

Abstract
Homocysteine, a sulfur-containing amino acid derived from methionine metabolism, is a known agonist of N-methyl-D-aspartate receptor (NMDAR) and is involved in neurotoxicity. Our previous findings showed that neuronal exposure to elevated homocysteine levels leads to sustained low-level increase in intracellular Ca2+, which is dependent on GluN2A subunit-containing NMDAR (GluN2A-NMDAR) stimulation. These studies further showed a role of ERK MAPK in homocysteine-GluN2A-NMDAR–mediated neuronal death. However, the intracellular mechanisms associated with such sustained GluN2A-NMDAR stimulation and subsequent Ca2+ influx have remained unexplored. Using live-cell imaging with Fluo3-AM and biochemical approaches, we show that homocysteine-GluN2A NMDAR-induced initial Ca2+ influx triggers sequential phosphorylation and subsequent activation of the proline rich tyrosine kinase 2 (Pyk2) and Src family kinases, which in turn phosphorylates GluN2A-Tyr1325 residue of GluN2A-NMDARs to maintain channel activity. The continuity of this cycle of events leads to sustained Ca2+ influx through GluN2A-NMDAR. Our findings also show that lack of activation of the regulatory tyrosine phosphatase STEP, which can limit Pyk2 and Src family kinase activity further contributes to the maintenance of this cycle. Additional studies using live-cell imaging of neurons expressing a redox-sensitive GFP targeted to the mitochondrial matrix show that treatment with homocysteine leads to a progressive increase in mitochondrial reactive oxygen species generation, which is dependent on GluN2A-NMDAR–mediated sustained ERK MAPK activation. This later finding demonstrates a novel role of GluN2A-NMDAR in homocysteine-induced mitochondrial ROS generation and highlights the role of ERK MAPK as the intermediary signaling pathway between GluN2A-NMDAR stimulation and mitochondrial reactive oxygen species generation.
Keywords: homocysteine, GluN2A-NMDA receptor, Pyk2, Src family kinase, Ca2+ influx, mitochondrial reactive oxygen species, ERK MAPK, neurotoxicity
The N-methyl-D-aspartate subtype of glutamate receptors (NMDARs) plays a critical role in promoting brain development, learning, and memory as well as in mediating the pathology of multiple neurological disorders. Earlier studies have shown that this opposing role of NMDARs is dependent on the subunit composition of the functional NMDAR complex at the neuronal surface. In the adult brain, functional NMDARs are predominantly composed of heteromeric assemblies of GluN1/GluN2A, GluN1/GluN2B, or GluN1/GluN2A/GluN2B subunits (1, 2, 3, 4, 5). Stimulation of GluN2A subunit containing NMDARs (GluN2A-NMDAR) by the NMDAR agonist and neurotransmitter glutamate leads to moderate and transient influx of Ca2+, resulting in activation of intracellular signaling pathways that promote synaptic plasticity, neuronal survival, and growth (2, 4, 6, 7, 8, 9), whereas stimulation of GluN2B subunit containing NMDARs (GluN2B-NMDAR) following excessive glutamate release results in large Ca2+ influx in neurons that leads to activation of several detrimental pathways and neuronal death, a phenomenon known as excitotoxicity (4, 9, 10, 11, 12). GluN2B-NMDAR–mediated excitotoxic cell death has been shown to be involved in the progression of multiple neurological disorders, including cerebral ischemia, traumatic brain injury, epilepsy, multiple sclerosis, and amyotrophic lateral sclerosis.
Homocysteine, a thiol-containing amino acid formed as an intermediate of the methionine cycle is another NMDAR agonist (13), and systemic elevation of plasma homocysteine also known as hyperhomocysteinemia is a metabolic disorder that is recognized as an independent risk factor for both acute and chronic neurological disorders (14, 15, 16, 17, 18, 19). In contrast to glutamate-mediated excitotoxicity, homocysteine induces neuronal cell death through a novel signaling pathway that involves GluN2A-NMDAR stimulation (13, 20, 21, 22). Studies in neuronal cultures have shown that homocysteine-mediated stimulation of GluN2A-NMDARs, but not GluN2B-NMDARs, leads to continuous and progressive increase in intracellular Ca2+, resulting in sustained increase in the extracellular signal-regulated kinase, a member of the mitogen activated protein kinase family (ERK MAPK) phosphorylation, which eventually leads to neuronal cell death (23). Using a rodent model of cerebral ischemic stroke, it has also been shown that predisposition to hyperhomocysteinemia exacerbates ischemic brain injury, which involves GluN2A-NMDAR–mediated sustained increase in ERK MAPK phosphorylation (24). Collectively, these findings suggest that sustained GluN2A-NMDAR stimulation plays a crucial role in homocysteine-mediated neuronal cell death. However, the precise molecular mechanism(s) underlying such unregulated GluN2A-NMDAR stimulation following exposure to elevated levels of homocysteine is not known. Furthermore, the signaling events downstream of GluN2A-NMDAR that are involved in neuronal cell death are also not well understood. In this context, although changes in cellular redox status are often linked to hyperhomocysteinemia-associated neurodegenerative disorders (25, 26), the causal link between homocysteine-induced GluN2A-NMDAR stimulation and the increase in intracellular oxidative stress is still not clear.
A significant goal of the current study is to unravel whether homocysteine-induced GluN2A-NMDARs activation is a critical determinant of mitochondrial reactive oxygen species (ROS) generation. To address this issue, we investigated whether homocysteine-GluN2A-NMDAR mediated initial Ca2+ influx and Ca2+-dependent ERK MAPK phosphorylation triggers a feedforward cycle to enhance the duration of GluN2A-NMDAR stimulation and ERK MAPK phosphorylation. Subsequent studies further examined whether such sustained increase in ERK MAPK phosphorylation plays a key role in mitochondrial ROS generation that results in neurotoxicity.
Results
Homocysteine-mediated phosphorylation of Pyk2 kinase and SFKs leads to sustained influx of Ca2+ through GluN2A-NMDAR in neurons
In an earlier study, we demonstrated that the treatment of neuronal cultures with homocysteine causes GluN2A-NMDAR stimulation (23). Using the high affinity Ca2+ dye Fura2-AM, the study further showed that homocysteine-induced GluN2A-NMDAR stimulation leads to a slow and progressive increase in Ca2+ influx over a period of 1 h. Although Fura-2 AM allows accurate quantification of intracellular Ca2+ level, it has a short half-life (27, 28, 29), thereby limiting its use for long-term evaluation of intracellular Ca2+. Therefore, in the current study, we used the single-wavelength Ca2+ indicator dye Fluo-3 AM to measure homocysteine-GluN2A-NMDAR–mediated long-term changes in intracellular Ca2+ dynamics monitored over a period of 4 h. Rat neuronal cultures were exposed to L-homocysteine (50 μM, 4 h) and changes in Fluo-3 AM fluorescent intensity in the cell soma was measured by live-cell imaging. The photomicrographs presented in Figure 1A exhibit large increase in fluorescent intensity over time on binding of the dye to intracellular Ca2+ (in false-color maps), following treatment with homocysteine. The line graph presented in Figure 1B shows the temporal profile of increase in fluorescent intensity, as an indicator of increasing intracellular Ca2+ levels in homocysteine treated cells when compared to vehicle-treated control cells. The corresponding bar graph at selective time points (15 min, 1 h, 2 h, and 4 h) representing Ca2+ changes in the soma of individual neurons shows significant increase in intracellular Ca2+ levels within 1 h of homocysteine treatment compared to time-matched vehicle-treated control (Fig. 1C). In subsequent studies, Fluo-3 AM fluorescent intensity was measured in neurons that were treated with homocysteine in the presence of NMDAR inhibitor DL-AP5 (200 μM) or selective GluN2A-NMDAR inhibitor NVP-AAM077 (30 nM). The findings (Fig. 1, D–G) show that pharmacological inhibition with either DL-AP5 or NVP-AAM077 blocks homocysteine-induced increase in intracellular Ca2+ levels, thus confirming the role of GluN2A-NMDAR in homocysteine-mediated sustained influx of Ca2+. Several lines of evidence indicate that Pyk2 and Src family kinases (SFKs) are part of the NMDAR complex and play a role in increasing surface expression and function of the receptor (30, 31, 32, 33, 34, 35, 36). Therefore, in additional studies, neurons were treated with L-homocysteine in the presence of Pyk2 inhibitor PF431396 (5 μM) or SFK inhibitor PP2 (5 μM), and Fluo3-AM fluorescence intensity was monitored for a period of 4 h. The results (Fig. 1, H–K) show that treatment with either PF431396 or PP2 blocks homocysteine-GluN2A-NMDAR–mediated increase in Fluo3-AM fluorescence intensity, indicating that homocysteine-GluN2A-NMDAR–mediated sustained influx of Ca2+ is dependent on the activation of Pyk2 and SFKs.
Figure 1.

Role of Pyk2 and SFK in homocysteine-induced changes in intracellular Ca2+in neurons.A, representative micrographs of Fluo3-AM loaded neurons showing changes in intracellular Ca2+ over time measured at 484 nm, following exposure to L-homocysteine (50 μM). Both black and white (4 h) and false-color images (0–4 h) are shown. B, temporal profile of intracellular Ca2+ increase over time was assessed by measuring increase in Fluo3-AM fluorescence intensity (mean ± SE) in soma of L-Hcy–treated and L-Hcy–untreated (control) neurons. C, individual responses in neuronal soma showing the range of increase in intracellular Ca2+ at specific time points, following exposure to L-Hcy, expressed as mean ± SD. Two-way ANOVA followed by Bonferroni’s multiple-comparisons test shows significant effect (treatment effect: F (1, 21) = 33.42; p < 0.0001, time effect: F (1.629, 34.21) = 32.49; p < 0.0001, interaction: F (4, 84) = 23.09; p < 0.0001). D, F, H, and J, temporal profile of changes in intracellular Ca2+ in neurons treated with L-Hcy (50 μM) in the presence or absence of (D) NMDAR inhibitor DL-AP5 (200 μM); (F) GluN2A-NMDAR inhibitor NVP-AAM077 (30 nM); (H) Pyk2 inhibitor PF431396 (5 μM); or (J) SFK inhibitor PP2 (5 μM) expressed as mean ± SE. E, G, I, and K, quantitative analysis of individual response in neuronal soma showing the range of increase in intracellular Ca2+ at the specified time points, following treatment with L-Hcy and in the presence or absence of pharmacological inhibitors expressed as mean ± SD. Two-way ANOVA followed by Bonferroni’s multiple-comparisons test shows significant effect of treatment, time, and interaction in the presence of (E) NMDAR inhibitor DL-AP5 (treatment effect: F (2, 33) = 36.83; p < 0.0001, time effect: F (1.803, 59.50) = 33.62; p < 0.0001, interaction: F (8, 132) = 29.75; p < 0.0001), (G) GluN2A-NMDAR inhibitor NVP-AAM077 (treatment effect: F (2, 31) = 33.16; p < 0.0001, time effect: F (1.701, 52.74) = 33.69; p < 0.0001, interaction: F (8, 124) = 27.59; p < 0.0001), (I) Pyk2 inhibitor PF431396 (treatment effect: F (2, 32) = 43.50; p < 0.0001, time effect: F (1.662, 53.18) = 32.93; p < 0.0001, interaction: F (8, 128) = 32.33; p < 0.0001), and (K) SFK inhibitor PP2 (treatment effect: F (2, 32) = 47.81; p < 0.0001, time effect: F (1.663, 53.21) = 33.83; p < 0.0001, interaction: F (8, 128) = 32.13; p < 0.0001). Post hoc analysis shows ∗p < 0.01 and ∗∗p < 0.0001 between the treatment groups at the given time points. Values are mean ± SD. Data points represent individual biological replicates. NMDAR, N-methyl-D-aspartate subtype of glutamate receptor; SFK, Src family kinase.
Considerable evidence from earlier studies suggest that the activity of GluN2A-NMDAR is dependent on its phosphorylation at the C-terminal Tyr1325 residue by SFKs (30, 31, 32, 33, 34). The activation of SFKs in turn is dependent on its phosphorylation at Tyr416, which is determined by activation of Pyk2 through Ca2+-dependent autophosphorylation of Pyk2 at Tyr402 (35, 36). As such, we next evaluated whether homocysteine-mediated GluN2A-NMDAR activation involves phosphorylation of GluN2A subunit at Tyr1325 through activation of Pyk2 and SFKs. For these experiments, in initial studies, neurons were treated with L-homocysteine (50 μM, 4 h) in the presence of Pyk2 inhibitor or SFK inhibitor, followed by immunoprecipitation of GluN2A-NMDAR from cell lysates with anti-GluN2A antibody. Immunoblot analysis of the immune complex with anti-phospho-GluN2AY1325 antibody show significant increase in the phosphorylation of Tyr1325 when treated with homocysteine alone (Fig. 2A), whereas pharmacological inhibition of either Pyk2 or SFKs attenuates the phosphorylation of Tyr1325 (Fig. 2A). To determine whether homocysteine treatment could also lead to an increase in phosphorylation of Pyk2 and SFKs, in subsequent studies neuronal cultures were treated with L-homocysteine (50 μM, 4 h) in the presence of either Pyk2 or SFK inhibitor. The extent of phosphorylation of Pyk2 at Tyr402 and SFK at Tyr416 in cell lysates was evaluated by immunoblotting with anti-pPyk2 or anti-pSrc kinase antibodies, respectively (Fig. 2, B and C). The results show that treatment with homocysteine alone leads to significant increase in the phosphorylation of both Pyk2 and SFKs, and inhibition of Pyk2 or SFKs blocks homocysteine-mediated increase in their respective phosphorylation (Fig. 2, B and C). However, inhibition of Pyk2 or SFKs in untreated neurons (controls) has no effect on the phosphorylation of Pyk2 or Src, respectively (Fig. 2, D and E). Since phosphorylation of SFKs at Tyr416 has been reported to be downstream of and dependent on the activation of Pyk2 (35, 36), we further evaluated whether inhibition of Pyk2 phosphorylation can block Src phosphorylation. For this experiment, neurons were treated with L-homocysteine (50 μM, 4 h) in the presence of the Pyk2 inhibitor. Immunoblot analysis with anti-pSrc antibody show that pharmacological inhibition of Pyk2 blocks homocysteine-induced phosphorylation of SFKs at Tyr416 (Fig. 2F, upper panel). Taken together, these findings suggest that homocysteine-GluN2A-NMDAR–mediated initial Ca2+ influx triggers activation of Pyk2, which in turn activates SFKs to help to maintain a feedforward cycle for prolonged GluN2A-NMDAR activation through phosphorylation of Tyr1325, resulting in sustained Ca2+ influx. To further confirm the role of GluN2A-NMDAR in the phosphorylation of Pyk2 and Src in additional studies, neuronal cultures were treated with L-homocysteine (50 μM, 4 h) in the presence of GluN2A-NMDAR inhibitor NVP-AAM077 (30 nM) or GluN2B-NMDAR inhibitor Ro25-6981 (5 μM). Immunoblot analysis show that inhibition of GluN2A-NMDAR blocks phosphorylation of both Pyk2 and Src, whereas inhibition of GluN2B-NMDARs fails to block phosphorylation of either Pyk2 or Src (Fig. 2, G and H).
Figure 2.

Homocysteine mediated increase in Pyk2 and SFK phosphorylation leads to GluN2A-NMDAR phosphorylation in neurons.A–F, rat neuron cultures were treated with 50 μM L-homocysteine (L-Hcy, 4 h) in the presence and absence of Pyk2 inhibitor PF431396 (5 μM) or SFK inhibitor PP2 (5 μM). A, GluN2A-subunit of NMDAR was immunoprecipitated from total cell lysate using anti-GluN2A antibody, and the immune complexes were processed for immunoblot analysis using anti-pGluN2AY1325 antibody (upper panel). Total cell lysates were immunoblotted with anti-β-tubulin antibody to ensure equal input protein for immunoprecipitation (lower panel). One-way ANOVA revealed significant main effect of treatment (F (3, 8) = 146.3; p < 0.0001). B–F, total cell lysates were processed for immunoblot analysis using (B and D) anti-phospho-Pyk2 Y402 (pPyk2) and anti-Pyk2 antibodies or (C, E, and F) anti-phospho-Src Y416 (pSrc) and anti-Src antibodies. B, C, and F, one-way ANOVA revealed significant main effect of treatment in the presence of (B) Pyk2 inhibitor: F (2, 6) = 50.27; p = 0.0002, (C) SFK inhibitor: F (2, 6) = 59.18; p = 0.0001, and (F) Pyk2 inhibitor: F (2, 6) = 34.94; p = 0.0005. G and H, rat neuron cultures were treated with 50 μM L-Hcy (4 h) in the presence and absence of GluN2A-NMDAR inhibitor NVP-AAM077 (30 nM) or GluN2B-NMDAR inhibitor Ro25-6981 (5 μM). Total cell lysates were processed for immunoblot analysis using (G) anti-pPyk2 and anti-Pyk2 antibodies or (H) anti-pSrc and anti-Src antibodies. I–L, rat neuron cultures were treated with either (I and J) 50 μM L-Glutamate (0 min, 5 min, and 30 min) or (K and L) 50 μM L-Hcy (0 min, 5 min, 0.5 h, 1 h, 2 h, and 4 h). Total cell lysates were processed for immunoblot analysis using (I and K) anti-STEP and anti-tubulin antibodies or (J and L) anti-pSrc and anti-Src antibodies. A, B, C, and E, post hoc analysis by Bonferroni’s multiple-comparisons test shows ∗p < 0.01, ∗∗p < 0.001, and ∗∗∗p < 0.001 between the treatment groups. Values are mean ± SD. Data points represent individual biological replicates. NMDAR, N-methyl-D-aspartate subtype of glutamate receptor; SFK, Src family kinase.
It is further evident from earlier studies that the activity of Pyk2 and SFKs is regulated by the brain-enriched and neuron-specific tyrosine phosphatase STEP (37, 38, 39, 40). STEP61, the membrane bound isoform of the STEP-family is ubiquitously expressed in the brain and is the predominant isoform expressed in cultured neurons (41, 42). STEP61 is a component of the NMDAR signaling pathway and its function is regulated through phosphorylation of a critical serine residue in the kinase-interacting motif (43, 44). STEP61 is basally phosphorylated at this residue, which renders it inactive in terms of its ability to bind to its substrates and dephosphorylate them (43, 44). Dephosphorylation of this serine residue after glutamate-mediated GluN2B-NMDAR stimulation renders STEP61 active (43), and active STEP61 (dephosphorylated) is typically detected by a downward shift in the mobility of the STEP61 protein band (43, 45, 46, 47). To evaluate the phosphorylation status of STEP61 after glutamate treatment, neuronal cultures were treated with glutamate (50 μM) for the specified time periods (5 and 30 min), and cell lysates were analyzed by immunoblotting with anti-STEP antibody. As previously reported, in the absence of any stimulation (control), STEP61 was found to be primarily phosphorylated (top band), (Fig. 2I, upper panel, lane 1), whereas treatment with glutamate decreases the intensity of the top band with a concomitant increase in the intensity of the lower band (Fig. 2I, upper panel, lane 3) within 30 min, confirming the dephosphorylation of STEP61. To evaluate the phosphorylation status of STEP61 following treatment with homocysteine, neuronal cultures were treated with L-homocysteine (50 μM) for specified time periods (5 min, 30 min, 1 h, 2 h, and 4 h). The findings show that the phosphorylated STEP61 (upper band) is the predominant isoform present under these conditions, which confirms that homocysteine treatment fails to dephosphorylate STEP61 (Figure 2K, upper panel). This finding suggests that the lack of activation of STEP61 following homocysteine-GluN2A NMDAR stimulation leads to persistent phosphorylation of Pyk2 and SFKs, which in turn helps to maintain prolonged GluN2A-NMDAR phosphorylation and channel activity. To further validate this hypothesis, in additional experiments, we evaluated the time course of SFK phosphorylation after treatment with glutamate (50 μM, 30 min) or L-homocysteine (50 μM, 4 h). The findings show that treatment with glutamate leads to a transient increase in SFK phosphorylation within 5 min of stimulation, whereas treatment with homocysteine leads to a sustained increase in SFK phosphorylation observed throughout the time period of the study (Fig. 2, J and L). Consistent with this finding, in earlier studies, we also observed that phosphorylation of ERK MAPK, another substrate of STEP, is also distinctly different after treatment of neurons with glutamate and homocysteine (21, 23). While treatment with glutamate (50 μM, 1 h) led to a rapid but transient increase in ERK MAPK phosphorylation within 5 min and returned to basal level by 30 min of stimulation, homocysteine-mediated ERK MAPK phosphorylation remained sustained for the entire period of the study.
Homocysteine-GluN2A-NMDAR–mediated sustained ERK MAPK phosphorylation is dependent on Pyk2 and SFK activation
The sustained increase in ERK MAPK phosphorylation following homocysteine-GluN2A-NMDAR–mediated Ca2+ influx observed in earlier studies is characterized by a rapid initial increase (2.5–5 min), followed by a delayed larger increase (4 h), and plays a role in homocysteine-mediated neuronal cell death (20, 21, 23). To evaluate the involvement of Pyk2 and SFKs in mediating the biphasic phosphorylation of ERK MAPK, neuronal cultures were treated with L-homocysteine (50 μM) for 5 min or 4 h in the presence of Pyk2 inhibitor or SFK inhibitor. Immunoblot analysis with anti-pERK antibody shows that treatment with homocysteine alone leads to increase in ERK MAPK phosphorylation at both 5 min and 4 h, which is consistent with the previous findings (20, 21, 23, 24). Pharmacological inhibition of Pyk2 or SFKs has no effect on the initial increase in ERK MAPK phosphorylation at 5 min (Fig. 3, A and B) but significantly reduces the delayed increase in ERK MAPK phosphorylation at 4 h (Fig. 3, C and D). This later finding raises the possibility that the initial increase in ERK MAPK phosphorylation leads to Pyk2-mediated SFK activation to maintain GluN2A-NMDAR stimulation, which is essential for sustained ERK MAPK phosphorylation. To test this hypothesis, neurons were treated with L-homocysteine (50 μM, 4 h) in the presence of ERK MAPK inhibitor PD98059 (15 μM), and immunoblot analysis was performed with anti-pPyk2 antibody. The findings show that inhibition of ERK MAPK phosphorylation leads to significant reduction in Pyk2 phosphorylation at 4 h (Fig. 3E). To further confirm the role of Pyk2 and SFKs in facilitating the delayed phosphorylation of ERK MAPK by 4 h, in subsequent studies, inhibitors of Pyk2 or SFKs was added 30 min after the onset of homocysteine treatment. Immunoblot analysis show (Fig. 3, F and G) that delayed administration of either the Pyk2 or SFK inhibitor significantly reduces homocysteine-induced ERK MAPK phosphorylation at 4 h. Taken together, the findings indicate the presence of a feedback loop triggered by the initial increase in ERK MAPK phosphorylation, to maintain Pyk2/Src/GluN2A-NMDAR–mediated sustained ERK MAPK phosphorylation following homocysteine treatment.
Figure 3.

Homocysteine-induced delayed phosphorylation of ERK MAPK is dependent on Pyk2 and SFK activation.A–G, rat neuron cultures were treated with 50 μM of L-homocysteine (L-Hcy) for (A and B) 5 min or (C–G) 4 h. L-Hcy treatment was carried out in the presence or absence of (A and C) Pyk2 inhibitor PF431396 (5 μM); (B and D) SFK inhibitor PP2 (5 μM); or (E) ERK MAPK inhibitor PD98059 (10 μM). In some experiments (F), Pyk2 inhibitor PF431396 or (G) SFK inhibitor PP2 was added 30 min after the onset of L-Hcy treatment. Immunoblot analysis was performed using (A, B, C, D, F, and G) anti-p-ERK and p-ERK antibodies or (E) anti-pPyk2 and anti-Pyk2 antibodies. One-way ANOVA revealed significant main effect of treatment in the presence of (A) Pyk2 inhibitor: F (2, 6) = 58.46; p = 0.0001 (B) SFK inhibitor: F (2, 9) = 471.2; p < 0.0001, (C) Pyk2 inhibitor: F (2, 6) = 54.12; p = 0.0001, (D) SFK inhibitor: F (2, 9) = 1038; p < 0.0001, (E) ERK MAPK inhibitor: F (2, 6) = 24.69; p = 0.0013, (F) Pyk2 inhibitor: F (2, 6) = 376.0; p < 0.0001, and (G) SFK inhibitor: F (2, 9) = 86.79; p < 0.0001. Post hoc analysis by Bonferroni’s multiple-comparisons test shows ∗p < 0.01, ∗∗p < 0.001, and ∗∗∗p < 0.0001 between the treatment groups. Values are mean ± SD. Data points represent individual biological replicates. SFK, Src family kinase.
Homocysteine-GluN2A-NMDAR–mediated ERK MAPK activation leads to sustained mitochondrial ROS generation that contributes to neuronal death
Previous studies have indicated that exposure to elevated levels of homocysteine leads to ROS generation in neuronal cells (13, 48). As mitochondria is a major source of ROS in brain cells, we next monitored long-term changes in oxidative status selectively in the mitochondria by live-cell imaging, utilizing a redox-sensitive fluorescent protein-based biosensor, mito-RoGFP targeted to the mitochondrial matrix (49). For these studies, neuronal cultures were infected with an adenovirus-encoding mito-RoGFP and the infected cells were treated with vehicle (saline) or L-homocysteine (50 μM), followed by assessment of fluorescent intensity of mito-RoGFP by live-cell imaging for 4 h, at specified intervals. The representative photomicrographs presented in Figure 4A shows that in vehicle-treated (control) cells, the fluorescence intensity of mito-RoGFP (in false-color maps) remains unchanged over time, whereas in homocysteine treated cells, there is a time-dependent decrease in mito-RoGFP fluorescence intensity, an indicator of increased mitochondrial ROS generation (49). Assessment of mito-RoGFP fluorescence intensity over time presented as a line graph (Fig. 4B) and the extent of mito-RoGFP oxidation at selective time points (15 min, 1 h, 2 h, and 4 h) presented in the corresponding bar graphs indicate that homocysteine treatment leads to progressive and significant increase in mitochondrial ROS generation, whereas no change in mitochondrial redox status is observed in neurons treated with vehicle. To ensure that the observed decrease in fluorescence intensity was due to generation of ROS, in subsequent studies, neurons infected with the adenovirus encoding mito-RoGFP were exposed to H2O2 (50 μM, 10 min), the most abundant form of ROS produced by mitochondria (50). The representative photomicrographs (Fig. 4D) and quantitative analysis (Fig. 4E) show that fluorescence intensity of mito-RoGFP decreases significantly within minutes of exposure to H2O2. To further confirm that mitochondria is the primary source of homocysteine-induced ROS generation, mito-RoGFP–expressing neurons were treated with L-homocysteine (50 μM, 4 h) in the presence or absence of MitoTEMPO (5 μM), a mitochondria-targeted antioxidant. The representative photomicrographs (Fig. 4F) and the quantitative analysis (Fig. 4, G and H) show that treatment with MitoTEMPO blocks homocysteine-induced reduction in fluorescence intensity of mito-RoGFP.
Figure 4.

Homocysteine leads to progressive increase in mitochondrial ROS generation.A–E, rat neuronal cultures expressing mito-RoGFP were treated with 50 μM of L-homocysteine (L-Hcy, 4 h) or 50 μM H2O2 (10 min), and loss of fluorescence signal was recorded (excitation 484 nm) at the specified times. A and D, representative photomicrographs (black and white, and false-color images) showing temporal loss of fluorescence signal of mito-RoGFP with (A) L-Hcy or (D) H2O2 treatment indicating generation of mitochondrial ROS. The representative photomicrographs displaying changes in fluorescence intensity of mito-RoGFP following L-Hcy treatment in Figure 4A has also been incorporated in (F), and Figure 5, A and D (as a positive control) for side-by-side visual comparison of the effects of pharmacological inhibitors on L-Hcy–induced changes in fluorescence intensity of mito-RoGFP. B, temporal changes in fluorescence intensity (arbitrary units) in control and L-Hcy–treated neurons-expressing mito-RoGFP (mean ± SE). C and E, quantitative analysis of the percentage decrease in fluorescence intensity of oxidized mito-RoGFP signal in individual cells following L-Hcy or H2O2 treatment compared to untreated control cells expressed as mean ± SD. Two-way ANOVA followed by Bonferroni’s multiple-comparisons test shows significant effect of treatment, time, and interaction in the presence L-Hcy (treatment effect: F (1, 44) = 192.2; p < 0.0001, time effect: F (1.891, 81.32) = 34.38; p < 0.0001, interaction: F (4, 172) = 66.21; p < 0.0001), and H2O2 (treatment effect: F (1, 30) = 77.37; p < 0.0001, time effect: F (3, 74) = 10.69; p < 0.0001, interaction: F (3, 74) = 15.09; p < 0.0001). F–H, neurons-expressing mito-RoGFP were treated with 50 μM of L-Hcy in the presence or absence of mitoTEMPO (5 μM) for 4 h. F, representative photomicrographs (black and white, and false-color images) showing temporal changes in fluorescence intensity of mito-RoGFP. G, temporal changes in mito-RoGFP fluorescence intensity (arbitrary units) over time and (H) quantitative analysis of percentage increase in reduced mito-RoGFP signal at the specified time points. Two-way ANOVA followed by Bonferroni’s multiple-comparisons test shows significant effect of treatment, time, and interaction (Treatment effect: F (1, 62) = 244.4; p < 0.0001, Time effect: F (1.826, 113.2) = 144.7; p < 0.0001, Interaction: F (4, 248) = 77.31; p < 0.0001). Post hoc analysis shows ∗p < 0.0001 between the treatment groups at the given time point. Data points represent individual biological replicates. ROS, reactive oxygen species; SFK, Src family kinase.
Next, we sought to evaluate the intracellular signaling pathway that is involved in homocysteine-induced mitochondrial ROS generation. To determine whether Ca2+ influx is necessary for ROS generation, mito-RoGFP–expressing neurons were treated with L-homocysteine (50 μM) and maintained in a calcium-free media containing 0.5 mM EGTA to chelate any residual extracellular calcium. The representative photomicrographs (Fig. 5A) and quantitative analysis of mito-RoGFP fluorescence intensity (Fig. 5, B and C) show that, in the absence of extracellular Ca2+, the decrease in homocysteine-induced fluorescence intensity of mito-RoGFP is completely blocked. To evaluate the role of GluN2A-NMDARs in homocysteine-induced mitochondrial ROS generation, mito-RoGFP–expressing neurons were treated with L-homocysteine (50 μM, 4 h) in the presence of NMDAR inhibitor or GluN2A-NMDAR inhibitor. The representative photomicrographs (Fig. 5D) and quantitative analysis of mito-RoGFP fluorescence intensity show that both DL-APV (Fig. 5, E and F) and NVP-AAM077 (Fig. 5, G and H) block homocysteine-induced decrease in mito-RoGFP fluorescence intensity. In subsequent studies, mito-RoGFP–expressing neurons were treated with L-homocysteine (50 μM, 4 h) in the presence of the GluN2B-NMDAR inhibitor. The analysis of mito-RoGFP fluorescent intensity show that GluN2B-NMDAR inhibition fails to block homocysteine-induced decrease in mito-RoGFP fluorescence intensity (Fig. 5, I and J), eliminating the role of GluN2B-NMDARs in homocysteine-induced mitochondrial ROS generation. To further confirm the specific role of GluN2A-NMDARs in homocysteine-induced mitochondrial ROS generation, in subsequent studies, mito-RoGFP was expressed in cortical neuronal cultures obtained from WT and GluN2A-KO mice. The cells were then treated with vehicle (saline) or L-homocysteine (50 μM, 4 h), followed by live-cell imaging to monitor changes in mito-RoGFP fluorescence intensity. The representative photomicrographs (Fig. 6A) and quantitative analysis (Fig. 6, B and C) show that the treatment of neuronal cultures obtained from WT mice with homocysteine leads to a significant decrease in mito-RoGFP fluorescence intensity over time, as compared to vehicle-treated WT control, which is consistent with our findings in neuronal cultures obtained from rat brain (Fig. 4A). However, neurons obtained form GluN2A-KO mice did not show significant decrease in mito-RoGFP fluorescence intensity, following homocysteine treatment (Fig. 6, D and E). Comparison of mito-RoGFP fluorescence intensity in neurons obtained from WT and GluN2A-KO mice treated with homocysteine shows significant decrease in fluorescence intensity in WT neurons over time (Fig. 2, F and G). Together, these findings confirm the role of GluN2A-NMDAR in homocysteine-induced mitochondrial ROS generation in neurons.
Figure 5.

Homocysteine-induced mitochondrial ROS generation is dependent on GluN2A-NMDAR.A–J, rat neuronal cultures expressing mito-RoGFP were treated with 50 μM of L-homocysteine (L-Hcy, 4 h) in the presence or absence of (A–C) EGTA (2.5 μM), (D–F) DL-AP5 (200 μM), (D, G, and H) NVP-AAM077 (30 nM), or (I and J) Ro25-6951 (5 μM). A and D, representative photomicrographs (black and white, and false-color images) showing temporal changes in fluorescence intensity of mito-RoGFP. B, E, and G, temporal changes in mito-RoGFP fluorescence intensity (arbitrary units) over time in the presence of (B) EGTA, (E) DL-APV, and (G) NVP-AAM077 expressed as mean ± SE. C, F, and H, quantitative analysis (two-way ANOVA followed by Bonferroni’s multiple-comparisons test) of the percentage increase in fluorescence intensity of reduced mito-RoGFP signal in the presence of (C) EGTA, (F) DL-APV, and (H) NVP-AAM077 expressed as mean ± SD. Data shows significant effect of treatment, time, and interaction in the presence EGTA (treatment effect: F (1, 41) = 125.8; p < 0.0001, time effect: F (2.026, 81.54) = 26.76; p < 0.0001, interaction: F (4, 161) = 30.22; p < 0.0001), DL-APV (treatment effect: F (1, 51) = 179.7; p < 0.0001, time effect: F (2.033, 102.2) = 81.99; p < 0.0001, interaction: F (4, 201) = 31.93; p < 0.0001) and NVP-AAM077 (treatment effect: F (1, 52) = 87.34; p < 0.0001, time effect: F (1.518, 78.93) = 34.10; p < 0.0001, interaction: F (4, 208) = 24.11; p < 0.0001). I, temporal profile of changes in mito-RoGFP fluorescence intensity (arbitrary units) in the presence of Ro25-6951 expressed as mean ± SE and (J) quantitative analysis of percentage change in fluorescence intensity of oxidized mito-RoGFP signal at the given time points does not show significant effect of treatment and interaction in the presence Ro25-6981(treatment effect: F (1, 38) = 0.0008; p = 0.9775, Time effect: F (1.604, 60.96) = 158.1; p < 0.0001, interaction: F (4, 152) = 1.09; p = 0.3593). Post hoc analysis shows ∗p < 0.01 and ∗∗p < 0.0001 between the treatment groups at the given time point. Data points represent individual biological replicates. Mito-RoGFP, mitochondria-targeted redox-sensitive GFP; NMDAR, N-methyl-D-aspartate subtype of glutamate receptor; ROS, reactive oxygen species.
Figure 6.

Knockdown of GluN2A subunit blocks homocysteine-induced mitochondrial ROS generation.A–G, neuronal cultures from WT and GluN2A KO mice–expressing mito-RoGFP were treated with or without 50 μM of L-homocysteine (L-Hcy, 4 h) and changes in fluorescence signal of mito-RoGFP was recorded at the specified time points. A, representative photomicrographs (black and white, and false-color images) showing temporal changes in fluorescence intensity of mito-RoGFP. B, D, and F, temporal profile of changes in mito-RoGFP fluorescence intensity (arbitrary units) over time in (B) WT control and L-Hcy–treated WT neurons, (D) GluN2A-KO control and L-Hcy–treated GluN2A-KO neurons, and (F) L-Hcy–treated WT and L-Hcy–treated GluN2A KO neurons expressed as mean ± SE. C, E, and G, quantitative analysis of the percentage change in fluorescence intensity of oxidized mito-RoGFP signal at the given time points in (C) L-Hcy–treated WT neurons compared to vehicle-treated WT neurons (control), (E) L-Hcy–treated GluN2A-KO neurons compared to vehicle-treated GluN2A-KO neurons (control), and (G) L-Hcy–treated WT neurons compared to L-Hcy–treated GluN2A KO neurons (control) expressed as mean ± SD. Two-way ANOVA followed by Bonferroni’s multiple-comparisons test shows significant effect of treatment, time, and interaction in (C) treatment effect: (F (1, 25) = 51.48; p < 0.0001), time effect: (F (2.744, 68.61) = 50.05; p < 0.0001), interaction: (F (4, 100) = 35.31; p < 0.0001) and (G) treatment effect: (F (1, 21) = 18.05; p = 0.0004), time effect: (F (2.924, 59.94) = 35.68; p < 0.0001), interaction: (F (4, 82) = 28.32; p < 0.0001). E, two-way ANOVA followed by Bonferroni’s multiple-comparisons test does not show significant effect of treatment and interaction (treatment effect: F (1, 18) = 0.498; p = 0.4894, time effect: F (4, 69) = 4.946; p = 0.0015, interaction: F (4, 69) = 2.412; p = 0.0572). Post hoc analysis shows ∗p < 0.001 and ∗∗p < 0.0001 between the treatment groups at the given time point. Data points represent individual biological replicates. Mito-RoGFP, mitochondria-targeted redox-sensitive GFP; ROS, reactive oxygen species.
To delineate the role of Pyk2 and SFKs in homocysteine-induced mitochondrial ROS generation, in subsequent studies, mito-RoGFP–expressing neurons obtained from rats were treated with L-homocysteine (50 μM) in the presence or absence of the Pyk2 inhibitor or SFK inhibitor. Quantitative analysis of mito-RoGFP fluorescence intensity shows that the inhibition of either Pyk2 or SFKs blocks homocysteine-induce decrease in mito-RoGFP fluorescence intensity (Fig. 7, A–D). To further evaluate the role of ERK MAPK in homocysteine-induced mitochondrial ROS generation, mito-RoGFP–expressing neurons were treated with L-homocysteine (50 μM) in the presence or absence of the ERK MAPK inhibitor. The findings show that treatment with ERK MAPK inhibitor also blocks homocysteine-induce decrease in mito-RoGFP fluorescence intensity (Fig. 7, E and F). Collectively, these findings demonstrate the role of GluN2A-NMDAR–mediated Pyk2, SFK, and ERK MAPK activation in homocysteine-induced mitochondrial ROS generation.
Figure 7.

Homocysteine-induced mitochondrial ROS generation comprises a feedback loop involving activation of ERK MAPK. Rat neuronal cultures expressing mito-RoGFP were treated with 50 μM of L-homocysteine (L-Hcy, 4 h) in the presence or absence (A and B) Pyk2 inhibitor PF431396, (C and D) SFK inhibitor PP2, or (E and F) ERK MAPK inhibitor PD98059 (15 μM). A, C, and E, temporal profile of changes in mito-RoGFP fluorescence intensity (arbitrary units) over time expressed as mean ± SE. B, D, and F, quantitative analysis (two-way ANOVA followed by Bonferroni’s multiple-comparisons test) of the percentage change in fluorescence intensity of oxidized mito-RoGFP signal at the given time points expressed as mean ± SD. Data shows significant effect of treatment, time, and interaction in the presence of PF431396 (treatment effect: F (1, 41) = 195.8; p < 0.0001, time effect: F (2.487, 102) = 74.82; p < 0.0001, interaction: F (4, 164) = 39.80; p < 0.0001), PP2 (treatment effect: F (1, 40) = 165.9; p < 0.0001, time effect: F (2.730, 109.2) = 62.13; p < 0.0001, interaction: F (4, 160) = 34.37; p < 0.0001) and PD98059 (treatment effect: F (1, 45) = 117.2; p < 0.0001, time effect: F (1.937, 84.74) = 81.53; p < 0.0001, interaction: F (4, 175) = 29.47; p < 0.0001). Post hoc analysis shows ∗p < 0.0001 between the treatment groups at the given time point. Data points represent individual biological replicates. Mito-RoGFP, mitochondria-targeted redox-sensitive GFP; ROS, reactive oxygen species; SFK, Src family kinase.
To evaluate the role of mitochondrial ROS generation in homocysteine-mediated neurotoxicity, neuronal cultures were treated with L-homocysteine (50 μM, 18 h) in the presence of mitoTEMPO, and cell death was assessed by Hoechst DNA staining. The representative photomicrographs and quantitative analysis of the pyknotic nuclei show significant reduction in homocysteine-induced neuronal cell death in the presence of mitoTEMPO (Fig. 8A). Since GluN2A-NMDAR–mediated activation of Pyk2/SFK/ERK MAPK signaling cascade contributes to homocysteine-induced mitochondrial ROS generation, we further evaluated their role in homocysteine-dependent neurotoxicity. For these studies, neuronal cultures were treated with L-homocysteine (50 μM) in the presence of GluN2A-NMDAR inhibitor, Pyk2 inhibitor, SFK inhibitor or ERK MAPK inhibitor followed by cell death assay. Figure 8B shows that the inhibition of both GluN2A-NMDAR and ERK MAPK leads to significant reduction in neuronal cell death, which is consistent with our earlier findings (21, 23). In addition, pharmacological inhibition of Pyk2 and SFK attenuates homocysteine-mediated neuronal cell death. In additional studies, neuronal cultures were treated with L-homocysteine (50 μM) in the presence of Ro25-6981 to evaluate the role of GluN2B-NMDAR in homocysteine-dependent neurotoxicity. The findings show that the inhibition of GluN2B-NMDARs fails to reduce homocysteine-induced neuronal cell death (Fig. 8B).
Figure 8.

Homocysteine-GluN2A-NMDAR–induced neurotoxicity involves Pyk2 and SFK activation and mitochondrial ROS generation.A and B, rat neuronal cultures were treated with 50 μM of L-homocysteine (L-Hcy) for 18 h in the presence of (A) mitoTEMPO (5 μM); (B) NVP-AAM077 (30 nM), PD98059 (15 μM), PF431396 (5 μM), PP2 (5 μM), or Ro25-6981 (5 μM). Representative photomicrographs of neurons stained with nuclear stain Hoechst 33342 (blue) showing pyknotic DNA (indicated with arrows). The percentage of neurons with pyknotic nuclei are expressed as mean ± SD. One-way ANOVA reveal significant main effect of treatment on cell death in the presence of (A) mitoTEMPO: F (2, 57) = 124.8; p < 0.0001 and (B) other pharmacological inhibitors: F (6, 123) = 55.95; p < 0.0001. Post hoc analysis by Bonferroni’s multiple-comparisons test shows ∗p < 0.0001 between the treatment groups. Data points represent individual biological replicates. NMDAR, N-methyl-D-aspartate subtype of glutamate receptor; ROS, reactive oxygen species; SFK, Src family kinase.
Discussion
The findings from the current study demonstrate for the first time the role of GluN2A-NMDARs in mitochondrial ROS generation and provide a critical insight into the molecular mechanisms underlying homocysteine-induced mitochondrial ROS generation and neurotoxicity (Fig. 9). We identified a unique signaling cascade where homocysteine-GluN2A-NMDAR–mediated initial Ca2+ influx and rapid activation of ERK MAPK leads to sequential phosphorylation and activation of Pyk2 kinase and SFKs. In contrast to the NMDAR agonist glutamate, homocysteine fails to activate the regulatory phosphatase STEP61, which is known to limit the duration of Pyk2 and SFKs activation upon NMDAR stimulation, thus enabling the activation of a positive feedback loop where unregulated Pyk2 and SFKs activation helps to prolong GluN2A-NMDAR stimulation and continued Ca2+ influx, resulting in sustained ERK MAPK phosphorylation (Fig. 9). Such sustained ERK MAPK phosphorylation is essential for homocysteine-GluN2A-NMDAR–mediated increased mitochondrial ROS generation. The study highlights the involvement of a complex signaling cascade in GluN2A-NMDAR–mediated mitochondrial ROS generation that results in neuronal death (Fig. 9).
Figure 9.
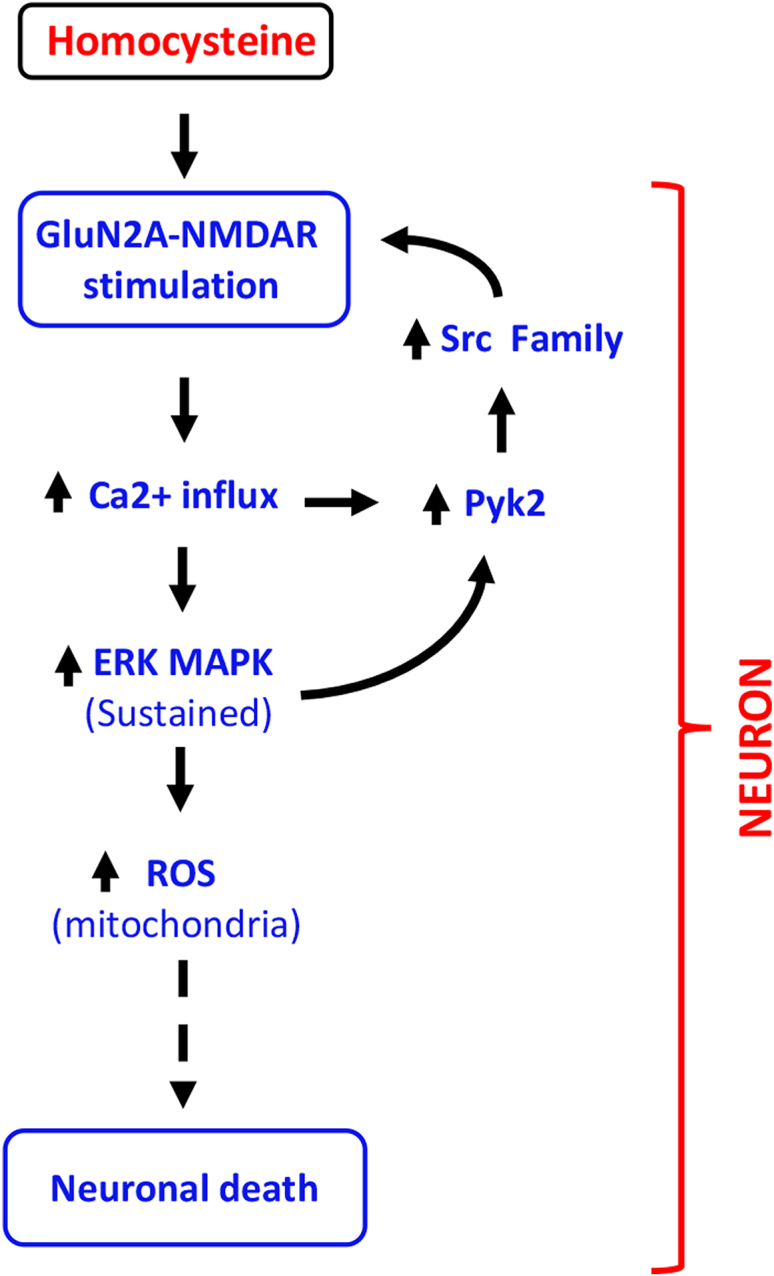
Schematic representation of homocysteine-GluN2A-NMDAR–mediated intracellular signaling. Homocysteine-mediated GluN2A-NMDAR stimulation leads to initial Ca2+ influx, triggering sequential activation of Pyk2 and Src family kinase. This in turn leads to phosphorylation of GluN2A-NMDAR-Tyr1325, thereby enhancing channel activity and further influx of Ca2+. The GluN2A-NMDAR–mediated initial influx of Ca2+ also leads to sustained activation of ERK MAPK that causes mitochondrial ROS generation. The sustained ERK MAPK activation also promotes Pyk2/Src kinase activation further contributing to the cycle of signaling events that eventually results to neuronal death. NMDAR, N-methyl-D-aspartate subtype of glutamate receptor; ROS, reactive oxygen species; SFK, Src family kinase.
Reversible phosphorylation of NMDAR subunits modulates channel function and its downstream signaling pathways (31). Phosphorylation of specific NMDAR subunits are known to upregulate, while dephosphorylation of these subunits downregulate NMDAR channel function. Specifically, the SFKs have been shown to play a critical role in upregulating glutamate-mediated NMDAR channel activity (31). Electrophysiological studies have showed that intracellular application of phosphorylated Src kinase during patch clamp recording enhanced NMDAR currents, while application of protein tyrosine kinase inhibitors depressed NMDAR currents and reduced intracellular Ca2+ concentrations (33, 51, 52, 53). Similarly, bath application of protein tyrosine phosphatase inhibitors enhanced NMDAR currents, while introducing exogenous protein tyrosine phosphatase suppressed NMDAR currents (54). Collectively, these findings indicate that any disbalance between the activity of the tyrosine kinases and tyrosine phosphatases could lead to dysregulation of NMDAR channel function, thereby triggering intracellular signaling cascades that are detrimental to neuronal survival. Biochemical studies have further shown that SFKs phosphorylate several tyrosine residues in the cytoplasmic tail of GluN2A-NMDAR (Tyr842, Tyr1292, Tyr1325, and Tyr1387) and GluN2B-NMDAR (Tyr1252, Tyr1336, and Tyr1472), resulting in receptor stabilization on the cell surface, channel activation, and influx of Ca2+ (31, 55). In this context, earlier studies have shown that while Fyn, a member of the SFKs can phosphorylate both GluN2A and GluN2B-NMDAR in the postsynaptic densities of rat forebrain, it selectively modulates the activity of GluN2B NMDAR (40, 55, 56, 57, 58, 59, 60, 61, 62, 63). On the other hand, Src, another member of the SFKs, phosphorylates multiple tyrosine residues in GluN2A-NMDAR to activate the receptor complex (64, 65, 66). In addition to SFKs, Pyk2 is also involved in upregulating NMDAR surface expression, phosphorylation of Tyr1472 in GluN2B-NMDAR, association of GluN2B-NMDAR subunits with postsynaptic density-95 (PSD-95) scaffolding protein, and enhancing NMDAR currents (35, 67, 68, 69, 70, 71). Evidence further suggest that Pyk2-mediated NMDAR activation requires Src kinase activation. Mutational studies along with electrophysiological analysis have shown that sequential activation of Pyk2 and SFKs leads to tyrosine phosphorylation of NMDARs and enhance its function (35). In the absence of a stimulus, Pyk2 remains in a closed conformation due to intramolecular interactions between different domains of Pyk2. Following glutamate-NMDAR stimulation, influx of Ca2+ leads to dissociation of the intramolecular interactions in Pyk2, allowing it to bind to Ca2+/calmodulin and facilitating its clustering to PSD-95. This results in autophosphorylation of Pyk2 at Tyr402, which in turn promotes high-affinity binding of the activated Pyk2 to the SH2 domain of SFKs. Such binding disrupts the intramolecular interactions in SFKs that helps maintain SFKs in a low-activity state, thereby allowing phosphorylation of SFKs at Tyr416. The activated SFKs subsequently phosphorylate NMDAR subunits to enhance NMDAR channel activity (72, 73, 74, 75). Although multiple evidence suggest that glutamate-mediated NMDAR stimulation leads to the formation of functional NMDAR complex between Pyk2/Src/PSD95 and either GluN2A- or GluN2B-NMDARs, most of the intracellular signaling cascades activated following binding of Pyk2/Src/PSD95 to NMDARs described till date involves GluN2B-NMDAR stimulation. Furthermore, several reports now demonstrate that although homocysteine is an agonist of NMDAR, the effect of homocysteine-mediated NMDAR stimulation is quite different from glutamate-mediated NMDAR stimulation (20, 21, 23). While glutamate mediates its effects by stimulating both GluN2A and GluN2B NMDARs, homocysteine mediates its effects by stimulating primarily the GluN2A-NMDARs. More importantly, in contrast to glutamate, homocysteine-induced neuronal death is mediated through GluN2A-NMDAR stimulation (20, 21, 23). In this context, the current study delineates the intracellular signaling mechanism involved in homocysteine-mediated sustained GluN2A-NMDAR stimulation. The findings show that homocysteine-mediated initial influx of Ca2+ through GluN2A-NMDARs triggers autophosphorylation and activation of Pyk2. Active Pyk2 facilitates increased phosphorylation of SFKs, which in turn leads to phosphorylation of GluN2A-NMDAR at Tyr1325, thereby increasing channel activity and further influx of Ca2+. The continuity of this cycle of events leads to sustained influx of Ca2+ through GluN2A-NMDAR and could be attributed to the lack of activation of an inhibitory tyrosine phosphatase that could counter Pyk2 and/or SFKs to decrease tyrosine phosphorylation of GluN2A-NMDARs.
Earlier studies indicate that several protein tyrosine phosphatases, including PTPα, PTPMEG, PTP1D, and STEP61, are components of the NMDAR complex and play a role in regulating NMDAR phosphorylation status and activity (39, 60, 76, 77, 78). Although the association of PTPα, PTPMEG, and PTP1D with the NMDAR complex has been correlated with tyrosine phosphorylation and upregulation of NMDAR activity (31), STEP61 is known to downregulate NMDAR function by opposing the activation of Pyk2 and SFKs. After the glutamate-mediated NMDAR stimulation, the activation of STEP61 in neurons leads to dephosphorylation of Tyr402 in Pyk2 (38), which in turn downregulates the activation of SFKs and NMDAR function. Consistent with this finding, genetic deletion of STEP in neurons has been shown to increase phosphorylation of Try402 in Pyk2 (79). In addition, active STEP has also been shown to inhibit glutamate-NMDAR–mediated activation of SFKs through dephosphorylation of Tyr420 (39, 40). However, our findings now show that homocysteine-mediated NMDAR activation fails to activate STEP61, which is not surprising as homocysteine preferentially stimulates the GluN2A-NMDAR (20, 22, 23), whereas activation of STEP has been shown to be downstream of glutamate-mediated GluN2B-NMDAR stimulation (9, 45). Therefore, in the absence of an active tyrosine phosphatase to limit the phosphorylation of Pyk2 and SFKs, exposure to homocysteine prolongs GluN2A-NMDAR channel activity resulting in sustained Ca2+ influx.
In addition to Pyk2 and SFKs, ERK MAPK is also a known substrate of STEP. It has been shown that the duration of ERK MAPK activation after glutamate-NMDAR stimulation is regulated through dephosphorylation of Tyr204 in ERK MAPK by active STEP, resulting in transient ERK MAPK activation (43). The magnitude and duration of ERK MAPK phosphorylation determines whether neurons survive or die (80). Glutamate-NMDAR–mediated transient ERK MAPK activation has been implicated in neuronal survival and synaptic plasticity (2, 43, 81, 82), whereas sustained ERK MAPK activation following exposure to a diverse array of extracellular stimuli has been shown to cause neuronal death (83, 84, 85). However, the precise role of NMDAR subunits in such sustained ERK MAPK activation is still not well understood. In this context, our earlier studies have demonstrated that homocysteine mediated GluN2A-NMDAR stimulation leads to sustained ERK MAPK phosphorylation that results in neuronal death (20, 21). The current study expands on these earlier findings to delineate the intracellular signaling pathway involved in such sustained ERK MAPK activation. The findings show that the activation of the upstream kinases Pyk2 and SFKs facilitates GluN2A-NMDAR–mediated Ca2+ influx, which in conjunction with the lack of activation of STEP61 contributes to the sustained ERK MAPK phosphorylation observed in our study.
Another potentially important finding from our study is the role of GluN2A-NMDARs in mitochondrial ROS generation. Earlier studies have implicated the role of ROS in homocysteine-induced neuronal death (13, 48, 86, 87). However, the precise mechanism and the cellular source of such ROS generation are still unclear. Using a redox-sensitive genetic probe targeted to the mitochondrial matrix (mito-RoGFP) and mitochondria-targeted superoxide scavenger (mitoTempo), our findings now demonstrate that neuronal exposure to elevated levels of homocysteine leads to mitochondrial ROS generation. The findings also demonstrate a crucial role of GluN2A-NMDAR in homocysteine-mediated mitochondrial ROS generation. Although the current study highlights the role of GluN2A-NMDARs in homocysteine-induced mitochondrial ROS generation, an earlier study has shown that inhibiting GluN2A-NMDAR function following an excitotoxic insult with glutamate increases endogenous ROS generation and oxidative stress (88). Since glutamate leads to sequential activation of both GluN2A- and GluN2B-NMDARs and the detrimental effects glutamate are primarily mediated through GluN2B-NMDARs, the earlier findings from Zhu et al. imply that upregulation of GluN2A-NMDAR could protect neurons against glutamate-induced oxidative stress. Collectively, these findings further confirm that glutamate and homocysteine mediated NMDAR stimulation can have opposing effects on GluN2A-NMDAR and neuronal function. We further show that the sustained activation of ERK MAPK serves as the intermediary signaling pathway between GluN2A-NMDAR stimulation and mitochondrial ROS generation. Although ROS have been suggested to have an important role in sustaining ERK MAPK signaling, the role of ERK MAPK in regulating mitochondrial ROS generation in neurons has not been reported earlier. As such, these findings not only provide insight into the signaling pathways involved in homocysteine-induced sustained GluN2A-NMDAR stimulation but also demonstrate a new relationship between mitochondrial ROS generation and GluN2A-NMDAR activation.
Experimental procedures
Materials and reagents
Pregnant female Sprague-Dawley rats were purchased from Envigo. GluN2A-subunit KO mice (GluN2A KO) obtained from Dr Andrew Holmes, NIH/NIAAA (89), were bred and made time-pregnant at the animal facility of University of New Mexico. The Institutional Animal Care and Use Committee of University of New Mexico, HSC, approved all animal procedures. L-homocysteine thiolactone hydrochloride, glycine, EGTA, Hoechst 33342, DL-AP5 (DL-2-amino-5-phosphopentanoic acid), NVP-AAM077 ([(R)-[(S)-1-(4-bromo-phenyl)-ethylamino]-(2,3-dioxo-1,2,3,4-tetrahydro-quinoxalin-5-yl)-methyl]phosphonic acid), Ro 25-6981, PF431396, PP2, and mitoTEMPO ((2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl)triphenylphosphonium chloride) were purchased from Millipore Sigma. Fluo-3 acetooxymethyl ester (Fluo3-AM) and all cell culture reagents were purchased from Invitrogen. Anti-phospho-ERK1/2 (Thr202/Tyr204) monoclonal antibody (pERK), anti-ERK2 polyclonal antibody (ERK), anti-phospho-Pyk2 (Tyr402) polyclonal antibody, anti-phospho-SFK (Tyr416) polyclonal antibody (p-Src), anti-SFK monoclonal antibody, and anti-rabbit and anti-mouse horseradish peroxidase–conjugated secondary antibodies were purchased from Cell Signaling Technology. Anti-Pyk2 polyclonal antibody was obtained from Abcam. The 35-mm tissue culture dishes were obtained from MatTek Corporation and the poly-D-lysine-coated 60 mm dishes, and 4-well culture slides were obtained from Corning BD Biocoat.
Neuron culture, L-homocysteine preparation, and stimulation
Embryos obtained from pregnant Sprague Dawley rats (16- to 17-day gestation), WT or GluN2A KO mice (15- to 16-day gestation) were used to establish primary cortical neuronal cultures as described earlier (20, 21, 90). For live-cell imaging involving assessment of Ca2+ and ROS levels, neurons were grown on glass-bottom 35 mm culture dishes coated overnight with poly-D-lysine (50 μg/ml) and Laminin (10 μg/ml). For biochemical studies, neurons were grown on poly-D-lysine-coated 60 mm dishes and for cell death assay, neurons were grown on poly-D-lysine-coated 4-well culture slides. Neuronal cultures were maintained in Modified Eagles Medium supplemented with 5% fetal bovine serum and antibiotic/antimycotic solution for 12 to 13 days. On the day of the experiment, the cells were treated with L-homocysteine (50 μM) in Hank’s balanced salt solution (HBSS) containing 50 μM of glycine as described previously (13, 20, 21, 90). L-homocysteine was prepared fresh each time from L-homocysteine thiolactone hydrochloride as previously described (20, 21). In some experiments, pharmacological inhibitors (DL-AP5, 200 μM; NVP-AAM077, 30 nM; Ro 25-6981, 5 μM; PF431396, 5 μM; PP2, 5 μM; or mitoTEMPO, 5 μM) were added 15 min prior to or 30 min after the onset of L-homocysteine treatment and maintained for the specified time periods. The cells were then subjected to either live-cell imaging for intracellular Ca2+ and ROS measurements or immunoblotting or cell death assay as described below.
Calcium measurements
Intracellular Ca2+ in neurons was determined using the fluorescent indicator, Fluo3-AM. Neuronal cultures (12–13 days) from rat embryos and maintained in phenol red free HBSS were loaded with Fluo3-AM (4 μM, solubilized in dimethyl sulfoxide) for 30 min at 37 °C. Neurons were then maintained in HBSS only (30 min, 37 °C) to allow complete de-esterification of intracellular AM esters according to manufacturer’s protocol. Fluo3-AM loaded cells were treated with vehicle (saline) or L-homocysteine (50 μM) in the absence or presence of pharmacological inhibitors as specified in each experiment. Time lapse live-cell imaging was performed using a Nikon Ti Eclipse inverted microscope equipped with Tokai Hit stage top incubator maintained at 37 °C and infused with 95% air and 5% CO2 mixture. Fields of 5 to 8 cells were imaged using a 40× oil immersion objective (Nikon). Fluorescence excitations (488 nm) were provided with Sutter LB-LS/30 Lambda xenon arc lamp, and fluorescence emissions were captured using a charged-coupled device (CCD) camera from Photometrics. NIS Elements AR software (https://www.nisoftware.net/NikonSaleApplication/) was used for data acquisition and analysis. Data from regions of interest were corrected for background.
Measurement of mitochondrial ROS
Primary neuronal cultures (8–9 days) from rat or mice embryos grown on glass-bottom 35-mm diameter culture dishes were transduced with adenovirus particles containing mito-RoGFP construct obtained from Viraquest Inc with permission from Dr Paul Schumacker, Northwestern University Feinberg School of Medicine (49). The RoGFP protein biosensor contains two engineered cysteine thiols created by introducing four mutations (C48S, Q80R, S147C, and Q204C) in the mammalian GFP expression vector (pEGFP-N1). The 48-bp region encoding the mitochondrial targeting sequence from cytochrome oxidase subunit IV was attached to the 5′ end of the coding sequence of RoGFP, followed by ligation into the VQ Ad5CMV K-NpA adenoviral shuttle vector to generate the mito-RoGFP construct (49). In normal conditions (reduced environment), the fluorescence intensity of the expressed mito-RoGFP is high at excitation maximum 484 nm, which decreases in the presence of ROS (oxidized environment) in the mitochondrial matrix (49). The adenoviral particles were transduced according to protocol suggested by Viraquest Inc. Briefly, 8 to 9 days old neuronal cultures were incubated with 2.5 μl of stock virus containing 1 × 109 particles in 1 ml of original growth media and incubated for 16 to 18 h. Following incubation, the media containing the viral particles were removed and replaced with the original growth media. The cells were maintained in the culture media for another 4 to 5 days for expression of mito-RoGFP. For live-cell imaging, the culture media were replaced with HBSS media without phenol red and subjected to fluorescence imaging at 484 nm wavelength using a Nikon Ti Eclipse inverted microscope equipped with Tokai Hit stage top incubator maintained at 37 °C and infused with 95% air and 5% CO2 mixture. Baseline photomicrographs were acquired for 5 to 8 cells per field with 40× oil immersion objective and at 484 nm. L-Homocysteine (50 μM) or vehicle (saline) was added to the cells, and time-lapse microscopy was performed every 15 min for the first 30 min and every half hour thereafter. As positive control, a subset of mito-RoGFP–expressing neurons from rat cultures were treated with H2O2 (50 μM) and time-lapse microscopy was performed every 30 s for 10 min. In some studies, selective pharmacological inhibitors were added to the cells either 15 min prior to or 30 min after onset of L-homocysteine treatment and maintained for the duration of time-lapse microscopy, performed as described above. Relative fluorescent intensity was estimated using NIS Elements AS software and expressed as arbitrary units at 484 nm. The decrease in mito-RoGFP fluorescence intensity was taken as a measure of mitochondrial ROS generation and expressed as percent oxidized relative to baseline control.
Immunoblotting
Primary neuronal cultures from rat and mice embryos were treated with or without L-homocysteine in the presence or absence of pharmacological inhibitors. The cells were harvested in SDS sample buffer (91). Equal protein from the cell lysates were resolved by SDS-PAGE (7.5%) and subjected to immunoblotting procedures as described earlier (20, 21, 43). The blots were analyzed with anti-pERK, anti-ERK, anti-pPyk2, anti-Pyk2, anti-pSrc family, and anti-Src family antibodies according to manufacturer’s protocol. Densitometric analyses of the images captured on X-ray films were performed using the Image J software (https://imagej.net/ij/).
Hoechst DNA staining
Neuronal cultures obtained from rat embryos were treated with L-homocysteine for 18 h in the presence or absence of mitoTEMPO (5 μM), PF431396 (5 μM), PP2 (5 μM) DL-AP5 (200 μM), NVP-AAM077 (30 nM), PD98059 (15 μM), or Ro 25-6951 (5 μM). Cells were fixed with 4% paraformaldehyde and incubated with Hoechst 33342 dye as described earlier (20). Percentage of pyknotic nuclei was quantitatively assessed by fluorescent microscopy to determine the extent of neuronal death. A total of 1500 cells were counted from at least 15 fields for each experimental condition (n = 4).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software (version 10.2.1; https://www.graphpad.com). Immunoblot data evaluating the effects of drug treatment on protein phosphorylation at a given time point were analyzed by computer assisted densitometry and Image J analysis. Quantitative effects of drug treatment were assessed by one-way ANOVA, followed by Bonferroni’s multiple comparison test. For evaluating the effects of drug treatment on changes in intracellular Ca2+ level and mitochondrial ROS generation over time, data were analyzed using two-way repeated measures ANOVA with drug treatment and time as the two factors, followed by Bonferroni’s post hoc test for detailed group comparisons. Effects of drug treatment on cell death were assessed by one-way ANOVA, followed by Bonferroni’s multiple comparison test. Greenhouse-Geisser correction was applied, where test of sphericity failed. Data are expressed as mean ± SD and significance was set at p <0.05.
Data availability
All data are contained within the article.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgments
We thank Dr Andrew Holmes for coordinating and providing us the GluN2A-KO mice.
Author contributions
S. N. D., S. S., and R. P. investigation; S. N. D., S. S., and R. P. methodology; S. N. D., S. S., S. P., and R. P. formal analysis; S. N. D., S. S., S. P., and R. P. writing-review and editing; S. P. and R. P. conceptualization; S. P. and R. P. validation; R. P. writing-original draft; R. P. supervision; R. P. funding acquisition.
Funding and additional information
This work was supported by the National Institute of Neurological Disorders and Stroke grants: R01 NS083914 (R. P.) and R01 NS059962 (S. P.).
Reviewed by members of the JBC Editorial Board. Edited by Roger Colbran
References
- 1.Hardingham G.E., Fukunaga Y., Bading H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 2.Kim M.J., Dunah A.W., Wang Y.T., Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- 3.Li B., Chen N., Luo T., Otsu Y., Murphy T.H., Raymond L.A. Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat. Neurosci. 2002;5:833–834. doi: 10.1038/nn912. [DOI] [PubMed] [Google Scholar]
- 4.Liu Y., Wong T.P., Aarts M., Rooyakkers A., Liu L., Lai T.W., et al. NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci. 2007;27:2846–2857. doi: 10.1523/JNEUROSCI.0116-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang S.J., Steijaert M.N., Lau D., Schutz G., Delucinge-Vivier C., Descombes P., et al. Decoding NMDA receptor signaling: identification of genomic programs specifying neuronal survival and death. Neuron. 2007;53:549–562. doi: 10.1016/j.neuron.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 6.Chen M., Lu T.J., Chen X.J., Zhou Y., Chen Q., Feng X.Y., et al. Differential roles of NMDA receptor subtypes in ischemic neuronal cell death and ischemic tolerance. Stroke. 2008;39:3042–3048. doi: 10.1161/STROKEAHA.108.521898. [DOI] [PubMed] [Google Scholar]
- 7.Riccio A., Ginty D.D. What a privilege to reside at the synapse: NMDA receptor signaling to CREB. Nat. Neurosci. 2002;5:389–390. doi: 10.1038/nn0502-389. [DOI] [PubMed] [Google Scholar]
- 8.Terasaki Y., Sasaki T., Yagita Y., Okazaki S., Sugiyama Y., Oyama N., et al. Activation of NR2A receptors induces ischemic tolerance through CREB signaling. J. Cereb. Blood Flow Metab. 2010;30:1441–1449. doi: 10.1038/jcbfm.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paul S., Connor J.A. NR2B-NMDA receptor-mediated increases in intracellular Ca2+ concentration regulate the tyrosine phosphatase, STEP, and ERK MAP kinase signaling. J. Neurochem. 2010;114:1107–1118. doi: 10.1111/j.1471-4159.2010.06835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch D.R., Guttmann R.P. Excitotoxicity: perspectives based on N-methyl-D-aspartate receptor subtypes. J. Pharmacol. Exp. Ther. 2002;300:717–723. doi: 10.1124/jpet.300.3.717. [DOI] [PubMed] [Google Scholar]
- 11.Scimemi A., Fine A., Kullmann D.M., Rusakov D.A. NR2B-containing receptors mediate cross talk among hippocampal synapses. J. Neurosci. 2004;24:4767–4777. doi: 10.1523/JNEUROSCI.0364-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vicini S., Wang J.F., Li J.H., Zhu W.J., Wang Y.H., Luo J.H., et al. Functional and pharmacological differences between recombinant N-methyl-D-aspartate receptors. J. Neurophysiol. 1998;79:555–566. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- 13.Lipton S.A., Kim W.K., Choi Y.B., Kumar S., D'Emilia D.M., Rayudu P.V., et al. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. U. S. A. 1997;94:5923–5928. doi: 10.1073/pnas.94.11.5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sharma M., Tiwari M., Tiwari R.K. Hyperhomocysteinemia: impact on neurodegenerative diseases. Basic Clin. Pharmacol. Toxicol. 2015;117:287–296. doi: 10.1111/bcpt.12424. [DOI] [PubMed] [Google Scholar]
- 15.Seshadri S., Beiser A., Selhub J., Jacques P.F., Rosenberg I.H., D'Agostino R.B., et al. Plasma homocysteine as a risk factor for dementia and Alzheimer's disease. N. Engl. J. Med. 2002;346:476–483. doi: 10.1056/NEJMoa011613. [DOI] [PubMed] [Google Scholar]
- 16.Zoccolella S., Martino D., Defazio G., Lamberti P., Livrea P. Hyperhomocysteinemia in movement disorders: current evidence and hypotheses. Curr. Vasc. Pharmacol. 2006;4:237–243. doi: 10.2174/157016106777698414. [DOI] [PubMed] [Google Scholar]
- 17.Hankey G.J., Eikelboom J.W. Homocysteine and stroke. Curr. Opin. Neurol. 2001;14:95–102. doi: 10.1097/00019052-200102000-00015. [DOI] [PubMed] [Google Scholar]
- 18.Zoccolella S., Bendotti C., Beghi E., Logroscino G. Homocysteine levels and amyotrophic lateral sclerosis: a possible link. Amyotroph. Lateral Scler. 2010;11:140–147. doi: 10.3109/17482960902919360. [DOI] [PubMed] [Google Scholar]
- 19.Ansari R., Mahta A., Mallack E., Luo J.J. Hyperhomocysteinemia and neurologic disorders: a review. J. Clin. Neurol. 2014;10:281–288. doi: 10.3988/jcn.2014.10.4.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poddar R., Paul S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 2009;110:1095–1106. doi: 10.1111/j.1471-4159.2009.06207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poddar R., Paul S. Novel crosstalk between ERK MAPK and p38 MAPK leads to homocysteine-NMDA receptor-mediated neuronal cell death. J. Neurochem. 2013;124:558–570. doi: 10.1111/jnc.12102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sibarov D.A., Abushik P.A., Giniatullin R., Antonov S.M. GluN2A subunit-containing NMDA receptors are the preferential neuronal targets of homocysteine. Front. Cell. Neurosci. 2016;10:246. doi: 10.3389/fncel.2016.00246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deep S.N., Mitra S., Rajagopal S., Paul S., Poddar R. GluN2A-NMDA receptor-mediated sustained Ca(2+) influx leads to homocysteine-induced neuronal cell death. J. Biol. Chem. 2019;294:11154–11165. doi: 10.1074/jbc.RA119.008820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jindal A., Rajagopal S., Winter L., Miller J.W., Jacobsen D.W., Brigman J., et al. Hyperhomocysteinemia leads to exacerbation of ischemic brain damage: role of GluN2A NMDA receptors. Neurobiol. Dis. 2019;127:287–302. doi: 10.1016/j.nbd.2019.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharjee N., Borah A. Oxidative stress and mitochondrial dysfunction are the underlying events of dopaminergic neurodegeneration in homocysteine rat model of Parkinson's disease. Neurochem. Int. 2016;101:48–55. doi: 10.1016/j.neuint.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 26.Bukharaeva E., Shakirzyanova A., Khuzakhmetova V., Sitdikova G., Giniatullin R. Homocysteine aggravates ROS-induced depression of transmitter release from motor nerve terminals: potential mechanism of peripheral impairment in motor neuron diseases associated with hyperhomocysteinemia. Front. Cell. Neurosci. 2015;9:391. doi: 10.3389/fncel.2015.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tombes R.M., Borisy G.G. Intracellular free calcium and mitosis in mammalian cells: anaphase onset is calcium modulated, but is not triggered by a brief transient. J. Cell Biol. 1989;109:627–636. doi: 10.1083/jcb.109.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poenie M., Tsien R. Fura-2: a powerful new tool for measuring and imaging [Ca2+]i in single cells. Prog. Clin. Biol. Res. 1986;210:53–56. [PubMed] [Google Scholar]
- 29.Malgaroli A., Milani D., Meldolesi J., Pozzan T. Fura-2 measurement of cytosolic free Ca2+ in monolayers and suspensions of various types of animal cells. J. Cell Biol. 1987;105:2145–2155. doi: 10.1083/jcb.105.5.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang M., Leonard J.P. Identification of mouse NMDA receptor subunit NR2A C-terminal tyrosine sites phosphorylated by coexpression with v-Src. J. Neurochem. 2001;77:580–588. doi: 10.1046/j.1471-4159.2001.00255.x. [DOI] [PubMed] [Google Scholar]
- 31.Salter M.W., Kalia L.V. Src kinases: a hub for NMDA receptor regulation. Nat. Rev. Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 32.Zheng Z.B., Nagai S., Iwanami N., Kobayashi A., Hijikata M., Natori S., et al. Selective inhibition of Src protein tyrosine kinase by analogues of 5-S-glutathionyl-beta-alanyl-L-dopa. Chem. Pharm. Bull. (Tokyo) 1998;46:1950–1951. doi: 10.1248/cpb.46.1950. [DOI] [PubMed] [Google Scholar]
- 33.Kohr G., Seeburg P.H. Subtype-specific regulation of recombinant NMDA receptor-channels by protein tyrosine kinases of the src family. J. Physiol. 1996;492:445–452. doi: 10.1113/jphysiol.1996.sp021320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen B.S., Roche K.W. Regulation of NMDA receptors by phosphorylation. Neuropharmacology. 2007;53:362–368. doi: 10.1016/j.neuropharm.2007.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang Y., Lu W., Ali D.W., Pelkey K.A., Pitcher G.M., Lu Y.M., et al. CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron. 2001;29:485–496. doi: 10.1016/s0896-6273(01)00220-3. [DOI] [PubMed] [Google Scholar]
- 36.Sheng M., Kim M.J. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298:776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- 37.Tian M., Xu J., Lei G., Lombroso P.J., Jackson M.F., MacDonald J.F. STEP activation by Galphaq coupled GPCRs opposes Src regulation of NMDA receptors containing the GluN2A subunit. Sci. Rep. 2016;6 doi: 10.1038/srep36684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J., Kurup P., Bartos J.A., Patriarchi T., Hell J.W., Lombroso P.J. Striatal-enriched protein-tyrosine phosphatase (STEP) regulates Pyk2 kinase activity. J. Biol. Chem. 2012;287:20942–20956. doi: 10.1074/jbc.M112.368654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pelkey K.A., Askalan R., Paul S., Kalia L.V., Nguyen T.H., Pitcher G.M., et al. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- 40.Nguyen T.H., Liu J., Lombroso P.J. Striatal enriched phosphatase 61 dephosphorylates Fyn at phosphotyrosine 420. J. Biol. Chem. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- 41.Fitzpatrick C.J., Lombroso P.J. The role of striatal-enriched protein tyrosine phosphatase (STEP) in cognition. Front. Neuroanat. 2011;5:47. doi: 10.3389/fnana.2011.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lombroso P.J., Ogren M., Kurup P., Nairn A.C. Molecular underpinnings of neurodegenerative disorders: striatal-enriched protein tyrosine phosphatase signaling and synaptic plasticity. F1000Res. 2016;5 doi: 10.12688/f1000research.8571.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paul S., Nairn A.C., Wang P., Lombroso P.J. NMDA-mediated activation of the tyrosine phosphatase STEP regulates the duration of ERK signaling. Nat. Neurosci. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 44.Paul S., Snyder G.L., Yokakura H., Picciotto M.R., Nairn A.C., Lombroso P.J. The dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J. Neurosci. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poddar R., Deb I., Mukherjee S., Paul S. NR2B-NMDA receptor mediated modulation of the tyrosine phosphatase STEP regulates glutamate induced neuronal cell death. J. Neurochem. 2010;115:1350–1362. doi: 10.1111/j.1471-4159.2010.07035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deb I., Manhas N., Poddar R., Rajagopal S., Allan A.M., Lombroso P.J., et al. Neuroprotective role of a brain-enriched tyrosine phosphatase, STEP, in focal cerebral ischemia. J. Neurosci. 2013;33:17814–17826. doi: 10.1523/JNEUROSCI.2346-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Poddar R., Rajagopal S., Shuttleworth C.W., Paul S. Zn2+-dependent activation of the trk signaling pathway induces phosphorylation of the brain-enriched tyrosine phosphatase STEP: molecular basis for Zn2+-induced Erk MAPK activation the. J. Biol. Chem. 2016;291:813–825. doi: 10.1074/jbc.M115.663468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kruman I.I., Culmsee C., Chan S.L., Kruman Y., Guo Z., Penix L., et al. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000;20:6920–6926. doi: 10.1523/JNEUROSCI.20-18-06920.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Waypa G.B., Marks J.D., Guzy R., Mungai P.T., Schriewer J., Dokic D., et al. Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ. Res. 2010;106:526–535. doi: 10.1161/CIRCRESAHA.109.206334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid. Redox Signal. 2005;7:1140–1149. doi: 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y.T., Salter M.W. Regulation of NMDA receptors by tyrosine kinases and phosphatases. Nature. 1994;369:233–235. doi: 10.1038/369233a0. [DOI] [PubMed] [Google Scholar]
- 52.Yu X.M., Askalan R., Keil G.J., 2nd, Salter M.W. NMDA channel regulation by channel-associated protein tyrosine kinase Src. Science. 1997;275:674–678. doi: 10.1126/science.275.5300.674. [DOI] [PubMed] [Google Scholar]
- 53.Salter M.W., Pitcher G.M. Dysregulated Src upregulation of NMDA receptor activity: a common link in chronic pain and schizophrenia. FEBS J. 2012;279:2–11. doi: 10.1111/j.1742-4658.2011.08390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y.T., Yu X.M., Salter M.W. Ca(2+)-independent reduction of N-methyl-D-aspartate channel activity by protein tyrosine phosphatase. Proc. Natl. Acad. Sci. U. S. A. 1996;93:1721–1725. doi: 10.1073/pnas.93.4.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trepanier C.H., Jackson M.F., MacDonald J.F. Regulation of NMDA receptors by the tyrosine kinase Fyn. FEBS J. 2012;279:12–19. doi: 10.1111/j.1742-4658.2011.08391.x. [DOI] [PubMed] [Google Scholar]
- 56.Nakazawa T., Komai S., Tezuka T., Hisatsune C., Umemori H., Semba K., et al. Characterization of Fyn-mediated tyrosine phosphorylation sites on GluR epsilon 2 (NR2B) subunit of the N-methyl-D-aspartate receptor. J. Biol. Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- 57.Nakazawa T., Tezuka T., Yamamoto T. [Regulation of NMDA receptor function by Fyn-mediated tyrosine phosphorylation] Nihon Shinkei Seishin Yakurigaku Zasshi. 2002;22:165–167. [PubMed] [Google Scholar]
- 58.Suzuki T., Okumura-Noji K. NMDA receptor subunits epsilon 1 (NR2A) and epsilon 2 (NR2B) are substrates for Fyn in the postsynaptic density fraction isolated from the rat brain. Biochem. Biophys. Res. Commun. 1995;216:582–588. doi: 10.1006/bbrc.1995.2662. [DOI] [PubMed] [Google Scholar]
- 59.Cheung H.H., Gurd J.W. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor by exogenous and postsynaptic density-associated Src-family kinases. J. Neurochem. 2001;78:524–534. doi: 10.1046/j.1471-4159.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 60.Tezuka T., Umemori H., Akiyama T., Nakanishi S., Yamamoto T. PSD-95 promotes Fyn-mediated tyrosine phosphorylation of the N-methyl-D-aspartate receptor subunit NR2A. Proc. Natl. Acad. Sci. U. S. A. 1999;96:435–440. doi: 10.1073/pnas.96.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dunah A.W., Sirianni A.C., Fienberg A.A., Bastia E., Schwarzschild M.A., Standaert D.G. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol. Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- 62.Hu J.L., Liu G., Li Y.C., Gao W.J., Huang Y.Q. Dopamine D1 receptor-mediated NMDA receptor insertion depends on Fyn but not Src kinase pathway in prefrontal cortical neurons. Mol. Brain. 2010;3:20. doi: 10.1186/1756-6606-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yaka R., Thornton C., Vagts A.J., Phamluong K., Bonci A., Ron D. NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc. Natl. Acad. Sci. U. S. A. 2002;99:5710–5715. doi: 10.1073/pnas.062046299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Groveman B.R., Xue S., Marin V., Xu J., Ali M.K., Bienkiewicz E.A., et al. Roles of the SH2 and SH3 domains in the regulation of neuronal Src kinase functions. FEBS J. 2011;278:643–653. doi: 10.1111/j.1742-4658.2010.07985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marin V., Groveman B.R., Qiao H., Xu J., Ali M.K., Fang X.Q., et al. Characterization of neuronal Src kinase purified from a bacterial expression system Protein. Expr. Purif. 2010;74:289–297. doi: 10.1016/j.pep.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu X.M., Groveman B.R. Src family kinases in the nervous system. FEBS J. 2012;279:1. doi: 10.1111/j.1742-4658.2011.08423.x. [DOI] [PubMed] [Google Scholar]
- 67.Macdonald D.S., Weerapura M., Beazely M.A., Martin L., Czerwinski W., Roder J.C., et al. Modulation of NMDA receptors by pituitary adenylate cyclase activating peptide in CA1 neurons requires G alpha q, protein kinase C, and activation of Src. J. Neurosci. 2005;25:11374–11384. doi: 10.1523/JNEUROSCI.3871-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heidinger V., Manzerra P., Wang X.Q., Strasser U., Yu S.P., Choi D.W., et al. Metabotropic glutamate receptor 1-induced upregulation of NMDA receptor current: mediation through the Pyk2/Src-family kinase pathway in cortical neurons. J. Neurosci. 2002;22:5452–5461. doi: 10.1523/JNEUROSCI.22-13-05452.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang J.Q., Guo M.L., Jin D.Z., Xue B., Fibuch E.E., Mao L.M. Roles of subunit phosphorylation in regulating glutamate receptor function. Eur. J. Pharmacol. 2014;728:183–187. doi: 10.1016/j.ejphar.2013.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Le H.T., Maksumova L., Wang J., Pallen C.J. Reduced NMDA receptor tyrosine phosphorylation in PTPalpha-deficient mouse synaptosomes is accompanied by inhibition of four src family kinases and Pyk2: an upstream role for PTPalpha in NMDA receptor regulation. J. Neurochem. 2006;98:1798–1809. doi: 10.1111/j.1471-4159.2006.04075.x. [DOI] [PubMed] [Google Scholar]
- 71.Giralt A., Brito V., Chevy Q., Simonnet C., Otsu Y., Cifuentes-Diaz C., et al. Pyk2 modulates hippocampal excitatory synapses and contributes to cognitive deficits in a Huntington's disease model. Nat. Commun. 2017;8 doi: 10.1038/ncomms15592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Park S.Y., Avraham H.K., Avraham S. RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner. J. Biol. Chem. 2004;279:33315–33322. doi: 10.1074/jbc.M313527200. [DOI] [PubMed] [Google Scholar]
- 73.Li X., Dy R.C., Cance W.G., Graves L.M., Earp H.S. Interactions between two cytoskeleton-associated tyrosine kinases: calcium-dependent tyrosine kinase and focal adhesion tyrosine kinase. J. Biol. Chem. 1999;274:8917–8924. doi: 10.1074/jbc.274.13.8917. [DOI] [PubMed] [Google Scholar]
- 74.de Pins B., Mendes T., Giralt A., Girault J.A. The non-receptor tyrosine kinase Pyk2 in brain function and neurological and psychiatric diseases. Front. Synaptic Neurosci. 2021;13 doi: 10.3389/fnsyn.2021.749001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dikic I., Tokiwa G., Lev S., Courtneidge S.A., Schlessinger J. A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature. 1996;383:547–550. doi: 10.1038/383547a0. [DOI] [PubMed] [Google Scholar]
- 76.Kalia L.V., Salter M.W. Interactions between Src family protein tyrosine kinases and PSD-95. Neuropharmacology. 2003;45:720–728. doi: 10.1016/s0028-3908(03)00313-7. [DOI] [PubMed] [Google Scholar]
- 77.Husi H., Ward M.A., Choudhary J.S., Blackstock W.P., Grant S.G. Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat. Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 78.Hironaka K., Umemori H., Tezuka T., Mishina M., Yamamoto T. The protein-tyrosine phosphatase PTPMEG interacts with glutamate receptor delta 2 and epsilon subunits. J. Biol. Chem. 2000;275:16167–16173. doi: 10.1074/jbc.M909302199. [DOI] [PubMed] [Google Scholar]
- 79.Venkitaramani D.V., Moura P.J., Picciotto M.R., Lombroso P.J. Striatal-enriched protein tyrosine phosphatase (STEP) knockout mice have enhanced hippocampal memory. Eur. J. Neurosci. 2011;33:2288–2298. doi: 10.1111/j.1460-9568.2011.07687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shaul Y.D., Seger R. The MEK/ERK cascade: from signaling specificity to diverse functions. Biochim. Biophys. Acta. 2007;1773:1213–1226. doi: 10.1016/j.bbamcr.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 81.Chandler L.J., Sutton G., Dorairaj N.R., Norwood D. N-methyl D-aspartate receptor-mediated bidirectional control of extracellular signal-regulated kinase activity in cortical neuronal cultures. J. Biol. Chem. 2001;276:2627–2636. doi: 10.1074/jbc.M003390200. [DOI] [PubMed] [Google Scholar]
- 82.Sweatt J.D. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J. Neurochem. 2001;76:1–10. doi: 10.1046/j.1471-4159.2001.00054.x. [DOI] [PubMed] [Google Scholar]
- 83.Colucci-D'Amato L., Perrone-Capano C., di Porzio U. Chronic activation of ERK and neurodegenerative diseases. Bioessays. 2003;25:1085–1095. doi: 10.1002/bies.10355. [DOI] [PubMed] [Google Scholar]
- 84.Zhuang S., Schnellmann R.G. A death-promoting role for extracellular signal-regulated kinase. J. Pharmacol. Exp. Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- 85.Stanciu M., Wang Y., Kentor R., Burke N., Watkins S., Kress G., et al. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J. Biol. Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- 86.Sibarov D.A., Boikov S.I., Karelina T.V., Antonov S.M. GluN2 subunit-dependent redox modulation of NMDA receptor activation by homocysteine. Biomolecules. 2020;10:1441. doi: 10.3390/biom10101441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kim W.K., Pae Y.S. Involvement of N-methyl-d-aspartate receptor and free radical in homocysteine-mediated toxicity on rat cerebellar granule cells in culture. Neurosci. Lett. 1996;216:117–120. doi: 10.1016/0304-3940(96)13011-1. [DOI] [PubMed] [Google Scholar]
- 88.Zhu J., Xu S., Li S., Yang X., Yu X., Zhang X. Up-regulation of GluN2A-containing NMDA receptor protects cultured cortical neuron cells from oxidative stress. Heliyon. 2018;4 doi: 10.1016/j.heliyon.2018.e00976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sakimura K., Kutsuwada T., Ito I., Manabe T., Takayama C., Kushiya E., et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature. 1995;373:151–155. doi: 10.1038/373151a0. [DOI] [PubMed] [Google Scholar]
- 90.Poddar R., Chen A., Winter L., Rajagopal S., Paul S. Role of AMPA receptors in homocysteine-NMDAR induced crosstalk between ERK and p38 MAP kinase. J. Neurochem. 2017;142:560–573. doi: 10.1111/jnc.14078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Laemmli U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are contained within the article.