency, all viruses should be clinical strains and should be engineered to express a fluorescent protein (i.e., GFP, RFP, mCherry) as a marker for infection. Retain approximately 50 million cells for a mock-infected control and for single-color sorting controls.
Count CD34+ cells from the overnight culture and pellet cells 450xg for 12 min.
Resuspend cells in infection media at a density of 2×107 cells/ml.
Seed cells into multi-well dishes appropriate for the cell numbers and volumes. The cells will not adhere but they will settle. To keep the settled cells in close proximity with the virus in solution, the media only needs to cover the bottom of the well. For example, use a 12 well plate for 0.5ml infection volume or a 6 well plate for 1.0 ml infection volume.
Thaw virus stock quickly in a 37°C bath. Sonicate (3 one-second pulses at 60% duty; empirically determined) to break aggregates and centrifuge at 10,000xg for 30 seconds to pellet debris. Add volume of virus equivalent to 2–5 PFU/cell to infection media (see Note 6).
Place cells in the 37°C + 5%CO2 incubator for the infection. Plates should be rocked periodically (2–3 times per hour) during infection to maximize cell contact with virus.
After 3–6 hrs of infection, centrifugally enhance the infection. Place the multi-well dish in the centrifuge and spin cells in dish at 450xg for 20 minutes at room temperature. After centrifugation, gently resuspend the cells in the media by pipetting the media over the surface of the dish. Do not remove the virus-containing media.
Virus infection will continue overnight in the 37°C+5%CO2 incubator. Incubate for at least 20–24 hr to achieve 50–70% infection.
3.4. Irradiation of stromal cells (see Note 7)
Collagen coat an appropriate amount of 6 well dishes that will be used for human long-term culture (hLTC). You will want 2–3 wells per infection. Distribute 1 ml of 1 mg/ml collagen solution per well and let sit for 2 min (see Note 8). Collect excess collagen for save for re-use. Allow the plates to dry, uncovered in the hood, for at least one hour. Rinse wells with PBS before seeding cells.
-
Prepare a 1:1 solution of MG3 and S1/S1 cells for irradiation.
3.0×105 cells (1.5×105 of each cell type) are required per well of a 6 well plate (0.9 ×106 of each cell type per 6-well plate). A confluent MG3 or S1/S1 10cm plate has approximately 4.0×106 cells.- Trypsinize and count the cells of each cell type. Allocate an appropriate number of cells to a round bottomed15 ml snap cap tube. See Example:
- Pellet cells 350xg for 7 min and re-suspend in 0.5 ml of hLTCM. The cells should be at 107-108 cells/ml. Keep cells on ice before irradiation.
- Example: For three 6-well dishes
Irradiate cells using a Gamma cell-40 (135Cesium) irradiator at 15 Gy. (see Note 7)
After irradiation, bring up the volume of the cells in the snap cap tube to 15 ml using PBS+0.5% FBS. Centrifuge 350xg for 7 min to pellet. Re-suspend the cells in 2 ml of complete hLTCM (w/ hydrocortisone) per well to be plated. Seed 2 ml of cell suspension per well into 6-well dishes.
Incubate cells at 37°C +5% CO2. After cells have attached, change media to remove dead cells and media containing free radicals as a byproduct of irradiation.
3.5. Purify infected CD34+ cells
Pure populations of infected (GFP+) cells that are CD34+ or CD34+CD38− are required for quantitative latency/reactivation assays; uninfected cells contaminating the population will lead to inaccurate quantitation.
Transfer the cells from the infection well into a 50 ml conical tube through a 70 μm cell strainer, being sure to wash the well with PBS+0.5% FBS to recover all cells. Pellet cells 12 minutes at 450xg. Aspirate media thoroughly from cell pellet. Resuspend cells in 50–100μl of DNAse I (1 mg/ml) by gently flicking the tube.
Add 200–500μl of Citric Acid Wash Solution (depending on the size of the cell pellet) to the cells to inactivate any remaining extracellular virus. Incubate cells at room temperature for exactly 1 min. During the incubation, transfer cells to a new 50ml conical tube to leave any residual active virus behind that may aberrantly contribute to the measurement of virus produced following reactivation. At the end of the 1 min incubation period, quickly bring up the volume with aMEM+2% FBS to 50 ml. The media color should change from yellow back to a light red indicating acid was neutralized. Pellet cells for 12 minutes at 450xg. Resuspend cells in 10 ml PBS+0.5% FBS to wash once more. Filter cells once or twice through a 70μm cell strainer and pellet.
- To stain cells for the CD34 cell surface marker, aspirate the media from the pellet and distribute cells for controls. All samples (controls and sorting tubes) are prepared in polystyrene 4 ml snap cap Falcon tubes for the flow cytometer. In addition to an unstained cell control, a single color control is needed for each fluorescent channel used. Typically, this includes PI, GFP, PE and sometimes APC.
- For the unstained and propidium iodide (PI) controls, transfer ~105 uninfected cells to each of two tubes in ~200 μl PBS+0.5% FBS. The unstained control is complete. Add PI to the PI single color control tube from a 100X stock.
- For the GFP alone control, transfer ~105 cells from an infected sample to a 4 ml polystyrene snap cap Falcon tube in 200 μl PBS+0.5% FBS.
- For each fluorescent single color control, transfer ~105 uninfected cells to a polystyrene 4 ml snap cap Falcon tube in ~200 μl PBS+0.5% FBS. Add 2 μl of the appropriate antibody to each tube.
Prepare experimental samples for sorting: Resuspend cells in PBS+0.5% FBS to a concentration of 2×106 cells/ml. Keep the cells in polypropylene tubes (fewer cells will be lost than in polystyrene tubes) that can accommodate large volumes. Add 20μl of the desired conjugated antibody (i.e., phycoerythrin conjugated anti-CD34) per 106 cells unless otherwise specified by the manufacturer.
Stain cells (controls and experimental samples) for 15 mins at 4°C (unless otherwise specified by the manufacturer); agitating occasionally to keep cells in suspension.
Add 10–20 volumes of PBS+0.5% FBS to wash and pellet 12 minutes at 450xg at 4°C.
Resuspend the single color controls in 250μl PBS+0.5% FBS (no PI). Resuspend the experimental samples to sort in FACS Buffer at a concentration of 1×107 cells/ml. All cells should be in polystyrene tubes appropriate for the flow cytometer. Keep all tubes on ice and in the dark.
Prepare two collection tubes per infection: 1ml of hLTCM in siliconized microcentrifuge tubes (SlickSeal microfuge tubes) to collect cells from flow cytometer.
Using FACSAria or similar cell sorting instrument, isolate GFP+/CD34+ cells. Yields will depend on the percentage of cells infected (typically 30% with FIX strain, 60% with TB40E, and 5% with AD169) and the gating.
3.6. Human CD34+ cells long-term culture
After the sort, pellet the cells at 450xg for 12 mins at 4°C in a microcentrifuge. Aspirate media carefully and re-suspend in 1ml of complete hLTCM (with hydrocortisone).
Add 1ml of complete hLTCM into each well that will have a transwell (do not remove the existing 2 ml in the well; total volume will be 3ml). Place transwells into the top of each well. Transfer cells resuspended in 2 ml into transwell. Cells densities should range between 104 and 105 cells per transwell; samples may have to be split cells between multiple transwells. The total volume of complete hLTCM in the well is 5ml (3ml in well + 2ml in transwell). Incubate at 37°C + 5%CO2.
Feed the cells every 5 days by aspirating approximately half of media from below the trans-well and replacing it with an equal volume of fresh complete hLTCM. If the stromal cell monolayer begins to lift or appear unhealthy, new stromal cells should be irradiated and the transwells transferred to fresh stromal cell monolayers.
3.7. Reactivation
Plate human primary fibroblasts in 96 well dishes at a cell density of 5×103 cells/well in reactivation medium (see Note 9) and incubate over night at 37°C + 5%CO2.
Collect the infected CD34+ cells from the trans-wells of the long term culture into a 15ml conical tube and wash the trans-wells 3x with PBS+0.5% FBS. Pool the cells and the washes. Count the total number of cells.
Take 1.2×106 cells for reactivation and 1.2×106 cells for lysate control in two separate siliconized tubes (see Note 10). Spin down the cells at 450xg for 12 mins. Resuspend the cells in 1.5 ml of reactivation medium.
For lysate control, lyse cells by freezing in liquid nitrogen and thawing at 37°C water bath (see Note 11). Mix 10 μl of lysate with 10 μl trypan blue and observe under light microscope to ensure the complete lysis of cells. Cell lysate should be in 1.5 ml total volume.
For each reactivation and lysate control sample, add 1.5 ml of cell slurry or cell lysate (both equivalent to 1.2×106 cells) into each well in row A of the deep well dish. Fill all other wells with 0.75 ml reactivation media.
Transfer 0.75 ml of each well in Row Z to the corresponding well in row B to perform a 1:2 dilution. Repeat for all rows to serially dilute cell slurry and lysate (Figure 3).
Once all dilutions have been made (row A-H), using a multichannel pipetman, transfer 50μl of each dilution series (one column of the 96 well dish, 8 dilutions) to each well in a row (12 total) of the 96 well dish containing human primary fibroblasts (see Note 12). Final volume will be 150 μl (50 μl of cell slurry + 100 μl of media from plating the fibroblasts)
Incubate cells at 37°C +5% CO2 for 15–20 days.
Score each well of 96 well dish for GFP expression using a fluorescence microscope. For each of the 8 dilutions, determine a fraction of wells that scored positively for GFP expression.
Enter the number of input infected CD34+ HPCs per well for each dilution and the fraction of GFP+ wells into the online shareware, Extreme limiting dilution analysis (ELDA, http://bioinf.wehi.edu.au/foftware/elda), following the direction of the website.
- Run the program to obtain an estimate of cells required to form an infectious center (see Note 13) based on the number of GFP+ cells at each dilution. See an example below.
Confidence Intervals Group Lower Estimate Upper WT X1 X2 X3 MUTANT 1 Y1 Y2 Y3 MUTANT 2 Z1 Z2 Z3
Figure 3. Schematic of Infectious Centers Assay.
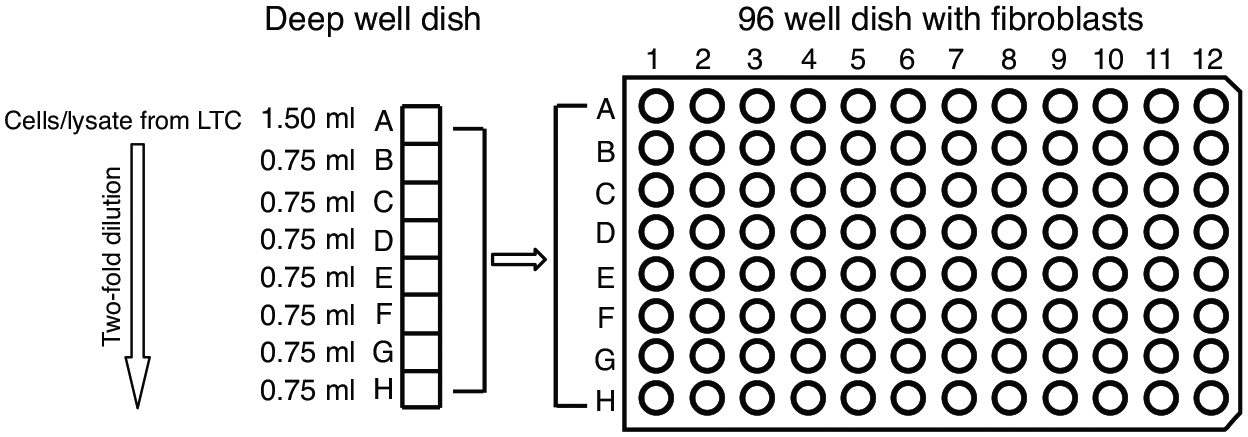
The infected CD34+ cells or the equivalent cell lysate from the long term culture are diluted two-fold in deep well dishes and transferred to monolayers of MRC5 cell in 96 well dishes for the infectious centers assay.
4. Notes
The M2–10B4 murine stromal cell line (MG3 cells) is engineered to express human interleukin-3 (IL-3) and granulocyte-colony stimulating factor (G-CSF). The S1/S1 cell line is engineered to express human IL-3 and stem cell factor (SCF). These cells are provided by Stem Cell Technologies Ltd. on behalf of D. Hogge, University of British Columbia (Vancouver, British Columbia). Other stromal cells systems have been used to effectively maintain latently infected CD34+ cells (5, 17); however, the ability of stromal cells to support HCMV latency must be empirically determined.
Hygromycin/G418 selection should be performed every 2nd-3rd passage. Complete selection requires ~1 week, so it is preferable to initially pass the cells sparsely (1:50) for selection as specified by the protocol from Stem Cell Technologies.
Isolate CD34+ cells within 36 hrs of harvesting the cord blood or bone marrow. Older samples yield fewer CD34+ cells. Typically, cord blood contains 0.5%−1.0% CD34+ cells and bone marrow contains 2% CD34+ cells.
For freezing, resuspend CD34+ cells in cold IMDM containing 20% high grade FBS, 100 U/ml penicillin and 100 μg/ml streptomycin at 1–2×107 cells/ml. Add an equal volume of ice cold solution of 80% high grade FBS and 20% cell culture grade DMSO drop wise, mix gently and aliquot into 1.8 ml cryogenic vials. Freeze the vials overnight in a rate-controlled cryopreservation cooler and on the next day, transfer the vials into liquid nitrogen for long-term storage.
Plan for 30–45 mins for the entire process. DMSO is toxic and makes cells somewhat fragile and hence, work quickly and gently while handling CD34+ Cells. Use of DNase I will prevent clumping of cells due to DNA released from broken cells. Multiple DNase I treatments and washes with large volumes are necessary to remove cells from DNA and cell debris. Usually, 50–60% of the cells should be viable at the end of thawing process. The overnight incubation period in infection media (low serum, low cytokine) enhances infectivity of thawed cells.
Virus stocks are concentrated stocks at 107-108 PFU/ml stored at −80°C. Viruses stocks are concentrated to high titer by sedimenting virus from infected culture supernatant through a 20% D-Sorbitol cushion. High titer virus is required for high titer infection in low volumes. Depending on virus strains, 2–5 PFU per cell as determined from titering on fibroblasts will infect 10–80% of CD34+ cells. Viruses used for infection should be marked with a fluorescent tag (i.e., GFP driven by the SV40 promoter) so that infected cells can be purified from uninfected cells. This is essential for accurate quantitation of latency/reactivation.
Stromal cells should be irradiated ~24 hours prior to setting up long-term cultures. This period of time allows the stromal cells to condition the media. Irradiation dose may need to be determined empirically based on the calibration of irradiator. With a well-calibrated irradiator, a dose of 15–20 Gy is adequate to arrest cell division.
Collagen solution is provided at a concentration of 3 mg/ml in acetic acid. Dilute in sterile water. Collagen can be saved for re-use up to 6 times as long as sterility is maintained.
Seed 100 μl cells per well of a 96 well dish. Plan for 10 ml per dish. 1 dish per reactivation and 1 dish per lysate control is required for each virus or condition to be tested (i.e., a minimum of two 96-well dishes per transwell of long-term culture)
1.2×106 cells will result in 40,000 cells (or lysate equivalent) per well for the first dilution (Row A). The cells numbers are for single replicates of reactivation and lysate controls. Many replicates of reactivation and lysate control can be done if the number of infected cells available from the long-term culture is not limiting. This protocol uses 40,000 cells per well of the 96 well dish to achieve optimal numbers to calculate infectious centers from latency and reactivation; however, the first dilution for reactivation can vary between 15,000 and 40,000 cells per well depending on the cell numbers available.
Lysate control provides an estimate of pre-formed virus prior to reactivation and hence it’s important to achieve complete cell lysis without damaging the virus to accurately quantify the virus pre-existing in the CD34+ cell cultures. Complete lysis of the cells can be analyzed using trypan blue. To estimate the loss of virus due to the freeze-thaw process, freeze and thaw an aliquot of virus of known concentration and then titer this virus (by plaque assay or TCID50) in parallel with an equivalent aliquot that was not frozen and thawed. From the titers, calculate the percentage of infectious virus remaining after the treatment.
Mix the cells intermittently in the dilution series to avoid settling of the cells at the bottom of the deep well dish and to obtain even distribution of cells across the 96 well dish.
The program performs a statistical analysis to show a goodness of fit accompanying graphs for the groups of data analyzed and a table showing confidence Intervals (21).
The number corresponding to the estimate is the number of cells required for one reactivation event. The inverse number of the estimate will give the frequency of infectious centers.
Acknowledgments
This work was supported by Public Health Service Grants CA11343 and AI079059 to F. G. from the National Cancer Institute (NCI) and the National Institute of Allergy and Infectious Disease (NIAID), respectively.
References
- 1.Sinzger C, Grefte A, Plachter B, Gouw AS, The TH, and Jahn G (1995) Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues, J Gen Virol 76, 741–750. [DOI] [PubMed] [Google Scholar]
- 2.Soderberg C, Larsson S, Bergstedt-Lindqvist S, and Moller E (1993) Definition of a subset of human peripheral blood mononuclear cells that are permissive to human cytomegalovirus infection, J Virol 67, 3166–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrier RD, Nelson JA, and Oldstone MB (1985) Detection of human cytomegalovirus in peripheral blood lymphocytes in a natural infection, Science 230, 1048–1051. [DOI] [PubMed] [Google Scholar]
- 4.Boeckh M, Hoy C, and Torok-Storb B (1998) Occult cytomegalovirus infection of marrow stroma, Clin Infect Dis 26, 209–210. [DOI] [PubMed] [Google Scholar]
- 5.Goodrum F, Jordan CT, Terhune SS, High KP, and Shenk T (2004) Differential outcomes of human cytomegalovirus infection in primitive hematopoietic subpopulations, Blood 104, 687–695. [DOI] [PubMed] [Google Scholar]
- 6.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, and Sinclair JH (2005) Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers, Proc Natl Acad Sci U S A 102, 4140–4145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sindre H, Tjoonnfjord GE, Rollag H, Ranneberg-Nilsen T, Veiby OP, Beck S, Degre M, and Hestdal K (1996) Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells, Blood 88, 4526–4533. [PubMed] [Google Scholar]
- 8.von Laer D, Meyer-Koenig U, Serr A, Finke J, Kanz L, Fauser AA, Neumann-Haefelin D, Brugger W, and Hufert FT (1995) Detection of cytomegalovirus DNA in CD34+ cells from blood and bone marrow, Blood 86, 4086–4090. [PubMed] [Google Scholar]
- 9.Hahn G, Jores R, and Mocarski ES (1998) Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells, Proc Natl Acad Sci U S A 95, 3937–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kondo K, Kaneshima H, and Mocarski ES (1994) Human cytomegalovirus latent infection of granulocyte-macrophage progenitors, Proc Natl Acad Sci U S A 91, 11879–11883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kondo K, Xu J, and Mocarski ES (1996) Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals, Proc Natl Acad Sci U S A 93, 11137–11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hargett D, and Shenk TE (2010) Experimental human cytomegalovirus latency in CD14+ monocytes, Proc Natl Acad Sci U S A 107, 20039–20044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith MS, Bentz GL, Alexander JS, and Yurochko AD (2004) Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence, J Virol 78, 4444–4453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soderberg-Naucler C, Fish KN, and Nelson JA (1997) Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors, Cell 91, 119–126. [DOI] [PubMed] [Google Scholar]
- 15.Soderberg-Naucler C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, and Nelson JA (2001) Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent, J Virol 75, 7543–7554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller CL, and Eaves CJ (2002) Long-term culture-initiating cell assays for human and murine cells, In Hematopoietic Stem Cell Protocols (Klug CA, and Jordan CT, Eds.), pp 123–141, Humana Press, Totowa. [DOI] [PubMed] [Google Scholar]
- 17.Goodrum FD, Jordan CT, High K, and Shenk T (2002) Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency, Proc Natl Acad Sci U S A 99, 16255–16260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodrum F, Reeves M, Sinclair J, High K, and Shenk T (2007) Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro, Blood 110, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petrucelli A, Rak M, Grainger L, and Goodrum F (2009) Characterization of a Novel Golgi-localized Latency Determinant Encoded by Human Cytomegalovirus, J Virol 83, 5615–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, Fleming WH, Streblow DN, and Goodrum F (2011) A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection, PLoS Pathog in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, and Smyth GK (2009) ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays, Journal of immunological methods 347, 70–78. [DOI] [PubMed] [Google Scholar]