


Abstract
The effect of mutations in the highly conserved Y-GG/A motif of B-type DNA polymerases was studied in the DNA polymerase from the hyperthermophilic euryarchaeon Thermococcus aggregans. This motif plays a critical role in the balance between the synthesis and degradation of the DNA chain. Five different mutations of the tyrosine at position 387 (Tyr387→Phe, Tyr387→Trp, Tyr387→His, Tyr387→Asn and Tyr387→Ser) revealed that an aromatic ring system is crucial for the synthetic activity of the enzyme. Amino acids at this position lacking the ring system (Ser and Asn) led to a significant decrease in polymerase activity and to enhanced exonuclease activity, which resulted in improved enzyme fidelity. Exchange of tyrosine to phenylalanine, tryptophan or histidine led to phenotypes with wild-type-like fidelity but enhanced PCR performance that could be related to a higher velocity of polymerisation. With the help of a modelled structure of T.aggregans DNA polymerase, the biochemical data were interpreted proposing that the conformation of the flexible loop containing the Y-GG/A motif is an important factor for the equilibrium between DNA polymerisation and exonucleolysis.
INTRODUCTION
DNA-dependent DNA polymerases exhibiting proofreading capability have to coordinate two opposite catalytic activities, a synthetic and a degradative one. For Escherichia coli Pol I-type DNA polymerase as well as for B-type DNA polymerases, these catalytic activities have been shown to reside in structurally distinct protein domains (1–3). It was described for DNA polymerase I Klenow fragment that this editing can be, dependent on the local context, an intramolecular process or an intermolecular one involving dissociation and reassociation of the DNA (4). In B-type DNA polymerases, including bacteriophage, archaeal and most eukaryotic DNA polymerases, the coordination of the two catalytic activities was proposed to take place intramolecularly, mediated by the conserved amino acid motif Y-GG/A located between the N-terminal 3′→5′ exonuclease and the C-terminal polymerisation domain (2). This motif, identified by multiple sequence alignment, was first investigated in the mesophile replicative B-type DNA polymerase of bacteriophage φ29 (φ29 pol) (2). It turned out that mutations in the Y-GG/A motif lead to phenotypes favouring either polymerisation or exonucleolysis compared to the wild-type enzyme. For φ29 pol, this effect was proven to be related to altered single-stranded (ss)DNA binding parameters influencing the stability of the primer terminus. Truniger and colleagues concluded that the motif should be important for the communication between the polymerase active site and the exonuclease active site in a combination of structural and functional roles.
In another approach, for the DNA polymerase of the hyperthermophilic archaeon Sulfolobus solfataricus (Sso pol) a region of 70 amino acids (region 1) involved in enzyme–DNA interaction was identified and proposed to be located in a solvent accessible loop (5). This loop turned out to be the connecting part between the exonuclease domain and the polymerase domain that contains the Y-GG/A motif. In the two crystal structures of B-type DNA polymerases available (6,7) the Y-GG/A motif is indeed located in a flexible loop and is not involved in secondary structure motifs. By mutational analysis of amino acids in the Y-GG/A motif, it was shown that amino acids in this part of the Sso pol enzyme determine the processivity of the proofreading function (3). This effect seems to be mediated via a Km effect rather than by a change in kcat, which is consistent with the observed enzyme–DNA interaction.
The studies on the Y-GG/A residues and neighbouring amino acids in φ29 and Sso pol have revealed that the clearest disturbances of the exonuclease versus polymerase equilibrium were provoked by mutation of the Tyr and Gly residues in the motif. We concentrated our mutational analysis on the Tyr in order to define the mechanism by which this residue takes part in influencing the equilibrium. As a model for our studies, we used the B-type DNA polymerase of the hyperthermophilic archaeon Thermococcus aggregans (Tag pol, GenBank accession no. Y13030) (8). This anaerobic organism was isolated from a deep-sea hydrothermal vent and grows optimally at a temperature of 88°C (9). The native gene of Tag pol contains three inteins that were removed for recombinant expression of the enzyme in E.coli (8). The resulting protein has a size of 94 kDa, a temperature optimum of 80°C and the ability to perform polymerase chain reaction (PCR). Whereas S.solfataricus belongs to the archaeal subdivision of Crenarchaea, T.aggregans is a Euryarchaeon. Crenarchaea possess two (or three) family B-DNA polymerases (10–12), whereas in euryarcheal genomes, only one B-type DNA polymerase was identified (13–16). Recently, a novel heterodimeric DNA polymerase unique to Euryarchaea was found (17–19). Therefore, different physiological roles of B-type DNA polymerases in Euryarchaea and Crenarchaea cannot be excluded.
Here, we present data on mutations in the highly-conserved Y-GG/A motif of Tag pol that were analysed in terms of their effect on enzyme activities, PCR performance, fidelity and kinetic parameters. The results are compared with data for the B-type DNA polymerases from the crenarchaeon S.solfataricus and the bacteriophage φ29. Our results show that mutations in the Y-GG/A motif of Tag pol lead to changes in PCR performance, a fact that was not detected previously since φ29 and Sso pol are not able to work in PCR.
Furthermore, the results were interpreted with the help of a three-dimensional model of Tag pol based on the recently solved X-ray structure of Thermococcus gorgonarius DNA polymerase (7).
MATERIALS AND METHODS
Materials
All chemicals used were of reagent grade. DNA restriction and modification enzymes (Taq DNA polymerase, Pwo DNA polymerase, ExpandTM High Fidelity PCR System) were from Roche Molecular Diagnostics unless otherwise stated. Column chromatography media were from Novagen or Pharmacia. Oligonucleotides were synthesised at ARK Scientific Biosystems (Darmstadt, Germany) or Primm S.R.L. (Milan, Italy). Radioactive reagents were purchased from Amersham Life Sciences Products.
Site-directed mutagenesis and expression of Tag DNA polymerase mutants
Cloning of the wild-type Tag DNA polymerase gene was described previously (8). The mutants presented in this study were prepared by PCR using primers containing the desired mutations as a mismatch. The forward primer common for all mutations was Kpn-fw, matching to a sequence ~100 bp upstream of the mutation site. It contained a natural Kpn-restriction site of the gene. The reverse primers contained a naturally occuring SnaBI restriction site and additionally the desired mutation. The sequences of the oligonucleotides were as follows (mismatch sites for mutagenesis underlined): Kpn-Fw, 5′-GCAACCTTGTAGAGTAGAGTGGTACCTGTTAAGGG-3′; TagY387F, 5′-GCCTCTTTCCGGCTCTTTTACGTATCCTCCCAGGAAAGTAGTCC-3′; TagY387H, 5′-GCCTCTTTCCGGCTCTTTTACGTATCCTCCCAGGTGAGTAGTCC-3′; TagY387N, 5′-GCCTCTTTCCGGCTCTTTTACGTATCCTCCCAGGTTAGTAGTCC-3′; TagY387S, 5′-GCCTCTTTCCGGCTCTTTTACGTATCCTCCCAGGGAAGTAGTCC-3′; TagY387W, 5′-GCCTCTTTCCGGCTCTTTTACGTATCCTCCCAGCCAAGTAGTCC-3′; TagG389A, 5′-GCCTCTTTCCGGCTCTTTTACGTATCCAGCCAGGTAAGTAGTCC-3′.
PCR reactions were carried out using the ExpandTM High Fidelity PCR System. The following program was used: 2 min 94°C, 30 cycles of 10 s at 94°C, 30 s at 55°C, 30 s at 72°C. The resulting 139 bp fragments were digested with KpnI and SnaBI yielding a 101 bp fragment that was ligated into the expression vector pTYPol linearised with the same enzymes. The cloned DNA segment was sequenced to confirm the presence of the desired mutation.
Proteins were expressed in E.coli BL21 (DE3) and purified as described previously (8).
DNA polymerase assay
The polymerase activity was determined by the incorporation of [α-32P]dCTP in DNA template (M13mp9 ssDNA) (20). Fifty microlitres test mix contained 5 µl 10× Tag-reaction buffer (100 mM Tris–HCl pH 8.9, 750 mM KCl, 15 MgCl2, 100 mM CHAPS), 200 µM each of dATP, dGTP, dTTP, 100 µM dCTP, 1 µCi [α-32P]dCTP and 1 µg of M13mp9 ssDNA annealed with 0.3 µg M13 primer. Assays were performed with various amounts of DNA polymerase (final amount of enzyme 2.5–15 fmol) yielding six reactions to calculate a mean value. As a reference enzyme, Pyrococcus woesei DNA polymerase (Pwo) was used. The DNA/primer mix was prepared by heating 277.2 µg M13mp9 ssDNA (Roche) and 156 µg M13 forward sequencing primer (17mer) for 30 min to 55°C and then cooling for 30 min to room temperature.
Assay reactions were incubated for 30 min at 65°C, stopped on ice by the addition of 500 µl of 10% TCA and kept on ice for another 10 min. Samples were filtered through GFC-filters (Whatman), the filters washed three times with 5% TCA, dried and counted in a β-counter in 2 ml of scintillation fluid. One unit is defined as the amount of enzyme necessary to incorporate 10 nmol dNTP into acid-insoluble form at 65°C in 30 min. Specific activities of the enzymes in U/pmol were: WT, 2.8; YF, 5.1; YW, 2.6; YH, 2.6; YN, 0.18; YS, 0.5; GA, 0.3.
Exonuclease assays
Three picomoles of enzyme was incubated with 5 µg of 3H-labelled calf thymus DNA for 4 h at 65°C in a buffer containing 10 mM Tris–HCl pH 8.9, 75 mM KCl, 1.5 MgCl2 and 10 mM CHAPS. Radioactivity liberated from ctDNA was measured in a scintillation counter. One unit of exonuclease activity is the amount of enzyme needed to release 1 nmol of acid-soluble nucleotide from sonicated ctDNA in 30 min at 65°C. Specific exonuclease activities of the Tag pol enzymes in U/pmol were: WT, 299; YF, 270; YW, 211; YH, 295; YN, 561; YS, 613; GA, 706. The assay used does not discriminate between the 3′→5′ exonuclease activity and the 5′→3′ exonuclease activity. As 5′→3′ exonuclease activity has not been detected in B-type polymerases of Thermococcales (21), the values obtained can be considered as 3′→5′ exonuclease activity.
Polymerase chain reactions (PCR)
The improvement of the PCR performance of the mutants was studied in a time-dependent PCR. A 2 kb fragment was amplified from λ DNA as described below. In consecutive experiments, the elongation time of the PCR was reduced to determine the minimal elongation time sufficient to amplify the 2 kb fragment for each enzyme. The following primers were used: λ 1 forward primer, 5′-GAT GAG TTC GTG TCC GTA CAA CA-3′; λ 6 reverse primer, 5′-CTT CAT CAT CGA GAT AGC TGT CG-3′.
PCR using wild-type and mutants of Tag pol was performed in a buffer optimised for this DNA polymerase: 10 mM Tris–HCl pH 8.9, 75 mM KCl, 1.5 MgCl2, 10 mM CHAPS and 200 µM dNTP. Ten nanograms of λ DNA was used as a template and 30 pmol of each primer. Template, primers and nucleotides were combined in a mixture called mix 1 in a volume of 25 µl. Twenty-five microlitres of mix 2 containing buffer and enzyme (1 pmol Tag pol wild-type or mutant; 2.5 U control enzyme) were added. All reactions were prepared in duplicate. The amplification was performed in a 2400 GeneAmp thermocycler (Perkin-Elmer). The cycle conditions were 2 min at 94°C, 30 cycles with 10 s denaturation at 94°C, 30 s annealing at 58°C and elongation at 72°C for the indicated times. The samples were stored at 4°C. Separation was performed by electrophoresis on 1% agarose gels.
lacI-based PCR Fidelity assay
We used the lacI-based PCR Fidelity assay described by Frey and Suppmann (22). This method is based on the amplification, circularisation and transformation of the pUC19 derivative pUCQ17, which contains a functional lacIq allele (23). PCR-derived mutations in lacI result in a derepression of the expression of lacZα and subsequent formation of a functional β-galactosidase enzyme, which can be easily detected on X-Gal indicator plates.
The PCR reactions were performed with 1 (wild-type, YF, YW, YH) or 5 pmol (for mutants YN and YS) of protein in the Tag-polymerase PCR-buffer described above or for the control reactions in the manufacturer’s PCR buffers with 2.5 U of enzyme. The cycle conditions were 10 s denaturation at 94°C, 30 s annealing at 57°C and 4 min elongation at 72°C for 18, 24 or 30 cycles depending on the enzyme.
After PCR, the yield of amplification product was determined (OD260 or in agarose gel) and the DNA fragments submitted to phenol/chloroform extraction to eliminate any protein. After digestion with ClaI, the DNA fragments were purified from a preparative agarose gel. Ligation reactions were carried out with the Rapid Ligation Kit (Roche Molecular Diagnostics), reactions containing 30 ng DNA. The resulting circular plasmids were transformed in E.coli DH5α as described by Hanahan (24) and plated on LB plates with 100 mg/ml ampicillin and 0.004% w/v X-Gal. After incubation overnight at 37°C, blue and white colonies were counted. The error rate, f, was calculated with a rearranged equation published by Keohavong and Thilly (25):
f = –ln F / d × b bp
where F is the fraction of white colonies (white colonies/total colonies), d is the number of DNA duplications (2d = output DNA/input DNA) and b is the effective target size (1080 bp) of the lacI gene, which is 349 bp according to Provost et al. (26). There are 349 phenotypically identified (by colour screening) single-base substitutions (nonsense and missense) at 179 codons (~50% of the coding region) within the lacI gene. Frameshift errors which may occur at every position in the 1080 bp open reading frame of lacI are not taken into account because little information is available for the specific polymerases used in PCR except for Taq DNA polymerase. Frey and Suppman showed that concatameric ligation products seem to be a very rare event as well as deleterious recombination of ligated DNA in DH5α.
Determination of the kinetic constants
A filter binding assay was used by combining the methods described in Kong et al. (27) and Polesky et al. (28). Fifty microlitre assays were carried out in the polymerase buffer described above containing variable amounts of dNTPs and primed M13mp18 DNA template. M13mp18 ssDNA was annealed with an equimolar amount of a 24mer oligonucleotide by heating to 80°C for 5 min followed by slow cooling to room temperature. In the case of DNA parameters, dNTP concentration of dATP, dTTP and dGTP were fixed at 200 µM, dCTP at 20 µM and [α-32P]dCTP at 3.5 µM (3 µCi/pmol). In the case of the dNTP parameters, DNA was present at 25 nM. Reactions were initiated by the addition of 1 pmol of enzyme to assay mixtures prewarmed to 70°C and performed in a thermocycler with heated lid to prevent evaporation. Ten microlitre samples were withdrawn as a function of time, the reaction stopped on ice and spotted on 2.4-cm diameter DE81 filters (Whatman). The values in Table 2 correspond to the activity of 0.2 pmol of enzyme (10 µl of 50 µl reaction). Unincorporated [α-32P]dCTP was removed by three washing steps in 0.3 M ammonium formate (pH 8.0), followed by a washing step in 95% ethanol and one washing step with diethylether. The dried filters were counted in vials with 2.5 ml scintillation fluid. Assays were calibrated with a known amount of [α-32P]dCTP on DE81 filters omitting the washing steps. Apparent Km and vmax values were calculated by regression analysis with the program GraFit (29).
Table 2. Comparison of kinetic parameters of DNA polymerases.
| Polymerase | Kmapp | vmax | ||||
|---|---|---|---|---|---|---|
| dNTPa | DNAb | |||||
| Average (µM) | Range | Average (nM) | Range | Average (fmol dCTP/s) | Range | |
| Tag wild-type | 0.3 | ± 0.016 (2) | 3.5 | ± 1.6 (2) | 0.11 | ± 0.010 (2) |
| Tag Y387F | 0.2 | ± 0.064 (2) | 5.5 | ± 5.5 (2) | 0.19 | ± 0.018 (2) |
| Tag Y387S | 0.56 | ± 0.153 (2) | 2.6 | ± 0.5 (2) | 0.02 | ± 0.001 (2) |
| Tli recombinantc | 57 | ± 30 (3) | 0.12 | ± 0.08 (4) | n.d.e | |
| T.sp. 9°N-7d | 75 | ± 36 | 0.05 | ± 0.03 | n.d. |
±, Standard errors. Numbers in parentheses indicate the number of independent experiments.
aMoles of each dNTP in an equimolar mixture of the four nucleotides in the presence of an excess of DNA.
bMoles of M13mp18 ss-template annealed with an equimolar amount of primer, in the presence of an excess of dNTP.
cThermococcus litoralis DNA polymerase, see Kong et al. (27).
dThermococcus sp. 9°N-7 DNA polymerase, see Southworth et al. (37).
en.d., not determined.
Homology based molecular modelling of Tag pol
For the modelling of the structure of Tag DNA polymerase, the crystal structure of the DNA polymerase from T.gorgonarius (Tgo) was used (PDB 1TGO) (7). Amino acid sequences (Tag pol, accession no. Y13030; Tgo pol, accession no. A79152) were aligned using the program CLUSTALX (30). The sequence alignment was taken as the basis for homology modelling with WHAT IF (31). The process was iterated to fit the geometric criteria of WHAT IF validation suite and the knowledge about conserved residues. Due to their high amino acid homology (89.7%), both proteins show essentially the same structure. In the alignment, two short insertions in the sequence of Tag pol, a single and a two amino acid insertion, were detected. The two amino acid insertion is located far away from the position of the tyrosine. The single amino acid insertion (i.e. Leu at position 383) is in direct proximity of the sequence motif that is the subject of this study (cf. Fig. 1). To model the loop containing the Y-GG/A motif, in-house software (K.Paliakasis and C.Vorgias, unpublished results) in combination with WHAT IF was used. After insertion of the amino acid leucine the conformation was modelled to fulfil the geometry and WHAT IF validation suite criteria.
Figure 1.

Multiple sequence alignment of a 24–25 amino acid region surrounding the Y-GG/A motif in archaeal B-type DNA polymerases. In addition to the conserved motif Y-GG/A itself, V, P and G residues (in bold) are also conserved throughout the archaeal enzymes. A clear distinction can be made between crenarchaeal and euryarchaeal sequences (above and beneath the horizontal line, respectively). Residues conserved in euryarchaeal B-type DNA polymerases are marked in green, residues characteristic for crenarchaeal B1-DNA polymerase sequences are shown in blue. Another interesting feature is a cluster of positively charged amino acids (red) preceding the Y-GG/A motif in both archaeal subdomains, being Arg (R) in the case of the euryarchaeal sequences and Lys (K) in the case of the crenarchaeal ones. This cluster of lysines can also be found in eukaryal α-DNA polymerases in this region of the enzyme. Note also the high number of Glu (E) residues in the euryarchaeal sequences. The created Tag pol mutants are indicated under the alignment. Asterisks mark analogous mutants investigated in φ29 and Sso pol (2,3). Accession numbers of the Thermococcus spec. DNA polymerases: T.aggregans, O33845; T.litoralis, M74198; T.spec. 9°N-7, Q56366; T.gorgonarius, P56689. Accession numbers of the Pyrococcus spec. proteins: P.spec KOD, S71551; P.abyssii, P77916; P.furiosus, P80061; P.horikoshii, O59610; Methanococcus jannaschii, Q58295. Crenarchaeal species and accession numbers of the DNA polymerases: Sulfolobus solfataricus, P26811; Sulfolobus acidocaldarius, P95690; Pyrodictium occultum, D38573; Aeropyrum pernix, AB017500; Sulfurisphaera ohwakuensis, O50607.
RESULTS AND DISCUSSION
Design and purification of T.aggregans DNA polymerase mutants
The aim of this study was to investigate the role of the Y-GG/A motif in the DNA polymerase of the hyperthermophilic euryarchaeon T.aggregans. This conserved sequence motif was reported to coordinate DNA synthesising versus DNA degrading enzymatic activities in B-type DNA polymerases from bacteriophage φ29 (2) and from the crenarchaeon Sso (3). Since a clear distinction can be made in the sequential context of the Y-GG/A motif of crenarchaeal versus euryarchaeal B-type DNA polymerases (Fig. 1), it was of interest to test the role of this motif also in an euryarchaeal family B-DNA polymerase. In Tag pol, motif Y-GG/A is located at positions 387–390. We started with the exchange of Tyr387 to Ser and to Phe (mutants will be addressed as YS and YF, respectively) and of the first Gly (Gly398) to Ala (GA) (Fig. 1). Analogous mutants have been investigated in φ29 pol and Sso pol (2,3). Additionally, we produced three more mutants in the position of Tyr387 inserting His, Trp and Asn (corresponding mutants addressed as YH, YW and YN, respectively). In this manner, we could test the effect of two more amino acids containing aromatic ring systems and another aliphatic amino acid containing a reactive group.
Wild-type and mutant Tag pol enzymes were expressed and purified as previously described (8). The purification scheme included three steps: a heat precipitation step at 80°C for 15 min followed by centrifugation; and two FPLC affinity chromatography steps on a Blue Sepharose column and a Ni2+-chelate column. All mutants were purified to near homogeneity and showed similar solubility and chromatographic behaviour compared to the wild-type enzyme, indicating that the mutations did not cause major structural changes (data not shown).
Enzymatic activities of wild-type and mutant Tag DNA polymerases
The polymerase activity of the wild-type and mutated enzymes was measured on primed M13mp9 DNA. According to their polymerase activity, mutants can be defined as: (i) mutants with enhanced polymerase activity (YF); (ii) mutants having polymerase activity almost unchanged compared to the wild-type (YW and YH); and (iii) mutants with clearly reduced polymerase activity (YN, YS and GA) (Table 1).
Table 1. DNA polymerase and exonuclease activities of Tag pol wild-type and its mutant derivatives on double-stranded DNA.
| Protein | WT | Y387F | Y387W | Y387H | Y387N | Y387S | G389A |
|---|---|---|---|---|---|---|---|
| Pol (%) | 100 | 160 | 92 | 93.6 | 6.4 | 17.8 | 10.7 |
| Exo (%) | 100 | 90 | 71 | 98 | 205 | 187 | 236 |
| Pol/Exo | 1 | 1.77 | 1.29 | 0.96 | 0.03 | 0.09 | 0.04 |
Data are given in percentage of wild-type enzyme activity (mean values from at least two independent measurements). Pol, DNA polymerase activity; Exo, 3′→5′-exonuclease activity.
As for the exonuclease activity, measured on 3H-labelled calf thymus DNA, mutants were divided into two groups: (i) mutants having wild-type-like exonucleolytic activity (YF, YW and YH) and (ii) mutants with enhanced exonuclease activity (YN, YS and GA, Table 1).
In order to be able to compare the results with the previous studies (2,3) and between the single mutants obtained in this study, we calculated the quotient of polymerase (pol) versus exonuclease (exo) activity. Based on this quotient, the Tag pol variants could be divided into two groups. The first group is characterised by a quotient of polymerase to exonuclease activity in the same order of magnitude as wild-type Tag pol (YF, YW, YH; pol/exo ratio wild-type = 1), the second group by a pol/exo quotient clearly <0.1 (YN, YS, GA) (Table 1). Thus, the latter group has an exonuclease function 10-fold favoured compared to the wild-type enzyme. Interestingly, all proteins in the first group contain a residue with aromatic ring system at position 387 (wild-type Tyr, Phe, Trp, His) while the members of the second group have non-aromatic amino acids at this position (Ser, Asn). We conclude that the electron cloud of the ring systems plays an important role for the synthetic function of the DNA polymerase.
Compared to this, the analogous mutants in Sso pol have pol/exo ratios of 2.3 for Y→F, 1.1 for Y→S (both enzymatic activities almost knocked out) and 0.6 for G→A (3). In the case of φ29 pol, using a different method, the following pol/exo values were obtained: 101 for Y→F, 0.002 for Y→S and 91 for G→A (2). Thus, mutations Y→F and Y→S led to deviations in the same direction from the wild-type pol/exo ratio in all three DNA polymerases tested (Tag pol, Sso pol, φ29 pol). On the other hand, Tag pol GA mutant possesses an exonucleolytic activity that is extremely favoured compared to the wild-type (pol/exo <0.1), while in the case of φ29 pol GA mutant, DNA synthesis is strongly enhanced over exonucleolysis (pol/exo >90). Strikingly, the phenotype displayed by Tag and Sso pol for the G→A exchange at the first glycine (pol/exo <<1) was observed in φ29 pol having the same mutation at the second glycine of the motif (pol/exo = 0.2). The dramatic effect obtained by substituting the glycine residues in the Y-GG motif is probably due to a reduced flexibility of the loop in which the above sequence is located.
PCR performance
As a functional test, we applied the Tag pol enzymes in PCR on λ DNA in a buffer optimised for this purpose. All mutants except for mutant GA were able to perform PCR, the latter being less thermostable than the wild-type Tag pol and the other mutants (K.Böhlke, unpublished observation). The five mutants with changes at position Y387 yielded different amounts of PCR product under identical conditions. Under these conditions, mutants YS and YN could amplify a maximum length of 2.5 kb (data not shown). Upon increasing the desired product length, the differences in PCR performance became more obvious (YH > YF > YW > WT). A maximum length of 7.5 kb was produced by proteins YH, YF and YW, but not by the wild-type and mutants YN and YS (data not shown).
The differences in PCR performance are also shown in an experiment we call ‘time-dependent’ PCR. In this experiment, a PCR product of constant length (2 kb) is amplified from λ DNA with subsequently reduced elongation times, keeping all other parameters constant. Under these conditions, YN was the ‘slowest’ mutant, requiring ~90 s to successfully amplify 2 kb from λ DNA (Fig. 2A), followed by mutant YS that is able to amplify 2 kb within 60 s (data not shown). Tag pol wild-type and mutant YW required at least 40 s to amplify 2 kb (Fig. 2B). Mutant YH could amplify 2 kb in only 30 s (Fig. 2C), the minimal elongation time of mutant YF for 2 kb in PCR lies between 30 and 40 s, the YF band in Figure 2C being very faint. The hypothesis, that the effect seen is related to a change in vmax, is supported by a kinetic analysis (cf. Table 2).
Figure 2.
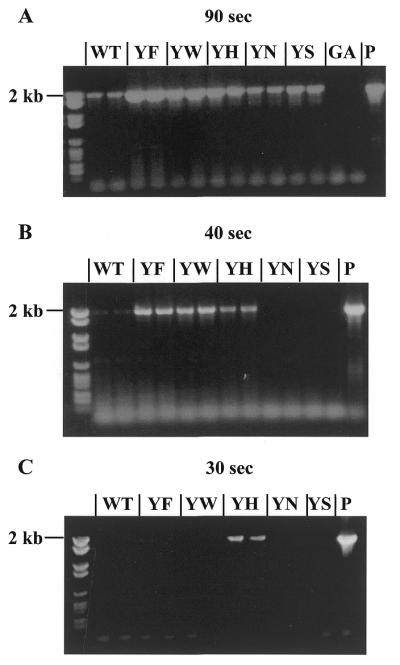
Time-dependent PCR. Agarose (1%) gels showing 2 kb PCR products from reactions performed with stepwise reduced elongation times of 90 (A), 40 (B) and 30 s (C). To determine minimal elongation time, 1 pmol of each Tag pol mutant or wild-type enzyme was added to a mix of 10 ng λ DNA and a primer pair designed to yield 2 kb DNA fragments. WT, T.aggregans wild-type DNA polymerase; YF, YW, YS, YN and YH are the corresponding mutants with an exchange in the position of tyrosine 387 to phenylalanine, tryptophan, serine, asparagine and histidine, respectively. GA, mutation of glycine 389 to alanine in the gene of the T.aggregans DNA polymerase. P, 2.5 U P.woesei DNA polymerase as control reaction. Left lane of each gel: molecular weight marker VI (Roche). In the 40 and 30 s reactions, mutant GA was omitted.
We can therefore assume that the extension rate of Tag pol is affected by the mutations created in the Y-GG/A motif described in this study. The mutants containing a non-aromatic amino acid instead of the tyrosine clearly needed more time to successfully amplify the 2 kb DNA fragment in comparison to wild-type, YW, YF and YH Tag pol. It is likely that due to the sequence similarity of archaeal DNA polymerases in this region, especially among the euryarchaeal ones (Fig. 1), our results may be valid also for DNA polymerases from other Thermococcus or Pyrococcus strains.
The extension rate of wild-type Tag pol in PCR calculated on the basis of the successful amplification of 2000 bp fragment in 40 s is in the order of 50 nt/s (3000 bp/min). This is comparable with values of ~1000 bp/min determined for the closely related DNA polymerase from Thermococcus litoralis (Vent DNA polymerase) with a different experimental approach (27).
Fidelity
As some of the mutations were shown to alter the exonucleolytic activity of the enzyme, we investigated the error rates of wild-type Tag pol and its mutants. This analysis was carried out using the PCR-based fidelity assay described by Frey and Suppmann (22), an assay suited to measure error rates in the range of 10–4–10–6 due to low background activity. In five independent runs, a mean error rate of 5.0 × 10–6 was found for wild-type Tag DNA polymerase. This value lies between the mean error rates of 1.8 × 10–6 for ExpandTM High Fidelity PCR System and 1.3 × 10–5 for Taq DNA polymerase determined in our experiments. Fidelity is one of the hardest biochemical properties to determine (21), and ‘absolute’ values determined are dependent on the assay used. We decided to present the fidelity data as a quotient to Taq DNA polymerase errors in the corresponding experiment in order to have a better possibility to compare the results by excluding influences of the individual experiment. Figure 3 shows the mean quotients of Tag pol variants error rates in relation to the relevant Taq DNA polymerase rates. The error rates of the mutants displaying improved synthetic activity did not differ significantly from the wild-type values (YF, YW and YH), while the mutants with enhanced exonuclease activity (YN and YS) revealed ~10-fold improved fidelity rates (mean values 0.63 × 10–6 and 0.62 × 10–6 for YN and YS, respectively). Fidelity values for mutant GA could not be determined since the protein was not able to synthesise the necessary PCR fragments.
Figure 3.

Fidelity of Tag pol and its mutants expressed in relation to the fidelity of Thermus aquaticus DNA polymerase (Taq, T). A quotient of 1 means that the polymerase has the same error rate as Taq DNA polymerase (mean value 1.3 × 10–5). Values >1 describe the factor by which the polymerase produces less errors than Taq DNA polymerase. The bars correspond to mean values calculated from 2–5 independent experiments; error bars missing are smaller than 0.36. Abbreviations for Tag pol mutants as indicated in legend to Figure 2. As controls P.woesei DNA polymerase (Pwo) and Expand High Fidelity PCR system (HiFi) were used.
The competition model between DNA polymerase catalytic sites argues that if the speed of polymerisation decreases, the probability of the primer end switching to the exonuclease site increases (32) and, accordingly, an enhanced exonucleolytic activity can be expected. Detailed kinetic experiments with T4 DNA polymerase proved that an increased transfer rate of duplex DNA from polymerase to exonuclease active site produces an antimutator phenotype (33). It seems that we have detected this phenomenon in our fidelity experiments with Tag pol in terms of a reduced error rate of YS and YN mutants.
Determination of kinetic constants
From the described function of the Y-GG/A motif in enzyme–DNA binding near the primer-template end (2,3), one would assume an effect of mutations in this region on Km for DNA. For the exonuclease function of Sso pol (ssDNA) this assumption was proven to be correct (3). The results of the PCR studies indicated that vmax could be influenced by the mutations in the Y-GG/A motif. Therefore, steady-state kinetic parameters were determined for wild-type Tag pol and two mutants (YF and YS). We chose YF representing the group of mutants with enhanced PCR performance and YS representing the mutants with lowered polymerase activity and enhanced exonuclease activity. Moreover, these were the mutants investigated also in Sso pol and φ29 pol. Pseudo-first-order conditions were created by determining constants for one substrate in the presence of an excess of the second substrate (see Materials and Methods). As Table 2 shows, the data indicate that a Km-effect for primer-template is rather negligible and for dNTP is small, whereas the effect on the kcat for the polymerisation reaction seems to be more important. We observed the following changes in vmax (determined with identical enzyme amounts): compared to wild-type Tag pol, the vmax of mutant YF was enhanced by a factor of 1.5–2, while mutant YS displays a 5-fold lower vmax. The difference between YF- and YS-values is almost 10-fold. These results show that the distinctions between the mutants observed in the time-dependent PCR are indeed related to altered values of vmax.
It is also interesting to note that the Km values for dNTP ranging from 0.2 to 0.56 µM are the lowest reported in literature. The lowest Km(dNTP) value reported was 1–2 µM for E.coli polymerase I (34). On the other hand, the Km values for DNA are high in comparison to the closely related polymerases (Table 2).
Structural considerations
Motif Y-GG/A in B-type DNA polymerases is located in a solvent accessible loop between the two catalytic domains (3,6,7). Based on a duplex DNA modelled into the crystal data of RB69 bacteriophage DNA polymerase, Truniger and colleagues proposed a direct interaction of the tyrosine with the phosphodiester bond between the two nucleotides preceding the one acting as template (35). Our results strongly suggest that the function of tyrosine 387 in Tag pol is defined mainly by its aromatic ring system indicating that stacking interactions are needed for a correct synthetic reaction. This can be deduced from the clear reduction of synthetic activity in mutants lacking this ring system (YS and YN). In this context it is worth considering that four arginine residues (R380–R382 and R384) precede the tyrosine in the loop. Analysis of the T.gorgonarius crystal structure suggests that this flexible loop becomes ordered in the presence of duplex DNA and probably binds the phosphate backbone of the primer strand, with the conserved arginines, as well as the minor groove, with the aromatic ring system of the tyrosine and the adjacent residues (K.-P.Hopfner, personal communication). Interestingly, this accumulation of Arg is conserved in all euryarchaeal B-type DNA polymerases, whereas in crenarchaeal B1- and eukaryal α-DNA polymerases, a cluster of lysine residues seems to contribute positive charge (Fig. 1).
The tyrosine at position 387 in our structural model of Tag pol is almost completely buried (Fig. 4). Phe, Trp or His could occupy the same position, whereas it is less clear if the environment would also accommodate the comparatively polar Asn and Ser residues. If not, this would lead to a different conformation of the loop taking part in the generation of the described phenotypes. Dong et al. (36) suggested for a glycine in human DNA polymerase α analogous to G398 in Tag pol a role as structural element in protein folding. Mutation of this glycine to alanine reduced polymerase activity (as in Tag pol) but did not change Km for DNA.
Figure 4.
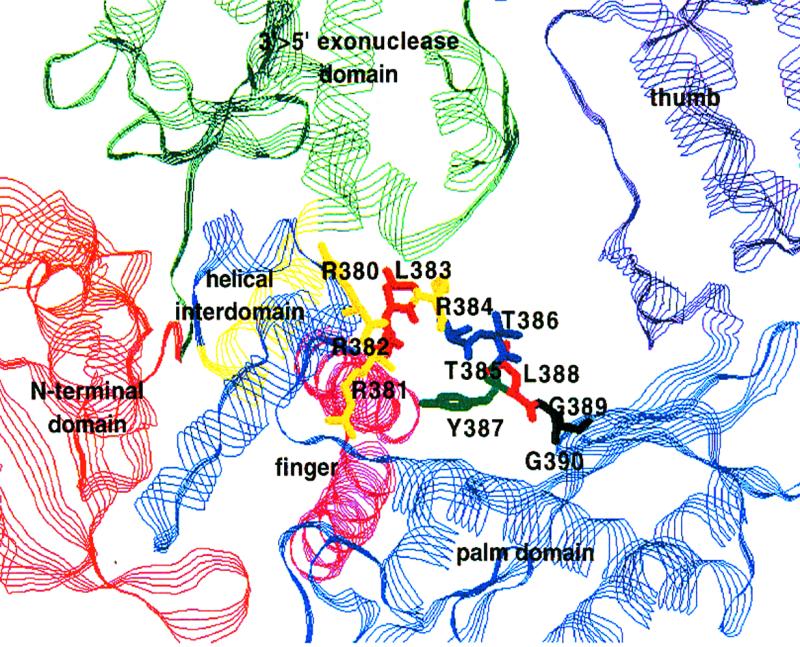
Model of Tag pol showing a view of the flexible loop containing the Y-GG/A motif. The loop follows the helical interdomain (yellow/blue) at the C-terminal end of the exonuclease domain (green) and runs out into a β-sheet of the active site (palm domain, blue). It is located close to the main groove on the surface of the protein, running from the upper left corner to the connection of palm and thumb domain in this presentation. The groove is known to harbour the template for DNA synthesis, the primer-end facing in the direction of the palm domain and the thumb (6). In this model derived from the crystal structure of T.gorgonarius DNA polymerase without ligands, Tyr387 is facing away from the groove and turned towards the fingers domain (pink) containing the dNTP binding motif B. It is reasonable to assume that the loop becomes reordered upon binding of the double-stranded DNA template (K.-P.Hopfner, personal communication).
It was noted that any factor causing difficulties in primer terminus translocation for the next round of replication will influence the equilibrium between DNA synthesis and degradation (2). The conformation and the flexibility of the loop seems to be such a factor. We propose that, in addition to template (de)stabilization effects of the amino acid residues, the fragile conformation of the loop is a factor that contributes to the diverse phenotypes seen after mutations in the Y-GG/A motif. Final evidence for the exact role of the tyrosine in the flexible loop will be accessible only with the help of a binary crystal structure from the relevant DNA polymerase with duplex DNA bound to its polymerase active site.
Acknowledgments
ACKNOWLEDGEMENTS
We thank M. Greif and M. Schmidt for experimental assistance with the activity and fidelity assays. The help of K. Paliakasis in the modelling process is kindly acknowledged. This work was supported by the DFG Graduiertenkolleg Biotechnologie (GRK 95/3-97), the Commission of European Union (Biotech Generic project Extremophiles as Cell Factories, contract BIO4CT975058), the Consiglio Nazionale delle Ricerche (Progetto Finalizzato Biotecnologie), the Ministero della Universita e Ricerca Scientifica e Tecnologica (Progetto ‘Biomolecole per la salute umana’) and the ‘Fonds der Chemischen Industrie’.
REFERENCES
- 1.Joyce C.M. and Steitz,T.A. (1994) Annu. Rev. Biochem., 63, 777–822. [DOI] [PubMed] [Google Scholar]
- 2.Truniger V., Lázaro,J., Salas,M. and Blanco,L. (1996) EMBO J., 15, 3430–3441. [PMC free article] [PubMed] [Google Scholar]
- 3.Pisani F.M., De Felice,M. and Rossi,M. (1998) Biochemistry, 37, 15005–15012. [DOI] [PubMed] [Google Scholar]
- 4.Joyce C.M. (1989) J. Biol. Chem., 254, 10858–10866. [PubMed] [Google Scholar]
- 5.Pisani F.M., Manco,G., Carratore,V. and Rossi,M. (1996) Biochemistry, 35, 9158–9166. [DOI] [PubMed] [Google Scholar]
- 6.Wang J., Sattar,A.K.M.A., Wang,C.C., Karam,J.D., Konigsberg,W.H. and Steitz,T.A. (1997) Cell, 89, 1087–1099. [DOI] [PubMed] [Google Scholar]
- 7.Hopfner K.-P., Eichinger,A., Engh,R.A., Laue,F., Ankenbauer,W., Huber,R. and Angerer,B. (1999) Proc. Natl Acad. Sci. USA, 96, 3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niehaus F., Frey,B. and Antranikian,A. (1997) Gene, 204, 153–158. [DOI] [PubMed] [Google Scholar]
- 9.Canganella F., Jones,W.J., Gambacorta,A. and Antranikian,G. (1998) Int. J. Syst. Bacteriol., 48, 1181–1185. [DOI] [PubMed] [Google Scholar]
- 10.Edgell D.R., Klenk,H.-P. and Doolittle,W.F. (1997) J. Bacteriol., 179, 2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cann I.K.O., Ishino,S., Nomura,Y., Sako,Y. and Ishino,Y. (1999) J. Bacteriol., 181, 5984–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uemori T., Ishino,Y., Doi,H. and Kato,I. (1995) J. Bacteriol., 177, 2164–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klenk H.P., Clayton,R.A., Tomb,J., White,O., Nelson,K.E., Ketchum,K.A., Dodson,R.J., Gwinn,M., Hickey,E.K., Peterson,J.D. et al. (1997) Nature, 390, 364–370. [DOI] [PubMed] [Google Scholar]
- 14.Bult C.J., White,O., Olsen,G.J., Zhou,R., Fleischmann,R.D., Sutton,G.G., Blake,J.A., Fitzgerald,L.M., Clayton,R.A., Gocayne,J.D. et al. (1996) Science, 273, 1058–1073. [DOI] [PubMed] [Google Scholar]
- 15.Kawarabayasi Y., Sawada,M., Horikawa,H., Haikawa,H., Hino,Y., Yamamoto,S., Sekine,M., Baba,S., Kosugi,H., Hosoyama,A. et al. (1998) DNA Res., 5, 55–76. [DOI] [PubMed] [Google Scholar]
- 16.Smith D.R., Doucette-Stamm,L.A., Deloughery,C., Lee,H.-M., Dubois,J., Aldredge,T., Bashirzadeh,R., Blakely,D., Cook,R., Gilbert,K. et al. (1997) J. Bacteriol., 179, 7135–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cann I.K.O., Komori,K., Toh,H., Kanai,S. and Ishino,Y. (1998) Proc. Natl Acad. Sci. USA, 95, 14250–14255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishino Y., Komori,K., Cann,I.K.O. and Koga,Y. (1998) J. Bacteriol., 180, 2232–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uemori T., Sato,Y., Kato,I., Doi,H. and Ishino,Y. (1997) Genes Cells, 2, 499–512. [DOI] [PubMed] [Google Scholar]
- 20.Lawyer F.C., Stoffel,S., Saiki,R.K., Chang,S.-Y., Landré,P.A., Abramson,R.D. and Gelfand,D.H. (1993) PCR Methods Appl., 2, 275–287. [DOI] [PubMed] [Google Scholar]
- 21.Perler F.B., Kumar,S. and Kong,H. (1996) Adv. Prot. Chem., 48, 377–435. [DOI] [PubMed] [Google Scholar]
- 22.Frey B. and Suppmann,B. (1995) Biochemica, 2, 34–35. [Google Scholar]
- 23.Barnes W.M. (1994) Proc. Natl Acad. Sci. USA, 91, 2216–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanahan D. (1983) J. Mol. Biol., 166, 557–580. [DOI] [PubMed] [Google Scholar]
- 25.Keohavong P. and Thilly,W.G. (1989) Proc. Natl Acad. Sci. USA, 86, 9253–9257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Provost G.S., Kretz,P.L., Hamner,R.T., Matthews,C.D., Rogers,B.J., Lundberg,K.S., Dycaico,M.J. and Short,J.M. (1993) Mutat. Res., 288, 133–149. [DOI] [PubMed] [Google Scholar]
- 27.Kong H., Kucera,R.B. and Jack,W.E. (1993) J. Biol. Chem., 268, 1965–1975. [PubMed] [Google Scholar]
- 28.Polesky A.H., Steitz,T.A., Grindley,N.D.F. and Joyce,C.M. (1990) J. Biol. Chem., 265, 14579–14591. [PubMed] [Google Scholar]
- 29.Leatherbarrow R.J. (1992) GraFit (version 3.0), Erithacus Software Ltd., Staines, UK.
- 30.Higgins D.G. and Sharp,P.M. (1988) Gene, 73, 237–244. [DOI] [PubMed] [Google Scholar]
- 31.Vried G. (1990) J. Mol. Graph., 8, 52–56. [DOI] [PubMed] [Google Scholar]
- 32.Freemont P.S., Friedman,J.M., Beese,L.S., Sanderson,M.R. and Steitz,T.A. (1988) Proc. Natl Acad. Sci. USA, 85, 8924–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu P., Nossal,N. and Benkovic,S.J. (1998) Biochemistry, 37, 14748–14755. [DOI] [PubMed] [Google Scholar]
- 34.Kornberg A. and Baker,T.A. (1992) DNA replication. Freeman, San Francisco, CA.
- 35.Truniger V., Blanco,L. and Salas,M. (1999) J. Mol. Biol., 286, 57–69. [DOI] [PubMed] [Google Scholar]
- 36.Dong Q., Copeland,W.C. and Wang,T.S.-F. (1993) J. Biol. Chem., 268, 24163–24174. [PubMed] [Google Scholar]
- 37.Southworth M.W., Kong,H., Kucera,R.B., Ware,J., Jannasch,W.H. and Perler,F.B. (1996) Proc. Natl Acad. Sci. USA, 93, 5281–5285. [DOI] [PMC free article] [PubMed] [Google Scholar]