

Abstract
Background:
Heterologous functional validation studies of putative long QT syndrome subtype 2 (LQT2)-associated variants clarifies their pathological potential and identifies disease mechanism(s) for most variants studied. The purpose of this study is to clarify the pathological potential for rare nonsynonymous KCNH2 variants seemingly associated with sudden infant death syndrome (SIDS).
Methods and Results:
Genetic testing of 292 SIDS cases identified nine KCNH2 variants: E90K, R181Q, A190T, G294V, R791W, P967L, R1005W, R1047L and Q1068R. Previous studies show R181Q-, P967L-, and R1047L-Kv11.1 channels function similar to wild-type (WT) Kv11.1 channels, whereas Q1068R-Kv11.1 channels accelerate inactivation gating. We studied the biochemical and biophysical properties for E90K-, G294V-, R791W- and R1005W-Kv11.1 channels expressed in HEK293 cells. Western blot and voltage-clamping analyses of cells expressing E90K-, G294V-, R791W- and R1005W-Kv11.1 channels demonstrated these variants express and generate peak Kv11.1 current levels similar to cells expressing WT-Kv11.1 channels, but R791W- and R1005W-Kv11.1 channels accelerated deactivation and activation gating, respectively. We examined the Electronic Health Record of patients who were genotype positive for these particular SIDS-linked KCNH2 variants and found that all of them had median heart rate corrected QT intervals <480 ms and none had been diagnosed with LQTS or suffered cardiac arrest. Simulating the impact of dysfunctional gating variants using computational models of the human ventricular action potential (AP) predicted they have little impact on AP duration.
Conclusion:
We conclude these rare Kv11.1 missense variants are not LQT2-causative variants and therefore do not represent the pathogenic substrate for SIDS in the variant-positive infants.
Keywords: Long QT syndrome, Sudden Infant Death Syndrome, KCNH2 (hERG), Variants of Uncertain Significance (VUS)
Subject Terms: Arrhythmias, Sudden Cardiac Death, Ventricular Fibrillation
Graphical Abstract
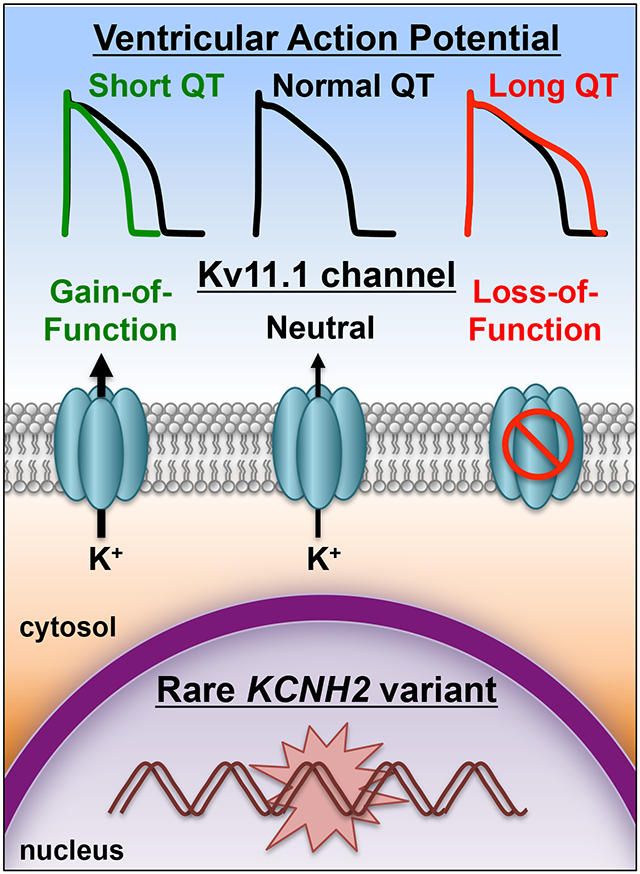
Introduction
The expert consensus is that sudden infant death syndrome (SIDS) is multifactorial. However, abnormalities in respiratory or cardiac dysfunction are suspected in some cases.1-3 Drs. Peter Schwartz and Stephen Epstein originally suggested that some cases of SIDS might represent infantile sudden cardiac death (SCD) stemming from primary or secondary abnormalities in ventricular repolarization.4, 5 This could occur by a number of mechanisms, including abnormal autonomic development, a secondary response to a medication or disease, or a genetic abnormality, specifically long QT syndrome (LQTS).6
The Schwartz hypothesis was tested in a 19-year study that examined electrocardiogram (ECG) data from over 33,000 infants during the first year of their life.2 Twenty four of the infants suffered SIDS and 10 others died from an attributable cause (not SIDS). The average heart rate corrected QT interval (QTc) as seen on ECG, for the healthy infants (using Bazett’s correction formula) was 400 ± 20 ms, whereas the average QTc for the SIDS cases was 435 ± 25 ms. Moreover, the SIDS infants had longer QTc than the infants who died from an attributable cause (392 ± 26 ms). Although half of the SIDS cases had a QTc > 440 ms, clinically screening neonates using such a QTc cutoff value is problematic because approximately 2.5% of the healthy neonates also had a QTc > 440 ms.
Since the major genes that are linked to abnormal QT prolongation have been identified,7 combining ECG testing with genetic screening holds the promise of potentially delineating vulnerable infants. To begin addressing the link between LQTS and SIDS, several groups have performed retrospective molecular autopsies (i.e. postmortem genetic testing) for the most common genetic subtypes of LQTS in SIDS cases, and found a rare nonsynonymous (amino acid altering) genetic variant in one of the major LQTS susceptibility genes 10-15% of the time. Unfortunately, genetic testing is limited by its probabilistic rather than deterministic nature for identifying a dysfunctional variant.8, 9
Loss-of-function mutations in KCNH2 underlies type 2 LQTS (LQT2) and is one of the most common forms of LQTS.10, 11 KCNH2 encodes the voltage-gated K+ channel α-subunit Kv11.1, which generates the rapidly activating delayed rectifier K+ current () in the heart.11, 12 Studying dozens of different suspected LQT2-associated variants using heterologous expression in mammalian cell systems has proven extremely effective for confirming true disease-causing variants.13 About 95% of LQT2-linked missense mutations have a dysfunctional phenotype. These studies show ~90% of the Kv11.1 missense mutations decrease the intracellular transport (trafficking) of Kv11.1 to the cell surface membrane, ~5% alter Kv11.1 channel gating and/or conduction, and ~5% traffic and function similar to wild-type Kv11.1 channels (WT).13-22 Many of the latter variants have been identified subsequently in exomic databases of non-LQTS subjects, suggesting that they are not pathogenic.23
Heterologous expression studies performed on a small number of KCNH2-linked SIDS variants suggest that several of them function normally. These are likely benign variants and not involved in the infant’s death.24, 25 The purpose of this study was to extend these findings and functionally investigate several additional KCNH2 variants that were identified previously in a cohort of 292 SIDS cases. What is novel in this study is that we perform electronic health care record (EHR) analysis of adult patients who had tested positive for any of the KCNH2 variants found among the deceased infants, and determine the physiological significance of dysfunctional KCNH2 variants using computational human action potential simulations.
Materials and Methods
Data, Materials, and Code Disclosure Statement
In order to minimize the possibility of unintentionally sharing information that can be used to re-identify private information, only the information for the functional data, methods used in the analysis, and materials used to conduct the research will be made available to researchers upon reasonable request (Brian P. Delisle; brian.delisle@uky.edu).
Sudden Infant Death Syndrome (SIDS) Cohort
Frozen necropsy tissue or autopsy blood from 292 sudden infant death syndrome (SIDS; 178 male, 114 female; average age at death = 2.9 ± 1.9 months, age range 0.1-12 months; 204 white, 75 black, 10 Hispanic, 2 Asian, 1 unknown ethnicity) cases derived from four population-based cohorts from the United States (US), representing the northeast, south, and southwestern portions of the US, were submitted by medical examiner offices to the Mayo Clinic Windland Smith Rice Sudden Death Genomics Laboratory for research-based postmortem genetic testing.26-33 The enrollment criterion was a comprehensive medico-legal, autopsy-negative sudden unexplained death of an infant < 1 year of age, including a negative toxicology screen and death scene investigation. Infants with asphyxia or specific disease causing death were excluded. This Mayo Foundation Institutional Review Board-approved, anonymous necropsy study only had limited medical information such as the sex, ethnicity and age at the time of death available. Time of day, medication use, infection, and position at death were unavailable.
Postmortem Mutational Analysis of the KCNH2-Encoded Kv11.1 K+ Channel
Genomic DNA was extracted using the Puregene DNA Isolation Kit (Qiagen, Inc, Valencia, Calif). Comprehensive coding region point-mutation analysis of KCNH2 was performed using PCR, DHPLC, and direct DNA sequencing. In this study, only non-synonymous variants (i.e. amino acid altering) were considered. Each variant was assessed for its presence in the most recent Genome Aggregation Database (GnomAD) (Http://exac.Broadinstitute.Org). Four different computational methods (KvSNP, Polyphen2, SNPs&Go, and SIFT) were used to predict whether or not the non-synonymous variants might be pathogenic.34-38 Briefly, KvSNP is a machine learning classifier that is applied to variants in Kv channels to predict the likelihood that the variant causes disease; PolyPhen2 is a tool which predicts possible impact of an amino acid substitution based on physical and comparative considerations; SNPs&GO predicts whether a variation is disease related based on evolutionary information; and SIFT prediction utilizes the degree of conservation from closely related sequences. Each computational approach that predicted a damaging or pathogenic mutation scored 1 point (combined score can range from 0-4). Since more than 97% of LQT2-associated dysfunctional Kv11.1 variants (n=184) have a score > 1 point (data not shown), we functionally characterized novel and/or previously uncharacterized KCNH2 variants with a score > 1.
Mutagenesis, Tissue Culture, and Transfection
The appropriate nucleotide changes of the Kv11.1 variants E90K, G294V, R791W and R1005W were engineered in the wild-type (WT) Kv11.1 cDNA cloned in the pcDNA3 vector using the QuickChange Site Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). The integrity of all the constructs was verified by DNA sequencing (AGTC, University of Kentucky, Lexington, KY). Human Embryonic Kidney 293 (HEK293) cells were cultured at 37°C (5% CO2) in MEM supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA). The cells were transfected using Lipofectamine (Life Technologies, Grand Island, NY) with WT, E90K, G294V, R791W and R1005W Kv11.1 plasmid DNA (3 μg). For electrophysiological studies, cells were also transfected with enhanced Green Fluorescent Protein (GFP) cDNA subcloned in pKR5 (0.3 μg). GFP positive cells were analyzed using for whole-cell patch clamp technique 46-54 hrs after transfection.
Western Blot
Cells for Western blotting were harvested with lysis buffer similar to that previously described.39 Equivalent amounts of total protein were electrophoresed on a 6.5% SDS-polyacrylamide gel, transferred electrophoretically to nitrocellulose, and probed with the anti-Kv11.1 (Santa Cruz Biotechnologies, Santa Cruz, CA) and anti-Na+/K+-ATPase (Abcam, Cambridge, MA) antibodies. The protein concentration was measured using the Bio-Rad DC Protein Assay (Bio-Rad, Hercules, CA). For each experiment, protein standards were created from 2 mg/mL albumin stock by serially diluting BSA stock in 1% NP-40 buffer. A standard concentration-absorbance curve was generated to calculate the lysate sample concentrations from linear regression. The immunoblots were also probed with a control antibody (anti-Na+/K+-ATPase) for protein loading and transfer. This allows for a better comparison when the loading and transfer might not be uniform. The anti-Kv11.1 and anti-Na+-K+-ATPase antibodies were detected using Odyssey Goat anti-Rabbit (LI-COR Biosciences, Lincoln, NE) and Odyssey Donkey anti-Mouse (LI-COR Biosciences, Lincoln, NE) antibodies, respectively. The LI-COR Odyssey infrared imaging system (Li-Cor Biosciences, Lincoln, NE) was used to image and quantify the immunolabeling.
Electrophysiology
Functional analyses were done using standard whole-cell patch clamp technique on GFP positive cells similar to that previously described.25, 40 The external solution contained (in mM) 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.4 with NaOH), and the internal pipette solution contained (in mM) 130 KCl, 1 MgCl2, 5 EGTA, 5 MgATP, 10 HEPES (pH 7.2 with KOH). An Axopatch-200B patch clamp amplifier (Axon Instruments, Union City, CA) was used to measure macroscopic currents and cell capacitance. The pipette resistances were 1-2 MΩ and series resistance was compensated up to 95%. The pCLAMP 10 software (Axon Instruments, Union City, CA) was used to generate the different voltage protocols, acquire current signals, and for data analyses. Origin 7.0 (Microcal, Northhampton, MA) was used for performing Boltzmann curve fitting to the current-voltage (I-V) relations and for generating graphs. The data were fit with the following Boltzmann equation:
where is the minimally activated current; is the maximally activated current; is the mid-point potential for half maximal activation; and is the slope factor. The holding potential was −80 mV and the dashed line in figures indicates zero current. Deactivation, inactivation, and recovery from inactivation kinetic measurements were performed similar to that described previously.25
Study Participants and Exome Sequencing
Whole exome sequencing was conducted as part of the DiscovEHR collaboration of the Geisinger Health System (GHS) and the Regeneron Genetics Center. The GHS Institutional Review Board approved the study. All clinical and genetic data for this study were de-identified by a data broker who was not involved in the study. The DiscovEHR study population included over 90,000 consented participants from the MyCode® Community Health Initiative of GHS, which consists of ~95% of individuals of European ancestry. The MyCode® cohort represents a cross section of Geisinger’s population (from sick to healthy) and it is representative of the local adult population. The average longitudinal EHR data for the non-transient Geisinger patient population is 15 years. The unselected nature of Geisinger’s cohort allows us to understand the full scope of phenotypic expression of various diseases. A de-identified EHR database is linked to WES and genotype data for the purpose of studying genotype-phenotype relationships. EHR data is maintained in a scalable enterprise data warehouse allowing access to various clinical measures including international classification of disease (ICD) codes, prescription information, lab results, procedure codes, imaging data, billing data etc. Exome capture, PCR amplification, and 75 bp paired-end sequencing on an Illumina v4 HiSeq 2500 have been described in detail elsewhere.41
ECG and EHR Data
ECG data maintained in the MUSE cardiology information system was used to automatically measure QT intervals. QTc values were calculated based on resting heart rate during the same ECG measurement. ICD9 codes for Long QT Syndrome (426.82) and syncope (780.2), as well as ICD10 codes for arrhythmias (I44 Atrioventricular and left bundle-branch block, I45 Other conduction disorders, I46 Cardiac arrest, I47 Paroxysmal tachycardia, I48 Atrial fibrillation and flutter, I49 Other cardiac arrhythmias) were extracted from Geisinger patients' EHR.
Computational Action Potential Modeling
All simulations were done using the Romero et al. modification of the O’Hara-Virag-Varro-Rudy (OVVR) human ventricular action potential (AP) model, which incorporates a linear Markovian IKr channel model that has three closed states (C3, C2, C1), one open (O), and one inactivated state (I).42, 43 The component was increased 2-fold to recapitulate AP duration/morphologies similar to the original O’Hara simulations (data not shown). We mimicked the gating effects of three different KCNH2 variants that were found in a SIDS victim by modifying individual rate constants to recapitulate the electrophysiological analysis. To simulate the functional impact of the R791W variant, we increased the deactivation transition rate (O to C1) 2-fold. To simulate the R1005W variant, we increased the three activation rate constants (C3 to C2, C2 to C1, and C1 to O) 2-fold (generating a −15 mV shift in the simulated I-V relation). To model the Q1068R variant, we increased the inactivation rate constant by a factor of 1.2. The duration of the AP to 90% repolarization (AP90) was calculated from the simulations similar to that described previously.21
Statistics
Data are reported as the mean ± standard error of the mean (SE) or standard deviation (SD). A one-way ANOVA was used to determine if there was difference among the groups, and post hoc analyses using the Dunnett’s test were performed to see which group(s) differed compared to cells expressing WT-Kv11.1 channels (GraphPad Prism software, La. Jolla, CA) Significance was determined at the P < 0.05 level.
Results
Following postmortem genetic testing, 8 rare KCNH2 (Kv11.1) missense variants (E90K, R181Q, A190T, G294V, R791W, P967L, R1005W, and Q1068R) were identified in 9/292 (3.1% overall; 4/204 [1.9%] white, 5/75 [6.7%] black) SIDS cases (Table 1). Of the 8 variants, only 2 variants (G294V and R1005W) were absent from GnomAD (Table 1). In addition, the common polymorphism R1047L was identified in 9/204 (4.4%) white and 0/75 (0%) black SIDS cases compared to 1717/33,584 (5.1%, p=0.87) European (non-Finnish) white and 50/7,646 (0.65%, p=1) black exomes in GnomAD.
Table 1.
Sudden Infant Death Syndrome Cases Identified With a Rare Non-Synonymous KCNH2 Variant
| Case# | Age (Months) |
Sex | Ethnicity | KCNH2 Variant |
Overall | European (non-Finish) |
European (Finnish) |
African | Latino | Ashkenaz i Jewish |
East Asian |
South Asian | Other |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 | M | White | E90K | 3/112,877 | 0/49,425 | 0/10,256 | 0/6,513 | 3/16,361 | 0/4669 | 0/8246 | 0/14,866 | 0/2541 |
| 2 | 2 | M | Black | R181Q | 90/50,191 | 0/21,119 | 0/3,904 | 85/4747 | 2/6,480 | 0/3,193 | 0/2,030 | 1/7,371 | 2/1,482 |
| 3 | 4 | F | Black | A190T | 25/15,223 | 0/7,440 | 0/1,643 | 24/4309 | 2/391 | 0/149 | 0/809 | NA | 0/482 |
| 4 | 4 | M | White | A190T | - | - | - | - | - | - | - | - | - |
| 5 | 6 | M | White | G294V | 0/526 | 0/248 | 0/35 | 0/20 | 0/105 | 0/29 | 0/22 | 0/46 | 0/21 |
| 6 | 1 | F | Black | R791W | 26/138,047 | 0/63,055 | 0/12,813 | 24/11,898 | 1/17,205 | 0/5,070 | 0/9,409 | 1/15,386 | 0/3,211 |
| 7 | 2 | M | Black | P967L | 30/127,481 | 5/57,328 | 0/11,423 | 21/10,714 | 0/16,630 | 0/4841 | 0/8,954 | 2/14,581 | 2/3,010 |
| 8 | 5 | F | White | R1005W | 0/86,582 | 0/36,106 | 0/8,044 | 0/4,561 | 0/12,870 | 0/4,206 | 0/6,363 | 0/12,349 | 0/2,083 |
| 9 | 8 | F | Black | Q1068R | 11/122,236 | 6/55,287 | 0/11,118 | 4/7,622 | 1/16,718 | 0/4872 | 0/8,612 | 0/15,293 | 0/2,714 |
A list of the sudden infant death syndrome (SIDS) cases with a rare non-synonymous KCNH2 variant. Also shown is the number of subjects and heterozygote frequency of each variant within Genome Aggregation Database (GenomAD) exome database. Because of exome sequencing coverage issues, the number of subjects with quality exome sequencing reads at a specific location is different when reporting data from the GenomAD database. Therefore the denominator differs for each of the variants listed. M = male, F=female.
We used four different bioinformatics methods (KvSNP, Polyphen2, Single Nucleotide Polymorphisms & Go, and SIFT) to predict whether or not the nonsynonymous variants may be pathogenic.34-38 Two or more computational methods predicted that the variant would be damaging or potentially disease causing for 7 of the 9 variants analyzed (Table 2). For two variants (R791W and R1005W), all four computational methods predicted the variant to be damaging or disease causing.
Table 2.
Summary of Computational Variant Prediction and Functional Analysis
| SIDS-linked Variants |
KvSNP | Polyphen2 | SNPs & Go | SIFT | Score | In controls |
Functional analysis |
|---|---|---|---|---|---|---|---|
| E90K | Neutral | Benign | Disease | Damaging | 2 | Y | normal |
| R181Q | Neutral | Damaging | Disease | Tolerated | 2 | Y | normal44 |
| A190T | Neutral | Benign | Disease | Tolerated | 1 | Y | ND |
| G294V | Disease | Benign | Disease | Tolerated | 2 | N | normal |
| R791W | Disease | Damaging | Disease | Damaging | 4 | Y | altered gating22 |
| P967L | Neutral | Benign | Neutral | Tolerated | 0 | Y | normal25 |
| R1005W | Disease | Damaging | Disease | Damaging | 4 | N | altered gating |
| R1047L | Disease | Benign | Disease | Tolerated | 2 | Y | normal25 |
| Q1068R | Benign | Damaging | Disease | Damaging | 3 | Y | altered gating25 |
The list of KCNH2 variants identified in the SIDS cohort along with the predicted effect of the variant on the function of the channel using KvSNP, Polyphen2, SNPs & Go or SIFT. The score is the number of computational predictions that predict the variant as damaging or disease. The table also summarizes whether or not the individual variants have been identified in control subjects (Y = yes, N = no) and the results of functional testing (normal = similar to WT, ND = not determined).
Of the 9 variants identified in our SIDS cases, 4 (R181Q, P967L, R1047L, and Q1068R) were characterized previously using heterologous expression (Table 2).22, 25, 44 Three of the variants (R181Q, P967L, and R1047L) were functionally similar to WT-Kv11.1 channels. However, Q1068R showed Kv11.1 channel gating with accelerated inactivation kinetics.25 We studied the remaining four Kv11.1 variants (E90K, G294V, R791W, and R1005W) that were suggested by two or more computational methods to be damaging or disease causing.
About 90% of Kv11.1 missense variants linked to LQT2 generate channels that do not traffic properly to the cell surface. Kv11.1 is modified co-translationally in the endoplasmic reticulum (ER) by the attachment of asparagine-linked (N-linked) core glycans at N598 to generate a 135 kDa immature glycoprotein.22 The glycan moiety undergoes further post-translational processing in the Golgi apparatus to generate the terminally-glycosylated or “mature” 155 kDa Kv11.1 α-subunit.45 Trafficking-deficient Kv11.1 mutations can be identified using Western blot analyses because of a decrease in the relative amount of 155 kDa mature Kv11.1 (mature Kv11.1/total Kv11.1).16, 21, 39, 46 Based on Western blot analyses, E90K-, G294V-, R791W-, or R1005W-Kv11.1 channels trafficked normally (Figure 1A). Using the whole-cell patch clamp technique, we next determined whether cells expressing E90K-, G294V-, R791W-, or R1005W-Kv11.1 channels altered the peak compared to cells expressing WT Kv11.1 channels. Cells were pre-pulsed from −80 mV to a maximally activating potential (50 mV) for 3 s, and then hyperpolarized to a test-pulse of −120 mV for 2 s. For cells expressing the Kv11.1 variant channels, the peak inward measured during the test-pulse was not different compared to cells expressing WT-Kv11.1 channels (Figure 1B). We also found that co-expressing WT-Kv11.1 channels with the variant-containing Kv11.1 channels did not negatively affect peak IKv11.1 (data not shown). Together these data demonstrate that the variant-containing Kv11.1 channels do not negatively affect either trafficking or peak IKv11.1.
Figure 1. Kv11.1 variants found in SIDS cases traffic and are expressed similar to WT.

A) Representative immunoblot analysis of lysates isolated from cells expressing WT, E90K, G294V, R791W, or R1005W. The immunoblots were probed with anti-Kv11.1 and anti-Na+-K+-ATPase as a loading/transfer control. The images are all from the same immunoblot and were cropped for presentation purposes. The relative amount of mature Kv11.1 protein (mature/total Kv11.1) based on immunoblot analyses for each set of experiments is plotted, as well as the relative amount of total Kv11.1 protein to Na+-K+-ATPase (n = 3-4, P > 0.05). B) The maximal amount of conducted by cells expressing WT-, E90K-, G294V-, R791W- and R1005W-Kv11.1 is also shown (n > 10 cells per group, P > 0.05). Error bars are SE.
Next, we determined whether E90K-, G294V-, R791W-, or R1005W-Kv11.1 channels altered the activation gating properties by applying step-like pulses from −80 mV to 70 mV in 10 mV increments for 5 s, followed by a “tail” pulse to −50 mV for 5 s (Figure 2A). The peak measured during the tail pulse was plotted as a function of the step-pulse potential and the corresponding I-V relations were fit with a Boltzmann function to calculate the mean , , and (Figure 2B). Compared to cells expressing WT-Kv11.1 channels, only R1005W-Kv11.1 channels altered activation properties by causing a ~15 mV shift of the to more negative potentials. There were no differences in the slope-factor. These data demonstrate that R1005W is unlikely to cause LQT2 because it alters Kv11.1 channel activation to increase at negative membrane potentials (gain-of-function). This raises the intriguing possibility this variant could cause pathogenicity by causing Short QT Syndrome (SQTS).47
Figure 2. The R1005W and R791W variants alter Kv11.1 channel activation and deactivation, respectively.

A) Representative families of currents measured from cells transiently expressing WT-, E90K-, G294V-, R791W-, or R1005W-Kv11.1 channels using the voltage protocol shown. B) The left graph shows the I-V relations of (normalized to the peak ) measured during the test-pulse and plotted as a function of the pre-pulse from cells expressing WT (solid squares), E90K (shaded downward triangles), G294V (shaded upward triangles), R791W (open circles), and R1005W (solid diamonds). C) The decay measured during the test-pulse following the pre-pulse to 70 mV was described as a double exponential process to calculate the fast and slow time-constants associated with Kv11.1 deactivation ( and ). The right graph shows the relative amplitude of the divided by the total amplitude of and () (n = 8-15 cells per group, *P < 0.05). Error bars are SE.
The decay of the during the tail-pulse to −50 mV reflects the kinetics of Kv11.1 channel deactivation gating. Acceleration of Kv11.1 deactivation kinetics has been implicated as a cause for LQT2.15, 48 To test if any of these variants altered Kv11.1 deactivation, we fit the decay phase of the during the test-pulse to −50 mV as a double exponential process with a fast and slow time component ( and ). Cells expressing R791W-Kv11.1 channels conducted that decayed with a faster compared to cells expressing WT-Kv11.1 channels (Figure 2C).
Since R791W-Kv11.1 channels accelerated deactivation rates at −50 mV, we tested whether these alterations persisted over a wider potential range. We pre-pulsed cells expressing WT- or R791W-Kv11.1 channels to 50 mV for 2 s to activate the channels, followed by a test-pulse from −120 to −60 mV for 10 s in 10 mV increments to measure the decay of at different potentials (Figure 3). Compared to cells expressing WT-Kv11.1 channels, cells expressing R791W-Kv11.1 channels showed faster and for many of the test-pulse potentials ≤ −60 mV. Also, the relative amplitude of the fast component was larger for several potentials tested. These data confirm that R791W-Kv7.11 accelerates Kv11.1 deactivation kinetics over a wide range of potentials and increases the fraction of channels that deactivate with the faster time component. These findings are similar to those reported previously.22
Figure 3. R791W-Kv11.1 speeds Kv11.1 channel deactivation over a wide range of voltages.

A) Representative families of currents measured from cells expressing WT or R791W using the voltage protocol shown. B-C) The left set of graphs show the mean (top), (bottom) plotted as a function of the test-pulse for WT (solid squares) or R791W (open circles), and the right graphs shows the corresponding fractional amplitude of (n = 4-6 cells per group, P < 0.05). Error bars are SE.
We next determined whether E90K, G294V, R791W, or R1005W altered the properties of Kv11.1 channel inactivation. The rate for the recovery of Kv11.1 channel inactivation was measured by pre-pulsing cells to 50 mV for 5 s followed by a test-pulse from −120 to −30 mV for 1.5 s (Figure 4A). To calculate the time constant for recovery from inactivation (), the rising phase of measured during the test-pulse was described as a single exponential process, and these rates were plotted as a function of the test-pulse potential (Figure 4B). Cells expressing E90K-, G294V-, R791W-, or R1005W-Kv11.1 channels did not alter the recovery of Kv11.1 inactivation kinetics for any of the potentials tested. We also measured the development of Kv11.1 channel inactivation by depolarizing cells to 60 mV for 300 ms, hyperpolarizing to −100 mV for 25 ms to re-open most channels, and then applied a test-pulse from −20 to 60 mV in 10 mV increments for 300 ms (Figure 4A). The decay measured during the test-pulse was described as a single-exponential process and used to calculate a time constant for the development of inactivation () (Figure 4B). Compared to cells expressing WT-Kv11.1 channels, cells expressing E90K-, G294V-, R791W-, or R1005W-Kv11.1 channels did not alter the .
Figure 4. KCNH2 variants found in SIDS cases do not alter Kv11.1 channel inactivation.

A) Representative families of currents measured from cells expressing WT using the voltage protocols to measure the recovery (left side) and development (right side) of inactivation are shown. The was calculated by describing the rising phase of measured during the test-pulse as a single exponential process, and similarly, the was calculated by describing the falling phase of measured during the test pulse as a single exponential process. B) The left and right sides of the graph shows the mean and , respectively, calculated from cells expressing WT- (solid squares), E90K- (shaded downward triangle), G294V- (shaded upward triangle), R791W- (open circle), or R1005W-Kv11.1 (solid diamond) plotted as a function of the test-pulse (n = 5-14 cells per group, P > 0.05). Error bars are SE.
Based on the functional analysis of the KCNH2 variants identified in this cohort of 292 SIDS cases, 3 of the 8 variants represent possible SIDS-associated mutations stemming from defective Kv11.1 channel gating while the others constitute background genetic noise (Figure 5A). We sought to confirm this using clinical data. The DiscovEHR collaboration is a joint effort of the GHS and Regeneron Genetics Center that uses WES data coupled to patient EHR to assess links between specific genetic variations and disease phenotypes.41 Using WES data in the first 92,455 subjects, we found over 60 individuals (1 per 1468 persons) who were positive for one of the six rare KCNH2 variants identified in the 292 SIDS cases (the R1047L variant is a common variant, with a MAF=0.02 in this cohort, and was therefore excluded from the analysis). ECG records were available for 32 of 63 rare variant-positive subjects and these were used to calculate median QTc for each patient. Summary of QTc values and history of syncope is shown in Figure 5B. Importantly, in these variant-positive individuals, none of the patients had an LQTS diagnosis or is suspected of having LQTS based on the expert consensus statement on the diagnosis of LQTS.49 The patients did not have a risk score > 3.5, and although several patients had been coded for episode(s) of unexplained syncope, the median QTc intervals were all < 480 ms. Only six of the 63 patients carrying these KCNH2 variants had an arrhythmia code in their EHR, which is within the normal prevalence in the general population. Additionally none of the patients harboring R1005W showed any signs or symptoms of SQTS. These clinical data demonstrate a lack of a positive link between these the KCNH2 variants, including the ones that alter Kv11.1 channel gating, and the manifestation of an inherited arrhythmia syndrome caused by mutations in KCNH2.
Figure 5. KCNH2 variants with altered gating properties in vitro do not associate with clinical phenotypes in vivo or predict ventricular AP prolongation in silico.

A) The schematic shows an individual Kv11.1 α-subunit with an intracellular amino terminus (N1), six transmembrane segments, and the carboxy terminus (C1159). The relative location of the Kv11.1 missense variants found in the SIDS cases is shown (squares). The solid red (R791W), green (R1005W), and blue (Q1068R) squares highlight the three variants with altered Kv11.1 channel gating. B) Shown is the median QTc of patients who were positive for one of the variants previously identified in our SIDS cohort. The patients’ genders are denoted as squares (male) and circles (female). Red indicates EHR history of unexplained syncope. Median QTc of the general population cohort (dashed line), as well as the QTc threshold for genetic testing (red line) is also shown. Error bars are SD. C) We used a modified version of the O’hara ventricular action potential (AP) model that included a Markovian model for . Shown are the rate transitions in the Markovian model that were adjusted to mimic the gating changes caused by R791W (red), R1005W (green), and Q1068R (blue), the corresponding steady-state AP waveforms stimulated at 0.5 Hz, and the respective steady-state APD90 for each of the simulations.
We were surprised the Kv11.1 variants that altered Kv11.1 channel gating did not associate with a clinical phenotype. To better understand how these gating changes might impact the ventricular AP duration (a cellular correlate of the QTc), we incorporated these gating changes using a modified OVVR AP model that includes a Markovian model for the component.42, 43 The gating changes caused by each of these Kv11.1 variants are different: R791W accelerates deactivation kinetics (loss-of-function), R1005W alters the voltage-dependence of Kv11.1 activation gating (gain-of-function), and Q1068R accelerates inactivation kinetics (loss-of-function). AP simulations at 1 Hz or 2 Hz showed that R791W, R1005W, or Q1068R predicted no change, a 6% shortening, or a 3% prolongation in the APD90, respectively (Figure 5C). These simulations suggest that KCNH2 variants that disrupt Kv11.1 channel gating minimally impact ventricular AP duration and are not pro-arrhythmic.
Discussion
Rare amino-acid altering genetic variants in KCNH2-encoded Kv11.1 K+ channel have been associated with SIDS previously. In 2007, Arnestad and colleagues reported finding 5 rare Kv11.1 missense variants in 5 out of 201 (2.5%) Norwegian SIDS cases. However, only two SIDS cases (1%) hosted variants that were functionally abnormal. Here, we identified rare Kv11.1 missense variants in 2% of white and nearly 7% of black cases among nearly 300 SIDS cases. However, similar to Arnstead and colleagues, the majority of rare Kv11.1 variants identified in our cohort were functionally normal. In fact, only 1/204 (0.5%) white and 2/75 (2.6%) black SIDS cases hosted a Kv11.1 variant with non-WT gating properties, at least in the standard heterologous expression system.
Our study suggests that the functionally abnormal KCNH2 variants generate distinct molecular phenotypes compared to LQT2-causative KCNH2 mutations.13-22, 25, 44 The majority of dysfunctional LQT2-causative mutations localize to Kv11.1 α-subunit regions of predicted secondary structures (i.e. α-helices or β-subunits) in the N-terminus, transmembrane segments, or C-terminus, whereas most of the KCNH2 variants identified in the SIDS cases localize to N- and C-terminal regions not predicted to have secondary structures. About 90% of LQT2-causative mutations disrupt Kv11.1 channel trafficking. In contrast, all of the variants in this study generated Kv11.1 channels that traffic normally. Only 5% of the LQT2-causative mutations alter gating, but about 30% of the variants in this study alter gating. Lastly, only ~5% of functionally characterized LQTS-linked KCNH2 mutations, versus ~70% of the variants identified in SIDS cases, do not have an overt dysfunctional phenotype. One possible conclusion is that in contrast to being an LQT2-causative mutation that precipitated an infantile sudden cardiac arrest, the majority of the KCNH2 variants found among the SIDS victims are innocuous variants that are irrelevant to the infant’s demise.
In fact, two of the three dysfunctional KCNH2 variants that alter gating are each present with a heterozygote allele frequency of approximately 0.1% among black reference samples. This raises the intriguing possibility that the KCNH2 variants that alter Kv11.1 channel gating are relatively benign. This possibility is corroborated by EHR analysis of genotype positive adult patients with these variants. They do not have an LQTS/SQTS diagnosis, had normal median QTc values, and did not have a higher frequency of arrhythmias. Moreover, human ventricular AP simulations using the OVVR model also support this concept. Two of the three KCNH2 variants that altered gating properties predicted changes in the AP duration, but the absolute magnitude of these changes was small and not expected to be arrhythmic.
With the rapid development of inexpensive genotyping, the number of post-mortem genetic autopsies in SIDS cases and other cases of sudden unexpected death is likely going to increase. This in combination with a cardiac evaluation of family members might help to identify pathogenic variants as well as other individuals at risk for sudden death. However, effectively using genotype data alone to determine the cause of death or for diagnostic purposes is problematic.9 Functional testing of individual KCNH2 mutations using mammalian heterologous expression is reliable at identifying variants that associate with LQT2.13, 22 We suggest strategies that lower the current cost, energy, and time to functionally study novel KCNH2 variants will improve the clinical value of genetic testing for LQT2. However, in vitro studies should not be considered the final determinant of potential pathology.
There are several limitations to this study. These data were obtained in a widely utilized heterologous expression system that might not completely recapitulate in vivo conditions. For example, we did not examine the impact of the KCNE2 K+ channel subunit50, we measured at room temperature, and we did not test the influence of autonomic or neurohormonal factors. Therefore caution must be used in classifying these sequence variants as strictly benign.
Additional family specific factors, in combination with dysfunctional KCNH2 variants, could have contributed to the SIDS. We analyzed all 292 SIDS cases for variants in LQTS-related genes (KCNQ1, KCNH2, SCN5A, KCNE1, KCNE2, SCN1B, SCN2B, SCN3B, SCN4B, CAV3 and SNTA1). Case #5 hosted the rare G294V-Kv11.1 variant and the common polymorphism R1047L-Kv11.1. Case #6 hosted R791W-Kv11.1 and the common SCN5A polymorphism S1103Y-Nav1.5.51 It is possible that the presence of these additional variants contributed to the infants’ outcome. However, G294V- or R1047L-Kv11.1 does not impact Kv11.1 channel function25 and none of the other cases hosted additional non-synonymous variants in these genes.
This study suggests there is a minimal role for dysfunctional KCNH2 mutations in SIDS, but this assertion is contracted by the observation that there is a higher frequency of nonsynonymous KCNH2 variants in SIDS cohorts as compared to control subjects. We suspect that this discrepancy is likely due to the large sample size difference between the number of genotyped SIDS cases and control subjects.
In conclusion, all of the putative KCNH2 variants identified in the SIDS cohort trafficked similar to WT-Kv11.1 channels and, although some modified Kv11.1 channel gating, they do not predict a significant prolongation (or shortening) in computational simulations of the human ventricular AP. EHR of adults harboring the particular KCNH2 variants found among the deceased infants showed the patients have QTc values in the normal range. However, it should be noted 9 subjects have QTc values in the 460 range or higher, where a large proportion of canonical LQT patients would be. We conclude that deceased infants who possessed one of these rare KCNH2 missense variants did not likely die suddenly and unexpectedly from an LQT2-triggered fatal arrhythmia. Instead, they hosted an ostensibly benign, albeit rare, KCNH2 variant and their cause of death remains elusive.
Supplementary Material
Acknowledgements
We thank Dr. Wayne Giles (University of Calgary) for consultation on the computational modeling of and the human ventricular (AP).
Funding Sources
This work was supported by a grant from the Saving tiny Hearts Society (BPD); the American Heart Association Cardiovascular Genome Phenome Discovery Grant (BPD) [15CVGPSD27580000]; and the Competitive Catalyst Renewal Grant [17CCRG33700289] (BPD). This work was also supported by Eunice Kennedy Shriver National Institute of Child Health & Human Development of the National Institutes of Health [R01HD042569 to MJA]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. DJT and MJA are also supported by the Mayo Clinic Windland Smith Rice Comprehensive Sudden Cardiac Death Program.
Footnotes
Disclosures
Dr. Michael J. Ackerman (MJA) is a consultant for Audentes Therapeutics, Boston Scientific, Gilead Sciences, Invitae, MyoKardia, Medtronic, and St. Jude Medical. Mayo Clinic and MJA are involved in an equity/royalty relationship with AliveCor, Blue Ox Health Corporation, and StemoniX. However, none of these entities were involved in this study in any manner. Dr. Brian Delisle has a research contract with Human BioMedical Research Institute (HBMRI) but HBMRI did not contribute to this study.
Bibliography & References Cited
- 1.Schwartz PJ. The quest for the mechanisms of the sudden infant death syndrome: doubts and progress. Circulation. 1987;75:677–83. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz PJ, Stramba-Badiale M, Segantini A, Austoni P, Bosi G, Giorgetti R, Grancini F, Marni ED, Perticone F, Rosti D, Salice P. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med. 1998;338:1709–14. [DOI] [PubMed] [Google Scholar]
- 3.The sudden infant death syndrome: cardiac and respiratory mechanisms and interventions. Proceedings. May 24–27, 1987, Como, Italy. Ann N Y Acad Sci. 1988;533:1–474. [PubMed] [Google Scholar]
- 4.Schwartz PJ. Cardiac sympathetic innervation and the sudden infant death syndrome. A possible pathogenetic link. Am J Med. 1976;60:167–72. [DOI] [PubMed] [Google Scholar]
- 5.Maron BJ, Clark CE, Goldstein RE, Epstein SE. Potential role of QT interval prolongation in sudden infant death syndrome. Circulation. 1976;54:423–30. [DOI] [PubMed] [Google Scholar]
- 6.Davis AM, Glengarry J, Skinner JR. Sudden Infant Death. QT or Not QT? That Is No Longer the Question. 2016;9. [DOI] [PubMed] [Google Scholar]
- 7.Moss AJ. Long QT Syndrome. Jama. 2003;289:2041–4. [DOI] [PubMed] [Google Scholar]
- 8.Kapa S, Tester DJ, Salisbury BA, Harris-Kerr C, Pungliya MS, Alders M, Wilde AA, Ackerman MJ. Genetic testing for long-QT syndrome: distinguishing pathogenic mutations from benign variants. Circulation. 2009;120:1752–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ackerman JP, Bartos DC, Kapplinger JD, Tester DJ, Delisle BP, Ackerman MJ. The Promise, Peril of Precision Medicine: Phenotyping Still Matters Most. Mayo Clin Proc. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. [DOI] [PubMed] [Google Scholar]
- 11.Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. [DOI] [PubMed] [Google Scholar]
- 12.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–5. [DOI] [PubMed] [Google Scholar]
- 13.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–73. [DOI] [PubMed] [Google Scholar]
- 14.Sanguinetti MC, Curran ME, Spector PS, Keating MT. Spectrum of HERG K+-channel dysfunction in an inherited cardiac arrhythmia. Proc Natl Acad Sci U S A. 1996;93:2208–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. J Biol Chem. 1999;274:10113–8. [DOI] [PubMed] [Google Scholar]
- 16.Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. J Biol Chem. 1998;273:21061–6. [DOI] [PubMed] [Google Scholar]
- 17.Furutani M, Trudeau MC, Hagiwara N, Seki A, Gong Q, Zhou Z, Imamura S, Nagashima H, Kasanuki H, Takao A, Momma K, January CT, Robertson GA, Matsuoka R. Novel mechanism associated with an inherited cardiac arrhythmia: defective protein trafficking by the mutant HERG (G601S) potassium channel. Circulation. 1999;99:2290–4. [DOI] [PubMed] [Google Scholar]
- 18.Zhou Z, Gong Q, January CT. Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome. Pharmacological and temperature effects. J Biol Chem. 1999;274:31123–6. [DOI] [PubMed] [Google Scholar]
- 19.Nakajima T, Furukawa T, Tanaka T, Katayama Y, Nagai R, Nakamura Y, Hiraoka M. Novel mechanism of HERG current suppression in LQT2: shift in voltage dependence of HERG inactivation. Circ Res. 1998;83:415–22. [DOI] [PubMed] [Google Scholar]
- 20.Nakajima T, Furukawa T, Hirano Y, Tanaka T, Sakurada H, Takahashi T, Nagai R, Itoh T, Katayama Y, Nakamura Y, Hiraoka M. Voltage-shift of the current activation in HERG S4 mutation (R534C) in LQT2. Cardiovasc Res. 1999;44:283–93. [DOI] [PubMed] [Google Scholar]
- 21.McBride CM, Smith AM, Smith JL, Reloj AR, Velasco EJ, Powell J, Elayi CS, Bartos DC, Burgess DE, Delisle BP. Mechanistic Basis for Type 2 Long QT Syndrome Caused by KCNH2 Mutations that Disrupt Conserved Arginine Residues in the Voltage Sensor. J Membr Biol. 2013;246:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP, January CT. Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nature communications. 2014;5:5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Refsgaard L, Holst AG, Sadjadieh G, Haunso S, Nielsen JB, Olesen MS. High prevalence of genetic variants previously associated with LQT syndrome in new exome data. European journal of human genetics : EJHG. 2012;20:905–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arnestad M, Crotti L, Rognum TO, Insolia R, Pedrazzini M, Ferrandi C, Vege A, Wang DW, Rhodes TE, George AL Jr., Schwartz PJ. Prevalence of long-QT syndrome gene variants in sudden infant death syndrome. Circulation. 2007;115:361–7. [DOI] [PubMed] [Google Scholar]
- 25.Anson BD, Ackerman MJ, Tester DJ, Will ML, Delisle BP, Anderson CL, January CT. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels. Am J Physiol Heart Circ Physiol. 2004;286:H2434–41. [DOI] [PubMed] [Google Scholar]
- 26.Tester DJ, Ackerman MJ. Sudden infant death syndrome: How significant are the cardiac channelopathies? Cardiovasc Res. 2005;67:388–396. [DOI] [PubMed] [Google Scholar]
- 27.Tester DJ, Dura M, Carturan E, Reiken S, Wronska A, Marks AR, Ackerman MJ. A mechanism for sudden infant death syndrome (SIDS): stress-induced leak via ryanodine receptors. Heart Rhythm. 2007;4:733–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng J, Van Norstrand DW, Medeiros-Domingo A, Valdivia C, Tan BH, Ye B, Kroboth S, Vatta M, Tester DJ, January CT, Makielski JC, Ackerman MJ. Alpha1-syntrophin mutations identified in sudden infant death syndrome cause an increase in late cardiac sodium current. Circ Arrhythm Electrophysiol. 2009;2:667–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tester DJ, Tan BH, Medeiros-Domingo A, Song C, Makielski JC, Ackerman MJ. Loss-of-function mutations in the KCNJ8-encoded Kir6.1 K(ATP) channel and sudden infant death syndrome. Circulation Cardiovascular genetics. 2011;4:510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Norstrand DW, Asimaki A, Rubinos C, Dolmatova E, Srinivas M, Tester DJ, Saffitz JE, Duffy HS, Ackerman MJ. Connexin43 mutation causes heterogeneous gap junction loss and sudden infant death. Circulation. 2012;125:474–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, Ackerman MJ. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. 2007;116:2253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, Ackerman MJ. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4:161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan BH, Pundi KN, Van Norstrand DW, Valdivia CR, Tester DJ, Medeiros-Domingo A, Makielski JC, Ackerman MJ. Sudden infant death syndrome-associated mutations in the sodium channel beta subunits. Heart Rhythm. 2010;7:771–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic acids research. 2012;40:W452–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stead LF, Wood IC, Westhead DR. KvSNP: accurately predicting the effect of genetic variants in voltage-gated potassium channels. Bioinformatics. 2011;27:2181–6. [DOI] [PubMed] [Google Scholar]
- 36.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–81. [DOI] [PubMed] [Google Scholar]
- 38.Calabrese R, Capriotti E, Fariselli P, Martelli PL, Casadio R. Functional annotations improve the predictive score of human disease-related mutations in proteins. Human mutation. 2009;30:1237–44. [DOI] [PubMed] [Google Scholar]
- 39.Smith JL, McBride CM, Nataraj PS, Bartos DC, January CT, Delisle BP. Trafficking-deficient hERG K channels linked to long QT syndrome are regulated by a microtubule-dependent quality control compartment in the ER. Am J Physiol Cell Physiol. 2011;301:C75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McBride CM, Smith AM, Smith JL, Reloj AR, Velasco EJ, Powell J, Elayi CS, Bartos DC, Burgess DE, Delisle BP. Mechanistic basis for type 2 long QT syndrome caused by KCNH2 mutations that disrupt conserved arginine residues in the voltage sensor. The Journal of membrane biology. 2013;246:355–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, O'Dushlaine C, Van Hout CV, Staples J, Gonzaga-Jauregui C, Metpally R, Pendergrass SA, Giovanni MA, Kirchner HL, Balasubramanian S, Abul-Husn NS, Hartzel DN, Lavage DR, Kost KA, Packer JS, Lopez AE, Penn J, Mukherjee S, Gosalia N, Kanagaraj M, Li AH, Mitnaul LJ, Adams LJ, Person TN, Praveen K, Marcketta A, Lebo MS, Austin-Tse CA, Mason-Suares HM, Bruse S, Mellis S, Phillips R, Stahl N, Murphy A, Economides A, Skelding KA, Still CD, Elmore JR, Borecki IB, Yancopoulos GD, Davis FD, Faucett WA, Gottesman O, Ritchie MD, Shuldiner AR, Reid JG, Ledbetter DH, Baras A, Carey DJ. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354. [DOI] [PubMed] [Google Scholar]
- 42.Romero L, Trenor B, Yang PC, Saiz J, Clancy CE. In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome. J Mol Cell Cardiol. 2014;72:126–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O'Hara T, Virag L, Varro A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mannikko R, Overend G, Perrey C, Gavaghan CL, Valentin JP, Morten J, Armstrong M, Pollard CE. Pharmacological and electrophysiological characterization of nine, single nucleotide polymorphisms of the hERG-encoded potassium channel. British journal of pharmacology. 2010;159:102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA, January CT. Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J. 1998;74:230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walker VE, Wong MJ, Atanasiu R, Hantouche C, Young JC, Shrier A. Hsp40 chaperones promote degradation of the HERG potassium channel. J Biol Chem. 2010;285:3319–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–5. [DOI] [PubMed] [Google Scholar]
- 48.Berecki G, Zegers JG, Verkerk AO, Bhuiyan ZA, de Jonge B, Veldkamp MW, Wilders R, van Ginneken AC. HERG channel (dys)function revealed by dynamic action potential clamp technique. Biophys J. 2005;88:566–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Priori SG, Wilde AA, Horie M, Cho Y, Behr ER, Berul C, Blom N, Brugada J, Chiang CE, Huikuri H, Kannankeril P, Krahn A, Leenhardt A, Moss A, Schwartz PJ, Shimizu W, Tomaselli G, Tracy C. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10:1932–63. [DOI] [PubMed] [Google Scholar]
- 50.Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SA. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–87. [DOI] [PubMed] [Google Scholar]
- 51.Splawski I, Timothy KW, Tateyama M, Clancy CE, Malhotra A, Beggs AH, Cappuccio FP, Sagnella GA, Kass RS, Keating MT. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297:1333–6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.