

Abstract
Background:
A large body of research has reported associations between depression and elevated interleukin-6 (IL-6), a cytokine with several roles including pro-inflammatory signaling. The nature and directionality of this relationship are not yet clear. In this study we use Mendelian Randomization to examine the possibility of a causal relationship between IL-6 and depressive symptoms, and to explore multiple signaling pathways that could serve as mechanisms for this relationship.
Methods:
This study uses a two-sample Mendelian Randomization design. Data come from the UK Biobank (n=89,119) and published summary statistics from six existing GWAS analyses. The primary analysis focuses on the soluble interleukin-6 receptor (sIL-6R), which is involved in multiple signaling pathways. Exploratory analyses use C-reactive protein (CRP) and soluble glycoprotein 130 (sgp130) to further examine potential underlying mechanisms.
Results:
Results are consistent with a causal effect of sIL-6R on depression (PCA-IVW Odds Ratio: 1.023 (95% Confidence Interval: 1.006–1.039), p=0.006). Exploratory analyses demonstrate that the relationship could be consistent with either decreased classical signaling or increased trans signaling as the underlying mechanism.
Discussion:
These results strengthen the body evidence implicating IL-6 signaling in depression. When compared with existing observational and animal findings, the direction of these results suggests involvement of IL-6 trans signaling. Further study is needed to examine whether IL6R genetic variants might influence IL-6 trans signaling in the brain, as well as to explore other potential pathways linking depression and inflammation.
Keywords: Depression, inflammation, Mendelian Randomization, interleukin-6, soluble interleukin-6 receptor, sIL-6R
1. Introduction
A large body of literature reports that depression is associated with elevated levels of inflammatory biomarkers.1–6 The reasons for this association are not yet fully understood, and the association could operate by way of several different neurobiological pathways. The cytokine interleukin-6 (IL-6) has a widely-replicated association with depressive symptoms, and may represent a plausible biological pathway through which inflammation could contribute to depressive symptoms.4–8
IL-6 is involved in brain signaling related to “sickness behavior”, an adaptive response to illness or injury that leads to behavioral changes such as reduced appetite and decreased activity.3,9,10 IL-6 signaling is also associated with reduced neurogenesis in the hippocampus,11,12 parallelling the reduced hippocampal volumes observed in individuals with depression.13,14 Experimental studies in humans and animals support the possibility of a causal relationship between IL-6 and depressive symptoms. A small human study (n=16) showed that injection with IL-6 produced short-term depression-like alterations in mood.15 In mice exposed to experimental stressors, IL-6 receptor blockade16 and IL-6 knockout mutations17 have been found to reduce development of depression-like behaviors. Similarly, in rats, blocking IL-6 receptors reduced sickness behavior after injection with lipopolysaccharide, an inflammation-provoking agent.10
IL-6 has two major signaling pathways, which have different mechanisms through which they could interact with the brain and influence risk of depression. In classical IL-6 signaling, IL-6 binds to a membrane-bound receptor (mIL-6R), which is found only on certain types of cells (such as liver cells and immune cells).18–20 In trans IL-6 signaling, IL-6 binds to a soluble IL-6 receptor (sIL-6R) in circulation, and the IL-6/sIL-6R complex is then capable of interaction with cells, including those that lack membrane IL-6 receptors.18,21 Much of the interaction between IL-6 and the brain occurs via the trans pathway,22,23 and animal models have confirmed an important role for the trans pathway in neuroinflammation24 and sickness behavior,25 suggesting that IL-6 trans signaling is plausible as the relevant pathway in depression.22 Additionally, animal studies have shown substantial reductions in inflammation-induced sickness behavior when using inhibitors specific to the trans pathway.25,26 However, there are also mechanisms through which classical IL-6 signaling could influence the brain, including activity of microglia (which have a membrane-bound receptor),27 and the effect of classical IL-6 signaling on immunoregulation and other cytokines,20,28 as shown in Figure 1. Identifying the specific pathway involved is of particular interest due to ongoing efforts to develop depression treatments which target IL-6 signaling.
Figure 1:
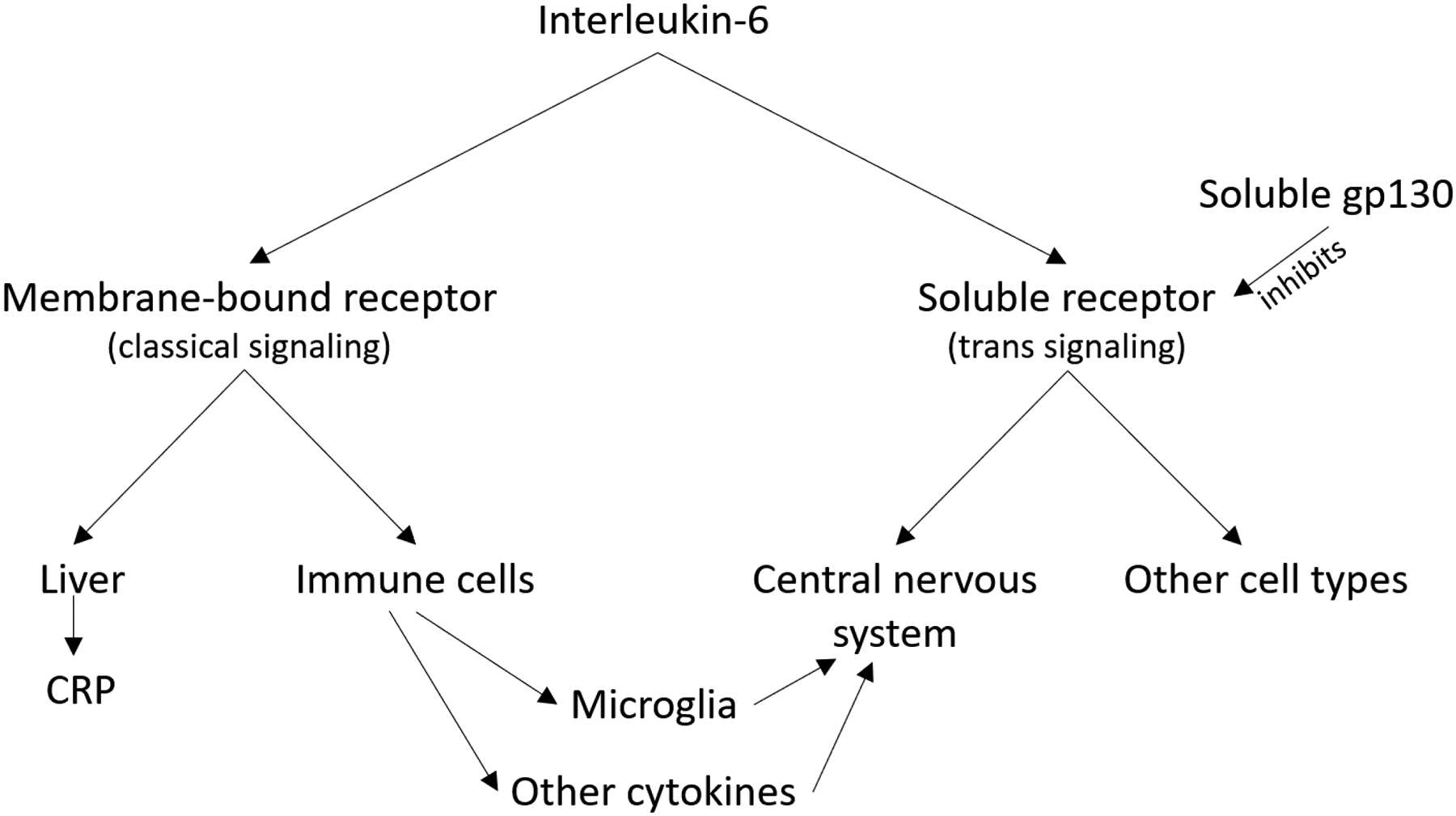
Interleukin-6 signaling pathways
Figure 1 illustrates the two IL-6 signaling pathways. The trans signaling pathway is regarded as the more plausible mechanism for a relationship between IL-6 and depression due to the important role of trans signaling in the central nervous system. However, mechanisms exist through which classical signaling could influence the central nervous system, including the role of classical IL-6 signaling on immune regulation (which may influence other inflammatory signaling chemicals that then interact with the brain) and the presence of membrane IL-6 receptors on some microglia.
Although it is plausible that IL-6 signaling plays a causal role in depression, alternative explanations for the association are also possible. These explanations include reverse causality, in which depression or depression-related health behaviors lead to increases in IL-6 signaling,29 or confounding by factors such as socioeconomic status30 that are associated with both depression and inflammatory signaling. The Mendelian Randomization study design is capable of distinguishing between such competing explanations. In Mendelian Randomization, a genetic variant with a known biological effect is used as an instrumental variable to assess the causal relationship between levels of an exposure or biomarker influenced by the genetic instrumental variable and an outcome of interest, as illustrated in Figure 2.31 Given certain assumptions such as the absence of population genetic stratification, genotype at the selected locus will be randomly distributed throughout the population, creating randomized “exposure groups” similar to those used in clinical trials.32
Figure 2:
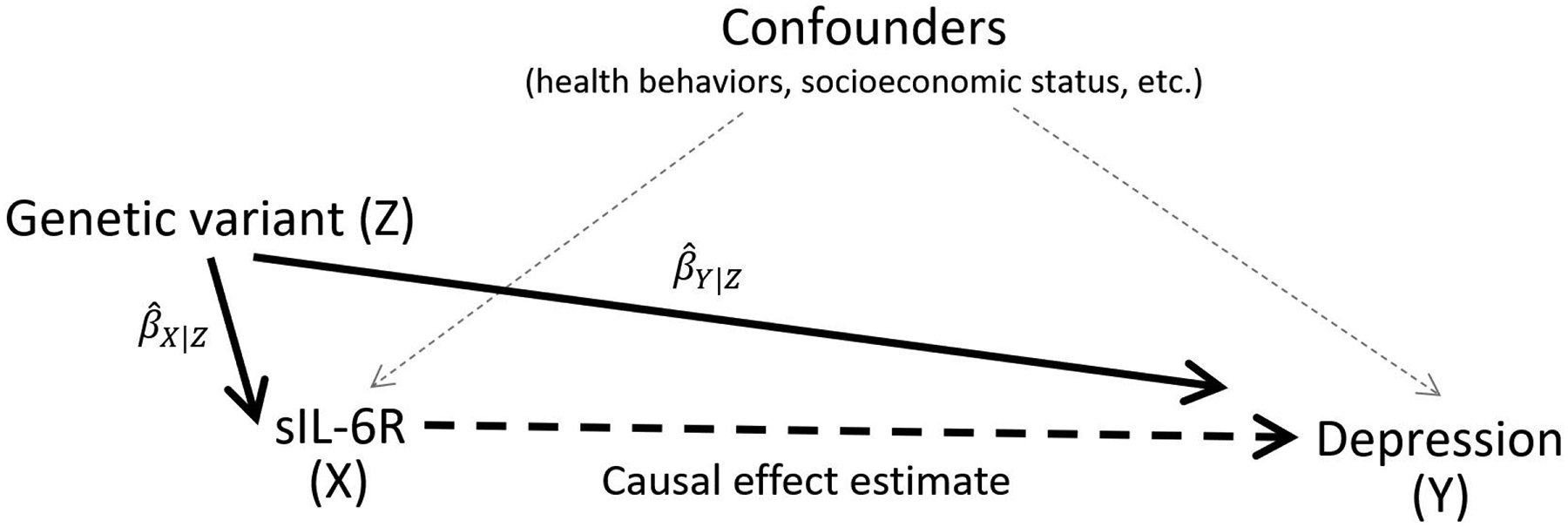
The Mendelian Randomization study design
Figure 2 illustrates the Mendelian Randomization study design. The relationship between a genetic variant and the exposure (βx|z) and the relationship between a genetic variant and the outcome (βxy|z) are measured and used to estimate the causal effect of the exposure on the outcome independent of confounders.
While the Mendelian Randomization design has major strengths as a means of testing causal hypotheses, the complexity of the IL-6 signaling system makes it a difficult target for Mendelian Randomization. Relatively few significant GWAS findings are available for levels of the IL-6 protein itself, and the identified SNPs do not meet the requirements for Mendelian Randomization.33 Although more promising genetic instruments are available for the IL-6 receptor, the IL6R gene encodes both the soluble and membrane-bound forms of the IL-6 receptor, and variants in this gene have the potential to influence both classical and trans signaling. For example, the SNP rs2228145 is a missense variant in a proteolytic cleavage site involved in releasing sIL-6R in its soluble form.34 The minor allele is associated with higher levels of sIL-6R, which results in reduction of IL-6 classical signaling (attributable to lower levels of mIL-6R or to buffering of IL-6 in circulation by excess sIL-6R and sgp130),34,35 and an increase in the levels and half-life of IL-6 itself.36,37 The effects of this variant on trans signaling are not as well understood. Although it increases both IL-6 and sIL-6R, it does not change levels of sgp130. The higher abundance of sgp130 relative to IL-6 and sIL-6R is predicted to prevent these changes from increasing trans signaling,35 although differing concentrations found in particular tissues35 or under certain physiological conditions38 may overcome this inhibition. Additionally, a recent study has suggested that sgp130 may have less than the expected ability to inhibit trans signaling at typical physiologic concentrations.39
Two recent publications have applied Mendelian Randomization using variants in the IL6R gene to examine the relationship between IL-6 signaling and depression. A study by Khandaker et al. (2019) used variants in the IL6R gene region as instrumental variables for interleukin-6 signaling, and found evidence consistent with a causal effect on depression.40 However, this study also noted that variants in the IL6R gene region had opposing effects (alleles that increased IL-6 levels also decreased classical signaling), and stated that further investigation was necessary to fully understand these findings. Another recent Mendelian Randomization used inversed values for SNP coefficients with sIL-6R to reflect its dampening effect on classical IL-6 signaling.41 This analysis reported an association between IL-6 signaling and suicidality, but found no overall association between IL-6 signaling and depression. These studies offer some evidence of a causal relationship, however further study is needed to confirm the relationship between IL-6 signaling and depression, and to determine which of the IL-6 signaling pathways is involved.
In this study we use Mendelian Randomization to test the hypothesis that sIL-6R levels have a causal relationship with depression. In additional exploratory analyses, we assess the robustness of the primary analysis and use Mendelian Randomization of other related proteins (C-reactive protein (CRP) and soluble glycoprotein 130 (sgp130)) to explore which IL-6 signaling pathway might explain the relationship.
2. Materials and Methods
2.1. Study design
This study uses a two-sample Mendelian Randomization design, in which information about the relationship between the genetic variants and the exposure (circulating levels of sIL-6R) is obtained from an existing published genome wide association study (GWAS).42 We then apply the regression coefficients and standard errors for the genotype-exposure variable relationship to the genotype and outcome (depression) data from the second sample with a similar ethnic background.42 This two-sample approach makes it possible to examine a relationship between an exposure and an outcome even when a large sample measuring both traits in the same individuals is not available.43
2.2. Main analysis samples
We obtained coefficients for the genotype/sIL-6R association from two studies, to allow for replication of results across samples. The first study was van Dongen et al 201444, a GWAS of 4,846 Dutch participants that measured sIL-6R using an ELISA assay. We also used the IMPROVE cohort GWAS45 of 3,394 participants from multiple European countries which measured several proteins using an Olink array. Both studies were selected because the samples did not contain British participants and were therefore unlikely to overlap with the UK Biobank sample.
We conducted GWAS to calculate coefficients for the genotype/depression association using data from the UK Biobank.46 Using this data, we defined the phenotype “recurrent depressive symptoms” based on self-reported symptoms. To allow for replication across depression samples, we obtained additional coefficients for the genotype/depression association from the Psychiatric Genomics Consortium meta-analysis of Major Depressive Disorder (PGC MDD 2018).47 The version of the summary statistics used in our analysis does not include data from 23andMe, producing a final sample size of 59,851 cases and 113,514 controls.
The PGC MDD 2018 analysis included individuals from a pilot release of UK Biobank genetic data, so we excluded participants in the pilot release from our UK Biobank analysis. Although the UK Biobank data and PGC MDD 2018 data are never used together as part of the same Mendelian Randomization (a scenario under which sample overlap would create bias), we still chose to exclude potential sample overlap to ensure that replication of results across samples could not be driven by individuals common to both samples. The supplemental note contains additional information about the phenotype definitions and GWAS methods used with the UK Biobank data.
The samples and phenotypes used in the analysis are shown in Table 1, and additional details are provided in Table S1 and Figures S1–S4.
Table 1:
Samples used in the analysis
| Sample | Phenotype | N | Age range | Gender composition | Format |
|---|---|---|---|---|---|
| van Dongen 201444 | sIL-6R blood levels | 4,846 | 18–90 | 61.3% female | GWAS coefficients |
| IMPROVE45 | sIL-6R blood levels | 3,394 | 55–79 | Includes males and females, percent unknown | GWAS coefficients |
| UK Biobank46 | Recurrent depressive symptoms | 89,119 | 40–80 | 53.1% female | Individual-level data |
| PGC MDD 201847 | Major depressive disorder | 173,005 | Adults | Includes males and females, percent unknown | GWAS coefficients |
| INTERVAL58 | C-reactive protein blood levels | 3,301 | Adults | 41% male | GWAS coefficients |
| KORA57 | C-reactive protein blood levels | 997 | 32–81 | Includes males and females, percent unknown | GWAS coefficients |
| Framingham56 | sgp130 blood levels | 5,257 | Adults | 53% female | GWAS coefficients |
2.3. Mendelian Randomization methods
We conducted Mendelian Randomization using several different methods: the Wald ratio of coefficients method,48,49 the two-sample maximum likelihood method,50 GSMR,51 and PCA-IVW.52 These methods differ in several important aspects including statistical power, requirements for instrumental SNP selection, and availability of diagnostic tests to check the Mendelian Randomization requirements, allowing the strengths and weaknesses of the selected methods to complement each-other. Consistency of results across multiple methods helps to confirm the robustness of the results and ensure that they do not result from biases particular to one Mendelian Randomization method.53,54
For the single-SNP analysis using the Wald ratio of coefficients method,48,49 we selected the biallelic SNP rs2228145, which explains approximately 51% of the variance in sIL-6R levels, making it a strong instrumental variable for Mendelian Randomization.44 In datasets where information for rs2228145 was not available, we used the SNPs rs4129267 and rs12126142 as proxies, because they have r2 values greater than 0.99 with rs2228145 in UK Biobank and in the 1000 Genomes EUR population. With the Wald ratio of coefficients method, the causal effect estimate is produced by dividing coefficient for the association between the instrumental SNP and the outcome (βY|Z) by the coefficient for the association between the instrumental SNP and the exposure (βX|Z),49 as shown in Figure 2.
We also used the two-sample maximum likelihood method,50 which combines information from multiple independent SNPs to produce a causal effect estimate. We used multiple methods to select independent SNPs, which are discussed further in the supplemental note. In order to make sure the effects of rs2228145 could be easily examined in visual plots, we ensured selection of this SNP (or its best-available proxy) by excluding other SNPs in close LD with it prior to SNP selection. All analyses were performed in R 3.6.0 using the package TwoSampleMR 0.5.4. We used additional diagnostics to ensure the quality and consistency of the results. These included MR-Egger regression to check for SNPs that had an association with the outcome through a mechanism other than the exposure,55 Cochran’s Q to test for heterogeneity in per-SNP estimates of the odds ratio, and leave-one-SNP-out analyses to confirm that no single SNP produced large changes in the estimated causal effect.
Mendelian Randomization analyses using selected independent SNPs are sensitive to the specific SNPs used in the analyses, particularly when only a small subset of all eligible SNPs can be selected.52 To address this limitation, we used two methods that can account for LD, PCA-IVW52 and GSMR,51 to allow for inclusion of a greater number of SNPs and to improve the statistical power of the analysis. GSMR can account for moderate levels of LD, allowing for a more lenient LD clumping threshold, while PCA-IVW uses principal components and eliminates the need for LD-based SNP selection entirely. The GSMR analysis was performed using GCTA 1.92.2 beta, with r2 clumping thresholds ranging from 0.05 to 0.2 (Table 2). During this analysis, we used the HEIDI-outlier test to exclude any SNPs detected to be outliers or to potentially violate the Mendelian Randomization pleiotropy assumption.51 The PCA-IVW analysis was performed in R 3.6.0 using code from Appendix A of Burgess (2017).52 For the PCA-IVW analysis, we included all SNPs having at least a suggestive association with the exposure (p < 1 * 10−6) and obtained data for the SNP correlation matrix using the UK Biobank sample and GCTA 1.92.2 beta.
Table 2:
Results from sIL-6R Mendelian Randomization analyses
| UK Biobank Sample | PGC MDD 2018 coefficients | ||||||
|---|---|---|---|---|---|---|---|
| Method / SNP selection | Odds ratio (95% CI) | P | # SNPs | Odds ratio (95% CI) | P | # SNPs | |
| van Dongen 2014 coefficients | Ratio of Coefficients (rs12126142) | 1.026 (1.009–1.042) | 0.002 | 1 | 1.014 (1.001–1.027) | 0.033 | 1 |
| Maximum Likelihood | |||||||
| Clumping at r2=0.001 | ** | ** | 2 | ** | ** | 2 | |
| Clumping at r2=0.01 | 1.024 (1.008–1.040) | 0.004 | 4 | 1.015 (1.002–1.027) | 0.024 | 4 | |
| GSMR | |||||||
| Clumping at r2=0.05 | 1.026 (1.010–1.043) | 0.001 | 14 | 1.015 (1.002–1.028) | 0.019 | 16 | |
| Clumping at r2=0.10 | 1.026 (1.010–1.042) | 0.001 | 23 | 1.017 (1.005–1.030) | 0.007 | 23 | |
| Clumping at r2=0.15 | 1.023 (1.007–1.038) | 0.004 | 26 | 1.016 (1.004–1.029) | 0.009 | 26 | |
| Clumping at r2=0.20 | 1.024 (1.008–1.040) | 0.002 | 28 | 1.016 (1.004–1.028) | 0.010 | 29 | |
| PCA-IVW | 1.023 (1.006–1.039) | 0.006 | 491 (4 PCs) | 1.016 (1.003–1.029) | 0.019 | 500 (4 PCs) | |
| PCA-IVW (conditional)* | 1.029 (0.994–1.065) | 0.107 | 275 (2 PCs) | 1.012 (0.985–1.040) | 0.387 | 280 (2 PCs) | |
| IMPROVE coefficients | Ratio of Coefficients (rs2228145) | 1.040 (1.014–1.066) | 0.002 | 1 | 1.021 (1.002–1.041) | 0.032 | 1 |
| Maximum Likelihood | |||||||
| Clumping at r2=0.001 | ** | ** | 2 | ** | ** | 2 | |
| Clumping at r2=0.01 | 1.045 (1.022–1.069) | < 0.001 | 4 | 1.023 (1.004–1.041) | 0.015 | 4 | |
| COJO at p=5e-8 | 1.016 (1.003–1.030) | 0.016 | 7 | 1.012 (1.001–1.023) | 0.026 | 7 | |
| COJO at p=1e-6 | 1.018 (1.005–1.032) | 0.009 | 9 | 1.013 (1.003–1.024) | 0.015 | 9 | |
| COJO at p=0.0001 | 1.013 (1.002–1.024) | 0.017 | 15 | 1.009 (1.000–1.017) | 0.040 | 14 | |
| GSMR | |||||||
| Clumping at r2=0.05 | 1.021 (1.004–1.039) | 0.016 | 11 | 1.008 (0.994–1.022) | 0.274 | 13 | |
| Clumping at r2=0.10 | 1.023 (1.006–1.040) | 0.009 | 15 | 1.008 (0.995–1.022) | 0.239 | 17 | |
| Clumping at r2=0.15 | 1.027 (1.010–1.044) | 0.002 | 22 | 1.009 (0.996–1.022) | 0.197 | 25 | |
| Clumping at r2=0.20 | 1.024 (1.008–1.041) | 0.003 | 25 | 1.012 (0.999–1.025) | 0.074 | 28 | |
| PCA-IVW | 1.023 (1.002–1.045) | 0.029 | 519 (7 PCs) | 1.021 (1.004–1.038) | 0.014 | 533 (7 PCs) | |
This analysis used coefficients from van Dongen 2014 supplementary table 3, a GWAS of sIL-6R conditional on rs2228145 genotype
Not enough SNPs to perform analysis
Table 2 shows the results of Mendelian Randomization analyses using two sIL-6R datasets (van Dongen 2014 and IMPROVE, shown on the left edge) and two outcome datasets (UK Biobank and PGC MDD 2018, shown across the top). The methods column shows both the analysis method (aligned left) and the SNP selection method (aligned right). For the Maximum Likelihood analysis, LD clumping was performed over a distance of 10,000 kilobases, and for the GSMR analysis clumping was performed using a 1 megabase window. For PCA-IVW analyses, the number appearing in parenthesis after the number of SNPs in the number of principle components (PCs) extracted from the SNP data to explain 99% of the variance.
2.4. Exploring potential mechanisms underlying IL-6 signaling and depression
The strongest SNP in the main analysis, rs2228145, has two potential effects on IL-6 signaling: the minor allele reduces signaling via the classical pathway, and under some circumstances might also be able to increase signaling via the trans pathway due to increasing levels of IL-6 and sIL-6R (Figure 1). There are two mechanisms through which rs2228145 could reduce classical signaling: buffering of IL-6 by increased availability of sIL-6R and reduced availability of mIL-6R due to increased shedding as sIL-6R.34,35 Other genetic variants that increase sIL-6R levels may have differing effects on classical signaling - while they could still contribute to buffering effects from sIL-6R, their mechanisms of action may or may not involve shedding and decreased availability of mIL-6R. Unfortunately, the extremely strong effects of rs2228145 allow it to act as a confounder for other nearby SNPs, even with relatively low levels of linkage disequilibrium, which can lead to difficulty examining effects of other IL6R SNPs independent from the effects of rs2228145.
2.5. Exploratory analysis: Samples
The exploratory analyses used samples from the main analysis and three additional studies. We used results from the Framingham Heart Study56 for sgp130, and results from the KORA study57 for CRP. Although both studies had results for both proteins available, neither had a sufficient number of eligible significant SNPs for the other protein to use for replication Mendelian Randomizations. For replication of both proteins, we selected the INTERVAL study.58 Because the INTERVAL study was based in the UK, there is potential for overlap with UK Biobank, which has the potential to bias Mendelian Randomization analysis.59 However any overlap (if present) is likely to be small, and any bias that resulted would not also occur in the analyses using the Framingham and KORA data.
2.6. Exploratory analysis: Approach
First, we conducted an analysis using CRP as a proxy for classical IL-6 signaling (i.e. signaling via the membrane receptor, which stimulates CRP production).35 We selected CRP as a proxy for classical IL-6 signaling because classical IL-6 signaling is the mechanism through which IL6R SNPs are likely to influence CRP levels.35 A similar approach has been used in other Mendelian Randomization studies, which confirmed that IL6R variants produce results which differ from those of CRP variants in the expected direction.41
Second, in addition to using r2 to assess LD between each SNP and rs2228145, we also examined Lewontin’s |D’| statistic60 because |D’| is not as severely affected by differences in allele frequency and may detect LD in some cases where r2 does not (illustrated in Figure S5).61 We then attempted to exclude the effects of rs2228145 by conducting additional Mendelian Randomization analyses using only SNPs having both r2 ≤ 0.01 and |D’| ≤ 0.15 with rs2228145. We checked whether these SNPs still showed signs of affecting classical signaling by using them to perform Mendelian Randomization for CRP.
Third, we conducted a Mendelian Randomization analysis examining soluble glycoprotein 130 (sgp130), a protein which inhibits only trans IL-6 signaling.18 If the trans signaling pathway were the mechanism for the causal relationship, higher levels of sgp130 might have a protective effect against depression. The gene that encodes sgp130 (IL6ST) also encodes a membrane-bound form of gp130 that is used in both IL-6 signaling pathways, so SNPs in IL6ST have the potential to influence both pathways. To examine this possibility, we created a scatter plot to check whether per-SNP associations with sgp130 and with depression differed for SNPs from non-IL6ST genes.
All exploratory analyses used the PCA-IVW method so that all eligible SNPs could be included.
2.7. Ethical approval
This analysis used only de-identified data (UK Biobank) and summary statistics (all other samples) and was therefore exempt from human subjects regulation.
3. Results
Table 1 provides details of the studies and samples used in the analyses. All study participants were adults of European ancestry and all studies included both males and females. All eligible significant SNPs for sIL-6R were located on chromosome 1.
In the main analysis (Table 2), across all combinations of samples and methods, the majority of associations were significant, indicating that higher levels of sIL-6R were associated with increased odds of depression. For example, using the PCA-IVW method with the van Dongen and UK Biobank samples, a 10−8 g/mL increase in sIL-6R was associated with 1.023 times higher odds of depression (95% Confidence Interval: 1.006 – 1.039, p=0.006). Furthermore, even analyses which did not reach significance produced odds ratios greater than 1.0 (consistent in direction with the significant results). The consistency of the findings across the various combinations of exposure and outcome samples and across analytic methods indicates that the results are robust to differences in samples and analytic methods. We then repeated the main analysis using the “recurrent DSM-V major depression” phenotype, which is a more stringent definition but produces a smaller sample size because it can only be evaluated in participants who completed the UK Biobank Online Mental Health supplement. Despite this smaller sample, most analyses still produced significant or near-significant results (Table S4), and the direction of all odds ratios remained consistent and greater than 1.0.
We obtained similar results when using SNP coefficients from the van Dongen (2014) conditional analysis that estimated SNP coefficients for sIL-6R while adjusting for rs2228145 (shown in Table 2 as “PCA-IVW (conditional)”). Although the effect estimates from the analyses using the conditional GWAS coefficients were not statistically significant, the direction and magnitude of the estimated ORs was consistent with the other results in Table 2. However, conditional GWAS coefficients may not fully account for LD with a SNP that has strong effects62,63 and Figure S10 shows patterns consistent with residual confounding by rs2228145.
We conducted a series of analyses to explore whether the identified causal relationship between IL-6 signaling and depression occurs via the classical or the trans signaling pathway. First, we performed Mendelian Randomization for the effects of IL6R variants on CRP, which demonstrated that increased sIL-6R is associated with decreased CRP (Table S6). Next, we used these same variants to perform Mendelian Randomization for the effects of classical IL-6 signaling on depression, using CRP as a proxy for classical signaling. The results show that when using IL6R variants, higher CRP is associated with lower odds of depression (Table S7). Previous studies have shown that this finding is specific to IL6R variants and does not replicate when using variants that affect CRP via mechanisms other than classical IL-6 signaling.41 These results demonstrate that reduced classical IL-6 signaling is one potential mechanism for the causal relationship sIL-6R and depression.
Table 3 extends the findings from Table 2 by repeating the Mendelian Randomization of sIL-6R with additional filtering to exclude the effects of rs2228145. Although the genetic instruments used in this exploratory analysis were not as strong as rs2228145, in several cases these analyses still suggested a relationship between sIL-6R and depression. In all filtered analyses producing significant or near-significant p-values, the effect estimates were above 1.0, illustrating that the relationship between sIL-6R and depression does not reverse direction when excluding the effect of rs2228145. We then used these same sets of filtered SNPs to perform Mendelian Randomization for the effects of sIL-6R on CRP (Table S6). The filtered variants no longer showed a significant causal effect on CRP, although it is not clear whether this results from a true lack of association or a reduction in statistical power from excluding the SNPs with the strongest effects.
Table 3:
Results from PCA-IVW analyses for sIL-6R and depression using SNPs filtered to exclude LD with rs2228145 (r2 ≤ 0.01 and |D’| ≤ 0.15)
| UK Biobank sample | PGC MDD 2018 coefficients | |||||
|---|---|---|---|---|---|---|
| Exposure sample | Odds ratio (95% CI) | P | # SNPs | Odds ratio (95% CI) | P | # SNPs |
| van Dongen 2014 | 0.991 (0.898–1.093) | 0.849 | 13 (2 PCs) | 1.109 (1.023–1.202) | 0.012 | 13 (2 PCs) |
| van Dongen 2014 (conditional)* | 0.991 (0.852–1.151) | 0.902 | 72 (2 PCs) | 1.157 (1.022–1.311) | 0.021 | 77 (3 PCs) |
| IMPROVE | 1.049 (0.991–1.110) | 0.099 | 89 (4 PCs) | 1.046 (0.999–1.095) | 0.056 | 91 (5 PCs) |
r2 refers to squared correlation between each SNP and rs2228145, |D’| refers to Lewontin’s d-prime statistic calculated between each SNP and rs2228145
This analysis used coefficients from van Dongen 2014 supplementary table 3, a GWAS of sIL-6R conditional on rs2228145 genotype.
The Mendelian Randomizations for sgp130 (Table S5) did not produce any statistically significant results, either when using variants only from the IL6ST gene or when including other variants associated with sgp130. Scatter plots (Figures S12 and S13) showed that the lack of correlation between SNP coefficients for sgp130 and for depression was not specific to the IL6ST gene, which suggests that the null finding is not attributable to opposing pleiotropic effects specific to IL6ST variants. The null result may indicate that the inhibitory effects of sgp130 on IL-6 trans signaling are not protective against depression. It is also possible that the null result occurred because the SNPs used in this analysis were not sufficiently strong instrumental variables. A recent paper has also suggested that rapid formation and dissociation of the IL-6/sIL-6R complex may limit sgp130’s ability to inhibit trans signaling at typical concentrations,39 which could mean that an sgp130 Mendelian Randomization may not reflect the effects of inhibiting the trans pathway.
4. Discussion
The results of the primary analysis are consistent with a causal effect of increased sIL-6R on risk of depression. This study used multiple Mendelian Randomization approaches to estimate this relationship, and the effect estimates were largely consistent regardless of the specific analytic approach used. These results build on existing cross-sectional4–7 and longitudinal8 studies suggesting a role for IL-6 signaling in depression. The results also strengthen evidence regarding the causal nature of the relationship between inflammation and depression by examining it in a manner that establishes both directionality and independence from environmental confounders.
While observational studies of inflammatory biomarkers and depression generally produce moderate effect estimates, the effect sizes estimated in this study were more modest. This discrepancy may reflect multiple factors. Observational studies often examine IL-6 itself, which may have larger and more direct effects than its receptor. In addition, depression is an etiologically heterogeneous condition, and inflammation may play a causal role in only a subset of cases.64,65 Without a means of separating out those depression cases with an inflammation-related etiology, this heterogeneity could result in under-estimation of effect sizes for IL6R variants in genetic studies of depression.66
The results of this study suggest that one or more of the effects of increased sIL-6R has a causal relationship with depression. The most well-known effect of increased sIL-6R is a reduction of signaling via the classical pathway (as shown by the decrease in production of CRP). The direction of this effect is not consistent with the results of human and animal studies which suggest that depression is associated with higher, not lower, IL-6 levels and signaling activity. This inconsistency makes classical signaling less convincing as a causal mechanism, and suggests that other potential explanations should be examined.
The second potential mechanism is an increase in trans signaling. Although it has been predicted that increased availability of the soluble receptor would not increase trans signaling, the effects may vary across tissues and locations.35 Typical concentrations of sIL-6R and sgp130 in cerebrospinal fluid (CSF) are considerably lower than those found in serum,67 and IL6R genetic variants affect sIL-6R in CSF as well as serum.68,69 When levels of sIL-6R and sgp130 are low, changes in sIL-6R might have the potential to influence trans signaling. rs2228145 explains 40% of the variance in sIL-6R in human CSF,68,69 and animal studies have shown that spinal injection of sIL-6R can affect pain sensitivity,70 which could support the idea that central nervous system concentrations of sIL-6R are low enough for receptor availability to influence trans signaling.71 In addition, both observational72,73 and experimental74 studies have reported that depression is associated with increased IL-6 in CSF. This increased availability of IL-6 may allow sIL-6R availability to influence trans signaling, a phenomenon already observed in other scenarios involving elevated IL-6.75,76 Although mechanisms involving the trans pathway offer an interesting explanation for our results, further study is needed to examine whether IL6R variants influence trans signaling in the brain and what conditions would be needed for such an influence to occur.
The complex biology of IL-6 signaling also introduces other possible explanations. Under certain circumstances the soluble IL-6 receptor may also act as a receptor for Ciliary Neurotrophic Factor,77 although this mechanism would not explain the observational associations between IL-6 and depression. Additionally, a recently-discovered third type of IL-6 signaling known as trans-presentation is involved in Th17 cell differentiation in mice,78 however little is currently known regarding this form of signaling in humans79 or how IL6R genetic variants might affect it. Finally, even if a causal effect could be isolated to one specific IL-6 signaling pathway, this would not necessarily indicate a direct causal effect, because IL-6 signaling also impacts other biological pathways which may mediate its relationship with depression. Thus we consider it most appropriate to describe our result as supporting “a causal effect of increased sIL-6R on depression” and to regard the specific underlying pathway as an area still requiring further study.
As discussed in Lawlor 2016,53 it is often helpful to use multiple study designs to “triangulate” an answer to complex causal questions. Animal studies have shown associations between IL-6 signaling and depression-like “sickness behaviors” as well as demonstrating that inhibition of trans signaling can reduce or prevent these symptoms.25,26 Meanwhile, numerous observational studies in humans have reported associations between elevated IL-6 and depression,4–6 and secondary analyses of clinical trials suggest that drugs which inhibit IL-6 signaling might have anti-depressant effects.80,81 However, animal studies are limited by the use of animal behaviors as models for human depression, and observational studies and secondary analyses of trials are not suitable for causal inference. Thus despite its inability to identify the specific underlying mechanism, Mendelian Randomization of sIL-6R contributes an important piece to the puzzle by offering evidence to support a causal effect of sIL-6R on depression in humans.
4.1. Strengths and limitations
The primary limitation of this study is the presence of multiple potential causal mechanisms that could exist between sIL-6R and depression. Although this limitation prevents us from definitively identifying one specific mechanism as causal, results can still be interpreted as supporting a causal relationship between sIL-6R and depression without specifying a specific pathway. Additionally, sample overlap is likely between the van Dongen (2014) coefficients and the PGC MDD 2018 coefficients because both studies include individuals drawn from the Netherlands Twin Register (NTR) cohort. Such overlap may lead to bias,59 however, we used multiple combinations of exposure and outcome samples to ensure that results were robust to overlap occurring between any specific pair of samples. Another limitation of this study is that the samples use similar, but not identical, phenotype definitions of depression. Our sIL-6R results replicated successfully in a supplementary analysis using a subset of UK Biobank participants who had data allowing for a phenotype definition that more closely resembled the clinical definitions used in PGC MDD 2018, but we did not use this subset as our main analytic sample due to the smaller sample size and resulting loss of statistical power. Finally, while of scientific interest, we did not attempt to use Mendelian Randomization to assess the potential causal effect of depression on IL-6 signaling for several reasons. Most salient is that unlike the direct effect of the IL6R variants on IL-6 receptors, variants associated with depression may have effects on the brain which simultaneously influence multiple psychiatric and behavioral phenotypes.82 As a result, the Mendelian Randomization requirement that “the effect of the genetic instrument on the outcome must be mediated exclusively by the exposure in question”33 precludes the examination of depression as an exposure until the biological mechanisms underlying genetic influences on depression are more fully understood.
This study also has several strengths, including the use of coefficients from large well-characterized samples (e.g., UK Biobank and PGC MDD 2018), analysis of multiple proteins and measures related to IL-6 signaling pathways, and consistency of results across multiple Mendelian Randomization approaches and samples. These findings help clarify the role of inflammation in the development of depression and suggest several avenues for future research that can inform efforts to both prevent and treat depression.
4.2. Conclusions
The findings from this study are consistent with a causal effect of increased sIL-6R on depression. Although we were not able to definitively isolate which of the IL-6 signaling pathways is the mechanism for the causal effect, examination of our results in combination with those produced by other studies provides some evidence to support the trans signaling pathway as a potential mechanism. These results should encourage further study of the effects of IL-6 signaling on depression, as well as encouraging exploration of drugs which modify IL-6 signaling as potential antidepressants.
Future research should examine the effects of rs2228145 and other IL6R variants on trans signaling in the human brain, and whether increasing sIL-6R levels in the brain can lead to increased susceptibility in animal models of sickness behavior or stress. Finally, the heterogeneous nature of depression65 introduces several additional questions, including examination of possible gender-specific effects, identification of a subset of depression cases for whom IL-6 signaling might play a causal role, and assessment of whether the association may be stronger for depressive symptoms that more closely resemble the ones found in sickness behavior.
Supplementary Material
Acknowledgements
KM Kelly was supported by the National Human Genome Research Institute (National Institutes of Health) Training Program in Genomic Science at the University of Michigan (T32-HG00040).
B Mezuk was supported by National Institute of Mental Health (National Institutes of Health) K01-MH093642-A1.
This research has been conducted using the UK Biobank Resource under Application Number 41812.
We would also like to thank the authors and cohorts from the following GWAS studies for generously making their GWAS coefficients available:
Folkersen et al 2017 (DOI: 10.1371/journal.pgen.1006706) (IMPROVE Consortium)
Sun et al 2018 (DOI: 10.1038/s41586-018-0175-2) (INTERVAL Study)
Suhre et al 2017 (DOI: 10.1038/ncomms14357) (KORA cohort)
van Dongen et al 2014 (DOI: 10.1007/s10519-014-9656-8) (Netherlands Twin Register)
Wray et al 2018 (DOI: 10.1038/s41588-018-0090-3) (Psychiatric Genomics Consortium)
Yao et al 2018 (DOI: 10.1038/s41467-018-05512-x) (Framingham Heart Study)
An earlier version of this analysis appeared in the poster Depression and the soluble interleukin-6 receptor: A Mendelian Randomization. The poster was presented at the 2019 World Congress of Psychiatric Genetics October 26–31 in Anaheim, California.
An earlier version of this analysis was included in KM Kelly’s dissertation, which was submitted to the University of Michigan on August 17, 2020.
KM Kelly would like to thank Drs. Pat Peyser, Michael Boehnke, and Laura Scott for their contributions to the development and refinement of this analysis as members of the dissertation committee, and to thank Drs. Minjung Kho and Wei Zhao for their assistance and guidance in working with tools and datasets used in the analysis.
Footnotes
Disclosures
KM Kelly, JA Smith and B Mezuk have no conflicts of interest to declare.
References
- 1.Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends in Immunology. 2006;27(1):24–31. doi: 10.1016/j.it.2005.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller AH, Maletic V, Raison CL. Inflammation and Its Discontents: The Role of Cytokines in the Pathophysiology of Major Depression. Biological Psychiatry. 2009;65(9):732–741. doi: 10.1016/j.biopsych.2008.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46–56. doi: 10.1038/nrn2297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ng A, Tam WW, Zhang MW, et al. IL-1β, IL-6, TNF- α and CRP in Elderly Patients with Depression or Alzheimer’s disease: Systematic Review and Meta-Analysis. Scientific Reports. 2018;8(1):12050. doi: 10.1038/s41598-018-30487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Köhler CA, Freitas TH, Maes M, et al. Peripheral cytokine and chemokine alterations in depression: a meta-analysis of 82 studies. Acta Psychiatrica Scandinavica. 2017;135(5):373–387. doi: 10.1111/acps.12698 [DOI] [PubMed] [Google Scholar]
- 6.Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Molecular Psychiatry. 2016;21(12):1696–1709. doi: 10.1038/mp.2016.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimäki M. Cumulative meta-analysis of interleukins 6 and 1β, tumour necrosis factor α and C-reactive protein in patients with major depressive disorder. Brain Behav Immun. 2015;49:206–215. doi: 10.1016/j.bbi.2015.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith KJ, Au B, Ollis L, Schmitz N. The association between C-reactive protein, Interleukin-6 and depression among older adults in the community: A systematic review and meta-analysis. Experimental Gerontology. 2018;102:109–132. doi: 10.1016/j.exger.2017.12.005 [DOI] [PubMed] [Google Scholar]
- 9.Dantzer R. Cytokine, Sickness Behavior, and Depression. Immunology and Allergy Clinics of North America. 2009;29(2):247–264. doi: 10.1016/j.iac.2009.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harden LM, du Plessis I, Poole S, Laburn HP. Interleukin-6 and leptin mediate lipopolysaccharide-induced fever and sickness behavior. Physiology & Behavior. 2006;89(2):146–155. doi: 10.1016/j.physbeh.2006.05.016 [DOI] [PubMed] [Google Scholar]
- 11.Monje ML, Toda H, Palmer TD. Inflammatory Blockade Restores Adult Hippocampal Neurogenesis. Science. 2003;302(5651):1760–1765. doi: 10.1126/science.1088417 [DOI] [PubMed] [Google Scholar]
- 12.Vallières L, Campbell IL, Gage FH, Sawchenko PE. Reduced Hippocampal Neurogenesis in Adult Transgenic Mice with Chronic Astrocytic Production of Interleukin-6. J Neurosci. 2002;22(2):486–492. doi: 10.1523/JNEUROSCI.22-02-00486.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Malykhin NV, Coupland NJ. Hippocampal neuroplasticity in major depressive disorder. Neuroscience. 2015;309:200–213. doi: 10.1016/j.neuroscience.2015.04.047 [DOI] [PubMed] [Google Scholar]
- 14.Santarelli L, Saxe M, Gross C, et al. Requirement of Hippocampal Neurogenesis for the Behavioral Effects of Antidepressants. Science. 2003;301(5634):805–809. doi: 10.1126/science.1083328 [DOI] [PubMed] [Google Scholar]
- 15.Späth-Schwalbe E, Hansen K, Schmidt F, et al. Acute Effects of Recombinant Human Interleukin-6 on Endocrine and Central Nervous Sleep Functions in Healthy Men. J Clin Endocrinol Metab. 1998;83(5):1573–1579. doi: 10.1210/jcem.83.5.4795 [DOI] [PubMed] [Google Scholar]
- 16.Zhang J c, Yao W, Dong C, et al. Blockade of interleukin-6 receptor in the periphery promotes rapid and sustained antidepressant actions: a possible role of gut–microbiota–brain axis. Transl Psychiatry. 2017;7(5):e1138–e1138. doi: 10.1038/tp.2017.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chourbaji S, Urani A, Inta I, et al. IL-6 knockout mice exhibit resistance to stress-induced development of depression-like behaviors. Neurobiology of Disease. 2006;23(3):587–594. doi: 10.1016/j.nbd.2006.05.001 [DOI] [PubMed] [Google Scholar]
- 18.Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: A highly regulated and dynamic system. Cytokine. 2014;70(1):11–20. doi: 10.1016/j.cyto.2014.05.024 [DOI] [PubMed] [Google Scholar]
- 19.Schaper F, Rose-John S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine & Growth Factor Reviews. 2015;26(5):475–487. doi: 10.1016/j.cytogfr.2015.07.004 [DOI] [PubMed] [Google Scholar]
- 20.Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2011;1813(5):878–888. doi: 10.1016/j.bbamcr.2011.01.034 [DOI] [PubMed] [Google Scholar]
- 21.Scheller J, Rose-John S. Interleukin-6 and its receptor: from bench to bedside. Med Microbiol Immunol. 2006;195(4):173–183. doi: 10.1007/s00430-006-0019-9 [DOI] [PubMed] [Google Scholar]
- 22.Maes M, Anderson G, Kubera M, Berk M. Targeting classical IL-6 signalling or IL-6 trans-signalling in depression? Expert Opinion on Therapeutic Targets. 2014;18(5):495–512. doi: 10.1517/14728222.2014.888417 [DOI] [PubMed] [Google Scholar]
- 23.März P, Otten U, Rose‐John S. Neural activities of IL-6-type cytokines often depend on soluble cytokine receptors. European Journal of Neuroscience. 1999;11(9). doi: 10.1046/j.1460-9568.1999.00755.x [DOI] [PubMed] [Google Scholar]
- 24.Campbell IL, Erta M, Lim SL, et al. Trans-Signaling Is a Dominant Mechanism for the Pathogenic Actions of Interleukin-6 in the Brain. Journal of Neuroscience. 2014;34(7):2503–2513. doi: 10.1523/JNEUROSCI.2830-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burton MD, Sparkman NL, Johnson RW. Inhibition of interleukin-6 trans-signaling in the brain facilitates recovery from lipopolysaccharide-induced sickness behavior. J Neuroinflammation. 2011;8(1):54. doi: 10.1186/1742-2094-8-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burton MD, Rytych JL, Freund GG, Johnson RW. Central inhibition of interleukin-6 trans-signaling during peripheral infection reduced neuroinflammation and sickness in aged mice. Brain, Behavior, and Immunity. 2013;30:66–72. doi: 10.1016/j.bbi.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2016;1863(6, Part A):1218–1227. doi: 10.1016/j.bbamcr.2016.03.018 [DOI] [PubMed] [Google Scholar]
- 28.Hoge J, Yan I, Jänner N, et al. IL-6 Controls the Innate Immune Response against Listeria monocytogenes via Classical IL-6 Signaling. The Journal of Immunology. 2013;190(2):703–711. doi: 10.4049/jimmunol.1201044 [DOI] [PubMed] [Google Scholar]
- 29.Duivis HE, de Jonge P, Penninx BW, Na BY, Cohen BE, Whooley MA. Depressive Symptoms, Health Behaviors, and Subsequent Inflammation in Patients With Coronary Heart Disease: Prospective Findings From the Heart and Soul Study. AJP. 2011;168(9):913–920. doi: 10.1176/appi.ajp.2011.10081163 [DOI] [PubMed] [Google Scholar]
- 30.Gruenewald TL, Cohen S, Matthews KA, Tracy R, Seeman TE. Association of socioeconomic status with inflammation markers in black and white men and women in the Coronary Artery Risk Development in Young Adults (CARDIA) study. Social Science & Medicine. 2009;69(3):451–459. doi: 10.1016/j.socscimed.2009.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–R98. doi: 10.1093/hmg/ddu328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davey Smith G Randomised by (your) god: robust inference from an observational study design. J Epidemiol Community Health. 2006;60(5):382–388. doi: 10.1136/jech.2004.031880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haycock PC, Burgess S, Wade KH, Bowden J, Relton C, Davey Smith G. Best (but oft-forgotten) practices: the design, analysis, and interpretation of Mendelian randomization studies. Am J Clin Nutr. 2016;103(4):965–978. doi: 10.3945/ajcn.115.118216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garbers C, Monhasery N, Aparicio-Siegmund S, et al. The interleukin-6 receptor Asp358Ala single nucleotide polymorphism rs2228145 confers increased proteolytic conversion rates by ADAM proteases. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2014;1842(9):1485–1494. doi: 10.1016/j.bbadis.2014.05.018 [DOI] [PubMed] [Google Scholar]
- 35.Ferreira RC, Freitag DF, Cutler AJ, et al. Functional IL6R 358Ala Allele Impairs Classical IL-6 Receptor Signaling and Influences Risk of Diverse Inflammatory Diseases. PLOS Genetics. 2013;9(4):e1003444. doi: 10.1371/journal.pgen.1003444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The Interleukin-6 Receptor Mendelian Randomisation Analysis (IL6R MR) Consortium. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. The Lancet. 2012;379(9822):1214–1224. doi: 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peters M, Jacobs S, Ehlers M, et al. The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. Journal of Experimental Medicine. 1996;183(4):1399–1406. doi: 10.1084/jem.183.4.1399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose‐John S The Soluble Interleukin 6 Receptor: Advanced Therapeutic Options in Inflammation. Clinical Pharmacology & Therapeutics. 2017;102(4):591–598. doi: 10.1002/cpt.782 [DOI] [PubMed] [Google Scholar]
- 39.Baran P, Hansen S, Waetzig GH, et al. The balance of interleukin (IL)-6, IL-6·soluble IL-6 receptor (sIL-6R), and IL-6·sIL-6R·sgp130 complexes allows simultaneous classic and trans-signaling. J Biol Chem. 2018;293(18):6762–6775. doi: 10.1074/jbc.RA117.001163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khandaker GM, Zuber V, Rees JMB, et al. Shared mechanisms between coronary heart disease and depression: findings from a large UK general population-based cohort. Mol Psychiatry. Published online March 19, 2019:1–10. doi: 10.1038/s41380-019-0395-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kappelmann N, Arloth J, Georgakis MK, et al. Dissecting the Association Between Inflammation, Metabolic Dysregulation, and Specific Depressive Symptoms. JAMA Psychiatry. Published online October 20, 2020. doi: 10.1001/jamapsychiatry.2020.3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pierce BL, Burgess S. Efficient Design for Mendelian Randomization Studies: Subsample and 2-Sample Instrumental Variable Estimators. Am J Epidemiol. 2013;178(7):1177–1184. doi: 10.1093/aje/kwt084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lawlor DA. Commentary: Two-sample Mendelian randomization: opportunities and challenges. Int J Epidemiol. 2016;45(3):908–915. doi: 10.1093/ije/dyw127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Dongen J, Jansen R, Smit D, et al. The Contribution of the Functional IL6R Polymorphism rs2228145, eQTLs and Other Genome-Wide SNPs to the Heritability of Plasma sIL-6R Levels. Behav Genet. 2014;44(4):368–382. doi: 10.1007/s10519-014-9656-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Folkersen L, Fauman E, Sabater-Lleal M, et al. Mapping of 79 loci for 83 plasma protein biomarkers in cardiovascular disease. PLOS Genetics. 2017;13(4):e1006706. doi: 10.1371/journal.pgen.1006706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wray NR, Ripke S, Mattheisen M, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nature Genetics. 2018;50(5):668–681. doi: 10.1038/s41588-018-0090-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawlor DA, Harbord RM, Sterne JAC, Timpson N, Davey Smith G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Statist Med. 2008;27(8):1133–1163. doi: 10.1002/sim.3034 [DOI] [PubMed] [Google Scholar]
- 49.Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Statistical Methods in Medical Research. Published online August 17, 2015:0962280215597579. doi: 10.1177/0962280215597579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. 2015;30(7):543–552. doi: 10.1007/s10654-015-0011-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu Z, Zheng Z, Zhang F, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun. 2018;9(1):1–12. doi: 10.1038/s41467-017-02317-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burgess S, Zuber V, Valdes-Marquez E, Sun BB, Hopewell JC. Mendelian randomization with fine-mapped genetic data: Choosing from large numbers of correlated instrumental variables. Genet Epidemiol. 2017;41(8):714–725. doi: 10.1002/gepi.22077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lawlor DA, Tilling K, Davey Smith G. Triangulation in aetiological epidemiology. Int J Epidemiol. 2016;45(6):1866–1886. doi: 10.1093/ije/dyw314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walker VM, Davies NM, Hemani G, et al. Using the MR-Base platform to investigate risk factors and drug targets for thousands of phenotypes. Wellcome Open Res. 2019;4. doi: 10.12688/wellcomeopenres.15334.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44(2):512–525. doi: 10.1093/ije/dyv080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao C, Chen G, Song C, et al. Genome‐wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat Commun. 2018;9(1):1–11. doi: 10.1038/s41467-018-05512-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Suhre K, Arnold M, Bhagwat AM, et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat Commun. 2017;8:14357–14357. doi: 10.1038/ncomms14357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558(7708):73–79. doi: 10.1038/s41586-018-0175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burgess S, Davies NM, Thompson SG. Bias due to participant overlap in two-sample Mendelian randomization. Genetic Epidemiology. 2016;40(7):597–608. doi: 10.1002/gepi.21998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lewontin RC. On measures of gametic disequilibrium. Genetics. 1988;120(3):849–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.VanLiere JM, Rosenberg NA. Mathematical properties of the r2 measure of linkage disequilibrium. Theor Popul Biol. 2008;74(1):130–137. doi: 10.1016/j.tpb.2008.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zeng B, Lloyd-Jones LR, Holloway A, et al. Constraints on eQTL Fine Mapping in the Presence of Multisite Local Regulation of Gene Expression. G3: Genes, Genomes, Genetics. 2017;7(8):2533–2544. doi: 10.1534/g3.117.043752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Becher H The concept of residual confounding in regression models and some applications. Statistics in Medicine. 1992;11(13):1747–1758. doi: 10.1002/sim.4780111308 [DOI] [PubMed] [Google Scholar]
- 64.Raison CL, Miller AH. Do Cytokines Really Sing the Blues? Cerebrum: the Dana Forum on Brain Science. 2013;2013. Accessed August 17, 2016. https://www-ncbi-nlm-nih-gov.proxy.library.vcu.edu/pmc/articles/PMC3788165/ [PMC free article] [PubMed] [Google Scholar]
- 65.Cai N, Choi KW, Fried EI. Reviewing the genetics of heterogeneity in depression: operationalizations, manifestations and etiologies. Hum Mol Genet. 2020;29(R1):R10–R18. doi: 10.1093/hmg/ddaa115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manchia M, Cullis J, Turecki G, Rouleau GA, Uher R, Alda M. The Impact of Phenotypic and Genetic Heterogeneity on Results of Genome Wide Association Studies of Complex Diseases. PLOS ONE. 2013;8(10):e76295. doi: 10.1371/journal.pone.0076295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lehtimäki KA, Keränen T, Huhtala H, et al. Regulation of IL-6 system in cerebrospinal fluid and serum compartments by seizures: the effect of seizure type and duration. Journal of Neuroimmunology. 2004;152(1):121–125. doi: 10.1016/j.jneuroim.2004.01.024 [DOI] [PubMed] [Google Scholar]
- 68.Sasayama D, Hattori K, Ogawa S, et al. Genome-wide quantitative trait loci mapping of the human cerebrospinal fluid proteome. Hum Mol Genet. 2017;26(1):44–51. doi: 10.1093/hmg/ddw366 [DOI] [PubMed] [Google Scholar]
- 69.Kauwe JSK, Bailey MH, Ridge PG, et al. Genome-wide association study of CSF levels of 59 alzheimer’s disease candidate proteins: significant associations with proteins involved in amyloid processing and inflammation. PLoS Genet. 2014;10(10):e1004758. doi: 10.1371/journal.pgen.1004758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kawasaki Y, Zhang L, Cheng JK, Ji RR. Cytokine Mechanisms of Central Sensitization: Distinct and Overlapping Role of Interleukin-1β, Interleukin-6, and Tumor Necrosis Factor-α in Regulating Synaptic and Neuronal Activity in the Superficial Spinal Cord. J Neurosci. 2008;28(20):5189–5194. doi: 10.1523/JNEUROSCI.3338-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Konig C, Morch E, Eitner A, et al. Involvement of Spinal IL-6 Trans-Signaling in the Induction of Hyperexcitability of Deep Dorsal Horn Neurons by Spinal Tumor Necrosis Factor-Alpha. Journal of Neuroscience. 2016;36(38):9782–9791. doi: 10.1523/JNEUROSCI.4159-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sasayama D, Hattori K, Wakabayashi C, et al. Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. Journal of Psychiatric Research. 2013;47(3):401–406. doi: 10.1016/j.jpsychires.2012.12.001 [DOI] [PubMed] [Google Scholar]
- 73.Kern S, Skoog I, Börjesson-Hanson A, et al. Higher CSF interleukin-6 and CSF interleukin-8 in current depression in older women. Results from a population-based sample. Brain, Behavior, and Immunity. 2014;41:55–58. doi: 10.1016/j.bbi.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 74.Engler H, Brendt P, Wischermann J, et al. Selective increase of cerebrospinal fluid IL-6 during experimental systemic inflammation in humans: association with depressive symptoms. Molecular Psychiatry. 2017;22(10):1448–1454. doi: 10.1038/mp.2016.264 [DOI] [PubMed] [Google Scholar]
- 75.Brenn D, Richter F, Schaible HG. Sensitization of unmyelinated sensory fibers of the joint nerve to mechanical stimuli by interleukin-6 in the rat: An inflammatory mechanism of joint pain. Arthritis & Rheumatism. 2007;56(1):351–359. doi: 10.1002/art.22282 [DOI] [PubMed] [Google Scholar]
- 76.Hurst SM, Wilkinson TS, McLoughlin RM, et al. IL-6 and Its Soluble Receptor Orchestrate a Temporal Switch in the Pattern of Leukocyte Recruitment Seen during Acute Inflammation. Immunity. 2001;14(6):705–714. doi: 10.1016/S1074-7613(01)00151-0 [DOI] [PubMed] [Google Scholar]
- 77.Schuster B, Kovaleva M, Sun Y, et al. Signaling of Human Ciliary Neurotrophic Factor (CNTF) Revisited THE INTERLEUKIN-6 RECEPTOR CAN SERVE AS AN α-RECEPTOR FOR CNTF. J Biol Chem. 2003;278(11):9528–9535. doi: 10.1074/jbc.M210044200 [DOI] [PubMed] [Google Scholar]
- 78.Heink S, Yogev N, Garbers C, et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T H 17 cells. Nature Immunology. 2017;18(1):74–85. doi: 10.1038/ni.3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Uciechowski P, Dempke WCM. Interleukin-6: A Masterplayer in the Cytokine Network. OCL. 2020;98(3):131–137. doi: 10.1159/000505099 [DOI] [PubMed] [Google Scholar]
- 80.Sun Y, Wang D, Salvadore G, et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain, Behavior, and Immunity. 2017;66:156–164. doi: 10.1016/j.bbi.2017.06.014 [DOI] [PubMed] [Google Scholar]
- 81.Tiosano S, Yavne Y, Watad A, et al. The impact of tocilizumab on anxiety and depression in patients with rheumatoid arthritis. European Journal of Clinical Investigation. 2020;50(9):e13268. doi: 10.1111/eci.13268 [DOI] [PubMed] [Google Scholar]
- 82.Ormel J, Hartman CA, Snieder H. The genetics of depression: successful genome-wide association studies introduce new challenges. Translational Psychiatry. 2019;9(1):1–10. doi: 10.1038/s41398-019-0450-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.