

Abstract
Cisplatin-induced hearing loss is a common side effect of cancer chemotherapy in clinics; however, the mechanism of cisplatin-induced ototoxicity is still not completely clarified. Cisplatin-induced ototoxicity is mainly associated with the production of reactive oxygen species, activation of apoptosis, and accumulation of intracellular lipid peroxidation, which also is involved in ferroptosis induction. In this study, the expression of TfR1, a ferroptosis biomarker, was upregulated in the outer hair cells of cisplatin-treated mice. Moreover, several key ferroptosis regulator genes were altered in cisplatin-damaged cochlear explants based on RNA sequencing, implying the induction of ferroptosis. Ferroptosis-related Gpx4 and Fsp1 knockout mice were established to investigate the specific mechanisms associated with ferroptosis in cochleae. Severe outer hair cell loss and progressive damage of synapses in inner hair cells were observed in Atoh1-Gpx4−/− mice. However, Fsp1−/− mice showed no significant hearing phenotype, demonstrating that Gpx4, but not Fsp1, may play an important role in the functional maintenance of HCs. Moreover, findings showed that FDA-approved luteolin could specifically inhibit ferroptosis and alleviate cisplatin-induced ototoxicity through decreased expression of transferrin and intracellular concentration of ferrous ions. This study indicated that ferroptosis inhibition through the reduction of intracellular ferrous ions might be a potential strategy to prevent cisplatin-induced hearing loss.
Keywords: cisplatin-induced ototoxicity, ferroptosis, Gpx4, luteolin, transferrin, Fsp1
Graphical abstract
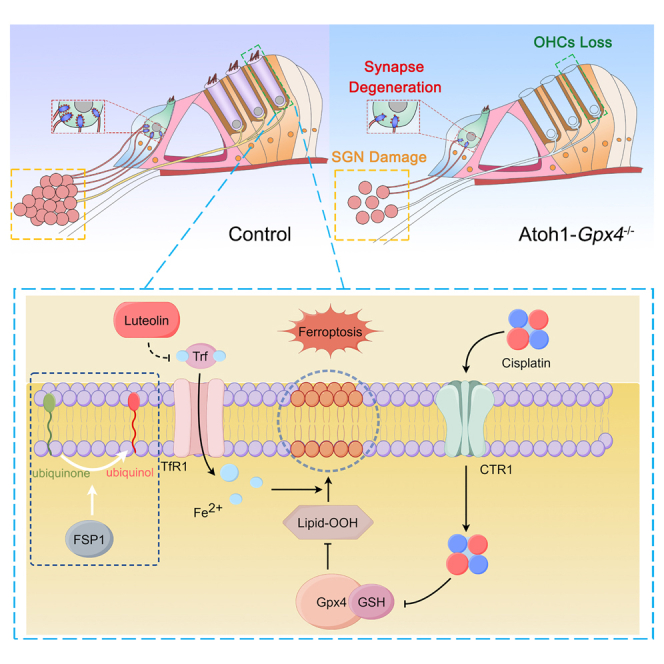
Chai and colleagues highlighted the critical role of Gpx4-mediated ferroptosis in cisplatin-induced hearing loss. Gpx4 deletion in the cochlear hair cells leads to OHC loss through ferroptosis pathway and impaired hearing function. Notably, luteolin could inhibit ferroptosis and protect hair cells, providing a potential strategy to prevent cisplatin-induced ototoxicity.
Introduction
Cisplatin is a highly effective agent that is widely used in the world for treating diverse malignancies, encompassing those affecting the bladder, endometrium, head, kidney, lung, neck, ovary, and testis.1,2,3,4 Nonetheless, cisplatin is associated with severe side effects, such as nephrotoxicity, peripheral neuropathy, and ototoxicity.5,6,7 Notably, cisplatin nephrotoxicity is mild and reversible8,9 and can be effectively prevented through hydration. However, cisplatin-induced ototoxicity is irreversible with its incidence ranging from 20% to 70%.10,11,12,13 Besides, few clinical, well-established, and physicochemical approaches can effectively mitigate and address cisplatin-induced hearing impairment. Moreover, the precise mechanism underlying cisplatin ototoxicity is unclear.
Cisplatin-induced hearing loss (CIHL) manifests as progressive, bilateral, and irreversible hearing loss. Ototoxicity is mainly characterized by deafness, earache,14 and tinnitus.15 Cisplatin primarily damages the outer hair cells (OHCs) and then damages the spiral ganglion neurons (SGNs).16 In addition, cisplatin can induce degeneration of the stria vascularis (SV).17 The primary etiology of CIHL involves the transportation of cisplatin into the endolymph via the copper transporter 1 (CTR1) and organic cation transporter-2 (OCT2) located on the SV.18,19,20,21 CTR1 can regulate homeostasis and transport divalent metal ions in adult mammals,22 while OCT2 primarily facilitates the uptake of drugs that are cations or weak bases under physiological pH conditions.20 Cisplatin accumulation occurs in the SV, leading to disruption of endolymph homeostasis and subsequent inflammation in the hair cells.23 However, hair cells might be more susceptible to cisplatin toxicity than the SV. Numerous studies have demonstrated that CTR1 and OCT2 actively facilitate cisplatin entry into OHCs in vitro and in vivo.24,25,26 In addition, the mechanoelectrical transduction channel and TRPV1, belonging to the transient receptor potential-vanilloid (TRPV) subfamily, mediate the entry of cisplatin into HCs.27,28,29 A persistent generation of reactive oxygen species (ROS) occurs after cisplatin entry into the OHCs due to the inflammatory response, DNA crosslinking, excessive oxidase production, and depletion of endogenous antioxidant systems. These cumulative effects can cause cellular demise, including apoptosis, necrosis, and autophagy.30,31,32,33 Nevertheless, attempts to restore auditory cell damage caused by cisplatin and the subsequent hearing impairment have been ineffective. Therefore, the causative factor and the underlying mechanisms for CIHL require further investigation.
Glutathione (GSH), as a key component of endogenous antioxidant systems, can be combined with cisplatin, and consumed by ROS, inducing the accumulation of lipid peroxide. Besides, the reduction of GSH participates in cisplatin-induced hair cell damage34 and lipid peroxide can trigger ferroptosis.35 Furthermore, Nrf2/HO-1, which is reported to protect against ferroptosis, could regulate the cellular response to oxidative stress and maintain redox homeostasis in cisplatin-induced ototoxicity.36,37 These findings indicate that ferroptosis may be involved in cisplatin-induced ototoxicity. Ferroptosis is a form of cell death dependent on ferrous ions and is characterized by the depletion of GSH and the inactivation of glutathione peroxidase 4 (GPX4).35,38,39,40 GPX4 is an intracellular antioxidant enzyme that inhibits lipid peroxidation in the cell membrane. Moreover, FSP1 has also been reported to be a GPX4-independent inhibitor of ferroptosis, which exerts its effects via reduction of ubiquinol/α-tocopherol on the level of lipid radicals.41 Ferroptosis exhibits distinct biochemical and morphological features that differentiate it from apoptosis, necroptosis, and autophagy.35 The inhibition of lipid peroxidation can hinder ferroptosis, providing a promising approach against cellular loss in various tissues, such as the brain, liver, and kidney, in the context of pathological conditions.38,42 In vivo studies have shown that inhibiting ferroptosis can mitigate cellular damage. However, only a few studies (limited to the verification of cells in vitro) have investigated the role of ferroptosis in cisplatin-induced ototoxicity.43,44 Besides, the detailed molecular mechanism underlying the role of ferroptosis in CIHL is unclear since the cochlear structure is very complex in vivo.
In this study, the mRNA expression level of ferroptosis-related critical genes, such as Gpx4 and Fsp1, were altered in the cochleae of cisplatin-treated mice. Therefore, Gpx4 and Fsp1 knockout mice were established to assess the role of ferroptosis in CIHL. The results showed that Gpx4 deficiency in hair cells, but not Fsp1, caused OHC loss and hearing impairment. Moreover, FDA-approved luteolin can specifically inhibit ferroptosis and alleviate cisplatin-induced ototoxicity by modulating the expression of transferrin (Trf). These findings demonstrate that Gpx4, but not Fsp1, play an important role in the functional maintenance of mouse HCs. In addition, inhibiting ferroptosis can alleviate CIHL, and luteolin can be used as a strong candidate protective drug for treating ototoxicity.
Results
Cisplatin-induced hair cell ferroptosis in both cochlear explants and in vivo
To gain insight into the CIHL, we developed a mouse model of cisplatin-induced ototoxicity that closely mimics clinical treatment regimens.10,45 Wild-type (WT) mice were exposed to two cycles of cisplatin treatment lasting 4 days each, with intervals for a 6-day recovery period between cycles (Figure 1A). In cisplatin-treated mice, the auditory brainstem response (ABR) thresholds were significantly elevated and the body weight was decreased compared with levels before cisplatin treatment (Figures 1B and 1C). These results confirmed that cisplatin induced ototoxicity in mice. The cochleae samples extracted from these mice were collected and stained for myosin7a and transferrin receptor 1 (TfR1), a biomarker for ferroptosis in multiple cell cultures and tissue contexts46 (Figure 1D). It was observed that the expression of TfR1 was increased in the OHCs of cisplatin-treated mice, indicating that cisplatin may induce ferroptosis in the OHCs of mice. Notably, the TfR1 expression was not increased in the inner hair cells (IHCs) of cisplatin-treated mice, suggesting that cisplatin-induced ferroptosis in IHCs may be significantly lower than that of OHCs. Besides, the expression of TfR1 was also not detected in other regions of paraffinized sections of cochleae in cisplatin-treated mice, such as SV and SGNs (data not shown). To exclude contributions from the microenvironment of endolymph to the induction of ferroptosis in OHCs, cochlear explant models were then used to identify ferroptosis-related potential targets. Consistent with previous studies,47 the cochlear explants from P3-P4 C57BL/6 mice were treated with 150 μM cisplatin for 12 and 24 h, which was sufficient to induce toxic effects (the OHCs were damaged, and the loss rate of about 50% after 24 h cisplatin treatment) on hair cells of cochlear explants (Figures 1E and 1F). Interestingly, the morphology of IHCs appeared normal. After 24 h cisplatin treatment, the RNA sequencing was performed in cisplatin or vehicle-treated cochlear explants to explore gene expression, and the results showed that 3,448 genes were upregulated and 7,053 genes were downregulated among 10,501 genes that were significantly altered (false discovery rate < 0.05) in cisplatin-treated cochlear explants compared with vehicle-treated groups (Figure 1G). To explore the ferroptosis biological processes and pathways involved in cisplatin-induced ototoxicity, we analyzed genes associated with the ferroptosis pathway. The results showed that the expression of several ferroptosis-related genes in cisplatin-treated cochlear explants was significantly altered, such as Gpx4, Trf, Slc7a11, and Slc40a1 (Figure 1H). Studies have demonstrated that GPX4 is a central repressor of ferroptosis,40 and its function is dependent on the production of GSH derived from the activation of the cystine-glutamate antiporter SLC7A11.38,48 In addition, Trf and its receptor synergize to promote ferroptosis by importing iron into cells.49 In mammalian cells, SLC40A1 is the sole iron exporter,50,51 and evidence has shown that the upregulation of SLC40A1 mitigates ferroptosis, whereas its downregulation exacerbates ferroptosis.52,53 The mRNA level of the above genes was further confirmed by qPCR in cochlear explants with cisplatin treatment for 12 and 24 h, and the results showed that the expression of Gpx4, Fsp1, Trf, and Slc7a11 increased at first and then decreased, while the expression of Slc40a1 decreased at first and then increased (Figure 1I). Furthermore, the protein levels of Gpx4 were also significantly upregulated in cochlear explants at 12 h after cisplatin treatment and downregulated at 24 h (Figures 1J and 1K). Finally, we investigated the level of GSH associated with cisplatin ototoxicity using ELISA kits, the result showed that the GSH content in cisplatin-treated cochlear explants was higher at 12 h, but significantly lower at 24 h compared with the vehicle-treated group (Figure 1L). These data implied that multiple ferroptosis-related genes, such as Gpx4 and Fsp1, may present a dynamically regulated process in the short-term cisplatin treatment course. However, Gpx4 may show a decreasing trend in the cochleae with long-term cisplatin treatment, which further leads to ferroptosis in OHCs.
Figure 1.

Cisplatin-induced ferroptosis on both cochlear explants and the inner ear of mice
(A) Schematic illustration of the experimental workflow of cisplatin-treated mice. ABR was measured in 6-week-old WT mice before and after cisplatin treatment. Cisplatin (4 mg/kg) was injected intraperitoneally for 4 days and then recovery for 6 days as a cycle (two cycles were performed in this study). (B and C) Cisplatin ototoxicity data showed that the hearing function and body weight of cisplatin-treated mice were decreased compared with the vehicle-treated group, n = 5 for each group. (D) Immunofluorescence analysis of TfR1 in the cochlea of WT mice after cisplatin treatment, the hair cells were labeled with anti-TfR1 (red) and anti-myosin7a (green) antibodies. TfR1-positive hair cells were found only in the cisplatin-treated mice. Scale bar, 20 μm. (E) Immunofluorescence analysis of myosin7a in vehicle-treated and 150 μM cisplatin-treated cochlear explants for 12 and 24 h. After 12 h, the morphology of cochlear explant hair cells altered slightly, but the number was comparable with the control group. In contrast, the morphology and number of OHCs were significantly damaged after 24 h. Scale bar, 20 μm. (F) Statistical analysis of the number of OHCs in (E), n = 4 for each group. (G) After 24 h of cisplatin treatment in cochlear explants, RNA sequencing (RNA-seq) was performed and revealed 10,501 differentially expressed genes compared with the vehicle-treated group. (H) RNA-seq results showed that ferroptosis-related genes were significantly altered. (I) The RNA-seq result of ferroptosis pathway-related genes were verified by qPCR from the cochlear explants treated with vehicle and cisplatin for 12 and 24 h, n = 5 for each group. (J) Western blot was performed to detect the protein level of Gpx4 in cochlear explants at different time points after cisplatin treatment. In the cisplatin-treated group, the Gpx4 content was increased at 12 h and significantly decreased at 24 h compared with the vehicle-treated group. (K) Quantitative analysis of the bands in (J), n = 4 for each group. (L) ELISA was used to detect the content of GSH in the cochlear explants of the vehicle- and cisplatin-treated groups at 12 and 24 h. The result showed that GSH content was higher at 12 h, but significantly lower at 24 h of cisplatin treatment compared with the vehicle-treated group, n ≥ 3 for each group. Statistical values are presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Activation of ferroptosis leads to OHC damage in Atoh1-Gpx4−/− mice
Considering that ferroptosis was involved in CIHL, we explored the effect of ferroptosis on cochlear hair cells. In brief, the cochlear explants from P3-P4 WT mice were treated with 10 μM RSL3, a specific ferroptosis inducer54,55 (Figure 2A). Interestingly, activation of ferroptosis-induced OHC loss after 24 h while the numbers of IHCs and supporting cells did not decrease. This result indicated that OHCs were more susceptible to ferroptosis compared with IHCs and supporting cells, which is consistent with the observations in cisplatin-treated mice. Since Gpx4 was the primary regulator of ferroptosis, and both mRNA and protein expressions of Gpx4 were altered in the cochleae of cisplatin-treated mice, we constructed Gpx4 knockout mice and used them to define the role of ferroptosis in hearing. First, immunofluorescence staining was performed to analyze the expression pattern of Gpx4 (Figure 2B). Results indicated that Gpx4 was expressed in OHCs, IHCs, Deiters’ cells, pillar cells, and Hensen cells, but not SV, suggesting that Gpx4 mainly functions in neurosensory cells. To further define the role of ferroptosis in cochlear hair cells, we generated Atoh1-Cre; Gpx4-floxed (termed Atoh1-Gpx4−/−) mice (Figure 2C). The homozygous Atoh1-Gpx4−/− mice were confirmed by PCR (Figure 2D). Then, the TfR1 expression was analyzed with an immunofluorescence assay in P1 whole-mount apical turn mouse cochlea. It was found to be significantly increased only in OHCs of Atoh1-Gpx4−/− mice (Figure 2E). This indicated that Gpx4 deletion resulted in the activation of ferroptosis of OHCs which caused OHC loss. Next, transmission electron microscopy (TEM) and scanning electron microscopy (SEM) were used to further examine the effect of ferroptosis on the subcellular structure of hair cells. On the TEM, Atoh1-Gpx4−/− mice showed reduced mitochondrial cristae and rupture of the outer mitochondrial membrane (red arrows) and cell membrane (yellow triangles) in OHCs (Figure 2F). This was consistent with the notion that both cell membrane rupture and mitochondrial damage are specific hallmarks of ferroptosis.56,57 However, the IHCs of Atoh1-Gpx4−/− mice exhibited a normal subcellular structure (Figure 2G), indicating that ferroptosis was not induced in IHCs with deletion of Gpx4, consistent with the exclusive expression of TfR1 in OHCs of Atoh1-Gpx4−/− mice (Figure 2E). Moreover, SEM analysis showed that the stereocilia of OHCs presented with an inward contraction shape and were surrounded with abnormal outer bulges (red dotted line) in the apical turn of Atoh1-Gpx4−/− mice (Figure 2H). Besides these, membrane blebbing was detected on cuticular plates and stereocilia of OHCs (yellow arrows) in the medial and basal turns of Atoh1-Gpx4−/− mice, which may be associated with the rupture of OHC membranes and cuticular plates induced by Gpx4 deletion. Collectively, these results indicated that ferroptosis did affect the membrane homeostasis of the OHCs. However, no abnormality was found in the stereocilia of the IHCs. Taken together, these results suggested that ferroptosis induced by Gpx4 deletion primarily affected the maintenance of OHCs in Atoh1-Gpx4−/− mice.
Figure 2.

Ferroptosis leads to loss of OHCs in Atoh1-Gpx4−/− mice at P1
(A) Immunofluorescence analysis of RSL3-treated cochlear explants showed that RSL3 caused the death of OHCs (myosin7a labeled) but not supporting cells (Sox2 labeled). Scale bar, 50 μm. (B) Representative images for Gpx4 (red), myosin7a (green), and DAPI (blue) immunofluorescence-stained cochlear sections from P1 WT mice. OHCs are identified with yellow arrows, IHCs are identified with white arrows, Deiters’ cells are circled with dotted yellow lines, pillar cells are circled with dotted white lines, and Hensen cells are denoted by the white triangles. Scale bar, 50 μm. (C) Schematic diagram showing the construction of Gpx4 conditional knockout mice. Gpx4-floxed mice were generated by adding loxP sequence on either side of 2–4 exons and then mated with Atoh1-cre mice to obtain Atoh1-cre:Gpx4-floxed mice (named Atoh1-Gpx4−/−). (D) Genotype identification of WT (+/+), heterozygous (f/+), homozygous (f/f) mice, and Cre by PCR and agarose gel. (E) Immunofluorescence images of TfR1 expression in the cochlear hair cells of Gpx4−/− mice. The result showed that TfR1 expression was significantly increased in the OHCs of Atoh1-Gpx4−/− mice. Scale bar, 50 μm. (F) Transmission electron micrograph of OHCs in cochlear sections from P1 WT and Atoh1-Gpx4−/− mice. Higher-magnification images of OHCs are shown on the right. In the OHC of Atoh1-Gpx4−/− mice, red arrows and yellow triangles indicate damaged mitochondria and cell membranes, respectively. Scale bar, 2 μm. (G) Transmission electron micrograph of IHCs in cochlear sections from P9 WT and Atoh1-Gpx4−/− mice, and no significant differences were found. Scale bar, 2 μm. (H) Scanning electron micrograph of hair cell stereocilia in the apical and medial turns from P1 Gpx4-floxed and Atoh1-Gpx4−/− mice. We found that knockout of Gpx4 altered the morphology of hair bundles and cuticular plates. The red line area represents the abnormal cuticular plates. Scale bar, 2 μm.
Given that Gpx4 was expressed in supporting cells, an inducible conditional supporting cells knockout mouse model (Plp-Gpx4Cre/ER) for the Gpx4 gene was established to dissect the role of Gpx4 in supporting cells of mice. Plp-Gpx4Cre/ER mice treated with tamoxifen did not show impaired hearing function (Figures S1A and S1B). Both the supporting cells (Figure S1C) and hair cells (Figure S1D) were normal, implying that the function of Gpx4 in supporting cells may not be as important as in OHCs.
Gpx4-mediated not Fsp1-mediated ferroptosis leads to hearing loss in mice
Atoh1-Gpx4−/− mice were viable and can live to adulthood. At P30, we conducted ABR measurements on both Gpx4-floxed and Atoh1-Gpx4−/− mice. Atoh1-Gpx4−/− mice showed a complete absence of ABR waves across all tested frequencies, indicating severe hearing impairment (Figures 3A and 3B). In addition, the increased distortion product otoacoustic emission (DPOAE) thresholds in Atoh1-Gpx4−/− mice also indicated damage to OHCs (Figure 3C). Myosin7a and phalloidin staining were performed to observe the hair cells and stereocilia in Gpx4-floxed and Atoh1-Gpx4−/− mice at P14 and P30 (Figure 3D). Contrary to the WT mice, Atoh1-Gpx4−/− mice showed severe loss of OHCs in all turns of the cochlea in both testing times (Figure 3E). Then, the morphology of the cochlea was analyzed by hematoxylin and eosin staining of paraffinized sections from the Gpx4-floxed and Atoh1-Gpx4−/− mice at P30 (Figure 3F), and Atoh1-Gpx4−/− mice showed severe atrophy of the SV and extensive loss of SGNs (Figures 3G and 3H). Given that Atoh1-cre is only expressed in hair cells and partial supporting cells, the abnormal SV and SGNs should be associated with secondary degeneration from loss of OHCs.58 Thus, considering the survival of IHCs in Atoh1-Gpx4−/− mice, we explored whether the ribbon synapses on the IHCs were affected. Specifically, we used the anti-CtBP2 and anti-GluR2 antibodies for immunofluorescence in Gpx4-floxed and Atoh1-Gpx4−/− mice at P14 and P30 (Figure 3I). Results showed that the number of synapses in Atoh1-Gpx4−/− mice was similar at P14 but decreased significantly at P30 relative to levels in Gpx4-floxed mice (Figure 3J). This was ascribed to the loss of SGNs or the effect of Gpx4 deletion on the function of IHCs and synapses. Meanwhile, considering that the Fsp1 expression was altered in cisplatin-treated cochlear explants, which has been reported to prevent lipid peroxidation and suppress ferroptosis via reduction of ubiquinol/α-tocopherol on the level of lipid radicals independent of GPX4/GSH.41 We developed Fsp1 knockout mice (Fsp1−/−) through CRISPR-Cas9 technology to further explore its function in hair cells (Figure 4A). The PCR and qPCR data indicated that the Fsp1 gene was deleted successfully in Fsp1−/− mice (Figures 4B and 4C). In addition, Fsp1 fluorescent signal was observed in the cochlear hair cells of the control mice, but not in the Fsp1−/− mice, indicating that Fsp1 was effectively deleted from the cochlear hair cells of the Fsp1−/− mice (Figure 4D). Moreover, the Fsp1 fluorescent signal were not found in SV of Fsp1−/− and WT mice (data not shown). Notably, Fsp1 knockout mice were viable and fertile, showing normal hearing function as determined by ABR and DPOAE tests (Figures 4E–4G). Moreover, the hair cells and the morphology of the cochlea of Fsp1−/− mice were normal at P90 (Figures 4H and 4I), and there was no significant difference in the number of SGNs and the thickness of SV between the two groups (Figures 4J–4L). Given that Fsp1 expressed in the cochlear hair cells, and that the expression level was altered after cisplatin treatment, we further evaluated the sensitivity of Fsp1−/− mice to cisplatin. WT and Fsp1−/− mice were exposed to two cycles of cisplatin (4 mg/kg) treatment for 4 days with intervals for a 6-day recovery period. The ABR technique was used to detect the hearing changes of mice before and after cisplatin treatment (Figure S2A). Firstly, the body weights of WT and Fsp1−/− mice were comparable before and after cisplatin treatment (Figure S2B). Then the ABR and immunofluorescence data also indicated that there were no significant differences between WT and Fsp1−/− mice before and after cisplatin treatment (Figures S2C–S2F). These results suggested that the role of Fsp1 in metabolic activities may not be as critical as Gpx4 in the inner ear of mice.
Figure 3.

Atoh1-induced Gpx4 knockout resulted in hearing loss
(A) Statistical analysis of the ABR threshold in Atoh1-Gpx4−/− and Gpx4-floxed mice at P30. The threshold at all frequencies is higher than that in Gpx4-floxed mice, n = 5 for each group. (B) Click-ABR waveforms in Gpx4-floxed and Atoh1-Gpx4−/− mice at P30, and the ABR traces were recorded at the same measure range of latency and amplitude. (C) At P30, DPOAE data showed that Atoh1-Gpx4−/− mice had impaired OHC function and hearing loss compared with the Gpx4-floxed mice, n = 5 for each group. (D) Representative confocal images for myosin7a (green) and phalloidin (red) staining in the cochlea of Gpx4-floxed and Atoh1-Gpx4−/− mice at P14 and P30. The result showed that the knockout of Gpx4 resulted in a massive loss of OHCs. Scale bar, 20 μm. (E) Quantification of the rate of OHC loss in (D), n = 5 for each group. (F) hematoxylin and eosin (H&E) staining of sections from the Gpx4-floxed and Atoh1-Gpx4−/− mice at P30. Scale bars, 50 μm. (G) Statistical analysis of the SV thickness in (F). Compared with Gpx4-floxed mice, the SV was thinner in the Atoh1-Gpx4−/− mice, n = 5 for each group. (H) SGN density result in (F) showed that the number of SGNs in Atoh1-Gpx4−/− mice was significantly less than Gpx4-floxed mice, n = 5 for each group. (I) Synapse staining by anti-CtBP2 (red) and anti-GluR2 antibodies in cochleae of Gpx4-floxed mice and Atoh1-Gpx4−/− mice at P14 and P30. Scale bar, 10 μm. (J) Quantification of the number of CtBP2-positive, GluR2-positive, and double-positive synapse puncta of the IHCs in (I), n = 3 for each group. Statistical values are presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Figure 4.

Fsp1−/− mice showed normal hearing function
(A) Schematic diagram showing the construction of knockout mice models, exon 4–8 was deleted in the region of the Fsp1 gene. (B) Genotype identification of Fsp1 WT, heterozygous (HET), and knockout (KO) mice by PCR. (C) Total RNA extracted from the mouse cochlea for qPCR analysis, and the Fsp1 mRNA level was significantly decreased in the cochlea of Fsp1−/− mice, n = 4 for each group. (D) Immunofluorescence analysis of Fsp1 (red) and myosin7a (green) in cochlear hair cells of WT and Fsp1−/− mice. Results showed that the Fsp1 signal was present in hair cells of WT mice but absent in Fsp1−/− mice, indicating that Fsp1 was effectively knocked out in hair cells of Fsp1−/− mice. OHC, dotted yellow lines; IHC, dotted white lines. Scale bar, 5 μm. (E–G) Analysis of auditory function in Fsp1−/− mice. We found no significant difference in ABR thresholds (E) and click-ABR waveforms (F) between WT and Fsp1−/− mice at P90, moreover, DPOAE results (G) also showed normal hearing function in the Fsp1−/− mice, n = 5. ns, no significance. (H) Representative confocal images for myosin7a (green) and phalloidin (red) staining in the apical, medial, and basal turn of cochlear regions from control and Fsp1−/− mice at P90, no differences were found between the WT and Fsp1−/− mice. Scale bar, 20 μm. (I) Quantification of the survival rate of OHC in (H), n = 5 for each group. (J–L) H&E staining was used to analyze the structure of the Fsp1−/− and WT cochlea at P90 (J), and there was no significant difference in the thickness of SV (K) and the number of SGNs (L) between the two groups, n = 5 for each group. Scale bars, 100 μm. SGNs, spiral ganglion neurons; SV, stria vascularis. Statistical values are presented as mean ± SEM, ∗∗∗p < 0.001.
Fer-1 alleviates OHC loss caused by ablation of Gpx4 and cisplatin treatment
To test whether ferrostatin-1 (Fer-1), a well-known lipophilic radical scavenger for ferroptosis,59 could specifically ameliorate hair cell dysfunction in Atoh1-Gpx4−/− mice, pregnant mice were intraperitoneally injected with 5 mg/kg Fer-1 once every other day from E15.5. Next, the Atoh1-Gpx4−/− mice at P1 with and without Fer-1 injection were observed for the OHCs through immunofluorescence staining. It was found that Atoh1-Gpx4−/− mice with Fer-1 injection from E15.5 showed significant improvements in the loss of OHCs both in apical and medial turns (Figures 5A and 5B). However, the loss of OHCs in the basal turn was not improved. This could potentially be attributed to factors such as the duration of the intervention, dosage of the injected drug, and the efficiency of Fer-1 entering the inner ear of embryonic mice. Given this, we also considered the impact of intervention duration and drug dosage. This led us to question whether Gpx4-mediated ferroptosis might affect matured hair cells. To achieve this, we crossed Gpx4flox/flox mice with inducible hair cell Cre mice (Pou4f3-Cre/ERT), and homozygous Pou4f3-Gpx4Cre/ER mice were induced with 10 mg/kg tamoxifen for five consecutive days at 6 weeks (Figure 5C). Meanwhile, some induced Pou4f3-Gpx4Cre/ER mice were intraperitoneally injected with 5 mg/kg Fer-1 once every other day after tamoxifen treatment for 3 weeks, and all groups of mice were subjected to the ABR and DPOAE tests. Analysis of the results revealed that deletion of Gpx4 in mature hair cells also caused hearing loss in mice, which was alleviated by Fer-1, this was consistent with observations in Atoh1-Gpx4−/− mice (Figures 5D and 5E). In agreement with the hearing test results, Pou4f3-Gpx4Cre/ER mice treated with tamoxifen showed significant OHC loss and normal IHCs at all turns of cochleae, and the OHC viability in the tamoxifen + Fer-1 group was significantly higher than that of the tamoxifen group, indicating that Fer-1 had a rescue effect on ferroptosis of mature hair cells with Gpx4 deletion (Figures 5F and 5G). Since cisplatin could alter with ferroptosis pathway and Fer-1 protected hair cells in Atoh1-Gpx4−/− mice by inhibiting ferroptosis, we further explored the effect of Fer-1 on CIHL in C57BL/6 WT mice. The protocol for cisplatin treatment was similar to that mentioned previously. Mice received 3 mg/kg Fer-1 pre-treatment 4 h before cisplatin injection every time (Figure 5H). Subsequently, all mice were subjected to the ABR and DPOAE tests. It was found that the thresholds of the click and tone burst ABR threshold were reduced in mice of the Fer-1 + cisplatin group compared with the cisplatin group (Figure 5I). In the DPOAE test, the threshold at 16 kHz was decreased in mice of the Fer-1 + cisplatin group compared with the cisplatin group (Figure 5J). The loss of OHCs was determined based on immunofluorescence in whole-mount turn mouse cochlea, and the OHC loss of basal turns was improved in the Fer-1 + cisplatin group relative to the cisplatin group (Figures 5K and 5L).
Figure 5.

Fer-1 alleviated the ferroptosis of OHCs induced by Gpx4 deletion
(A) Representative images of myosin7a (red) and DAPI (blue) staining of cochlear hair cells from the Gpx4-floxed, Atoh1-Gpx4−/−, and Atoh1-Gpx4−/− with Fer-1-treated mice. Scale bar, 20 μm. (B) Quantification of OHC number in (A), and the data showed that the Fer-1 could partially rescue the OHC loss caused by Gpx4 deficiency, n = 5 for each group. (C) Schematic of the experimental workflow showing Fer-1 injection in Pou4f3-Gpx4Cre/ER mice with tamoxifen induction. The Pou4f3-Gpx4Cre/ER mice were injected with 10 mg/kg tamoxifen for five consecutive days at 6 weeks, and treated with Fer-1 (5 mg/kg) once every other day for 21 consecutive days. (D and E) ABR and DPOAE techniques were used to detect the hearing function of Pou4f3-Gpx4Cre/ER, Pou4f3-Gpx4Cre/ER with tamoxifen, and Pou4f3-Gpx4Cre/ER with tamoxifen and Fer-1 mice, the results showed that Fer-1 treatment could rescue hearing loss caused by Gpx4 deficiency, n = 5 for each group. (F) Representative confocal images of myosin7a (green) and phalloidin (red) staining in the cochleae of Pou4f3-Gpx4Cre/ER, Pou4f3-Gpx4Cre/ER with tamoxifen, and Pou4f3-Gpx4Cre/ER with tamoxifen + Fer-1 mice. Scale bar, 20 μm. (G) Quantification of the OHC loss in (F), n = 5 for each group. (H) Schematic illustration of the experimental workflow showing Fer-1 injection in cisplatin-treated mice. The WT mice pre-treated with Fer-1 (3 mg/kg) were injected with cisplatin (4 mg/kg) every day at 6 weeks for 4 days and recovery for 6 days as a cycle (two cycles were performed). (I and J) ABR and DPOAE techniques were used to detect the hearing function of control, cisplatin-treated mice, and cisplatin-treated mice with Fer-1 pre-treatment. The results showed that Fer-1 treatment could rescue hearing loss caused by Gpx4 deficiency, n = 5 for each group. (K) Representative images of myosin7a (green) and phalloidin (red) staining in the cochlea of WT mice treated with vehicle, cisplatin, and cisplatin + Fer-1. Scale bar, 20 μm. (L) Quantification of OHC loss in (K), n = 5 for each group. Statistical values are presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
FDA-approved luteolin effectively alleviates cisplatin-induced ferroptosis in vitro
Although the results revealed that Fer-1 conferred protection against the loss of OHCs induced by cisplatin, Fer-1 is not a drug that can be directly used in clinical practice. Therefore, to find a drug that can be applied in clinical practice to alleviate cisplatin-induced ototoxicity by inhibiting ferroptosis, we screened a library containing 235 FDA-approved drugs targeting ferroptosis in Selleck small-molecule compound libraries. Cell viability assays were performed using the CCK-8 assay kit to identify drugs against 50 μm cisplatin ototoxicity using the immortalized inner ear cell line HEI-OC1 derived from mouse cochlea (Figure 6A). In this analysis, the cell activity of untreated cells was designated as 100%. Among the compounds tested in the FDA-approved and ferroptosis-associated library, the relatively effective compounds include luteolin (Figure 6B), daidzin, and sodium danshensu. These compounds were further characterized via CCK-8 viability assay to determine whether they could protect against 3 μM RSL3-induced ferroptosis in HEI-OC1 cells, and the results showed that only luteolin ameliorated RSL3-induced ferroptosis (Figure 6C). Consequently, luteolin was selected for further characterization because it can act as an anticancer agent against various types of human malignancies,60,61,62,63 combine with cisplatin to sensitize the antitumor effect in drug-resistant cancer cell lines,64 and cross the blood-brain barrier.65 The protective effect of luteolin against cisplatin was further analyzed with CCK-8 assay in HEI-OC1 cells. As the results show, 1 and 2 μM luteolin exhibited the best protective effects (Figure 6D), which were further confirmed through light microscopy in HEI-OC1 cells. The density of viable cells in the group treated with 50 μM cisplatin combined with 1 μM luteolin was significantly higher compared with the group treated with cisplatin alone (Figure 6E). To assess the influence of luteolin on the protection of cochlear hair cells following cisplatin-induced damage, cochlear explants from P2 mice were subjected to 150 μM cisplatin for 24 h with or without pre-treatment with 2 μM luteolin for 8 h. Analysis of the results indicated that luteolin mitigated the OHC loss induced by cisplatin in both medial and basal turns of cochlear explants (Figures 6F and 6G). ROS accumulation is strongly associated with ferroptosis,39,66 and we next examined whether cisplatin induced ROS accumulation in HEI-OC1 cells, and cochlear explants were altered by luteolin pre-treatment. Using DCFH-DA dye, we found that the intensity of the ROS fluorescence signal in both HEI-OC1 cells and cochlear explants was higher than that in the control group when treated with cisplatin alone, and luteolin pre-treatment could significantly attenuate the ROS fluorescence signal (Figures 6H–6K). Meanwhile, results of immunofluorescence staining for TfR1 showed that luteolin treatment reduced the expression of TfR1 in RSL3-treated HEI-OC1 cells (Figures 6L and 6M), which further verified the specific inhibitory effect of luteolin on ferroptosis.
Figure 6.

Luteolin alleviated ferroptosis caused by cisplatin in HEI-OC1 cells
(A) Cell-based ferroptosis associated small-molecule screening using the HEI-OC1 cell line; medium alone (green dots), cisplatin alone (blue dots), compound + cisplatin (gray dots), and luteolin + cisplatin (red dots). (B) Diagram of the molecular structure of luteolin. (C) Dose-response curve for RSL3 of luteolin, daidzin, and sodium danshensu based on results of the CCK-8 assay in HEI-OC1 cell treated with 3 μM RSL3, RSL3 + luteolin, RSL3 + daidzin, and RSL3 + sodium danshensu, n = 5 for each group. (D) Luteolin dose-response curve for cisplatin based on results of the CCK-8 assay in HEI-OC1 cells treated with 50 μM cisplatin, and the data showed that HEI-OC1 cells pre-treated with 1 and 2 μM luteolin had a significant higher cell viability after cisplatin treatment, n = 5 for each group. (E) Light microscopy analysis of HEI-OC1 cells treated with medium alone, 50 μM cisplatin, and 50 μM cisplatin + 1 μM luteolin for 24 h. Scale bar, 200 μm. (F) Representative images of myosin7a-staining of whole-mount middle turn cochlear explants treated with medium alone, 150 μM cisplatin, and 150 μM cisplatin + 2 μM luteolin for 24 h. Scale bar, 50 μm. (G) Quantification the survival rate of OHC in (F), and the results indicate that luteolin could reduce the damage of hair cells induced by cisplatin, n = 5 for each group. (H and I) DCFH-DA dye was used to detect ROS level generated by cisplatin, and the fluorescence signal of ROS in HEI-OC1 cells (H) and cochlear explants (I) treated with cisplatin alone was higher than that in the control group, while the fluorescence signal was significantly attenuated by luteolin pre-treatment. Scale bars, 100 μm (H) and 20 μm (I). (J and K) Statistical analyses of the relative fluorescence intensity in (H) and (I), respectively, n = 5 for each group. (L) Representative images of TfR1 (green)- and phalloidin (red)-stained HEI-OC1 cells treated with medium, 1 μM RSL3, and 1 μM RSL3 + 5 μM luteolin for 12 h. The intensity of TfR1 fluorescence in the RSL3-treated group was higher than that in the control group, but decreased significantly with luteolin pre-treatment. Scale bar, 100 μm. (M) Statistical analysis of the relative intensity of TfR1 fluorescence in (L), n = 5 for each group. Statistical values are presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Luteolin alleviates ferroptosis by reducing Trf and increasing HO-1 expression
We next tested whether luteolin could protect against cisplatin-induced ototoxicity in vivo. C57BL/6 WT mice at P30 were measured by ABR before injection, and then divided into three groups: vehicle, cisplatin, and cisplatin + luteolin treatment group. Luteolin (1 mg/kg) was intraperitoneally injected for 4 h every time before cisplatin (4 mg/kg) treatment in luteolin + cisplatin-treated groups. Consistent with the treatment above, ABR was measured again after two injection cycles and hearing differences were analyzed (Figure 7A). Mice treated with vehicle or luteolin alone showed no significant side effects (data not shown). Cisplatin-treated mice had significantly elevated threshold shifts at 16-, 24-, and 32-kHz frequencies and mice pre-treated with luteolin had significantly reduced threshold shifts at 16- and 24-kHz frequencies (Figure 7B). Moreover, OHC loss was analyzed using immunofluorescence in mouse cochlea. It was observed that the OHC loss of basal turns was improved in the luteolin + cisplatin group compared with the cisplatin group (Figures 7C and 7D). Furthermore, the effect of luteolin on ferroptosis in Pou4f3-Gpx4Cre/ER mice induced by tamoxifen was also tested by administering 6 mg/kg luteolin using a similar protocol to Fer-1 (Figure 5C), the mice were measured by ABR before injection, and then divided into three groups: Pou4f3-Gpx4Cre/ER-, Pou4f3-Gpx4Cre/ER + tamoxifen-, and Pou4f3-Gpx4Cre/ER + tamoxifen + luteolin-treated groups (Figure 7E). The results of ABR showed that Pou4f3-Gpx4Cre/ER mice pre-treated with luteolin had significantly reduced threshold shifts at 4-, 8-, and 16-kHz frequencies (Figure 7F) and reduced the loss rate of OHCs at medial and basal turns based on the myosin7a staining assay (Figures 7G and 7H). In addition, SEM was used to check the structure of the stereocilia in groups of Pou4f3-Gpx4Cre/ER mice treated with vehicle, tamoxifen alone, and tamoxifen + luteolin, and no difference of stereociliary structure of IHCs in the three group of mice was found (Figure S3), which further validates that ferroptosis does not affect IHCs. Moreover, we detected the ROS level in Pou4f3-Gpx4Cre/ER mice after tamoxifen treatment using dihydroethidium (DHE) dye (Figures S4A and S4B). The results were similar to the HEI-OC1 and cochlear explants data (Figures 6H and 6I). Compared with Pou4f3-Gpx4Cre/ER mice treated with vehicle, the ROS level was significantly increased in the OHCs of Pou4f3-Gpx4Cre/ER mice treated with tamoxifen alone and decreased when tamoxifen and luteolin were co-treated.
Figure 7.

Luteolin alleviated the ferroptosis by downregulating Trf and upregulating HO-1
(A) Schematic illustration of the experimental workflow for treatment of cisplatin and luteolin. ABR was measured in C57BL/6 WT mice before and after cisplatin treatment, and the luteolin (1 mg/kg) was administered before cisplatin injection every time, followed by injection of cisplatin (4 mg/kg) for 4 days and recovery for 6 days as a treatment cycle (two cycles in total). (B) ABR was performed to detect the hearing of vehicle-, cisplatin-, and luteolin + cisplatin-treated mice, and the results showed that luteolin could reduce the ototoxicity of cisplatin and protect hearing, n = 5 for each group. (C) Representative images of myosin7a (green) and phalloidin (red) staining in the cochlea of vehicle, cisplatin, and luteolin + cisplatin mice. Scale bar, 20 μm. (D) Quantification of OHC loss in (C), the data indicated that luteolin could partially protect hair cells against the ototoxicity of cisplatin, n = 5 for each group. (E) Schematic illustration of the experimental workflow for treatment of tamoxifen and luteolin. ABR was measured in Pou4f3-Gpx4Cre/ER mice before and after tamoxifen treatment, and the tamoxifen (10 mg/kg every day for 5 days) was administered before luteolin (6 mg/kg once every other day for 21 days) injection. (F) ABR analysis for the Pou4f3-Gpx4Cre/ER mice treated with vehicle, tamoxifen, and tamoxifen + luteolin, and the results showed that luteolin could partially prevent deafness caused by Gpx4 deficiency, n = 5 for each group. (G) Representative images of myosin7a (green) and phalloidin (red) staining in the cochlea of the Pou4f3-Gpx4Cre/ER mice treated with vehicle, tamoxifen, and tamoxifen + luteolin. Scale bar, 20 μm. (H) Quantification of OHC loss in (G), n = 5 for each group. (I) Western blot was used to analyze the expression of Trf, HO-1, and Nrf2 in the cochlea of WT mice treated with vehicle, cisplatin, and cisplatin + luteolin. (J) Statistical analysis of the protein level in (I), and the result showed that the expression of Trf and HO-1 in the cochlea of the cisplatin-treated cochlea were higher than that of the vehicle group, and luteolin pre-treatment reduced Trf content but not HO-1. In addition, the expression of Nrf2 was consistent among the three groups, n = 3 for each group. (K) Western blot was used to analyze the expression of Trf, TfR1, HO-1, and Nrf2 from vehicle, RSL3, and luteolin + RSL3-treated HEI-OC1 cells. (L) Statistical analysis of the protein level in (K), and the protein level of Trf and TfR1 in RSL3-treated HEI-OC1 cells was increased compared with that in vehicle-treated HEI-OC1 cells, which was downregulated by luteolin pre-treatment, n = 4 for each group. (M) Analysis of intracellular ferrous ions using FerroOrange staining in vehicle-, 50 μM cisplatin-, and 50 μM cisplatin + 1 μM luteolin-treated HEI-OC1 cells for 24 h. Scale bar, 10 μm. (N) Statistical analysis of the mean FerroOrange fluorescence relative intensity in (M). The intensity FerroOrange fluorescence signal in the cisplatin-treated group was significantly higher than that of the control group, while the fluorescence intensity was reduced by luteolin pre-treatment, n = 10 for each group. (O) Analysis of intracellular ferrous ions using FerroOrange staining in vehicle-, 1 μM RSL3-, and 1 μM RSL3 + 5 μM luteolin-treated HEI-OC1 cells for 12 h. Scale bar, 10 μm. (P) Statistical analysis of the mean FerroOrange fluorescence relative intensity in (O). The intensity FerroOrange fluorescence signal in the RSL3-treated group was significantly higher than that of the control group, while the fluorescence intensity was reduced by luteolin pre-treatment, n = 10 for each group. Statistical values are presented as mean ± SEM, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Next, we explored the mechanisms of luteolin-induced ferroptosis resistance and protection against cisplatin-induced ototoxicity. Firstly, qPCR was performed to detect the expression of ferroptosis-related genes in cochleae of WT mice treated with 10 mg/kg cisplatin for 1 day with or without 1 mg/kg luteolin pre-treatment for 4 h. Results showed that the increased expression of Trf in the cisplatin-treated group was downregulated in the luteolin + cisplatin-treated group (data not shown). Similar results were obtained in the western blot (Figure 7I). Furthermore, reports are linking luteolin with the Nrf2/HO-1 signaling pathway in cisplatin-induced cardiac dysfunction, a pathway also implicated in ferroptosis.67,68,69 Therefore, we examined the Nrf2/HO-1 signaling pathway in the cochleae treated with luteolin and cisplatin. The results revealed an upregulation of HO-1 protein levels in both the cisplatin-treated and luteolin + cisplatin-treated groups compared with control groups (Figures 7I and 7J). However, no significant difference was observed in the levels of both HO-1 and Nrf2 between the cisplatin-treated group and the luteolin + cisplatin-treated group (Figures 7I and 7J). We further corroborated these protein level findings in HEI-OC1 cells treated with 1 μM RSL3 (Figure 7K) for 24 h. Interestingly, the protein expression of HO-1 demonstrated a significant increase in RSL3-treated HEI-OC1 cells, an effect that was further augmented with pre-treatment of 5 μM luteolin. The different results between cisplatin-treated cochleae and RSL3-treated cells indicated that the Nrf2/HO-1 pathway may not be the primary mediator of the protective effect of luteolin on CIHL. However, the protein level of Trf in RSL3-treated HEI-OC1 cells was consistent with that in cisplatin-treated cochlea, which was also downregulated by luteolin pre-treatment. Besides, the changes in protein expression of TfR1 were consistent with that of Trf (Figure 7L). These results implied that luteolin protected the cochleae against cisplatin-induced damage by inhibiting ferroptosis mediated by reduced Trf and TfR1. In addition, since luteolin inhibits the increase of Trf and TfR1, which is mainly responsible for importing iron into the cell during ferroptosis, we next performed FerroOrange staining to detect the level of intracellular concentration of ferrous ions in cisplatin-treated (Figures 7M and 7N) and RSL3-treated (Figures 7O and 7P) HEI-OC1 cells. As expected, additional pre-treatment with luteolin significantly reduced FerroOrange fluorescence in both the luteolin + cisplatin and luteolin + RSL3 groups compared with that in vehicle-treated control groups, indicating a decrease in ferrous ions. These findings provide further evidence of luteolin’s effectiveness in inhibiting ferroptosis. This effect is achieved by decreasing Trf protein levels, ultimately leading to the attenuation of cisplatin-induced ototoxicity.
Discussion
In this study, we found that ferroptosis mediated by Gpx4 was involved in cisplatin-induced ototoxicity through in vitro and in vivo experiments. Gpx4 was observed to play an important role in the maintenance of auditory function in mice. Besides, through screening a library containing FDA-approved and ferroptosis-associated bioactive compounds, luteolin, a multifunctional natural compound, was found to specifically inhibit ferroptosis and alleviate cisplatin-induced ototoxicity. The protective effects of luteolin appeared to be mediated by the reduction of cisplatin-induced activation of ferroptosis and a decrease in Trf activity.
Deletion of Gpx4 induced ferroptosis of OHCs but not IHCs or supporting cells, which provided the first direct evidence for the specific sensitivity of ferroptosis in OHCs in vivo. In the cochlea, a noticeable trend is observed in the susceptibility and depletion of hair cells, depending on their type and position along the tonotopic axis. Specifically, OHCs and high-frequency hair cells are more prone to injury from various ototoxic agents such as aminoglycoside antibiotics and cisplatin, as opposed to IHCs and those responsible for low-frequency hair cells.70,71,72 The underlying reasons for the selective vulnerability of OHCs are not fully understood. Nevertheless, there is evidence that disparities in hair cell metabolism, free radical-induced damage, or (Ca2+) regulation may contribute to this phenomenon, and all of them are implicated in mitochondrial regulation.73,74,75 Hair cells require a significant amount of energy, and the connection between impaired mitochondrial function and hearing loss has been a subject of research because it has been observed in various forms of hair cell damage. Typically, dying hair cells display enlarged mitochondrial structure and produce ROS in response to ototoxic substances.76,77,78 Specific mutations in mitochondrial genes have been identified to be potential causes of both syndromic and non-syndromic deafness.79,80 Previous studies have demonstrated that the susceptibility of hair cells to the toxic drug neomycin did correspond to the cumulative history of mitochondrial activity in zebrafish mechanosensory hair cells.81 Studies have reported that the variation in mitochondrial metabolism of high-frequency OHCs was greater compared with that in IHCs, supporting cells, and pillar cells of gentamicin-treated mice.82 In our study, TEM results showed that the number of mitochondria in OHCs was significantly higher than that in IHCs, implying the mitochondrial metabolism in OHCs was more active than that in IHCs (Figures 2F and 2G). Besides, ferroptosis has been linked to severe damage in mitochondrial morphology and metabolism.83 Although the expression of Gpx4 was observed in both OHCs and IHCs and indeed ablated in IHCs of Atoh1-Gpx4−/− mice, it appears that GSH metabolism regulated by Gpx4 may not be dominant or may be replaceable. This hypothesis needs to be validated in future studies. In summary, the resistance to ferroptosis varies among IHCs, supporting cells, and OHCs. Notably, OHCs displayed a significant susceptibility to ferroptosis in both the hair cell-specific conditional Gpx4-knockout and cisplatin-treated mice. These findings offer potential insights into why OHCs are the primary cellular targets most affected by cisplatin-induced damage.
In this study, we generated Gpx4 and Fsp1 knockout mice. Interestingly, only deletion of Gpx4 induced ferroptosis in OHCs of mice and the auditory function of Fsp1−/− mice was normal. Moreover, given that cisplatin treatment affects both Gpx4 and Fsp1 in mouse cochleae, we also evaluated the sensitivity of Fsp1−/− mice to cisplatin. However, Fsp1−/− mice proved to be equally vulnerable to cisplatin compared with WT mice, suggesting that the role of Fsp1 in metabolic activities may not be as critical as Gpx4 in the inner ear of cisplatin-treated mice. GPX4 is widely recognized as the principal regulator that prevents the occurrence of ferroptosis. However, in certain cell types or cell lines, inhibition of GPX4 does not induce ferroptosis, suggesting the existence of alternative mechanisms. Subsequently, FSP1 has been confirmed to be another inhibitor of ferroptosis, which exerts its effects through a GPX4-independent pathway.41,84 The N terminus of FSP1 contains a canonical myristoylation motif, which mediates its ability to inhibit ferroptosis by promoting the localization of FSP1 to the plasma membrane.41,85,86 Previous studies demonstrated that FSP1 may function as an NADP-dependent coenzyme Q oxidoreductase in vitro,87 acting as a mobile lipophilic electron carrier that synthesizes lipid-soluble antioxidants and lipophilic free radical-trapping agents in the plasma membrane.88 Simultaneously inhibiting both FSP1 and GPX4 shows promise in increasing cancer cell susceptibility. However, it is important to note that FSP1 does not play a dominant role in hair cells. Previous studies have shown that cisplatin mainly accumulates in the SV of the cochlea.23 However, in this study, neither Gpx4 nor Fsp1 expression was found in the SV, indicating that the ferroptosis mediated by Fsp1 and Gpx4 did not affect the SV directly. However, we cannot rule out the possibility that the whole ferroptosis pathway plays a role in the SV, which needs to be further explored.
Moreover, we found that luteolin can specifically inhibit ferroptosis and alleviate cisplatin-induced ototoxicity. These results showed that luteolin treatment reduced CIHL and OHC loss in mice. Luteolin, a flavone derived from celery, green pepper, and chamomile, has been documented to display anti-inflammatory, antioxidant, and anticarcinogenic properties.89,90,91 Luteolin can cross the blood-brain barrier and has a significant anticancer effects against human breast, ovarian, and colorectal cancer cell lines. Furthermore, luteolin has been observed to synergistically enhance the anticancer effects of cisplatin by stabilizing p53.92,93 Recent investigations have uncovered that a diet rich in flavonoids, particularly luteolin, is linked to reduced serum levels of inflammatory cytokines.94 However, few investigations have explored the involvement of luteolin in the therapeutic effects of cisplatin-induced ototoxicity and ferroptosis. This study demonstrates that the protective effects of luteolin may be attributed to its ability to mitigate the heightened ferroptosis induced by cisplatin, which involves the reduction in Trf activity. These results demonstrate that a combination of cisplatin and luteolin at lower dosages might be effective in alleviating ototoxicity and enhancing cytotoxic on cancer cells.
The underlying mechanism linking the effect of luteolin to the reduction in Trf expression remained unclear. The serum iron carrier protein Trf can be internalized into the cell through receptor-mediated endocytosis,95 exclusively interact with TfR, and enter the cell when it is bound to iron. In the extracellular environment, the serum constituents L-glutamine and Trf were implicated in the regulation of ferroptosis. Within the intracellular milieu, the specific metabolic pathway known as glutaminolysis and the components mediating the importation of Trf contribute to the occurrence of ferroptosis.49 Therefore, the mechanisms that govern the iron-loading process of Trf have the potential to influence the occurrence of ferroptosis. The carrier protein TfR1, crucial for transporting Trf and recognized as a specific marker for intracellular ferroptosis, played an essential role in cellular iron uptake.96 In this study, we observed positive TfR1 staining in OHCs of cisplatin-treated mice. Furthermore, luteolin was able to reduce the elevated expression of TfR1 in RSL3-treated HEI-OC1 cells. We did not confirm the elevated expression of TfR1 in the cochleae of cisplatin-treated mice. This might be attributed to the complex cell types present in mouse cochleae, where the increased TfR1 expression in OHCs constitutes only a small fraction of the overall cell population, making detection challenging. Iron maintains the viability of almost all organisms, as it serves as a cofactor in numerous biochemical processes, including oxygen storage, oxidative phosphorylation, and essential enzymatic reactions for cellular growth.97 However, maintaining strict control over free iron levels within cells is crucial to prevent the generation of ROS through processes such as the Fenton reaction,98 which could potentially trigger ferroptosis. In this study, the results of FerroOrange staining showed that the intracellular concentration of ferrous ions was increased both in cisplatin-treated and RSL3-treated cells, and that this was reversed by luteolin treatment. Consequently, based on these findings, we propose that luteolin may be regarded as a promising pharmacological candidate for ameliorating chemotherapy-induced ototoxicity.
Materials and methods
Study approval
This study followed the guidelines for the welfare of laboratory animals and was approved by the Institutional Animal Care and Use Committee of Shandong First Medical University.
Mice and genotyping
C57BL/6 background mice used in the cisplatin-treated model were bought from Beijing Vital River Laboratory. Atoh1-Cre mice (no. 011104) were obtained from The Jackson Laboratory. Gpx4-floxed mice were kindly gifted by Prof. Bo Chu (School of Basic Medical Sciences, Shandong University, Jinan, China), and Pou4f3-CreER and Plp-CreER mice were kindly gifted by Prof. Guoqiang Wan (Model Animal Research Center of Medical School, Nanjing University, Nanjing, China). All mice in this study were raised in a specific pathogen-free-grade environment. And the type and number of mice were listed (Table S1). Primers used for mouse genotyping included: Gpx4-floxed: 5′-CTGCAACAGCTCCGAGTTC-3′ (forward) and 5′-CGGTGCCAAAGAAAGAAAGT-3′ (reverse). All Atoh1-Cre, Pou4f3-CreER, and Plp-CreER mice were genotyped using generic Cre primers: 5′-AGCTAAACATGCTTCATCGTCGGTC-3′ (forward) and 5′-TATCCAGGTTACGGATATAGTTCATG-3′ (reverse). Fsp1 knockout mice were generated via CRISPR-Cas9 technology. The Fsp1 guide RNA targeting from 4 to 8 exons with gRNA1 (matching forward strand of gene) were: TCATGGCCACCGGTGTCCTGAGG, gRNA2 (matching reverse strand of gene); AAGCATCTACTAGGAGAGTAAGG, gRNA3 (matching reverse strand of gene); ACACCTAAGGAAACATGGCGTGG, gRNA4 (matching reverse strand of gene); CAGGCCTCATCTCTTAAAGTTGG. The Fsp1 guide RNA and the Cas9 RNA were co-microinjected into zygotes of C57BL/6 mice, then transferred into pseudopregnant CD1 female mice. The following primer sequences were used for confirming the knockout mice: 5′-GATTCTAAACTGGAGGTGTGAG-3′ (forward); 5′- TTTAAAGAGGCTTTGTGCGGAGAC-3′ (reverse for mutant); 5′- AGTGGATAAGAAATAGGAGAGTAG-3′ (reverse for WT).
Cell viability assay
CCK8 Assay Kit (Vazyme, A311-02) was used for cell viability assay. In brief, mouse-derived HEI-OC1 cells were seeded (5 × 103 cells/well) in a 96-well plate containing 100 μL DMEM medium overnight, then pre-treated with different FDA-approved compounds (Drug Library from Selleck, L1300,) in a final concentration of 1 μM for 24 h. The cells were treated with cisplatin (Selleck, S1166) to a final concentration of 50 μM for another 24 h, as described previously,35,40 then incubated with 10 μL of CCK-8 at 37°C for 45–60 min. Finally, a microtitration plate reader (Molecular Devices) was used to measure the absorbance at 450 nm.
Cochlear explants
Post-natal 3-day (P3) C57BL/6 mouse cochleae were dissected in cold phosphate-buffered saline (PBS) and attached to a confocal dish through Cell-Tak (Corning, 354242). Cochlear explants were maintained in DMEM/F12 medium (HyClone, SH30022.01) containing 1% N-2/2% B-27 (STEMCELL, 07152; Invitrogen, 17504044) and 100 μg/mL Primocin (InvivoGen, ant-pm) at 37°C with 5% CO2 for 24 h. The explants were then pre-incubated with a culture medium with or without 2 μM luteolin (Selleck, S2320) at 37°C in 5% CO2 for 8 h, followed by co-incubation with 150 μM cisplatin (Selleck, S1166) for another 24 h. The cochlear explants were fixed with 4% paraformaldehyde (PFA) at room temperature for 1 h and stained with myosin7a (1:800, Proteus bioscience, 25–6790) to determine the viability of hair cells.
GSH assay of cochlear explants
GSH was detected using a GSH ELISA kit (Elabscience, E-EL-0026). In brief, 12 cochlear explants were attached and culture to a 6-cm cell culture dish for 24 h as one group. Each group of cochlear explants was treated with vehicle or 150 μM cisplatin for 12 and 24 h. Then cochlear explants were harvested and broken by sonication in PBS at 4°C according to the manufacturer’s instructions. Total protein amounts were estimated using a BCA kit (Thermo Fisher Scentific, 23225). The absorbance of products at 450 nm was detected using a microplate reader (Tecan Spark).
RNA extraction and sequencing
Four cochleae without vestibular parts from two mice in different experimental group groups were pooled for RNA extraction, while ten cultured cochlear explants from WT mice treated with vehicle or cisplatin were pooled for RNA extraction. TRIzol reagent (Ambion, 15596018) was used to extract total RNA following the manufacturer’s instructions. The extracted RNA of cochlear explants was sent to the Annoroad Gene Technology Corporation (Beijing) for next-generation sequencing. In brief, RNA was identified using the BioAnalyzer 2100, while sequencing libraries were generated using a NEBNext Ultra RNA Library Prep Kit for Illumina (no. E7530L, NEB, USA). The index-coded samples were clustered following the manufacturer’s instructions. Differentially expressed genes were detected at q ≤ 0.05 and |log2_ratio| ≥ 1.
qPCR
ReverTra Ace (Toyobo, FSQ-301) was used to reverse transcribe total RNA into cDNA following the manufacturer’s instructions. Real-time PCR system of QuantStudio 1 Plus (Applied Biosystems, USA) and SYBR Green Real-time PCR Master Mix (Toyobo, QPK-201) were used for qPCR. Primers used for real-time qPCR included: m-Gpx4: 5′-TGCATCGTCACCAACGTGGC-3’ (forward); 5′-CTTCACCACGCAGCCGTTCT-3′ (reverse); m-β-Actin: 5′-ACTGCCGCATCCTCTTCCT-3′ (forward); 5′-TCAACGTCACACTTCATGATGGA-3’ (reverse); m-Ncoa4: 5′-GAAAAGAGGCTATATCCAGGTGC-3′ (forward); 5′-AAGAAGCCACTCACTCAGAGA-3′ (reverse); m-Slc40a1: 5′-GCGATCACAATCCAAAGGGAC-3′ (forward); 5′-TTGGTTAGCTGGTCAATCCTTC-3′ (reverse); m-Slc7a11: 5′-AGGGCATACTCCAGAACACG-3′ (forward); 5′-GGACCAAAGACCTCCAGAATG-3′ (reverse); m-Trf: 5′-GCTTCCGTGACCACATGAAGA-3′ (forward); 5′-CCGGCATCGTACACCCAAC-3′ (reverse); m-Fsp1: 5′-ACCGCAGTGCATTTGAGAGT-3′ (forward) 5′-GGCCTCTGCTTCATGGAGTT-3′ (reverse).
Immunofluorescence staining
Paraffin-embedded cochleae tissue sections, cochlear explants, and basilar membranes of mice cochleae were prepared for immunofluorescence staining as described previously.99 The samples were permeabilized with 0.1% Triton X-100 for 30 min and blocked with 10% goat serum at room temperature for 1 h, then incubated with primary antibodies (rabbit anti-Gpx4 (1:200, Abcam, UK, ab12506), rabbit anti-Sox2 (1:200 Abcam, ab97959), mouse anti-TfR1 (1:200, Invitrogen, 13-6800), rabbit anti-myosin7a (1:800, Proteus BioSciences, CA, 25-6790), and mouse anti-Ctbp2 (1:400, BD Biosciences, 612044) at 4°C overnight. The samples were incubated with Alexa Fluor 488/594-labeled secondary antibody at room temperature for 1 h, then labeled with phalloidin-iFluor 594 (Abcam, ab176757). The samples were then washed with PBS and mounted with a medium containing DAPI (Abcam, ab104139) for visualization using confocal fluorescence (Zeiss, LSM 980) and fluorescence microscopy (Leica, DMi8).
ROS detection
In vitro
DCFH-DA (Solarbio, CA1410) was used to evaluate intracellular ROS generation in cisplatin-treated HEI-OC1 cells and cochlear explants. HEI-OC1 cells and cochlear explants were cultured and subjected to the specified treatments. Then, the medium of each dish was replaced with 1 mL of fresh culture medium without FBS containing DCFH-DA (10 μM) and the cells were further incubated at 37°C in 5% CO2 for 20 min. After washing thrice with culture medium without FBS, the fluorescence of DCF (λex = 488 nm, λem = 525 nm) in each dish was immediately captured using an inverted fluorescence microscope (Leica, DMi8) and a confocal microscope (Zeiss, Celldiscoverer 7). Images were analyzed for fluorescence intensity using ImageJ software.
In vivo
The detection of endogenous ROS level in OHCs in vivo was performed as described previously. In vivo ROS levels were determined using the ROS-sensitive dye, hydroethidine.100,101 The Pou4f3-Gpx4Cre/ER mice subjected to the specified treatments were intraperitoneally injected with hydroethidine (10 μg/g, Sigma, 37291) 6 h before the mice were sacrificed. The cochleae tissues were fixed in 4% PFA overnight and decalcified in 10% EDTA at 4°C. The basilar membrane was isolated from the cochlea and mounted with DAPI Fluorescence Mounting Medium (Abcam, ab104139). The fluorescence of DHE (λex = 532 nm) in OHCs was measured using confocal microscopes (Zeiss, LSM 980, and Leica, SP8). Images were analyzed for fluorescence intensity using ImageJ software.
Western blot
HEI-OC1 cells were cultured in 10-cm plates, and whole cochleae homogenates without vestibular parts were prepared in RIPA lysis buffer (Thermo Fisher, 89901). The blots were incubated with the corresponding primary antibodies as follows, rabbit anti-Gpx4 (1:1,000, Abcam, ab125066), rabbit anti-Trf (1:1,000, Abcam, ab313583), mouse anti-TfR1 (1:1,000, Invitrogen, 13-6800), rabbit anti-HO-1 (1:1,000, ABclonal, Chinal, A1346), rabbit anti- Nrf2 (1:1,000, ABclonal, A1244), and mouse anti-β-actin (1:6,000, Proteintech, Chinal, 81115-1-RR). The blots were also incubated with secondary horseradish peroxidase-conjugated anti-mouse (Zhongshan Golden Bridge Biotechnology, ZB-2305) and anti-rabbit (Zhongshan Golden Bridge Biotechnology, ZB-2301) antibodies (1:5,000 dilution). Finally, a chemiluminescent substrate kit (Tanon, China, 4600SF) and a chemiluminescence image analysis system (Tanon, 5200) were used to visualize protein bands.
SEM
First, P1 mice were anesthetized with sodium pentobarbitone and decapitated. The cochleae were then removed and fixed with 2.5% glutaraldehyde at 4°C for 24 h. The basilar membranes of cochleae were microdissected and postfixed in 1% OsO4 at 4°C for 4 h. The basilar membranes were dehydrated using ethanol (critical point dried from liquid CO2). The samples were then mounted onto an SEM stub for analysis using a Hitachi Regulus 8100 field emission SEM at 5 kV.
TEM
In brief, the basilar membranes from P1 mice were prefixed with a 3% glutaraldehyde, then postfixed in 1% OsO4, dehydrated using acetone, infiltrated in Epox 812, and embedded. The semi-thin sections were stained with methylene blue for tissue orientation. Ultrathin sections were obtained using a diamond knife and then stained with uranyl acetate and lead citrate for subcellular structure analysis using a JEM-1400-FLASH transmission electron microscope.
Cisplatin and luteolin treatment
Cisplatin (0.5 mg/mL, Selleck, S2320) was freshly prepared using sterile physiological saline and intraperitoneally injected into mice (4 mg/kg for continuous injections). Luteolin (0.5 mg/mL, Selleck, S2320) was freshly prepared using 5% DMSO + 40% PEG300 + 5% Tween 80 + 50% ddH2O.
ABR and DPOAE measurements
ABR and DPOAE measurements were conducted as described previously.102 In brief, the mice were anesthetized with pentobarbital sodium (100 mg/kg) and kept warm during the experiments. The mice were then put in a soundproof chamber and disinfected using a 75% alcohol solution. An active electrode was positioned at the vertex between the eyes, the ground electrode was positioned beneath the ear, and the reference electrode was positioned toward the posterior region, adjacent to the tail, of the anesthetized mice. The hearing threshold of ABR was evaluated at 4, 8, 16, 24, and 32 kHz and for a broadband click using the TDT System III (Tucker-Davis Technologies). The stimulus intensity was reduced from 90 to 10 dB at each frequency to determine the hearing threshold. DPOAEs were also recorded using the TDT hardware and software. The levels were swept from 80 to 10 dB SPL (for f2) for each f2/f1 main pair. The f2 levels sufficient for inducing a DPOAE at 5 dB SPL were used to establish hearing thresholds.
Intracellular iron assay
FerroOrange dye (no. F374, Dojindo Laboratories) was used to assess the concentration of intracellular ferrous ions. The cells were cultured and subjected to the specified treatments. The cells were then treated with FerroOrange (1:2,000, v/v) and incubated at 37°C for 30 min. Fluorescence images were captured using a confocal microscope (Zeiss, LSM 980).
Statistical analysis
GraphPad Prism 8.0 software was used for statistical analysis in this study. All experiments were performed at least three times independently (n ≥ 3), and the number of mice in each group was greater than three. Statistical significance was determined using unpaired Student’s t test between groups. All quantitative data are expressed as mean ± SEM, and ∗p < 0.05 was considered a statistically significant difference.
Data and code availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Acknowledgments
This work was supported by grants from National Key R&D Program of China (nos. 2019YFA0111400, 2021YFA1101300, 2021YFA1101800, and 2020YFA0112503), the National Natural Science Foundation of China (nos. 82271175, 82192863, 82001204, 82201294, 82201296, 82301305, 82330033, 82030029, and 92149304), the Natural Science Foundation of Shandong Province (nos. ZR2022QH338, ZR2021QH269, and ZR2022QH205), the Science and Technology Department of Sichuan Province (no. 2021YFS0371), Shenzhen Science and Technology Program (JCYJ20190814093401920 and JCYJ20210324125608022), the 2022 Open Project Fund of Guangdong Academy of Medical Sciences (YKY-KF202201). We acknowledge the support of the Medical Science and Technology Innovation Center to supply the Celldiscoverer 7 confocal fluorescent microscope and Regulus 8100 scanning electron microscope for the observation of samples. An abbreviation list is provided (Table S2).
Author contributions
X.F. and R.C. designed and supervised the research. Z.L., G.H., and X.F. wrote the manuscript and interpreted the data. Z.L., H.Z., G.H., and X.B. conducted most experiments and analyzed data with the help of J.H., T.Z., Y.A., N.G., and F.D. Y.X., W.L., X.Z., B.C., S.G., and X.Z. provided technical support for this research.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2024.02.029.
Contributor Information
Renjie Chai, Email: renjiac@seu.edu.cn.
Xiaolong Fu, Email: 210112@sdfmu.edu.cn.
Supplemental information
References
- 1.Li Y., Chen X., He W., Xia S., Jiang X., Li X., Bai J., Li N., Chen L., Yang B. Apigenin Enhanced Antitumor Effect of Cisplatin in Lung Cancer via Inhibition of Cancer Stem Cells. Nutr. Cancer. 2021;73:1489–1497. doi: 10.1080/01635581.2020.1802494. [DOI] [PubMed] [Google Scholar]
- 2.Meng F., Sun G., Zhong M., Yu Y., Brewer M.A. Anticancer efficacy of cisplatin and trichostatin A 5-aza-2′-deoxycytidine on ovarian cancer. Br. J. Cancer. 2013;108:579–586. doi: 10.1038/bjc.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coen J.J., Zhang P., Saylor P.J., Lee C.T., Wu C.L., Parker W., Lautenschlaeger T., Zietman A.L., Efstathiou J.A., Jani A.B., et al. Bladder Preservation With Twice-a-Day Radiation Plus Fluorouracil/Cisplatin or Once Daily Radiation Plus Gemcitabine for Muscle-Invasive Bladder Cancer: NRG/RTOG 0712A Randomized Phase II Trial. J. Clin. Oncol. 2019;37:44–51. doi: 10.1200/Jco.18.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Du J., Wang X., Li Y., Ren X., Zhou Y., Hu W., Zhou C., Jing Q., Yang C., Wang L., et al. DHA exhibits synergistic therapeutic efficacy with cisplatin to induce ferroptosis in pancreatic ductal adenocarcinoma via modulation of iron metabolism. Cell Death Dis. 2021;12 doi: 10.1038/s41419-021-03996-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dugbartey G.J., Peppone L.J., de Graaf I.A.M. An integrative view of cisplatin-induced renal and cardiac toxicities: Molecular mechanisms, current treatment challenges and potential protective measures. Toxicology. 2016;371:58–66. doi: 10.1016/j.tox.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galluzzi L., Senovilla L., Vitale I., Michels J., Martins I., Kepp O., Castedo M., Kroemer G. Molecular mechanisms of cisplatin resistance. Oncogene. 2012;31:1869–1883. doi: 10.1038/onc.2011.384. [DOI] [PubMed] [Google Scholar]
- 7.Shen D.W., Pouliot L.M., Hall M.D., Gottesman M.M. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012;64:706–721. doi: 10.1124/pr.111.005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Finley R.S., Fortner C.L., Grove W.R. Cisplatin nephrotoxicity: a summary of preventative interventions. Drug Intell. Clin. Pharm. 1985;19:362–367. doi: 10.1177/106002808501900505. [DOI] [PubMed] [Google Scholar]
- 9.Davoudi M., Jadidi Y., Moayedi K., Farrokhi V., Afrisham R. Ameliorative impacts of polymeric and metallic nanoparticles on cisplatin-induced nephrotoxicity: a 2011-2022 review. J. Nanobiotechnology. 2022;20:504. doi: 10.1186/s12951-022-01718-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Breglio A.M., Rusheen A.E., Shide E.D., Fernandez K.A., Spielbauer K.K., McLachlin K.M., Hall M.D., Amable L., Cunningham L.L. Cisplatin is retained in the cochlea indefinitely following chemotherapy. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-01837-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao D., Yu H., Li B., Chen L., Li X., Gu W. Cisplatin Toxicology: The Role of Pro-inflammatory Cytokines and GABA Transporters in Cochlear Spiral Ganglion. Curr. Pharm. Des. 2019;25:4820–4826. doi: 10.2174/1381612825666191106143743. [DOI] [PubMed] [Google Scholar]
- 12.Ding D., Zhang J., Jiang H., Xuan W., Qi W., Salvi R. Some Ototoxic Drugs Destroy Cochlear Support Cells Before Damaging Sensory Hair Cells. Neurotox. Res. 2020;37:743–752. doi: 10.1007/s12640-020-00170-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paken J., Govender C.D., Pillay M., Sewram V. A Review of Cisplatin-Associated Ototoxicity. Semin. Hear. 2019;40:108–121. doi: 10.1055/s-0039-1684041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddel R.R., Kefford R.F., Grant J.M., Coates A.S., Fox R.M., Tattersall M.H. Ototoxicity in patients receiving cisplatin: importance of dose and method of drug administration. Cancer Treat. Rep. 1982;66:19–23. [PubMed] [Google Scholar]
- 15.Kaltenbach J.A., Rachel J.D., Mathog T.A., Zhang J., Falzarano P.R., Lewandowski M. Cisplatin-induced hyperactivity in the dorsal cochlear nucleus and its relation to outer hair cell loss: relevance to tinnitus. J. Neurophysiol. 2002;88:699–714. doi: 10.1152/jn.2002.88.2.699. [DOI] [PubMed] [Google Scholar]
- 16.Wu X., Li X., Song Y., Li H., Bai X., Liu W., Han Y., Xu L., Li J., Zhang D., et al. Allicin protects auditory hair cells and spiral ganglion neurons from cisplatin - Induced apoptosis. Neuropharmacology. 2017;116:429–440. doi: 10.1016/j.neuropharm.2017.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Tange R.A., Vuzevski V.D. Changes in the stria vascularis of the guinea pig due to cis-platinum. Arch. Otorhinolaryngol. 1984;239:41–47. doi: 10.1007/BF00454261. [DOI] [PubMed] [Google Scholar]
- 18.Blair B.G., Larson C.A., Safaei R., Howell S.B. Copper transporter 2 regulates the cellular accumulation and cytotoxicity of Cisplatin and Carboplatin. Clin. Cancer Res. 2009;15:4312–4321. doi: 10.1158/1078-0432.CCR-09-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciarimboli G., Deuster D., Knief A., Sperling M., Holtkamp M., Edemir B., Pavenstädt H., Lanvers-Kaminsky C., am Zehnhoff-Dinnesen A., Schinkel A.H., et al. Organic Cation Transporter 2 Mediates Cisplatin-Induced Oto- and Nephrotoxicity and Is a Target for Protective Interventions. Am. J. Pathol. 2010;176:1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciarimboli G., Deuster D., Knief A., Sperling M., Holtkamp M., Edemir B., Pavenstädt H., Lanvers-Kaminsky C., am Zehnhoff-Dinnesen A., Schinkel A.H., et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am. J. Pathol. 2010;176:1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waissbluth S., Daniel S.J. Cisplatin-induced ototoxicity: Transporters playing a role in cisplatin toxicity. Hear. Res. 2013;299:37–45. doi: 10.1016/j.heares.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 22.Wee N.K.Y., Weinstein D.C., Fraser S.T., Assinder S.J. The mammalian copper transporters CTR1 and CTR2 and their roles in development and disease. Int. J. Biochem. Cell B. 2013;45:960–963. doi: 10.1016/j.biocel.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 23.Hibino H., Nin F., Tsuzuki C., Kurachi Y. How is the highly positive endocochlear potential formed? The specific architecture of the stria vascularis and the roles of the ion-transport apparatus. Pflugers Arch. 2010;459:521–533. doi: 10.1007/s00424-009-0754-z. [DOI] [PubMed] [Google Scholar]
- 24.Kuo M.T., Chen H.H.W., Song I.S., Savaraj N., Ishikawa T. The roles of copper transporters in cisplatin resistance. Cancer Metast Rev. 2007;26:71–83. doi: 10.1007/s10555-007-9045-3. [DOI] [PubMed] [Google Scholar]
- 25.Wakai E., Ikemura K., Mizuno T., Takeuchi K., Tamaru S., Okuda M., Nishimura Y. Repositioning of Lansoprazole as a Protective Agent Against Cisplatin-Induced Ototoxicity. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.896760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fox E., Levin K., Zhu Y., Segers B., Balamuth N., Womer R., Bagatell R., Balis F. Pantoprazole, an Inhibitor of the Organic Cation Transporter 2, Does Not Ameliorate Cisplatin-Related Ototoxicity or Nephrotoxicity in Children and Adolescents with Newly Diagnosed Osteosarcoma Treated with Methotrexate, Doxorubicin, and Cisplatin. Oncologist. 2018;23:762–e79. doi: 10.1634/theoncologist.2018-0037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Giese A.P.J., Tang Y.Q., Sinha G.P., Bowl M.R., Goldring A.C., Parker A., Freeman M.J., Brown S.D.M., Riazuddin S., Fettiplace R., et al. CIB2 interacts with TMC1 and TMC2 and is essential for mechanotransduction in auditory hair cells. Nat. Commun. 2017;8 doi: 10.1038/s41467-017-00061-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramkumar V., Sheth S., Dhukhwa A., Al Aameri R., Rybak L., Mukherjea D. Transient Receptor Potential Channels and Auditory Functions. Antioxid. Redox Signal. 2022;36:1158–1170. doi: 10.1089/ars.2021.0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mukherjea D., Jajoo S., Whitworth C., Bunch J.R., Turner J.G., Rybak L.P., Ramkumar V. Short interfering RNA against transient receptor potential vanilloid 1 attenuates cisplatin-induced hearing loss in the rat. J. Neurosci. 2008;28:13056–13065. doi: 10.1523/JNEUROSCI.1307-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee C.H., Lee D.H., Lee S.M., Kim A.S.Y. Otoprotective Effects of Zingerone on Cisplatin-Induced Ototoxicity. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21103503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang W., Xiong H., Pang J., Su Z., Lai L., Lin H., Jian B., He W., Yang H., Zheng Y. Nrf2 activation protects auditory hair cells from cisplatin-induced ototoxicity independent on mitochondrial ROS production. Toxicol. Lett. 2020;331:1–10. doi: 10.1016/j.toxlet.2020.04.005. [DOI] [PubMed] [Google Scholar]
- 32.Yang Q., Sun G., Yin H., Li H., Cao Z., Wang J., Zhou M., Wang H., Li J. PINK1 Protects Auditory Hair Cells and Spiral Ganglion Neurons from Cisplatin-induced Ototoxicity via Inducing Autophagy and Inhibiting JNK Signaling Pathway. Free Radic. Biol. Med. 2018;120:342–355. doi: 10.1016/j.freeradbiomed.2018.02.025. [DOI] [PubMed] [Google Scholar]
- 33.Rosati R., Shahab M., Neumann W.L., Jamesdaniel S. Inhibition of protein nitration prevents cisplatin-induced inactivation of STAT3 and promotes anti-apoptotic signaling in organ of Corti cells. Exp. Cell Res. 2019;381:105–111. doi: 10.1016/j.yexcr.2019.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mei H., Zhao L., Li W., Zheng Z., Tang D., Lu X., He Y. Inhibition of ferroptosis protects House Ear Institute-Organ of Corti 1 cells and cochlear hair cells from cisplatin-induced ototoxicity. J. Cell. Mol. Med. 2020;24:12065–12081. doi: 10.1111/jcmm.15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma Q. Role of Nrf2 in Oxidative Stress and Toxicity. Annu. Rev. Pharmacol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.So H., Kim H., Kim Y., Kim E., Pae H.O., Chung H.T., Kim H.J., Kwon K.B., Lee K.M., Lee H.Y., et al. Evidence that cisplatin-induced auditory damage is attenuated by downregulation of pro-inflammatory cytokines via Nrf2/HO-1. Jaro-j Assoc. Res. Oto. 2008;9:290–306. doi: 10.1007/s10162-008-0126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angeli J.P.F., Schneider M., Proneth B., Tyurina Y.Y., Tyurin V.A., Hammond V.J., Herbach N., Aichler M., Walch A., Eggenhofer E., et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014;16:1180–U1120. doi: 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Imai H., Matsuoka M., Kumagai T., Sakamoto T., Koumura T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top Microbiol. 2017;403:143–170. doi: 10.1007/82_2016_508. [DOI] [PubMed] [Google Scholar]
- 40.Yang W.S., SriRamaratnam R., Welsch M.E., Shimada K., Skouta R., Viswanathan V.S., Cheah J.H., Clemons P.A., Shamji A.F., Clish C.B., et al. Regulation of Ferroptotic Cancer Cell Death by GPX4. Cell. 2014;156:317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bersuker K., Hendricks J.M., Li Z., Magtanong L., Ford B., Tang P.H., Roberts M.A., Tong B., Maimone T.J., Zoncu R., et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–692. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Linkermann A., Skouta R., Himmerkus N., Mulay S.R., Dewitz C., De Zen F., Prokai A., Zuchtriegel G., Krombach F., Welz P.S., et al. Synchronized renal tubular cell death involves ferroptosis. P Natl. Acad. Sci. USA. 2014;111:16836–16841. doi: 10.1073/pnas.1415518111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jian B., Pang J., Xiong H., Zhang W., Zhan T., Su Z., Lin H., Zhang H., He W., Zheng Y. Autophagy-dependent ferroptosis contributes to cisplatin-induced hearing loss. Toxicol. Lett. 2021;350:249–260. doi: 10.1016/j.toxlet.2021.07.010. [DOI] [PubMed] [Google Scholar]
- 44.Cho S., Hong S.J., Kang S.H., Park Y., Kim S.K. Alpha-Lipoic Acid Attenuates Apoptosis and Ferroptosis in Cisplatin-Induced Ototoxicity via the Reduction of Intracellular Lipid Droplets. Int. J. Mol. Sci. 2022;23 doi: 10.3390/ijms231810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Roy S., Ryals M.M., Van den Bruele A.B., Fitzgerald T.S., Cunningham L.L. Sound preconditioning therapy inhibits ototoxic hearing loss in mice. J. Clin. Invest. 2013;123:4945–4949. doi: 10.1172/JCI71353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng H., Schorpp K., Jin J., Yozwiak C.E., Hoffstrom B.G., Decker A.M., Rajbhandari P., Stokes M.E., Bender H.G., Csuka J.M., et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020;30:3411–3423.e7. doi: 10.1016/j.celrep.2020.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ingersoll M.A., Malloy E.A., Caster L.E., Holland E.M., Xu Z., Zallocchi M., Currier D., Liu H., He D.Z.Z., Min J., et al. BRAF inhibition protects against hearing loss in mice. Sci. Adv. 2020;6 doi: 10.1126/sciadv.abd0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu S.C. Regulation of glutathione synthesis. Mol. Aspects Med. 2009;30:42–59. doi: 10.1016/j.mam.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gao M., Monian P., Quadri N., Ramasamy R., Jiang X. Glutaminolysis and Transferrin Regulate Ferroptosis. Mol. Cell. 2015;59:298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shang Y., Luo M., Yao F., Wang S., Yuan Z., Yang Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell. Signal. 2020;72 doi: 10.1016/j.cellsig.2020.109633. [DOI] [PubMed] [Google Scholar]
- 51.Tuo Q.Z., Lei P., Jackman K.A., Li X.L., Xiong H., Li X.L., Liuyang Z.Y., Roisman L., Zhang S.T., Ayton S., et al. Tau-mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry. 2017;22:1520–1530. doi: 10.1038/mp.2017.171. [DOI] [PubMed] [Google Scholar]
- 52.Ma S., Henson E.S., Chen Y., Gibson S.B. Ferroptosis is induced following siramesine and lapatinib treatment of breast cancer cells. Cell Death Dis. 2016;7:e2307. doi: 10.1038/cddis.2016.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Geng N., Shi B.J., Li S.L., Zhong Z.Y., Li Y.C., Xua W.L., Zhou H., Cai J.H. Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells. Eur. Rev. Med. Pharmacol. Sci. 2018;22:3826–3836. doi: 10.26355/eurrev_201806_15267. [DOI] [PubMed] [Google Scholar]
- 54.Dixon S.J., Patel D.N., Welsch M., Skouta R., Lee E.D., Hayano M., Thomas A.G., Gleason C.E., Tatonetti N.P., Slusher B.S., Stockwell B.R. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife. 2014;3 doi: 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vibert J., Watson S. Drug-Tolerant Persister Cancer Cells Are Vulnerable to GPX4 Inhibition. Oncologie. 2018;20:140–141. [Google Scholar]
- 56.Neitemeier S., Jelinek A., Laino V., Hoffmann L., Eisenbach I., Eying R., Ganjam G.K., Dolga A.M., Oppermann S., Culmsee C. BID links ferroptosis to mitochondrial cell death pathways. Redox Biol. 2017;12:558–570. doi: 10.1016/j.redox.2017.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xie Y., Hou W., Song X., Yu Y., Huang J., Sun X., Kang R., Tang D. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dodson H.C., Mohuiddin A. Response of spiral ganglion neurones to cochlear hair cell destruction in the guinea pig. J. Neurocytol. 2000;29:525–537. doi: 10.1023/a:1007201913730. [DOI] [PubMed] [Google Scholar]
- 59.Shah R., Margison K., Pratt D.A. The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem. Biol. 2017;12:2538–2545. doi: 10.1021/acschembio.7b00730. [DOI] [PubMed] [Google Scholar]
- 60.Lin Y., Shi R., Wang X., Shen H.M. Luteolin, a Flavonoid with Potential for Cancer Prevention and Therapy. Curr. Cancer Drug Tar. 2008;8:634–646. doi: 10.2174/156800908786241050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lim D.Y., Jeong Y., Tyner A.L., Park J.H.Y. Induction of cell cycle arrest and apoptosis in HT-29 human colon cancer cells by the dietary compound luteolin. Am. J. Physiol. Gastrointest. Liver Physiol. 2007;292:G66–G75. doi: 10.1152/ajpgi.00248.2006. [DOI] [PubMed] [Google Scholar]
- 62.Osman N.H.A., Said U.Z., El-Waseef A.M., Ahmed E.S.A. Luteolin supplementation adjacent to aspirin treatment reduced dimethylhydrazine-induced experimental colon carcinogenesis in rats. Tumour Biol. 2015;36:1179–1190. doi: 10.1007/s13277-014-2678-2. [DOI] [PubMed] [Google Scholar]
- 63.Imran M., Rauf A., Abu-Izneid T., Nadeem M., Shariati M.A., Khan I.A., Imran A., Orhan I.E., Rizwan M., Atif M., et al. Luteolin, a flavonoid, as an anticancer agent: A review. Biomed. Pharmacother. 2019;112 doi: 10.1016/j.biopha.2019.108612. [DOI] [PubMed] [Google Scholar]
- 64.Wang H., Luo Y., Qiao T., Wu Z., Huang Z. Luteolin sensitizes the antitumor effect of cisplatin in drug-resistant ovarian cancer via induction of apoptosis and inhibition of cell migration and invasion. J. Ovarian Res. 2018;11:93. doi: 10.1186/s13048-018-0468-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sawmiller D., Li S., Shahaduzzaman M., Smith A.J., Obregon D., Giunta B., Borlongan C.V., Sanberg P.R., Tan J. Luteolin reduces Alzheimer's disease pathologies induced by traumatic brain injury. Int. J. Mol. Sci. 2014;15:895–904. doi: 10.3390/ijms15010895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stockwell B.R. Ferroptosis: Death by lipid peroxidation. Free Radic. Biol. Med. 2018;120:S7. doi: 10.1016/j.freeradbiomed.2018.04.034. [DOI] [Google Scholar]
- 67.Qi Y., Fu S., Pei D., Fang Q., Xin W., Yuan X., Cao Y., Shu Q., Mi X., Luo F. Luteolin attenuated cisplatin-induced cardiac dysfunction and oxidative stress via modulation of Keap1/Nrf2 signaling pathway. Free Radic. Res. 2022;56:209–221. doi: 10.1080/10715762.2022.2067042. [DOI] [PubMed] [Google Scholar]
- 68.Adedoyin O., Boddu R., Traylor A., Lever J.M., Bolisetty S., George J.F., Agarwal A. Heme oxygenase-1 mitigates ferroptosis in renal proximal tubule cells. Am. J. Physiol-renal. 2018;314:F702–F714. doi: 10.1152/ajprenal.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Anandhan A., Dodson M., Schmidlin C.J., Liu P., Zhang D.D. Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell Chem. Biol. 2020;27:436–447. doi: 10.1016/j.chembiol.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Forge A., Schacht J. Aminoglycoside antibiotics. Audiol. Neurootol. 2000;5:3–22. doi: 10.1159/000013861. [DOI] [PubMed] [Google Scholar]
- 71.Kopke R., Allen K.A., Henderson D., Hoffer M., Frenz D., Van de Water T. A radical demise. Toxins and trauma share common pathways in hair cell death. Ann. N. Y. Acad. Sci. 1999;884:171–191. doi: 10.1111/j.1749-6632.1999.tb08641.x. [DOI] [PubMed] [Google Scholar]
- 72.Alharazneh A., Luk L., Huth M., Monfared A., Steyger P.S., Cheng A.G., Ricci A.J. Functional hair cell mechanotransducer channels are required for aminoglycoside ototoxicity. PLoS One. 2011;6 doi: 10.1371/journal.pone.0022347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jensen-Smith H.C., Hallworth R., Nichols M.G. Gentamicin rapidly inhibits mitochondrial metabolism in high-frequency cochlear outer hair cells. PLoS One. 2012;7 doi: 10.1371/journal.pone.0038471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sha S.H., Taylor R., Forge A., Schacht J. Differential vulnerability of basal and apical hair cells is based on intrinsic susceptibility to free radicals. Hear. Res. 2001;155:1–8. doi: 10.1016/s0378-5955(01)00224-6. [DOI] [PubMed] [Google Scholar]
- 75.Engel J., Braig C., Rüttiger L., Kuhn S., Zimmermann U., Blin N., Sausbier M., Kalbacher H., Münkner S., Rohbock K., et al. Two classes of outer hair cells along the tonotopic axis of the cochlea. Neuroscience. 2006;143:837–849. doi: 10.1016/j.neuroscience.2006.08.060. [DOI] [PubMed] [Google Scholar]
- 76.Esterberg R., Hailey D.W., Coffin A.B., Raible D.W., Rubel E.W. Disruption of intracellular calcium regulation is integral to aminoglycoside-induced hair cell death. J. Neurosci. 2013;33:7513–7525. doi: 10.1523/JNEUROSCI.4559-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Esterberg R., Hailey D.W., Rubel E.W., Raible D.W. ER-mitochondrial calcium flow underlies vulnerability of mechanosensory hair cells to damage. J. Neurosci. 2014;34:9703–9719. doi: 10.1523/JNEUROSCI.0281-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Esterberg R., Linbo T., Pickett S.B., Wu P., Ou H.C., Rubel E.W., Raible D.W. Mitochondrial calcium uptake underlies ROS generation during aminoglycoside-induced hair cell death. J. Clin. Invest. 2016;126:3556–3566. doi: 10.1172/JCI84939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kokotas H., Petersen M.B., Willems P.J. Mitochondrial deafness. Clin. Genet. 2007;71:379–391. doi: 10.1111/j.1399-0004.2007.00800.x. [DOI] [PubMed] [Google Scholar]
- 80.Yamasoba T., Someya S., Yamada C., Weindruch R., Prolla T.A., Tanokura M. Role of mitochondrial dysfunction and mitochondrial DNA mutations in age-related hearing loss. Hear. Res. 2007;226:185–193. doi: 10.1016/j.heares.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 81.Pickett S.B., Thomas E.D., Sebe J.Y., Linbo T., Esterberg R., Hailey D.W., Raible D.W. Cumulative mitochondrial activity correlates with ototoxin susceptibility in zebrafish mechanosensory hair cells. Elife. 2018;7 doi: 10.7554/eLife.38062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zholudeva L.V., Ward K.G., Nichols M.G., Smith H.J. Gentamicin differentially alters cellular metabolism of cochlear hair cells as revealed by NAD(P)H fluorescence lifetime imaging. J. Biomed. Opt. 2015;20 doi: 10.1117/1.JBO.20.5.051032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Battaglia A.M., Chirillo R., Aversa I., Sacco A., Costanzo F., Biamonte F. Ferroptosis and Cancer: Mitochondria Meet the "Iron Maiden" Cell Death. Cells. 2020;9:1505. doi: 10.3390/cells9061505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doll S., Freitas F.P., Shah R., Aldrovandi M., da Silva M.C., Ingold I., Goya Grocin A., Xavier da Silva T.N., Panzilius E., Scheel C.H., et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–698. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 85.Meinnel T., Dian C., Giglione C. Myristoylation, an Ancient Protein Modification Mirroring Eukaryogenesis and Evolution. Trends Biochem. Sci. 2020;45:619–632. doi: 10.1016/j.tibs.2020.03.007. [DOI] [PubMed] [Google Scholar]
- 86.Borgese N., Aggujaro D., Carrera P., Pietrini G., Bassetti M. A role for N-myristoylation in protein targeting: NADH-cytochrome b5 reductase requires myristic acid for association with outer mitochondrial but not ER membranes. J. Cell Biol. 1996;135:1501–1513. doi: 10.1083/jcb.135.6.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marshall K.R., Gong M., Wodke L., Lamb J.H., Jones D.J.L., Farmer P.B., Scrutton N.S., Munro A.W. The human apoptosis-inducing protein AMID is an oxidoreductase with a modified flavin cofactor and DNA binding activity. J. Biol. Chem. 2005;280:30735–30740. doi: 10.1074/jbc.M414018200. [DOI] [PubMed] [Google Scholar]
- 88.Crane F.L. Discovery of ubiquinone (coenzyme Q) and an overview of function. Mitochondrion. 2007;7(Suppl):S2–S7. doi: 10.1016/j.mito.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 89.Choi J.S., Choi Y.J., Park S.H., Kang J.S., Kang Y.H. Flavones mitigate tumor necrosis factor-alpha-induced adhesion molecule upregulation in cultured human endothelial cells: role of nuclear factor-kappa B. J. Nutr. 2004;134:1013–1019. doi: 10.1093/jn/134.5.1013. [DOI] [PubMed] [Google Scholar]
- 90.Shi R.X., Ong C.N., Shen H.M. Luteolin sensitizes tumor necrosis factor-alpha-induced apoptosis in human tumor cells. Oncogene. 2004;23:7712–7721. doi: 10.1038/sj.onc.1208046. [DOI] [PubMed] [Google Scholar]
- 91.Ju W., Wang X., Shi H., Chen W., Belinsky S.A., Lin Y. A critical role of luteolin-induced reactive oxygen species in blockage of tumor necrosis factor-activated nuclear factor-kappaB pathway and sensitization of apoptosis in lung cancer cells. Mol. Pharmacol. 2007;71:1381–1388. doi: 10.1124/mol.106.032185. [DOI] [PubMed] [Google Scholar]
- 92.Chiang C.T., Way T.D., Lin J.K. Sensitizing HER2-overexpressing cancer cells to luteolin-induced apoptosis through suppressing p21(WAF1/CLP1) expression with rapamycin. Mol. Cancer Ther. 2007;6:2127–2138. doi: 10.1158/1535-7163.Mct-07-0107. [DOI] [PubMed] [Google Scholar]
- 93.Shi R., Huang Q., Zhu X., Ong Y.B., Zhao B., Lu J., Ong C.N., Shen H.M. Luteolin sensitizes the anticancer effect of cisplatin via c-Jun NH2-terminal kinase-mediated p53 phosphorylation and stabilization. Mol. Cancer Ther. 2007;6:1338–1347. doi: 10.1158/1535-7163.Mct-06-0638. [DOI] [PubMed] [Google Scholar]
- 94.Fan X., Du K., Li N., Zheng Z., Qin Y., Liu J., Sun R., Su Y. Evaluation of Anti-Nociceptive and Anti-Inflammatory Effect of Luteolin in Mice. J. Environ. Pathol. Toxicol. Oncol. 2018;37:351–364. doi: 10.1615/JEnvironPatholToxicolOncol.2018027666. [DOI] [PubMed] [Google Scholar]
- 95.Andrews N.C., Schmidt P.J. Iron homeostasis. Annu. Rev. Physiol. 2007;69:69–85. doi: 10.1146/annurev.physiol.69.031905.164337. [DOI] [PubMed] [Google Scholar]
- 96.Qian Z.M., Li H., Sun H., Ho K. Targeted drug delivery via the transferrin receptor-mediated endocytosis pathway. Pharmacol. Rev. 2002;54:561–587. doi: 10.1124/pr.54.4.561. [DOI] [PubMed] [Google Scholar]
- 97.Pantopoulos K., Porwal S.K., Tartakoff A., Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51:5705–5724. doi: 10.1021/bi300752r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Muckenthaler M.U., Galy B., Hentze M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- 99.Li P., Liu Z., Wang J., Bi X., Xiao Y., Qiao R., Zhou X., Guo S., Wan P., Chang M., et al. Gstm1/Gstt1 is essential for reducing cisplatin ototoxicity in CBA/CaJ mice. FASEB J. 2022;36 doi: 10.1096/fj.202200324R. [DOI] [PubMed] [Google Scholar]
- 100.Le Belle J.E., Orozco N.M., Paucar A.A., Saxe J.P., Mottahedeh J., Pyle A.D., Wu H., Kornblum H.I. Proliferative Neural Stem Cells Have High Endogenous ROS Levels that Regulate Self-Renewal and Neurogenesis in a PI3K/Akt-Dependant Manner. Cell Stem Cell. 2011;8:59–71. doi: 10.1016/j.stem.2010.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kunz A., Anrather J., Zhou P., Orio M., Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J. Cerebr Blood F Met. 2007;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- 102.Schwander M., Sczaniecka A., Grillet N., Bailey J.S., Avenarius M., Najmabadi H., Steffy B.M., Federe G.C., Lagler E.A., Banan R., et al. A forward genetics screen in mice identifies recessive deafness traits and reveals that pejvakin is essential for outer hair cell function. J. Neurosci. 2007;27:2163–2175. doi: 10.1523/JNEUROSCI.4975-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.