

Abstract
Charcot–Marie‐Tooth type 2A (CMT2A) is an inherited sensorimotor neuropathy associated with mutations within the Mitofusin 2 (MFN2) gene. These mutations impair normal mitochondrial functioning via different mechanisms, disturbing the equilibrium between mitochondrial fusion and fission, of mitophagy and mitochondrial axonal transport. Although CMT2A disease causes a significant disability, no resolutive treatment for CMT2A patients to date. In this context, reliable experimental models are essential to precisely dissect the molecular mechanisms of disease and to devise effective therapeutic strategies. The most commonly used models are either in vitro or in vivo, and among the latter murine models are by far the most versatile and popular. Here, we critically revised the most relevant literature focused on the experimental models, providing an update on the mammalian models of CMT2A developed to date. We highlighted the different phenotypic, histopathological and molecular characteristics, and their use in translational studies for bringing potential therapies from the bench to the bedside. In addition, we discussed limitations of these models and perspectives for future improvement.
Keywords: animal model, Charcot–Marie‐Tooth type 2A, mitofusin 2, mouse models
1. INTRODUCTION
Charcot–Marie‐Tooth (CMT) type 2A (CMT2A; OMIM 609260) is an inherited sensorimotor axonal neuropathy and is the most frequent subtype of axonal CMT. 1 , 2 , 3 , 4 , 5 , 6 , 7 The disease is associated with mutations in the r Mitofusin 2 (MFN2) gene, which encodes for the protein Mitofusin 2 (MFN2). 1 MFN2 is a GTPase that is anchored to the outer mitochondrial membrane which is involved in the maintenance of the equilibrium between mitochondrial fusion and fission, as well as other mitochondrial processes. 3 CMT2A is clinically defined by marked neuropathy, primarily affecting motor function, and in certain instances, accompanied by substantial proprioception loss. This results in progressive muscle weakness and atrophy in the legs and arms, typically onset during childhood and leading to considerable disability. 6 , 7 , 8 , 9 More than 100 MFN2 mutations have been described in affected subjects, 10 , 11 , 12 , 13 Nevertheless, although CMT2A is generally associated with autosomal dominant or de novo dominant inheritance, few recessive and semidominant forms have been reported. 1 , 2 , 3 , 5 , 6 , 7 , 10 , 11 , 13 MFN2 mutations seem to induce the disease through a ‘dominant‐negative’ mechanism, where the expression of the wild‐type (WT) MFN2 allele is negatively controlled by the mutant protein. 6 , 14 , 15 , 16 , 17 To date, no clinical phenotype has been associated with certainty to MFN2 variants resulting in haploinsufficiency. The molecular processes underlying neuronal degeneration associated with MFN2 mutations are not entirely clear. Recent advancements have shed light on the various mechanisms underlying MFN2‐related axonal degeneration, which include disruptions in mitochondrial transport and positioning 8 , 18 , 19 as well as impairment in the communication between mitochondria and the endoplasmic reticulum. 20 , 21 Despite the efforts of researchers so far, no Food and Drug Administration (FDA) or European Medical Agency (EMA)‐approved curative or symptomatic treatments are currently available, and patients' management rely only on supportive care. The reasons for this lack of treatments, despite a known genetic basis, lie in several factors: firstly, the rarity of the disease, which makes it less appealing to both investors and the research community. In the second place, the absence of a clear gain‐of‐function or haploinsufficiency makes envisioning an effective curative strategy more complex. To precisely dissect and unravel the pathogenesis of CMT2A and to devise effective therapeutic strategies, reliable experimental models are essential. Here, we provide an overview and an update of the in vivo disease models with a focus on mouse models developed to date, 22 highlighting similarities and differences of their phenotypic and molecular characterization and discussing limitations and perspectives for future improvement (Table 1).
TABLE 1.
Comparison between available CMT2A mouse models.
| HB9 Mfn2*T105M 33 | Eno MFN2*R94Q 41 | Nestin‐cre MFN2T105M 46 | Thy1.2 MFN2R94Q 51 | Mfn2*K357T 64 | ENU‐induced MFN2*L643P 66 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | +/+ | +/− | +/+ MitoCharc2 |
+/− MitoCharc1 |
+/+ | +/− | Y‐linked | +/+ | +/− | +/+ |
| Transgene |
Promoter: Hb9 Mutation: T105M |
Promoter: Eno Mutation: R94Q |
+/+ Promoter: Rosa‐STOP/CAG‐CreERT2 Mutation: T105M |
+/− Promoter: Nestin‐Cre Mutation: T105M |
Promoter: Thy1.2 Mutation: R94Q |
Promoter: N/A Mutation: K357T |
Promoter: N/A Mutation: L643P |
|||
| Phenotype onset | Severe congenital | Mild congenital | Mild late | Mild late | 6 weeks post‐tamoxifen induction | Mild late | Early | Postnatally letal | Mild late | Early severe |
| Motor performance | Hindlimb muscles weakness | No alteration | Frequent fall off at Rotarod test | Frequent fall off at Rotarod test | N/A | No alteration | Frequent fall off at Rotarod and Grip tests | N/A | No alteration | Decreased latency to fall at Rotarod and Grip tests |
| Gait | Defect in dorsi‐flexion but no gaiting alteration | No alteration | Abnormal print length | No alteration | N/A | Abnormal print length | Progressive gaiting worsening | N/A | N/A | Decreased locomotor activity and rearing events |
| Axon | 40% fewer axons in motor roots | No alteration | 55% increase of <3,5 μm axons | 40% increase of <3,5 μm axons; Aδ fibres altered in the sciatic nerves | N/A | No alteration | Degeneration in tibialis muscle | N/A | No alteration | Reduced myelinated axons in the motor branch of the femoral nerve |
| Muscle fibre | Smaller anterior hindlimb muscles | No alteration | No alteration | No alteration | N/A | Smaller tibialis and soleus muscles | N/A | N/A | N/A | No alteration |
| Mitochondria | Highly aggregation and clusters | Highly aggregation and clusters | 28% increase in mitochondria in <3,5 μm axons | 34% increase in mitochondria in <3,5 μm axons | Reduced number in tibialis axons | No alteration | Clusters and morphology abnormalities. No mitophagy alteration | N/A | Clusters and morphology abnormalities in sciatic and optic nerves | Reduction in the average mitochondrial diameter |
| Sensoryphenotype | No alteration | No alteration | N/A | No alteration | N/A | No alteration | Precocious onset of severe sensorimotor symptoms | N/A | N/A | Impaired sensory responses |
2. EXPERIMENTAL MODELS OF CMT2A
Experimental models of CMT2A have played a key role in elucidating underlying disease mechanisms. In addition, reliable models are essential to screen novel potential therapeutic compounds and to assess their efficacy and safety. In vitro works were performed first in non‐human primary cell lines (e.g. mouse embryonic fibroblasts (MEFs) and mouse dorsal root ganglia (DRGs)) 3 or in neuronal cell lines in which MFN2 function was silenced through RNA interference or modulated by overexpression of mutated MFN2. 14 , 16 , 23 , 24 These knock‐down or mutant cells show significant impairment of mitochondrial axonal transport and heightened mitochondrial clustering. Subsequently, induced pluripotent stem cells (iPSCs) derived from CMT2A subjects have been used to generate motor or sensory neurons with the same genetic background as donor patients, 18 , 19 , 25 , 26 offering a more accurate representation of the disease compared to previous models based on non‐human cell types or less disease‐relevant cell types (like fibroblasts). Overall, these works highlighted the role of mutated MFN2 in abnormal mitochondrial shape, motility, clustering and transport along the axons.
Concerning in vivo models, transgenic models in Drosophila and Zebrafish have proven invaluable in elucidating the role of MFN2 in mitochondrial dynamics and its contribution to the gradual decline of motor function. 27 , 28 , 29 , 30 The Drosophila model described by El Fissi et al. 31 involved the incorporation of four CMT2A‐associated mutations (R364W, R94Q, T105M and L76P) into the marf gene, the Drosophila counterpart of the human MFN2 gene. While these mutated flies exhibited comparable phenotypic and pathological aspects s, a notable distinction emerged when examining mitochondrial morphology. Clusters of unfused mitochondria were observed in mutations located within the GTPase domain (R94Q and T105M). The diminished fusion rate likely resulted from the loss of GTPase activity, while mitochondrial tethering leading to aggregate formation might have been triggered by unique features acquired by MFN2 mutants. This suggests a loss of function due to a dominant negative effect rather than haploinsufficiency. This finding in vivo supported the dominant negative effect of MFN2 mutants previously observed in DRG cultures. 14 In contrast, R364W and L76P mutations induced opposing effects: the increased fusion rate resulted in large, round‐shaped mitochondria, suggesting that specific CMT2A mutations might have a dominant positive impact on MFN2 activity. In summary, this data demonstrated that both MFN2 loss and gain of function lead to disease traits in Drosophila models. 31
Two groups evaluated the effects of MFN2 dysfunction in Zebrafish models. Vettori and colleagues generated a loss‐of‐function model using a knock‐down approach with a morpholino oligonucleotides (mfn2‐MO) directed against the acceptor splice site of intron 2 of mfn2, which introduces a premature stop codon. 28 A control morpholino (nc‐MO) with no complementary RNA target was also concomitantly used. Knock‐down of mfn2 reduced the rate of survival of embryos, since 90% of nc‐MO‐injected embryos survived 24 h post‐fertilization, in contrast with only 60% of mfn2‐MO injected embryos. Mfn2 depletion also caused phenotypic alterations starting from 48 h post‐fertilization, which could be mild (curved tail, irregular somites, facial prognathism, underdeveloped eyes and brain ventricles edema) or severe (shortened body axis, little head with encephalic necrosis, ventral expansion of the yolk and trunk oedema) (mfn2‐MO vs. nc‐MO, p < 0.001). Mfn2 deficient larvae also displayed a significantly impaired escape response to physical touch (mfn2‐MO vs. nc‐MO, p < 0.001), indicative of motility alterations (mfn2‐MO vs. nc‐MO, p < 0.001). Immunofluorescence performed 48 h post‐fertilization revealed a significant difference in motor neuron (MN) organization between mfn2‐MO and nc‐MO treated embryos (p < 0.0001), together with a reduction of neuromuscular junctions (NMJs) was observed in mfn2‐MO‐treated embryos compared to nc‐MO controls.
Some years later, Chapman and colleagues published a study detailing a Zebrafish model carrying the nonsense mfn2 L285X mutation. 27 They observed an adult and slowly progressive phenotype, with mfn2L285X/L258X mutants displaying a significant progressive increase in mortality starting from day 175 (p < 0.0001) and a reduction of weight loss (p < 0.0001) and nose to tail length (p < 0.001) starting from day 60. Mutants also developed alterations in muscular function, that were tested by measuring critical swimming velocity in a manner similar to Rotarod test, through a swim tunnel apparatus which determines the maximum flow rate which fish could swim against for a sustained period. The results demonstrated a reduction of critical swimming velocity at 100 (mfn2WT/WT vs. mfn2L285X/L258X, 28 cm/s vs. 12.11 ± 7.55 cm/s (mean ± SD), p < 0.01) and 200 (28 cm/s vs. 4.55 ± 3.23 cm/s, p < 0.001) days of life. At the same days, assessment of NMJs revealed an alteration in homozygous mutants compared with controls. The authors also reported a selective reduction in mitochondrial retrograde transport along the axons in mfn2L285X/L258X neurons (p < 0.001).
Overall, these models provided the first evidence for a role of MFN2 in MN function and in mitochondrial axonal transport. Nevertheless, the complexity of mammalian physiology makes it difficult for them to faithfully replicate human disease.
3. MOUSE MODELS OF CMT2A
Mammalian models may offer a more pertinent approach to understanding human disease, although they come with physiological, genetic and technical constraints when attempting to model genetic neuropathy. 22 A meaningful disease model relies on factors such as the mouse strain and its genetic background, which should not interfere with the observed traits, as well as the choice of a proper promoter used for the transgene delivery. Additionally, the development and assessment of such models are time‐consuming, and the attainment of the anticipated phenotype is not guaranteed.
So far, different murine disease models have been generated for CMT2A. Since KO of mfn1 or mfn2 resulted in murine embryonal death, possibly due to a placental defect, 3 transgenic or mutagenized mice currently represent the most faithful in vivo disease models. A summary of available CMT2A mouse models is provided in Table 1.
3.1. HB9 mfn2T105M transgenic model
In 2008, Detmer and his research team pioneered the creation of the first transgenic mouse model for CMT2A. They engineered this model to express the mfn2T105M mutation, located within the GTPase G1 motif of MFN2, using the MN‐specific promoter HB9 which selectively target mfn2 expression in transgenic embryonic mice spinal cord. 32 HB9 is an MN‐specific promoter with proven activity in the developing and postnatal spinal cord, which holds two regulatory sequences in its distal region with a size of 313 and 125 base pairs that are sufficient for targeting gene expression exclusively to the spinal cord in transgenic embryonic mice. 33 MFN2T105M had been previously linked to early‐onset lower limb muscle atrophy, scoliosis and ataxia in unrelated CMT2A patient groups. 1 , 34 Intriguingly, MFN2T105M protein correctly localized to mitochondria but exhibited defects in mitochondrial fusion and prompted the aggregation of mitochondria when in excess. 5 , 16 Given these findings in cell cultures, it was reasonable to anticipate that introducing these mutated mfn2 alleles into mice's MNs could yield a reliable animal model replicating clinical motor impairments observed in CMT2A.
Heterozygous and homozygous Mfn2T105M transgenic mice displayed varying levels of transgene expression, with heterozygous animals manifesting a notably less severe phenotype. This suggested that the phenotype was primarily influenced by the extent of transgene expression. Concerning motor symptoms, this model effectively mirrored some of the features characteristics in CMT2A patients. Homozygous transgenic mice exhibited critically deformed and substantially shorter tails, along with evident gait abnormalities from birth. Most of these animals (86%, n = 85) could not perform ankle dorsiflexion and showed hindlimb dragging when walking, reflecting tibialis anterior weakness, a typical clinical sign of CMT2A subjects, resulting in gait problems related to foot drop. 35 Additionally, homozygous animals often presented clenched hind paws, seemingly unable to spread their toes. Nevertheless, homozygous mice managed to walk with short strides of their hindlimbs and performed adequately in rotarod and beam walking assessments. Differently from CMT2A patients, these hindlimb issues remained constant despite ageing.
Given that CMT2A patients typically exhibit distal muscle wasting in both the anterior and posterior compartments of their lower limbs, Detmer's group assessed this aspect in their model. Homozygous mice displayed a significant reduction in the muscle mass of the legs, particularly in the distal anterior regions, compared to wt and heterozygous mice; the muscle mass in the posterior hindlimb was similar among all genotypes. Microscopic examination of L4 and L5 motor roots revealed that homozygous transgenic animals had 40% fewer motor axons compared to wt mice, with more pronounced loss in the smaller‐calibre axons. Notably, in this model, the forelimbs remained unaffected, differently from CMT2A patients who often experience upper limb involvement in 50% of cases. 36 In terms of sensory symptoms, both homozygous and heterozygous Mfn2T105M transgenic mice did not display any sensory issues, even after 1 year of age.
Previously, ultrastructural examinations of the peripheral nerves of affected patients had disclosed anomalous mitochondrial clustering in the distal axon segments, 4 , 37 , 38 accompanied by disruptions in the mitochondrial network and organization associated with MFN2 mutations. 16 , 39 Clustered mitochondria and irregular distribution along the axons were also seen in homozygous animals. It is plausible that this improper mitochondrial recruitment could contribute to axonopathy.
In summary, these findings underscore the significance of this initial mammalian model for replicating several, though not all, clinical aspects of the disease. Nevertheless, certain limitations exist, for instance, the presence of a pathological phenotype only in a homozygous genotype (while human disease shows a dominant pattern of transmission) and the restricted expression of the mutated MFN2 exclusively in MNs whereas in affected subjects, it is expressed ubiquitously.
3.2. Eno MFN2 R94Q transgenic model
In 2010, Cartoni and his group introduced a novel CMT2A transgenic mouse model named MitoCharc1 (heterozygous) and MitoCharc2 (homozygous), which carried the R94Q mutation in the human MFN2 gene. 40 This genetic alteration was controlled by a neuron‐specific enolase (Eno) promoter, ensuring widespread expression throughout the central nervous system (CNS) and peripheral nervous system (PNS). 41 , 42 Neuron‐specific enolase is a dimeric isoform of the glycolytic enzyme enolase found mainly in neurons. The 1.8‐kb genomic region upstream of the Eno gene is widely used as a neuron‐specific promoter.
For comparative purposes, they also generated Eno‐MFN2 WT mice expressing the wt MFN2 (MFN2 WT ) under the same promoter. At 5 months of age, both mutant lines displayed statistically significant locomotor deficiencies and gait abnormalities compared to MFN2WT mice, as shown by the Rotarod test data (MitoCharc1 vs. MFN2WT, p < 0.001; and MitoCharc2 vs. MFN2WT, p = 0.001)t. Notably, the motor phenotype in MitoCharc1 animals appeared more consistent than that in MitoCharc2, possibly due to incomplete penetrance. MitoCharc2 mice exhibited even more pronounced phenotypic characteristics, including a lowered body posture, paw eversion and tail dragging, mirroring the walking difficulties and tendency to fall observed in CMT2A patients. 4 Lately, Bernard‐Marissal's group deepened the characterization of this mouse model, performing multiple behavioural tests in MitoCharc1 mice at early (6 months) and late (12 months) time points. 43 In addition to the confirmation of locomotor dysfunction via the Rotarod test, they found more comprehensive walking difficulties, with symptoms showing slight progression between 6 and 12 months. Specifically, MitoCharc1 mice exhibited alterations in paw pressure, contact surface, gait, posture and coordination as assessed by the CatWalk test (MitoCharc1 vs. MFN2WT, p < 0.05). Despite the locomotor impairments, no changes in muscle strength were found at either the 6‐ or 12‐month mark according to the grid test (6 months, WT: 2308 ± 96.2 and Mitocharc1: 2328 ± 144.8; 12 months, WT: 1886 ± 107.7 and Mitocharc1: 1852 ± 155.2). However, the model did demonstrate the loss of NMJs at 12 months of age, in absence of MN death. In terms of sensory function, this model did not exhibit significant impairments. 43
Regarding other notable human disease features, such as abnormalities in mitochondrial structure and distribution, 4 , 44 5‐month‐old MitoCharc1 mice showed an increase in number of mitochondria (roughly 30%), together with reduced mitochondrial diameters, in the distal portion of small calibre axons of the sciatic nerve. Bernard‐Marissal and his team also observed mitochondrial alterations in live imaging studies, 43 revealing a lower proportion of moving mitochondria in the sciatic nerve of 1‐month‐old MitoCharc1 mice compared to WT (32% retrograde and 68% anterograde moving mitochondria in WT, 55% retrograde and 44% anterograde moving mitochondria in Mitocharc1). These morphological and speed changes in mitochondria could be attributed to reduced contacts between mitochondria and the endoplasmic reticulum (ER), particularly at the mitochondria‐associated membranes (MAM), as also observed in MNs' soma in the lumbar spinal cord of 12‐month‐old MitoCharc1 mice. 43
Nerve conduction studies performed in MitoCharc1 mice showed alterations in the Aδ fibres component in the sciatic nerves, specifically an increase in the area/amplitude ratio of the compound action potential. This finding confirmed the observed axonal functional defects, which appeared to match the higher proportion of small to medium‐calibre axons, as the compound action potential area is influenced by the number and diameters of activated fibres.
In summary, Cartoni's mouse model seemed a significant advancement from the HB9 mfn2 T105M transgenic model, as it expands MFN2 transgene expression to the entire nervous system and replicates several characteristic disease symptoms, including locomotor impairment and axonopathy associated with changes in mitochondrial content and transport. However, a notable limitation of this model is the mild‐to‐late onset of pathological phenotypes, in contrast to the severe‐early clinical presentation typically seen in CMT2A patients with the R94Q MFN2 mutation.
3.3. Nestin‐cre MFN2T105M transgenic model
In 2016, Bannerman and his team introduced two additional mouse models for CMT2A, both hemizygous and carrying the human T105M missense mutation in the MFN2 gene. 45 In a departure from previous models, Bannerman employed a knock‐in approach (Rosa‐STOP‐MFN2T105M) to extend the expression of the mutated MFN2 transgene to all cell types. 46 , 47 Homozygous mice exhibited a broader clinical spectrum characterized by multiorgan dysfunction, severe motor function loss and the accumulation of abnormal mitochondria in Schwann cells within the sciatic nerve, which compromised their survival.
To address this issue, Bannerman and colleagues planned to restrict the expression of the MFN2 transgene to neural and certain non‐neural cells, such as myosatellite cells, using the nestin‐cre strategy. 48 , 49 Given that a hallmark clinical feature of CMT2A patients is length‐dependent motor neuropathy, assessing variations in the muscle‐nerve interactions induced by mutant MFN2 is of great interest for translational applications.
Tests of MN function revealed no long‐term differences between hemizygous nestin‐cre MFN2T105M mutant mice and control nestin‐cre mice, as evidenced by the normal resistance, equilibrium and coordination observed at the Rotarod test. However, the Noldus Catwalk system pointed out gait abnormalities, disclosing a statistically significant reduction in the print length of MFN2 mutant mice (control = 0.71 ± 0.072 cm, mutant = 0.58 ± 0.038 cm; p < 0.0003). These findings may be associated with one of the key CMT2A clinical signs, namely pes cavus.
These mice displayed a lower number of mitochondria in peripheral nerves, but they did not form clusters nor aggregates within MN soma, differently from findings reported in other models. 14 , 16 , 18 , 19 Additionally, the expression of the MFN2 transgene did not alter axon numbers, sizes or g‐ratios. Nevertheless, it did lead to significant changes in muscle fibre diameter, particularly in the soleus and tibialis anterior muscles, which are involved in plantar flexion and dorsiflexion actions, respectively. Reduced expression of actin in the sarcomere, augmented levels of satellite cells and anomalies of striatal mitochondrial organization were also observed.
In summary, the nestin‐cre MFN2T105M mouse model extends the expression of mutant MFN2 to all cell types, and presents critical phenotypic features of CMT2A patients, such as gait disturbances and reduction in the number of mitochondria in the tibialis muscle. On the contrary, certain findings appear to conflict with results from previous research works, and two limitations prevent it from being a suitable platform for drug testing, namely the late onset and low severity of symptoms and the absence of peripheral sensory impairment.
3.4. Thy1.2 MFN2R94Q transgenic model
In 2019, Zhou and collaborators developed a new model of CMT2A, (Tg(Thy1‐MFN2R94Q)44Balo/J), characterized by hemizygous expression of the MFN2‐R94Q transgene under the thymocyte differentiation antigen 1, allele 2 (Thy1.2) promoter integrated into the Y chromosome. 50 The murine Thy1.2 expression cassette derives from the mouse endogenous Thy1.2 gene. Thy1.2, together with Thy1.1, is one of the two alleles of the Thy1 membrane glycoprotein gene. The mThy1.2 expression cassette was engineered by eliminating all its exons, along with the second and third introns (which include the thymus‐specific enhancer). However, the first intron, housing the neural enhancer element, and the noncoding exons (Ia, Ib, II and IV) were preserved. The c.1070A > C mutation was then inserted within the MFN2WT coding sequence in the mThy1.2‐flag‐MFN2WT plasmid, in order to create the mThy1.2‐flag‐MFN2R94Q plasmid. The animals show symptoms and neuropathological characteristics with earlier onset and greater severity, thus making it a much more reliable model than the MitoCharc1 model, especially for studies evaluating the effectiveness of a therapeutic strategy. In particular, the Thy1.2‐MFN2R94Q transgenic model shows signs of muscle weakness from the second month of life which become progressively more pronounced in the subsequent period in association with a reduction in visual capacity. Affected mice are significantly smaller than healthy pups and mortality is observed around 150 days of life. Due to the severity of the phenotype, Thy1.2‐MFN2R94Q mice are not commercially available, while C57BL/6J‐Tg(Prnp‐MFN1)1Balo Tg(Thy1‐MFN2*R94Q)44Balo/ mice, which express in hemizygosis in addition to the Tg‐Th1MFN2‐R94Q also the Tg(Prnp‐MFN1) transgene under the control of the prion (Prnp) promoter in the CNS, are available for purchase. The overexpression of MFN1 through the Tg(Prnp‐MFN1) transgene in the nervous system improves the growth curve (at 5 months, Thy1.2‐MFN2R94Q vs. Thy1.2‐MFN2R94Q:MFN1, p < 0.0001; WT vs. Thy1.2‐MFN2R94Q:MFN1, ns) and increases survival (Thy1.2‐MFN2R94Q vs. Thy1.2‐MFN2R94Q:MFN1, p < 0.01; WT vs. Thy1.2‐MFN2R94Q:MFN1, ns) compared to mice hemizygous for the Thy1‐MFN2*R94Q transgene alone.
In the Cartoni mouse model, the Eno promoter ensures gene expression primarily in mature neurons, leading to the late appearance of neuromotor symptoms (after 5 months). 40 Conversely, the transgene under the Thy1.2 promoter is expressed very early, from the first days after birth, 51 , 52 , 53 and restricted to the CNS, targeting projection neurons, which are primarily affected in CMT2A patients. 54 This promoter has been previously utilized in studies related to sensory nerve function and synaptic plasticity in both the CNS and PNS. 55 , 56 , 57
Thy1.2‐MFN2R94Q transgenic mice exhibited a significant reduction in body weight starting from the second month of life and reduced survival compared to nontransgenic and Thy1.2‐MFN2WT mice. Behavioural assessments showed an impairment of neuromotor functions at the Rotarod test (at 6 months, Thy1.2‐MFN2R94Q vs. WT, p < 0.0001) and grip strength test compared to WT or nontransgenic mice (at 6 months, Thy1.2‐MFN2R94Q vs. WT, p < 0.0001; Thy1.2‐MFN2R94Q vs. nTg, p < 0.0001 for both tests). In addition, sensory functions were also assessed. Visual acuity was measured using the optokinetic response test and optic nerve analysis, which revealed severe vision loss associated with significant reduction of neurofilaments and axonal spheroids.
Since some CMT2A patients demonstrate spinal cord and brain white matter changes, 9 , 58 , 59 neuronal and axonal alterations in both the PNS and CNS were investigated in Thy1.2‐MFN2R94Q mice, revealing axonal damage and loss in absence of neuronal death in the pyramidal tracts at the age of 5 months. 2 , 60 , 61
On neuropathological analysis of nerve biopsies, aggregates of damaged mitochondria could be observed in the cytoplasm of neurons and along the axons of Thy1.2‐MFN2R94Q mice. Similar observations have been reported in CMT2A patient nerve biopsies 4 , 44 and iPSC‐ and primary cell based in vitro models 5 , 19 , 24 but have not been previously demonstrated in vivo.
Overall, this model accurately replicated crucial steps of CMT2A pathogenesis, including the precocious onset of severe sensorimotor symptoms, visual disturbances, diffuse axonal degenerations and mitochondrial abnormalities. It's worth noting that the disease phenotype was only limited to male mice due to the MFN2 transgene insertion into the Y chromosome, although no sex differences in phenotypes have been highlighted in clinical data from CMT2A patients thus far.
3.5. Mfn2K357T transgenic model
In 2021, a new model carrying the previously undescribed p.K357T mutation found in a 4‐year‐old CMT boy was developed. 62 The knock‐in mouse model was generated using the CRISPR/Cas9 genome editing and single‐stranded oligodeoxynucleotide (ssODN)‐mediated repair approach. Mfn2K357T/K357T mouse pups did not survive after birth, while Mfn2WT/K357T heterozygous mice showed significantly improved survival up to 10 months (Mfn2K357T/K357T vs. Mfn2WT/K357T, p < 0.0001; Mfn2K357T/K357T vs. Mfn2WT/WT, p < 0.0001), showing no symptoms and exhibiting normal histopathological features in their sciatic nerves. Voltage‐dependent anion channel 1 (VDAC1) is a protein located in the outer mitochondrial membrane. Immunofluorescence revealed VDAC1+ clusters in the spinal cords of Mfn2K357T/K357T mouse pups, and, to a lesser degree, of Mfn2WT/K357T, suggesting aberrant mitochondrial clustering (Mfn2K357T/K357T vs. Mfn2WT/K357T, p < 0.01; Mfn2K357T/K357T vs. Mfn2WT/WT, p < 0.01; Mfn2WT/K357T vs. Mfn2WT/WT, ns). Given the reduced survival of homozygous transgenic mice, subsequent analyses were performed only on hemizygous mice. VDAC+ clusters were also detected in the lumbar spinal cord white matter of 10‐month‐old Mfn2WT/K357T mice (Mfn2WT/K357T vs. Mfn2WT/WT, P < 0.05). Although histological and morphometric analyses revealed no differences between Mfn2WT/K357T and Mfn2WT/WT mice, ultrastructural analysis of sciatic and optic nerve axons of 8‐month‐old Mfn2WT/K357T mice showed an altered distribution of axonal mitochondria with aberrant clustering and abnormal morphology, with disorganized or absent cristae, detachment of mitochondrial membranes and increased mitochondrial diameter.
Given the known anti‐inflammatory properties of MFN2, 63 6‐month‐old mice were exposed to lipopolysaccharide (LPS) in an attempt to uncover MFN2‐related changes following a systemic inflammatory stimulus. Four hours after LPS injection, Mfn2WT/K357T mice exhibited a significant decline in the Rotarod test (Mfn2WT/K357T *p < 0.05, **p < 0.01), and in the hindlimb grip strength test, while 48 and 96 h later, performances in the two tests did not differ significantly between the two groups. In addition, the levels of interleukin‐6 (IL‐6), a pro‐inflammatory cytokine, increased significantly in the serum of Mfn2WT/K357T mice 4 h after LPS exposure (Mfn2WT/K357T vs. Mfn2WT/WT, p < 0.0001), decreasing in the subsequent hours. The researchers also assessed CNS microgliosis 96 h after LPS exposure, showing that, in LPS‐treated Mfn2WT/K357T mice, IBA1 intensity and area were significantly higher compared both to untreated hemizygous mice (Mfn2WT/K357T vs. Mfn2WT/WT, p < 0.0001), and to Mfn2+/+ LPS mice at the level of the optic nerves (IBA1 fluorescence intensity: LPS Mfn2WT/K357T vs. Mfn2WT/K357TT, p < 0.0001; LPS Mfn2WT/K357T vs. LPS Mfn2WT/WT, p < 0.01; IBA1 area: LPS Mfn2WT/K357T vs. Mfn2WT/K357TT, p < 0.01; LPS Mfn2WT/K357T vs. LPS Mfn2WT/WT, p < 0.05) and lumbar spinal cord (IBA1 fluorescence intensity: LPS Mfn2WT/K357T vs. Mfn2WT/K357TT, p < 0.0001; LPS Mfn2WT/K357T vs. LPS Mfn2WT/WT, p < 0.01; IBA1 area: LPS Mfn2WT/K357T vs. Mfn2WT/K357TT, p < 0.0001; LPS Mfn2WT/K357T vs. LPS Mfn2WT/WT, p < 0.001). Analysis of cell infiltrates within the spinal cord revealed a significantly higher number of CD45+ (a general marker of overall cell infiltration) and CD68+ (macrophages) cells in the spinal cord of LPS‐treated Mfn2WT/K357T mice compared to untreated hemizygous mice (cell number in whole lumbar spinal cord area, LPS Mfn2WT/K357T vs. Mfn2WT/K357TT, CD45+ p < 0.0001, CD68+ p < 0.01) and to LPS‐treated WT mice (cell number in whole lumbar spinal cord area, LPS Mfn2WT/K357T vs. LPS Mfn2WT/WT, CD45+ p < 0.001, CD68+ p < 0.05).
Overall, this model showed a late‐onset, subclinical phenotype with no overt disease signs. However, pathological analysis revealed aberrant mitochondrial clustering in their sciatic and optic nerves, altered mitochondrial morphology and localization and microglial activation in the CNS. Induction of stress with injection of LPS triggered a more severe peripheral inflammatory response and a significantly decreased motor performance 4 h after injection, as well as increased CNS infiltration and microglial activation after 96 h compared to treated WT mice.
3.6. ENU‐induced MFN2L643P transgenic model
In 2023, Hines and colleagues published their study related to a novel N‐Ethyl‐N‐nitrosourea (ENU)‐induced mutant mouse model displaying a severe neuromuscular phenotype. 64 ENU is a mutagen commonly used to generate mouse models of human diseases. 63 Analysing the genomes of these mice can unveil the genetic mechanisms behind the observed disease features. This approach, termed forward genetic screening, is valuable for discovering novel disease‐related genes and understanding the biological pathways involved. Researchers at the Genomics Institute of the Novartis Research Foundation (GNF) utilized ENU‐induced mutagenesis, employing high‐throughput phenotyping methods to assess behavioural, metabolic and immunological characteristics. This effort led to the generation and identification of a mouse line carrying a homozygous mutation in exon 18 of MFN2 (c.T1928C; p.L643P), within the highly conserved transmembrane domain. This mutation has not been described in patients so far, even though another missense mutation in the same transmembrane domain (p.R632W) was previously identified as potentially pathogenic. 65 Behavioural evaluations on MFN2L643P/L643P mice demonstrated progressive weight loss starting from 10 weeks of age and peaking at 16 weeks of age (MFN2L643P/L643P vs. MFN2WT/WT, p < 0.0001 both females and males). They also showed a fast neuromuscular decline starting at 8 weeks of age, with decreased performance at the rotarod test (MFN2L643P/L643P vs. MFN2WT/WT, p < 0.0001) and open field test (MFN2L643P/L643P vs. MFN2WT/WT, distance travelled p < 0.0001, number of rearing events p < 0.01). Electrophysiological testing revealed no differences in motor and sensory NCV and CMAP amplitude in the sciatic nerve between affected and WT mice, but a significant increase to react to a wire filament poke or an elevated temperature stimulus in the Von Frey test and hot plate assay for mutant mice (Von Frey test: MFN2L643P/L643P vs. MFN2WT/WT, p < 0.0001; hot plate assay: MFN2L643P/L643P vs. MFN2WT/WT, p < 0.001), suggesting decreased sensitivity to mechanical stimulus and heat. Histopathological analysis of whole brains and lumbar spinal cords of mutant and WT mice at 12 weeks of age showed no gross abnormalities in cortical, midbrain or brainstem structures and no differences in the number of motor neurons in the lumbar spinal cord. Histopathological analysis of motor and sensory branches of the femoral nerve revealed a reduction in the number of myelinated axons in the motor branch (MFN2L643P/L643P vs. MFN2WT/WT, p < 0.05). Histopathological analysis of gastrocnemius proved a slight, but nonsignificant increase in the number of partially innervated neuromuscular junctions and no signs of denervation, but ultrastructural assessment highlighted a significant reduction in the average mitochondrial diameter in mutant mice compared to WT (MFN2L643P/L643P vs. MFN2WT/WT, p < 0.0001). Since reduced neuromuscular loading was shown to impair bone thickness and density, the authors analysed the tibiae and femurs of mutant and WT mice with micro computed tomography (CT), finding thinner bone cortex (MFN2L643P/L643P vs. MFN2WT/WT, p < 0.05) and a reduced bone area fraction (MFN2L643P/L643P vs. MFN2WT/WT, p < 0.05) in affected mice. These findings suggest potential defects in bone remodelling, in line with recent studies that demonstrated a role of Mfn2 in bone development. 66 , 67
Overall, the ENU‐induced MFN2L643P transgenic model presented a recessive mild neuromuscular phenotype in mice, coupled with mild skeletal and mitochondrial abnormalities.
4. LIMITATIONS OF MURINE MODELS
The primary technical limitations of existing mouse models revolve around the level and pattern of expression of the MFN2 gene, the causative factor in the disease. These models are transgenic, often involving the overexpression of mutant MFN2, typically through a human transgene. Some models restrict this expression to the CNS using promoters, while others have ubiquitous transgene expression. The critical question is whether a physiological level of expression of the mutant MFN2 is needed in order to accurately represent the pathogenic mechanism, or overexpression might be able to faithfully replicate it. This complex interplay between genetic expression, regulatory mechanisms, and the effect of a given mutation necessitates a nuanced approach when interpreting the results from animal models, highlighting the need for more refined and sophisticated methodologies to truly grasp the underlying mechanisms of neurological diseases.
In addition, it is worth pointing out that, among models reproducing autosomal dominant CMT2A, motor deficits were only observed in homozygous mice. Their heterozygous counterparts, with genetics similar to human patients, failed to develop a disease phenotype. This might be ascribed to differences between mice and humans in life span, disease duration and environmental exposure. It is also surprising that heterozygous mice displayed no disease progression or increasing severity during ageing. Such an observation raises several questions: is murine life expectancy too short to develop late‐onset neuropathy? Or is it possible that known pathogenic mutations act synergistically with other unknown genetic and epigenetic modifiers to accelerate or delay symptom onset? The genetic conundrum of CMT2A is exemplified by the wide phenotypic spectrum of human disease and by the impossibility to draw precise genotype–phenotype correlations. Indeed, the same mutations often result in different onset ages and disease severity. One open question is whether it is possible to develop an in vivo model that would collectively reflect all the features described in patients with various mutations. These findings notwithstanding, a unique mouse model overexpressing a single MFN2 mutation and with a severe, early‐onset phenotype might be enough to model the most severe hallmarks and as a platform for therapeutic assessment.
In the authors' experience, a more severe phenotype allows for easier testing of therapeutic compounds and stronger evidence of efficacy. In this perspective, the model Thy1.2‐MFN2R94Q by Zhou and colleagues 50 offers an early and severe sensorimotor phenotype and is commercially available, making it a potential choice for translational studies.
5. THERAPEUTIC IMPLICATIONS
No effective therapy has reached the clinic so far, although some strategies have obtained promising results in animal models.
Rocha and colleagues investigated mitofusin agonists, small molecules mimicking the MFN2 peptide–peptide interface, activating the protein allosterically and promoting mitochondrial fusion. Their study successfully reversed static mitochondrial clusters in cultured primary murine neurons carrying R94Q or T105M mutations and restored mitochondrial axonal transport in peripheral nerves of mice with the T105M mutation. 68
Promising findings also emerged from studies involving histone deacetylase (HDAC) inhibitors, enzymes that deacetylate conserved serine residues in histones and nonhistone proteins. HDAC6, a cytoplasmic member, deacetylates alpha‐tubulin, a component of the cytoskeletal network. HDAC6 inhibitors demonstrated efficacy in HSPB1‐associated CMT2F transgenic mouse models, correcting axonal transport defects and reversing the CMT phenotype caused by HSPB1 mutations. 69 , 70 , 71 , 72 Following these results, HDAC6 inhibitors were tested in CMT2A models. A recent study explored the impact of pharmacological HDAC6 inhibition using the compound SW‐100 on MitoCharc1 mice. This treatment prevented α‐tubulin hypoacetylation and improved motor performance both at presymptomatic and postsymptomatic stages. 73 The same research group also investigated new compounds derived from SW‐100, particularly SW‐101, that showed a significantly enhanced drug exposure in plasma and the brain in comparison with SW‐100. Administration of SW‐101 once daily to Mitocharc1 mice increased acetylated α‐tubulin levels in the distal sciatic nerve, counteracted progressive motor dysfunction, and ameliorated motor and sensory symptoms. 74
Given that CMT2A primarily appears to be caused by a dominant negative effect rather than a toxic gain of function, 2 , 6 , 10 , 13 exploring the effectiveness of MFN2 overexpression alone is relevant because dominant‐negative activities can be overcome by the overexpression of wild‐type MFN2. Indeed, the overexpression of MFN2 also showed promising results in murine models. Increased expression of MFN2WT was indeed able to decrease the amount of mitochondrial aggregates induced by mutant MFN2 in a genetically modified primary neuronal cell line, and to rescue phenotypical and ultrastructural alterations in the Thy1.2‐MFN2R94Q mouse model. 50
Recently, our group tested a new approach based on the combination of gene replacement and gene silencing via RNA interference (RNAi). 75 In this study, both the mutant and wild‐type MFN2 mRNA are inhibited by a short hairpin RNA (shRNA), while the wild‐type protein is restored by overexpressing a cDNA encoding functional MFN2 modified to be resistant to RNAi. We tested this approach in an in vitro IPSC‐derived motor neuronal CMT2A model 19 and in the Mitocharc1 mouse model. 40 In vitro, we observed a rescue of the cellular and molecular alterations in CMT2A MNs, more specifically we observed a reversal of altered mitochondrial distribution along the axons and a reduction of increased levels of mitophagy. We then delivered an AAV9 encoding the combination of shRNA/MFN2 cDNA (AAV9‐KD‐rMFN2) via intracerebroventricular injection in newborn Mitocharc1 mice, and observed a correct MFN2 molecular correction, with silencing of endogenous MFN2 and overexpression of exogenous MFN2 in their CNS. Further studies are needed to validate the therapeutic benefit of this strategy in animal models; however, this work provides a proof‐of‐principle of the feasibility of MFN2 silencing and replacement via AAV9 delivery in CMT2A mice.
On the contrary, also the overexpression of MFN1 would therapeutically benefit CMT2A models. Indeed, in vitro studies have demonstrated that MFN1 has the capacity to compensate for the loss of MFN2, safeguarding against mitochondrial fusion and transport defects induced by mutations in MFN2. 5 , 76 These data have been also confirmed in the Thy1.2‐MFN2R94Q transgenic mice expressing the point mutant R94Q on MFN2 specifically in neurons under the Thy1.2 promoter. 50 , 77 These mice recapitulate a range of neurological features observed in CMT2A. Notably, increased expression of MFN1 as a transgene (PrP‐MFN1) in the nervous system is well‐tolerated and results in nearly complete alleviation of phenotypic impairments in Thy1.2‐MFN2R94Q mice. 50 Indeed, while mutant MFN2R94Q seems to initiate the tethering process with MFN1 or MFN2 on neighbouring mitochondria, it fails to execute full fusion, contributing to mitochondrial aggregation and to axonal degeneration. Furthermore, low levels of endogenous MFN1 in neurons promote a conformation of MFN2R94Q that allows MFN1‐MFN2 heterodimer tethering, enhancing the frequency of failed fusion events and mitochondrial clustering.
Overexpression of MFN1 in the Thy1.2‐MFN2R94Q mouse model resulted in the normalization of the phenotype and of the transcriptomic signature and in the rescue of mitochondrial aggregation and axon degeneration. In a subsequent work using the same transgenic animals, Shahin and colleagues observed that increasing MFN1 levels successfully restored vision and retinal structure to normal levels by balancing the MFN1/MFN2 ratio and promoting PINK1‐dependent mitophagy. 78 Thus, increasing MFN1 levels should restore fusion, improving the pathological phenotype.
Overall, these studies highlight the importance of animal models in the development of new therapeutic strategies.
6. CONCLUSIONS
As mentioned above, CMT2A is a hereditary disease which causes a significant disability. Presently, there is no effective treatment which is able to revert or halt the progression of the underlying pathogenic mechanisms. Developing reliable animal models that accurately mimic genetic features and progress towards progressive, length‐dependent axonal neuropathy remains a significant challenge in understanding the disease and developing treatments. The existing mouse models mostly overexpress mutant MFN2 through a human transgene, some restricted to the nervous system via specific promoters while others exhibit ubiquitous expression (Figure 1). Questions persist about whether mimicking physiological mutant MFN2 levels is essential for understanding the disease mechanism and if humanized mice mirror patient regulatory mechanisms. The complexities of CMT2A, evidenced by diverse human clinical presentations and limited phenotype–genotype correlations, pose challenges in modelling mutations effectively. Integrating diverse model data—mice, humans, in vivo, in vitro—remains pivotal in constructing a reliable disease model. Despite the complexity, available models have provided vital insights into CMT2A, raising hope for the discovery and testing of potential therapeutic strategies.
FIGURE 1.
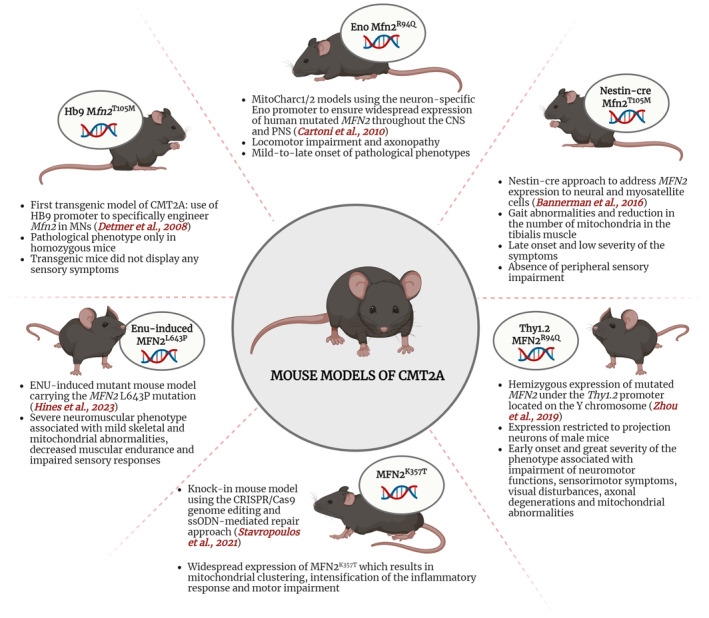
Schematic description of the characteristics of available CMT2A mouse models.
AUTHOR CONTRIBUTIONS
Elena Abati: Conceptualization (lead); investigation (lead); methodology (equal); project administration (equal); supervision (lead); writing – original draft (lead); writing – review and editing (equal). Mafalda Rizzuti: Conceptualization (equal); visualization (equal); writing – review and editing (equal). Alessia Anastasia: Data curation (equal); investigation (equal); writing – review and editing (equal). Giacomo Pietro Comi: Funding acquisition (equal); project administration (equal); resources (equal); supervision (equal); writing – review and editing (supporting). Stefania Corti: Funding acquisition (lead); investigation (equal); project administration (equal); resources (lead); supervision (equal); writing – review and editing (equal). Federica Rizzo: Conceptualization (equal); data curation (equal); formal analysis (equal); funding acquisition (lead); supervision (equal); writing – review and editing (equal).
FUNDING INFORMATION
This study was funded by the Italian Ministry of Health grant GR‐2018‐12365358 to FR (2018–2021).
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We thank Associazione Progetto Mitofusina 2 ONLUS and Associazione Amici del Centro Dino Ferrari for their support. Figures were created with BioRender.com.
Abati E, Rizzuti M, Anastasia A, Comi GP, Corti S, Rizzo F. Charcot–Marie‐Tooth type 2A in vivo models: Current updates. J Cell Mol Med. 2024;28:e18293. doi: 10.1111/jcmm.18293
Stefania Corti and Federica Rizzo equally contributed to the work.
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analysed in this study.
REFERENCES
- 1. Züchner S, Mersiyanova IV, Muglia M, et al. Mutations in the mitochondrial GTPase mitofusin 2 cause Charcot‐Marie‐Tooth neuropathy type 2A. Nat Genet. 2004;36:449‐451. doi: 10.1038/ng1341 [DOI] [PubMed] [Google Scholar]
- 2. Cartoni R, Martinou J‐C. Role of mitofusin 2 mutations in the physiopathology of Charcot‐Marie‐Tooth disease type 2A. Exp Neurol. 2009;218:268‐273. doi: 10.1016/j.expneurol.2009.05.003 [DOI] [PubMed] [Google Scholar]
- 3. Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189‐200. doi: 10.1083/jcb.200211046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Verhoeven K, Claeys KG, Züchner S, et al. MFN2 mutation distribution and genotype/phenotype correlation in Charcot‐Marie‐Tooth type 2. Brain. 2006;129:2093‐2102. doi: 10.1093/brain/awl126 [DOI] [PubMed] [Google Scholar]
- 5. Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176:405‐414. doi: 10.1083/jcb.200611080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feely SME, Laura M, Siskind CE, et al. MFN2 mutations cause severe phenotypes in most patients with CMT2A. Neurology. 2011;76:1690‐1696. doi: 10.1212/WNL.0b013e31821a441e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pipis M, Feely SME, Polke JM, et al. Natural history of Charcot‐Marie‐Tooth disease type 2A: a large international multicentre study. Brain. 2021;143:3589‐3602. doi: 10.1093/brain/awaa323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stuppia G, Rizzo F, Riboldi G, et al. MFN2‐related neuropathies: clinical features, molecular pathogenesis and therapeutic perspectives. J Neurol Sci. 2015;356:7‐18. doi: 10.1016/j.jns.2015.05.033 [DOI] [PubMed] [Google Scholar]
- 9. Abati E, Manini A, Velardo D, et al. Clinical and genetic features of a cohort of patients with MFN2‐related neuropathy. Sci Rep. 2022;12:6181. doi: 10.1038/S41598-022-10220-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tomaselli PJ, Rossor AM, Polke JM, Poh R, Blake J, Reilly MM. Semi‐dominant mutations in MFN2‐related neuropathy and implications for genetic counselling. J Peripher Nerv Syst. 2016;21:52‐54. doi: 10.1111/jns.12155 [DOI] [PubMed] [Google Scholar]
- 11. Piscosquito G, Saveri P, Magri S, et al. Mutational mechanisms in MFN2‐related neuropathy: compound heterozygosity for recessive and semidominant mutations. J Peripher Nerv Syst. 2015;20:380‐386. doi: 10.1111/jns.12145 [DOI] [PubMed] [Google Scholar]
- 12. Polke JM, Laurá M, Pareyson D, et al. Recessive axonal Charcot‐Marie‐Tooth disease due to compound heterozygous mitofusin 2 mutations. Neurology. 2011;77:168‐173. doi: 10.1212/WNL.0b013e3182242d4d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicholson GA, Magdelaine C, Zhu D, et al. Severe early‐onset axonal neuropathy with homozygous and compound heterozygous MFN2 mutations. Neurology. 2008;70:1678‐1681. doi: 10.1212/01.wnl.0000311275.89032.22 [DOI] [PubMed] [Google Scholar]
- 14. Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30:4232‐4240. doi: 10.1523/JNEUROSCI.6248-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amiott EA, Lott P, Soto J, et al. Mitochondrial fusion and function in Charcot‐Marie‐Tooth type 2A patient fibroblasts with mitofusin 2 mutations. Exp Neurol. 2008;211:115‐127. doi: 10.1016/j.expneurol.2008.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot‐Marie‐Tooth disease from Mitofusin 2 mutations. J Neurosci. 2007;27:422‐430. doi: 10.1523/JNEUROSCI.4798-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Loiseau D, Chevrollier A, Verny C, et al. Mitochondrial coupling defect in Charcot‐Marie‐Tooth type 2A disease. Ann Neurol. 2007;61:315‐323. doi: 10.1002/ana.21086 [DOI] [PubMed] [Google Scholar]
- 18. Saporta MA, Dang V, Volfson D, et al. Axonal Charcot‐Marie‐Tooth disease patient‐derived motor neurons demonstrate disease‐specific phenotypes including abnormal electrophysiological properties. Exp Neurol. 2015;263:190‐199. doi: 10.1016/J.EXPNEUROL.2014.10.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rizzo F, Ronchi D, Salani S, et al. Selective mitochondrial depletion, apoptosis resistance, and increased mitophagy in human Charcot‐Marie‐Tooth 2A motor neurons. Hum Mol Genet. 2016;25:4266‐4281. doi: 10.1093/hmg/ddw258 [DOI] [PubMed] [Google Scholar]
- 20. Filadi R, Greotti E, Turacchio G, Luini A, Pozzan T, Pizzo P. Mitofusin 2 ablation increases endoplasmic reticulum–mitochondria coupling. Proc Natl Acad Sci. 2015;112:E2174‐E2181. doi: 10.1073/pnas.1504880112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cosson P, Marchetti A, Ravazzola M, Orci L. Mitofusin‐2 independent juxtaposition of endoplasmic reticulum and mitochondria: an ultrastructural study. PLoS One. 2012;7:e46293. doi: 10.1371/journal.pone.0046293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. De Gioia R, Citterio G, Abati E, et al. Animal models of CMT2A: state‐of‐art and therapeutic implications. Mol Neurobiol. 2020;57:5121‐5129. doi: 10.1007/S12035-020-02081-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pich S, Bach D, Briones P, et al. The Charcot‐Marie‐Tooth type 2A gene product, Mfn2, up‐regulates fuel oxidation through expression of OXPHOS system. Hum Mol Genet. 2005;14:1405‐1415. doi: 10.1093/HMG/DDI149 [DOI] [PubMed] [Google Scholar]
- 24. Misko AL, Sasaki Y, Tuck E, Milbrandt J, Baloh RH. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J Neurosci. 2012;32:4145‐4155. doi: 10.1523/JNEUROSCI.6338-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Lent J, Verstraelen P, Asselbergh B, et al. Induced pluripotent stem cell‐derived motor neurons of CMT type 2 patients reveal progressive mitochondrial dysfunction. Brain. 2021;144:2471‐2485. doi: 10.1093/BRAIN/AWAB226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ohara R, Imamura K, Morii F, et al. Modeling drug‐induced neuropathy using human iPSCs for predictive toxicology. Clin Pharmacol Ther. 2017;101:754‐762. doi: 10.1002/CPT.562 [DOI] [PubMed] [Google Scholar]
- 27. Chapman AL, Bennett EJ, Ramesh TM, de Vos KJ, Grierson AJ. Axonal transport defects in a Mitofusin 2 loss of function model of Charcot‐Marie‐Tooth disease in zebrafish. PLoS One. 2013;8:e67276. doi: 10.1371/JOURNAL.PONE.0067276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vettori A, Bergamin G, Moro E, et al. Developmental defects and neuromuscular alterations due to mitofusin 2 gene (MFN2) silencing in zebrafish: a new model for Charcot‐Marie‐Tooth type 2A neuropathy. Neuromuscul Disord. 2011;21:58‐67. doi: 10.1016/j.nmd.2010.09.002 [DOI] [PubMed] [Google Scholar]
- 29. Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW II. Mitochondrial contagion induced by parkin deficiency in drosophila hearts and its containment by suppressing mitofusin. Circ Res. 2014;114:257‐265. doi: 10.1161/CIRCRESAHA.114.302734/-/DC1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. El FN, Rojo M, Aouane A, et al. Mitofusin gain and loss of function drive pathogenesis in drosophila models of CMT2A neuropathy. EMBO Rep. 2018;19:e45241. doi: 10.15252/EMBR.201745241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. El Fissi N, Rojo M, Aouane A, et al. Mitofusin gain and loss of function drive pathogenesis in drosophila models of CMT2A neuropathy. EMBO Rep. 2020;21:e50703. doi: 10.15252/embr.202050703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Detmer SA, Vande VC, Cleveland DW, Chan DC. Hindlimb gait defects due to motor axon loss and reduced distal muscles in a transgenic mouse model of Charcot–Marie–Tooth type 2A. Hum Mol Genet. 2008;17:367‐375. doi: 10.1093/HMG/DDM314 [DOI] [PubMed] [Google Scholar]
- 33. Nakano T, Windrem M, Zappavigna V, Goldman SA. Identification of a conserved 125 base‐pair Hb9 enhancer that specifies gene expression to spinal motor neurons. Dev Biol. 2005;283:474‐485. doi: 10.1016/J.YDBIO.2005.04.017 [DOI] [PubMed] [Google Scholar]
- 34. Lawson VH, Graham BV, Flanigan KM. Clinical and electrophysiologic features of CMT2A with mutations in the mitofusin 2 gene. Neurology. 2005;65:197‐204. doi: 10.1212/01.WNL.0000168898.76071.70 [DOI] [PubMed] [Google Scholar]
- 35. Neves EL d A, Kok F. Clinical and neurophysiological investigation of a large family with dominant Charcot‐Marie‐Tooth type 2 disease with pyramidal signs. Arq Neuropsiquiatr. 2011;69:424‐430. doi: 10.1590/S0004-282X2011000400003 [DOI] [PubMed] [Google Scholar]
- 36. Gemignani F, Marbini A, Teunissen LL, et al. Disease course of Charcot‐Marie‐Tooth disease type 2 and comorbidity. Arch Neurol. 2004;61:1470. doi: 10.1001/ARCHNEUR.61.9.1470-A [DOI] [PubMed] [Google Scholar]
- 37. Barbullushi K, Abati E, Rizzo F, Bresolin N, Comi GP, Corti S. Disease modeling and therapeutic strategies in CMT2A: state of the art. Mol Neurobiol. 2019;56:6460‐6471. doi: 10.1007/s12035-019-1533-2 [DOI] [PubMed] [Google Scholar]
- 38. Calvo J, Funalot B, Ouvrier RA, et al. Genotype‐phenotype correlations in Charcot‐Marie‐Tooth disease type 2 caused by Mitofusin 2 mutations. Arch Neurol. 2009;66:1511‐1516. doi: 10.1001/ARCHNEUROL.2009.284 [DOI] [PubMed] [Google Scholar]
- 39. Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858‐862. doi: 10.1126/SCIENCE.1099793 [DOI] [PubMed] [Google Scholar]
- 40. Cartoni R, Arnaud E, Médard J‐J, et al. Expression of mitofusin 2(R94Q) in a transgenic mouse leads to Charcot‐Marie‐Tooth neuropathy type 2A. Brain. 2010;133:1460‐1469. doi: 10.1093/brain/awq082 [DOI] [PubMed] [Google Scholar]
- 41. Levy YS, Merims D, Panet H, Barhum Y, Melamed E, Offen D. Induction of neuron‐specific enolase promoter and neuronal markers in differentiated mouse bone marrow stromal cells. J Mol Neurosci. 2003;21:121‐132. doi: 10.1385/JMN:21:2:121/METRICS [DOI] [PubMed] [Google Scholar]
- 42. Forss‐Petter S, Danielson PE, Catsicas S, et al. Transgenic mice expressing β‐galactosidase in mature neurons under neuron‐specific enolase promoter control. Neuron. 1990;5:187‐197. doi: 10.1016/0896-6273(90)90308-3 [DOI] [PubMed] [Google Scholar]
- 43. Bernard‐Marissal N, Van Hameren G, Juneja M, et al. Altered interplay between endoplasmic reticulum and mitochondria in Charcot‐Marie‐Tooth type 2A neuropathy. Proc Natl Acad Sci USA. 2019;116:2328‐2337. doi: 10.1073/PNAS.1810932116/SUPPL_FILE/PNAS.1810932116.SAPP.PDF [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vallat JM, Ouvrier RA, Pollard JD, et al. Histopathological findings in hereditary motor and sensory neuropathy of axonal type with onset in early childhood associated with Mitofusin 2 mutations. J Neuropathol Exp Neurol. 2008;67:1097‐1102. doi: 10.1097/NEN.0B013E31818B6CBC [DOI] [PubMed] [Google Scholar]
- 45. Bannerman P, Burns T, Xu J, Miers L, Pleasure D. Mice Hemizygous for a pathogenic Mitofusin‐2 allele exhibit hind limb/foot gait deficits and phenotypic perturbations in nerve and muscle. PLoS One. 2016;11:e0167573. doi: 10.1371/JOURNAL.PONE.0167573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70‐71. doi: 10.1038/5007 [DOI] [PubMed] [Google Scholar]
- 47. Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen‐inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol. 2002;244:305‐318. doi: 10.1006/DBIO.2002.0597 [DOI] [PubMed] [Google Scholar]
- 48. Jiang Y, Tong D, Hofacer RD, Loepke AW, Lian Q, Danzer SC. Long‐term fate mapping to assess the impact of postnatal isoflurane exposure on hippocampal progenitor cell productivity. Anesthesiology. 2016;125:1159‐1170. doi: 10.1097/ALN.0000000000001358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Michalczyk K, Ziman M. Nestin structure and predicted function in cellular cytoskeletal organisation. Histol Histopathol. 2005;20:665‐671. doi: 10.14670/HH-20.665 [DOI] [PubMed] [Google Scholar]
- 50. Zhou Y, Carmona S, Muhammad AKMG, et al. Restoring mitofusin balance prevents axonal degeneration in a Charcot‐Marie‐Tooth type 2A model. J Clin Invest. 2021;131:e147307. doi: 10.1172/JCI147307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xue G‐P, Morris RJ. Evidence for cell‐type differences in the regulation of neuronal expression of Thy‐1. Biochem Soc Trans. 1990;18:441‐442. doi: 10.1042/bst0180441 [DOI] [PubMed] [Google Scholar]
- 52. Evans GA, Chen S. Expression of the thy‐1 gene and thy‐1‐derived fusion genes in transgenic mice. Mol Biol Med. 1989;6:549‐556. [PubMed] [Google Scholar]
- 53. Marinković P, Godinho L, Misgeld T. Generation and screening of transgenic mice with neuronal labeling controlled by Thy1 regulatory elements. Cold Spring Harb Protoc. 2015;2015:875‐882. doi: 10.1101/PDB.TOP087668 [DOI] [PubMed] [Google Scholar]
- 54. Klein CJ, Kimmel GW, Pittock SJ, et al. Large kindred evaluation of Mitofusin 2 novel mutation, extremes of neurologic presentations, and preserved nerve mitochondria. Arch Neurol. 2011;68:1295‐1302. doi: 10.1001/ARCHNEUROL.2011.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Taylor‐Clark TE, Wu KY, Thompson JA, Yang K, Bahia PK, Ajmo JM. Thy1.2 YFP‐16 transgenic mouse labels a subset of large‐diameter sensory neurons that lack TRPV1 expression. PLoS One. 2015;10:e0119538. doi: 10.1371/JOURNAL.PONE.0119538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Belle MD, Pattison EF, Cheunsuang O, et al. Characterization of a thy1.2 GFP transgenic mouse reveals a tissue‐specific organization of the spinal dorsal horn. Genesis. 2007;45:679‐688. doi: 10.1002/DVG.20331 [DOI] [PubMed] [Google Scholar]
- 57. Yan Y, Sun HH, Mackinnon SE, Johnson PJ. Evaluation of peripheral nerve regeneration via in vivo serial transcutaneous imaging using transgenic Thy1‐YFP mice. Exp Neurol. 2011;232:7‐14. doi: 10.1016/J.EXPNEUROL.2011.06.013 [DOI] [PubMed] [Google Scholar]
- 58. Chung KW, Suh BC, Cho SY, et al. Early‐onset Charcot‐Marie‐Tooth patients with mitofusin 2 mutations and brain involvement. J Neurol Neurosurg Psychiatry. 2010;81:1203‐1206. doi: 10.1136/jnnp.2009.181669 [DOI] [PubMed] [Google Scholar]
- 59. Del Bo R, Moggio M, Rango M, et al. Mutated mitofusin 2 presents with intrafamilial variability and brain mitochondrial dysfunction. Neurology. 2008;71:1959‐1966. doi: 10.1212/01.wnl.0000327095.32005.a4 [DOI] [PubMed] [Google Scholar]
- 60. Seburn KL, Nangle LA, Cox GA, Schimmel P, Burgess RW. An active dominant mutation of Glycyl‐tRNA Synthetase causes neuropathy in a Charcot‐Marie‐Tooth 2D mouse model. Neuron. 2006;51:715‐726. doi: 10.1016/j.neuron.2006.08.027 [DOI] [PubMed] [Google Scholar]
- 61. Lee J, Jung SC, Joo J, et al. Overexpression of mutant HSP27 causes axonal neuropathy in mice. J Biomed Sci. 2015;22:1‐7. doi: 10.1186/S12929-015-0154-Y/FIGURES/5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Stavropoulos F, Sargiannidou I, Potamiti L, et al. Aberrant mitochondrial dynamics and exacerbated response to neuroinflammation in a novel mouse model of cmt2a. Int J Mol Sci. 2021;22:11569. doi: 10.3390/IJMS222111569/S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Harland M, Torres S, Liu J, Wang X. Neuronal mitochondria modulation of LPS‐induced Neuroinflammation. J Neurosci. 2020;40:1756‐1765. doi: 10.1523/JNEUROSCI.2324-19.2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hines TJ, Bailey J, Liu H, et al. A novel ENU‐induced Mfn2 mutation causes motor deficits in mice without causing peripheral neuropathy. Biology (Basel). 2023;12:953. doi: 10.3390/BIOLOGY12070953/S1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hikiami R, Yamashita H, Koita N, et al. Charcot‐Marie‐Tooth disease type 2A with an autosomal‐recessive inheritance: the first report of an adult‐onset disease. J Hum Genet. 2018;63:89‐92. doi: 10.1038/S10038-017-0353-3 [DOI] [PubMed] [Google Scholar]
- 66. Ballard A, Zeng R, Zarei A, et al. The tethering function of mitofusin2 controls osteoclast differentiation by modulating the Ca2+‐NFATc1 axis. J Biol Chem. 2020;295:6629‐6640. doi: 10.1074/JBC.RA119.012023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zarei A, Ballard A, Cox L, et al. Osteolineage depletion of mitofusin2 enhances cortical bone formation in female mice. Bone. 2021;148:115941. doi: 10.1016/J.BONE.2021.115941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rocha AG, Franco A, Krezel AM, et al. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot‐Marie‐Tooth disease type 2A. Science. 2018;360:336‐341. doi: 10.1126/science.aao1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. D'Ydewalle C, Krishnan J, Chiheb DM, et al. HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1‐induced Charcot‐Marie‐Tooth disease. Nat Med. 2011;17:968‐974. doi: 10.1038/nm.2396 [DOI] [PubMed] [Google Scholar]
- 70. Adalbert R, Kaieda A, Antoniou C, et al. Novel HDAC6 inhibitors increase tubulin acetylation and rescue axonal transport of mitochondria in a model of Charcot‐Marie‐Tooth type 2F. ACS Chem Neurosci. 2020;11:258‐267. doi: 10.1021/acschemneuro.9b00338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kim JY, Woo SY, Hong YB, et al. HDAC6 inhibitors rescued the defective axonal mitochondrial movement in motor neurons derived from the induced pluripotent stem cells of peripheral neuropathy patients with HSPB1 mutation. Stem Cells Int. 2016;2016:9475981. doi: 10.1155/2016/9475981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Guedes‐Dias P, de Proença J, Soares TR, et al. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim Biophys Acta. 2015;1852:2484‐2493. doi: 10.1016/J.BBADIS.2015.08.012 [DOI] [PubMed] [Google Scholar]
- 73. Picci C, Wong VSC, Costa CJ, et al. HDAC6 inhibition promotes α‐tubulin acetylation and ameliorates CMT2A peripheral neuropathy in mice. Exp Neurol. 2020;328:113281. doi: 10.1016/j.expneurol.2020.113281 [DOI] [PubMed] [Google Scholar]
- 74. Shen S, Picci C, Ustinova K, et al. Tetrahydroquinoline‐capped histone deacetylase 6 inhibitor SW‐101 ameliorates pathological phenotypes in a Charcot‐Marie‐Tooth type 2A mouse model. J Med Chem. 2021;64:4810‐4840. doi: 10.1021/ACS.JMEDCHEM.0C02210 [DOI] [PubMed] [Google Scholar]
- 75. Rizzo F, Bono S, Ruepp MD, et al. Combined RNA interference and gene replacement therapy targeting MFN2 as proof of principle for the treatment of Charcot‐Marie‐Tooth type 2A. Cell Mol Life Sci. 2023;80:373. doi: 10.1007/S00018-023-05018-W [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Stavropoulos F, Georgiou E, Schiza N, et al. Mitofusin 1 overexpression rescues the abnormal mitochondrial dynamics caused by the Mitofusin 2 K357T mutation in vitro. J Peripher Nerv Syst. 2023;28:329‐340. doi: 10.1111/JNS.12564 [DOI] [PubMed] [Google Scholar]
- 77. Shahin S, Lu B, Zhou Y, et al. MFN1 augmentation prevents retinal degeneration in a Charcot‐Marie‐Tooth type 2A mouse model. iScience. 2023;26:106270. doi: 10.1016/J.ISCI.2023.106270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Figley MD, DiAntonio A. The SARM1 axon degeneration pathway: control of the NAD+ metabolome regulates axon survival in health and disease. Curr Opin Neurobiol. 2020;63:59‐66. doi: 10.1016/J.CONB.2020.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analysed in this study.