

Abstract
Extracellular vesicles (EVs) are nanoscale particles that facilitate intercellular communication. They are regarded as a promising natural drug delivery system for transporting and delivering bioactive macromolecules to target cells. Recently, researchers have engineered EVs with FKBP12/FRB heterodimerization domains that interact with rapamycin to load and deliver exogenous proteins for both in vitro and in vivo applications. In this study, we examined the tissue distribution of EVs using near‐infrared fluorescent imaging. We evaluated the effectiveness of EV‐mediated delivery of Cre recombinase specifically to hepatocytes in the livers of Ai9 Cre‐loxP reporter mice. Intravenous injection resulted in more efficient Cre protein delivery to the liver than intraperitoneal injections. Depleting liver‐resident macrophages with clodronate‐encapsulated liposome pre‐treatment did not enhance EV‐mediated Cre delivery to hepatocytes. Moreover, we demonstrated that multiple intravenous injections of Cre‐EVs facilitated functional Cre delivery to hepatocytes. To the best of our knowledge, this is the first study to simultaneously investigate the tissue distribution of FKBP12/FRB‐engineered EVs and their subsequent intracellular protein delivery in Ai9 Cre‐loxP reporter mice. These insights can inform preclinical research and contribute to developing next‐generation EV‐based platforms for delivering therapeutic proteins or genome editing technologies targeting the liver.
Keywords: drug delivery, EV uptake, EV‐mediated drug delivery, exosomes, extracellular vesicles, microvesicles, protein delivery
1. INTRODUCTION
Extracellular vesicles (EVs) have emerged as an attractive drug delivery vehicle due to their natural properties for delivering bioactive cargo to recipient cells (de Jong et al., 2019). EVs are cell‐derived nanoparticles consisting of a lipid bilayer encapsulating biomolecules from their parent cell, including nucleic acids and proteins (Raposo & Stoorvogel, 2013; van Dommelen et al., 2012). Their inherent function is to foster intercellular communication by transporting biomolecules between cells, leading to phenotypical changes in the recipient cell upon delivery (Al‐Nedawi et al., 2008; Ratajczak et al., 2006; Skog et al., 2008). Compared to synthetic drug delivery vehicles, such as liposomes, EVs have favorable characteristics, including low toxicity, low immunogenicity, high stability in circulation, biocompatibility, and biological barrier permeability (Alvarez‐Erviti et al., 2011; Escudier et al., 2005; Marcus & Leonard, 2013; Tsui et al., 2002). These unique features have sparked interest in their utilization as drug delivery vehicles. Over the past decade, most preclinical studies investigated EV‐based delivery of nucleic acids (±47%) and small molecule drugs (±40%) (de Castilla et al., 2021). Only ±18% of preclinical studies examined EV‐mediated delivery of peptides or proteins, suggesting that this is technically more challenging and there is a need for a better understanding of EV‐mediated delivery of protein/peptide‐based payloads (de Castilla et al., 2021).
Recently, the rapamycin‐interacting heterodimer complex K506‐binding protein (FKBP12) and the T82L mutant FKB12‐rapamycin binding (FRB) domain have been demonstrated to be an effective method to load and deliver proteins through EVs (Campbell et al., 2019; Gee et al., 2020; Ilahibaks et al., 2023; Montagna et al., 2018; Somiya & Kuroda, 2021). Various target proteins can be coupled to the FRB domain, whereas the FKBP12 domain is anchored to the cellular membrane via an N‐myristoylation sequence. Upon exposing FKBP12/FRB‐engineered producer cells to a rapamycin orthologue, dimerization is induced between FRKBP12 and FRB, which promotes FRB‐anchored protein to be endogenously loaded into the EV lumen. To further facilitate an effective protein delivery to recipient cells, vesicular stomatitis virus glycoprotein (VSV‐G) is often additionally expressed on EVs (Campbell et al., 2019; Gee et al., 2020; Montagna et al., 2018; Somiya & Kuroda, 2021).
Although the FKBP12/FRB loading method has been investigated for EV‐mediated delivery of different protein payloads in vitro and in vivo, little is known about the tissue distribution of such engineered EVs. Currently, only Gee et al. (2020) demonstrated that a single intramuscular injection of FKBP12/FRB engineered EVs, loaded with the CRISPR/Cas9 ribonucleoprotein complex, induced permanent exon skipping in a luciferase reporter‐ and mdx mice with 6.9% and 1.1%–1.6% efficacy, respectively.
After systemic administration, nanomedicines tend to accumulate in the liver because of discontinuous vasculature and decreased blood flow rates (Tsoi et al., 2016). Here, we studied the optimal administration method for EV‐mediated protein delivery to hepatocytes because of a growing interest in cholesterol‐lowering gene therapy with the liver as the primary target organ (Chadwick et al., 2018; Ding et al., 2014; Musunuru et al., 2021; Rothgangl et al., 2021). By labeling Cre recombinase loaded FKBP12/FRB‐engineered EVs (Cre‐EVs) with a fluorescent dye and administrating them to Ai9 Cre‐loxP reporter mice, we could simultaneously study EV tissue distribution and functional EV‐mediated intracellular delivery of Cre recombinase.
We observed that intravenous injection (i.v.) was a more effective systemic administration route than intraperitoneal (i.p) injection to deliver EV‐based protein payloads to the liver. Furthermore, depleting Kupffer cells through clodronate‐encapsulated liposomes did not improve EV‐mediated protein delivery to hepatocytes. Finally, we demonstrated that repeated i.v. injections improved EV‐mediated delivery of Cre recombinase in hepatocytes. In conclusion, EVs can be used as vehicles for the functional intracellular delivery of exogenous proteins in hepatocytes. These findings can potentially be applied in the further development of gene therapy or therapeutic protein delivery strategies using EVs.
2. MATERIAL AND METHODS
2.1. Cell culture
Human embryonic kidney 293FT cells (HEK293FT, ThermoFisher Scientific, R70007), Cre‐loxP T47D stoplight reporter cells (Zomer et al., 2016), HepG2 and NIH3T3 were cultured in Dulbecco's Modified Eagle Medium (DMEM) (Gibco, 41965039) supplemented with 10% fetal bovine serum (FBS, Corning, 27419001) and 1% penicillin‐streptomycin (P/S, Gibco, 15140‐122). All cells were kept in a 5% CO2 atmosphere at 37°C.
2.2. Plasmids
The N‐Myriostolated (N‐Myr)‐FKBP12/2xFRB‐Cre recombinase plasmid was created by subcloning the N‐Myr signal, FKB12, and T82L mutant FRB sequence into the expression vector pcDNA5/FRT/TO (ThermoFisher Scientific, K65001) according to the manufacturer's protocol for the master mix NEBuilder HiFi DNA assembly (NEB, E2621L). Subsequently, the Cre recombinase sequence was cloned at the N‐terminus of the T82L mutant FRB sequence separated by a GGSGG linker. See Table S1 for the complete sequence.
2.3. EV production
Per T175 flask, 1 × 1010 HEK293FT cells were seeded and cultured overnight. For Cre‐EV production, HEK293FT cells were transfected with the N‐Myr‐FKBP12/2xFRB‐Cre plasmid (30 μg) together with the VSV‐G plasmid (15 μg) in a 1:2 ratio of DNA:Lipofectamine 3000 transfection reagent (w:w) (Invitrogen Corp., L3000008), according to the manufacturer's protocol. After 6 h, the medium was replaced with DMEM (Gibco, 41965039) supplemented with 10% exosome‐depleted FBS (Gibco, A2720801), 1% P/S, and 500 nM A/C heterodimer ligand (Takara Bio, 635055). After 72 h, the conditional medium was isolated and centrifuged at 2000 x g for 15 min at 4°C. Next, the conditioned medium was concentrated via tangential flow filtration using a Minimate Tangential Flow Filtration Capsule 500 kDa MWCO (Pall, OA500C12). The conditional medium was concentrated to a final volume of 5 mL and loaded onto an S400 high prep column (GE Healthcare) using an AKTAStart (GE Healthcare) containing a UV 280 nm flow cell. The EV‐containing fractions were pooled together, filtered through a 0.45 μm syringe SFCA membrane filter (Corning, 516–1954), and concentrated using a 100‐kDa MWCO Amicon spin filter (Millipore, Z648043‐24EA).
2.4. EV labeling and purification
Cre‐EVs were labeled with Alexa Fluor 647 NHS ester dye (Invitrogen, A20006) for in vitro uptake assays and Alexa Fluor 790 NHS ester (Invitrogen, A37569) for in vivo tracking. Cre‐EVs were suspended in 0.05 nM Alexa Fluor NHS ester in 0.1 M NaHCO3 in PBS and incubated for 30 min at 37°C while shaking. Next, the dye was quenched by incubating the sample with 0.1 M Tris‐HCl for 30 min at room temperature. Subsequently, the labeled Cre‐EVs were loaded on a Sepharose CL‐4B column (GE Healthcare) using an AKTA start system (GE Healthcare) containing a UV 280 nm flow cell. EV‐containing fractions were concentrated using a 100‐kDa MWCO Amicon spin filter (Merck Millipore).
2.5. Transmission electron microscopy
The Cre‐EVs were adsorbed to carbon‐coated grids (75−200 mesh, Sigma–Aldrich) for 15 min at room temperature. Subsequently, the grids were washed with PBS, fixed with 2% PFA, and 0.2% glutaraldehyde in PBS for 30 min at room temperature. Next, uranyl‐oxalate was used for staining, and the grids were embedded with 1.8% methylcellulose and 0.4% uranyl acetate for 10 min on ice. Cre‐EVs were visualized with a JEOL 3 microscope at 29.000x magnification.
2.6. Cre‐loxP reporter assay
In a 96‐well plate, T47D Cre‐loxP stoplight reporter cells were seeded at a density of 10.000 cells/well and incubated overnight. The next day, cells were exposed to Cre‐EVs and EV‐mediated Cre delivery inside T47D Cre‐loxP stoplight reporter cells was visualized using an EVOS cell imaging system 3 days following Cre‐EV administration (M5000, Invitrogen). Subsequently, the cells were rinsed in PBS and dissociated with 0.25% Trypsin‐EDTA solution (Sigma–Aldrich, T4049) for flow cytometric analyses. The cells were then centrifuged at 500 x g for 3 min in a 96‐well round‐bottom plate (Greiner CELLSTAR, 650185) and resuspended in 250 μL PBS supplemented with 2% FBS. A CytoFLEX (Beckman Coulter) was used to quantify the number of recombined cells, and Kaluza software v2.1 was used to process the data (Beckman Coulter Inc.).
2.7. Nanoparticle tracking analysis
EV size and particle concentrations were determined using a Nanosight NS500 system (Malvern Technologies) equipped with a 405 nm laser with Nanosight NTA 3.3 software (Malvern Technologies).
EVs were analyzed through 3 × 30‐s videos using a camera level of 15 and a detection threshold of 5. EVs were diluted in PBS to a concentration of between 30 and 100 tracks per frame.
2.8. Western blot
For western blot analysis, cell lysate (CL) and EV were dispersed in RIPA lysis buffer (Merck, 20–188). Protein levels were measured with microBCA (ThermoFisher, 23235). Samples were denatured with NuPAGE Sample Reducing Agent (Invitrogen, NP0004), mixed with NuPAGE LDS Sample Buffer (Invitrogen, NP0007), and boiled at 95°C for 10 min. The samples were loaded onto Bolt 4%–12% Bis‐Tris Plus Gel (Thermo Fischer Scientific, NW04125BOX) at 130 V for 75 min and transferred to PVDF membranes (Millipore, IPVH00010). The membranes were blocked with 5% (w/v) bovine serum albumin (BSA, Roche, 10735086001) in Tri‐Buffered saline (TBS) for 1 h at room temperature. Subsequently, the membranes were incubated with primary antibodies and incubated overnight. The primary antibodies used in this study were anti‐Cre recombinase (1 μg/mL, Millipore, MAB3120), anti‐β‐Actin (0.2 μg/mL, Sigma–Aldrich, A5441), anti‐VSVG epitope tag (1 μg/mL, Novus Biologicals, NB100‐2485), anti‐DmrA (1 μg/mL Takara, 635089), anti‐CD81 (1 μg/mL, Santa Cruz, SC‐166029) and anti‐syntenin‐1 (1 μg/mL, Origene, TA504796). Proteins were visualized with secondary Alexa Fluor 680 conjugated antimouse antibody (0.1 μg/mL, LI‐COR Bioscience, A‐21057) and IRDye 800CW anti‐rabbit antibody (0.1 μg/mL, LI‐COR Biosciences, 1926−322) were used when imaging was performed on an Odyssey Infrared Imager (LI‐COR Biosciences) at 700 and 800 nm. Western blot analysis in Figure S1 was performed similarly, while CD63 samples were prepared without the NuPAGE Sample Reducing Agent (Invitrogen, NP0004). The primary antibodies used were anti‐Alix (0.1 μg/mL, ab117600, clone 3A9, Abcam), anti‐calnexin (1:500, GTX101676, GeneTex), and anti‐CD63 (0.2 μg/mL, Abcam, 8219). Secondary antibodies used were goat anti‐rabbit (0.05 μg/mL, P0448, Dako) or goat anti‐mouse (0.05 μg/mL, P0447, Dako). The proteins were visualized with Western‐Ready ECL Substrate (426,303, Biolegend) and imaged with Chemi Doc XRS + system (Bio‐Rad).
2.9. In vivo experiments
The Animal Ethical Experimentation Committee of Utrecht University approved the animal experiments and were executed according to the Guide for the Care and Use of Laboratory Animals. We used Ai9 Cre‐loxP reporter mice (B6;129S6‐Gt(ROSA)26Sortm9(CAG‐tdTomato+)Hze/J, The Jackson Laboratory) which received standard food and water ad libitum and were housed under standard conditions with 12 h light/dark cycles until experimental procedures.
2.10. Intravenous and intraperitoneal injection
Ai9 Cre‐loxP reporter mice were divided between an i.v. injection group (n = 3) and an i.p. injection group (n = 3). Since we aimed to exclude functional EV uptake differences due to direct EV labeling, NHS‐Ester790‐labeled and unlabeled Cre‐EVs were mixed in a 1:100 ratio before administration. Either 5 × 1011 particles (200 μL) were administered by tail‐vein injection to the i.v. injection group, or via i.p. injection to the mice from the i.p. injection group. The tissue distribution of Cre‐EVs was followed over time with the Pearl Imager (LI‐COR Biosciences) and processed with the Pearl Cam software. After a follow‐up of 6 days, the mice were sacrificed and organs were harvested for immunofluorescent‐ and histological analyses.
2.11. Treatment with clodronate liposomes
Ai9 Cre‐loxP reporter mice were divided between a non‐clodronate (n = 2) and a clodronate pretreated group (n = 2). The latter group was injected with 100 μL of suspension/10 grams of animal weight injected with clodronate‐encapsulated liposomes (Liposoma B.V., SKU C010, 5 mg/mL). After 24 h, equal amounts of NHS‐Ester790‐labeled and unlabeled Cre‐ EVs were mixed, and 4 × 1011 particles (100 μL) of the NHS‐Ester790‐labeled/unlabeled Cre‐EVs were injected per mouse. The tissue distribution of Cre‐EVs was followed over time with the Pearl Imager (LI‐COR Biosciences) and processed with Pearl Cam software. The mice were sacrificed 5 days post‐injection.
2.12. Single versus multiple injections of Cre‐EVs
In a dose‐dependency study, Ai9 Cre‐loxP reporter mice were divided into three groups, including a single dose (100 μL, 5 × 1011 particles, D1, n = 3), high dose (200 μL, 1 × 1012 particles, D2, n = 2) and repeated dosing (100 μL, 5 × 1011 particles, 3xD1, n = 3) group. Before administrating the Cre‐EVs, the NHS‐Ester790‐labeled and unlabeled Cre‐EVs were mixed in a 1:500 ratio. The mice receiving repeated injections with 5 × 1011 Cre‐EVs were administered within a 24‐h interval for 3 days (3xD1 group). After 7 days, the mice were sacrificed. As a negative control, 100 μL PBS was administrated via tail‐vein injection in Ai9 Cre‐loxP reporter mice. The tissue distribution of Cre‐EVs was followed over time with the Pearl Imager (LI‐COR Biosciences) and processed with the Pearl Cam software.
2.13. Near‐infrared fluorescent (NIRF) imaging
The localization and tissue distribution of NHS‐Esther790‐labeled Cre‐ EVs were examined using NIRF imaging in Ai9 Cre‐loxP reporter mice at various time points, whereby the mice were shortly anesthetized with isoflurane. Images were acquired on a Pearl Impulse Imaging system (LI‐COR Biosciences) in living animals before and after EV administration. At the final time point, mice were terminated, and their organs were harvested for immunofluorescent‐ and histological analysis.
2.14. Immunofluorescence and histological staining
The obtained tissues were routinely fixed in formalin for 24 h and subsequently embedded in paraffin. Paraffin‐embedded livers were cut into 5 μm sections, fixated on slides, and dried at 55°C for 1 h. Sections were deparaffinized and rehydrated (3x Tissue Clear, 5 min, 2 × 99% EtOH for 5 min, 2 × 96% EtOH for 5 min, 2 × 70% EtOH for 5 min, 3x demi water for 5 min). Next, antigen retrieval was obtained by submerging the sections in sodium citrate (10 mM, pH = 6.0), supplemented with 0.5 % (v/v) Tween20, and followed by cooking in the autoclave. The tissue sections were washed in PBS (3 × 5 min) and blocked with 3% BSA (Roche, 10735086001) for 1 h at room temperature. Next, the tissue sections were incubated with a primary antibody RFP Antibody Pre‐adsorbed (2 μg/mL, Rockland Immunochemicals Inc., 600‐401‐379) overnight. Subsequently, the tissue sections were washed with PBS (3 × 5 min) and incubated with a secondary Alexa Fluor 555 antibody (5 μg/mL, Invitrogen, A‐21428). The nuclei were stained with Hoechst (0.1 μg/mL, Invitrogen, H3570) for 10 min at room temperature, and washed with PBS (3 × 5 min). The sections were enclosed with Fluoromount‐G (Southern Biotech, 0100‐01). Tissue section images were captured with a BX53 microscope (Olympus). To visualize macrophages, the Tyramide SuperBoost Kit with Alexa Fluor Tyramides (Thermo Fisher Scientific, B40923) was used. After antigen retrieval, tissue sections were blocked with 10% goat serum for 1 h at room temperature. Subsequently, the sections were incubated overnight with the preabsorbed primary antibody RFP Antibody Pre‐adsorbed (2 μg/mL, 600‐401‐379, Rockland Immunochemicals, Inc.). Next, the sections were washed with PBS (3 × 5 min) and incubated with poly‐HRP‐conjugated secondary antibody from the Tyramide SuperBoost Kit for 1 h at room temperature. Subsequently, the sections were washed and incubated with the Tyramide working solution for 10 min at room temperature, followed by washing the tissue sections in PBS (3 × 5 min). Next, tissue sections were incubated with CD163 (10 μg/mL, SC‐33560, Santa Cruz) for 1 h at room temperature, followed by a PBS wash (3 × 5 min), incubated with Alexa Fluor 488 diluted in 1% BSA in PBS (2.5 μg/mL, A11034, ThermoFisher) for 1 h at room temperature and washed with PBS+0.5% Tween20 (3 × 5 min). The sections were incubated with Hoechst (0.1 μg/mL, Invitrogen, H3570) for 10 min at room temperature and washed with PBS afterward (3 × 5 min). The tissue sections were enclosed in Fluoromount‐G (0100‐01, SouthernBiotech). Tissue section images for macrophages were captured with aTunder. To visualize endothelial cells or hepatocytes, tissue sections were blocked with 5% BSA for 1 h at room temperature upon antigen retrieval. Sections were incubated overnight with the RFP Antibody (2 μg/mL, HPA004712, Atlas Antibodies). For co‐staining with an endothelial cell or hepatocyte marker, the RFP antibody was co‐incubated with either the primary antibody PECAM‐1 (2 μg/mL, sc‐1506, Santa Cruz) or HNF4A (2 μg/mL, HPA004712, Atlas Antibodies) in 1% BSA in PBS overnight at 4°C, respectively. Next, sections were washed with PBS (3 × 5 min) and incubated for 1 h at room temperature with secondary antibody conjugated to Alexa 568 conjugate (5 μg/mL, A11057, ThermoFisher Scientific) and secondary antibody conjugated to Alexa 488 conjugate (5 μg/mL, A21206, ThermoFisher Scientific). Subsequently, tissue sections were washed in PBS (3 × 5 min) and incubated with Hoechst (0.1 μg/mL, Invitrogen, H3570) for 15 min at room temperature. Afterward, sections were washed with PBS (3 × 5 min) and mounted using Mowoil (9002‐89‐5, Merck). Tissue section images were captured with a Leica Tunder.
2.15. Statistical analysis
GraphPad PRISM v.9 was used for the statistical analysis. The two‐tailed unpaired t‐test was used to assess the comparisons between the two groups. Two one‐way analyses of variance (ANOVA) with Šídák's multiple comparisons test were used to determine comparisons between several groups. The data is presented as mean ± SEM, with p‐values *<0.05, **<0.01, ***<0.001, and ****<0.0001 indicating statistical significance.
3. RESULTS
3.1. EVs can effectively deliver Cre recombinase in vitro
We transiently transfected HEK293FT cells with N‐Myr‐2xFKBP12/FRB‐Cre‐ and VSV‐G plasmids to produce Cre‐EVs. Six hours post‐transfection, cells were exposed to EV‐depleted media with the rapamycin‐orthologue. After 72 hours, conditioned medium was collected, concentrated with tangential flow filtration with a 500 kDa molecular weight cutoff membrane (MWCO) and Cre‐EVs were separated from free contaminants by size‐exclusion chromatography (SEC) (Figure 1a). Nanoparticle tracking analysis (NTA) displayed that Cre‐EVs had a size distribution around 80–120 nm (Figure 1b). Transmission electron microscopy demonstrated that Cre‐EVs have a typical EV membranous structure and morphology (Figure 1c). To verify the presence of Cre, VSV‐G, FKBP12, β‐actin, and EV‐marker proteins, we compared the protein composition of Cre‐EVs with the CL of their producer cells. Western blot analysis showed the presence of Cre, FKBP12, and VSV‐G in both CL and isolated EVs (Figure 1d). Cre‐EVs were enriched for EV‐marker proteins Syntenin‐1, CD81, Alix, CD63 and negative for the endoplasmic reticulum marker calnexin compared to the CL (Figure 1d, Figure S1). These observations demonstrated that we successfully isolated nano‐sized EVs containing Cre recombinase.
FIGURE 1.
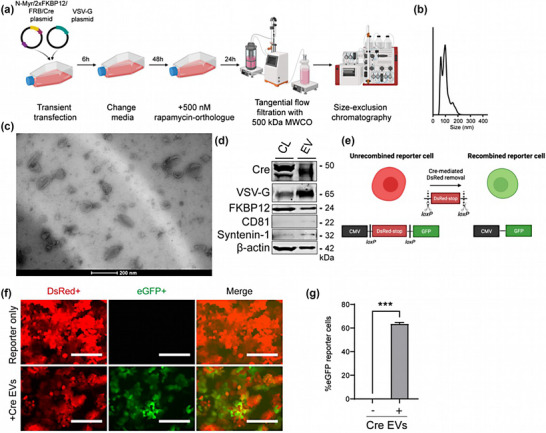
Characterization of Cre‐EVs. (a) Schematic illustration of Cre‐EV production, the concentration of conditional media with tangential flow filtration, and purification with size‐exclusion chromatography. (b) Nanoparticle Tracking Analysis showed Cre‐EVs had a size distribution between 50–200 nm. (c) Transmission electron microscopy showed that Cre‐ EVs have a typical ‘cup‐shaped’ morphology. (d) Western blot analysis of Cre‐EVs and cell lysate of their donor cells (CL) for Cre, VSV‐G, FKBP12, CD81, Syntenin‐1, and β‐actin. (e) Schematic illustration of Cre‐mediated recombination in Cre‐loxP stoplight reporter cells. The administration of Cre‐EVs to Cre‐loxP stoplight reporter cells resulted in intracellular delivery of Cre recombinase, leading to Cre‐recombined eGFP+ reporter cells as determined by (f) microscopic and (g) flow cytometric analysis. Scale bar = 200 μm. Representative data of three individual experiments. Data are represented as mean ± SEM. Statistical analysis was performed with an unpaired t‐test, p‐values *<0.05, **<0.01, ***<0.001, and ****<0.0001 are considered statistically significant. Representative images from three individual experiments.
Next, we investigated EVs' ability to deliver Cre recombinase to T47D Cre‐loxP stoplight reporter cells. Intracellular delivery of Cre recombinase to Cre‐loxP reporter cells will result in Cre‐recombined LoxP removal leading to a color‐switch from DsRed+ reporter cells to eGFP+ reporter cells (Figure 1e). Administration of 1 × 1010 Cre‐EV particles recombined 63.6 ± 1.2% eGFP+ reporter cells as determined by microscopic‐ (Figure 1f) and flow cytometric analysis (Figure 1g). These observations showed that Cre‐EVs could successfully deliver Cre recombinase with high efficacy in vitro.
3.2. Assessing Alexa NHS Ester as an EV labeling tool for in vitro uptake
To explore tissue distribution upon systemic administration in mice, EVs were fluorescently labeled with the AlexaFluor647 NHS‐ester and separated from the free dye (Kooijmans et al., 2018) (Figure 2a; Lai et al., 2014). AlexaFluor647‐labeling did not affect EV size distribution (Figure 2b). Next, AlexaFluor647‐labeled Cre‐EV uptake in HepG2 and NIH3T3 cells was confirmed by fluorescent microscopic analysis 6 hours after administering 1 × 109 particles (Figure 2c,d).
FIGURE 2.
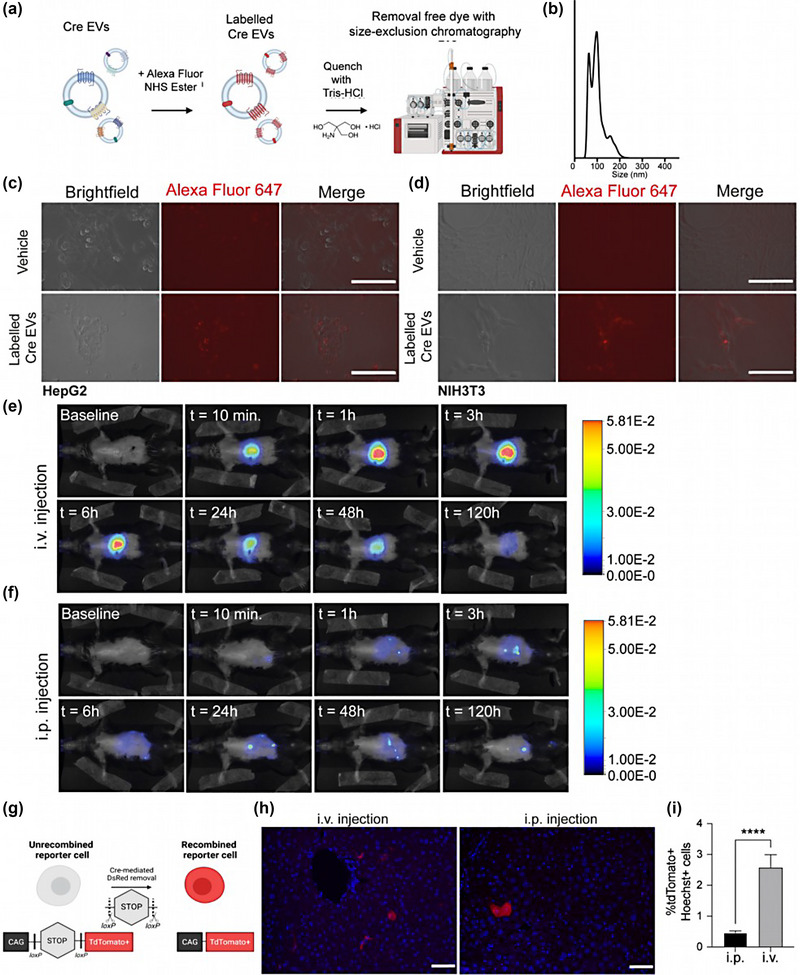
Higher liver protein delivery upon intravenous administration of Cre‐EVs as compared to intraperitoneal injection. (a) Schematic illustration of labeling EVs with an Alexa Fluor NHS ester, followed by dye quenching and separating the free dye from the labeled EVs through SEC. (b) NTA analysis of AlexaFluor647‐labeled EVs showed size distribution between ± 50–200 nm. The uptake of AlexaFluor647‐labeled EVs (in red) was confirmed in (c) HepG2 and (d) NIH3T3 cells by fluorescent microscopic analysis. Representative live images of in vivo tracking of AlexaFluor790‐labeled EVs in Ai9 Cre‐loxP reporter mice that received Cre‐EVs through (e) i.v.‐or (f) i.p. injection over 120 h by NIRF imaging. (g) Schematic illustration of Cre‐mediated recombination in Ai9 Cre‐loxP reporter mice, wherein the functional intracellular delivery of Cre will lead to Cre‐recombined tdTomato+ cells. (h) Immunofluorescence images from liver sections. Blue = Hoechst, Red = tdTomato+, scale bar = 50 μm. (i) Immunofluorescence quantification of total tdTomato+Hoechst+ cells in liver tissue sections from Ai9 Cre‐loxP reporter mice that received Cre‐EVs either through i.v. or i.p. injection. Statistical analysis was performed via an unpaired t‐test with p‐values *<0.05, **<0.01, ***<0.001, and ****<0.0001 considered statistically significant. Per group n = 3, total n = 6.
3.3. EV tissue distribution pattern is affected by the administration method
To identify the most optimal route of administration, we compared i.v.‐ and i.p. injection of the Cre‐EVs. Previous studies have mainly focused on the tissue distribution and intracellular delivery of natural EVs that lack targeting‐ or delivery mechanisms (Gupta et al., 2020; Wiklander et al., 2015). Regarding targeting, the low‐density lipoprotein receptor abundantly expressed in hepatocytes is an entry receptor for VSV‐G present on Cre‐EVs (Finkelshtein et al., 2013). To simultaneously study in vivo tissue distribution and EV‐mediated intracellular protein delivery, Cre‐EVs were labeled with AlexaFluor 790 NHS Ester to enable near‐infrared fluorescent (NIRF) imaging in living animals and administered to Ai9 Cre‐loxP reporter mice. EV membrane proteins, particularly VSV‐G (Mangeot et al., 2011), are important for the intracellular delivery of EVs' intraluminal cargo. Since AlexaFluor 790 NHS Ester is a protein‐binding dye that might block or mask membrane protein function, we mixed labeled and unlabeled Cre‐EVs before in vivo administration.
Via either i.v.‐ or i.p. injection, 5 × 1011 particles of AlexaFluor790‐labeled/unlabeled Cre‐EVs were administered to Ai9 Cre‐loxP reporter mice, NIRF images were taken of living animals at various time points over 120 h (Figure 2e,f). The localization of the fluorescent signal over time following i.p. injection suggests that the EVs are excreted more rapidly than i.v. injection. We subsequently visualized Cre‐mediated recombination in the liver to investigate how the route of the systemic injection method affected EV‐mediated intracellular protein delivery of Cre recombinase. A color switch occurs upon successful intracellular Cre‐mediated recombination, leading to tdTomato+ cells (Figure 2g). Immunofluorescence microscopic analysis showed that i.v. administration led to significantly more tdTomato+Hoechst+ cells compared to i.p. injection (i.v.: 2.6 ± 0.4%; i.p.: 0.5 ± 0.1%) (Figure 2h,i). Both administration methods resulted in EV‐mediated delivery to hepatocytes, as confirmed by immunofluorescence histological analysis with an antibody against hepatocyte nuclear factor 4 alpha (HNF4A) (Figure S2). Taken together, i.v. injection led to a higher accumulation of AlexaFluor790‐labeled Cre‐EVs and significantly more EV‐mediated delivery of Cre recombinase to liver cells than i.p. injection.
3.4. Pretreatment with clodronate encapsulated liposomes did not improve Cre‐EV delivery to hepatocytes
Although i.v. injection led to higher accumulation and uptake of Cre‐EVs in the liver, the tdTomato+ cells resembled more the morphology of non‐parenchymal cells, such as Kupffer‐ and endothelial cells, rather than the typical octagon shape of hepatocytes. These findings are suggestive of a preferred absorbance of Cre‐EVs by Kupffer cells. Since liver pathologies arise through hepatocyte dysfunction, we specifically aimed to improve EV‐mediated delivery to hepatocytes. Consequently, we hypothesized that pretreating the Ai9 Cre‐loxP reporter mice with clodronate‐encapsulated liposomes (0.05 mg/g of animal body weight) to deplete the Kupffer cells in the liver would improve EV‐mediated Cre delivery in hepatocytes (Samuelsson et al., 2017; Sun et al., 2021; Tavares et al., 2017).
To investigate this hypothesis, Ai9 Cre‐loxP reporter mice were injected with clodronate‐encapsulated liposomes to deplete the Kupffer cells. After 24 hours, we administered 4 × 1011 particles of AlexaFluor790‐labeled/unlabeled Cre‐EVs via i.v. injection (Figure 3a). Immunofluorescent histological evaluation revealed a lack of CD163+ cells, indicating successful depletion of Kupffer cells in Ai9 Cre‐loxP reporter mice pretreated with clodronate‐encapsulated liposomes (Figure S4B). Live in vivo tracking of AlexaFluor790‐labeled Cre‐EVs in Ai9 Cre‐loxP reporter mice could only be used to explore direct tissue distributions. Although signals after pretreatment with clodronate‐encapsulated liposomes tend to indicate a higher fluorescent signal than mice that did not receive clodronate liposomes over 120 hours following i.v. injection, one should realize that within 24 h, EVs are being metabolized and the Alexa Fluor labels do not directly reflect the EV retention (Figure 3b,c, Figure S3). Immunohistochemical‐ and immunofluorescent histological examinations revealed more tdTomato+Hoechst+ cells in the Ai9 Cre‐loxP reporter mice administered with Cre‐EVs (Figure 3d,e). The quantification of immunofluorescent images demonstrated that pretreatment with clodronate‐encapsulated liposomes notably decreased the functional delivery of Cre to the liver via EVs, as evidenced by a reduction of tdTomato+Hoechst cells (Figure 3f). The functional delivery of Cre through EVs occurred within the characteristic octagonal‐shaped hepatocytes in both groups, as indicated by the hepatocyte‐specific marker HNF4A (Figure S4A). Surprisingly, the Alexa fluorescent signals (Figure 3) tended to be higher in the group pretreated with clodronate‐encapsulated liposomes. At the same time, the EV‐mediated functional delivery of Cre was more efficient without the pretreatment. These results imply no direct correlation between fluorochrome tissue distribution outcomes and functional delivery, suggesting that both tissue distribution and functional delivery should be examined concurrently to explore the potential of EVs for in vivo drug delivery applications.
FIGURE 3.
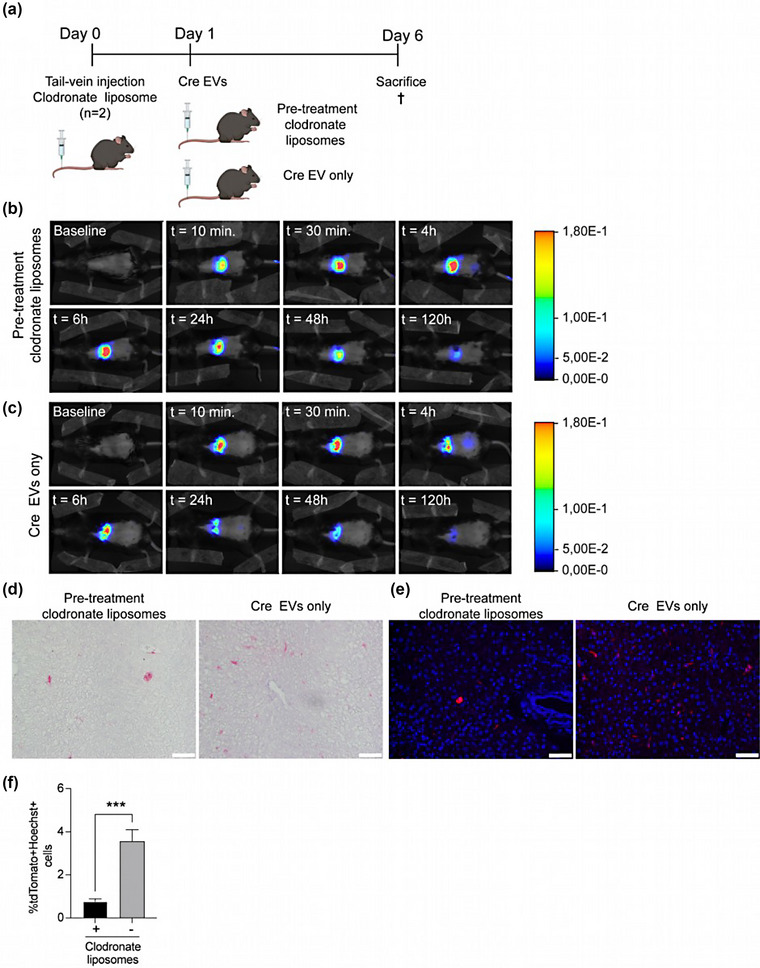
Pretreatment with clodronate‐encapsulated liposomes does not improve EV‐mediated intracellular protein delivery. (a) Schematic illustration of study design comparing the effect of pretreatment with clodronate‐encapsulated liposomes of Cre‐EV i.v. injection. (b) Representative images from the live in vivo tracking by NIRF imaging of AlexaFluor790‐labeled Cre‐EVs in Ai9 mice that received (c) pretreatment with clodronate‐encapsulated liposomes or (d) only Cre‐EVs. (d) Immunohistochemistry‐ and (e) immunofluorescent images of liver sections from mice that received were pretreated with or without clodronate‐encapsulated liposomes. Red = tdTomato, blue = Hoechst, scale bar = 50 μm. (f) Immunofluorescence quantification of total tdTomato+Hoechst+ cells in liver tissue sections from Ai9 Cre‐loxP reporter mice that received either Cre‐EVs or pretreatment with clodronate encapsulated liposomes. Statistical analysis was performed with an unpaired t‐test, p‐values *<0.05, **<0.01, ***<0.001, and ****<0.0001 are considered statistically significant. Per group n = 2, total n = 4.
Immunofluorescent histological analysis showed functional Cre‐EVs delivery in both Kupffer‐ and endothelial cells in the liver of the Ai9 Cre‐loxP mice that only received Cre‐EVs (Figure S4B,C). In contrast, we could only observe functional EV‐mediated transfer in PECAM1‐positive endothelial cells in the Ai9 Cre‐loxP mice that received pretreatment with clodronate‐encapsulated liposomes (Figure S4C). These observations suggest that pre‐treatment with clodronate‐encapsulated liposomes did not enhance EV‐mediated delivery to hepatocytes after a single i.v. injection.
3.5. Repeated injections improved EV‐mediated delivery of Cre recombinase in hepatocytes
Since depleting Kupffer cells with clodronate‐encapsulated liposomes did not improve Cre‐EVs uptake by hepatocytes, we investigated if repeated i.v. injections or a higher dose of Cre‐EVs could improve hepatic delivery. Ai9 Cre‐loxP reporter mice received an i.v. injection of a single dose of 5 × 1010 particles AlexaFluor790‐labeled/unlabeled Cre‐EVs (D1) or multiple i.v. injections of the same dosage within 3 days over a 24‐hour interval (3xD1). Additionally, a group of Ai9 Cre‐loxP reporter mice received a single i.v injection of 1 × 1012 particles of AlexaFluor790‐labeled/unlabeled Cre‐EVs (D2) (Figure 4a).
FIGURE 4.
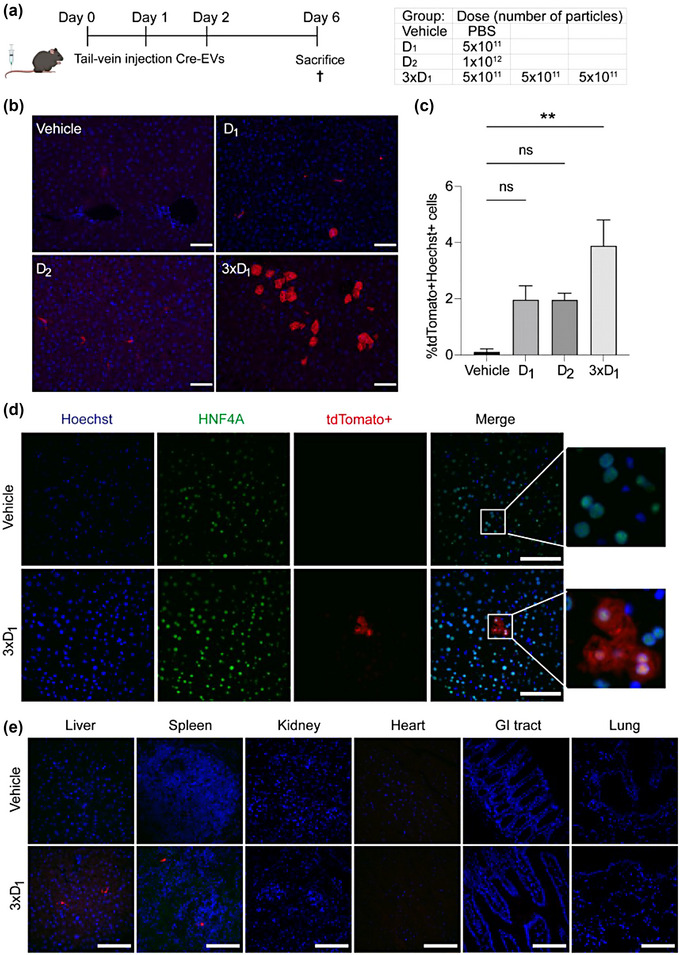
Repeated injections of Cre‐EVs improved delivery to hepatocytes. (a) Schematic illustration of study design administrating a PBS (Vehicle), single dose‐ (D1), higher dose‐ (D2) or multiple doses (3xD1) or Cre‐EVs in Ai9 mice. (b) Immunofluorescence images from liver sections of Ai9 mice that received i.v. injection of the vehicle, D1, D2 or 3xD1. Red = tdTomato+, Blue = Hoechst. Scale bar = 50 μm. (c) Quantification of liver sections showed 3xD1 resulted in more tdTomato+Hoechst+ cells. (d) Immunofluorescence images of liver tissue section of Ai9 Cre‐loxP reporter mice from the 3xD1 group with antibodies against tdTomato+, HNF4A and Hoechst. Green = HNF4A, Red = tdTomato+, Blue = Hoechst, Scale bar = 100 μm. (e) Immunofluorescence images from different organs of Ai9 mice that received i.v. injection of the vehicle or 3xD1. Scale bar = 100 μm. Vehicle n = 3, D1 n = 3, D2 n = 2, 3xD1 n = 3, total n = 11.
To assess if a higher dose or repeated i.v. injections translated into increased EV‐mediated Cre delivery, immunofluorescent staining was performed. We found that multiple injections of Cre‐EVs resulted in more tdTomato+Hoechst+ cells with a typical octagon hepatocyte morphology as determined by microscopy analysis (Figure 4b). We observed a significant increase of tdTomato+Hoechst+ cells in the 3xD1 group (Vehicle: 0.0%; D1 : 2.0 ± 0.5%; D2 : 2.0 ± 0.2%; 3xD1: 3.9 ± 0.9% %) (Figure 4c). The increased uptake within hepatocytes was observed only in local clusters and not consistently throughout the liver (Figure S5). Immunofluorescence histological analysis with an antibody against HNF4A confirmed that the tdTomato+Hoechst+ cells in the 3xD1 group were hepatocytes (Figure 4d). Additionally, we detected EV‐mediated delivery in the liver and spleen of the 3xD1 group, with no evidence of such delivery in other organs (Figure 4e). These findings suggest that multiple injections can promote EV‐mediated delivery of Cre recombinase in hepatocytes.
4. DISCUSSION
Therapeutic proteins are emerging as a vital drug class, and EV‐based protein delivery presents novel opportunities for modulating intracellular targets. Despite this potential, only ±18% of preclinical studies on EVs as drug delivery vehicles have focused on protein and peptide‐based payloads in the past decade (de Castilla et al., 2021). Although EVs are biocompatible, non‐immunogenic, and possess intrinsic targeting abilities, more effective payload engineering strategies are needed. The FKBP12/FRB rapamycin interacting system in donor cells has been shown to facilitate efficient loading and delivery of various target proteins through released EVs (Campbell et al., 2019; Gee et al., 2020; Montagna et al., 2018; Somiya & Kuroda, 2021). However, studies exploring the in vivo application of these engineered EVs have been relatively limited (Campbell et al., 2019; de Castilla et al., 2021; Gee et al., 2020; Ilahibaks et al., 2023; Montagna et al., 2018; Somiya & Kuroda, 2021).
Since most liver pathologies are manifested in hepatocytes, our study aimed to identify the optimal in vivo delivery strategy for EV‐mediated protein delivery to hepatocytes (Chadwick et al., 2018; Ding et al., 2014; Musunuru et al., 2021; Rothgangl et al., 2021). To the best of our knowledge, this is the first study to currently investigate EV tissue distribution and EV‐mediated intracellular protein delivery through NIRF imaging and Cre‐mediated recombination, respectively. Although the fluorescent signal appeared higher in the group pretreated with clodronate‐encapsulated liposomes compared to the Cre‐EV only group, the efficiency of EV‐mediated functional Cre delivery was greater in the Ai9 Cre‐loxP reporter mice that were given only Cre‐EVs. This finding indicates that tissue distribution outcomes do not necessarily correspond with functional delivery, highlighting the importance of investigating both tissue distribution and functional delivery simultaneously to better understand the potential of EVs for in vivo drug delivery applications.
Within the drug delivery field, the natural tropism of EVs and lipid nanoparticles (LNPs) toward the liver is a well‐recognized phenomenon (Andaloussi et al., 2013; Kooijmans et al., 2016). This tropism is influenced by numerous factors, including the physicochemical properties of EVs/LNPs, the role of hepatocytes in clearing circulating particles, and the metabolic demands of the liver (Mulcahy et al., 2014; Wiklander et al., 2015). Notwithstanding, it's crucial to remember that natural tropism does not equate to targeted or efficient delivery. Several hurdles, including enzymatic degradation, immune clearance, and non‐specific uptake by other cells, challenge the successful intracellular delivery of EVs payloads in vivo (Mulcahy et al., 2014; Vader et al., 2016). In our study, we employed VSV‐G to enhance EV uptake through the LDL‐R present on hepatocyte membranes and to facilitate their subsequent endosomal release (Finkelshtein et al., 2013; Mangeot et al., 2011). This two‐tiered approach, which takes advantage of the innate liver‐specific attraction of EVs while enhancing their delivery efficiency, presents a promising new direction for targeted liver therapeutics. Nonetheless, to optimize EV‐mediated uptake in hepatocytes, it would be beneficial for future research to investigate the effectiveness of VSV‐G compared to other targeting entities, such as GalNac. While VSV‐G has been known to facilitate endosomal escape (Mangeot et al., 2011), it is important to acknowledge the potential complications that may arise, such as lysosomal degradation. A common fate for many endocytic cargoes, the acidic lysosomal environment can induce enzymatic degradation of the delivered contents, potentially impacting the overall efficiency of EV‐mediated delivery (Huotari & Helenius, 2011). Moreover, we recognize the challenge of non‐specific uptake by off‐target cell types. Given the widespread presence of endocytic pathways across various cell types, Cre‐EVs may inadvertently be taken up by cells not intended for targeting (Mulcahy et al., 2014). These considerations underscore the necessity for more advanced EV engineering strategies and rigorous cell type‐specific validation methods to optimize targeting specificity and delivery efficiency. The intricate interplay of these factors highlights the complexity of EV‐mediated delivery. It stresses the importance of a thorough understanding of EV biology for its successful implementation in therapeutic applications.
In the past decade years, i.v. injections have been the dominant route of administration for EVs, accounting for approximately 88% of published preclinical investigations (de Castilla et al., 2021). Therefore, we compared i.v.‐ and i.p.‐mediated injections as systemic administration methods. Our findings revealed that Cre‐EV administration via i.p. injection resulted in a lower fluorescent signal in the live in vivo tracking images and fewer tdTomato+Hoechst+cells than i.v. injection. In line with the observations of Wiklander et al. (Wiklander et al., 2015) and Gupta et al. (Gupta et al., 2020), we found that i.p. injection of HEK293 EVs led to less accumulation and functional delivery in the liver compared to i.v. injection. These results highlight the importance of carefully considering the administration route for EV‐based drug delivery, as it may greatly impact EVs' tissue distribution, clearance, and therapeutic efficacy. Future research should explore administration routes and their potential advantages for specific therapeutic applications to optimize EV‐based drug delivery strategies.
We hypothesized that Cre‐EV delivery to hepatocytes could be improved by depleting the Kupffer cells through pretreatment with clodronate‐encapsulated liposomes (Samuelsson et al., 2017; Sun et al., 2021; Tavares et al., 2017). Although Cre‐EVs’ tissue distribution differed between both groups, microscopic analysis revealed fewer tdTomato+Hoechst+ cells were observed in the mice that received the pretreatment compared to those that did not. The difference between tissue distribution and functional delivery highlights the importance of studying both simultaneously to assess EVs’ efficacy as an intracellular delivery vehicle in vivo. This observation aligns with previous research highlighting the complex interplay between EV tissue distribution, cellular uptake, and functional delivery of their payloads (Lai et al., 2014; Smyth et al., 2015). The discrepancy between tissue distribution and functional delivery may be attributed to various factors, including cell type‐specific barriers to EV internalization and intracellular trafficking, differences in payload release mechanisms and activity within the cytosol (Andaloussi et al., 2013; Sahay et al., 2010). Further investigation is needed to elucidate the factors influencing the relationship between tissue distribution and functional delivery of EVs. A better understanding of these processes will inform the development of more effective strategies for engineering and delivering EV‐based therapeutics, particularly for liver‐targeted applications.
Kupffer cells, the body's primary population of mononuclear phagocytes, constitute between 80% and 90% of all natural macrophages and 20% of the nonparenchymal cells in the liver (Duarte et al., 2015). Hence, we hypothesized that depleting Kupffer cells within the liver could improve EV‐mediated delivery of Cre in the liver. Immunofluorescence images showed functional EV‐mediated transfer in Kupffer and endothelial cells of Ai9 Cre‐loxP reporter mice administered with only Cre‐EVs. Conversely, in the Ai9 Cre‐loxP reporter mice pretreated with clodronate‐encapsulated liposomes, only endothelial cells exhibited tdTomato+ cells. Additionally, significantly fewer tdTomato+Hoechst+ cells were present in the clodronate liposome pretreated group, suggesting that other nonparenchymal cells, such as hepatic stellate, biliary epithelial or resident immune cells (dendritic cells, natural killer cells, and lymphocytes), may have taken up Cre‐EVs but did not exhibit functional Cre delivery within the cytosol. These findings are consistent with the study by Reshke et al., which reported that the internalization of small EVs carrying small interfering RNA in liver cells does not always result in cytosolic delivery of the EV content (Reshke et al., 2020). Our observations suggest that pretreatment with clodronate‐encapsulated liposomes did not improve EV‐mediated delivery of Cre to hepatocytes.
Previous studies have indicated that the internalization of EVs by liver cells may vary depending on the cell type. Németh et al. observed that HEK293T‐palmGFP‐derived medium and small EVs were preferentially internalized by Kupffer‐ and liver sinusoidal endothelial cells in an in vitro uptake assay with ex vivo isolated murine liver cells (Németh et al., 2021). Additionally, Hayashi et al. showed that inhibition of scavenger receptors on the liver sinusoidal endothelial cells increased nanoparticle uptake in Kupffer cells, indicating a competitive nature between the two cell types (Hayashi et al., 2020). In a study by Sago et al., LNP delivery of Cre mRNA was found to have the highest functional Cre delivery in Kupffer cells, followed by endothelial cells, after a single i.v. (Sago et al., 2019). These findings highlight the differential nature of functional EV‐mediated delivery across various cell types in the liver. To comprehensively assess the tissue distribution of EVs, further research incorporating quantitative flow cytometric analysis is warranted. This approach will provide a more precise evaluation of EVs' presence in various tissues. Moreover, it will enable us to elucidate the cell‐specific uptake dynamics and intracellular cargo release mechanisms in greater detail. A refined understanding of these intricate processes holds great potential for advancing the development of targeted and efficient EV‐based drug delivery systems, tailored specifically to address liver‐related diseases.
Zhang et al. found that multiple injections of engineered EVs, loaded with Cas9 orthologue through split GFP complementation, could effectively inactive the Mus Musuculs Pcks9 gene leading to reduced circulating low‐density lipoprotein cholesterol levels by 18% in 28 days (Zhang et al., 2020). Based on these findings, we hypothesized that a higher dose or multiple injections could improve EV‐mediated delivery of Cre recombinase to hepatocytes. Our results showed that repeated i.v.‐injections improved EV‐mediated delivery of Cre recombinase to hepatocytes, where we observed local clusters of HNF4A+tdTomato+Hoechst+ cells. Several studies demonstrated that phospholipids could reversibly block the reticuloendothelial system (RES) phagocytosis, thereby increasing the half‐life of the injected liposomes (Allen et al., 1984; Johnston et al., 2007; Kanada et al., 2015; Proffitt et al., 1983; Souhami et al., 1981). Henceforth, prior studies employed the systemic injection of empty liposomes followed by the administration of drug‐loaded liposomes to improve the drugs' therapeutic efficacy (Lewis et al., 2007; Liu et al., 2015). Repeated i.v. injection of Cre‐EVs may have exerted a similar effect by ‘saturating’ the phagocytic cells and temporarily blocking RES phagocytosis. By reducing the ability of RES phagocytosis to clear EVs from the bloodstream, Cre‐EVs potentially circulate longer, increasing their chances for functional delivery to hepatocytes. Although Kupffer cells are the main cell type responsible for RES phagocytosis in the liver, other cells, such as liver sinusoidal endothelial cells and hepatic stellate cells (Elvevold et al., 2008; Zhan et al., 2006), can also play a role in this process. Therefore, depleting Kupffer cells alone may not be sufficient to improve the functional delivery of EVs to hepatocytes. This could explain our observation that depletion of Kupffer cells did not improve EV‐mediated delivery of Cre to hepatocytes, while repeated injections that potentially ‘saturate’ RES phagocytosis did. Further research is necessary to investigate the cell‐specific involvement of Cre‐EV uptake in the liver, particularly in relation to RES phagocytosis (Allen et al., 1984; Lewis et al., 2007; Liu et al., 2015; Souhami et al., 1981).
Furthermore, repeated i.v. injections resulted in functional EV‐mediated delivery in the liver and spleen, aligning with previous observations (El‐Andaloussi et al., 2012; Lai et al., 2014), which also reported the successful delivery of EV‐associated cargo to these organs. This suggests that repeated EV administration may enhance the therapeutic efficacy of EV‐based treatments for liver and spleen‐related disorders. While the liver, lungs, and spleen are the common destinations due to their role in metabolism and clearance, modern techniques offer the possibility to expand the reach of EVs (Wang et al., 2021). Notably, research by Alvarez‐Erviti et al., demonstrated the successful delivery of siRNA to the mouse brain by systemic injection of targeted exosomes (Alvarez‐Erviti et al., 2011). In another study, Tian et al. leveraged the inherent ability of dendritic cell‐derived exosomes to home to lymph nodes to deliver an anti‐cancer drug for lymphoma treatment via i.v. injection (Tian et al., 2014). The potential of EV engineering to boost organ‐specific targeting is a compelling prospect. Fine‐tuning the surface of EVs with tailored ligands (Roefs et al., 2022), proteins (Ohno et al., 2013; Zhang et al., 2019), or antibodies (Li et al., 2018) presents a promising avenue in this direction. Together, these strategies underscore the remarkable versatility of EVs as drug delivery vehicles, demonstrating their potential to reach an expanded range of tissues and organs.
It is important to note that repeated EV i.v. injections have been linked to accelerated blood clearance, potentially due to EV‐specific IgGs (Driedonks et al., 2022). Additionally, extremely high EV doses potentially cause adverse reactions and impact tissue distribution or cellular uptake resulting from EV aggregation. Smyth et al. reported that tumor‐derived EVs caused asphyxiation after i.v. injection in mice, likely because of EV aggregation causing microvasculature obstruction in the lungs (Smyth et al., 2015). SEC purification of EVs is associated with lower EV aggregation and higher EV functionality than differential ultracentrifugation (Mol et al., 2017). Moreover, electron microscopy analysis of Cre‐EVs did not show significant EV aggregates. Yet, we cannot entirely exclude that our fresh SEC‐isolated Cre‐EVs at the injected dose concentration of ∼1013 particles/ml did not result in EV aggregation, which may affect its tissue distribution or cellular uptake in the liver. Hence, future in vivo studies studying EV‐mediated drug delivery should consider the accelerated blood clearance phenomena and adverse effects resulting from EV aggregating while optimizing the EV administration method to have maximum efficacy in the target organ.
EV tissue distribution and targeted delivery may be affected by their producer cells. HEK293 cells are easy‐to‐transfect cells and, over the past decade, commonly used (±22%) in preclinical EV drug delivery studies, next to cancer‐ (±24%) and stem cell lines (±23%) (de Castilla et al., 2021). One study limitation is that we only studied EV‐mediated delivery from HEK293FT cells. Wiklander et al. (Wiklander et al., 2015) found that EVs derived from C2C12, a melanoma cell line, displayed a significantly higher liver accumulation than EVs derived from dendritic cells. In contrast, EVs derived from dendritic cells accumulated to a considerably higher degree in the spleen. Hence, future research into EVs from different donor cells, such as C2C12 or hepatoma cell lines, could give new insights into their tissue distribution and delivery efficacies.
5. LIMITATIONS OF THE STUDY
In this study, we have demonstrated that repeated i.v. injections of Cre‐EVs can enhance functional EV‐mediated delivery of Cre in hepatocytes. However, the frequency of tdTomato+ recombined cells remained low, even after high or repeated dosages of Cre‐EVs. Compared to in vitro experiments, the number of Cre‐EVs administered in vivo was relatively low, given the large number of cells within the liver of a whole animal. The number of Cre‐EVs administered per in vivo experiment was limited by the amount of fresh SEC‐isolated Cre‐EVs that could be purified prior to the experiment. This constraint highlights the need for additional studies to overcome this limitation, including scaling up Cre‐EV production to investigate the efficacy of functional EV‐mediated Cre delivery at higher doses (Van Niel et al., 2018). Furthermore, optimizing the production and purification process may enable more efficient utilization of Cre‐EVs for in vivo applications (Lener et al., 2015). By addressing these challenges, future research can further elucidate the potential of EV‐mediated protein delivery to the liver and potentially enhance the therapeutic outcomes of this promising approach.
Additionally, the low frequency of tdTomato+ recombined cells could indicate the involvement of other immune cells in the uptake and clearance of Cre‐EVs besides Kupffer cells. Although clodronate liposome‐mediated depletion of Kupffer cells is a well‐established method (Van Rooijen & Sanders, 1994), it does not exclude the participation of other immune cells in Cre‐EV clearance or uptake. Future studies should consider alternative approaches for selectively depleting or labeling other immune cell types to address this limitation, providing a more comprehensive understanding of the cellular mechanisms involved in Cre‐EV uptake and clearance (A‐Gonzalez et al., 2013). Despite this limitation, our study offers valuable insights into engineered EVs' intracellular delivery to the liver, which is crucial for developing liver‐targeted therapies using EVs (Andaloussi et al., 2013; Yáñez‐Mó et al., 2015).
A limitation of our study was that we did not investigate the half‐life of AlexaFluor790 labeled EVs in circulation. Analyzing the clearance rate of EVs by immune cells in circulation could have offered valuable insights into the mechanism of EV‐mediated protein delivery (Hoshino et al., 2015; Wiklander et al., 2015). Additionally, utilizing flow cytometric analysis to identify EV tissue distribution and quantify EV‐mediated delivery in different cell types would have provided a more comprehensive understanding of functional EV delivery in the liver (Gimona et al., 2017; Németh et al., 2017). Despite this limitation, we successfully determined functional EV‐mediated delivery of Cre in Kupffer cells, endothelial cells, and hepatocytes through immunofluorescent analysis.
6. CONCLUSION
EVs hold great promise as a next‐generation drug delivery system for protein/peptide‐based therapeutics. It is essential for preclinical studies investigating EV‐based drug delivery to examine both EV tissue distribution and their capacity to deliver exogenous payloads in relevant preclinical models using appropriate read‐outs. In the current study, we demonstrated that multiple i.v. injections of FKBP12/FRB‐engineered EVs represented the most efficient administration regimen for EV‐mediated delivery of Cre recombinase to hepatocytes. These findings lay the groundwork for future investigations exploring the potential of EVs in the intracellular delivery of therapeutic proteins and genome editing technologies specifically targeting the liver.
AUTHOR CONTRIBUTIONS
Nazma F. Ilahibaks: Data curation, formal analysis, investigation, validation, visualization, writing—original draft and writing—review and editing. Marieke T. Roefs: Methodology, validation, and writing—review and editing. Maike A. D. Brans: Investigation. Christian Snijder Blok: Investigation. Saskia C. A. de Jager: Funding acquisition and resources. Raymond M. Schiffelers: Funding acquisition and resources. Pieter Vader: Funding acquisition, resources, supervision, and writing—review and editing. Zhiyong Lei: Conceptualisation, funding acquisition, project administration, supervision, and writing—review and editing. Joost P. G. Sluijter: Conceptualisation, funding acquisition, resources, supervision, and writing—review and editing.
CONFLICT OF INTEREST STATEMENT
Pieter Vader serves as an advisor for Evox Therapeutics.
Supporting information
Supplementary Figure 1. Cre EVs characterization Western blot analysis showed that Cre‐EVs are positive for the tetraspanin CD63 and Alix, while negative for the endoplasmic reticulum marker calnexin.
Supplementary Figure 2. i.v. and i.p. injection led to EV‐mediated delivery of Cre to hepatocytes in Ai9 Cre‐loxP reporter mice Immunofluorescence images of liver tissue sections fro Ai9 Cre‐loxP reporter mice that received vehicle (PBS), Cre EVs through i.v. injection or i.p. injection.
Supplementary Figure 3. Live in vivo tracking by NIRF imaging of AlexaFluor790‐labeled Cre‐EVs in Ai9 mice that received (A) pretreatment with clodronate‐encapsulated liposomes or (B) only Cre‐EVs.
Supplementary Figure 4. Functional delivery of Cre via EVs in hepatocytes, endothelial cells, and Kupffer cells.
Supplementary Figure 5. Local clusters of hepatocytes in Ai9 Cre‐loxP reporter mice that received multiple injections of Cre‐EVs (A) Immunofluorescence images of liver tissue sections from the three Ai9 Cre‐loxP reporter mice within the 3xD1 group with antibodies against tdTomato+ and Hoechst (Red = tdTomato+, Blue = Hoechst. Scale bar = 50 μm).
ACKNOWLEDGEMENTS
We thank Petra van der Kraak and Cor van Seinen for their experimental support. Funding for the present work was provided by Project EVICARE [No. 725229] of the European Research Council (ERC) to J.P.G.S, co‐funded by the Project SMARTCARE‐II of the BioMedicalMaterials institute to J.P.G.S, the ZonMw‐TAS program [No. 116002016] to J.P.G.S./Z.L, PPS grant [No. 2018B014] to J.P.G.S./Z.L/P.V./S.J., the Dutch Ministry of Economic Affairs, Agriculture and Innovation and the Netherlands CardioVascular Research Initiative (CVON): the Dutch Heart Foundation to J.P.G.S, Dutch Federations of University Medical Centers, the Netherlands Organization for Health Research and Development and the Royal Netherlands Academy of Sciences.
Ilahibaks, N. F. , Roefs, M. T. , Brans, M. A. D. , Blok, C. S. , de Jager, S. , Schiffelers, R. , Vader, P. , Lei, Z. , & Sluijter, J. P. G. (2023). Extracellular vesicle‐mediated protein delivery to the liver. Journal of Extracellular Biology, 2, e97. 10.1002/jex2.97
Zhiyong Lei and Joost P.G. Sluijter contributed equally to this work.
REFERENCES
- A‐Gonzalez, N. , Guillen, J. A. , Gallardo, G. , Diaz, M. , De La Rosa, J. V. , Hernandez, I. H. , Casanova‐Acebes, M. , Lopez, F. , Tabraue, C. , Beceiro, S. , Hong, C. , Lara, P. C. , Andujar, M. , Arai, S. , Miyazaki, T. , Li, S. , Corbi, A. L. , Tontonoz, P. , Hidalgo, A. , & Castrillo, A. (2013). The nuclear receptor LXRα controls the functional specialization of splenic macrophages. Nature Immunology, 14, 831–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Nedawi, K. , Meehan, B. , Micallef, J. , Lhotak, V. , May, L. , Guha, A. , & Rak, J. (2008). Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nature Cell Biology, 10, 619–624. [DOI] [PubMed] [Google Scholar]
- Allen, T. M. , Murray, L. , MacKeigan, S. , & Shah, M. (1984). Chronic liposome administration in mice: Effects on reticuloendothelial function and tissue distribution. Journal of Pharmacology and Experimental Therapeutics, 229, 267–275. [PubMed] [Google Scholar]
- Alvarez‐Erviti, L. , Seow, Y. , Yin, H. , Betts, C. , Lakhal, S. , & Wood, M. J. A. (2011). Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotechnology, 29, 341–345. [DOI] [PubMed] [Google Scholar]
- Andaloussi, S. E. L. , Lakhal, S. , Mäger, I. , & Wood, M. J. A. (2013). Exosomes for targeted siRNA delivery across biological barriers. Advanced Drug Delivery Reviews, 65, 391–397. [DOI] [PubMed] [Google Scholar]
- Campbell, L. A. , Coke, L M. , Richie, C T. , Fortuno, L. V. , Park, A. Y. , & Harvey, B. K. (2019). Gesicle‐mediated delivery of CRISPR/Cas9 ribonucleoprotein complex for inactivating the HIV provirus. Molecular Therapy, 27(1), 151–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick, A. C. , Evitt, N. H. , Lv, W. , & Musunuru, K. (2018). Reduced blood lipid levels with in vivo CRISPR‐Cas9 base editing of ANGPTL3. Circulation, 137, 975–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castilla, P. E. M. , Tong, L. , Huang, C. , Sofias, A. M. , Pastorin, G. , Chen, X. , Storm, G. , Schiffelers, R. M. , & Wang, J.‐W. (2021). Extracellular vesicles as a drug delivery system: A systematic review of preclinical studies. Advanced Drug Delivery Reviews, 175, 113801. [DOI] [PubMed] [Google Scholar]
- de Jong, O. G. , Kooijmans, S. A. A. , Murphy, D. E. , Jiang, L. , Evers, M. J. W. , Sluijter, J. P. G. , Vader, P. , & Schiffelers, R. M. (2019). Drug delivery with extracellular vesicles: From imagination to innovation. Accounts of Chemical Research, 52(7), 1761–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, Q. , Strong, A. , Patel, K M. , Ng, S.‐L. , Gosis, B. S. , Regan, S. N. , Cowan, C. A. , Rader, D. J. , & Musunuru, K. (2014). Permanent alteration of PCSK9 with in vivo CRISPR‐Cas9 genome editing. Circulation Research, 115, 488–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driedonks, T. , Jiang, L. , Carlson, B. , Han, Z. , Liu, G. , Queen, S. E. , Shirk, E. N. , Gololobova, O. , Liao, Z. , Nyberg, L. H. , Lima, G. , Paniushkina, L. , Garcia‐Contreras, M. , Schonvisky, K. , Castell, N. , Stover, M. , Guerrero‐Martin, S. , Richardson, R. , Smith, B. , … Witwer, K W. (2022). Pharmacokinetics and biodistribution of extracellular vesicles administered intravenously and intranasally to Macaca nemestrina. Journal of Extracellular Biology, 1, e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte, N. , Coelho, I C. , Patarrão, R S. , Almeida, J I. , Penha‐Gonçalves, C. , & Macedo, M. P (2015). How inflammation impinges on NAFLD: A role for Kupffer cells. BioMed Research International, 2015, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EL Andaloussi, S. , Mäger, I. , Breakefield, X. O. , & Wood, M. J. (2013). Extracellular vesicles: Biology and emerging therapeutic opportunities. Nature Reviews Drug Discovery, 12, 347–357. [DOI] [PubMed] [Google Scholar]
- El‐Andaloussi, S. , Lee, Y. , Lakhal‐Littleton, S. , Li, J. , Seow, Y. , Gardiner, C. , Alvarez‐Erviti, L. , Sargent, I. L. , & Wood, M. J. A. (2012). Exosome‐mediated delivery of siRNA in vitro and in vivo . Nature Protocols, 7, 2112–2126. [DOI] [PubMed] [Google Scholar]
- Elvevold, K. , Smedsrød, B. , & Martinez, I. (2008). The liver sinusoidal endothelial cell: A cell type of controversial and confusing identity. American Journal of Physiology‐Gastrointestinal and Liver Physiology, 294, G391–G400. [DOI] [PubMed] [Google Scholar]
- Escudier, B. , Dorval, T. , Chaput, N. , André, F. , Caby, M.‐P. , Novault, S. , Flament, C. , Leboulaire, C. , Borg, C. , Amigorena, S. , Boccaccio, C. , Bonnerot, C. , Dhellin, O. , Movassagh, M. , Piperno, S. , Robert, C. , Serra, V. , Valente, N. , Le Pecq, J.‐B. , … Zitvogel, L. (2005). Vaccination of metastatic melanoma patients with autologous dendritic cell (DC) derived‐exosomes: Results of thefirst phase I clinical trial. Journal of Translational Medicine, 3, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein, D. , Werman, A. , Novick, D. , Barak, S. , & Rubinstein, M. (2013). LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proceedings of the National Academy of Sciences, 110, 7306–7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee, P. , Lung, M. S. Y. , Okuzaki, Y. , Sasakawa, N. , Iguchi, T. , Makita, Y. , Hozumi, H. , Miura, Y. , Yang, L. F. , Iwasaki, M. , Wang, X. H. , Waller, M. A. , Shirai, N. , Abe, Y. O. , Fujita, Y. , Watanabe, K. , Kagita, A. , Iwabuchi, K. A. , Yasuda, M. , … Hotta, A. (2020). Extracellular nanovesicles for packaging of CRISPR‐Cas9 protein and sgRNA to induce therapeutic exon skipping. Nature Communications, 11, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimona, M. , Pachler, K. , Laner‐Plamberger, S. , Schallmoser, K. , & Rohde, E. (2017). Manufacturing of human extracellular vesicle‐based therapeutics for clinical use. International Journal of Molecular Sciences, 18, 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, D. , Liang, X. , Pavlova, S. , Wiklander, O. P. B. , Corso, G. , Zhao, Y. , Saher, O. , Bost, J. , Zickler, A. M. , Piffko, A. , Maire, C. L. , Ricklefs, F. L. , Gustafsson, O. , Llorente, V. C. , Gustafsson, M. O. , Bostancioglu, R. B , Mamand, D. R. , Hagey, D W. , Görgens, A. , … El Andaloussi, S. (2020). Quantification of extracellular vesicles in vitro and in vivo using sensitive bioluminescence imaging. Journal of Extracellular Vesicles, 9, 1800222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi, Y. , Takamiya, M. , Jensen, P. B. , Ojea‐Jiménez, I. , Claude, H. , Antony, C. , Kjaer‐Sorensen, K. , Grabher, C. , Boesen, T. , Gilliland, D. , Oxvig, C. , Strähle, U. , & Weiss, C. (2020). Differential nanoparticle sequestration by macrophages and scavenger endothelial cells visualized in vivo in real‐time and at ultrastructural resolution. ACS Nano, 14, 1665–1681. [DOI] [PubMed] [Google Scholar]
- Hoshino, A. , Costa‐Silva, B. , Shen, T.‐L. , Rodrigues, G. , Hashimoto, A. , Tesic Mark, M. , Molina, H. , Kohsaka, S. , Di Giannatale, A. , Ceder, S. , Singh, S. , Williams, C. , Soplop, N. , Uryu, K. , Pharmer, L. , King, T. , Bojmar, L. , Davies, A. E. , Ararso, Y. , … Lyden, D. (2015). Tumour exosome integrins determine organotropic metastasis. Nature, 527, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huotari, J. , & Helenius, A. (2011). Endosome maturation. The EMBO Journal, 30, 3481–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilahibaks, N. F. , Ardisasmita, A. I. , Xie, S. , Gunnarsson, A. , Brealey, J. , Vader, P. , de Jong, O. G. , de Jager, S. , Dekker, N. , Peacock, B. , Schiffelers, R. M. , Sluijter, J. P. G. , & Lei, Z. (2023). TOP-EVs: Technology of Protein delivery through Extracellular Vesicles is a versatile platform for intracellular protein delivery. Journal of Controlled Release, 355, 579–592. [DOI] [PubMed] [Google Scholar]
- Johnston, M. J. , Semple, S. C. , Klimuk, S. K. , Ansell, S. , Maurer, N. , & Cullis, P. R. (2007). Characterization of the drug retention and pharmacokinetic properties of liposomal nanoparticles containing dihydrosphingomyelin. Biochimica et Biophysica Acta, 1768(5), 1121–1127. [DOI] [PubMed] [Google Scholar]
- Kanada, M. , Bachmann, M H. , Hardy, J W. , Frimannson, D. O. , Bronsart, L. , Wang, A. , Sylvester, M. D. , Schmidt, T. L. , Kaspar, R. L. , Butte, M. J. , Matin, A. C. , & Contag, C. H. (2015). Differential fates of biomolecules delivered to target cells via extracellular vesicles. Proceedings of the National Academy of Sciences, 112(12), E1433–E1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans, S. A. A. , Gitz‐Francois, J. J. J. M. , Schiffelers, R. M. , & Vader, P. (2018). Recombinant phosphatidylserine‐binding nanobodies for targeting of extracellular vesicles to tumor cells: A plug‐and‐play approach. Nanoscale, 10, 2413–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooijmans, S. A. A. , Schiffelers, R. M. , Zarovni, N. , & Vago, R. (2016). Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacological Research, 111, 487–500. [DOI] [PubMed] [Google Scholar]
- Lai, C. P. , Mardini, O. , Ericsson, M. , Prabhakar, S. , Maguire, C. A. , Chen, J. W. , Tannous, B. A. , & Breakefield, X. O. (2014). Dynamic biodistribution of extracellular vesicles in vivo using a multimodal imaging reporter. ACS Nano, 8, 483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lener, T. , Gimona, M. , Aigner, L. , Börger, V. , Buzas, E. , Camussi, G. , Chaput, N. , Chatterjee, D. , Court, F. A. , Portillo, H. A. D , O'driscoll, L. , Fais, S. , Falcon‐Perez, J M. , Felderhoff‐Mueser, U. , Fraile, L. , Gho, Y. S. , Görgens, A. , Gupta, R. C. , Hendrix, A. , … Giebel, B. (2015). Applying extracellular vesicles based therapeutics in clinical trials–an ISEV position paper. Journal of Extracellular Vesicles, 4, 30087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, R. E. , Chamilos, G. , Prince, R. A. , & Kontoyiannis, D. P. (2007). Pretreatment with empty liposomes attenuates the immunopathology of invasive pulmonary aspergillosis in corticosteroid‐immunosuppressed mice. Antimicrobial Agents and Chemotherapy, 51, 1078–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Gao, Y. , Gong, C. , Wang, Z. , Xia, Q. , Gu, F. , Hu, C. , Zhang, L. , Guo, H. , & Gao, S. (2018). A33 antibody‐functionalized exosomes for targeted delivery of doxorubicin against colorectal cancer. Nanomedicine, 14, 1973–1985. [DOI] [PubMed] [Google Scholar]
- Liu, T. , Choi, H. , Zhou, R. , & Chen, I.‐W. (2015). RES blockade: A strategy for boosting efficiency of nanoparticle drug. Nano Today, 10, 11–21. [Google Scholar]
- Mangeot, P.‐E. , Dollet, S. , Girard, M. , Ciancia, C. , Joly, S. , Peschanski, M. , & Lotteau, V. (2011). Protein transfer into human cells by VSV‐G‐induced nanovesicles. Molecular Therapy, 19, 1656–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus, M. E. , & Leonard, J. N. (2013). FedExosomes: Engineering therapeutic biological nanoparticles that truly deliver. Pharmaceuticals, 6, 659–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mol, E. A. , Goumans, M. J. , Doevendans, P. A. , Sluijter, J. P. G , & Vader, P. (2017). Higher functionality of extracellular vesicles isolated using size‐exclusion chromatography compared to ultracentrifugation. Nanomedicine, 13, 2061–2065. [DOI] [PubMed] [Google Scholar]
- Montagna, C. , Petris, G. , Casini, A. , Maule, G. , Franceschini, G. M. , Zanella, I. , Conti, L. , Arnoldi, F. , Burrone, O. R. , Zentilin, L. , Zacchigna, S. , Giacca, M. , & Cereseto, A. (2018). VSV‐G‐enveloped vesicles for traceless delivery of CRISPR‐Cas9. Molecular Therapy‐Nucleic Acids, 12, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy, L. A. , Pink, R. C. , & Carter, D. R. F. (2014). Routes and mechanisms of extracellular vesicle uptake. Journal of Extracellular Vesicles, 3, 24641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musunuru, K. , Chadwick, A. C. , Mizoguchi, T. , Garcia, S. P. , Denizio, J. E. , Reiss, C. W. , Wang, K. , Iyer, S. , Dutta, C. , Clendaniel, V. , Amaonye, M. , Beach, A. , Berth, K. , Biswas, S. , Braun, M. C. , Chen, H.‐M. , Colace, T. V. , Ganey, J. D. , Gangopadhyay, S. A. , … Kathiresan, S. (2021). In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature, 593, 429–434. [DOI] [PubMed] [Google Scholar]
- Németh, A. , Orgovan, N. , Sódar, B. W. , Osteikoetxea, X. , Pálóczi, K. , Szabó‐Taylor, K. É. , Vukman, K. V. , Kittel, Á. , Turiák, L. , Wiener, Z. , Tóth, S. , Drahos, L. , Vékey, K. , Horvath, R. , & Buzás, E. I. (2017). Antibiotic‐induced release of small extracellular vesicles (exosomes) with surface‐associated DNA. Scientific Reports, 7, 8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Németh, K. , Varga, Z. , Lenzinger, D. , Visnovitz, T. , Koncz, A. , Hegedűs, N. , Kittel, Á. , Máthé, D. , Szigeti, K. , Lőrincz, P. , O'neill, C. , Dwyer, R. , Liu, Z. , Buzás, E. I. , & Tamási, V. (2021). Extracellular vesicle release and uptake by the liver under normo‐and hyperlipidemia. Cellular and Molecular Life Sciences, 78, 7589–7604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno, S. , Takanashi, M. , Sudo, K. , Ueda, S. , Ishikawa, A. , Matsuyama, N. , Fujita, K. , Mizutani, T. , Ohgi, T. , Ochiya, T. , Gotoh, N. , & Kuroda, M. (2013). Systemically injected exosomes targeted to EGFR deliver antitumor microRNA to breast cancer cells. Molecular Therapy, 21, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proffitt, R. T. , Williams, L. E. , Presant, C. A. , Tin, G W. , Uliana, J. A. , Gamble, R. C. , & Baldeschwieler, J. D. (1983). Liposomal blockade of the reticuloendothelial system: Improved tumor imaging with small unilamellar vesicles. Science, 220, 502–505. [DOI] [PubMed] [Google Scholar]
- Raposo, G. , & Stoorvogel, W. (2013). Extracellular vesicles: Exosomes, microvesicles, and friends. Journal of Cell Biology, 200, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak, J. , Miekus, K. , Kucia, M. , Zhang, J. , Reca, R. , Dvorak, P. , & Ratajczak, M. Z. (2006). Embryonic stem cell‐derived microvesicles reprogram hematopoietic progenitors: Evidence for horizontal transfer of mRNA and protein delivery. Leukemia, 20, 847–856. [DOI] [PubMed] [Google Scholar]
- Reshke, R. , Taylor, J. A. , Savard, A. , Guo, H. , Rhym, L. H. , Kowalski, P. S. , Trung, M. T. , Campbell, C. , Little, W. , Anderson, D. G. , & Gibbings, D. (2020). Reduction of the therapeutic dose of silencing RNA by packaging it in extracellular vesicles via a pre‐microRNA backbone. Nature Biomedical Engineering, 4, 52–68. [DOI] [PubMed] [Google Scholar]
- Roefs, M. T. , Heusermann, W. , Brans, M A. D. , Snijders Blok, C. , Lei, Z. , Vader, P. , & Sluijter, J. P. G. (2022). Evaluation and manipulation of tissue and cellular distribution of cardiac progenitor cell‐derived extracellular vesicles. Front Pharmacol, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothgangl, T. , Dennis, M. K. , Lin, P. J. C. , Oka, R. , Witzigmann, D. , Villiger, L. , Qi, W. , Hruzova, M. , Kissling, L. , Lenggenhager, D. , Borrelli, C. , Egli, S. , Frey, N. , Bakker, N. , Walker, J. A. , Kadina, A. P. , Victorov, D. V. , Pacesa, M. , Kreutzer, S. , … Schwank, G. (2021). In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nature Biotechnology, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sago, C. D. , Krupczak, B. R. , Lokugamage, M. P. , Gan, Z. , & Dahlman, J. E. (2019). Cell subtypes within the liver microenvironment differentially interact with lipid nanoparticles. Cellular and Molecular Bioengineering, 12, 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay, G. , Alakhova, D. Y. , & Kabanov, A. V. (2010). Endocytosis of nanomedicines. Journal of Controlled Release, 145, 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson, E. , Shen, H. , Blanco, E. , Ferrari, M. , & Wolfram, J. (2017). Contribution of Kupffer cells to liposome accumulation in the liver. Colloids Surf B Biointerfaces, 158, 356–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skog, J. , Würdinger, T. , Van Rijn, S. , Meijer, D H. , Gainche, L. , Curry, W. T. , Carter, B. S. , Krichevsky, A. M. , & Breakefield, X. O. (2008). Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nature Cell Biology, 10, 1470–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, T. , Kullberg, M. , Malik, N. , Smith‐Jones, P. , Graner, M. W. , & Anchordoquy, T. J. (2015). Biodistribution and delivery efficiency of unmodified tumor‐derived exosomes. Journal of Controlled Release, 199, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somiya, M. , & Kuroda, S. (2021). Engineering of extracellular vesicles for small molecule‐regulated cargo loading and cytoplasmic delivery of bioactive proteins. Biorxiv, 19(7), 2495–2505. [DOI] [PubMed] [Google Scholar]
- Souhami, R. L. , Patel, H. M. , & Ryman, B. E. (1981). The effect of reticuloendothelial blockade on the blood clearance and tissue distribution of liposomes. Biochimica Et Biophysica Acta (BBA)‐General Subjects, 674, 354–371. [DOI] [PubMed] [Google Scholar]
- Sun, L. , Fan, M. , Huang, D. , Li, B. , Xu, R. , Gao, F. , & Chen, Y. (2021). Clodronate‐loaded liposomal and fibroblast‐derived exosomal hybrid system for enhanced drug delivery to pulmonary fibrosis. Biomaterials, 271, 120761. [DOI] [PubMed] [Google Scholar]
- Tavares, A. J. , Poon, W. , Zhang, Y.‐N. , Dai, Q. , Besla, R. , Ding, D. , Ouyang, B. , Li, A. , Chen, J. , Zheng, G. , Robbins, C. , & Chan, W. C. W. (2017). Effect of removing Kupffer cells on nanoparticle tumor delivery. Proceedings of the National Academy of Sciences, 114, E10871–E10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Y. , Li, S. , Song, J. , Ji, T. , Zhu, M. , Anderson, G J. , Wei, J. , & Nie, G. (2014). A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials, 35(7), 2383–2390. [DOI] [PubMed] [Google Scholar]
- Tsoi, K. M. , Macparland, S A. , Ma, X.‐Z. , Spetzler, V N. , Echeverri, J. , Ouyang, B. , Fadel, S M. , Sykes, E A. , Goldaracena, N. , Kaths, J M. , Conneely, J B. , Alman, B. A. , Selzner, M. , Ostrowski, M. A. , Adeyi, O. A. , Zilman, A. , Mcgilvray, I. D. , & Chan, W. C. W. (2016). Mechanism of hard‐nanomaterial clearance by the liver. Nature Materials, 15, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui, N. B. Y. , Ng, E. K. O. , & Lo, Y. M. D. (2002). Stability of endogenous and added RNA in blood specimens, serum, and plasma. Clinical Chemistry, 48, 1647–1653. [PubMed] [Google Scholar]
- Vader, P. , Mol, E. A. , Pasterkamp, G. , & Schiffelers, R. M. (2016). Extracellular vesicles for drug delivery. Advanced Drug Delivery Reviews, 106, 148–156. [DOI] [PubMed] [Google Scholar]
- van Dommelen, S. M. , Vader, P. , Lakhal, S. , Kooijmans, S. A. A. , Van Solinge, W. W. , Wood, M. J. A. , & Schiffelers, R. M. (2012). Microvesicles and exosomes: Opportunities for cell‐derived membrane vesicles in drug delivery. Journal of Controlled Release, 161, 635–644. [DOI] [PubMed] [Google Scholar]
- Van Niel, G. , d'Angelo, G. , & Raposo, G. (2018). Shedding light on the cell biology of extracellular vesicles. Nature Reviews Molecular Cell Biology, 19, 213–228. [DOI] [PubMed] [Google Scholar]
- Van Rooijen, N. , & Sanders, A. (1994). Liposome mediated depletion of macrophages: Mechanism of action, preparation of liposomes and applications. Journal of Immunological Methods, 174, 83–93. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Maimaitiaili, R. , Yao, J. , Xie, Y. , Qiang, S. , Hu, F. , Li, X. , Shi, C. , Jia, P. , Yang, H. , Wei, M. , Zhao, J. , Zhou, Z. , Xie, J. , Jiang, J. , Cai, H. , Sluijter, J. P. G. , Xu, Y. , Zhang, Y. , & Xiao, J. (2021). Percutaneous intracoronary delivery of plasma extracellular vesicles protects the myocardium against ischemia‐reperfusion injury in Canis. Hypertension, 78, 1541–1554. [DOI] [PubMed] [Google Scholar]
- Wiklander, O. P. B. , Nordin, J. Z. , O'loughlin, A. , Gustafsson, Y. , Corso, G. , Mäger, I. , Vader, P. , Lee, Y. , Sork, H. , Seow, Y. , Heldring, N. , Alvarez‐Erviti, L. , Smith, C. E. , Le Blanc, K. , Macchiarini, P. , Jungebluth, P. , Wood, M. J. A. , & Andaloussi, S. E. (2015). Extracellular vesicle in vivo biodistribution is determined by cell source, route of administration and targeting. Journal of Extracellular Vesicles, 4, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yáñez‐Mó, M. , Siljander, P. R.‐M. , Andreu, Z. , Bedina Zavec, A. , Borràs, F E. , Buzas, E. I. , Buzas, K. , Casal, E. , Cappello, F. , Carvalho, J. , Colás, E. , Cordeiro‐Da Silva, A. , Fais, S. , Falcon‐Perez, J. M. , Ghobrial, I. M. , Giebel, B. , Gimona, M. , Graner, M. , Gursel, I. , … De Wever, O. (2015). Biological properties of extracellular vesicles and their physiological functions. Journal of Extracellular Vesicles, 4, 27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, S. , Jiang, J X. , Wu, J. , Halsted, C. , Friedman, S L. , Zern, M. A. , & Torok, N. J. (2006). Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo . Hepatology, 43, 435–443. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Wu, J. , Wu, J. , Fan, Q. , Zhou, J. , Wu, J. , Liu, S. , Zang, J. , Ye, J. , Xiao, M. , Tian, T. , & Gao, J. (2019). Exosome‐mediated targeted delivery of miR‐210 for angiogenic therapy after cerebral ischemia in mice. Journal of Nanobiotechnology, 17, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Xu, Q. , Zi, Z. , Liu, Z. , Wan, C. , Crisman, L. , Shen, J. , & Liu, X. (2020). Programmable extracellular vesicles for macromolecule delivery and genome modifications. Developmental Cell, 55, 784–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zomer, A. , Steenbeek, S. C. , Maynard, C. , & van Rheenen, J. (2016). Studying extracellular vesicle transfer by a Cre‐loxP method. Nature Protocols, 11, 87–101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Cre EVs characterization Western blot analysis showed that Cre‐EVs are positive for the tetraspanin CD63 and Alix, while negative for the endoplasmic reticulum marker calnexin.
Supplementary Figure 2. i.v. and i.p. injection led to EV‐mediated delivery of Cre to hepatocytes in Ai9 Cre‐loxP reporter mice Immunofluorescence images of liver tissue sections fro Ai9 Cre‐loxP reporter mice that received vehicle (PBS), Cre EVs through i.v. injection or i.p. injection.
Supplementary Figure 3. Live in vivo tracking by NIRF imaging of AlexaFluor790‐labeled Cre‐EVs in Ai9 mice that received (A) pretreatment with clodronate‐encapsulated liposomes or (B) only Cre‐EVs.
Supplementary Figure 4. Functional delivery of Cre via EVs in hepatocytes, endothelial cells, and Kupffer cells.
Supplementary Figure 5. Local clusters of hepatocytes in Ai9 Cre‐loxP reporter mice that received multiple injections of Cre‐EVs (A) Immunofluorescence images of liver tissue sections from the three Ai9 Cre‐loxP reporter mice within the 3xD1 group with antibodies against tdTomato+ and Hoechst (Red = tdTomato+, Blue = Hoechst. Scale bar = 50 μm).