

Abstract

Allosteric regulation of protein dynamics infers a long-range deliberate propagation of information via micro- and macroscale interactions. The Y220C structural mutant is one of the most frequent cancerous p53 mutants. The mutation is distally located from the DNA-binding site of the p53 DNA-binding domain yet causes changes in DNA recognition. This system presents a unique opportunity to examine the allosteric control of mutated proteins under a drug design paradigm. We focus on the key case study of p53 Y220C mutation restoration by a series of new compounds suggested to have Y220C reactivation properties in comparison to our previous findings on the restorative potential of PK11000, a compound studied extensively for reactivation in vitro and in vivo. Previously, we implemented all-atom molecular dynamics (MD) simulations and our lab’s techniques of MD-Sectors and MD-Markov state models on the wild type, the Y220C mutant, and Y220C with PK11000 to characterize the effector’s restorative properties in terms of conformational dynamics and hydrogen bonding. In this study, we turn to probing the effects made by docking the battery of a new but less well-tested set of aminobenzothiazole derivative compounds reported by Baud et al., which show promise of Y220C rescue. We find that while complete and precise reconstitution of p53 WT molecular dynamics may not be observed as was the case with PK11000, dispersed local reconstitution of loop dynamics provides evidence of rescuing effects by aminobenzothiazole derivative N,2-dihydroxy-3,5-diiodo-4-(1H-pyrrol-1-yl)benzamide, Effector 22, like what we observed for PK11000. Generalizable insights into the mutation and allosteric reactivation of p53 by various effectors by reconstitution of WT dynamics observed in statistical conformational ensemble analysis and network inference are discussed, considering the development of allosteric drug design rooted in first principles.
Background and Introduction
Allosteric regulation refers to the modulation of protein activity by binding of a ligand, also known as an effector, to a site topographically distal from the protein active site.1,2 Allostery was first coined in 1904 by Bohr et al. when upon discovering that carbon dioxide affects the binding affinity of oxygen by inducing a conformation change in the quaternary structure of hemoglobin.3 Cooperative binding of ligands to distinct protein sites and various allosteric effects have subsequently been studied under various biological settings such as transcription modification and DNA binding and repair.4,5 Early studies were riveted by conformational change as a means to capture allostery, and these works were highly contingent on a static view of protein dynamics.6 The model often draws upon the idea of a pathway of residues operating by physical contact to propagate the signal. However, inconsistencies arise when systems known to operate allosterically fail to demonstrate a change in the average structure. An alternate model based on energetics has also been proposed. Protein conformation, dynamic and transient in nature, comprises various conformations dispersed in an energetically coupled landscape in a continuous time-dependent ensemble.7,8 Within such a landscape, Cooper and Dryden propose that allostery without an average conformational change is possible through the long-range influence of kinetic processes.9 Thus, a difference in dispersion about a common mean posits one way in which an energetic landscape could be borne out.
Moreover, many methods for analyses of MD (molecular dynamics) conformational ensembles provide information about paths of long-range communications such as the ones based on network theory. Network approaches can complement the overall process by illustrating the structural pathways from which one site communicates with the distal one. The manifestation of conformational changes from the subtle localized ones can be understood as a mechanical domino effect, where a perturbation at the binding site of the allosteric effector causes sequential effects to subsequent neighboring residues, constituting a path between the allosteric and active sites. A structure-based statistical mechanical model by Guarnera et al. and an improved statistical coupling analysis (SCA) by Rivoire et al, a sequence-based method identifying allosteric networks through coevolved residues, have shown considerable promise in detecting residue-to-residue allosteric.10,11 We modified the method to track the covariance in the motion of the residues from MD simulations in place of the evolutionary covariance to ground it in the theory of statistical mechanics, which then allows us to infer energetic weights between the conformational populations.
Allostery within the context of drug design holds great promise because allosteric effectors modulate an active site while leaving it fully unobstructed to carry out a function. This new drug modality may prove important for curing currently uncurable diseases.12 While some allosteric drugs have been discovered, many have been found via high-throughput screens or serendipitous routes. Lacking the description of first principles as to how allosteric signals propagate, a de novo drug design proves quite difficult. To this end, our long-term goal is to develop both the requisite basic knowledge and a generalizable pipeline to design this new drug modality. Molecular dynamics simulations13 provide an atomistic level detail of the ensemble dynamics of intermolecular interactions needed to gain insight into the mechanisms. The large number of snapshots generated from molecular dynamics simulations captures the motion of the protein systems. Thus, comparative studies involving proteins with and without ligands and allosteric effectors provide insights into the population shift of the protein conformational ensemble.
The model system for our studies is the allosteric protein, p53. p53 has long been referred to as the guardian of the human genome, but its function is far more nuanced and intricate as more studies uncover its omnipresent role in maintaining cell homeostasis. It acts as a regulator or “orchestrator” of manifold processes in cells to maintain a properly functioning environment. As a transcription factor, p53 stability is highly mediated by post-translational modification, which also induces long-range changes via the N- and C-terminal regions. We consider the binding interface with the DNA as the active site and the binding of an allosteric drug in response to allosteric perturbation as a paradigm for exploring not just allostery itself but also their reactivating potential. The wild-type protein consists of 393 amino acid residues. The p53 DBD folds into an immunoglobulin-like β-sandwich structure with an extended DNA-binding surface, which is formed by a loop-sheet-helix motif (including loop L1, F113 to T123) and two large loops (L2, i.e., K164–C176 and L3, i.e., M237–P250) that are held together by zinc coordination, which has been crystallized (PDB ID: 1TUP).14 However, the N- and C-terminal regions, accounting for about half of the protein’s total length, are intrinsically disordered.15 For this reason, their structural determination has proven exceedingly difficult, and many studies of the protein work with constructs excluding these regions, including this study.
The L1 loop can adopt an extended conformation and interacts directly with DNA via Lysine-120 (K120). Our previous molecular dynamics (MD) studies have explored the local conformational changes of the p53 DBD in complex with DNA and have shown L1 and L3 as the most critical regions undergoing conformational changes.16 The most common alterations of p53 in cancer are missense mutations that can result in gain-of-function (GOF) that triggers aggressive phenotypes and even loss of transcriptional activity. About 75% of all p53 alterations in tumors are missense mutations, suggesting that cancer cells expressing mutant p53 have a distinct advantage over cells that lack p53 altogether.17 The cell type and context in which p53 is extremely specific, and activation varies widely, make it crucial to understand how and where it is controlled. A recent study draws a parallel between post-translational modifications (PTMs) and allosteric perturbation, suggesting that any perturbations, both naturally occurring and artificial, that are distal from the active site of a protein are allosteric in principle.18 We then draw analogous conclusions with structural mutants that are distal from the active site of p53, where the Y220C mutant in particular fits that description, a structural mutant that is responsible for about 100,000 new cancer cases every year.20 Mutant p53 proteins have been for a long time expected to be “undruggable”, but recent studies suggest the opposite, as recapitulated by small molecules such as PK11000 and aminobenzothiazole derivatives.19−21 These studies provide an important proof of concept that (i) structural mutants can behave like allosteric effectors, and (ii) it is possible to rescue allosterically dysregulated structural mutants of p53 by allosteric modulation. In this context, the identification of the structural mechanisms that trigger mutant p53 activities becomes fundamental to provide tailored solutions for new treatments to undermine mutant p53 activities. However, a detailed investigation in the context of the p53 conformational ensemble and its interfaces for allosteric signaling is still missing for the mutant variants, suggesting that apart from local effects elicited by the mutations, more complex and long-range mechanisms are at work. A recent report of the only known allosteric effector, PK1100, 5-chloro-2-methanesulfonylpyrimidine-4-carboxylic acid, PK11000, exhibited promising potential in chemoprevention due to its ability to rescue mutant Y220C p53 with specificity and without affecting the DNA-binding affinity.16 The study offers findings that show evidence of rescue by reaching wild-type level thermostability and also presents a unique example of p53 Y220C rescue by allosteric modulation. The ability to target the Y220C mutation by PK11000 is unique because the Y220C mutant does not directly participate in DNA binding. The Y220C mutation’s distal placement from the active site and its significant disturbance in p53-DNA binding make for an ideal system to study allosterism for targeted drug therapy.
Despite its therapeutic potential, PK1100 has not been able to pass clinical trials, which is common among many of the hits discovered through high-throughput screening, further stressing the emergent need for a comprehensive theory on allosteric signaling to better recapitulate protein function. As an extension of our previously reported proof-of-concept study with PK11000 on the dynamics of the Y220C p53 mutant DNA-binding domain (DBD), in this study, we have included a cohort of 27 more compounds that specifically target Y220C mutant p53 expressing cell lines as reported via BindingDB.22 These compounds come from a collection of two studies, one from a study by Pfizer Inc. on allosteric inhibitors that target protein misfolding and the other by the University of Southampton that reports the reactivating potential of aminobenzothiazole derivatives. The same collaborators in the study also reported on the reactivating potential of PK11000. Both studies report comparable binding properties of these Y220C-specific reactivators, noting their affinities for cysteine residues C182 and C277 as probable binding sites.19−21 With the arsenal of compounds that reportedly target Y220C specifically, we performed docking studies using average ensemble structures of 1 μs wild-type p53 and Y220C mutant p53 DBD simulations. Our previous clustering studies on p53 WT and Y220C mutant dynamics reveal that 15% of Y220C mutant p53 DBD simulations cluster with that of WT simulations, suggesting that Y220C simulations undergo conformational transformations that resemble WT dynamics. In theory, these drugs must then also be able to recognize conformations of Y220C mutant p53 that transiently resemble some of the wild-type dynamics. With this knowledge at hand, we wanted to account for the bidirectionality of Y220C p53 dynamics, oscillating between WT-like and Y220C-like behaviors, and the potential ability of these compounds to recognize in concert both transitorily WT-like and Y220C-like conformations.
Our research advances the understanding of p53 Y220C mutant dynamics and allosteric modulation, distinguishing itself from the studies that incorporate traditionally extensive screening processes that necessitates it.25 While both works explore p53 dynamics, our study uniquely employs molecular dynamics simulations to investigate the allosteric effects of various effectors on the Y220C mutant on a phenomenological level, providing insights into their potential for therapeutic targeting through theory and computation. We demonstrate the nuanced impact of these effectors on protein dynamics, offering a more detailed picture of p53’s conformational landscape emphasizing the importance of understanding the intricate interplay between mutations and allosteric sites at the molecular level.
In this study, we embark on an exploratory journey through the molecular landscape of the Y220C p53 mutant, leveraging computational methods to understand its allostery and potential for therapeutic intervention. The Y220C mutation, notorious in oncology, presents unique challenges and opportunities in drug design due to its intricate influence on p53’s structure and function. Our research delves into the allosteric modulation of this mutant and other effectors in the presence of the mutant, aiming to contribute to the development of novel therapeutic strategies that can counteract its detrimental effects in cancer. This endeavor not only enriches our understanding of p53 allostery but also underscores the vital role of computational approaches in unraveling the complexities of protein dynamics and drug interactions.
Results
Docking
A total of 27 compounds that specifically target Y220C mutant expressing p53 cell lines were screened via the BindingDB online database.21 Using the global average structure of the Y220C mutant and WT 1 μs simulations previously reported by Han et al., docking was performed using Autodock Vina 1.20 to achieve optimal binding states with p53 DBD.16,23,24 Our previous results strongly suggested that the DBD of Y220C adopts WT dynamics in approximately 20% of its simulations when even accompanied by sufficient equilibration.16 To account for the bivalent WT-like conformational dynamics of Y220C mutant p53, we docked these compounds using the WT structure as well as the Y220C structure as a means to identify compounds that can recognize Y220C favored conformations when undergoing dynamic changes. Among the 27 compounds docked, four of the compounds successfully docked globally and allosterically (distal from the DNA-binding sites) to both the Y220C and WT structures (Figure 1). The four effectors are named by their CHEMBLs, and their structural and binding information are given in Table 1. Information about the other docked complexes is available in Table S1.
Figure 1.
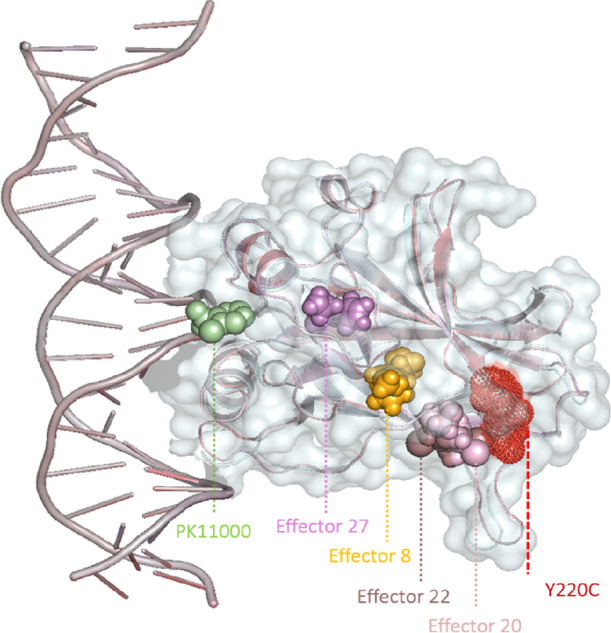
Cartoon representation of the Y220C mutant DBD with docked effectors. Docked effectors are represented in spheres, PK11000 in green, Effector 8 in yellow, Effector 20 in light pink, Effector 22 in brown, Effector 27 in violet, and the Y220C mutant in red dots.
Table 1. Table Showing Four Effectors, Along with Structural and Binding Affinity Data, That Were Identified and Allosterically Docked to the Average Global Structures of 1 μs WT p53 and y220c p53 Simulations.

Global and Local Topological Measures
Root-Mean-Square Measures and Principle Components
RMSD and RMSF were performed to assess the general stability of the simulations and to extrapolate the fluctuations of individual residues with reference to the average structure of the starting heating stage (Figure 2). RMSD results show that our simulations undergo dynamics within 2–3 Å with the wild type displaying the lowest RMSD among all the cohorts, and with Effector 8 bound Y220C mutant p53 showing the highest RMSD at 4 Å as it approaches 9000 frames (0.9 μs). While the Y220C mutant p53 is relatively higher in RMSD, the system still falls within 2–3 Å, adding credence to the stability of our systems. Dynamic cross-correlation and principle component analysis of various distance measures were also performed to assess the topological distributions of the trajectories (Supporting Information). The solvent-accessible surface area (SASA) and the radius of gyration (Rg) of each simulation were also assessed to ascertain the generalized stability and dynamics of the systems (Supporting Information). In both analyses, the Y220C mutant had the lowest SASA and the lowest Rg.
Figure 2.

One shows the RMSD and RMSF of α carbon atoms and heavy backbone atoms, respectively. Top: RMSD, locally weighted scatterplot smoothing plot over 10,000 frames of 1 μs simulation of WT, Y220C mutant, and effector bound simulations. WT in blue, Y220C in red, PK11000 in green, Effector 8 in orange, Effector 20 in brown, Effector 22 in magenta, and Effector 27 in pink. Bottom: RMSF plot of heavy backbone atoms. RMSF of each effector is plotted with the WT and Y220C p53 simulation RMSF plots. WT is shown as a blue line with connecting dots, Y220C as an orange dotted line, and PK11000 and other effectors in green. The plots for WT and Y220C are overlaid with newly identified compounds in grayscale.
RMSF results show instances of rescue by the allosteric effectors in various residue regions with a residue-specific resolution. PK1100 RMSF plots show that the peaks that span residues 112–120 (Lysine 120, DNA-binding region), 196–204, and 220–238 (Y220C mutation hot spot) have dampened fluctuations and resemble WT-like behavior. The extra peak that appears in the Y220C mutant’s K120 region is also quenched in the presence of PK1100, and similar changes are observable with Effectors 20 and 22 bound Y220C mutant simulations. While Effector 8 still harbors the same extra peak around K120, its peak is significantly dampened. In the same region, the peak is bifurcated in the Y220C mutant; however, the presence of Effector 27 results in a singular but broader peak. Regions 182–187 are cysteine-rich and the binding site of PK11000 and other effectors. Interestingly, this peak is only slightly damped and shifted in the presence of Effectors 8, 20, and 22 when compared to the peak in the Y220C mutant; the same peak is unaffected in this region for both PK11000 and Effector 27. In the region that spans residues 204–216, Effector 27 is the only effector that shows a more mutant-like peak, while the other allosteric effectors display peaks exactly those of the WT. Notably, the peak containing the Y220C mutant, residues 220–238, is reconstituted to WT levels with the presence of all effectors.
Hydrogen Bonding
We also conducted global hydrogen bond measures between the protein and the DNA for all cohorts. A global hydrogen bond count between the protein and DNA is presented in Figure 3 (left panel). For both the wild-type and the Y220C mutant, the number of hydrogen bonds nearly doubles from the first half to the second half of the simulation. The presence of PK11000 significantly decreases the number of hydrogen bonds made in the second half of its simulation. A similar dampening effect is observed in the presence of Effector 22 with the Y220C mutant. The number of hydrogen bonds in the Effector 22 system nearly halves when compared to its lone Y220C mutant counterpart all the while preserving a similar trend in the doubling of the total hydrogen bond count from the first half to the second half of the microsecond simulation. The presence of Effectors 8, 20, and 27 had significantly more hydrogen bonds, nearly doubling that of wild-type levels. We also compared how a specific and extensively studied DNA-binding residue may behave differently among all our systems, specifically Lysine-120 (Figure 3, right panel). While the global hydrogen bond count shows similar trends between WT and Y220C mutant simulations, the opposite is observed at a more site-specific level. K120-DNA-binding events only occurred in approximately 3% of the simulations in WT, and nearly 30% of the simulations occurred in the Y220C mutant simulation. K120 binding events are drastically dampened in the presence of the allosteric effectors with the Y220C mutant.
Figure 3.

Left: Global total hydrogen bond count 1–500 ns vs 501–1000 ns for each system. Black is depicted as hydrogen bonds from 1 to 500 ns, while gray shows bonds from 501 to 1000 ns. Right: Percentage of simulation with hydrogen bonding between Lysine 120 and the DNA consensus sequence. Black is depicted as DNA as the hydrogen bond donor and gray as the acceptor.
Thus far, we have considered structural dynamics globally (RMSD) and by local sequence (RMSF) and also compared between the constructs by hydrogen bonding. We turned to methods to extricate how allosteric signaling within the protein in these various states may propagate through the protein. For this, we turn to tools we developed addressing the networks of residues within the protein achieved through pairwise decomposition based on their cooperative distance (MD-Sectors) and global and local K-means clustering (MDMSM).
MD-Sectors
Spectral analysis of the MD calculated motional covariance matrix resulted in an MD-Sector of 39 residue positions. The sectors were also taken every 200 ns for each 1 μs cohort (1–200, 201–400, 401–600, 601–800, and 801–1 μs) showing the top 39 correlated sectors for each consecutive 200 ns block. (Table S2). Sectors are residues that frequently coexist in movement with other residues. The top 20% of the sectors that are highly correlated are projected on a structural map in Figure 4. The cartoon figure with CPK representation of the sector residues is represented by color at two different timestamps at 0.2 and 1 μs for WT (Figure 4A, upper left), Y220C (Figure 4A, upper right), PK1100 + Y220C (lower left), and Effector 22 + Y220C (lower right). The darker colors represent the sectors present in the first 20% of the simulation, while the lighter colors represent the sectors of 100% of the simulation. While the positions of the sectors do not vary greatly between the two timestamps for Y220C (orange) and PK11000 present systems (green), the Effector 22 present Y220C simulation shows an overlap in sectors with sectors present in the WT simulation. Brown residues appear to share sectors that are present in both the WT and Y220C mutant (Figure 4B, left). Upon closer inspection between just the WT and Effector 22 sectors, we observe salient similarity in the changes in sectors identified between the two timestamps (Figure 4B, right) where sectors at 0.2 μs are localized in the β sheet surrounding the Y220C mutation site and the later sectors migrating toward the DNA-binding site. Zooming back to a more global overview, we observe various regions in the p53 DBD in which sectors are highly conserved among different systems (Figure 4B). For instance, we observe multiple regions of shared sectors between the WT system (blue) and the Effector 22 present system (sand), spanning regions that include residues 154–158, 212–226, and 258–266. Some overlap is present between WT and PK1100 in the regions spanning residues 154–158 and 272–280 (Figure 5).
Figure 4.

MD-Sectors at 200 and 1000 ns. Yellow cartoon components represent the N- and C-terminal regions in the full-length protein. (A) Upper left: WT sectors at 200 ns in baby blue and 1 μs in dark blue. Upper right: Y220C mutant sectors at 200 ns in light yellow-orange and 1 μs in orange. Lower left: PK11000 bound Y220C mutant sectors at 200 ns in light green and 1 μs in dark green. Lower right: Effector 22, N,2-dihydroxy-3,5-diiodo-4-(1H-pyrrol-1-yl)benzamide docked Y220C mutant sectors at 200 ns in sand and 1 μs in chocolate. (B) Top: 1 μs WT sector residues in blue, Y220C in orange, and compound 22 in chocolate. Bottom: 200 ns and 1 μs WT sector residues in comparison to 2000 ns and 1 μs compound 22 bound sector residues. Same coloring scheme as that depicted in previous panels.
Figure 5.

System-wide MD-Sector comparison per residue. The occurrence count of MD-Sectors is displayed as a stacked bar graph with each stack represented by the color of the relevant cohort. WT in blue, Y220C in orange, PK11000 in green, and Effectors 8, 20, 22, and 27 bound Y220C in red, purple, sand, and pink, respectively.
MD-Markov State Models (MDMSM)
In the global MDMSM measure, the optimal clustering is at K = 4, WT, PK11000 + Y220C, and Effector 22 + Y220C where simulations cluster together in cluster 1 (Figure 7, left, blue, green, and brown). The optimal clustering was determined by assessing the rate of decay of the weighted average of each cluster up to K = 7 (Figure S2A). Effectors 20 and 27 present Y220C simulation clusters in cluster 2, in purple and pink, respectively, while the Effector 8 system clusters on its own in cluster 3, in red. The Y220C mutant simulation (73.6%) appears in its own cluster in cluster 4, in orange. The centroids were overlaid to observe structural differences between them (Figure 6, left). While centroids 1, 2, and 3 from clusters 1, 2, and 3, respectively, remain structurally alike one another, centroid 4 from cluster 4 varies in the Y220C mutation site loop (in cartoon yellow). The loop containing the mutation clearly deviates from the other centroids, suggesting a potential rescue of that region by the allosteric effectors. Further analysis of the centroid structures shows a 45.5° shift in DNA-binding residue K120 in centroid 4 as well as a 41.4° shift in the Y220C loop region between Glutamate-219 of centroid 1 in blue and centroid 4 in orange. A closer look at the Lysine-120 shows a shift in 45.5° between the two centroids (Figure 6).
Figure 7.

Left: Global MDMSM measure. K-means clustering is shown at K = 4. Each cluster represents the frequency of frames that reside in each cluster. The legend follows a similar coloring scheme as previous figures with Effector 22 replaced for brown. Middle: MDMSM measure with local alignment to WT sector residues. Right: MDMSM measure with reference to DNA-binding residues, K120, S241, R248, R273, A276, C277, R280, and R283.
Figure 6.

Centroid structures of each cluster are overlaid in cartoon representation. Cluster 1 in metallic blue, cluster 2 in purple, cluster 3 in pink, and cluster 4 in mustard yellow. Smaller panels show the angular shift. In the left, the Y220C mutation hotspot at 41.4° between the Glutamate-219 α carbon atom of centroid 1 in blue and centroid 4 in orange with loop residue Glycine-225. The right shows Lysine-120 shifted by 45.5° between the two centroids.
Local measures using MDMSMs were performed by local alignment to two different sets of residues: (i) WT p53 MD-Sector residues (ii) and DNA direct binding residues. WT p53 sectors at 1 μs are listed in Table S2, and the hydrogen bonding residues, K120, S241, R248, R273, A276, C277, R280, and R283, were previously identified by Cho et al. in the original 1TUP crystal structure.14 These local alignments were implemented to extrapolate the contributions of specific local residues to the overall centroid structures from our clustering analyses. Cluster 3 of MDMSMs in reference to WT sector residues shows the Effector 22 simulation and 24.6% of the WT simulation sharing the same cluster residency, showing the preference of the Effector 22 present system for adopting a WT-like centroid structure (Figure 7, middle). MDMSM measure with reference to DNA-binding residues shows trends similar to those of the global measure with WT, PK11000, and Effector 22 simulations sharing residency in cluster 1 (Figure 7, right).
Discussion and Conclusions
Our local and global perspectives reveal structural variation and the dynamicity of allosteric effectors on the Y220C p53 structural mutant. MD simulations of the major four reported Y220C targeting drugs including wild-type, Y220C mutant, and PK11000 present Y220C mutant provide molecular-level insight into the process of Y220C dysregulation and rescue. We have explored the effects of the combination of allosteric effectors that confer on the p53 DBD as an ideal test case to study the effect of allosteric disruption and points of control for rescue. We have tackled this assessment in terms of atomic flux and population dynamics (RMSD/RMSF and MDMSM, respectively) and measures of allosteric communication networks by statistical coupling analysis of “motionally” coupled residues (MD-Sectors).
Each effector was docked to the average structure of both 1 WT p53 and Y220C p53 DBD 1 μs simulations, and effectors for all-atom molecular dynamics simulations were chosen based on their bimodality—their ability to dock to both metastable WT and Y220C structures due to the chimeric WT-like and mutant-like conformation population of previous MSM studies on the Y220C mutant.16 Our simulations characterize the β barrel structure of p53 and binding with the consensus sequence consistent with the well-established Levant characterization and time resolution at the atomic level. Local regions exhibit high fluctuations, and much of the β barrel remains stably connected by highly flexible loop regions. The RMSF results identified several segments of residues whose dynamics are disrupted by the Y220C mutation and restored by the presence of allosteric effectors. This is demonstrated by the recapitulation of local WT-like dynamics, specifically in the loop regions in which hot spot mutant Y220C and DNA-binding residue K120 reside. Many of these, interestingly, correspond to the location of the binding sites of antibodies capable of distinguishing between wild-type and mutant p53. These features are distinctive in the principal component contributions of residues in flux, displaying high peaks in regions that are WT-specific and mutant-specific antibody epitopes, suggesting that they are provoked by the dynamics in the region and that some similarities in the role of destabilization may be shared between mutants.
Hydrogen bonding results also show quenching of the exaggerated Lysine-120 (K120) mobility and global hydrogen bonding in Y220C mutant p53 by the docked allosteric effectors, suggesting a long-range signal between the Y220C mutation hotspot, allosteric effector that is bound distally from the active site, and the active site itself at Lysine-120. Vainer et al.’s 2016 study, published in the Journal of Molecular Biology, demonstrates that the acetylation of K120 in p53’s DNA-binding domain expands L1 conformation space and DNA-binding specificity and ultimately influences proapoptotic gene transcription.44 This modification leads to conformational changes, allowing p53 to adapt its DNA-binding modes based on the DNA sequence. The increased hydrogen bonding at Lysine 120 in the Y220C mutant p53 perhaps is a result of this adaptive behavior of K120 in p53, albeit the mechanism in this case differs from acetylation. The increased hydrogen bonding at this site in the presence of the Y220C mutant might be a compensatory mechanism to maintain DNA-binding affinity or specificity despite the Y220C-induced destabilization. The most surprising aspect of this result is the drastic quenching of the 10-fold increase in hydrogen bonding in Y220C mutant p53 when allosteric effectors are introduced into the system without directly disturbing the active site itself. This finding suggests a nuanced and variable adaptation of p53 to retain or even modify its DNA-binding behavior. Further structural and functional analyses, perhaps through techniques such as X-ray crystallography or nuclear magnetic resonance (NMR) spectroscopy, could provide deeper insights into these compensatory mechanisms and their biological significance. This could have significant implications for how Y220C mutant p53 interacts with DNA and carries out its transcriptional regulation roles and could be pivotal for therapeutic strategies targeting p53 mutations in cancer.
With further inspection into the trends of global measures, we assessed the solvent-accessible surface area (SASA) and the radius of gyration of each system (Rg). The Y220C mutant shows the lowest SASA and Rg among all the cohorts, telling of the protein’s relative compactness throughout the simulation. Lower Rg and lower compactness infer the protein’s aggregation-prone and align with experimental reports of Y220C’s tendency to destabilize p53 signaling in tumors via aggregation.26 In the presence of the effectors, both SASA and Rg are amplified in all effector present systems to a WT-like level. Y220C also exhibits aggregative properties through amplified hydrogen bonding with the consensus DNA sequence, and much of the hydrogen bonds present in Y220C are significantly diminished by the presence of allosteric effectors.
Markov state models reveal that four clusters with PK11000 and Effector 22 present the Y220C mutant system in the same cluster as the WT simulation and the Y220C mutant system in its own cluster, suggesting that PK1100 bound and Effector 22 present Y220C p53 that is similar in dynamics to that of wild-type p53. Dissimilarity between WT and rescued p53 simulations and Y220C mutant is evident upon further inspection of local structural changes. Loop 1 and Loop 6, where K120 and Y220C hotspot mutations reside, respectively, exhibit angular shifts in the “rescued” and mutant centroidal structures. Statistical coupling analysis (SCA) by pairwise decomposition of distance-based variables from our simulations reveals an allosteric hub of amino acids. The sectors present a subnetwork of residues where the kinetic signals from the allosteric effector binding site and the allosteric disruptor site (Y220C hotspot) undergo structural convergence toward DNA activation. With this assessment, we identified 39 residues that are motionally covariant at various timestamps. These residues reveal how allosteric signaling propagates between the Y220C hotspot and the DNA-binding active site with the element of time. Many of the WT sector residues are in regions of low atomic fluctuations along the β sheet, while conversely, the Y220C mutant sectors are crowded in highly mobile loop regions in the proximity of the mutation hotspot and drug binding site. This suggests that the concerted β sheet region is involved in allosteric motioning, mediating kinetic motioning between the hotspot mutation and the DNA-binding site, therefore indirectly modulating p53’s recognition of the DNA. Evidence from our previous studies on the energetic network of the p53 DBD strongly suggests that much of the structural changes seen globally are driven by local van der Waals and electrostatic interactions.16 Among the cohorts and their time-dependent multiplicity of sectors, Effector 22 bound Y220C shows the most promising indication of rescue. Multiple shared sectors that span regions 154–158, 212–226, and 258–266 are observed where the coupled movements of residues traverse from the Y220C hotspot region to the active site. When Effector 22 is introduced to the mutant, we see that some of the sectors begin to cluster in regions involved in DNA contact in the C terminus α helix and the L1 loop. Furthermore, we observe that many sector residues coincide with regions of highly scrutinized sites such as hotspot mutations, zinc-binding residues, and DNA contact residues, verifying that our pipeline can identify functionally crucial residues and the variability of those regions between cohort systems. The finding resonates with the observation from RMS measures that restoration needs not be wholly the same and that the level of resolution by which MDMSMs and MD-Sectors reveal similar overall conformations is suitable for capturing allosteric activity and their vicissitudes.
Much of the evidence gathered in this study aligns with previous studies on the molecular dynamics of the infamous mutant Y220C p53 structure, specifically in the communication between Loop 1, the binding site, and Loop 6, the mutation hotspot. However, this is the first of its kind where in silico reactivation of the Y220C mutant by allosteric effectors on a residue-by-residue basis is demonstrated by molecular dynamics simulations. Taken together, our findings reveal an allosteric signaling hub at play in the presence of allosteric disruptor Y220C, Y220C reactivator, PK11000, and aminobenzothiazole derivative N,2-dihydroxy-3,5-diiodo-4-(1H-pyrrol-1-yl)benzamide (Effector 22). The Y220C mutant is aggregation-prone, where hydrogen bonds are made extensively and indiscriminately with the DNA interface as evidenced by the divergent behavior of K120. Effector 22 and with PK11000 at a close second recapitulates both local and global wild-type p53 dynamics. Protein-wide and local measures were taken to examine the extent to which communication between the distal points of the mutation and binding could be linked to the active site, as indicated by some level of interaction via an allosteric signaling network. Furthermore, the diversified dynamics in the presence of allosteric effectors bears particular importance on the development of allosteric modulators of p53. Naturally, it only seems fitting to counter the tempestuous consequences of allosteric disrupters such as the case of the p53 Y220C mutation by harnessing the power of their innate temper, with allostery itself.
The choice of a computational approach was driven by the need to deeply understand the intricate dynamics and structural nuances of the Y220C mutant, which are difficult to capture experimentally. While our results provide theoretical insights, they pave the way for future experimental studies, particularly in investigating the interaction of allosteric effectors with the Y220C mutant. These findings contribute meaningfully to the field of cancer research, particularly in understanding p53 mutations. We believe that this work is a significant advance in understanding the molecular inner workings of p53.
Methods
To address the key questions regarding the effects of allosteric reactivators on an allosterically compromised protein, the Y220C p53 mutant, we have undertaken molecular dynamics studies on docked structures of various molecules that target Y220C mutant p53 using AutoDock Vina v1.20.23,24 To examine our simulated dynamics, we have analyzed our trajectories using well-established techniques and an emergent analytical pipeline from our lab.
Simulation Specifications and Trajectory Analysis
All-atom 1 μs MD simulations on the p53 DNA-binding domain, residues 96–290, with an explicit solvent were performed using the standard lab protocol with the AMBER14.0 and AMBER16.0 simulation packages and AMBERTOOLS14 suite.27−29 FF19SB force fields for the protein and the TIP3P potential for solvent water were used.30−32 p53 DBD starting configurations of the wild-type (PDB ID: 1TUP) and Y220C + PK11000 (PDB ID: 5LAP) systems were obtained from the Protein Data Bank (PDB).14,19,20 Using the average structure of both our wild-type and mutant simulations, we performed a global docking of reported Y220C targeting molecules as aforementioned in this paper, selected four molecules with allosteric binding modes, and simulated low binding affinity (ΔG, kcal/mol).22 Mutagenesis of residues of interest was performed using the molecular visualization program PyMol Molecular structure of ligand PK11000 (PDB ID: 6SM) that was parametrized using AMBER’s Antechamber suite and covalently bonded to Cysteine-182 using t-LeaP.33 The zinc coordination parametrization was achieved using a zinc AMBER force field (ZAFF).34 The simulation system was treated under particle mesh Ewald periodic boundary conditions with a 10 Å Lennard-Jones cutoff in a truncated octahedral box. Na+ counterions were added to the system for electroneutrality, and SHAKE was applied for hydrogen bond motions. Energy minimization with decreasing constraints on the protein solute was carried out followed by heating to 300 K, and temperature was maintained using the Berendsen algorithm.35,36 Simulations were run using the parallelized CUDA version of the pmemd routine on NVIDIA Graphical processing units (GPUs).37,38 All MD trajectories were analyzed with the AMBER utility “cpptraj” in AMBERTOOLS14 and VMD to calculate the root-mean-square deviation (RMSD) and root-mean-square fluctuations (RMSF) of the C-α backbone atoms in AmberTools16.29,39 Hydrogen bonding analysis was carried out using cpptraj from the Amber14 package, with standard distance and angle cutoff set to 3.5 Å and 30°, respectively.
Molecular Dynamics Markov State Models (MDMSM)
Our MDMSM study of p53 DBD is a statistically driven illustration of allostery and based on a previously done study on the CRIB-PDZ in our lab.40 Conformational selections may occur when an ensemble of p53 DBD structures of protein, in the absence of ligand, clusters with those of a ligand bound trajectory or, in this case, small molecules such as PK11000. The categorization into discrete probabilistic states indicates the protein’s innate predisposition to adopt specific conformations in the presence or absence of an allosteric effector or a mutation. MSMs were constructed in terms of the nodes and links of a complex network, with the nodes obtained by clustering the microstates using K-means clustering using Amber’s native cpptraj analysis toolkit.41,42 In all calculations, atom-based quantities obtained from MD were merged to present the results for each residue.
MD-Sectors
Motional and energy covariance matrices were computed from MD trajectories by standard methods using the AMBER utility cpptraj in AmberTools16.28,29 Spectral analysis was applied to the covariance matrices. The first eigenvalue of a spectral decomposition is unity, and the remaining eigenvalues define modes that comprise a series expansion of the matrix in the covariance space. The leading terms make the largest contributions to the expansion, and to be reasonably inclusive, MD-Sectors were defined based on the eigenvectors of the leading 20% of the eigenvalues of the spectral decomposition. Distance covariance was used to measure the correlations of the joint independence of any two vectors. By computing the pairwise covariance between all residues over the simulation time, pairwise covariance was mapped for all residues as defined by Lakhani et al.27
Molecular Visualization
Molecular dynamics simulations were animated for visual inspection using the visual molecular dynamics (VMD) software package. All structural figures were rendered using PyMOL or VMD.34,43
Acknowledgments
We would like to thank Molecules to Medicine consortium for fruitful discussion, especially Prof. David L. Beveridge, Prof. Michael P. Weir, and their lab members. We gratefully acknowledge Bharat Lakhani and Jonathan Fabry for providing programming structures to successfully perform MDMSM and MD-Sectors. We thank Henk Meij for HPC's technical assistance. This work was supported by NIH R15 GM128102 to K.M.T. and by NSF grants CNS-0619508 and CNS-0959856 to Wesleyan University for the development of the High-Performance Computing Cluster.
Data Availability Statement
A GitHub page for ongoing updates on relevant code is maintained at https://github.com/iarmourgarb/p53-isoforms.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c08509.
The authors declare no competing financial interest.
Supplementary Material
References
- Koshland D. E.; Némethy G.; Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry 1966, 5, 365–385. 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- Changeux J.-P.; Edelstein S. Conformational Selection or Induced Fit? 50 Years of Debate Resolved. F1000. Biol. Reprod. 2011, 3, 19. 10.3410/B3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohr C.; Hasselbalch K.; Krogh A. Ueber einen in biologischer beziehung wichtigen Einfluss, den die kohlensäurespannung des blutes auf dessen sauerstoffbindung übt. Skand. Arch. Physiol. 1904, 16, 402–412. 10.1111/j.1748-1716.1904.tb01382.x. [DOI] [Google Scholar]
- Garcia-Pino A.; Balasubramanian S.; Wyns L.; Gazit E.; de Greve H.; Magnuson R. D.; Charlier D.; van Nuland N. A. J.; Loris R. Allostery and Intrinsic Disorder Mediate Transcription Regulation by Conditional Cooperativity. Cell 2010, 142 (1), 101–111. 10.1016/j.cell.2010.05.039. [DOI] [PubMed] [Google Scholar]
- del Sol A.; Tsai C. J.; Ma B.; Nussinov R. The Origin of Allosteric Functional Modulation: Multiple Pre-Existing Pathways. Structure 2009, 17 (8), 1042–1050. 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monod J.; Wyman J.; Changeux J. P. On the Nature of Allosteric Transitions: A Plausible Model. J. Mol. Biol. 1965, 12 (1), 88–118. 10.1016/S0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- Zhuravlev P. I.; Papoian G. A. Protein Functional Landscapes, Dynamics, Allostery: A Tortuous Path towards a Universal Theoretical Framework. Q. Rev. Biophys. 2010, 43 (3), 295–332. 10.1017/S0033583510000119. [DOI] [PubMed] [Google Scholar]
- McLeish T. C. B.; Cann M. J.; Rodgers T. L. Dynamic Transmission of Protein Allostery without Structural Change: Spatial Pathways or Global Modes?. Biophys. J. 2015, 109 (6), 1240–1250. 10.1016/j.bpj.2015.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A.; Dryden D. T. F. Allostery without Conformational Change - A Plausible Model. Eur. Biophys. J. 1984, 11 (2), 103–109. 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- Guarnera E.; Berezovsky I. N. Structure-Based Statistical Mechanical Model Accounts for the Causality and Energetics of Allosteric Communication. PLoS computational biology. 2016, 12 (3), e1004678 10.1371/journal.pcbi.1004678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivoire O.; Reynolds K. A.; Ranganathan R. Evolution-Based Functional Decomposition of Proteins. PLoS computational biology. 2016, 12 (6), e1004817 10.1371/journal.pcbi.1004817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Chung D. J.; Abramson D.; Langley D. R.; Thayer K. M. Mutagenic Activation of Glutathione Peroxidase-4: Approaches toward Rational Design of Allosteric Drugs. ACS omega. 2022, 7 (34), 29587–29597. 10.1021/acsomega.2c01289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheatham T. E. III; Miller J. L.; Fox T.; Darden T. A.; Kollman P. A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The Particle Mesh Ewald Method Leads to Stable Trajectories of DNA, RNA, and Proteins. J. Am. Chem. Soc. 1995, 117 (14), 4193–4194. 10.1021/ja00119a045. [DOI] [Google Scholar]
- Cho Y.; Gorina S.; Jeffrey P. D.; Pavletich N. P. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science. 1994, 265 (5170), 346–355. 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Wells M.; Tidow H.; Rutherford T. J.; Markwick P.; Jensen M. R.; Mylonas E.; Svergun D. I.; Blackledge M.; Fersht A. R. Structure of tumor suppressor p53 and its intrinsically disordered N-terminal transactivation domain. Proceedings of the National Academy of Sciences of the United States of America. 2008, 105 (15), 5762–5767. 10.1073/pnas.0801353105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han I. S. M.; Abramson D.; Thayer K. M. Insights into Rational Design of a New Class of Allosteric Effectors with Molecular Dynamics Markov State Models and Network Theory. ACS omega. 2022, 7 (3), 2831–2841. 10.1021/acsomega.1c05624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Agostino S.; Strano S.; Emiliozzi V.; Zerbini V.; Mottolese M.; Sacchi A.; Blandino G.; Piaggio G. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer cell. 2006, 10 (3), 191–202. 10.1016/j.ccr.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Stetz G.; Tse A.; Verkhivker G. M. Dissecting Structure-Encoded Determinants of Allosteric Cross-Talk between Post-Translational Modification Sites in the Hsp90 Chaperones. Scientific reports. 2018, 8 (1), 6899. 10.1038/s41598-018-25329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M. R.; Joerger A. C.; Fersht A. R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proceedings of the National Academy of Sciences of the United States of America. 2016, 113 (36), E5271–E5280. 10.1073/pnas.1610421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud M. G. J.; Bauer M. R.; Verduci L.; Dingler F. A.; Patel K. J.; Horil Roy D.; Joerger A. C.; Fersht A. R. Aminobenzothiazole derivatives stabilize the thermolabile p53 cancer mutant Y220C and show anticancer activity in p53-Y220C cell lines. Eur. J. Med. Chem. 2018, 152, 101–114. 10.1016/j.ejmech.2018.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrin L. K.; Denny R. A.; Saiah E. Small molecules that target protein misfolding. J. Med. Chem. 2012, 55 (24), 10823–10843. 10.1021/jm301182j. [DOI] [PubMed] [Google Scholar]
- Liu T.; Lin Y.; Wen X.; Jorissen R. N.; Gilson M. K. BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities. Nucleic acids Res. 2007, 35, D198–D201. 10.1093/nar/gkl999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt J.; Santos-Martins D.; Tillack A. F.; Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 3891. 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry 2010, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Wang Z.; Chen Y.; Petersen R. B.; Zheng L.; Huang K. Salvation of the fallen angel: Reactivating mutant p53. Br. J. Pharmacol. 2019, 176 (7), 817–831. 10.1111/bph.14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.; Fersht A. R. First-order rate-determining aggregation mechanism of p53 and its implications. Proc. Natl. Acad. Sci. U.S.A. 2012, 109 (34), 13590–13595. 10.1073/pnas.1211557109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhani B.; Thayer K. M.; Black E.; Beveridge D. L. Spectral analysis of molecular dynamics simulations on PDZ: MD sectors. Journal of biomolecular structure & dynamics. 2020, 38 (3), 781–790. 10.1080/07391102.2019.1588169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Cheatham T. E. III; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr.; Onufriev A.; Simmerling C.; Wang B.; Woods R. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11 (8), 3696–713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Quantum and statistical mechanical studies of liquids. 10. Transferable intermolecular potential functions for water, alcohols, and ethers. Application to liquid water. J. Am. Chem. Soc. 1981, 103 (2), 335–340. 10.1021/ja00392a016. [DOI] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Cho Y.; Gorina S.; Jeffrey P. D.; Pavletich N. P. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1984, 265 (5170), 346–355. 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- Bauer M. R.; Joerger A. C.; Fersht A. R. 2-Sulfonylpyrimidines: Mild alkylating agents with anticancer activity toward p53-compromised cells. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (36), E5271–E5280. 10.1073/pnas.1610421113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L.The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA; 2002.
- Peters M. B.; Yang Y.; Wang B.; Füsti-Molnár L.; Weaver M. N.; Merz K. M. Jr. Structural Survey of Zinc Containing Proteins, and the Development of the Zinc AMBER Force Field (ZAFF). Journal of chemical theory and computation. 2010, 6 (9), 2935–2947. 10.1021/ct1002626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98 (12), 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. Journal of Computational Physics. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular Dynamics with Coupling to an External Bath. Journal of Chemical Physics. 1984, 81 (8), 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Thayer K. M.; Lakhani B.; Beveridge D. L. Molecular Dynamics-Markove State Model of Protein Ligand Binding and Allostery in CRIB-PDZ: Conformational Selection and Induced Fit. journal of physical chemistry. 2017, 121 (22), 5509–5514. 10.1021/acs.jpcb.7b02083. [DOI] [PubMed] [Google Scholar]
- Prinz J. H.; Wu H.; Sarich M.; Keller B.; Senne M.; Held M.; Chodera J. D.; Schütte C.; Noé F. Markov models of molecular kinetics: generation and validation. J. Chem. Phys. 2011, 134 (17), 174105. 10.1063/1.3565032. [DOI] [PubMed] [Google Scholar]
- Pieniazek S. N.; Hingorani M. M.; Beveridge D. L. Dynamical allosterism in the mechanism of action of DNA mismatch repair protein MutS. Biophysical journal 2011, 101 (7), 1730–1739. 10.1016/j.bpj.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer P. L.; De Boer D.; Alvarado W.; Martinez X.; Sorin E. J. Overcoming the Heuristic Nature of k-Means Clustering: Identification and Characterization of Binding Modes from Simulations of Molecular Recognition Complexes. J. Chem. Inf. Model. 2020, 60 (6), 3081–3092. 10.1021/acs.jcim.9b01137. [DOI] [PubMed] [Google Scholar]
- Hsin J.; Arkhipov A.; Yin Y.; Stone J. E.; Schulten K. Using VMD: An Introductory Tutorial. Curr. Protoc. Bioinf. 2008, 24, 5.7.1. 10.1002/0471250953.BI0507S24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainer R.; Cohen S.; Shahar A.; Zarivach R.; Arbely E. Structural Basis for p53 Lys120-Acetylation-Dependent DNA-Binding Mode. Journal of molecular biology. 2016, 428 (15), 3013–3025. 10.1016/j.jmb.2016.06.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A GitHub page for ongoing updates on relevant code is maintained at https://github.com/iarmourgarb/p53-isoforms.