

Abstract
The workhorse of developmental biology is the confocal microscope, which allows researchers to determine the three-dimensional localization of tagged molecules within complex biological samples. While traditional confocal microscopes allow one to resolve two adjacent fluorescent point sources located a few hundred nanometers apart, observing the finer details of subcellular biology requires the ability to resolve signals in the order of tens of nanometers. Numerous hardware-based methods for super-resolution microscopy have been developed to allow researchers to sidestep such resolution limits, although these methods require specialized microscopes that are not available to all researchers. An alternative method for increasing resolving power is to isotropically enlarge the sample itself through a process known as expansion microscopy (ExM), which was first described by the Boyden group in 2015. ExM is not a type of microscopy per se but is rather a method for swelling a sample while preserving the relative spatial organization of its constituent molecules. The expanded sample can then be observed at an effectively increased resolution using a traditional confocal microscope. Here, we describe a protocol for implementing ExM in whole-mount Drosophila embryos, which is used to examine the localization of Par-3, myosin II, and mitochondria within the surface epithelial cells. This protocol yields an approximately four-fold increase in sample size, allowing for the detection of subcellular details that are not visible with conventional confocal microscopy. As proof of principle, an anti-GFP antibody is used to distinguish distinct pools of myosin-GFP between adjacent cell cortices, and fluorescently labeled streptavidin is used to detect endogenous biotinylated molecules to reveal the fine details of the mitochondrial network architecture. This protocol utilizes common antibodies and reagents for fluorescence labeling, and it should be compatible with many existing immunofluorescence protocols.
Introduction
In cell and developmental biology, seeing is believing, and the ability to accurately determine the localization patterns of proteins is fundamental to many types of experiments. Laser-scanning confocal microscopy is the standard tool for imaging fluorescently labeled proteins in three dimensions within intact samples. Conventional confocal microscopes are incapable of distinguishing (resolving) adjacent fluorescent signals that are separated by less than one-half of the wavelength of the light they emit1. In other words, two point sources must be separated by at least 200–300 nm in the lateral direction (500–700 nm in the axial direction) to resolve them as two distinct signals. This technical barrier is known as the diffraction limit, and it is a fundamental hurdle to studies of complex subcellular structures (e.g., the actomyosin cytoskeletal or mitochondrial networks) with spatial features below the diffraction limit. Therefore, techniques for increasing the resolving power of conventional confocal microscopes are of general interest to the biological community.
To sidestep the diffraction limit, a number of different super-resolution microscopy technologies have been developed that allow for resolution in the order of tens of nanometers or less1,2,3, revealing a world of biological complexity that was previously only accessible via electron microscopy. Despite the obvious advantages of these hardware-based methods, super-resolution microscopes often have specific sample labeling requirements and long acquisition times, limiting their flexibility, or they may simply be too expensive for some labs to access. An alternative to microscope-based super-resolution is expansion microscopy (ExM), which is not a type of microscopy per se but is rather a method for swelling a sample while preserving the relative spatial organization of its constituent molecules4. The isotropically expanded samples can then be observed at an effectively increased resolution using a traditional fluorescence confocal microscope. ExM was first described by the Boyden group in 20155, and the basic technique has since been adapted for use in a variety of experiments6,7,8. ExM has also been adapted for use in whole-mount embryos, notably in Drosophila9,10,11, C. elegans12, and zebrafish13, making it a powerful tool for developmental biologists.
ExM is based on two different hydrogel chemistries: 1) swellable polyelectrolyte hydrogels, which greatly increase in size when soaked in water14, and 2) polyacrylamide hydrogels, which have extremely small polymer spacing to allow for isotropic sample expansion15. While there are many published ExM protocols, they generally share the following steps: sample fixation, labeling, activation, gelation, digestion, and expansion4. The fixation conditions and fluorescence labeling strategies will of course vary based on the needs of the experiment and system, and in some protocols, labeling occurs after expansion. The target molecules in the sample must be primed (activated) for binding to the hydrogel, which can be achieved using different chemistries4. During the gelation steps, the sample is saturated with monomers of the future hydrogel (sodium acrylate, acrylamide, and the crosslinker bisacrylamide), and the hydrogel is then formed by free-radical polymerization catalyzed by an initiator, such as ammonium persulfate (APS), and an accelerator, such as tetramethylenediamine (TEMED)4. After gelation, the sample is enzymatically digested to homogenize sample resistance to swelling and ensure the isotropic expansion of the hydrogel4. Finally, the digested hydrogel is placed in water, which causes it to expand to approximately four times its original linear size4.
This protocol describes how to perform ExM on whole-mount early- to mid-stage Drosophila embryos16 to visualize subcellular protein localization patterns at super-resolution (Figure 1). This method uses methylacrylic acid N-hydroxysuccinimidyl ester (MA-NHS) chemistry to activate and anchor protein molecules to the hydrogel17, and it is a modification of a previously published ExM protocol for use in late-stage Drosophila embryos and tissues11. This protocol uses polydimethylsiloxane (PDMS) wells to mold the hydrogels and facilitate solution exchange during activation and gelation. An alternative method that does not require the creation of PDMS wells involves lowering embryos attached to coverslips into drops of monomer solution sitting on a piece of laboratory sealing film22. In addition, this protocol describes a method for manually removing the impermeable vitelline membrane that surrounds Drosophila embryos, which is a prerequisite for immunofluorescence staining. Importantly, this method of hand-peeling embryos can be used to select only properly staged Drosophila embryos prior to sample labeling, which greatly increases the likelihood of ending up with expanded samples of the correct stage and orientation and, thus, makes the downstream data collection much more efficient.
Figure 1: Overview of expansion microscopy in Drosophila embryos.
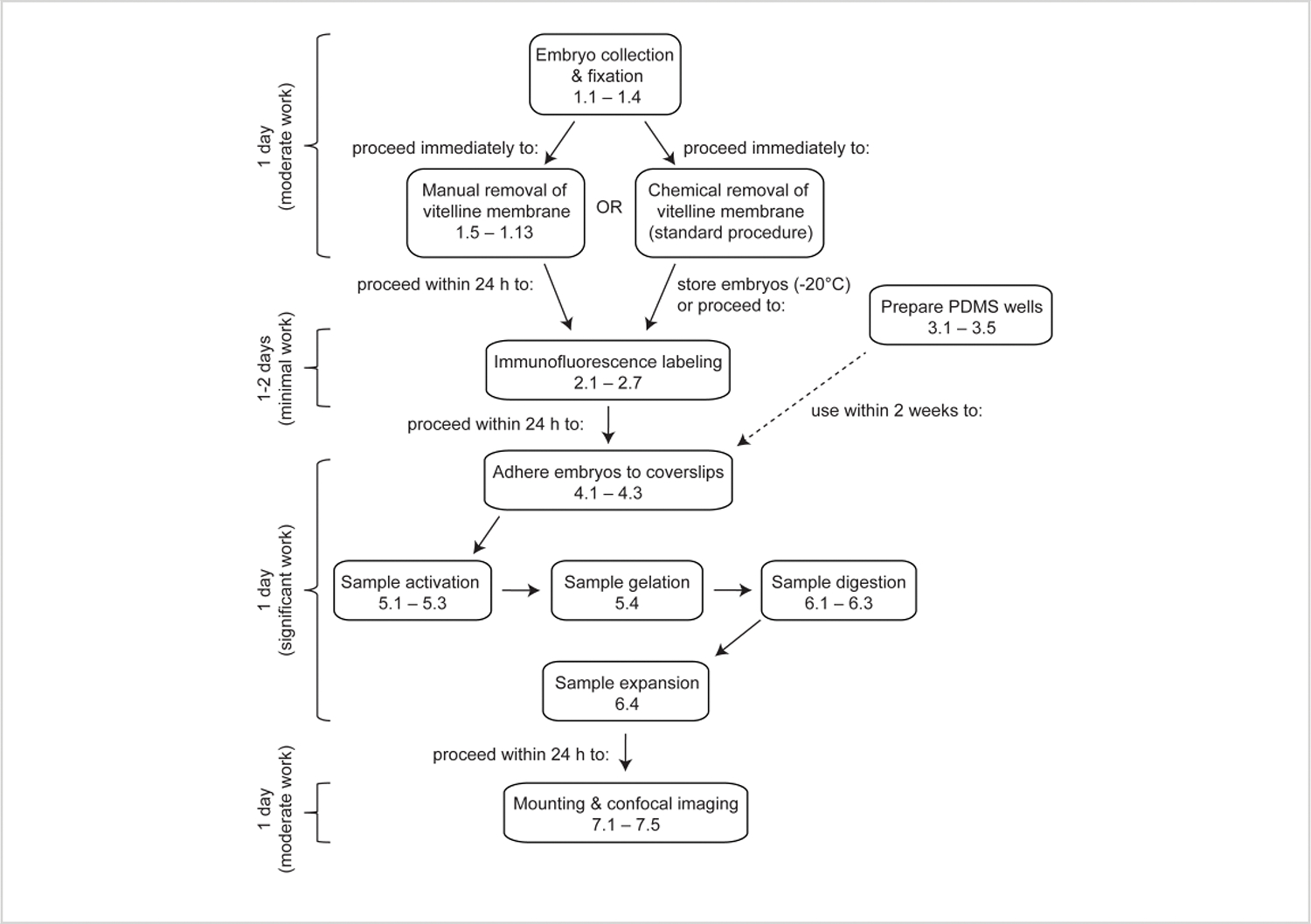
ExM is a multi-step protocol that takes at least 4 days to complete. Embryo collection, fixation, and devitellinization take 1 day or more depending on whether embryos from multiple collections are pooled. Immunofluorescence labeling takes 1 day or 2 days depending on whether the embryos are incubated overnight with the primary antibodies. Embryo activation, gelation, digestion, and expansion can be performed in a single day. The gels can be mounted and imaged immediately after expansion, although for practical reasons it is often desirable to start imaging the following day.
Protocol
This protocol follows the University of Arkansas (UARK) guidelines for research on invertebrate animals, such as Drosophila melanogaster, and was approved by the UARK Institutional Biosafety Committee (protocol #20001).
1. Drosophila embryo fixation and devitellinization
NOTE: Step 1 describes a procedure (hand-peeling) for the manual removal of the vitelline membrane, a transparent impermeable membrane that surrounds the embryo. Importantly, hand-peeling allows for the selection of properly staged embryos at the start of the ExM protocol, thus greatly enhancing the likelihood of obtaining embryos in a useable orientation at the end of the ExM protocol. However, this ExM protocol is completely compatible with bulk embryo collection and standard procedures for methanol-based removal of the vitelline membrane, in which case one can skip directly to step 2 (immunofluorescence labeling).
Prepare or purchase a number of fine glass needles. The actual dimensions of the needle tip are not critical, but ensure the needles are rigid and sharp enough to pierce the vitelline membranes of fixed embryos. Make needles from glass capillary tubes (1 mm outer diameter, 0.75 mm inner diameter) using a micropipet puller, as one would prepare needles for embryo microinjections18; alternatively, purchase pre-pulled needles.
Collect embryos using standard Drosophila techniques19 by placing >100 adult Drosophila in a vented plastic cup sealed with a fruit-juice/agar plate20. Use timed collection windows to enrich for embryos of the proper stage16. For example, to enrich for the gastrulation (stage 6) and convergent-extension (stage 7) stages, change the fruit-juice plate, collect the embryos for 2 h at 25 °C, then remove the plate, and age it for a further 2 h at 25 °C to obtain embryos that are ~2–4 h old.
To remove the eggshell-like chorion from the embryos, cover the surface of the fruit-juice plates with 50% bleach (Table 1), release the embryos from the surface of the agar by agitating them with a small paintbrush, and wait 3 min for the chorion to dissolve.
-
Transfer the dechorionated embryos using a paintbrush to a 30 mL scintillation vial containing 4 mL of heptane (organic top phase) and 4 mL of fixation buffer (aqueous bottom phase; Table 1). Freshly dilute the formaldehyde from a freshly prepared or recently opened stock, and mix with 10x PBS and deionized water immediately before adding the embryos.
NOTE: Prepare the formaldehyde from paraformaldehyde power or 16% EM-grade formaldehyde in glass ampules. Stocks of concentrated formaldehyde (e.g., 37% formaldehyde) can be used, although the results may be less consistent.
The embryos will accumulate at the interface between the organic and aqueous phases. Add as many embryos as will form a single layer at the interface. If too many embryos are added to a vial, they will not fix as well.
Using strong tape, immobilize the scintillation vials on their sides on a tabletop shaker, and agitate them for 20 min at 220 rpm. For optimal fixation, maintain a vigorous emulsion between the organic and aqueous phases during the entire fixation.
- During the fixation time, prepare one of the following for each of the samples.
- Take a plastic 6 cm Petri dish base filled halfway with 3% agar and score a ~5 cm × 3 cm rectangle in the agar with a razor blade or scalpel. Fruit juice/agar plates can also be used for this purpose.
- Using a small lab spatula, remove the agar slab. Invert the base of the Petri dish, and set it on the bench. Place the agar slab on top of the inverted dish (Figure 2A).
- Take the lid of the Petri dish and make sure it is dry. Wearing gloves, place a piece of double-sided tape inside of the lid (the piece of tape should be slightly larger than the agar slab; Figure 2B).
Remove the vials from the shaker, set them upright on the bench, and allow the organic and aqueous phases to separate. Properly fixed embryos will remain at the interface between the two phases.
Transfer the fixed embryos onto the agar slab using a glass Pasteur pipet fitted with a latex bulb. To prevent the embryos from adhering to the inside of the pipet, try to keep the embryos within the narrow neck of the pipet, and transfer the embryos in multiple small batches rather than all at once. Once all the embryos are on the agar slab, remove most of the residual heptane from around the embryos using a P200 pipettor. Perform this step as quickly as possible (<3 min) to avoid drying out the fixed embryos, which can negatively affect the morphology.
From a height of ~2 cm, drop the lid with the double-sided tape onto the agar slab to adhere the embryos to the tape (Figure 2C). Gently remove the lid from the agar slab, place it upside down on the bench, and then add enough PBS-Tween (Table 1) to cover the embryos in the lid.
- Using a stereo dissecting microscope at approximately 100x magnification with indirect lighting, identify properly staged embryos using morphological markers. For stage 6 embryos, use markers like a visible cephalic furrow and invaginated mesoderm; for stage 7 embryos, use markers like an extended germband; for stage 11 embryos, use markers like a fully extended germband and visible segmentation along the head-to-tail axis16.
- To collect the desired embryos, first prick the vitelline membrane (a transparent oval membrane around the embryo) near the anterior or posterior end of the embryo with a fine glass needle; the membrane will deflate a bit as the pressure releases. Then, using fine forceps or a metal probe, gently push the embryo on the other end through the hole; the vitelline membrane will remain adhered to the double-sided tape. Leave undesired embryos adhered to the tape.
Periodically collect the floating devitellinized embryos with a glass Pasteur pipet, and move them to a 1.5 mL microfuge tube.
- At this point, perform one of the following steps.
- Continue directly to the immunofluorescence labeling steps. The devitellinized embryos can go directly into blocking solution (step 2.2). Do not allow the embryos to remain in PBS-Tween or blocking solution (Table 1) for longer than 16 h before proceeding to the next step.
- Move the embryos into methanol for storage. Remove as much of the PBS-Tween as possible, and then add 1 mL of methanol. Once the embryos have settled, remove as much methanol as possible, and add 1 mL of fresh methanol. Store the embryos at −20 °C indefinitely. Methanol storage also allows for the pooling of embryos from multiple collections.
Table 1. Solution recipes.
Composition for the solutions used in this protocol in order of appearance. All the stocks are liquids unless otherwise noted. The chemicals were resuspended or diluted in autoclaved filtered water unless otherwise noted.
| RECIPES (in order of appearance in protocol) | ||
|---|---|---|
| All stocks are liquids unless otherwise noted | ||
| Chemicals are resuspended or diluted in autoclaved filtered water unless otherwise noted | ||
| 50% bleach | ||
| 1 part standard (i.e., non-concentrated) commercial bleach (6% sodium hypochlorite) | ||
| 1 part tap water | ||
| Fixation buffer | stock | comments |
| 4% paraformaldehyde | 16% | stock stored in small glass ampules |
| 1× phosphate-buffered saline | 10× | |
| Blocking solution | stock | comments |
| 1× phosphate-buffered saline | 10× | |
| 10% bovine serum albumin | 30% | |
| 0.1% Tween-20 | 10% (v/v) | stock diluted from original 100% liquid |
| 0.1% sodium azide | 1% (w/v) | stock prepared from powder; TOXIC |
| Antibody solution | stock | comments |
| 1× phosphate-buffered saline | 10× | |
| 5% bovine serum albumin | 30% | |
| 0.1% Tween-20 | 10% (v/v) | stock diluted from original 100% liquid |
| 0.1% sodium azide | 1% (w/v) | stock prepared from powder; TOXIC |
| PBS-Tween | stock | comments |
| 1× phosphate-buffered saline | 10× | |
| 1% bovine serum albumin | 30% | |
| 0.1% Tween-20 | 10% (v/v) | stock diluted from original 100% liquid |
| PDMS | ||
| 10 parts Sylgard silicone elastomer base | ||
| 1 part Sylgard elastomer curing agent | ||
| Activation solution | stock | comments |
| 1 mM MA-NHS* | 1 M | stock resuspended in DMSO |
| 1× phosphate-buffered saline | 10× | |
| * methylacrylic acid N-hydroxysuccinimidyl ester | ||
| Monomer solution* | stock | comments |
| 2 M NaCl | 5 M | |
| 1× phosphate-buffered saline | 10× | |
| 8.625% (w/v) sodium acrylate | powder | TOXIC |
| 2.5% (w/v) acrylamide | powder | TOXIC |
| 0.15% (w/v) bisacrylamide | powder | TOXIC |
| *Monomer solution can be stored at 4°C for up to two weeks | ||
| Gelation solution | stock | comments |
| Add ingredients in this order: | ||
| 3920 μl monomer solution (above) | ||
| 60 μl TEMED* | 10% (v/v) | diluted in H2O; keep stock at -20°C |
| 20 μl TEMPO** | 1% (w/v) | diluted in H2O; prepare fresh from powder |
| Add last: | ||
| 80 μl ammonium persulfate | 10% (w/v) | diluted in H2O; keep stock at -20°C |
| *N,N,N',N'-tetramethylethane-1,2-diamine | ||
| ** (2,2,6,6-tetramethylpiperidin-1-yl)oxyl | ||
| Digestion buffer | stock | comments |
| 1× TAE | 10× | |
| 80 mM guanidine HCl | powder | TOXIC |
| 0.1% Tween-20 | 10% (v/v) | stock diluted from original 100% liquid |
| Add last: | ||
| 250 μl proteinase K ( 20 mg/ml, 600 U/ml) to 20 ml digestion buffer |
Figure 2: Manual devitellinization and working with hydrogels.

(A) Cutting an agar slab from an agar/fruit-juice plate. (B) Placing double-sided tape inside the lid of a 6 cm Petri dish. (C) Adhering embryos to the taped lid. (D) A PDMS slab with a square well adhered to a 22 mm x 22 mm coverslip. (E) Coverslip with a PDMS well inside a 6-well plate.
2. Immunofluorescence labeling
NOTE: Aside from the antibody incubation steps, exact liquid amounts and times are not critical in this section. To perform a rinse or wash, allow the embryos to settle to the bottom of the tube, remove as much liquid as possible without sucking up embryos, and then add ~1 mL of new liquid; use a glass Pasteur pipet fitted with a latex bulb for optimal clarity and control. For the rinse step, the embryos are not rocked, just allowed to settle; for the wash step, the embryos are rocked on a nutator for the indicated amount of time and then allowed to settle.
If embryos were not stored in methanol, proceed to step 2.2. If the embryos were stored in methanol, rinse twice with PBS-Tween, and then wash for 20 min twice with PBS-Tween.
Wash the embryos for 30–60 min in 1 mL of blocking solution.
- Incubate the embryos with primary antibodies diluted in antibody solution (Table 1) for 2 h at room temperature or preferably overnight at 4 °C. Perform this step in as small a volume as possible (50–300 μL) to conserve the primary antibodies; rocking on a nutator is not strictly required.
- Increase the amount of primary antibody used in a typical immunofluorescence experiment by at least 50% for ExM. Use the following primary antibody concentrations: 1:200 for anti-Par-3 guinea pig polyclonal21 and 1:100 for anti-GFP rabbit polyclonal.
Remove the primary antibody solution (save at 4 °C if desired), rinse twice with PBS-Tween, and then wash for 15 min four times with PBS-Tween.
- Incubate the embryos with fluorescent secondary antibodies in a final volume of 300 μL (diluted in antibody solution) for 1 h at room temperature on a nutator. Fluorescently labeled streptavidin can be added during this step. From this step onward, protect the embryos from excessive and prolonged light exposure when possible, for example by covering the tubes with an opaque box lid or keeping the samples in a drawer.
- Use the following concentrations: 1:500 for anti-rabbit IgG goat polyclonal fused to Alexa Fluor 488; 1:500 for anti-guinea pig IgG goat polyclonal fused to Alexa Fluor 568; and 1:1000 for streptavidin-Alexa Fluor 488.
Remove and dispose of the secondary antibody solution. Rinse the embryos twice with PBS-Tween, and wash for 15 min four times with PBS-Tween.
At this point, the embryos can be stored at 4 °C in the dark but process the samples as quickly as possible (<24 h).
3. Preparing PDMS wells
NOTE: The PDMS wells can be made up to 2 weeks in advance.
Set an incubator or hot plate to 55 °C, and set a centrifuge that can spin conical tubes to 15 °C.
To prepare the PDMS solution (Table 1), place a 50 mL conical tube in a secondary container on a scale, and add 10 g of silicone elastomer base to the tube using a syringe. Then, add 1 g of silicone elastomer curing agent, and invert the tube several times to mix.
Create a balance tube by adding an appropriate amount of water to a second 50 mL conical tube. Centrifuge the PDMS solution at 500 × g for 3 min at 15 °C, and then pour it into a 10 cm Petri dish to a depth of ~1 mm. If necessary, remove bubbles by blowing gently on the solution with an air hose. Allow the PDMS solution to solidify overnight at 55 °C.
Once the PDMS slab is solidified, using a scalpel, score square areas that are slightly smaller than a 22 mm × 22 mm coverslip. Inside each square, score and remove an ~8 mm-wide square well.
Transfer each square PDMS well onto a 22 mm × 22 mm coverslip, and firmly adhere it (Figure 2D). Preparing six or more coverslips should yield a good number of expanded embryos to image.
4. Adhering the embryos to the coverslips
Apply enough 0.1% poly-L-lysine to cover the coverslip surface inside of each well (~50 μL), and place them in a 55 °C incubator to air dry. Repeat this step to increase adhesiveness.
Briefly rinse the embryos once in 1x PBS to remove the Tween detergent, and then transfer >10 embryos into each of the poly-L-lysine-coated wells.
Allow the embryos to settle to the bottom of the wells. Remove the excess liquid from the adhered embryos using a Pasteur pipet. Immediately proceed to the next step.
5. Activation and gelation
NOTE: Activation refers to the addition of MA-NHS to the embryos, which will modify the sample proteins and antibodies so they can bind to the hydrogel. Gelation refers to the generation of a hydrogel in and around the embryos in each well. During gelation, the embryos are permeated with a monomer solution and then treated with a gelation solution to form the hydrogel.
Activate the embryos for 1 h at room temperature by filling the wells with activation solution (1 mM MA-NHS freshly diluted in 1x PBS; Table 1). Change this solution approximately every 10 min over the course of 1 h.
Rinse the embryos with 1x PBS three times. Incubate the embryos in monomer solution (Table 1) for 45 min at 4 °C.
- While the embryos are sitting in the monomer solution, prepare the gelation solution (Table 1). Preparing ~2 mL of gelation solution is enough to cover the PDMS wells from an entire 10 cm Petri dish. Be sure to add the APS last, as it will initiate polymerization and begin the gelation.
- Dilute the catalytic oxidant freshly from powder (e.g., 1% TEMPO w/v in water). Combine 1,960 μL of monomer solution with 30 μL of 10% TEMED and 10 μL of 1% TEMPO.
- To avoid polymerizing the entire batch of gelation solution at once, work in small batches. Split the gelation solution (without APS) into 125 μL aliquots between the eight tubes of a PCR strip.
- Remove the monomer solution from the three PDMS wells using a vacuum while being careful not to disrupt the embryos. Add 5 μL of APS to one of the PCR tubes containing gelation solution to initiate polymerization. Quickly distribute the polymerizing gelation solution amongst the wells (~40 μL per well). Repeat this until all of the wells and embryos are covered.
Let the samples gel for 1.5–2.5 h at 37 °C. Agitate the hydrogels every so often to monitor the polymerization. Solidified hydrogels will not wiggle. Thicker hydrogels will take longer to complete polymerization and solidify.
6. Digestion and expansion
NOTE: Thicker and larger gels will take longer to expand, and the center of the gels may take several hours to completely expand; this can be sped up by trimming the edges of the gel. As the gels expand, their refractive index will become nearly identical to that of water, and they will become very hard to see.
After gelation is complete, peel the PDMS wells from the coverslip while trying not to disturb the hydrogels. Cut away excess hydrogel material, if desired.
Transfer the hydrogels (still attached to the coverslips) individually into the wells of a six-well plate (Figure 2E). Note that the hydrogels may slightly expand during digestion.
Cover the gels completely with digestion buffer (Table 1) for 1 h at 37 °C. In general, 30 mL of digestion buffer is sufficient to cover the gels in a 6-well plate.
After digestion, transfer each hydrogel individually into a 6 cm Petri dish by sliding them off the coverslip. It may be necessary to use a second coverslip to dislodge the gel. Fill each Petri dish with deionized water to expand the gel. Change the water three to four times over the course of 1–2 h until the gels are fully expanded (expect an approximate four-fold increase in width).
7. Mounting and imaging
NOTE: Expanded hydrogels are composed almost entirely of water, making them nearly transparent and extremely fragile. The gels can be manipulated using long coverslips to move them around and pick them up. Mount and image only one or two gels at a time, as gels will gradually release water and begin to slide around the coverslip.
Using a Pasteur pipet, remove as much excess water from the Petri dish as possible to minimize the gels moving around when handled.
Maneuver each expanded gel, with the embryos on the bottom surface, onto a large coverslip (e.g., 24 mm × 40 mm) for imaging.
Mount each coverslip with gel over the objective of an inverted laser-scanning confocal microscope. Locate properly staged and oriented specimens using the epifluorescence or brightfield microscopy modes on the microscope with low-magnification (5x or 10x) or medium-magnification (20x) air objectives.
To image at high resolution, switch to a high-magnification (60x, 63x, or 100x) oil- or water-immersion objective. The surface of the embryos must be within the focusing range of the objective (<300 μm from the coverslip for a 63x objective) in order to be imaged.
Collect data from samples using the laser-scanning confocal mode on the microscope. Be sure to collect non-saturated images with good dynamic range, and use an appropriate number of pixels per image to capture the maximal possible information about the sample23.
Representative Results
To characterize the general efficacy of ExM in whole-mount Drosophila embryos, embryo length along the head- to-tail axis was measured in unexpanded control embryos versus expanded embryos (Figure 3A–C). The unexpanded control embryos were subjected to the same fixation conditions and immunofluorescence labeling steps as the expanded embryos, except they were mounted using a solidified mounting medium prior to imaging. The individual unexpanded embryos spanned approximately one-half of a field of view when using a 10x objective (Figure 3A). By contrast, the expanded embryos spanned approximately two full fields of view when using the same 10x objective (Figure 3B). To assess how the degree of expansion varied both within and between experiments, the same ExM protocol was performed on three separate occasions, and embryo length was measured in three different gels within each individual experiment. The average head-to-tail length of the unexpanded control embryos was 398.8 μm (standard deviation [SD] = 22.93 μm; n = 74; Figure 3C). For experiment 1, experiment 2, and experiment 3, the average embryo lengths were 1,596 μm (SD = 159.9 μm; n = 57), 1,868 μm (SD = 150.5 μm; n = 51), and 1,954 μm (SD = 120.3 μm; n = 44), respectively, representing expansion factors of 4.0-fold, 4.7-fold, and 4.9-fold, respectively (Figure 3C). The intra-experimental variation between the gels was much less noticeable than the inter-experimental variation, which was approximately 20% (Figure 3C). To assess the effects of ExM on cell and embryo morphology, an antibody against the adherens junction component Par-3 (Bazooka)21 was used to label the apical cell membranes, and we imaged the developing mouth segments of stage 11 Drosophila embryos —a stage with a complex segmented structure (Figure 3D–F). In the control sample, the cells in the maxillary segment had an average width of 4.76 μm (SD = 1.053 μm, n = 25; Figure 3D,F). In the expanded samples imaged using the same 40x objective and zoom factor (1x), the cells in the maxillary segment had an average width of 19.10 μm (SD = 3.966 μm, n = 18; Figure 3E,F), representing a 4.0-fold expansion. Therefore, consistent with previous reports11, we were able to expand whole-mount Drosophila embryos approximately four-fold in linear dimensions using ExM without sample tearing or obvious distortions in the cellular or tissue morphology.
Figure 3: Four-fold expansion of Drosophila embryos.
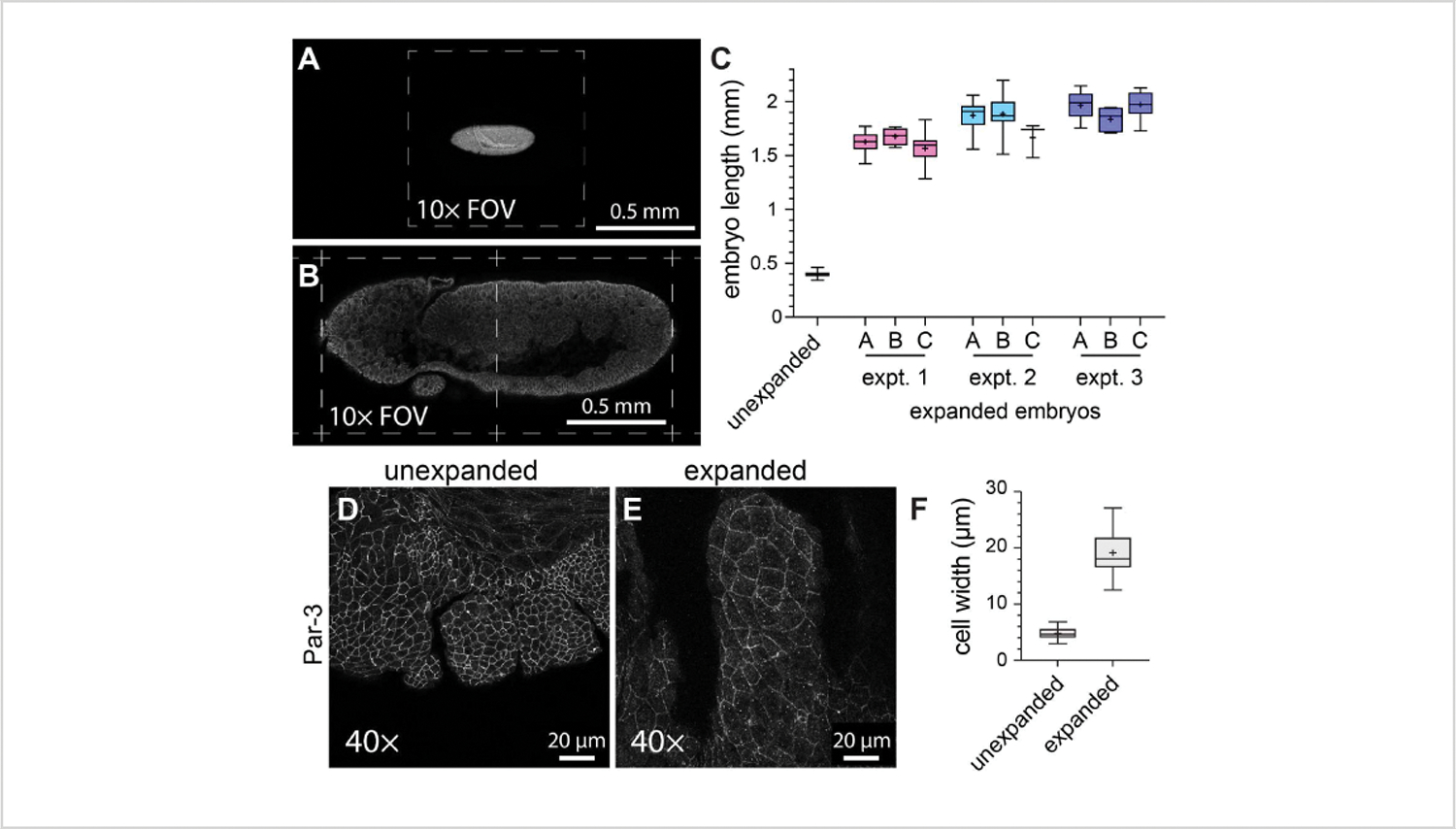
(A) Unexpanded and (B) expanded Drosophila embryos imaged using a 10x objective (0.3 NA) at 1x zoom. Individual fields of view (FOV) are indicated with dashed lines. The embryos expressed a GFP-tagged version of myosin light chain and were stained with an anti-GFP antibody. (C) Quantification of embryo length (along the head-to-tail axis) in three hydrogels per experiment and from three separate ExM experiments compared with unexpanded controls. (D,E) Maxillary segments from (D) unexpanded and (E) expanded stage 11 Drosophila embryos imaged using a 40x objective (1.3 NA) at 1x zoom. The cell outlines (adherens junctions) were detected with an anti Par-3/Bazooka antibody (white). (F) Quantification of the cell width (long axis) from equivalent groups of cells from (D) and (E). The box plots in (C) and (F) show the 25th, 50th, and 75th percentile ranges; the whiskers indicate the minimum and maximum values; the “+” symbols indicate the mean.
To demonstrate that ExM can be used to resolve subcellular details below the typical diffraction limit, the actomyosin cytoskeleton was imaged in unexpanded control versus expanded embryos undergoing convergent extension (stage 7). The tissue remodeling events of gastrulation and convergent extension are largely controlled by changes in the localization of the motor protein myosin II24. However, in the densely packed columnar epithelium of the early Drosophila ectoderm, it is difficult to observe many fine details of the myosin II localization pattern, even when imaged at 158x magnification (63x objective with a 2.5x optical zoom)—a typical maximal resolving power for a laser-scanning confocal microscope. For example, because myosin II is a cortical protein (located directly beneath the plasma membrane), pools of myosin II25 located on either side of cell-cell contacts were not resolvable in stage 7 embryos, and they appeared as a single line where neighboring cells met (Figure 4A). By contrast, in expanded stage 7 embryos, parallel lines of myosin II could be observed at cell-cell junctions, representing cortical protein pools in adjacent cells (Figure 4B). The distance between parallel myosin II lines in expanded samples was 892.7 nm (SD = 0.171 nm, n = 12); when divided by four, this yields a predicted distance of ~220 nm between the myosin lines in adjacent cells in unexpanded embryos, which is indeed just below the diffraction limit for a signal detected with Alexa 488 (peak emission of ~520 nm/2 = 260 nm).
Figure 4: Details of actomyosin cytoskeleton and mitochondria revealed by expansion microscopy.
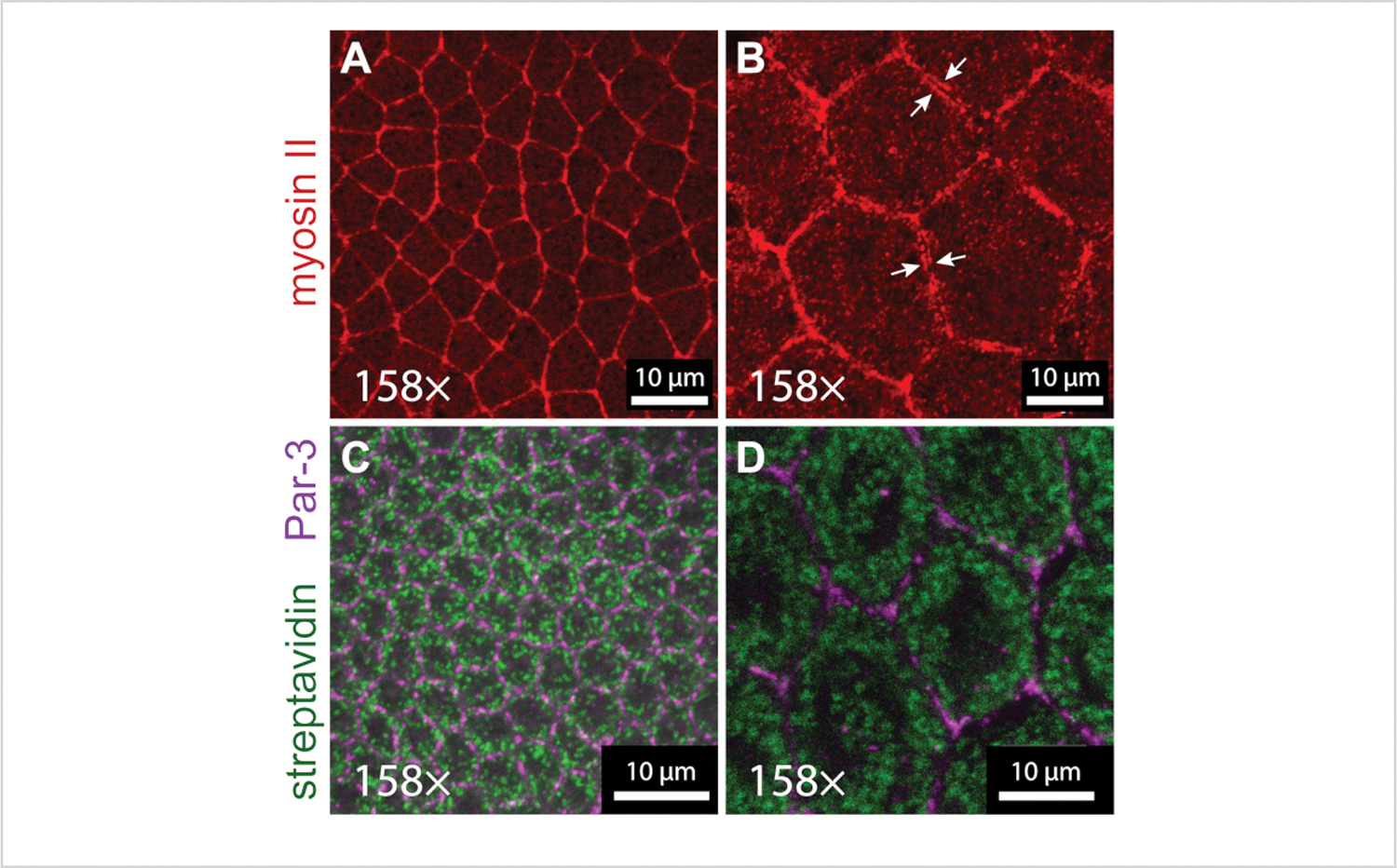
(A,B) Myosin II localization in neuroectoderm (germband) cells imaged with a 63x objective (1.4 NA) at 2.5x zoom in stage 7 (A) unexpanded and (B) expanded embryos. Myosin II was detected in the embryos expressing a transgenic GFP-tagged version of the myosin II regulatory light chain (sqh-GFP), which was detected with an anti-GFP antibody (red). Distinct pools of cortical myosin located in adjacent cells can be resolved in the expanded embryo (white arrows). (C,D) Mitochondrial networks in neuroectoderm cells imaged with a 63× objective (1.4 NA) at 2.5x zoom in stage 6 unexpanded (C) and expanded (D) embryos. The mitochondria were detected with streptavidin-Alexa 488 (green), and the cell outlines were detected with an anti-Par-3/Bazooka antibody (magenta). The experiments were performed with a laser-scanning confocal microscope.
In addition, we also tested whether ExM could be used to resolve the mitochondrial network architecture in densely packed cells of gastrulating Drosophila embryos (stage 6). Mitochondrial function is closely linked to network structure (i.e., fused vs. fragmented organelles), but the details of mitochondrial network organization are hard to visualize using conventional confocal microscopy in cell types that are not flat and/or thin. Mitochondria are naturally rich in biotinylated molecules, and, thus, mitochondria can be labeled in the early Drosophila embryo using fluorescently labeled streptavidin26. In the unexpanded stage 6 embryos labeled with streptavidin-Alexa 488, the signal appeared as cytoplasmic puncta that were often overlapping and difficult to resolve (Figure 4C). By contrast, in the expanded stage 6 embryos, many more fine details of the mitochondrial network were visible and puncta were more easily resolvable (Figure 4D)26, 27. These results indicate that ExM can be used to study mitochondrial network organization in cell types not traditionally suited for mitochondrial analysis.
Discussion
Manual devitellinization
Most Drosophila embryo fixation protocols involve removing vitelline membrane by shaking fixed embryos in an emulsion of methanol and heptane, which causes the membranes to burst off via osmotic rupture26. While methanol-based devitellinization (methanol popping) is effective and appropriate for many applications, manual devitellinization (hand-peeling) offers some significant advantages. First, hand-peeling allows one to choose precisely staged embryos to devitellinize and collect, greatly increasing the likelihood of obtaining expanded embryos in a useable orientation at the end of the experiment. This enrichment is critical when studying specific aspects of rapid developmental processes (e.g., mesoderm invagination or convergent extension), for which appropriately staged embryos may represent only a few percent of all embryos, even within a tightly timed collection window. Of course, for many applications, the more traditional bulk methanol popping of embryos from a timed collection window will be sufficient, and hand-peeling may not be worth the extra effort. Second, the binding of certain primary antibodies and dyes is negatively affected by previous exposure of the sample to methanol. For this reason, hand-peeling can yield significant increases in the immunofluorescence signal quality compared with methanol-popped samples, making it a useful general technique for Drosophila developmental biologists.
High-resolution confocal microscopy in expanded whole-mount Drosophila embryos
While performing high-resolution confocal microscopy on expanded samples is conceptually the same as on unexpanded samples, ExM does introduce some technical hurdles. Notably, embryo orientation, which is random, becomes even more important as the sample size increases, because high-magnification, high-NA objectives are only able to focus light from sample regions that are very close to the coverslip27. Therefore, it is usually only possible to focus on the cells at or near the surface of the embryo that ended up adjacent to the coverslip when the gel was formed. The best way to ensure there are specimens of the correct orientation at the end is to start the ExM protocol with a tightly staged collection of fixed embryos (e.g., by using hand-peeling) and to seed many embryos in each well (>10). To visualize cells deep in the interior of the embryo, it may be necessary to utilize more specialized imaging setups, such as light-sheet microscopy28. Additionally, we find that image quality can be improved by opening the confocal pinhole to a size greater than one airy unit. Of course, an increased pinhole size will come at the cost of decreased maximal resolution, but in practice, even small increases in pinhole size can significantly boost the signal intensity (data not shown). Future studies should systematically address pinhole size and effective resolution in ExM samples.
Variations on basic ExM
The protocol described here is a relatively simple example of ExM that should work for many applications and be easy to implement in most developmental biology labs. However, there are numerous variations on the basic concept of ExM4,5,7 that can be used to increase the signal intensity, achieve even further degrees of expansion, and detect nucleic acid molecules as well as proteins. In this protocol, the embryos are incubated with antibodies prior to gelation and expansion. Alternatively, the samples can be treated with antibodies after they are expanded6,30, which can increase signal intensity due to increased epitope accessibility and decreased loss of bound antibodies during the expansion steps. In addition, specific crosslinker molecules can be used to attach RNA molecules to the hydrogel to allow the detection of RNA in expanded gels using the hybridization chain reaction method30. Finally, the samples can be subjected to multiple rounds of expansion, as in iterative expansion microscopy (iExM)31, pan-ExM32, and expansion revealing (ExR)31, to achieve even higher degrees of increased resolution.
Supplementary Material
Acknowledgments
We would like to thank Dr. Jennifer Zallen for providing the guinea pig anti-Par-3 primary antibody. This work was supported by generous funding (1R15GM143729-01 and 1P20GM139768-01 5743) from the National Institute of General Medical Science (NIGMS), one of the members of National Institutes of Health (NIH), as well as the Arkansas Biosciences Institute (ABI), which provided partial funding for the purchase of our confocal microscope.
Footnotes
A complete version of this article that includes the video component is available at http://dx.doi.org/10.3791/64662.
Disclosures
The authors have no conflicts of interest to declare.
References
- 1.Klar TA, Jakobs S, Dyba M, Egner A, Hell SW Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proceedings of the National Academy of Sciences of the United States of America. 97 (15), 8206–8210 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rust MJ, Bates M, Zhuang X Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nature Methods. 3 (10), 793–796 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betzig E et al. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 313 (5793), 1642–1645 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Wassie AT, Zhao Y, Boyden ES Expansion microscopy: principles and uses in biological research. Nature Methods. 16 (1), 33–41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen F, Tillberg PW, Boyden ES Optical imaging. Expansion microscopy. Science. 347 (6221), 543–548 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen F et al. Nanoscale imaging of RNA with expansion microscopy. Nature Methods. 13 (8), 679–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tillberg PW et al. Protein-retention expansion microscopy of cells and tissues labeled using standard fluorescent proteins and antibodies. Nature Biotechnology. 34 (9), 987–992 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang J-B et al. Iterative expansion microscopy. Nature Methods. 14 (6), 593–599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cahoon CK et al. Superresolution expansion microscopy reveals the three-dimensional organization of the Drosophila synaptonemal complex. Proceedings of the National Academy of Sciences of the United States of America. 114 (33), E6857–E6866 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mosca TJ, Luginbuhl DJ, Wang IE, Luo L Presynaptic LRP4 promotes synapse number and function of excitatory CNS neurons. eLife. 6, e27347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang N et al. Superresolution imaging of Drosophila tissues using expansion microscopy. Molecular Biology of the Cell. 29 (12), 1413–1421 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu C-CJ et al. Expansion microscopy of C. elegans. eLife. 9, e46249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freifeld L et al. Expansion microscopy of zebrafish for neuroscience and developmental biology studies. Proceedings of the National Academy of Sciences of the United States of America. 114 (50), E10799–E10808 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka T et al. Phase transitions in ionic gels. Physical Review Letters. 45 (20), 1636–1639 (1980). [Google Scholar]
- 15.Hausen P, Dreyer C The use of polyacrylamide as an embedding medium for immunohistochemical studies of embryonic tissues. Stain Technology. 56 (5), 287–293 (1981). [DOI] [PubMed] [Google Scholar]
- 16.Campos-Ortega JA, Hartenstein V The Embryonic Development of Drosophila melanogaster. Springer Berlin Heidelberg. Berlin, Germany: (2013). [Google Scholar]
- 17.Chozinski TJ et al. Expansion microscopy with conventional antibodies and fluorescent proteins. Nature Methods. 13 (6), 485–488 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller DFB, Holtzman SL, Kaufman TC Customized microinjection glass capillary needles for P-element transformations in Drosophila melanogaster. BioTechniques. 33 (2), 366–367, 369–370, 372 passim (2002). [DOI] [PubMed] [Google Scholar]
- 19.Rothwell WF, Sullivan W Drosophila embryo dechorionation. CSH Protocols. 2007, db.prot4826 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Cold Spring Harbor Protocols. Drosophila apple juice-agar plates. Cold Spring Harbor Laboratory Press. (2011). [Google Scholar]
- 21.de Matos Simões S et al. Rho-kinase directs bazooka/Par-3 planar polarity during drosophila axis elongation. Developmental Cell. 19 (3), 377–388 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gambarotto D et al. Imaging cellular ultrastructures using expansion microscopy (U-ExM). Nature Methods. 16 (1), 71–74 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paddock SW, Fellers TJ, Davidson MW Specimen Preparation and Imaging. Nikon MicroscopyU. at <https://www.microscopyu.com/techniques/confocal/specimen-preparation-and-imaging>. (2023).
- 24.Paré AC, Zallen JA Cellular, molecular, and biophysical control of epithelial cell intercalation. Current Topics in Developmental Biology. 136, 167–193 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Royou A, Field C, Sisson JC, Sullivan W, Karess R Reassessing the role and dynamics of nonmuscle myosin II during furrow formation in early Drosophila embryos. Molecular Biology of the Cell. 15 (2), 838–850 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chowdhary S, Tomer D, Dubal D, Sambre D, Rikhy R Analysis of mitochondrial organization and function in the Drosophila blastoderm embryo. Scientific Reports. 7 (1), 5502 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chowdhary S, Madan S, Tomer D, Mavrakis M, Rikhy R Mitochondrial morphology and activity regulate furrow ingression and contractile ring dynamics in Drosophila cellularization. Molecular Biology of the Cell. 31 (21), 2331–2347 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stelzer EHK et al. Light sheet fluorescence microscopy. Nature Reviews Methods Primers. 1, 73 (2021). [Google Scholar]
- 29.Ku T et al. Multiplexed and scalable super-resolution imaging of three-dimensional protein localization in size-adjustable tissues. Nature Biotechnology. 34 (9), 973–981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wen G et al. A Universal labeling strategy for nucleic acids in expansion microscopy. Journal of the American Chemical Society. 143 (34), 13782–13789 (2021). [DOI] [PubMed] [Google Scholar]
- 31.M’Saad O, Bewersdorf J Light microscopy of proteins in their ultrastructural context. Nature Communications. 11 (1), 3850 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarkar D et al. Expansion revealing: Decrowding proteins to unmask invisible brain nanostructures. bioRxiv. (2020). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.