

Abstract

Unprecedented MsOH-promoted diastereoselective cascade dimerization and intramolecular lactonization of readily accessible α,β-unsaturated γ-ketoesters are presented. The results obtained in this work, control experiments, and density functional theory (DFT) calculations suggested that the initial enolization and E to Z isomerization/equilibration of olefin (C=C) of substrate α,β-unsaturated γ-ketoesters give a Z-isomer preferentially over an E-isomer. Subsequently, the Z-isomer undergoes intermolecular annulation with α,β-unsaturated γ-ketoesters via domino Michael addition/ketalization/lactonization steps to furnish fused tetracyclic pyrano-ketal-lactone. However, the Z-isomer prefers intramolecular trans-esterification in a competing pathway and gives bicyclic γ-ylidene-butenolide. The key features of this work include simple Brønsted acid catalysis, the formation of three bonds, two rings, and three contiguous stereogenic centers in a single step, DFT calculations, and the assignment of relative stereochemistry through X-ray diffraction (XRD) and two-dimensional (2D) nuclear magnetic resonance (NMR) analyses.
Introduction
The biological activity, stereochemical and structural complexity of natural and unnatural molecules is a longstanding motivation for organic chemists to develop novel synthetic methodologies. In recent times, the expansion of the chemical space in drug discovery has encountered the inclusion of diverse three-dimensional small molecules possessing sp3-rich scaffolds.1 Major problems associated with the sp3-rich molecules-based drug discovery include the lack of efficient synthetic methods, stereochemical selectivity, and supply. In the last four decades, an upsurge in the development of cascade/domino reactions enabling the construction of intricate molecular scaffolds from simple building blocks with brevity has been witnessed to address these concerns.2 Notably, dimerization reactions represent synthetically powerful strategies for rapidly constructing molecular complexity in organic synthesis.3
So far, various dimerization reactions have been disclosed for the facile stereoselective construction of natural and unnatural molecules, which include [1 + 1] radical dimerization of resveratrol,4 oxidation of isatins,5 alkynols,6 the [4 + 4]-cycloaddition reaction to give eight-membered rings, and others.7 Moreover, diverse domino dimerization reactions involving the initial addition of p-quinol alcohol onto an electrophilic enone followed by 5-exo-trig ring-closure to give fused oxa-heterocycles are well-documented, and Carreño’s quadruple domino reaction to afford a pentacyclic trimer is one of the notable examples.8
Recently disclosed Duarte and Lawrence’s dimerization (domino [1,4]/[1,4]/[1,4]/[1,4] addition) of p-quinols to construct tetracyclic caged oxa-heterocycles (via the construction of four new bonds, four rings, and eight stereogenic centers) is considered as a most complex example of the dimerization reaction ever disclosed.9 However, dimerization cascades of unsaturated ketoesters involving Brønsted acid catalysis and carbon-based nucleophiles remain elusive.
Fused pyrano-ketal scaffolds found in numerous bioactive natural products (for instance, alboartins, xyloketals A–D and H, hyperaspidinol, myxostiolides, spicatolide-C, guaianolide, and others)10 and fused pyrano-ketal-lactones are key structural units in diverse bioactive natural products phomoidride B and D,11 acylophlorocinols,12 and artemisinin degradation products (Figure 1).13 Inspired by the exciting structural and biochemical features of furo-pyran-containing molecules, our research group recently reported novel synthetic methodologies for the facile construction of furo-pyranones (fused pyrano-ketal-lactones),14a chromanol-lactones,14b and other oxygen-heterocycles via intermolecular cascade annulation reactions.14 Herein, we disclose our fortuitous findings of MsOH (methanesulfonic acid)-mediated enolization and E to Z isomerization/equilibration of α,β-unsaturated γ-ketoesters 1,15 wherein the Z-isomer undergoes domino Michael addition/ketalization/lactonization with 1 to furnish complex pyrano-ketal-lactones 2, and also delivers the bicyclic γ-ylidene-butenolide 3 through competing intramolecular lactonization (vide infra) (Scheme 1).
Figure 1.

Natural products containing fused pyrano-ketal-lactones and pyrano-ketals.
Scheme 1. Initial Synthesis of the Fused Pyrano-ketal-lactone (2a) and γ-Ylidene-butenolide (3a).

Results and Discussion
The initial scouting reaction was performed using a known α,β-unsaturated γ-ketoester (1a)14b as a model substrate. The first experiment using 10 mol % MsOH as a promoter in dichloroethane (DCE) solvent at room temperature (rt) for 24 h did not lead to any conversion (Table 1, entry 1). To our delight, when the reaction temperature was increased to 60 °C, cyclodimerization product 2a was obtained as a single diastereomer and also γ-ylidene-butanolide 3a in 20 and 35% yield, respectively, in a long reaction time of 24 h (Table 1, entry 2). Nuclear magnetic resonance (NMR), mass spectrometry (MS), and single-crystal X-ray diffraction analyses unambiguously established the structure and relative stereochemistry of 2a. Product 3a was confirmed by comparison of its NMR data with the reported data.16 The imparted exclusive diastereoselectivity of the dimerized product 2a could be attributed to the extra stability of the axial ketal-lactone group by anomeric and associated effects (Scheme 1; entries 1 and 2 of Table 1).17
Table 1. Reaction Optimization Studiesa.

| entry | catalyst (mol %) | solvent, temp | 2ab | 3ab |
|---|---|---|---|---|
| 1c,d | MsOH (10 mol %) | DCE, rt | - | - |
| 2d | MsOH (10 mol %) | DCE, 60 °C | 20 | 35 |
| 3c,d | p-TSA (10 mol %) | DCE, rt | - | - |
| 4d | p-TSA (10 mol %) | DCE, 60 °C | 8 | 25 |
| 5c,d | PPTS (10 mol %) | DCE, rt | - | - |
| 6c,d | PPTS (10 mol %) | DCE, 60 °C | - | - |
| 7d | TFA (10 mol %) | DCE, 60 °C | 10 | 32 |
| 8d | TfOH (10 mol %) | DCE, 60 °C | 10 | 25 |
| 9c,d | AcOH (10 mol %) | DCE, 60 °C | - | - |
| 10c,d | CSA (50 mol %) | DCE, rt | - | - |
| 11c,d | CSA (50 mol %) | DCE, 60 °C | - | - |
| 12c,d | l-proline (10 mol %) | DCE, 60 °C | - | - |
| 13c,d | (R)-BINOL-phosphoric acid (10 mol %) | DCE, 60 °C | - | - |
| 14e | MsOH (50 mol %) | DCE, 60 °C | 24 | 70 |
| 15e | MsOH (100 mol %) | DCE, 60 °C | 20 | 68 |
| 16c,d | MsOH (10 mol %) | CH3CN, rt | - | - |
| 17c,d | MsOH (10 mol %) | CH3CN, 60 °C | - | - |
| 18c,d | MsOH (100 mol %) | CH3CN, rt | - | - |
| 19c,d | MsOH (100 mol %) | CH3CN, 60 °C | - | - |
| 20c,d | no catalyst | DCE, 60 °C | - | - |
Reaction conditions unless otherwise specified: 1a (0.5 mmol) was used in 2 mL of solvent.
Isolated % yields.
No conversion was observed.
The reaction time is 24 h.
The reaction time is 5 h. DCE = dichloroethane.
Encouraged by these initial results and to improve the outcome of the reaction, we further verified the effect of other Brønsted acid catalysts and reaction parameters (Table 1). The reaction using para-toluenesulfonic acid (p-TSA), pyridinium p-toluenesulfonate (PPTS), trifluoromethanesulfonic acid (TfOH), and trifluoroacetic acid (TFA) at room temperature did not lead to any conversion. However, products 2a and 3a were formed at 60 °C in good yields with moderate selectivity and close to MsOH in 24 h (entries 1–8). No conversion was observed using AcOH and CSA at rt and in DCE at 60 °C even in a prolonged reaction time of 24 h (entries 9–11). It was also found that increased catalyst loading, reaction temperature, and reaction time had a detrimental effect on the outcome using these Brønsted acid catalysts (entries 3–11). Next, we verified the influence of chiral Brønsted acid catalysts l-proline and (R)-BINOL-derived phosphoric acid and found that they are incompatible with this cascade reaction (entries 12 and 13). As an ultimate option, MsOH was chosen as a reliable promoter due to its clean reaction profile and the effects of its loading (using 30, 40, 50, and 100 mol %) and solvents (DCE, ACN) at 60 and 100 °C (entries 14 and 15) were further verified. To our delight, the reaction using 50 mol % MsOH in DCE at 60 °C for 5 h was found to be optimal for this transformation by giving 2a and 3a in 24 and 70% isolated yields, respectively (Table 1, entry 14).16
Having optimal reaction conditions in hand, we set out to synthesize diverse α,β-unsaturated γ-ketoesters 1 using our earlier optimized reaction conditions (aldol reaction of cyclohexanones with ethyl glyoxylate, mesylation, and elimination sequence)14b,16 and then embarked onto the substrate scope studies.
Substrates possessing 2-methyl cyclohexanone, 3-methyl cyclohexanone, 4-methyl cyclohexanone, and 4-ethyl cyclohexanone participated well in this transformation and delivered anticipated products 2b–2e (2b with a 2:1 diastereomeric ratio and 2c–2e with 1:1 dr) and 3b–3e in good yields (entries 2–4, Scheme 2). Switching of enone-ethyl esters to methyl, isopropyl, allyl, and benzyl esters delivered corresponding products 2f, 2g, 2h, and 2i and butenolide 3a with equal ease and outcome (entry 5). Cycloheptane-tethered enone also participated well and produced the corresponding dimer 2m and butenolide 3m (entry 6). However, cyclooctane-tethered enone-ester gave the dimer 2n exclusively (with a 2:1 diastereomeric ratio), and no trace amount of butenolide 3n was noticed (entry 7). X-ray diffraction analyses rigorously assigned products 2a, 2f, and 2i; the analogy confirmed the remaining products (Scheme 2 and Figure 2).
Scheme 2. Synthesis of Fused Pyrano-ketal-lactones 2 and γ-Ylidene-butenolides 3,

See the Supporting Information (SI) for the synthesis of starting materials 1.
Isolated yield.
Figure 2.

ORTEP diagrams of fused pyrano-ketal-lactones 2f and 2i.
In contrast to these results, 3,3-dimethyl- and 3,3,5-trimethyl-cyclohexanone-derived substrates (1j and 1k) failed to produce dimers and instead furnished corresponding butenolides 3j and 3k exclusively in good yields (Scheme 3, entries A and B).18,19 In contrast, cyclopentane-derived enone-ester underwent inward double-bond isomerization and gave 4a instead of lactonization or dimerization products (Scheme 3, entry C).14b
Scheme 3. Reaction Profiles of Other α,β-Unsaturated γ-Ketoesters,

See the Supporting Information (SI) for the synthesis of starting materials 1.
Reaction conditions: MsOH (50 mol %), DCE, 60 oC, Isolated yield.
Next, we were curious to verify the reactivity of α,β-unsaturated γ-ketoesters 1 possessing an acyclic ketone moiety, for which the E → Z isomerization profile can be quite different, leading to differential selectivity over the formation of products 2 and 3. Accordingly, a series of substrates 1 containing methyl, isopropyl, cyclobutyl, cyclopentyl, cyclohexyl, chloromethyl, and ethyl-tethered ketones were synthesized and subjected to the annulation reaction using optimized conditions, which delivered corresponding γ-ylidene-butenolides18,193o–3u in moderate to good yields (61–98%) exclusively (entries 1–7, Scheme 4). This outcome could be attributed to the preferential E → Z isomerization of enone 1 and subsequent enolization-induced lactonization (in previous reports, similar products were obtained through the ring-closure of corresponding keto-acids).18 Ketoester 1v possessing trisubstituted olefin gave the ketal-lactone 3v (entry 8) instead of the expected γ-ylidene-butenolide. The structure and stereochemistry of butenolides 3u and 3t were established by comparison of NMR data with the reported data19 and nuclear over-Hauser effect spectroscopy (NOESY) correlations.16 Further, the utility of this protocol was exemplified by performing gram-scale experiments and preparing products 2a/3a (Scheme 2) and 3p (Scheme 4) with good yields.
Scheme 4. Synthesis of γ-Ylidene-butenolides 3,

See the Supporting Information (SI) for the synthesis of starting materials 1.
Isolated yield.
To gain insight into the mechanistic sequence of this protocol, we performed a reaction of 3a with MsOH (using optimized conditions) to verify the probable ring opening and dimerization, where we did not observe any change and the conversion of 3a into starting material 1a or dimer 2a (entry A, Scheme 5). Next, we tested the reaction between butenolide 3b and ketoester 1a under optimal reaction conditions to verify the probable inverse electron-demand Diels–Alder reaction, which delivered butenolide 3a, homodimer 2a, and unreacted butenolides 3b, providing evidence for the absence of the suspected [4 + 2]-cycloaddition pathway (entry B, Scheme 5).
Scheme 5. Supporting Experiments for the Mechanism.

In order to provide a comprehensive analysis of the formation of products γ-ylidene-butenolide (3a) and pyrano-ketal-lactones (2a), we employed density functional theory (DFT) to perform full quantum chemical calculations. In the presence of MsOH, the α,β-unsaturated γ-ketoester (1a) undergoes a tautomerization process, resulting in the formation of E and Z isomers, denoted as T1 and T2, respectively. Despite this process being endergonic, there exists a thermodynamic preference favoring the Z-isomer (T2) over the E-isomer (T1) by 1.7 kcal/mol. This preference can be attributed to the presence of a hydrogen bond in the T2 isomer, providing increased stability compared to T1, as illustrated in Figure 3A. In the subsequent step, the Z-isomer (T2) undergoes lactonization with the assistance of MsOH acid, forming major product 3a through an eight-membered cyclic transition state (TS). The lactonization is an exergonic step, with a free-energy change (ΔGsol) of 1.4 kcal/mol and a corresponding free-energy barrier (ΔGsol#) of 21.4 kcal/mol, as illustrated in Figure 3B.
Figure 3.
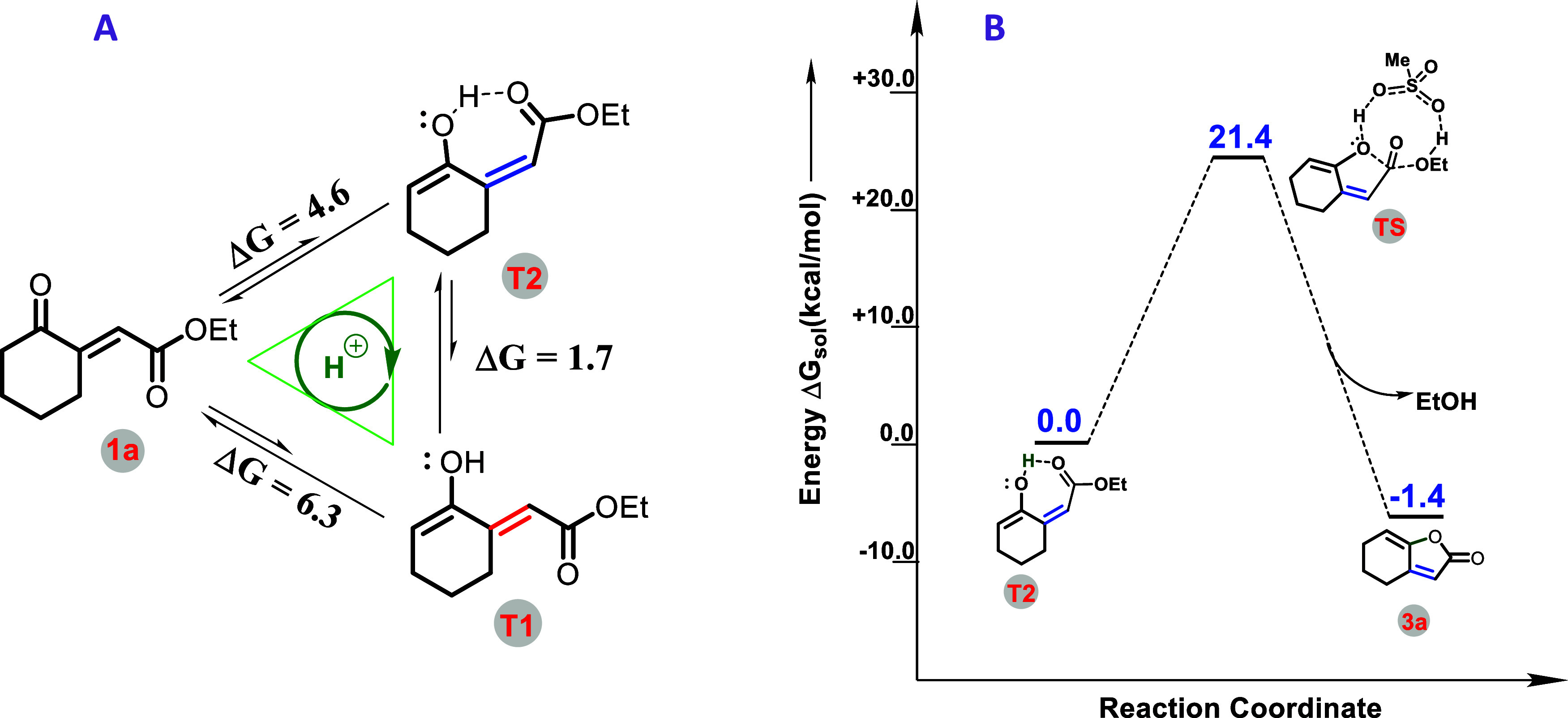
(A) Tautomerization of the α,β-unsaturated γ-ketoester (1a) and (B) free-energy profile for the formation of product γ-ylidene-butenolide (3a) have been shown here. The DFT calculations have been done at the PBE-D3/def2-TZVP level of theory using DCE as the solvent (ε = 10.36). All of the values are in kcal/mol.
Furthermore, to gain a deeper comprehensive understanding of the formation of the minor product [pyrano-ketal-lactones (2a)], we have investigated a mechanism where the α,β-unsaturated γ-ketoester (1a) reacts with the more thermodynamically favorable tautomer, the Z-isomer (T2). The nucleophilic carbon of the Z-isomer (T2) initiates a Michael addition reaction by attacking the electrophilic carbon of the α,β-unsaturated γ-ketoester (1a). This reaction leads to the formation of an intermediate T3, which contains a newly formed C–C bond. This exergonic addition reaction releases 8.4 kcal/mol of energy and occurs via a six-membered transition state TS1, with an activation energy of 26.9 kcal/mol. The next step involves the enolization process in which intermediate T3 generates intermediate T4 with the assistance of MsOH acid. This enolization process is highly endergonic, with a ΔGsol of 6.3 kcal/mol, and occurs through an eight-membered transition state TS2, which presents an energy barrier of 18.7 kcal/mol. Subsequently, intermediate T4 initiates a hemiketalization reaction, progressing through the eight-membered transition state TS3 (ΔGsol# = 10.7 kcal/mol), which releases a modest 0.1 kcal/mol of free energy and results in the formation of intermediate T5. It is important to note that this step is reversible, allowing for the conversion of T5 back to T4 with an equivalent energy barrier of 10.8 kcal/mol (Figure 4).
Figure 4.

Free-energy profiles with volume correction, for the formation of product 2a has been shown here. The DFT calculations have been done at the PBE-D3/def2-TZVP level of theory using dichloroethane (DCE) as a solvent (ε = 10.36). All of the values are in kcal/mol.
In the final stage of the reaction mechanism, intermediate T5 undergoes lactonization to yield the desired product 2a. This lactonization process is exergonic, releasing 4.3 kcal/mol of free energy. However, it is marked by a notable free-energy barrier (TS4) of 19.3 kcal/mol, which stands 8.5 kcal/mol higher than the reverse energy barrier for T5 to revert to T4.
Upon a thorough analysis of the energy profiles for the formation of 3a and 2a, a noticeable difference emerges. The overall free-energy barrier for the formation of 2a is measured at 26.9 kcal/mol, involving four sequential steps. Conversely, the formation of 3a from 1a presents a significantly lower free-energy barrier of 26.0 kcal/mol, requiring only a single-step process. In light of these findings and in accordance with the Eyring equation, it is reasonable to anticipate the coexistence of both products, with 3a being the major product and 2a the minor one, under the specified reaction conditions.
Based on results obtained in this work, control experiments, DFT calculations, and previous reports,14,15 a plausible mechanistic sequence is presented in Scheme 6. The reaction is initiated by the Brønsted acid (MsOH)-mediated equilibration of substrate 1 into its E (T1) and Z (T2, favored and stabilized by intramolecular hydrogen bonding) reaction intermediates, which would diversify the outcome. The Z-isomer T2 would lead to the formation of γ-ylidene-butenolide 3 through lactonization (Scheme 6, Path A). In contrast, the E-isomer 1 would undergo dimerization reaction via MsOH-promoted Michael addition (conjugate addition) of T2 on to 1 to give T3. Subsequent enolization of T3 to form T4 (via TS2) followed by the intramolecular hemiketalization (to give T5) and lactonization cascade delivers fused pyrano-ketal-lactone 2 (Scheme 6, Path B).
Scheme 6. Plausible Reaction Mechanism.

Conclusions
In conclusion, we have identified the first MsOH-promoted divergent access to complex fused pyrano-ketal-lactones by forming three new bonds, two rings, and three contiguous stereocenters, and γ-ylidene-butenolides related to bioactive natural products from readily accessible α,β-unsaturated γ-ketoesters. Cycloalkanone-derived substrates furnished both products (pyrano-ketal-lactones and γ-ylidene-butenolides), whereas substrates containing acyclic ketones delivered exclusively known γ-ylidene-butenolides. Products were unambiguously confirmed by NMR and single-crystal X-ray analyses and analogy. DFT calculations supported the experimental outcome and provided insights into the most probable mechanistic sequences. This protocol provided novel chemical entities with structural and stereochemical complexity in good yields, employing a simple Brønsted acid as a promoter. This protocol provided novel chemical entities with structural and stereochemical complexity, employing a simple Brønsted acid as a promoter. This work may provide solutions for designing unique cascade dimerization reactions and pave the way for building a complex three-dimensional chemical space.
Experimental Section
General Information
All reactions were performed under an argon atmosphere with an oven (80 °C) or flame-dried glassware with a septum seal. Tetrahydrofuran (THF) was distilled from sodium-benzophenone under an argon atmosphere immediately before use. Anhydrous dichloromethane, dichloroethane, methanol, and fluorobenzene were purchased from commercial sources and used without further treatment. Reaction temperatures are reported as the bath temperature surrounding the reaction vessel, and 30 °C corresponded to the laboratory’s room temperature (rt) when the experiments were carried out. Analytical thin-layer chromatography (TLC) was performed on TLC Silica gel 60 F254. Visualization was accomplished with short-wave UV light, anisaldehyde, or KMnO4 staining solutions followed by heating. Chromatography was performed on silica gel (100–200 mesh) by standard techniques eluting with solvents as indicated. 1H and 13C NMR spectra were recorded on Bruker AV 200, 400, and 500 in solvents, as shown. Chemical shifts (δ) are given in ppm. The residual solvent signals were used as references, and the chemical shifts converted to the TMS scale (CDCl3: δH = 7.27 ppm, δC = 77.00 ppm). The following abbreviations were used: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; dd, doublet of doublet; td, triplet doublet; and br, broad. Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. High-resolution mass spectrometry (HRMS) data were recorded on a Thermo Scientific Q-Exactive, Accela 1250 pump, FT-IR instrument (Bruker α Model), at normal temperature with a KBr pellet (IR grade). Experimental procedures for all new and known compounds without published experimental procedures are described below.
Synthesis of α,β-Unsaturated γ-Ketoesters (1) (Method A)16

Ethyl (E)-2-(4-Methyl-2-oxocyclohexylidene)acetate (1c)
3-Methyl cyclohexanone (A, 10.9 g, 97 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture, DABCO (1.31 g, 11 mmol) was added slowly over 10 min at rt, followed by ethyl glyoxalate (1 g, 9 mmol) dropwise. Then, the reaction mixture was stirred overnight at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl 2-hydroxy-2-(4-methyl-2-oxocyclohexyl)acetate (S3c) (0.92 g, 44%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). Compound S3c (0.92 g, 4.29 mmol) was taken in a flame-dried two-neck 100 mL round-bottom flask, dissolved in anhydrous dichloromethane (DCM, 40 mL) under an argon atmosphere, and the mixture was cooled to 0 °C. Then, Et3N (0.72 mL, 5.11 mmol) was added slowly, followed by MeSO2Cl (0.39 mL, 5.1 mmol) dropwise at 0 °C. The reaction mixture was allowed to stir at rt overnight. Then, it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl (E)-2-(4-methyl-2-oxocyclohexylidene)acetate (1c) (0.54 g, 40%), overall yield (17.6%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.47 (br. s., 1H), 4.34–4.05 (m, 2H), 3.60 (d, J = 17.55 Hz, 1H), 2.77–2.48 (m, 2H), 2.12–1.98 (m, 2H), 1.62 (br. s., 2H), 1.32–1.26 (m, 4H), 1.05 (dd, J = 6.10, 3.05 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 201.3, 166.1, 150.7, 122.1, 60.6, 49.1, 31.5, 30.4, 27.5, 21.7, 14.2; HRMS (ESI) m/z calcd for C11H16O3Na [M + Na]+, 219.0992; found, 219.0986.
Ethyl (E)-2-(5-Methyl-2-oxocyclohexylidene)acetate (1d)
4-Methyl cyclohexanone (10.99 g, 97 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture, DABCO (1.31 g, 11.6 mmol) was added slowly over 10 min at rt, followed by ethyl glyoxylate (1 g, 9.8 mmol) dropwise. Then, the reaction mixture was stirred overnight at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl 2-hydroxy-2-(5-methyl-2-oxocyclohexyl)acetate (S3d) (1.33 g, 64%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). Next, S3d (1.33 g, 6.2 mmol) was forwarded to the next step without purification. Anhydrous DCM (40 mL) was added S3d under an argon atmosphere and the mixture was cooled to 0 °C. Then, Et3N (1.04 mL, 7.4 mmol) was added slowly, followed by MeSO2Cl (0.57 mL, 7.4 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at 0 °C to rt. After completion of the reaction, it was quenched with saturated aqueous NH4Cl solution, and extraction with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to obtain ethyl (E)-2-(5-methyl-2-oxocyclohexylidene)acetate (1d) (1.33 g, 40%), overall yield (25.6%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.46–6.43 (m, 1H), 4.19 (q, J = 7.12 Hz, 2H), 3.67–3.48 (m, 1H), 2.68–2.60 (m, 1H), 2.49–2.35 (m, 1H), 2.28 (ddd, J = 16.98, 11.25, 3.05 Hz, 1H), 2.03–1.86 (m, 2H), 1.62–1.50 (m, 1H), 1.33–1.23 (m, 3H), 1.07 (d, J = 6.87 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 201.2, 166.1, 150.6, 122.2, 60.5, 39.7, 36.7, 31.3, 29.9, 21.5, 14.1; HRMS (ESI) m/z calcd for C11H16O3Na [M + Na]+, 219.0992; found, 219.0986.
Ethyl (E)-2-(5-Ethyl-2-oxocyclohexylidene)acetate (1e)
4-Ethyl cyclohexanone (12.36 g, 97 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture, DABCO (1.31 g, 11.6 mmol) was added slowly over 10 min at rt, followed by ethyl glyoxylate (1 g, 9.8 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl 2-(5-ethyl-2-oxocyclohexyl)-2-hydroxyacetate (S3e) (1.2 g, 54%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). This product S3e was forwarded to the next step, S3e (1.2 g, 5.2 mmol) was added to a flame-dried two-neck 100 mL round-bottom flask and dissolved in anhydrous DCM (40 mL) under an argon atmosphere, and the mixture was cooled to 0 °C. Then, Et3N (1.39 mL, 6.2 mmol) was added slowly, followed by MeSO2Cl (0.48 mL, 6.28 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at 0 °C to rt. After completion of the reaction, it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to obtain ethyl (E)-2-(5-ethyl-2-oxocyclohexylidene)acetate (1e) (0.66 g 55%), overall yield (29.7%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.41 (dd, J = 1.53, 3.05 Hz, 1H), 4.16 (q, J = 6.87, 13.73 Hz, 2H), 3.65–3.40 (m, 1H), 2.68–2.49 (m, 1H), 2.43–2.18 (m, 2H), 1.99 (m, 1H), 1.62 (m, 1H), 1.52–1.33 (m, 3H), 1.25 (t, J = 7.63 Hz, 3H), 0.92 (t, J = 7.63 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 201.6, 166.3, 150.9, 122.4, 77.6, 60.7, 39.8, 36.6, 34.7, 28.9, 28.8, 14.01, 13.95, 11.7; HRMS (ESI) m/z calcd for C12H18O3 Na [M + Na]+, 233.1148; found, 233.1141.
Methyl (E)-2-(2-Oxocyclohexylidene)acetate (1f)
Cyclohexanone (13.38 g, 136 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture was added DABCO (1.8 g, 16.04 mmol) at rt, followed by methyl glyoxylate (1.2 g, 11.7 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford methyl 2-hydroxy-2-(2-oxocyclohexyl)acetate (S3f) (1.4 g, 56%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). This product S3f (1.4 g, 7.5 mmol) was forwarded to the next step, dissolved in anhydrous DCM (40 mL) under an argon atmosphere, and the mixture was cooled to 0 °C. Then, Et3N (1.26 mL, 8.9 mmol) was added, followed by MeSO2Cl (0.69 mL, 9 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at rt. After completion of the reaction (1 h), it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford methyl (E)-2-(2-oxocyclohexylidene)acetate (1f) (0.58 g, 38%), overall yield (21.28%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.44–6.17 (m, 1H), 3.67–3.58 (m, 3H), 2.98 (ddd, J = 7.63, 5.34, 2.29 Hz, 2H), 2.48–2.37 (m, 2H), 1.90–1.74 (m, 3H), 1.72–1.60 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 200.8, 166.1, 151.4, 121.3, 51.3, 40.7, 28.5, 23.1; HRMS (ESI) m/z calcd for C9H13O3 [M + H]+, 169.0859; found, 169.0855.
Isopropyl (E)-2-(2-Oxocyclohexylidene)acetate (1g)
Cyclohexanone (16.9 g, 172.2 mmol) was taken in a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture, DABCO (2.31 g, 20.5 mmol) was added at rt, followed by isopropyl glyoxylate (2 g, 17.2 mmol) dropwise. Then, the reaction mixture was stirred overnight at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford isopropyl 2-hydroxy-2-(2-oxocyclohexyl)acetate (S3g) (3 g, 83%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). Isopropyl 2-hydroxy-2-(2-oxocyclohexyl)acetate (S3g) (3 g, 14.0 mmol) was forwarded to the next step, taken in a flame-dried two-neck 100 mL round-bottom flask, dissolved in anhydrous DCM (40 mL) under an argon atmosphere, and the mixture was cooled to 0 °C. Then, Et3N (2.2 mL, 15.8 mmol) was added slowly, followed by MeSO2Cl (1.2 mL, 16.7 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at 0 °C to rt. After completion of the reaction, it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford isopropyl (E)-2-(2-oxocyclohexylidene)acetate (1g) (0.56 g, 47%), overall yield (39.01%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.38 (d, J = 2.29 Hz, 1H), 5.07–4.83 (m, 1H), 3.03 (td, J = 6.29, 1.91 Hz, 3H) 2.51–2.44 (m, 2H), 1.91–1.83 (m, 2H),1.79–1.70 (m, 3H), 1.23–1.22 (m, 4H), 1.2 (d, J = 2.29 Hz, 4H); 13C{1H} NMR (101 MHz, CDCl3): δ 201.5, 165.8, 151.1, 122.8, 77.6, 68.1, 41.1, 28.9, 23.6, 22.0; HRMS (ESI) m/z calcd for C11H16O3Na [M + Na]+, 219.0892; found, 219.0987.
Allyl (E)-2-(2-Oxocyclohexylidene)acetate (1h)
Cyclohexanone (7.8 g, 79 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture, DABCO (1.07 g, 9.5 mmol) was added at rt, followed by allyl 2-oxoacetate (1 g, 8.7 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to allyl 2-hydroxy-2-(2-oxocyclohexyl)acetate (S3h) (0.61 g, 64%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). Allyl 2-hydroxy-2-(2-oxocyclohexyl)acetate (S3h) was forwarded to the next step, taken in a flame-dried two-neck 100 mL round-bottom flask (0.61 g, 2.87 mmol), and dissolved in anhydrous DCM (40 mL) under an argon atmosphere and the mixture was cooled to 0 °C. Then, Et3N (0.48 mL, 3.4 mmol) was added, followed by MeSO2Cl (0.26 mL, 3.4 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at 0 °C to rt. After completion of the reaction, it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford allyl (E)-2-(2-oxocyclohexylidene)acetate (1h) (1.6 g, 59%), overall yield (37.76%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.47 (t, J = 2.29 Hz, 1H), 6.02–5.74 (m, 1H), 5.47–5.26 (m, 1H), 5.22 (d, J = 11.44 Hz, 1H), 4.71–4.41 (m, 2H), 3.21–2.94 (m, 2H), 2.56–2.39 (m, 2H), 1.94–1.81 (m, 2H), 1.80–1.67 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 201.0, 165.6, 151.8, 131.8, 121.6, 118.3, 65.1, 40.9, 28.8, 23.3; HRMS (ESI) m/z calcd for C11H15O3 [M + H]+, 195.2300; found, 195.2301.
Benzyl (E)-2-(2-Oxocyclohexylidene)acetate (1i)
Cyclohexanone (4j) (6.18 g, 62 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture was added DABCO (0.84 g, 7.4 mmol) at rt, followed by benzyl 2-oxoacetate (1 g, 6 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to access benzyl 2-hydroxy-2-(2-oxocyclohexyl)acetate (S3i) (3.17 g, 64%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). The S3i (0.31 g, 1.46 mmol) was forwarded to the next step, dissolved in anhydrous DCM (40 mL) under an argon atmosphere, and the mixture was cooled to 0 °C. Then, Et3N (2.03 mL, 13.8 mmol) was added, followed by MeSO2Cl (1.12 mL, 14.4 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at rt. After completion of the reaction, it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to give benzyl (E)-2-(2-oxocyclohexylidene)acetate (1i) (0.15 g, 53%), overall yield (33.92%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 7.37 (br. s., 5H), 6.54 (br. s., 1H) 5.20, (s, 2H), 3.21–3.04 (m, 2H), 2.53 (t, J = 6.28 Hz, 2H), 2.00–1.72 (m, 4H); 13C{1H} NMR (101 MHz, CDCl3): δ 201.1, 165.8, 152.0, 135.6, 128.5.
Ethyl (E)-2-(4,4-Dimethyl-2-oxocyclohexylidene)acetate (1j)
3,3-Dimethyl cyclohexanone (12.3 g, 97.6 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture was added DABCO (1.31 g, 11.6 mmol) at rt, followed by ethyl glyoxylate (1 g, 9.8 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl 2-(4,4-dimethyl-2-oxocyclohexyl)-2-hydroxyacetate (S3j) (1.07 g, 48%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). Ethyl 2-(4,4-dimethyl-2-oxocyclohexyl)-2-hydroxyacetate (S3j) (1.07 g, 4.6 mmol) was forwarded to the next step, taken in a flame-dried two-neck 100 mL round-bottom flask, dissolved in anhydrous DCM (40 mL) under an argon atmosphere and the mixture was cooled to 0 °C. Then, Et3N (0.78 mL, 5.5 mmol) was added slowly, followed by MeSO2Cl (0.43 mL, 5.5 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at rt. After completion of the reaction, it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl (E)-2-(4,4-dimethyl-2-oxocyclohexylidene)acetate (1j) (0.98 g, 53%), overall yield (25.44%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.46 (t, J = 2.31 Hz, 1H), 4.19 (q, J = 7.13 Hz, 2H), 3.11 (td, J = 6.88, 2.38 Hz, 2H), 2.31 (s, 2H), 1.65 (t, J = 6.88 Hz, 2H) 1.28 (t, J = 7.13 Hz, 3H), 1.01 (s, 6H).13C{1H} NMR (101 MHz, CDCl3): δ 201.4, 166.1, 150.1, 122.1, 60.5, 54.2, 36.0, 32.6, 28.2, 24.7, 14.1; HRMS (ESI) m/z calcd for C12H19O3 [M + H]+, 211.1329; found, 211.1321.
Ethyl (E)-2-(2,4,4-Trimethyl-6-oxocyclohexylidene)acetate (1k)
3,3,5-Trimethyl cyclohexanone (13.7 g, 97.7 mmol) was taken in a single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL) at room temperature. To this mixture was added DABCO (1.31 g, 11.6 mmol) at rt, followed by ethyl glyoxylate (1 g, 9.8 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at rt. After completion of the reaction, neutralization with aqueous 2 N HCl, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl 2-hydroxy-2-(2,4,4-trimethyl-6-oxocyclohexyl)acetate (S3k) (0.9 g, 48%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane). Ethyl 2-hydroxy-2-(2,4,4-trimethyl-6-oxocyclohexyl)acetate (S3k) was forwarded to the next step. Product S3k (0.9 g, 3.7 mmol) was dissolved in anhydrous DCM (40 mL) under an argon atmosphere and the mixture was cooled to 0 °C. Then, Et3N (0.62 mL, 4.4 mmol) was added, followed by MeSO2Cl (0.34 mL, 4.4 mmol) dropwise at 0 °C. The reaction mixture was stirred overnight at rt. After completion of the reaction, it was quenched with saturated aqueous NH4Cl solution and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to obtain ethyl (E)-2-(2,4,4-trimethyl-6-oxocyclohexylidene)acetate (1k) (0.9 g, 91%), overall yield (43.68%), as a yellow oil. TLC: Rf = 0.3 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.44 (d, J = 2.00 Hz, 1H), 4.37–4.08 (m, 2H), 3.90–3.76 (m, 1H), 2.33–2.24 (m, 2H), 2.12 (s, 1H), 1.86 (dd, J = 13.88, 7.75 Hz, 1H), 1.34–1.28 (m, 4H), 1.19 (d, J = 7.00 Hz, 3H), 1.05 (s, 3H), 0.99 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 202.5, 165.9, 155.5, 122.9, 60.6, 52.9, 44.2, 31.3, 31.1, 29.3 27.4, 22.9, 14.2; HRMS (ESI) m/z calcd for C13H21O3 [M + H]+, 225.1329; found, 225.1321.
Synthesis of Open-Chain α,β-Unsaturated γ-Ketoesters (Method B)16

Ethyl (E)-4-Oxopent-2-enoate (1o)
Acetone (1 g, 17.2 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL). To this mixture, para-toluenesulfonic acid (PTSA) (2.9 g, 17.2 mmol) was added at rt, followed by glyoxalate in toluene (1.75 g, 17.2 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at 100 °C. After completion of the reaction, neutralization with aqueous NaHCO3, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl (E)-4-oxopent-2-enoate (1o) (0.64 g, 58%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 7.01 (d, J = 16.13 Hz, 1H), 6.64 (d, J = 16.13 Hz, 1H), 4.27 (q, J = 7.13, 14.26 Hz, 2H), 2.35 (s, 3H), 1.32 (t, J = 7.13 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 197.6, 165.4, 139.9, 131.6, 61.4, 28.1, 14.1; HRMS (ESI) m/z calcd for C7H11O3 [M + H]+, 142.1540; found, 142.1542.
Ethyl (E)-5-Chloro-4-oxopent-2-enoate (1t)
Chloroacetone (1 g, 10.8 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL). To this mixture, PTSA (1.8 g, 10.8 mmol) was added slowly at rt, followed by glyoxalate (1.1 g, 10.8 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at 100 °C. After completion of the reaction, neutralization with aqueous NaHCO3, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl (E)-5-chloro-4-oxopent-2-enoate (1t) (0.3 g, 60%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 7.64–7.46 (m, 2H), 7.10 (d, J = 16.01 Hz, 1H), 4.61–4.51 (m, 5H), 1.61 (t, J = 7.13 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 190.7, 164.8, 135.2, 133.3, 61.6, 47.3, 14.1; HRMS (ESI) m/z calcd for C7H10ClO3 [M + H]+, 176.5960; found, 176.5965.
Ethyl (E)-3-Methyl-4-oxohex-2-enoate (1u)
3-Pentanone (1 g, 11 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL). To this mixture, PTSA (1.89 g, 11 mmol) was added at rt, followed by glyoxalate (1.12 g, 11 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at 100 °C. After completion of the reaction, neutralization with aqueous NaHCO3, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl (E)-3-methyl-4-oxohex-2-enoate (1u) (0.15 g, 52%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 6.52 (s, 1H), 4.21 (q, J = 7.13, 14.26 Hz, 2H), 2.71 (q, J = 7.25, 14.51 Hz, 2H), 2.20 (s, 3H), 1.29 (t, J = 7.25 Hz, 3H), 1.09 (t, J = 7.25 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 202.7, 166.3, 150.4, 125.1, 60.7, 31.4, 14.2, 13.4, 8.1; HRMS (ESI) m/z calcd for C9H14O3 [M + Na]+, 193.0835; found, 193.0830.
Ethyl (E)-3-Methyl-4-oxopent-2-enoate (1v)
Methyl ethyl ketone (1 g, 13.8 mmol) was added to a flame-dried single-neck round-bottom flask (250 mL) followed by anhydrous toluene (10 mL). To this mixture, p-TSA (2.3 g, 13.8 mmol) was added at rt, followed by glyoxalate (1.41 g, 13.8 mmol) dropwise. Then, the reaction mixture was stirred for 24 h at 100 °C. After completion of the reaction, neutralization with aqueous NaHCO3, and extraction with EtOAc (3 × 20 mL), the combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The resulting crude product was purified by silica gel column chromatography (SiO2, 20% EtOAc/hexane) to afford ethyl (E)-3-methyl-4-oxopent-2-enoate (1v) (0.2 g, 41%) as a colorless liquid. TLC: Rf = 0.2 (SiO2, 30% EtOAc/hexane); 1H NMR (400 MHz, CDCl3): δ 5.82 (s, 1H), 3.51–3.39 (m, 1H), 3.20 (dd, J = 7.13, 14.26 Hz, 1H), 1.98 (s, 4H), 1.57 (s, 4H), 1.16 (t, J = 7.1 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 199.6, 161.0, 150.2, 126.3, 60.6, 26.0, 14.0, 12.8.
Synthesis of Open-Chain α,β-Unsaturated γ-Ketoesters (Method C)
Ketoesters 1q and 1r were synthesized in three linear steps (as described below) following
the known literature procedures.20
General Procedure for the Synthesis of Fused Pyrano-ketal-lactones (2) and Bicyclic γ-Ylidene-butenolides (3) from α,β-Unsaturated γ-Ketoesters (1)
Ethyl (E)-2-(2-oxocyclohexylidene)acetate 1 (1.01 mmol) was taken into a single-neck 10 mL round-bottom flask equipped with positive argon flow and dissolved in 2 mL of anhydrous DCE. Then, MsOH (0.101 mmol) was added under an argon atmosphere at room temperature (rt), then the reaction mixture was moved to 60 °C. The resulting reaction mixture was stirred at 60 °C for 4–5 h. After completion of the reaction (monitored by TLC, visualized using UV, anisaldehyde, and KMnO4 staining solutions), it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified using silica gel column chromatography (100–200 mesh) to afford the corresponding complex pyrano-ketal-lactones 2 and bicyclic γ-ylidene-butenolide.
Synthesis of Fused Pyrano-ketal-lactones and Bicyclic γ-Ylidene-butenolides from α,β-Unsaturated γ-Ketoesters16
Ethyl 2-Oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2a) and 5,6-Dihydrobenzofuran-2(4H)-one (3a)
Following the General Procedure, ethyl (E)-2-(2-oxocyclohexylidene)acetate (1a) (0.1 g, 0.5 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.026 g, 0.27 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was heated in an oil bath with temperature 60 °C. The resulting reaction mixture was stirred at 60 °C for 5 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford ethyl 2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2a) (0.042 g, 24%) as a white crystal (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)) and 5,6-dihydrobenzofuran-2(4H)-one (3a) (0.052 g, 70%) as a yellow liquid (TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes)).
Gram-Scale Synthesis of 2a and 3a
Following the General Procedure, ethyl (E)-2-(2-oxocyclohexylidene)acetate (1a) (1 g, 5.4 mmol) treated with MsOH (0.263 g, 2.7 mmol) in anhydrous ClCH2CH2Cl (10 mL) to obtain 2a with a yield of 22% (0.384 g) and 3a with a yield of 70% (0.523 g).
2a
1H NMR (CDCl3, 400 MHz): δ 5.75 (d, J = 1.53 Hz, 1H), 4.30–4.09 (m, 2H), 2.79–2.71 (m, 1H), 2.57 (s, 1H), 2.49–2.29 (m, 3H), 2.18–1.98 (m, 3H), 1.84–1.64 (m, 6H), 1.56–1.46 (m, 2H), 1.28 (m, 3H); 13C{1H} NMR (CDCl3, 101 MHz): δ 171.9, 169.7, 167.3, 144.5, 114.7, 102.5, 101.9, 61.2, 45.2, 42.2, 28.6, 28.5, 26.6, 26.2, 25.6, 22.8, 22.6, 14.1; IR (KBr, cm–1): υ 3685, 3022, 1768, 1596, 1575, 1217, 1020, 921, 767, 670; HRMS (ESI) m/z calcd for C18H22O5Na [M + Na]+, 341.1359; found, 341.1350.
3a
1H NMR (400 MHz, CDCl3): δ 5.87 (t, J = 4.63 Hz, 1H), 5.75 (s, 1H), 2.71 (t, J = 6.63, Hz, 2H), 2.39 (q, J = 5.50, 10.76 Hz, 2H), 1.86 (quin, J = 6.00, 12.13 Hz, 2H); 13C{1H} (101 MHz, CDCl3): δ 170.1, 155.7, 150.3, 111.0, 24.2, 23.7, 22.6; IR (KBr, cm–1): υ 3685, 3022, 1720, 1595, 1520, 1216, 927, 768, 671; HRMS (ESI) m/z calcd for C8H8O2 [M + H]+, 137.0597; found, 137.0592.
Ethyl 6a,11-Dimethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2b) and 7-Methyl-5,6-dihydrobenzofuran-2(4H)-one (3b)
Following the General Procedure, to ethyl (E)-2-(3-methyl-2-oxocyclohexylidene)acetate (1b) (0.1 g, 1 mmol) in 2 mL of anhydrous DCE, MsOH (0.048 g, 0.49 mmol) was added under an argon atmosphere at room temperature (rt) and then the reaction mixture moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 5 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford ethyl 6a,11-dimethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2b) as a colorless liquid (0.039 g, 26%) in d.r. ratio 2:1 (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)) and 7-methyl-5,6-dihydrobenzofuran-2(4H)-one (3b) (0.032 g, 41%) as a yellow liquid (TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes)).
2b
1H NMR (400 MHz, CDCl3): δ 5.75 (t, J = 2.29 Hz, 1H), 5.31 (minor) (s, 0.23H), 4.31–4.21 (minor) (m, 1H), 4.11–4.05 (m, 2H), 2.83–2.76 (m, 2H), 2.73–2.65 (m, 2H), 2.58–2.40 (m, 3H), 2.30–2.17 (m, 3H), 1.97–1.76 (m, 7H), 1.74–1.61 (m, 4H), 1.35–1.28 (m, 3H), 1.27–1.24 (m, 4H), 1.24–1.21 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 172.1, (minor) 172.0, 171.9, 169.9, (minor) 169.7, 169.7, (minor) 167.0, 165.6, (minor) 144.3, 144.2, (minor) 144.1, (minor) 116.9, 116.8, (minor) 114.6, 102.6, (minor) 102.5, (minor) 102.4, 102.2, 101.1, (minor) 101.0, (minor) 100.9, 61.2, 45.7, (minor) 45.6, 44.4, (minor) 44.3, 41.4, (minor) 41.1, 37.3, 37.1, (minor) 37.1, 36.8, (minor) 36.7, 34.4, (minor) 34.2, 34.2, 33.4, 32.3, 30.9, 30.5, 29.7, 28.8, (minor) 28.7, 28.7, (minor) 26.6, (minor) 26.3, 21.5, (minor) 21.4, 20.8, 18.2, 14.1; IR (KBr, cm–1): υ 3685, 3022, 2940, 2403, 1732, 1673, 1519, 1217, 1030, 921, 769, 671; HRMS (ESI) m/z calcd for C20H26O5Na [M + Na]+, 369.4231; found, 369.4226.
3b
1H NMR (400 MHz, CDCl3): δ 5.70 (s, 1H), 2.72–2.64 (m, 2H), 2.33–2.30 (m, 2H), 1.97 (s, 3H), 1.90–1.84 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 175.8, 170.5, 155.5, 123.0, 109.3, 30.0, 24.1, 22.6, 16.9; IR (KBr, cm–1): υ 3685, 3022, 2928, 2402, 1760, 1600, 1520, 1216, 923, 772, 672; HRMS (ESI) m/z calcd for C9H11O2 [M + H]+, 151.0754; found, 151.0751.
Ethyl 4,10-Dimethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2c) and 6-Methyl-5,6-dihydrobenzofuran-2(4H)-one (3c)
Following the General Procedure, ethyl (E)-2-(4-methyl-2-oxocyclohexylidene)acetate (1c) (0.2 g, 1.01 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.048 g, 0.49 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 5 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford ethyl 4,10-dimethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2c) (0.072 g, 20%) as a colorless liquid in d.r. ratio 1:1 (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)) and 6-methyl-5,6-dihydrobenzofuran-2(4H)-one (3c) (0.066 g, 46%) as a yellow liquid (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)).
2c
1H NMR (400 MHz, CDCl3): δ 5.75 (d, J = 1.53 Hz, 1H), 4.31–4.21 (m, 1H), 4.19–4.11 (m, 1H), 2.89 (s, 1H), 2.76–2.71 (m, 1H),2.60 (s, 1H), 2.51–2.10 (m, 1H), 2.20–2.10 (m, 2H), 2.05–1.96 (m, 2H), 1.85–1.58 (m, 6H), 1.27 (d, J = 6.87 Hz, 6H), 1.01–0.98(m, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 172.5, 172.4, 169.7, 167.3, 144.3, 144.2, 114.2, 102.6, 102.5, 101.3, 61.2, 48.3, 48.0, 41.9, 40.7, 34.8, 34.7, 34.7, 31.2, 31.1, 30.8, 30.7, 29.7, 29.3, 28.7, 28.5, 28.2, 25.8, 21.4, 21.2, 18.4, 14.1; IR (KBr, cm–1): υ 3021, 2926, 2403, 1765, 1449, 1377, 1217, 927, 767, 762, 669; HRMS (ESI) m/z calcd for C20H26O5Na [M + Na]+, 369.1785; found, 369.1780.
3c
1H NMR (400 MHz, CDCl3): δ 5.75 (s, 1H), 5.73 (br. s., 1H), 2.87–2.80 (m, 1H), 2.62–2.58 (m, 1H), 1.98 (dd, J = 8.85, 13.38 Hz, 1H), 1.51–1.44 (m, 1H), 1.28 (d, J = 10.38 Hz, 1H), 1.15 (d, J = 7.0 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.2, 155.5, 150.0, 116.6, 110.9, 31.0, 29.8, 29.7, 23.1, 201.0; IR (KBr, cm–1): υ 3383, 3023, 2932, 1720, 1648, 1520, 1216, 767, 671; HRMS (ESI) m/z calcd for C9H11O2 [M + H]+, 151.0754; found, 151.0751.
Ethyl 5,9-Dimethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2d) and 5-Methyl-5,6-dihydrobenzofuran-2(4H)-one (3d)
Following the General Procedure, ethyl (E)-2-(5-methyl-2-oxocyclohexylidene)acetate (1d) (0.2 g, 1.01 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.048 g, 0.101 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 5 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford ethyl 5,9-dimethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2d) (0.068 g, 19%) as a colorless liquid in d.r. ratio 1:1 (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)) and 5-methyl-5,6-dihydrobenzofuran-2(4H)-one (3d) (0.071 g, 46%) as a yellow liquid (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)).
2d
1H NMR (400 MHz, CDCl3): δ 5.82 (d, J = 1.53 Hz, 1H), 5.74 (d, J = 1.53 Hz, 0.31H), 4.26–4.14 (m, 3H), 2.66–2.60 (m, 2H), 2.49–2.45 (m, 2H), 2.41–2.34 (m, 2H), 2.22–2.03 (m, 4), 1.88–1.69 (m, 6H), 1.53–1.45 (m, 2H), 1.32–1.24 (m, 8H), 1.02–1.00 (m, 3H), 0.99–0.98 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 172.1, 172.0, 169.7, 169.7, 165.6, 144.3, 144.2, 116.9, 116.8, 102.6, 102.5, 101.0, 100.9, 61.2, 45.6, 44.3, 37.3, 37.1, 37.1, 36.8, 36.7, 34.4, 34.2, 34.2, 33.4, 32.3, 30.9, 30.5, 28.8, 28.7, 28.7, 26.6, 26.3, 21.5, 21.4, 20.8, 18.2, 14.1; IR (KBr, cm–1): υ 3687, 3022, 2402, 1727, 1520, 1424, 1215, 92, 764, 671; HRMS (ESI) m/z calcd for C20H26O5Na [M + Na]+, 369.1621; found, 369.1616.
3d
1H NMR (400 MHz, CDCl3): δ 5.82–5.76 (m, 1H), 5.72 (s, 1H), 2.83–2.78 (m, 1H), 2.47–2.34 (m, 1H), 2.27 (dd, J = 16.78, 9.92 Hz, 2H), 2.10–1.98 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.2, 155.8, 150.0, 110.8, 110.0, 32.0, 31.8, 30.1 20.7; IR (KBr, cm–1): υ 3687, 3022, 2402, 1769, 1595, 1520, 1215, 927, 772, 672; HRMS (ESI) m/z calcd for C9H10O2 [M + H]+, 151.0754; found, 151.0751.
5-Ethyl 5,9-Diethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2e) and 5-Ethyl-5,6-dihydrobenzofuran-2(4H)-one (3e)
Following the General Procedure, ethyl (E)-2-(5-ethyl-2-oxocyclohexylidene)acetate (1e) (0.2 g, 0.95 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.045 g, 0.46 mmol) was added under an argon atmosphere at room temperature (rt) and then the reaction mixture moved v. The resulting reaction mixture was stirred at 60 °C for 6 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure, and purification by column chromatography (SiO2, 5% EtOAc/hexanes) afforded 5-ethyl 5,9-dimethyl-2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2e) (0.085 g, 24%) as a colorless liquid in d.r. ratio 1:1 (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)) and 5-ethyl-5,6-dihydrobenzofuran-2(4H)-one (3e) (0.091 g, 57%) as a yellow liquid (TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes)).
2e
1H NMR (400 MHz, CDCl3): δ 5.82 (d, J = 1.53 Hz, 1H), 5.74 (d, J = 1.53 Hz, 0.33H), 4.26–4.14 (m, 3H), 2.66–2.60 (m, 2H), 2.51–2.44 (m, 3H), 2.42–2.34 (m, 2H), 2.25–2.2.00 (m, 6H), 1.86–1.67 (m, 8H), 1.54–1.45 (m, 3H), 1.32–1.20 (m, 10H), 1.02–0.96 (m, 10H); 13C{1H} NMR (101 MHz, CDCl3): δ 172.1, 172.0, 169.7, 169.7, 165.6, 144.3, 144.2, 116.9, 116.8, 102.6, 102.5, 101.0, 100.1, 61.2, 45.6, 44.3, 37.3, 37.1, 36.8, 36.7, 34.4, 34.2, 34.2, 33.4, 32.4, 31.0, 30.5, 29.7, 28.8, 28.7, 28.7, 26.6, 26.3, 21.5, 21.4, 20.8, 18.2, 14.1; IR (KBr, cm–1): υ 3686, 3022, 2402, 1746, 1520, 1425, 1216, 927, 768, 671; HRMS (ESI) m/z calcd for C22H31O5 [M + H]+, 375.2155; found, 375.2161.
3e
1H NMR (400 MHz, CDCl3): δ 5.85–5.80 (m, 1H), 5.74 (s, 1H), 2.87 (dd, J = 3.81, 16.78 Hz, 1H), 2.47 (m, 1H), 2.27 (ddd, J = 3.05, 5.34, 16.78 Hz, 1H), 1.90–1.78 (m, 1H), 1.89–1.78 (m, 1H), 1.45–1.39 (m, 2H), 0.95 (t, J = 7.63 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3): δ 170.3, 155.9, 150.3, 111.1, 110.1, 36.9, 30.0, 298, 28.2, 11.3; IR (KBr, cm–1): υ 3687, 3022, 2929, 2402, 1755, 1519, 1427, 1216, 927, 768, 671; HRMS (ESI) m/z calcd for C10H13O2 [M + H]+, 165.0910; found, 165.0910.
Methyl 2-Oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2f) and 5,6-Dihydrobenzofuran-2(4H)-one (3a)
Following the General Procedure, methyl (E)-2-(2-oxocyclohexylidene)acetate (1f) (0.2 g, 1.19 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.057 g, 0.59 mmol) was added under an argon atmosphere at room temperature (rt) and then the reaction mixture moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 5 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford methyl 2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2f) (0.089 g, 25%) as a colorless liquid. TLC: Rf = 0.50 (SiO2, 15% EtOAc/hexanes). 2f was confirmed by 1H, 13C, DEPT, HRMS, and X-ray analysis and 5,6-dihydrobenzofuran-2(4H)-one (3a) (0.085 g, 52%) was obtained as a colorless liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
2f
1H NMR (400 MHz, CDCl3): δ 5.77 (d, J = 2.29 Hz, 1H), 3.74 (s, 3H), 2.80–2.71 (m, 1H), 2.60 (s, 1H), 2.49–2.35 (m, 3H), 2.15–2.11 (m, 1H), 2.08–2.01 (m, 2H), 1.85–1.78(m, 2H), 1.70–1.62 (m, 5H), 1.55–1.47 (m, 1H); 13C{1H} NMR (101 MHz, CDCl3): δ 171.7, 169.7, 167.3, 144.6, 114.6, 102.4, 101.7, 52.4, 45.1, 42.3, 28.6, 28.5, 26.7, 26.2, 25.6, 22.8, 22.6; IR (KBr, cm–1): υ 3685, 3022, 2403, 1731, 1519, 1439, 1216, 1028, 768, 671; HRMS (ESI) m/z calcd for C17H20O5Na [M + Na]+, 327.1203; found, 327.1198.
Isopropyl 2-Oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2g) and 5,6-Dihydrobenzofuran-2(4H)-one (3a)
Following the General Procedure, isopropyl (E)-2-(2-oxocyclohexylidene)acetate (1g) (0.2 g, 1.02 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.049 g, 0.50 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 4 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford isopropyl 2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2g) (0.095 g, 28%) as a colorless liquid (TLC: Rf = 0.50 (SiO2, 15% EtOAc/hexanes)) and 5,6-dihydrobenzofuran-2(4H)-one (3a) (0.091 g, 65%) as a yellow liquid (TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes)).
2g
1H NMR (400 MHz, CDCl3): δ 5.75 (d, J = 2.29 Hz, 1H), 5.11–5.03 (m, 1H), 2.81–2.69 (m, 1H), 2.54 (s, 1H), 2.45–2.38 (m, 2H), 2.15–2.09 (m, 1H), 2.06–1.99 (m, 2H), 1.83–1.72 (m, 3H), 1.69–1.63 (m, 4H), 1.59–1.46 (m, 2H), 1.29–1.24 (m, 6H); 13C{1H} NMR (101 MHz, CDCl3): δ 171.4, 169.7, 167.3, 144.4, 114.7, 102.5, 102.1, 68.7, 45.3, 42.3, 28.6, 28.5, 26.7, 26.2, 25.6, 22.8, 22.6, 21,64, 21.59; IR (KBr, cm–1): υ 3685, 3022, 2403, 2403, 1770, 1611, 1519, 1217, 965, 768, 670; HRMS (ESI) m/z calcd for C19H24O5Na [M + Na]+, 355.1506; found, 355.1501.
Allyl 2-Oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2h) and 5,6-Dihydrobenzofuran-2(4H)-one (3a)
Following the General Procedure, allyl (E)-2-(2-oxocyclohexylidene)acetate (1h) (0.150 g, 0.77 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.037 g, 0.38 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 4 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford allyl 2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2h) (0.069 g, 27%) as a colorless liquid (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)) and 5,6-dihydrobenzofuran-2(4H)-one (3a) (0.051 g, 48%) as a yellow liquid (TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes)).
2h
1H NMR (400 MHz, CDCl3): δ 5.99–5.82 (m, 1H), 5.47–5.19 (m, 2H), 4.78–4.54 (m, 2H), 3.04 (s, 1H), 2.52–2.27 (m, 2H), 2.13 (br. s., 3H), 1.86–1.59 (m, 8H), 1.31–1.22 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 171.6, 169.7, 167.2, 144.7, 132.2,118.3, 114.7, 102.4, 101.7, 65.9, 45.2, 42.2, 28.6, 28.5, 26.7, 26.2, 25.6, 22.8, 22.7, 14.1; IR (KBr, cm–1): υ 3687, 3022, 2402, 1764, 1596, 1520, 1220, 1055, 926, 768, 671; HRMS (ESI) m/z calcd for C19H22O5Na [M + Na]+, 353.1359; found, 353.1343.
Benzyl 2-Oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2i) and 5,6-Dihydrobenzofuran-2(4H)-one (3a)
Following the General Procedure, benzyl (E)-2-(2-oxocyclohexylidene)acetate (1i) (0.5 g, 2.04 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.098 g, 0.101 mmol) was added under an argon atmosphere at room temperature (rt) and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 4 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford benzyl 2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2i) (0.2 g, 21%) as a colorless liquid. TLC: Rf = 0.50 (SiO2, 15% EtOAc/hexanes). 2i was confirmed by 1H, 13C, DEPT, HRMS, and X-ray analysis, and 5,6-dihydrobenzofuran-2(4H)-one (3a) (0.230 g, 78%) was obtained as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
2i
1H NMR (400 MHz, CDCl3): δ 7.38–7.35 (m, 5H), 5.77 (d, J = 2.00 Hz, 1H), 5.27 (d, J = 12.26 Hz, 1H), 5.11 (d, J = 12.38 Hz, 1H), 2.78–2.72 (m, 1H), 2.64 (s, 1H), 2.51–2.33 (m, 4H), 2.13–1.99 (m, 4H), 1.85–1.78 (m, 2H), 1.70–1.63 (m, 4H); 13C{1H} NMR (101 MHz, CDCl3): δ 171.7, 169.6, 167.2, 144.7, 135.9,128.5, 128.2, 128.1 114.7, 102.4, 101.7, 67.0, 45.2, 42.1, 28.6, 28.5, 26.7, 26.2, 25.5, 22.8, 22.6; IR (KBr, cm–1): υ 3687, 3022, 2402, 1764, 1594, 1519, 1216, 928, 765, 669.
Ethyl 2-Oxo-2,4,5,6,7,7a,8,9,10,11,12,13-dodecahydrocyclohepta[b]furo[2′,3′:2,3]cyclohepta[1,2-e]pyran-8-carboxylate (2m) and 4,5,6,7-Tetrahydro-2H-cyclohepta[b]furan-2-one (3m)
Following the General Procedure, ethyl (E)-2-(4-methyl-2-oxocycloheptylidene)acetate (1m) (0.2 g, 1.01 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.048 g, 0.49 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 5 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 1% EtOAc/hexanes) to give ethyl 2-oxo-2,4,5,6,7,7a,8,9,10,11,12,13-dodecahydrocyclohepta[b]furo[2′,3′:2,3]cyclohepta[1,2-e]pyran-8-carboxylate (2m) (0.093 g, 26%) as a colorless liquid (TLC: Rf = 0.90 (SiO2, 10% EtOAc/hexanes)) and 4,5,6,7-tetrahydro-2H-cyclohepta[b]furan-2-one (3m) (0.091 g, 59%) as a yellow liquid (TLC: Rf = 0.90 (SiO2, 10% EtOAc/hexanes)).
2m
1H NMR (500 MHz, CDCl3): δ 5.84 (s, 1H), 3.59–3.50 (m, 1H), 3.36–3.27 (m, 1H), 2.79–2.67 (m, 1H), 2.58–2.45 (m, 1H), 2.39–2.26 (m, 1H), 1.87–1.61 (m, 12H), 1.50–1.38 (m, 2H), 1.28–1.20 (m, 6H); 13C{1H} NMR (101 MHz, CDCl3): δ 171.9, 169.7, 167.3, 144.5, 114.7, 102.5, 101.9, 61.2, 45.2, 42.2, 31.9, 29.7, 28.6, 28.5, 26.7, 26.2, 25.6, 22.8, 22.7, 14.1; IR (KBr, cm–1): υ 3686, 3022, 2927, 2402, 1747, 1632, 1520, 1215, 1022, 925, 770, 671; HRMS (ESI) m/z calcd for C20H26O5Na [M + Na]+, 369.1672; found, 369.1661.
3m
1H NMR (400 MHz, CDCl3): δ 5.98–5.95 (m, 1H), 5.88–5.87 (m, 1H), 2.78–2.75 (m, 2H), 2.44 (d, J = 5.75, 11.26 Hz, 2H), 1.82–1.75 (m, 4H); 13C{1H} NMR (101 MHz, CDCl3): δ 169.6, 158.6, 150.2, 117.8, 116.8, 29.5, 28.0, 27.5, 24.6; IR (KBr, cm-1): υ 3687, 3023, 2402, 1770, 1597, 1520, 1216, 928, 765, 671; HRMS (ESI) m/z calcd for C9H11O2 [M + H]+, 151.0754; found, 151.0750.
Ethyl 2-Oxo-4,5,6,7,7a,8,9,10,11,12,13,14-dodecahydro-2H-cycloocta[b]furo[2′,3′:2,3]cycloocta[1,2-e]pyran-9-carboxylate (2n)
Following the General Procedure, ethyl (E)-2-(4-methyl-2-oxocycloheptylidene)acetate (1n)(0.2 g, 1.01 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.048 g, 0.49 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 5 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 1% EtOAc/hexanes) to afford ethyl 2-oxo-4,5,6,7,7a,8,9,10,11,12,13,14-dodecahydro-2H-cycloocta[b]furo[2′,3′:2,3]cycloocta[1,2-e]pyran-9-carboxylate (2n) (0.108 g, 32%) as a colorless liquid in d.r. ratio 2:1. TLC: Rf = 0.90 (SiO2, 10% EtOAc/hexanes).
2n
1H NMR (500 MHz, CDCl3): δ 5.96 (s, 1H), 3.51–3.45 (m, 1H), 3.25–3.20 (m, 1H), 2.88–2.85 (m, 1H), 2.66–2.49 (m, 3H), 2.41–2.35 (m, 1H), 2.25–2.09 (m, 3H), 1.85–1.79 (m, 2H), 1.78–1.73 (m, 2H), 1.69–1.61 (m, 6H), 1.57–1.51 (m, 3H), 1.48–1.38 (m, 3H), 1.31–1.25 (m, 2H), 1.19 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.2, 170.0, 169.8, 157.7, 153.6, 121.0, 117.5, 112.0, 111.4, 58.7, 34.4, 28.0, 27.4, 26.9, 26.8, 25.8, 25.0, 24.8, 23.3, 21.6, 20.7, 15.1; IR (KBr, cm–1): υ 3686, 3022, 2927, 2402, 1747, 1632, 1520, 1215, 1022, 925, 770, 671; HRMS (ESI) m/z calcd for C21H29O5 [M + H]+, 375.2166; found, 375.2159.
Synthesis of γ-Ylidene-butenolides from α,β-Unsaturated γ-Ketoesters16
6,6-Dimethyl-5,6-dihydrobenzofuran-2(4H)-one (3j)
Following the General Procedure, ethyl (E)-2-(4,4-dimethyl-2-oxocyclohexylidene)acetate (1j) (0.2 g, 0.95 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.045 g, 0.46 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 4 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford 6,6-dimethyl-5,6-dihydrobenzofuran-2(4H)-one (3j) (0.131 g, 83%) as a yellow liquid. TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 5.79–5.72 (m, 1H), 5.65 (d, J = 1.53 Hz, 1H), 2.80–2.70 (m, 2H), 1.69 (t, J = 6.48 Hz, 2H), 1.16 (s, 6H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.3, 155.0, 148.9, 120.4, 111.0, 36.5, 32.9, 28.6, 21.2; IR (KBr, cm–1): υ 3687, 3022, 2929, 2402, 1727, 1598, 1520, 1216, 912, 769, 671; HRMS (ESI) m/z calcd for C10H13O2 [M + H]+, 165.0910; found, 165.0904.
4,6,6-Trimethyl-5,6-dihydrobenzofuran-2(4H)-one (3k)
Following the General Procedure, ethyl (E)-2-(2,4,4-trimethyl-6-oxocyclohexylidene)acetate (1k) (0.2 g, 0.89 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.042 g, 0.43 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 4 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford 4,6,6-trimethyl-5,6-dihydrobenzofuran-2(4H)-one (3k) (0.12 g, 75%) as a yellow liquid. TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 5.78 (t, J = 2.20 Hz, 1H), 5.69–5.61 (m, 1H), 2.89 (m, 1H), 1.71–1.63 (m, 1H), 1.44 (t, J = 13.13 Hz, 1H), 1.29 (d, J = 6.63 Hz, 3H), 1.18 (s, 3H), 1.15 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.2, 160.5, 148.8, 120.1, 110.1, 45.9, 33.3, 31.1, 27.4, 26.6, 18.2; IR (KBr, cm–1): υ 3686, 3022, 2926, 2402, 1729, 1599, 1521, 1216, 925, 768, 671; HRMS (ESI) m/z calcd for C11H14O2Na [M + Na]+, 201.0886; found, 201.0879.
Ethyl 2-(5-Oxocyclopent-1-en-1-yl)acetate (4a)
Following the General Procedure, ethyl (E)-2-(2-oxocycloheptylidene)acetate (1l) (0.25 g, 1.48 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.057 g, 0.59 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 4 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 1% EtOAc/hexanes) to afford ethyl 2-(5-oxocyclopent-1-en-1-yl)acetate (4a) (0.15 g, 60%) as a yellow liquid. TLC: Rf = 0.90 (SiO2, 10% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 6.26 (s, 1H), 4.16–3.96 (m, 2H), 3.00–2.86 (m, 2H), 2.30–2.17 (m, 2H), 1.91–1.78 (m, 2H), 1.19–1.09 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3): δ 206.7, 165.8, 150.5, 119.0, 60.7, 37.4, 28.9, 19.1, 13.7; IR (KBr, cm–1): υ 3636, 2986, 2014, 1749, 1451, 1373, 1226, 1047, 930, 847, 670; HRMS (ESI) m/z calcd for C9H13O3 [M + H]+, 169.0859; found, 169.0855.
5-Methylenefuran-2(5H)-one (3o)
Following the General Procedure, ethyl (E)-4-oxopent-2-enoate (1o) (0.1 g, 0.7 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.033 g, 0.35 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) affording 5-methylenefuran-2(5H)-one (3o) (0.045 g, 67%) as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
3o
1H NMR (500 MHz, CDCl3): δ 7.41 (d, J = 5.72 Hz, 1H), 6.29–6.25 (m, 1H), 5.25 (t, J = 2.10 Hz, 1H), 4.93 (d, J = 2.67 Hz, 1H); 13C{1H} NMR (126 MHz, CDCl3): δ 169.8, 154.9, 143.4, 128.3, 121.7, 98.1; IR (KBr, cm–1): υ 3682, 3022, 2933, 1746, 1546, 1514, 1216, 908, 767, 668; HRMS (ESI) m/z calcd for C5H5O2 [M + H]+, 97.0850; found, 97.0851.
5-(Propan-2-ylidene)furan-2(5H)-one (3p)
Following the General Procedure, ethyl (E)-5-methyl-4-oxohex-2-enoate (1p) (0.1 g, 0.58 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.028 g, 0.29 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) affording 5-(propan-2-ylidene)furan-2(5H)-one (3p) (0.058 g, 98%) as a white crystal. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
Gram-Scale Synthesis of 3p
Following the General Procedure, ethyl (E)-5-methyl-4-oxohex-2-enoate (1a)(1 g, 5.87 mmol) was treated with MsOH (0.282 g, 2.93 mmol) in anhydrous ClCH2CH2Cl (10 mL) to obtain 3p in 90% yield (0.656 g).
3p
1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 5.50 Hz, 1H), 6.10 (d, J = 5.50 Hz, 1H), 2.02 (s, 3H), 1.97–1.94 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3): 170.6, 146.1, 139.7, 123.6, 117.9, 18.70, 18.66; IR (KBr, cm–1): υ 3682, 3022, 2933, 1746, 1546, 1514, 1216, 908, 767, 668; HRMS (ESI) m/z calcd for C7H9O2 [M + H]+, 125.0597; found, 125.0594.
5-Cyclobutylidenefuran-2(5H)-one (3q)
Following the General Procedure, ethyl (E)-4-cyclohexyl-4-oxobut-2-enoate (1q) (0.1 g, 0.4 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.022 g, 0.2 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. Purification of the crude product by column chromatography (SiO2, 5% EtOAc/hexanes) afforded 5-cyclobutylidenefuran-2(5H)-one (3q) (0.085 g, 94%) as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.38 (d, J = 5.38 Hz, 1H), 6.07 (d, J = 5.38 Hz, 1H), 3.02–2.93 (m, 4H), 2.25–2.17 (m, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.5, 143.1, 139.8, 132.3, 117.2, 29.1, 27.7, 17.7; IR (KBr, cm–1): υ 3682, 3022, 2933, 1746, 1546, 1514, 1216, 908, 767, 668; HRMS (ESI) m/z calcd for C8H9O2 [M + H]+, 137.1499; found, 137. 1496.
5-Cyclopentylidenefuran-2(5H)-one (3r)
Following the General Procedure, ethyl (E)-4-cyclohexyl-4-oxobut-2-enoate (1r) (0.1 g, 0.4 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.022 g, 0.2 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford 5-cyclopentylidenefuran-2(5H)-one (3r) (0.078 g, 95%) as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes); 1H NMR (400 MHz, CDCl3): δ 7.49 (d, J = 5.38 Hz, 1H), 6.07 (d, J = 5.38 Hz, 1H), 2.64 (m, Hz, 2H), 2.58–2.52 (m, 2H), 1.83–1.78 (m, 4H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.6, 143.3, 140.8, 134.8, 117.2, 30.4, 29.6, 26.6, 25.9; IR (KBr, cm–1): υ 3682, 3022, 2933, 1746, 1546, 1514, 1216, 908, 767, 668; HRMS (ESI) m/z calcd for C9H11O2 [M + H]+, 150.1775; found, 150.1762.
5-Cyclohexylidenefuran-2(5H)-one (3s)
Following the General Procedure, ethyl (E)-4-cyclohexyl-4-oxobut-2-enoate (1s) (0.1 g, 0.4 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.022 g, 0.2 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to obtain 5-cyclohexylidenefuran-2(5H)-one (3s) (0.048 g, 61%) as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
3s
1H NMR (400 MHz, CDCl3): δ 7.62 (d, J = 5.50 Hz, 1H), 6.11 (d, J = 5.50 Hz, 1H), 2.02 (s, 3H), 2.52 (m, 2H), 2.39–2.31 (m, 2H), 1.66–1.64 (m, 2H), 1.25 (s, 2H); 13C{1H} NMR (101 MHz, CDCl3): δ 170.7, 143.5, 139.4, 131.7, 117.9, 28.8, 28.1, 27.1, 26.1; IR (KBr, cm–1): υ 3682, 3022, 2933, 1746, 1546, 1514, 1216, 908, 767, 668; HRMS (ESI) m/z calcd for C10H12O2Na [M + Na]+, 187.0730; found, 187.0722.
(Z)-5-(Chloromethylene)furan-2(5H)-one (3t)
Following the General Procedure, ethyl (E)-5-chloro-4-oxopent-2-enoate (1t) (0.1 g, 0.5 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.027 g, 0.28 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford (Z)-5-(chloromethylene) furan-2(5H)-one (3t) (0.055 g, 75%) as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
3t
1H NMR (500 MHz, CDCl3): δ 7.42 (d, J = 5.50 Hz, 1H), 6.29, (dd, J = 5.50, 0.75 Hz, 1H), 5.99 (s, 1H); 13C{1H} NMR (126 MHz, CDCl3): δ 172.9, 141.6, 120.3, 104.1; IR (KBr, cm–1): υ 3687, 3022, 2923, 2402, 1595, 1520, 1215, 920, 766, 671; HRMS (ESI) m/z calcd for C5H4ClO2 [M + H]+, 131.5270; found, 131.5267.
(Z)-5-Ethylidene-4-methylfuran-2(5H)-one (3u)
Following the General Procedure, ethyl (E)-3-methyl-4-oxohex-2-enoate (1u) (0.1 g, 0.58 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.028 g, 0.29 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure, and purification by column chromatography (SiO2, 5% EtOAc/hexanes) afforded (Z)-5-ethylidene-4-methylfuran-2(5H)-one (3u) (0.064 g, 89%) as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
3u
1H NMR (500 MHz, CDCl3): δ 5.87 (s, 1H), 5.35 (q, J = 7.32, 14.65 Hz, 1H), 2.13 (d, J = 0.92 Hz, m, 3H), 1.92 (d, J = 7.32 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3): δ 169.5, 154.5, 151.4, 115.8, 107.9, 11.64, 11.60; IR (KBr, cm–1): υ 3686, 3022, 2924, 2402, 1758, 1605, 1521, 1427, 1215, 1022, 926, 769, 672; HRMS (ESI) m/z calcd for C7H9O2 [M + H]+, 125.0597; found, 125.0595.
5-Ethoxy-4,5-dimethylfuran-2(5H)-one (3v)
Following the General Procedure, ethyl (E)-3-methyl-4-oxopent-2-enoate (1v) (0.1 g, 0.64 mmol) was dissolved in 2 mL of anhydrous DCE. MsOH (0.030 g, 0.32 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was moved to a 60 °C oil bath. The resulting reaction mixture was stirred at 60 °C for 3 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, extracted with CH2Cl2 (2 × 5 mL), and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford 5-ethoxy-4,5-dimethylfuran-2(5H)-one (3v) (0.065 g, 65%) as a yellow liquid. TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes).
3v
1H NMR (500 MHz, CDCl3): δ 5.86 (q, J = 1.58 Hz, 1H), 3.49 (dd, J = 9.01, 7.13 Hz, 1H), 3.23 (dd, J = 8.88, 7.00 Hz, 1H), 2.01 (d, J = 1.63 Hz, 3H), 1.60 (s, 3H), 1.20 (t, J = 7.07 Hz, 3H); 13C{1H} NMR (126 MHz, CDCl3): δ 169.9, 166.5, 118.8, 109.1, 77.3, 76.7, 58.9, 29.7, 22.8, 15.1, 12.5; IR (KBr, cm-1): υ 3687, 3022, 2924, 2402, 1595, 1521, 1426, 1215, 1023, 924, 770, 671; HRMS (ESI) m/z calcd for C8H13O3 [M + H]+, 156.1811; found, 156.1810.
Ethyl 2-Oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2a), 5,6-Dihydrobenzofuran-2(4H)-one (3a), and 7-Methyl-5,6-dihydrobenzofuran-2(4H)-one (3b)
Following the General Procedure, ethyl (E)-2-(2-oxocyclohexylidene)acetate (1a) (0.1 g, 0.54 mmol) and 7-methyl-5,6-dihydrobenzofuran-2(4H)-one (3b) (0.082 g, 0.54 mmol) were dissolved in 2 mL of anhydrous DCE. MsOH (0.026 g, 0.27 mmol) was added under an argon atmosphere at room temperature (rt), and then the reaction mixture was heated (oil bath temperature 60 °C). The resulting reaction mixture was stirred at 60 °C for 6 h. After completion of the reaction, it was quenched with saturated aqueous NaHCO3 solution, then extracted with CH2Cl2 (2 × 5 mL) and washed with brine solution (10 mL). The combined organic layers were dried over anhydrous Na2SO4 and filtered through a sintered glass funnel. The filtrate was concentrated under reduced pressure and purified by column chromatography (SiO2, 5% EtOAc/hexanes) to afford ethyl 2-oxo-2,4,5,6,6a,7,8,9,10,11-decahydrofuro[3,2-d]xanthene-7-carboxylate (2a) (0.039 g, 22%) as a white crystal (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)), 5,6-dihydrobenzofuran-2(4H)-one (3a) (0.055 g, 74%) as a yellow liquid (TLC: Rf = 0.70 (SiO2, 15% EtOAc/hexanes)), and 7-methyl-5,6-dihydrobenzofuran-2(4H)-one (3b) (0.080 g, 97%) as a yellow liquid (TLC: Rf = 0.60 (SiO2, 15% EtOAc/hexanes)).
2a
1H NMR (400 MHz, CDCl3): δ 5.75 (d, J = 1.53 Hz, 1H), 4.32–4.09 (m, 2H), 2.83–2.67 (m, 1H), 2.57 (s, 1H), 2.49–2.29 (m, 3H), 2.18–1.98 (m, 4H), 1.84–1.64 (m, 6H), 1.56–1.46 (m, 2H), 1.30–1.26 (m, 3H).
3a
1H NMR (400 MHz, CDCl3): δ 5.87 (t, J = 4.63 Hz, 1H), 5.75 (s, 1H), 2.71 (t, J = 6.63 Hz, 2H), 2.39 (q, J = 5.50, 10.76 Hz, 2H), 1.89–1.83 (m, 2H).
3b
1H NMR (400 MHz, CDCl3): δ 5.70 (s, 1H), 2.70–2.65 (m, 2H), 2.32 (t, J = 5.34 Hz, 2H), 1.97 (s, 3H), 1.87 (quin, J = 6.10, 12.21 Hz, 2H).
Computational Details
Our protocol began with an initial conformational sampling using Grimme’s semiempirical Conformer–Rotamer Ensemble Sampling Tool (CREST)21 at xTB-GFN222−24 level of theory. This step aimed to identify potential conformers of various E/Z reactants, intermediates, and transition states involved in the chemical reaction. To enhance the accuracy of our conformational search, we performed geometry optimizations on all structures obtained from CREST at the Perdew–Burke–Ernzerhof (PBE) density functional method25 and def2-TZVP basis set,26 facilitated by the Turbomole 7.5.0 suite of programs.27 Long-range interactions were taken into account through the application of Grimme’s dispersion correction (D3).28 To ensure precise and efficient treatment of the electronic Coulomb term in the DFT calculations, we employed the resolution of identity (RI)29 and multipole-accelerated resolution of identity (Marij)30 approximations. Furthermore, solvent corrections were incorporated by conducting optimization calculations using the COSMO model.31 In our DFT calculation, dichloroethane (DCE; ε = 10.36) was used as the explicit solvent. To validate whether the stationary points were local minima or transition-state structures, harmonic frequency calculations were conducted at a temperature of 298.15 K. The reported values correspond to ΔG values, incorporating zero-point energy corrections as well as internal energy and entropic contributions through frequency calculations on the optimized minima. The absence of imaginary frequencies confirmed the minima, while the presence of a single imaginary frequency verified the transition states. Additionally, intrinsic reaction coordinate (IRC)32 calculations were performed on all transition states to further validate their authenticity and confirm the correct determination of reactant and product structures. The translational entropy term in the calculated structures was corrected through a free volume correction introduced by Mammen et al.33
Acknowledgments
Funding from the SERB (Science & Engineering Research Board), New Delhi, India (Grant No. CRG/2020/001875), is gratefully acknowledged. R.V. thanks CSIR—India for the award of Senior Research Fellowship (SRF), and A.K.N. thanks DST—India for the INSPIRE Fellowship. K.V. is grateful to DST-SERB (CRG/2021/003255) and CSIR-FIRST scheme for providing financial assistance. H.S. and K.V. also acknowledge the Multi-Scale Simulation and Modeling project (MSM) for the computational facilities. The support and resources provided by “PARAM Brahma Facility” under the National Supercomputing Mission, Government of India, at the Indian Institute of Science Education and Research (IISER), Pune, are gratefully acknowledged.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c08873.
Copies of 1H and 13C NMR spectra for all new compounds, X-ray crystallography data, and computational details (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]; b Wei W.; Cherukupalli S.; Jing L.; Liu X.; Zhan P. Fsp3: A new parameter for drug-likeness. Drug Discovery Today 2020, 25, 1839. 10.1016/j.drudis.2020.07.017. [DOI] [PubMed] [Google Scholar]; c Meyers J.; Carter M.; Mok N. Y.; Brown N. On the origins of three-dimensionality in drug-like molecules. Future Med. Chem. 2016, 8, 1753–1767. 10.4155/fmc-2016-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Düfert A.; Werz D. B. Carbopalladation cascades using carbon–carbon triple bonds: recent advances to access complex scaffolds. Chem. – Eur. J. 2016, 22, 16718–16732. 10.1002/chem.201603044. [DOI] [PubMed] [Google Scholar]; b Dömling A.; Ugi I. Multicomponent reactions with isocyanides. Angew. Chemie. Int. Ed. 2000, 39, 3168–3210. . [DOI] [PubMed] [Google Scholar]; c Bonne D.; Dekhane M.; Zhu J. Exploiting the dual reactivity of o-Isocyanobenzamide: three-component synthesis of 4-imino-4H-3,1-benzoxazines. Org. Lett. 2005, 7, 5285–5288. 10.1021/ol052239w. [DOI] [PubMed] [Google Scholar]; d Dömling A. Recent developments in isocyanide based multicomponent reactions in applied chemistry. Chem. Rev. 2006, 106, 17–89. 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; e Ruijter E.; Scheffelaar R.; Orru R. V. A. Multicomponent reaction design in the quest for molecular complexity and diversity. Angew. Chem., Int. Ed. 2011, 50, 6234–6246. 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]; f de Graaff C.; Ruijter E.; Orru R. V. A. Recent developments in asymmetric multicomponent reactions. Chem. Soc. Rev. 2012, 41, 3969–4009. 10.1039/c2cs15361k. [DOI] [PubMed] [Google Scholar]; g Dömling A.; Wang W.; Wang K. Chemistry and biology of multicomponent reactions. Chem. Rev. 2012, 112, 3083–3135. 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tietze L. F.; Brasche G.; Gericke K. M. Domino reactions in organic synthesis. Domino React. Org. Synth. 2006, 1–617. 10.1002/9783527609925.ch. [DOI] [Google Scholar]; b Yang H.; Sun H. R.; Xue R. Di; Wu Z. B.; Gou B. B.; Lei Y.; Chen J.; Zhou L. Selectfluor-mediated stereoselective [1 + 1 + 4 + 4] dimerization of styrylnaphthols. Org. Lett. 2019, 21, 9829–9835. 10.1021/acs.orglett.9b03587. [DOI] [PubMed] [Google Scholar]; c Keylor M. H.; Matsuura B. S.; Stephenson C. R. J. Chemistry and biology of resveratrol-derived natural products. Chem. Rev. 2015, 115, 8976–9027. 10.1021/cr500689b. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Matsuura B. S.; Keylor M. H.; Li B.; Lin Y.; Allison S.; Pratt D. A.; Stephenson C. R. J. A Scalable biomimetic synthesis of resveratrol dimers and systematic evaluation of their antioxidant activities. Angew. Chem. 2015, 127, 3825–3828. 10.1002/ange.201409773. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Keylor M. H.; Matsuura B. S.; Griesser M.; Chauvin J. P. R.; Harding R. A.; Kirillova M. S.; Zhu X.; Fischer O. J.; Pratt D. A.; Stephenson C. R. J. Synthesis of resveratrol tetramers via a stereoconvergent radical equilibrium. Science 2016, 354, 1260–1265. 10.1126/science.aaj1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Li X. M.; Huang K. S.; Lin M.; Zhou L. X. Studies on formic acid-catalyzed dimerization of isorhapontigenin and of resveratrol to tetralins. Tetrahedron 2003, 59, 4405–4413. 10.1016/S0040-4020(03)00623-9. [DOI] [Google Scholar]; b Panzella L.; De Lucia M.; Amalfitano C.; Pezzella A.; Evidente A.; Napolitano A.; d’Ischia M. Acid-promoted reaction of the stilbene antioxidant resveratrol with nitrite ions: mild phenolic oxidation at the 4′-hydroxystiryl sector triggering nitration, dimerization, and aldehyde-forming routes. J. Org. Chem. 2006, 71, 4246–4254. 10.1021/jo060482i. [DOI] [PubMed] [Google Scholar]
- Wang C. M.; Xia P. J.; Xiao J. A.; Li J.; Xiang H. Y.; Chen X. Q.; Yang H. Photoredox-catalyzed reductive dimerization of isatins and isatin- derived ketimines: diastereoselective construction of 3,3′-disubstituted bisoxindoles. J. Org. Chem. 2017, 82, 3895–3900. 10.1021/acs.joc.6b03056. [DOI] [PubMed] [Google Scholar]
- Pertschi R.; Wagner P.; Ghosh N.; Gandon V.; Blond G. Gold(I)-catalyzed synthesis of furopyrans: insight into hetero- Diels–Alder reactions. Org. Lett. 2019, 21, 6084–6088. 10.1021/acs.orglett.9b02228. [DOI] [PubMed] [Google Scholar]
- a Sieburth S. M.; Chen J. L. A photochemical [4+4] method for the construction of Annulated eight-membered rings. J. Am. Chem. Soc. 1991, 113, 8163–8164. 10.1021/ja00021a050. [DOI] [Google Scholar]; b Sieburth S. M.; Chen J.; Ravindran K.; Chen J. A 2-Pyridone photo-[4 + 4] approach to the taxanes. J. Am. Chem. Soc. 1996, 118, 10803–10810. 10.1021/ja962207r. [DOI] [Google Scholar]; c Lee Yg.; McGee K. F.; Chen J.; Rucando D.; Sieburth S. M. 2-Pyridone approach to taxol. 3. stereocontrol during elaboration of the cyclooctane. J. Org. Chem. 2000, 65, 6676–6681. 10.1021/jo005532c. [DOI] [PubMed] [Google Scholar]
- Carreño M. C.; Ribagorda M. Stereoselective trimerization of [(S) R]-[(p-Tolylsulfinyl) Methyl]-p-quinols and p-quinamines. Org. Lett. 2003, 5, 2425–2428. 10.1021/ol034589t. [DOI] [PubMed] [Google Scholar]
- Green N. J.; Connolly C. A.; Rietdijk K. P.; Nichol G. S.; Duarte F.; Lawrence A. L. Bio-inspired Domino oxa-Michael/Diels–Alder/oxa-Michael Dimerization of para-Quinols. Angew. Chem. 2018, 130, 6306–6310. 10.1002/ange.201802125. [DOI] [PubMed] [Google Scholar]
- a Biswas B.; Sarkar D.; Venkateswaran R. V. Total synthesis of alboatrin, a phytotoxic metabolite from Verticillium alboatrum. Tetrahedron 2008, 64, 3212–3216. 10.1016/j.tet.2008.01.097. [DOI] [Google Scholar]; b Ichihara A.; Nonaka M.; Sakamura S.; Sato R.; Tajimi A. Structure and synthesis of alboatrin, a novel phytotoxic metabolite from verticillium albo-atrum. Chem. Lett. 1988, 17, 27–30. 10.1246/cl.1988.27. [DOI] [Google Scholar]; c Xu Z.; Li Y.; Xiang Q.; Pei Z.; Liu X.; Lu B.; Chen L.; Wang G.; Pang J.; Lin Y. Design and synthesis of novel xyloketal derivatives and their vasorelaxing activities in rat thoracic aorta and angiogenic activities in zebrafish angiogenesis screen. J. Med. Chem. 2010, 53, 4642–4653. 10.1021/jm1001502. [DOI] [PubMed] [Google Scholar]; d Kimura Y.; Shimada A.; Kusano M.; Yoshii K.; Morita A.; Nishibe M.; Fujioka S.; Kawano T. Myxostiolide, myxostiol, and clavatoic acid, plant growth regulators from the fungus myxotrichum stipitatum. J. Nat. Prod. 2002, 65, 621–623. 10.1021/np010600r. [DOI] [PubMed] [Google Scholar]; e Paterson L. D.; Barker D. Synthesis of the furo[2,3-b]chromene ring system of hyperaspindols A and B. J. Org. Chem. 2015, 11, 265–270. 10.3762/bjoc.11.29. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Issa H. H.; Chang S. M.; Yang Y. L.; Chang F. R.; Wu Y. C. New sesquiterpene lactones from the aerial parts of pseudoelephantopus spicatus. Chem. Pharm. Bull. 2006, 54, 1599–1601. 10.1248/cpb.54.1599. [DOI] [PubMed] [Google Scholar]; g Phan M. G.; Tran T. T. N.; Phan T. S.; Matsunami K. Corrigendum to “Guaianolides from Curcuma kwangsiensis” [Phytochem. Lett. 9 (2014) 137–140]. Phytochem. Lett. 2014, 9, 174. 10.1016/j.phytol.2014.06.013. [DOI] [Google Scholar]
- a Leung J. C.; Bedermann A. A.; Njardarson J. T.; Spiegel D. A.; Murphy G. K.; Hama N.; Twenter B. M.; Dong P.; Shirahata T.; McDonald I. M.; Inoue M.; Taniguchi N.; McMahon T. C.; Schneider C. M.; Tao N.; Stoltz B. M.; Wood J. L. Total Synthesis of (±)-Phomoidride D. Angew. Chem. 2018, 130, 2009–2012. 10.1002/ange.201712369. [DOI] [Google Scholar]; b Spiegel D. A.; Wiberg K. B.; Schacherer L. N.; Medeiros M. R.; Wood J. L. Deoxygenation of alcohols employing water as the hydrogen atom source. J. Am. Chem. Soc. 2005, 127, 12513–12515. 10.1021/ja052185l. [DOI] [PubMed] [Google Scholar]; c Malihi F.; Clive D. L. J.; Chang C. C.; Minaruzzaman Synthetic studies on CP-225,917 and CP-263,114: access to advanced tetracyclic systems by intramolecular conjugate displacement and [2,3]-wittig rearrangement. J. Org. Chem. 2013, 78 (3), 996–1013. 10.1021/jo302467w. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Zhang N.; Sun W.; Duan X.; Zhang Q.; Zhou Q.; Chen C.; Zhu H.; Luo Z.; Liu J.; Li X. N.; Xue Y.; Zhang Y. Bioactive polycyclic polyprenylated acylphloroglucinols from hypericum perforatum. Org. Biomol. Chem. 2018, 16, 8130–8143. 10.1039/C8OB02067A. [DOI] [PubMed] [Google Scholar]
- a Haynes R. K.; Chan H. W.; Lung C. M.; Ng A. C.; Wong H. N.; Shek L. Y.; Williams I. D.; Cartwright A.; Gomes M. F. Artesunate and dihydroartemisinin (DHA): unusual decomposition products formed under mild conditions and comments on the fitness of DHA as an antimalarial drug. ChemMedChem. 2007, 2, 1448–1463. 10.1002/cmdc.200700064. [DOI] [PubMed] [Google Scholar]; b Zeng M.-Y.; Li L.-N.; Chen S.-F.; et al. Chemical transformations of qinchaosu, a peroxidic antimalarial. Tetrahedron 1983, 39 (18), 2941–2946. 10.1016/s0040-4020(01)92155-6. [DOI] [Google Scholar]
- a Thorat S. S.; Kataria P.; Kontham R. Synthesis of Furo[2,3b]pyran-2-ones through Ag(I)- or Ag(I)– Au(I)-catalyzed cascade annulation of alkynols and α ketoesters. Org. Lett. 2018, 20, 872–875. 10.1021/acs.orglett.7b04027. [DOI] [PubMed] [Google Scholar]; b Borade B. R.; Nomula R.; Gonnade R. G.; Kontham R. Fe(III)-catalyzed diastereoselective friedel – crafts alkylation–hemiketalization–lactonization cascade for the synthesis of polycyclic bridged 2-chromanol lactones. Org. Lett. 2019, 21, 2629–2633. 10.1021/acs.orglett.9b00614. [DOI] [PubMed] [Google Scholar]; c Kambale D. A.; Thorat S. S.; Pratapure M. S.; Gonnade R. G.; Kontham R. Lewis acid catalyzed cascade annulation of alkynols with α-ketoesters: A facile access to γ-spiroketal-γ–lactones. Chem. Commun. 2017, 53, 6641–6644. 10.1039/C7CC03668J. [DOI] [PubMed] [Google Scholar]; d Nakate A. K.; Pratapure M. S.; Kontham R. Bismuth(III)-catalyzed cycloisomerization and (hetero)arylation of alkynols: simple access to 2-(hetero)aryl tetrahydrofurans and tetrahydropyrans. Org. Biomol. Chem. 2018, 16, 3229–3240. 10.1039/C8OB00368H. [DOI] [PubMed] [Google Scholar]; e Palange M. N.; Gonnade R. G.; Kontham R. TiCl4-n-Bu3N-mediated cascade annulation of ketones with α-ketoesters: a facile synthesis of highly substituted fused γ-alkylidene-butenolides. Org. Biomol. Chem. 2019, 17, 5749–5759. 10.1039/C9OB00649D. [DOI] [PubMed] [Google Scholar]; f Thorat S. S.; Kontham R. Strategies for the synthesis of furo-pyranones and their application in the total synthesis of related natural products. Org. Chem. Front. 2021, 8, 2110–2162. 10.1039/D0QO01421D. [DOI] [Google Scholar]
- a Lienhard G. E.; Wang T. C. Mechanism of acid-catalyzed enolization of ketones. J. Am. Chem. Soc. 1969, 91, 1146–1153. 10.1021/ja01033a019. [DOI] [Google Scholar]; b Neveselý T.; Molloy J. J.; McLaughlin C.; Brüss L.; Daniliuc C. G.; Gilmour R. Leveraging the n→π* interaction in alkene isomerization by selective Energy transfer catalysis. Angew. Chem., Int. Ed. 2022, 61 (2), e202113600 10.1002/anie.202113600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for details.
- a Thatcher G. R. J.The Anomeric Effect and Associated Stereoelectronic Effects; American Chemical Society, WA, 1993. [Google Scholar]; b Freitas M. P. The anomeric effect on the basis of natural bond orbital analysis. Org. Biomol. Chem. 2013, 11, 2885–2890. 10.1039/c3ob40187a. [DOI] [PubMed] [Google Scholar]; and references cited therein.
- a Zhejiang U.; Chao Q.; Shizhen N.; Xinzhi C.. Method for preparing 2-hydroxycyclohexenylacetic acid enol lactone under catalysis of base. Patent No. CN106986848.; b Bonadies F.; Scarpati M. Gazz. Chim. Ital. 1983, 113, 421–426. [Google Scholar]; c Tada M.; Ohtsu K.; Chiba K. Synthesis of patulin and its cyclohexane analogue from furan derivatives. Chem. Pharm. Bull. 1994, 42, 2167–2169. 10.1248/cpb.42.2167. [DOI] [Google Scholar]; d Goh W. K.; Black D. S. C.; Kumar N. Synthesis of Novel 7-Substituted 5,6-Dihydroindol-2-Ones via a Suzuki-Miyaura Cross-Coupling Strategy. Tetrahedron Lett. 2007, 48, 9008–9011. 10.1016/j.tetlet.2007.10.093. [DOI] [Google Scholar]; e Michael S.; Peter H.; Curt Z.; Marianne C.. Method for the production of 2-coumarone and substituted 2-coumarones. Patent No. WO0194329A1.
- Knight D. W.; Pattenden G. Synthetic approaches towards 4-ylidenebutenolides and 4-ylidenetetronic acids. Regioselective nucleophilic additions to unsymmetrically substituted maleic anhydrides. J. Chem. Soc., Perkin Trans. 1 1979, 62–69. 10.1039/p19790000062. [DOI] [Google Scholar]
- Nair R. N.; Bannister T. D. Grubbs Cross-Metathesis Pathway for a Scalable Synthesis of γ-Keto-α,β-Unsaturated Esters. J. Org. Chem. 2014, 79, 1467–1472. 10.1021/jo4023606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pracht P.; Bohle F.; Grimme S. Automated exploration of the low-Energy chemical space with fast quantum chemical methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. 10.1039/C9CP06869D. [DOI] [PubMed] [Google Scholar]
- Pracht P.; Caldeweyher E.; Ehlert S.; Grimme S. A Robust Non-self-consistent tight-binding quantum chemistry method for large molecules computational chemistry at frustrated Lewis pair systems view project. ChemRxiv 2019, 8326202. [Google Scholar]
- Bannwarth C.; Ehlert S.; Grimme S. GFN2-XTB - An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. 10.1021/acs.jctc.8b01176. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Bannwarth C.; Shushkov P. A Robust and accurate tight-binding quantum chemical method for structures, Vibrational frequencies, and Noncovalent interactions of large molecular systems parametrized for all Spd-block elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. 10.1021/acs.jctc.7b00118. [DOI] [PubMed] [Google Scholar]
- a Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]; b Ernzerhof M.; Scuseria G. E. Assessment of the perdew–burke–ernzerhof exchange- correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. 10.1063/1.478401. [DOI] [PubMed] [Google Scholar]
- a Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and Quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]; b Schäfer A.; Horn H.; Ahlrichs R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. 10.1063/1.463096. [DOI] [Google Scholar]
- TURBOMOLE V7.5.0 2016, a development of University of Karlsruhe and ForschungszentrumKarlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007.
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Eichkorn K.; Treutler O.; Ohm H.; Haser M.; Ahlrichs R. Auxiliary basis sets to approximate coulomb potentials (Chem. Phys. Letters 240 (1995) 283-290). Chem. Phys. Lett. 1995, 242, 652–660. 10.1016/0009-2614(95)00838-U. [DOI] [Google Scholar]
- Sierka M.; Hogekamp A.; Ahlrichs R. Fast evaluation of the coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. 10.1063/1.1567253. [DOI] [Google Scholar]
- Klamt A.; Schüürmann G. J. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, 5, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Fukui K. The path of chemical reaction-the IRC approach. Acc. Chem. Res. 1981, 14 (12), 363–368. 10.1021/ar00072a001. [DOI] [Google Scholar]
- Mammen M.; Shakhnovich E. I.; Deutch J. M.; Whitesides G. M. Estimating the Entropic Cost of Self-Assembly of Multiparticle Hydrogen-Bonded Aggregates Based on the Cyanuric Acid·Melamine Lattice. J. Org. Chem. 1998, 63 (12), 3821–3830. 10.1021/jo970944f. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.