

Abstract
Introduction:
The standard of care for patients with infantile-onset Pompe disease (IOPD) is enzyme replacement therapy (ERT), which does not cross the blood brain barrier. While neuromuscular manifestations of IOPD are well-described, central nervous system (CNS) manifestations of this disorder are far less characterized. Here we describe severe CNS-related neurological manifestations including seizures and encephalopathy in six individuals with IOPD.
Method:
We identified six children with IOPD who developed CNS manifestations such as seizures and/or encephalopathy. We studied their brain magnetic resonance imaging scans (MRIs) and graded the severity of white matter hyperintensities (WMHI) using the Fazekas scale scoring system as previously published. Longitudinal cognitive measures were available from 4/6 children.
Results:
All six IOPD patients (4 males/2 females) had been treated with ERT for 12–15 years. Seizures and/or encephalopathy were noted at a median age at onset of 11.9 years (range 9 – 15 years). All were noted to have extensive WMHI in the brain MRIs and very high Fazekas scores which preceded the onset of neurological symptoms. Longitudinal IQ scores from four of these children suggested developmental plateauing.
Discussion:
Among a subset of IOPD patients on long-term ERT, CNS manifestations including hyperreflexia, encephalopathy and seizures may become prominent, and there is likely an association between these symptoms and significant WMHI on MRI. Further study is needed to identify risk factors for CNS deterioration among children with IOPD and develop interventions to prevent neurological decline.
Keywords: Pompe disease, epilepsy, white matter disease, case series
Introduction:
Pompe disease (glycogen storage disease type II) is an autosomal recessive disorder caused by deficiency of the lysosomal enzyme acid alpha-glucosidase (GAA). This results in the accumulation of glycogen in the heart, skeletal and smooth muscles, and the nervous system 1,2. Pompe disease is broadly classified into two subtypes – infantile onset Pompe disease (IOPD) and late-onset Pompe disease (LOPD). Treatment with enzyme replacement therapy (ERT), available since 2006, earlier diagnosis via newborn screening, higher doses of ERT, and immune modulation have changed the landscape for many children with IOPD, who are now entering adulthood 3–7. However, there are unmet needs and an emerging phenotype in these older patients.
Next generation ERTs that can better target the skeletal muscles are beneficial for patients with IOPD 8, yet ERT does not fully correct the skeletal muscle manifestations and does not cross the blood-brain barrier. ERT-treated individuals with IOPD have neuromuscular manifestations such as scoliosis, obstructive sleep apnea, restrictive lung disease, sensorineural hearing loss, dysarthria, dysphonia, dysphagia, ptosis, lingual weakness, small fiber neuropathy, and a foot-slapping gait 9. On standardized measures of cognition and language, children with IOPD may exhibit relative weaknesses in processing speed, fluid reasoning, visual perception, and receptive vocabulary 10,11. Autopsy reports from ERT-treated individuals with IOPD showed glycogen accumulation in the neurons of the brain and spinal cord 1. Brain imaging studies (MRI, MRS, and CT) on ERT-treated individuals with IOPD have shown that some children have early and progressive white matter hyperintense foci, and neuronal and myelination loss 12–17. We have previously described the use of the Fazekas scoring system for WMHI in children with IOPD and noted that a Fazekas score of >15 is indicative of severe white matter (WM) involvement 18.
In this case series, we describe six long-term ERT-treated children with IOPD who developed seizures and/or encephalopathy. These cases provide new insights to the understanding of the wide clinical spectrum among survivors of IOPD.
Materials and methods:
Participants:
Our cohort consisted of children with biochemically and genetically confirmed IOPD enrolled in research studies at Duke University aimed at understanding the long-term outcomes of individuals with Pompe disease (Pro00001562; ClinicalTrials.gov: NCT01665326) and/or understanding the extent and severity of central nervous system (CNS) involvement (Pro00072329; ClinicalTrials.gov: NCT04639336). Brain MRIs in children on long-term ERT were obtained. This cohort was reviewed for children with evidence of progressive CNS disease, including seizures, hyperreflexia and/or neurodevelopmental deterioration. We conducted a retrospective and prospective review of medical records of these six patients to explore patient characteristics and contributing factors for the presence of CNS-related disease manifestations.
Informed consent:
Patients 1–5 were consented to the natural history study and/or the CNS study at Duke University. Written consent was obtained from parents of these children, as per the study protocols and Duke Institutional Review Board. Patient 6 was consented to a local research study abiding by their local institutional review board with deidentified clinical data shared.
Medical records:
All six children with IOPD were under the clinical care of their local geneticists at the time of their most recent assessments. We received clinical information from each patient’s most recent clinic visit, and relevant interim history. We reviewed medical records to capture each patient’s pathogenic variants, cross reactive immunologic material (CRIM) status, age at diagnosis, treatment with ERT, immune tolerance induction (ITI) protocol if any, dose of ERT, and baseline and follow up echocardiography findings for left ventricular mass index (LVMI) and ejection fraction (EF). Past history was reviewed for vision, hearing, speech, feeding difficulties and developmental trajectory along with clinical details on seizure semiology, onset, neurological examination findings, family history of seizures and related conditions, medications given, electroencephalogram (EEG ) reports, whole genome sequencing (WGS) and whole exome sequencing (WES) reports, if available. We also reviewed standardized laboratory biomarkers such as creatine phosphokinase (CK), AST and urinary glucose tetrasaccharide (Glc4).
Neuroimaging:
Brain MRI scans were acquired on a Siemens (Munich, Germany) 3.0T MRI Scanner (MAGNETOM Trio) using an 8-channel head coil at Duke University. All scans at Duke were performed on the same scanner without contrast administration or sedation. T1-weighted, T2-weighted, and fluid-attenuated inversion recovery (FLAIR) images were obtained. We also received brain MRI scans through collaborative efforts from local geneticists. We followed our previously described methodology using a grading system to quantify WMHI in children with IOPD 11,19. Three experienced neuroradiologists (J.M.P., M.M., and W.W.) examined the brain MRIs in this current study. All three neuroradiologists were blinded to the patients’ clinical information. Two neuroradiologists (M.M. and W.W.) graded the severity of the WMHI using a Fazekas scale scoring system and J.M.P. adjudicated the scores if there were any discrepancies, except for patients 5 and 6 who were scored by only 1 rater. The brain was systematically divided into 9 brain areas, and each area was assigned a score ranging from 0 (absent WMHI ) to 3 (severe WMHI). One brain area (brainstem decussation, previously described in our earlier publication) was excluded due to technical considerations. These individual area scores were summed to obtain a total score (possible range = 0 to 27). A score of 15 or more was considered severe WM involvement. J.M.P. also provided scan reports describing the white and gray matter areas, ventricular size, vascular structures, and abnormalities in adjacent structures.
Cognitive assessments:
A clinical child psychologist (G.A.S.) with expertise in Pompe disease completed psychological evaluations on 4 children enrolled in the Duke research studies. The assessments were completed at multiple time points using standardized measures, including the Leiter International Performance Scale-Third Edition (Leiter-3), age-appropriate Wechsler scales, the Beery-Buktenica Developmental Test of Visual-Motor Integration, 6th Edition (Beery-Buktenica), and the Peabody Picture Vocabulary Test (PPVT, one-word receptive vocabulary). The methodology was detailed in our most recent publication 11,19. These assessments yielded standard scores with a mean of 100 and a standard deviation (SD) of 15. Scores between 85 and 115 were considered to be in the average range in comparison to same-aged, typically developing peers. In the current study, we report the Leiter-3 Nonverbal IQ, the Wechsler Full Scale IQ, the PPVT standard score, and age-based standard scores for the Beery VMI, Visual Perception, and Motor Coordination subtests of the Beery-Buktenica. We also included the raw scores (total number of correct items) for each of the four subtests on the Leiter-3: Figure Ground, Form Completion, Classification/Analogies, and Sequential Order.
Data availability:
Anonymized data not published within this article will be made available by request from any qualified investigator.
Results:
Of 33 children with IOPD in our cohort, 6 (18%) (4 males/2 females) developed seizures or encephalopathy at a median age at onset of 11.9 years (age range 9 – 15 years). All presented with the typical features of Pompe disease and also had disease progression as previously described in IOPD until the onset of seizures/ encephalopathy. Details are available in Table 1. One patient (Patient 1) was cross-reactive immunologic material negative (CRIM−); all others (5/6) were cross-reactive immunologic material positive (CRIM+). Median age at diagnosis of Pompe disease was 3.75 months (range prenatal - 6 months). Median age at ERT initiation was 5 months (range 18 hours of life – 6 months), and all six children were on long-term ERT for 12–15 years (20–40 mg/kg every other week or weekly regimen). Among those children who developed seizures, the median age at seizure onset was 12.5 years (range 10–15 years). All six children had extensive WMHI on brain MRI, primarily in the supratentorial regions of the brain with extension to subcortical regions. Total Fazekas scale scores were between the range of 18 and 24, with a median of 20, which are indicative of severe WM abnormalities.
Table 1:
Patient characteristics of six children with IOPD who developed seizures/neurological manifestations
| Patient ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 |
|---|---|---|---|---|---|---|
| Sex | M | F | M | F | M | M |
| History of consanguinity | No | No | first degree | first degree | No | No |
| Pathogenic variants in the GAA gene (NM_000152.5) |
c.[546+2T>C];[546+2T>C] | c.[1210G>A];[2481+110_2646+39del] | c.[1327-A>G];[1327-2A>G] | c.[2104C>T];[2104C>T] | c.[1221C>A;1281G>T];[1564C>T;2296T>A] | c.[1935C>A],[1935C>A] |
| Age at diagnosis | 3.5 months | 5.5 months | Prenatally | At birth | 6 months | 4 months |
| Age at ERT initiation a | 4 months b | 6 months | 18 hours of life | 1.5 months | 6 months | 6 months |
| ERT Dose at most recent visit (age at dose increase) | 40 mg/kg EOW (8.3 years) | 40 mg/kg/week (9.5 years) | 40 mg/kg weekly (10 years) | 40 mg/kg/week (4.8 years) | 40 mg/kg/week (10 years) | 20 mg/kg EOW |
| WES/genom e sequencing or other gene panelc | Not done | WES - Negative | WGS-Negative | Autism/intellectual disability gene panel negative | Not done | Not done |
| Family history of seizures or related conditions | No | No | No | NA | No | No |
M- male, F- female, ERT- enzyme replacement therapy, EOW- every other week, IVIG- Intravenous immunoglobulin, PUO- Pyrexia of unknown origin and infectious causes were ruled out, WMHI- white matter hyperintensities, NA-unavailable.
All children received ERT dose of 20 mg/kg EOW at initiation.
Patient 1 was the only CRIM- in the cohort. He received rituximab, methotrexate, and IVIG (4 months age), as per the Immune Tolerance Induction (ITI) protocol developed at Duke University. All other patients did not receive immune modulation and were all CRIM+.
To explore other causes of seizures, WES/WGS was done on genetic variants other than Pompe disease-causing GAA variant.
Table 1 outlines the patient characteristics, and provides a summary of the onset of seizures, brain MRIs and scoring of WMHI as well as possible contributing factors.
Neuroimaging:
Brain MRIs from all six patients were noted to have bilateral, extensive WMHI in the supratentorial WM involving most lobes, and extending to the subcortical WM (Table 2 and Figure 1). All six patients received high total Fazekas scale scores (≥15) (Table 2).
Table 2:
Baseline and most recent MRI characteristics of six children with IOPD presenting with a history of seizures/neurological manifestations. MRI was graded according to a modified Fazekas score (maximum score = 27).
| Patient ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MRI findings (age at assessment) | Bilateral, extensive WMHI throughout supratentorial areas; involvement of subcortical and cerebellar WM (7 years) | MRI not done prior to seizure onset | Bilateral, extensive WMHI throughout supratentorial areas; involvement of basal ganglia and thalami, volume loss, ventricular enlargement (13 years) | Bilateral, extensive WMHI throughout supratentorial areas; thinning of corpus callosum, ventricular enlargement* (8 years) | Bilateral, extensive WMHI throughout supratentorial areas (6.5 years) | Bilateral, extensive WMHI throughout supratentorial areas; basal ganglia involvement, severe cortical atrophy d (12 years) | ||||||
| total FSS (age at assessment) | NA | MRI not done prior to seizure onset | 24 | 19 | 20 | NA | ||||||
| Most recent MRI characteristics | ||||||||||||
| Patient ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | ||||||
| Changes in MRI findings from baseline (age at assessment) | Substantial involvement of the brainstem (10 years) | first MRI - bilateral, extensive WMHI in all lobes of the supratentorial WM (15 years) | Involvement of brainstem and cerebellum (13 years) | Progression of WMHI, marked involvement of the basal ganglia (11 years) | Lateral and third ventricles mildly dilated* (11 years) | Progressive WM hyperintense foci, cortical atrophy, cystic WM degeneration, volume loss, cerebellar microhemorrhages (16 years) | ||||||
| total FSS | 18 | 21 | 24 | 21e | 22e | 23 | ||||||
WMHI- white matter hyperintensities, FSS - Fazekas scale scores to grade the severity of white matter hyperintense foci in brain MRI. NA-unavailable.
as per reports in the medical records from outside Duke scans.
The Fazekas scores for these studies were assessed by a single radiologist.
Fig. 1.

Brain MRIs from five children with infantile onset Pompe disease with evidence of progressive CNS disease. All six patients had bilateral extensive hyperintense foci in all the lobes of the supratentorial brain as seen on brain MRI and five are shown here. Additionally, Patients 1 and 3 had brainstem involvement, and Patients 3, 4, and 6 had basal ganglia involvement. Patient 1 (CRIM−): Red arrows show CNS involvement in the three axial T2-FLAIR images and a sagittal T1 at the level of (a) centrum semiovale, (b) cerebellum, and (c) posterior limb of internal capsule at age 7 years. (d) New involvement of the brainstem region at age 10 years post-seizures. Patient 2–6 were CRIM+. Patient 2: Orange arrows in an axial T2-FLAIR image at level of lateral ventricles involving the periventricular and subcortical areas at 15 years. Patient 3: Green arrows in the two axial T2-FLAIR images at the level of centrum semiovale (a) at 13.1 years, and at deep gray structures (b – lentiform nucleus and thalamus) at 13.25 years age. Patient 4: Yellow arrows in two axial T2-FLAIR images at the level of (a) centrum semiovale at age 8 years, and (b) involvement of both basal ganglia and internal capsules at age 11 years. Patient 5: Blue arrows in the axial T2-FLAIR images at the level of corona radiata at age 11 years. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Cognition:
A graphical representation of pertinent neuropsychological measures over time from patients 2–5 can be seen in Figure 2. We report the Leiter-3 Nonverbal IQ, the Wechsler Full Scale IQ, the PPVT standard score, and standard scores for the Beery VMI, Visual Perception, and Motor Coordination subtests (Figure 2 and Supplemental Table 1). Figure 2 also includes the raw scores (total number of correct items) for each of the four subtests on the Leiter-3: Figure Ground, Form Completion, Classification/Analogies, and Sequential Order, showing scores on the Leiter-3 plateauing without regression among this cohort.
Fig. 2.
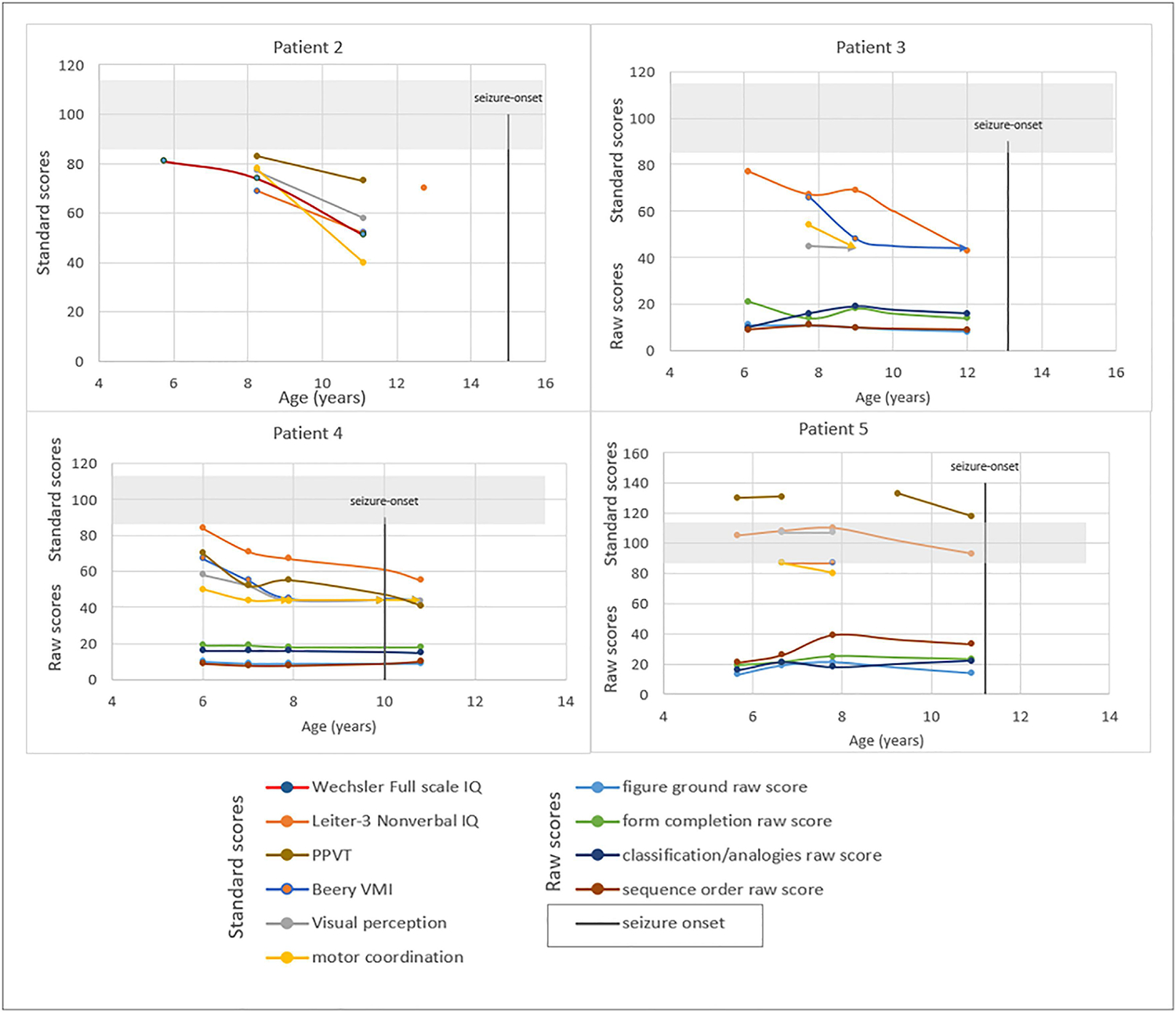
Neuropsychological findings over time in children with infantile onset Pompe disease who developed neurological changes (Patient 2–5). Age based standard scores (mean = 100, SD = 15) are plotted for each administration of the Wechsler scale, the Leiter-3, the PPVT, and subtests of the Beery-Buktenica (Beery VMI, Visual Perception, and Motor Coordination) for patients 2–5 at increased ages. The light gray shaded area on each graph represents the average range. The raw scores (total number of items correct) for each subset of the Leitter-3 are also plotted to illustrate each child’s performance over time on this measure.
Cases:
To systematically understand the natural disease progression, we describe each case (Patient IDs 1–6) with a brief history at the time of diagnosis of Pompe disease and subsequent neurologic course.
Patient 1:
Patient 1 was a CRIM− male child with IOPD who was born preterm at 30.6 weeks of gestational age. His echocardiogram at age 20 days showed an LVMI of 78.46 g/m2 (normative range of mean LVMI for preterm babies=37.08–48.57)20. At age 3 months, CK and transaminases were elevated, and urinary glucose tetrasaccharide (Glc4) was elevated at 59.7 mmol/mol creatinine (normal upper limit was 19). At age 4 months, he was noted to have head lag, skeletal muscle weakness and hypotonia. ERT (20 mg/kg every other week) and ITI with rituximab, methotrexate and IVIG were initiated 4, with good CD19 recovery, low antibody titers and no untoward immune response. At 11 months, there was no head lag when he was pulled to sit but he remained unable to sit unsupported. At age 2 years, he was able to sit unsupported and had pincer and raking grasps; however, he only pointed to objects with no intelligible words and could not recognize his body parts or animal sounds. At age 6 years, he was able to move his arms against gravity but had no leg movements. At age 7 years, he was hospitalized with an altered level of consciousness, and had severe neurological decline, evolving to ventilator dependence. Local brain MRI showed bilateral extensive WMHI within the centrum semiovale, internal capsules, and temporal WM, extending to subcortical areas and the bilateral cerebellum (Figure 1). The ERT dose was increased from 20 to 40 mg/kg every other week.
At 10 years, he began to have seizures with a semiology of intermittent right gaze deviation and abnormal tongue movements. An EEG showed right temporal epileptiform discharges. He was started on anticonvulsant therapy with good seizure control; however, he continued to have a fluctuating level of consciousness. At 11 years, he presented with respiratory infection, fever and lethargy which progressed to worsened encephalopathy. Follow-up EEG showed generalized slowing (more severe in the left posterior and parasagittal regions). Follow-up brain MRI at age 11.5 years showed global volume loss along with interval progression of WM involvement, with bilateral subcortical WMHI noted in the basal ganglia, thalamus, internal capsule and brainstem. The total Fazekas score was 18 (severe). The child is currently 12 years of age, and continues to be ventilator dependent and nonambulatory with severe encephalopathy.
Patient 2:
Patient 2 was a CRIM+ female patient with IOPD born at term by C-section. At age 6 weeks, she had dyspnea, feeding difficulties, slow weight gain, failure to thrive, and generalized hypotonia. At age 5.5 months, she had head lag and was unable to roll onto her back. Lab studies showed an elevated CK and transaminitis. ERT was initiated at 20 mg/kg every other week. At age 6 months, a brain CT scan was normal for age. At age 1.8 years, she was noted to have mild hypotonia and hypernasal speech and was able to sit but not stand independently. At age 2.5 years, she was walking with a wide-based gait using ankle-foot orthoses. She could with difficulty go from squatting to standing. At age 7 years, the ERT dose was increased to 20 mg/kg weekly with subsequent improvements in her strength. At age 8 years, she was doing well at school with support services, and was physically active. A neuropsychological assessment completed locally revealed a below-average Full-Scale IQ on the Wechsler Intelligence Scale for Children-Fourth Edition, with weaknesses in visual-motor integration, visual perception, and motor coordination on the Beery-Buktenica VMI. At age 9.5 years, the dose of ERT was increased to 40 mg/kg weekly, and the family reported improvements in ptosis, energy levels, and speech. At age 11 years, a follow-up neuropsychological evaluation showed significantly below-average Full-scale IQ (Wechsler scale) and nonverbal intellectual functioning. By age 13 years the patient was using a walker and wheelchair. A speech-language evaluation showed hypernasality, moderate-severe dysarthria and severe impairments in receptive, expressive and pragmatic language [CELF-5 Core Language Index=57 (average 86–114)]. The Leiter-3 nonverbal IQ administered locally was also significantly below average.
At age 15 years, she developed recurrent electrographic right temporal seizures with a focal dyscognitive semiology. Anticonvulsant therapy was initiated. At age 15 years, brain MRI showed global diffuse WMHI with extension to the subcortical WM. The total Fazekas scale score (FSS) was 21. WES showed no genetic cause for leukodystrophy or seizures. By age 16 years, the seizures had become medically refractory to anticonvulsants. Laser ablation of a presumed epileptogenic focus was planned; however, prior to this procedure the patient died after a brief intercurrent illness at age 16 years.
Patient 3
Patient 3 was born to first-degree consanguineous parents, and IOPD was diagnosed in utero based on a known family history. ERT (20 mg/kg/every other week) was initiated at 18 hours of life. He was rolling by 4–5 months, sitting without support by 6 months, and walking by 13 months. At age 6 years, he was seen at Duke University. At that time, his Nonverbal IQ on the Leiter-3 was assessed as being below average although parents reported good academic achievement. At 9 years, his Nonverbal IQ score was significantly below average. The ERT dose was increased to 40 mg/kg weekly. At 10 years, he had worsening weakness, fatigue and frequent falls. By evaluation at age 12 years he had regressed academically, with slurred and difficult to understand speech and myopia that had progressed to legal blindness. Neurological examination showed a foot slapping, Trendelenburg gait, global hyperreflexia and bilateral ankle clonus. Brain MRI showed extensive symmetric T2 hyperintensity throughout the supratentorial WM, basal ganglia, and thalami with a Fazekas scale score of 25. His Nonverbal IQ on the Leiter-3 had continued to decline since the previous evaluation at age 9 years. An examination of the raw scores showed that his skills had regressed on all the Leiter-3 subtests. At age 13 years, he developed aspiration pneumonia, progressing to respiratory failure with eventual gastrostomy and ventilator dependence. At about this time, he developed pyrexia to 39 C, the origin of which remained obscure despite an infectious disease evaluation.
At age 13 years, he started anticonvulsants following a presentation with focal status epilepticus with right-sided motor seizures. Exam showed generalized hypotonia with global areflexia aside from minimal reflexes in the left upper limb. Response to painful stimulus was limited to blinking. Brain MRI showed bihemispheric, frontally-predominant progression of diffuse WMHI (Figure 1) with involvement of descending WM tracts in the brainstem and cerebellum. The total FSS was 24. The striatum, thalamus and midbrain showed T1 hyperintensity. MRS obtained at the local hospital showed decreased NAA peak, increased choline peak, and decreased creatinine peak. EEG showed generalized slowing and left hemispheric epileptiform discharges. WGS report was negative for other causes of seizures. A month later, he developed mild pyrexia unexplained despite infectious workup. CK at that time was elevated at 1360 U/L. The child died at age 14 years.
Patient 4
Patient 4 was the term product of first-degree consanguineous relations. She presented at birth with hypotonia, poor feeding, respiratory distress, and cardiomegaly. A diagnosis of CRIM positive Pompe disease was confirmed, and ERT was initiated at 1.5 months of life at 20 mg/kg every other week. She had developmental delays from the first months of life. At age 5 years, the dose of ERT was increased to 40 mg/kg weekly. Her Leiter-3 nonverbal IQ was only slightly below average at 5 years but was significantly below average by age 7 years. By 8 years she was noted to have generalized muscle weakness, foot drop, wide based gait, lumbar lordosis, scoliosis, scapular winging, hypotonia, hyporeflexia, and bilateral sensorineural hearing loss. Her Leiter-3 Nonverbal IQ score remained stable but significantly below average. At school, she was sociable, and met all goals included in her special education program despite frequent falls and headaches. Brain MRI at age 8 years showed diffuse supratentorial WMHI with extension to the subcortical WM (Figure 1).
At age 9 years, she developed a gait instability and balance problems. An EMG showed an axonal motor neuropathy in the lower extremities. She developed a tremor in all four extremities during weight-bearing and exertion, which was attributed to muscle weakness. She also developed episodes of loss of muscle tone and falling without loss of consciousness triggered by laughter. EEG showed generalized, frontally-predominant background slowing and bifrontal epileptiform discharges. She was evaluated by neurologists who did not feel her episodes were clearly epileptic in nature, though she may be at risk for seizures based on her EEG. Her local geneticist evaluated her for other neurological conditions using an autism/intellectual disability gene panel which was unremarkable. Brain MRI at age 11 years showed progression of supratentorial WMHI extending to subcortical areas, along with a marked involvement of the basal ganglia. At age 11 years, clinical evaluation revealed worsened muscle weakness and hypotonia, dysphagia, bowel and bladder incontinence, and severe gait instability with increasing wheelchair dependence. On examination, she had significant weakness and global hyporeflexia. She had muffled speech, producing one-word responses, and demonstrated significant impairment in receptive language. There was a significant decline in her Leiter-3 Nonverbal IQ score in comparison to age 7 years (Supplemental Table 1).
Patient 5
Patient 5 was born full term to nonconsanguineous parents. At birth, he presented with dyspnea, feeding difficulties, and hypotonia. At 6 months of age, he was diagnosed with CRIM+ IOPD and was started on ERT at 20 mg/kg every other week. By age 3 years, he was unable to sit up by himself; by 4.5 years, he could scoot and use a wheelchair but remained unable to walk or stand and had dysarthria and expressive language delay. At 6 years, ERT was increased to 40 mg/kg every other week. At age 6.5 years, baseline brain MRI showed diffuse WMHI with relative sparing of the temporal lobes. The total FSS was 20 (severe). A follow-up brain MRI at 8 years continued to show WMHI with a total FSS of 21 (severe). Nevertheless, his Leiter-3 Nonverbal IQ remained at the upper end of the average range. At age 10 years his ERT dose was increased from 40 mg/kg every other week to weekly. At 11 years, when evaluated at Duke, he was globally hypotonic and areflexic, with only antigravity movement of the upper extremities and no movement of the lower extremities. He had decreased facial muscle strength and palate elevation, with severe impairment of speech and a Leiter-3 Nonverbal IQ at the lower end of the average range. Follow-up brain MRI showed no changes from previous scans.
At age 11 years, he developed spells of dizziness. He was taken to the local ED when he had a 30-second focal seizure (semiology of dizziness, followed by unresponsiveness and left arm twitching). A follow-up brain MRI showed no changes from previous scans. EEG showed excessive background slowing and intermittent polymorphic delta activity.
Patient 6
Patient 6 presented in the neonatal period with tachypnea and heart failure. He was diagnosed with CRIM+ IOPD at age 4 months. At age 6 months, ERT was initiated. He was areflexic, immobile due to severe muscle atrophy, and ventilator and gastrostomy tube dependent. In early childhood, he was able to look around, and would communicate with his parents using tongue gestures. No formal cognitive assessments were conducted.
At 12.5 years of age, the child presented with sudden-onset lethargy. Brain MRI showed severe cortical atrophy along with extensive non-enhancing, patchily diffusion-restricted signal abnormalities in the central regions of the brain, including internal and external capsules and basal ganglia. Evidence of chronic left parietal and right thalamic ischemic stroke were noted. Etiology for these infarcts could not be identified. An EEG identified right posterotemporal electrographic seizures. Anticonvulsant therapy was initiated with eventual return of mental status to his pre-hospitalization baseline. At 16 years of age, he presented to the hospital with altered level of consciousness, and was admitted to the ICU after identification of acute on chronic renal failure, likely due to neurogenic bladder and subsequent obstructive uropathy and concurrent urinary tract infection. Follow-up brain MRI identified interval progression of cortical atrophy along with diffuse, bilateral supratentorial WMHI with involvement of the basal ganglia. Brainstem atrophy was particularly pronounced. Cystic changes were noted within the left posterior frontal lobe and left superior parietal lobule as well as a tiny acute cerebellar infarct together with bilateral cerebellar petechial microhemorrhages. The total FSS was 23. After two weeks of inpatient treatment, all electrolyte derangements had been corrected; however, the patient was still severely encephalopathic. By age 16.5 years, he was non-responsive without voluntary movements, had occasional eye-opening but did not fix or follow, and could no longer make gestures with his tongue. He continued on ERT (20 mg/kg every other week). EEG showed profound background slowing.
The patient expired at 18 years after a three-month hospitalization following a hypotensive event, bacteremia, and multi-organ failure.
Discussion
In this case series, we describe severe neurological manifestations in a subset of patients with IOPD. In the current study, patients with IOPD developed seizures (n=5), hyperreflexia or clonus (n=1) and/or pyrexia of unknown origin (n=1) (Table 1). These neurological presentations have been previously described as case studies (n=4 with seizures, n=1 with hyperreflexia, and n=2 with pyrexia of unknown origin) (Table 2). Cognition among our cohort was also affected, with a decline in IQ score over time (due primarily to developmental plateauing) among three of the four patients who were longitudinally assessed. Of note, the presentation of each of these patients was consistent with the typical features of Pompe disease until the onset of CNS symptoms.
In the current study, all six patients had extensive WMHI on brain MRIs, with corresponding high (≥15) total Fazekas scale scores (median score 21; range 18–24) (Table 1). The WMHI seen on brain MRI preceded CNS involvement in this cohort. WMHI have been described in brain MRIs of children with IOPD 4, consistent with evidence of involvement of the corticospinal tract and deep gray structures (e.g., basal ganglia) 1,12,15,16,18,22,24 based on radiologic and autopsy findings 17. It has been conjectured that some of these hyperintense foci could relate to water following an osmotic gradient into cells in which glycogen has accumulated 22. Converging lines of evidence including DTI and biomarkers such as plasma neurofilament light chain suggest CNS injury including axonal destruction and neuronal damage 24,28 among at least a subset of individuals with IOPD. However, it must be borne in mind that the relationship between clinical status and the CNS radiologic findings among individuals with IOPD is still being assessed. To that end, radiologic grading tools (e.g., the Fazekas scoring system) can provide an objective and quantifiable measurement of radiologic progression in IOPD and other disorders18,24.
Among our cohort, radiologic and clinical findings suggest a predilection for the involvement of not only WM but also deep gray structures, particularly the hypothalamus and basal ganglia. Radiologic involvement of deep gray structures (particularly the basal ganglia) has been recently described in various different publications in 11 other patients15,16,18,22,24. The basal ganglia serve a variety of functions, including control of voluntary motor movements, procedural learning, habit forming, cognition, behavior and emotions; consistent with the clinical manifestations in patients with Pompe disease in varying degrees.
Two children presented with fever around the onset of seizures (Table 1). Patient 1’s fever could be related to an ongoing respiratory infection. However, Patient 3 had pyrexia of unknown origin, with infectious causes ruled out. The presence of intractable fever of unknown origin or hyperthermia has been previously described in three children with IOPD (Table 2). Two children had evidence of brainstem involvement, and the other was suspected to have dysregulated thermoregulation at the level of the CNS. An autopsy report from 1 of these 3 children confirms widespread brain involvement. In our cohort, Patients 1, 3, and 6 had findings of brainstem involvement on MRI. While it may be speculated that otherwise-unexplained fever among children with IOPD can be associated with significant CNS involvement affecting autonomic control of thermoregulation, this association awaits further corroboration.
Based on the radiological and clinical evidence described above, we feel there is ground to propose that Pompe disease be considered a mixed disorder with potential to involve both CNS and muscle. It seems likely that with the advent of ERT and resulting prolonged survivorship among children with IOPD, a phenotype with potential for significant CNS involvement has been revealed. Ideal management of children with IOPD will need to take this potential comorbidity into account. Ethical considerations and counseling regarding the continuation of ERT in the context of neurodegeneration may also need to be addressed, depending on the clinical context.
While it seems clear that there is a phenotype of IOPD that despite ERT is associated with CNS dysfunction, including radiologic markers of WM injury, intellectual and motoric deterioration and seizures, many questions remain. It is important to note that the clinical significance of WMHI among children with IOPD remains unclear and is a topic of active clinical investigation. In addition, elucidation of the pathophysiology of neurodegeneration among some children with IOPD, modifiable risk factors, early biomarkers of CNS injury and therapeutic trials remain as important future directions for research in this space. Indeed, the variable pattern of progression among our cohort with respect to seizures and/or cognitive decline suggests the possibility of unknown protective/risk factors. We evaluated 3 cases with WES, WGS or CNS-related targeted gene panel, which returned normal. Further systematic investigation of possible disease-modifying factors is an area under active investigation by our own and other groups.
The current study has certain limitations, primarily ascribable to its being a case series. However, the potential for ascertainment bias is somewhat mitigated by the size of the database from which these cases were drawn. As children with IOPD are living longer, CNS manifestations of the disease are being uncovered. Further study is needed in a larger cohort to better understand the percentage of patients with treated IOPD who develop neurodegeneration and to determine which patients are at greatest risk. Similarly, it is not possible in the context of this study to establish genotype-phenotype correlation, although it is interesting to note that all but one of these patients was CRIM+.
This study demonstrates that severe neurological involvement is a risk for a subset of long-term survivors of IOPD; furthermore, that radiologic involvement (as quantified by elevated Fazekas scores) and rate of progression of the Fazekas score deserves exploration as a potential biomarker that appears to precede clinical deterioration. In addition, our study suggests that deep gray structures (particularly the hypothalamus and basal ganglia) may be at particular risk among this subset of patients. These findings, taken together, lend credence to the notion that IOPD is not merely a myoneuropathy but rather constitutes a mixed CNS/PNS disorder. Glycogen accumulation with subsequent axonal degeneration and neuronal loss are likely to represent the overarching driver of neurological decline in IOPD (Table 2)17,24. Monitoring for radiologic progression with MRI as well as clinically supporting patients with CNS-oriented diagnostics and therapies (including PT, OT and neuropsychological assessments and a detailed neurological assessment) are indicated for children with IOPD. Future directions will involve understanding how best to stratify risk for deterioration among children with IOPD and determining which risk factors might be modifiable, in hopes of better understanding, development of therapies that target the CNS and staving off CNS progression in IOPD.
Supplementary Material
Supplemental table 1: Age-based standard scores on neuropsychological measures from children with IOPD over time (Patient IDs 2–5)
Table 3:
Literature review of individuals with IOPD with a history suggestive of central nervous system involvement
| # | Publication, (n-value) |
Patient characterization | Clinical history of seizures, EEG, and neurological decline | Additional data (radiological findings/autopsy data) |
|---|---|---|---|---|
| IOPD with history of seizures (n=4) | ||||
| 1 | n=1 21 | CRIM- girl, ERT+ ITI regimen initiated at 6 months; had high sustained antibody titers against ERT | Onset of seizures at 5.5 years with semiology of staring and eye rolling, facial pallor, and unresponsiveness to stimuli lasting for 10–30 minutes | –Brain MRI was not done –First report of seizures in Pompe disease |
| 2 | n=1 22 | CRIM+, ERT initiated at 4 months (40 mg/kg EOW) At age 12–16 years, progressive cognitive decline |
Subject had a history of seizures (presumably around 14–16 years) Treated with levetiracetam EEG repeatedly reported diffuse slowing with sharp waves in the anterior derivations, which was interpreted as consistent with diffuse leukoencephalopathy |
–Longitudinal brain MRI (ages 1.9 years-16.5 years) showed progressive extensive WMHI with progressive ventricular enlargement – MRS- Progressive reduction in N-acetyl-aspartate (NAA) (ages 14 – 16.5 years) suggests neuronal loss in the CNS –Etiology of seizures: could not be determined |
| 3 | n=1 23 | IOPD (c.2066_2069delAGCC/p.Glu689Glyfs*6), CRIM status-unavailable, ERT+ITI was initiated at 6 months (20 mg/kg weekly) |
At 3 age months, neurological decline At age 9 months, developed convulsions and progressive neurologic deterioration, and passed away |
Autopsy report: –Severe cerebral and cerebellar atrophy –hypoxic cortical neuronal changes throughout (frontal lobes to cerebellum), enlarged neurons, cytoplasmic vacuolization of lysosomes filled with glycogen (neurons in brainstem, mesencephalon, and basal ganglia and glial cells) |
| 4 | n=1 24 | , CRIM+, identified via NBS and ERT initiated at 29 days | At 12.9 years age, generalized tonic clonic seizures for 2 minutes with hypoxia Resolved with a double dose of anti-convulsant therapy EEG showed epileptiform discharges |
–DTI showed axonal degeneration –Brain MRI showed extensive hyperintense foci at the levels of pons, periventricular WM, corona radiata, and centrum semiovale in the frontal, parietal, and occipital areas |
| IOPD with history of intractable fever (n=3) | ||||
| 5 | n=2 25 | 1.5-year-old and 1.6-year-old children with IOPD in the pre-ERT era (variants – unavailable) | both presented terminal hyperthermia triggered by episodes of urinary and respiratory infections Progress: Intractable fever (for 3-4 weeks) unresponsive to medications progressing to coma and death |
Autopsy on 1 patient: -Moderate edema of brain parenchyma with leptomeningeal congestion; Extensive glycogen in subcortical neurons. EM showed lysosomes of neurons filled with glycogen; Glycogen accumulation in cerebral blood vessel wall, and in the neurons of brainstem, cerebellum, and anterior horn cells in spinal cord – hyperthermia ascribed to injury to autonomic regulatory systems at the level of the CNS in Pompe disease |
| 6 | n=1 26,27 | CRIM-, (c.525delT in homozygosity); ERT initiated at 3 months age | At 4.3 years, patient reported to have died after a period of unexplained hyperpyrexia (intractable fever >42 degrees C, possibly due to brainstem dysfunction) progressing to unstable hypertension, coma, and death. Previously showed low scores on cognitive assessments | Brain MRI, and autopsy data — not available |
| IOPD with hyperreflexia | ||||
| 7 | n=1 14 | IOPD, CRIM- girl | At 4 years age - gait changes, hyperreflexia of the ankles, lower limb spasticity, cognitive impairment. | MRI showed rapidly progressive CNS involvement |
EEG - eletroencephalography, IOPD - infantile onset Pompe disease, CRIM- cross-reactive immunologic material, ERT- enzyme replacement therapy, EOW - every other week, NBS- newborn screening, ITI- immune tolerance induction, MRS - Magnetic resonance spectroscopy, CNS- central nervous system
Funding
This study was funded in part by Sanofi-Genzyme, MA and the Lysosomal Disease Network (LDN). The LDN (U54NS065768) is a part of the National Institutes of Health (NIH) Rare Diseases Clinical Research Network (RDCRN), an initiative of the Office of Rare Diseases Research (ORDR) at the National Center for Advancing Translational Sciences (NCATS). This consortium is funded through a collaboration between NCATS, the National Institute of Neurological Disorders and Stroke (NINDS), and the National Institute of Diabetes and Kidney Diseases (NIDDK). The authors acknowledge the generous support of The Lucas Garrett Pompe Foundation, Inc., which provided philanthropic funding for this research.
Study Funding:
Supported by Sanofi-Genzyme, MA and the Lysosomal Disease Network
Footnotes
DISCLOSURES
Competing interests
D. Kenney-Jung, A. Korlimarla, G. A. Spiridigliozzi, M. Malinzak, G. Nichting, S.-H. Jung, C. Phornphutkul and J. Owen report no disclosures.
W. F. Wiggins is a Strategic Advisor to Qure.ai. He has served on the Medical Advisory Board of the University of Wisconsin-GE CT Protocols Partnership.
A. Sun receives research funding and clinical trial support from Ultragenyx, LogicBio, BioMarin, Aeglea, and Takeda.
R. Wang receives research/grant support from Biomarin Pharmaceuticals and Ultragenyx, has received consulting fees / honoraria from Biomarin Pharmaceuticals, Takeda, and Regenxbio Inc., and owns equity in Biomarin Pharmaceuticals and Regenxbio, Inc.
J. M. Provenzale receives research funding from Bayer, Inc.
P. S. Kishnani has received research/grant support from Sanofi Genzyme and Amicus Therapeutics. She has received consulting fees and honoraria from Sanofi Genzyme, Amicus Therapeutics, Maze Therapeutics, Bayer and Asklepios Biopharmaceutical, Inc. (AskBio). She is a member of the Pompe and Gaucher Disease Registry Advisory Board for Sanofi Genzyme, Pompe Disease Advisory Board for Amicus Therapeutics, and Advisory Board for Baebies. P. S. Kishnani has equity in Asklepios Biopharmaceutical, Inc. (AskBio) and Maze Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pena LD, Proia AD, Kishnani PS. Postmortem Findings and Clinical Correlates in Individuals with Infantile-Onset Pompe Disease. JIMD Reports. 2015;23:45–54. doi: 10.1007/8904_2015_426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genetics in Medicine. 2006;8(5):267–288. doi: 10.1097/01.gim.0000218152.87434.f3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desai AK, Kazi ZB, Bali DS, Kishnani PS. Characterization of immune response in Cross-Reactive Immunological Material (CRIM)-positive infantile Pompe disease patients treated with enzyme replacement therapy. Mol Genet Metab Rep. 2019;20:100475. doi: 10.1016/j.ymgmr.2019.100475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Desai AK, Baloh CH, Sleasman JW, Rosenberg AS, Kishnani PS. Benefits of Prophylactic Short-Course Immune Tolerance Induction in Patients With Infantile Pompe Disease: Demonstration of Long-Term Safety and Efficacy in an Expanded Cohort. Frontiers in Immunology. 2020;11(1727). doi: 10.3389/fimmu.2020.01727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li C, Desai AK, Gupta P, et al. Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefits of early treatment with enzyme replacement therapy and immune tolerance induction. Genetics in Medicine. Published online January 25, 2021. doi: 10.1038/s41436-020-01080-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chien YH, Tsai WH, Chang CL, et al. Earlier and higher dosing of alglucosidase alfa improve outcomes in patients with infantile-onset Pompe disease: Evidence from real-world experiences. Mol Genet Metab Rep. 2020;23:100591–100591. doi: 10.1016/j.ymgmr.2020.100591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khan AA, Case LE, Herbert M, et al. Higher dosing of alglucosidase alfa improves outcomes in children with Pompe disease: a clinical study and review of the literature. Genetics in medicine : official journal of the American College of Medical Genetics. 2020;22(5):898–907. doi: 10.1038/s41436-019-0738-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kronn D, Davison J, Brassier A, et al. OP016: Mini-COMET: Safety and efficacy of ≥97 weeks’ avalglucosidase alfa in infantile-onset Pompe disease participants previously treated with alglucosidase alfa. Genetics in Medicine. 2022;24(3, Supplement):S348–S349. doi: 10.1016/j.gim.2022.01.566 [DOI] [Google Scholar]
- 9.Prater SN, Banugaria SG, DeArmey SM, et al. The emerging phenotype of long-term survivors with infantile Pompe disease. Genetics in medicine : official journal of the American College of Medical Genetics. 2012;14(9):800–810. doi: 10.1038/gim.2012.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spiridigliozzi GA, Keeling LA, Stefanescu M, Li C, Austin S, Kishnani PS. Cognitive and academic outcomes in long-term survivors of infantile-onset Pompe disease: A longitudinal follow-up. Molecular Genetics and Metabolism. 2017;121(2):127–137. doi: 10.1016/j.ymgme.2017.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korlimarla A, Spiridigliozzi GA, Crisp K, et al. Novel approaches to quantify CNS involvement in children with Pompe disease. Neurology. 2020;95(6):e718–e732. doi: 10.1212/WNL.0000000000009979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chien YH, Lee NC, Peng SF, Hwu WL. Brain development in infantile-onset Pompe disease treated by enzyme replacement therapy. Pediatric research. 2006;60(3):349–352. doi: 10.1203/01.pdr.0000233014.84318.4e [DOI] [PubMed] [Google Scholar]
- 13.Burrow TA, Bailey LA, Kinnett DG, Hopkin RJ. Acute progression of neuromuscular findings in infantile Pompe disease. Pediatric neurology. 2010;42(6):455–458. doi: 10.1016/j.pediatrneurol.2010.02.006 [DOI] [PubMed] [Google Scholar]
- 14.Broomfield A, Fletcher J, Hensman P, et al. Rapidly Progressive White Matter Involvement in Early Childhood: The Expanding Phenotype of Infantile Onset Pompe? JIMD Reports. 2018;39:55–62. doi: 10.1007/8904_2017_46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McIntosh PT, Hobson-Webb LD, Kazi ZB, et al. Neuroimaging findings in infantile Pompe patients treated with enzyme replacement therapy. Molecular Genetics and Metabolism. 2018;123(2):85–91. doi: 10.1016/j.ymgme.2017.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ebbink BJ, Poelman E, Aarsen FK, et al. Classic infantile Pompe patients approaching adulthood: a cohort study on consequences for the brain. Developmental medicine and child neurology. 2018;60(6):579–586. doi: 10.1111/dmcn.13740 [DOI] [PubMed] [Google Scholar]
- 17.Korlimarla A, Lim JA, Kishnani PS, Sun B. An emerging phenotype of central nervous system involvement in Pompe disease: from bench to bedside and beyond. Annals of Translational Medicine. 2019;7(13):289. doi: 10.21037/atm.2019.04.49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Korlimarla A, Stefanescu E, Austin S, Chen S, Provenzale JM, Kishnani PS. Quantitative evaluation of white matter hyperintensities in the central nervous system in infantile Pompe disease. Molecular Genetics and Metabolism. 2019;126(2):S87. doi: 10.1016/j.ymgme.2018.12.214 [DOI] [Google Scholar]
- 19.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR American journal of roentgenology. 1987;149(2):351–356. doi: 10.2214/ajr.149.2.351 [DOI] [PubMed] [Google Scholar]
- 20.Abushaban L, Rathinasamy J, Sharma PN, Vel MT. Normal reference ranges for the left ventricular mass and left ventricular mass index in preterm infants. Ann Pediatr Cardiol. 2020;13(1):25–30. doi: 10.4103/apc.APC_171_18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rairikar M, Kazi ZB, Desai A, Walters C, Rosenberg A, Kishnani PS. High dose IVIG successfully reduces high rhGAA IgG antibody titers in a CRIM-negative infantile Pompe disease patient. Mol Genet Metab. 2017;122(1–2):76–79. doi: 10.1016/j.ymgme.2017.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paoletti M, Pichiecchio A, Colafati GS, et al. Multicentric Retrospective Evaluation of Five Classic Infantile Pompe Disease Subjects Under Enzyme Replacement Therapy With Early Infratentorial Involvement. Front Neurol. 2020;11:569153. doi: 10.3389/fneur.2020.569153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cerón-Rodríguez M, Castillo-García D, Acosta-Rodríguez-Bueno CP, et al. Classic infantile-onset Pompe disease with histopathological neurologic findings linked to a novel GAA gene 4 bp deletion: A case study. Molecular Genetics & Genomic Medicine. 2021;n/a(n/a):e1957. doi: 10.1002/mgg3.1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsu YK, Chien YH, Shinn-Forng Peng S, et al. Evaluating brain white matter hyperintensity, IQ scores, and plasma neurofilament light chain concentration in early-treated patients with infantile-onset Pompe disease. Genet Med. 2023;25(1):27–36. doi: 10.1016/j.gim.2022.10.005 [DOI] [PubMed] [Google Scholar]
- 25.Martini C, Ciana G, Benettoni A, et al. Intractable fever and cortical neuronal glycogen storage in glycogenosis type 2. Neurology. 2001;57(5):906–908. doi: 10.1212/wnl.57.5.906 [DOI] [PubMed] [Google Scholar]
- 26.van Capelle CI, Poelman E, Frohn-Mulder IM, et al. Cardiac outcome in classic infantile Pompe disease after 13 years of treatment with recombinant human acid alpha-glucosidase. Int J Cardiol. 2018;269:104–110. doi: 10.1016/j.ijcard.2018.07.091 [DOI] [PubMed] [Google Scholar]
- 27.Van den Hout JMP, Kamphoven JHJ, Winkel LPF, et al. Long-Term Intravenous Treatment of Pompe Disease With Recombinant Human α-Glucosidase From Milk. Pediatrics. 2004;113(5):e448–e457. doi: 10.1542/peds.113.5.e448 [DOI] [PubMed] [Google Scholar]
- 28.van den Dorpel JJA, Dremmen MHG, van der Beek N, et al. Diffusion tensor imaging of the brain in Pompe disease. J Neurol. 2023;270(3):1662–1671. doi: 10.1007/s00415-022-11506-z [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental table 1: Age-based standard scores on neuropsychological measures from children with IOPD over time (Patient IDs 2–5)
Data Availability Statement
Anonymized data not published within this article will be made available by request from any qualified investigator.