

Abstract
Protein acetylation is one of the extensively studied post-translational modifications (PTMs) due to its significant roles across a myriad of biological processes. Although many computational tools for acetylation site identification have been developed, there is a lack of benchmark dataset and bespoke predictors for non-histone acetylation site prediction. To address these problems, we have contributed to both dataset creation and predictor benchmark in this study. First, we construct a non-histone acetylation site benchmark dataset, namely NHAC, which includes 11 subsets according to the sequence length ranging from 11 to 61 amino acids. There are totally 886 positive samples and 4707 negative samples for each sequence length. Secondly, we propose TransPTM, a transformer-based neural network model for non-histone acetylation site predication. During the data representation phase, per-residue contextualized embeddings are extracted using ProtT5 (an existing pre-trained protein language model). This is followed by the implementation of a graph neural network framework, which consists of three TransformerConv layers for feature extraction and a multilayer perceptron module for classification. The benchmark results reflect that TransPTM has the competitive performance for non-histone acetylation site prediction over three state-of-the-art tools. It improves our comprehension on the PTM mechanism and provides a theoretical basis for developing drug targets for diseases. Moreover, the created PTM datasets fills the gap in non-histone acetylation site datasets and is beneficial to the related communities. The related source code and data utilized by TransPTM are accessible at https://www.github.com/TransPTM/TransPTM.
Keywords: Non-histone acetylation, deep learning, transformer, protein language model
INTRODUCTION
Protein post-translational modification (PTM) is a fundamental mechanism where chemical groups are added to amino acid chains. It is widely reported that PTMs modulate protein functions, physicochemical properties, conformation, stability and interactions between proteins [1–3]. The most prominent PTMs include methylation, phosphorylation, glycosylation, ubiquitination and acetylation [4–7]. In particular, protein acetylation, a type of covalent PTM, involves the bonding of an acetyl group to the amino group of a lysine residue within a protein [8]. It is suggested that lysine acetylation can be categorized into histone acetylation and non-histone protein acetylation, playing distinct roles in cellular functions. Histone acetylation typically occurs in the context of chromatin structure and gene regulation, which have been widely studied [9]. It reduces the electrostatic attraction between histones and DNA, thereby loosening chromatin structure and facilitating transcriptional activation [10]. These reversible regulation processes are catalyzed by histone acetyltransferases and histone deacetylases [11]. On the other hand, non-histone protein acetylation encompasses a broader range of enzymes and target proteins. A notable example is the acetylation of 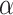 -tubulin at the lysine 40 (K40) position, which is catalyzed by the acetyltransferase
-tubulin at the lysine 40 (K40) position, which is catalyzed by the acetyltransferase  TAT1 [12]. Moreover, non-histone protein acetylation plays diverse roles in cellular signaling, DNA damage repair, autophagy, messenger RNA stability and protein–protein interactions [13–15]. Furthermore, many non-histone protein acetylations are associated with various diseases, such as heart failure, Alzheimer’s disease and cancers [16–18].
TAT1 [12]. Moreover, non-histone protein acetylation plays diverse roles in cellular signaling, DNA damage repair, autophagy, messenger RNA stability and protein–protein interactions [13–15]. Furthermore, many non-histone protein acetylations are associated with various diseases, such as heart failure, Alzheimer’s disease and cancers [16–18].
In recent years, with the growth of acetylome databases, a large number of computational tools for acetylation site identifications have emerged. For example, Wu et al. [19] employed deep learning to create DeepAcet, a new model for acetylation site prediction. This prediction model merges various feature extraction methods and utilizes a multilayer perceptron (MLP) for classification. Upon evaluating the model’s prediction performance via 10-fold cross-validation and independent testing set, the accuracies of 84.95% and 84.87% were reported, respectively. Meanwhile, Muhammad et al. [20] proposed Histone-Net, a novel deep learning predictor capable of predicting three types of histone makers (histone occupancy, acetylation and methylation levels) across multiple datasets in intra-domain and cross-domain binary classification paradigms. Within the study, ‘intra-domain’ means the model is trained and tested on the same type of histone marker, while ‘cross-domain’ means it is trained on one type of histone marker and tested on another type of histone marker. This model outperforms state-of-the-art approaches by an average accuracy of 7% across 10 different datasets. However, the landscape of lysine acetylation remains incomplete. The existing in silico protein acetylation tools, whether they are traditional machine learning models or deep learning models, are applied only to histone acetylation. Despite the identification of a substantial number of lysine-acetylated proteins, the development of predictors for non-histone acetylation site identification has significantly lagged behind.
Hence, we present a deep model called TransPTM (Transformer PTM) for non-histone protein lysine acetylation site prediction. First, we have meticulously constructed an unprecedented dataset of non-histone acetylation site, NHAC (Non-Histone Acetylation Collection), incorporating experimentally identified site obtained from a comprehensive review conducted by Narita et al. [15]. Secondly, to effectively transform amino acid sequence character signals into numerical signals, we utilize embeddings extracted from the protein language model (pLM), ProtT5 (based on the NLP seq2seq model, ProtT5 [21]). This method is employed to gather multiple residue information and generate feature vector. After that, the data are represented by graph, which consists of a pLM embedded sequences as node features and amino acids interactions as edge features. To detect acetylation, graphs are then fed into a transformer-based [22] graph neural network (GNN) architecture, which comprises a transformer module and an MLP module. Its performance on the non-histone acetylation independent testing set outperforms three existing acetylation site predictors, in terms of accuracy, area under the receiver operating characteristics (ROC) curve (AUC) and area under precision-recall curve (AUPRC) (0.88, 0.83 and 0.51), respectively. Finally, amino acid distribution analysis [23], attention maps of positions and dimensionality reduction visualization [24] are conducted to illustrate the interpretability of TransPTM. The results indicate that non-histone acetylation tends to occur on the downstream regions with specific positions of protein sequences; TransformerConv layers in our model can transform the original data vectors into separable space before MLP module classification.
METHODS
Benchmark dataset construction
Non-histone acetylation sites refer to a broad concept that includes all acetylation events on lysine residues within proteins, aside from those in histones. Given this concept’s extensive scope, it is challenging to directly obtain accurate statistics on non-histone acetylation sites from existing databases. To construct the benchmark dataset, NHAC (Non-Histone Acetylation Collection) (Figure 1), we leveraged Narita et al.’s [15] review article as our original data source. To the best of our knowledge, this study provides the most comprehensive collection of non-histone protein acetylation site information, including 379 full-length protein sequences with 1100 acetylated positions. These long sequences were first downloaded in bulk from the UniProt [25] database. Then, peptide sequences of length 2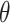 +1 were truncated from full-length proteins. In this context, ‘1’ represents our target amino acid lysine (K), and the symbol
+1 were truncated from full-length proteins. In this context, ‘1’ represents our target amino acid lysine (K), and the symbol 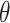 is an integer serving as an indexing notation for the sequence. This approach enables a standardized representation of varying peptide sequence lengths as derived from Chou’s formulation [26]. The peptide sequence samples can be described by the following equation:
is an integer serving as an indexing notation for the sequence. This approach enables a standardized representation of varying peptide sequence lengths as derived from Chou’s formulation [26]. The peptide sequence samples can be described by the following equation:
Figure 1.

Workflow of the non-histone acetylation benchmark dataset (NHAC) construction.
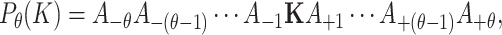 |
(1) |
where the character  denotes lysine at the center position of the sequence and
denotes lysine at the center position of the sequence and 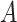 s represent neighboring amino acids of the
s represent neighboring amino acids of the 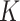 site.
site. 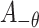 represents the
represents the  th upstream amino acid residue from the center, and
th upstream amino acid residue from the center, and 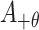 represents the
represents the 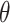 th downstream amino acid residue.
th downstream amino acid residue.
We designate the peptide sequences that contain acetylated central K as positive samples, while the sequences that encompass non-acetylated central K are referred as negative samples. Quantitatively, 1100 positive samples and 11 784 negative samples are obtained from the original full-length protein sequences. In order to examine the impact of window size diversity on performance, we varied the value of 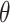 across 11 increments from 5 to 30. For each sequence length, we obtained a total of 1100 positive samples and 11 784 negative samples.
across 11 increments from 5 to 30. For each sequence length, we obtained a total of 1100 positive samples and 11 784 negative samples.
To minimize redundancy and alleviate data imbalance, both positive and negative sequences with more than 40% homology were cut off using the CD-HIT software [27, 28]. Finally, the obtained 886 acetylated and 4707 non-acetylated peptide samples were randomly split to construct the training dataset, independent testing dataset and validation set. The ratio of samples allocated to each set was maintained at 7:2:1. Table 1 tabulates the information of our benchmark dataset, which is provided in the github page.
Table 1.
Statistics of the benchmark dataset NHAC
| Data type | Positive samples | Negative samples | Total | Sequence length |
|---|---|---|---|---|
| Training | 637 | 3387 | 4024 | 11, 15, 21, 25, 31, 35, 41, 45, 51, 55, 61 |
| Validation | 76 | 401 | 477 | |
| Testing | 173 | 919 | 1092 | |
| Total | 886 | 4707 | 5593 |
Amino acid encoding
To convert each amino acid residue in sequence into a numerical vector, we apply two amino acid encoding methods: (1) one-hot encoding and (2) ProtT5 embedding (Figure 2), as described below.
Figure 2.

Extraction of embeddings from ProtT5 language model (L = 2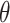 +1 (
+1 (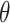 = 5, 7, 10, 12, 15, 17, 20, 22, 25, 27, 30)).
= 5, 7, 10, 12, 15, 17, 20, 22, 25, 27, 30)).
One-hot encoding processes non-histone protein sequence data by converting each amino acid in the protein sequence into a 20-dimensional vector. We use it for two main reasons: One is that it serves as a straightforward and effective feature representation for biological sequences, such as proteins [29] and RNAs [30]. More importantly, earlier studies [7] have frequently employed one-hot as a baseline encoding scheme for amino acid chain, such as MusiteDeep [31], one of the most cited tools in this field. Therefore, we also employ one-hot encoding to represent our peptide chains. The 20 distinct amino acids (as there are a total of 20 natural amino acids) is encoded into a 20-dimensional vector, consisting solely of 0s and 1s. For example, amino acid alanine (A) was encoded as (0,1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0), and lysine (K) was encoded as (1,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0,0). As a result, each sequence with a window size of 2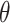 +1 (
+1 (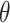 = 5, 7, 10, 12, 15, 17, 20, 22, 25, 27, 30) was transformed into a 20
= 5, 7, 10, 12, 15, 17, 20, 22, 25, 27, 30) was transformed into a 20 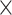 (2
(2 +1)-dimensional feature vector.
+1)-dimensional feature vector.
Recently, language models (LMs) have been given attention for their capacity to derive contextualized embeddings from large unlabeled language datasets, in contrast to static and context-insensitive word embeddings. This progress is now applied to proteins via pLMs [32]. Due to the abundance of protein sequence databases, many pLMs have been created to extract useful information from those resources [33]. This information can then be repurposed for other tasks, such as protein property prediction [34]. Notably, these pLMs have demonstrated their capacity to better understand sequence relationships. In our research, we utilize the encoder output of the pre-trained model ProtT5-XL-U50 [35] to extract the embedding feature. ProtT5-XL-U50 is a transformer-based LM with 3 billion parameters. It is initially trained on the Big Fantastic Database (BFD) [36], which contains 65 million protein families cataloged using multiple sequence alignments (MSAs) and hidden Markov models. And subsequently fine-tuned on the UniRef50 [37] database, which provides clustered sets of sequence data from UniProtKB and selected UniParc records [25]. The embedding feature for a peptide chain is acquired by inputting its sequence into ProtT5-XL-U50 to enable the encoder output, yielding the embedding with 1024 embedded dimensions for each residue. Consequently, this embedding feature is position-dependent, capturing each residue’s contextual information.
Graph representation
A graph  consists of a set of nodes (also known as vertices)
consists of a set of nodes (also known as vertices) 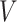 and a set of edges
and a set of edges  . In this context, node
. In this context, node 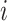 is represented by a vector
is represented by a vector 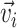 . The edges can be represented as an adjacency matrix
. The edges can be represented as an adjacency matrix 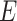 , in which, if
, in which, if 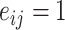 , it indicates that nodes
, it indicates that nodes  and
and  are connected by an edge. In a protein sequence graph, each node represents an amino acid residue, and an edge defines the relationship between protein pairs (residual contacts). In our setting, protein sequences have been embedded by pre-trained pLM ProtT5 as mentioned above, which is a
are connected by an edge. In a protein sequence graph, each node represents an amino acid residue, and an edge defines the relationship between protein pairs (residual contacts). In our setting, protein sequences have been embedded by pre-trained pLM ProtT5 as mentioned above, which is a 
 1024 matrix where
1024 matrix where  is length of protein sequence. Each residue is considered as a node and edges are defined between any two residues, which means
is length of protein sequence. Each residue is considered as a node and edges are defined between any two residues, which means 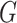 is a complete graph.
is a complete graph.
Non-histone acetylation site prediction model based on transformer
We present a computation model, TransPTM, for non-histone protein acetylation site prediction, utilizing a transformer (TransformerConv) [22] based GNN to extract information from protein sequence. The overall design of TransPTM is outlined and depicted in Figure 3. Specifically, pLM-embedded protein sequences (11 sets of data according to length difference) are the input node data, which capture the relationships among different amino acids. Here, GNN is a widely used approach for graph data feature learning. It aggregates information from neighboring regions via convolution, exhibiting significant performance in graph representation learning [38]. First, the model leverages three transformer graph convolutional layers to encode and propagate node features throughout the graph. Each TransformerConv layer employs self-attention mechanism, enabling the model to capture both local and global graph patterns. Subsequently, the self-attention coefficient was computed, embodying the similarity between the query and the key. This self-attention coefficient served as the weight, and the output vector of the layer was determined as the weighted sum of the values. The TransformerConv layer calculates the output feature for each node using the following equation:
Figure 3.

The overall architecture of the TransPTM. Protein sequence data are encoded by one-hot and ProtT5. Node features (ProtT5-XL-U50 embedding) and edge features (adjacency matrix) were compiled to construct the graphs. The graphs were then processed through three TransformerConv layers and input into an MLP to produce the final prediction. L = 2 +1, which is the length of input protein sequence.
+1, which is the length of input protein sequence.
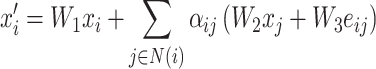 |
(2) |
In this equation,  represents the input feature vector of node
represents the input feature vector of node 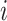 ,
,  signifies the output feature vector for node
signifies the output feature vector for node  and
and 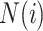 denotes the set of neighboring nodes for node
denotes the set of neighboring nodes for node 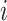 . The attention matrix
. The attention matrix 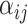 is calculated using the following equation:
is calculated using the following equation:
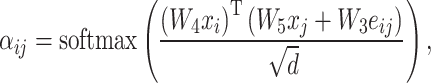 |
(3) |
where  refers to the input feature of node
refers to the input feature of node 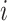 ,
, 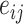 represents the edge feature of edge
represents the edge feature of edge 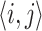 and
and  corresponds to the hidden size of the node feature. All
corresponds to the hidden size of the node feature. All 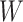 s are weight matrices that can be adjusted or optimized during training. The attention matrix
s are weight matrices that can be adjusted or optimized during training. The attention matrix 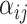 assists the network in determining the importance of different neighbors for each residue. For our objective, given that the per-residue embeddings are contextualized features, we only picked the 64-length embeddings for the site under investigation, namely the lysine (K). Following these layers, an MLP module with three dropout layers and two ReLU layers is applied. This module predicts the acetylation probability, denoted as S, using equation (4):
assists the network in determining the importance of different neighbors for each residue. For our objective, given that the per-residue embeddings are contextualized features, we only picked the 64-length embeddings for the site under investigation, namely the lysine (K). Following these layers, an MLP module with three dropout layers and two ReLU layers is applied. This module predicts the acetylation probability, denoted as S, using equation (4):
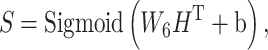 |
(4) |
where 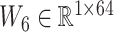 represents the weight matrix and
represents the weight matrix and  stands for the bias term.
stands for the bias term.  is the output from the last layer. The sigmoid function is used to map the value into the range (0,1). Output values that exceed the threshold of 0.5 are classified as acetylation, while output values that are below 0.5 are classified as non-acetylation. This architecture blends the capabilities of transformer-based feature learning and neural network-based classification, utilizing complex graph structures to achieve accurate node classification.
is the output from the last layer. The sigmoid function is used to map the value into the range (0,1). Output values that exceed the threshold of 0.5 are classified as acetylation, while output values that are below 0.5 are classified as non-acetylation. This architecture blends the capabilities of transformer-based feature learning and neural network-based classification, utilizing complex graph structures to achieve accurate node classification.
Model training
Deep learning models here were trained to minimize the binary cross-entropy (BCE) loss, which is showed by equation (5):
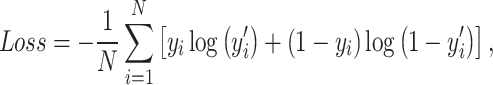 |
(5) |
where 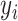 represents the actual value, and
represents the actual value, and 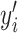 corresponds to the predicted probability for the
corresponds to the predicted probability for the 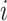 th instance among
th instance among 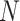 data points. In particular, we employed a batch size of 64, set an initial learning rate of 0.00003 and used a weight decay of 0.0001, all in conjunction with the Adam optimizer. Subsequently, the model was trained on an independent validation set, with the BCE function serving as the loss function. We use 5-fold stratified cross-validation to select the best protein sequence encoding method. In 5-fold cross-validation, the original dataset is evenly divided into five subsets. The model is then iteratively trained on four of these subsets and validated on the remaining subset. This process is repeated five times, and the average of the validation results from the five iterations is calculated to evaluate the performance of the model and reduce the risk of overfitting. In addition, we compare our approach with the other seven benchmark methods, including four baseline models and three existing acetylation site prediction tools. They are Random Forest (RF) [39], Support Vector Machine (SVM) [40] Convolutional Neural Network (CNN) [41] Long Short-Term Memory (LSTM) [42], GPS-PAIL [11], Musitedeep [31] and Deep-PLA [43]. In addition, we also fine-tuned the ProtT5 model, employing it as a baseline for comparison.
data points. In particular, we employed a batch size of 64, set an initial learning rate of 0.00003 and used a weight decay of 0.0001, all in conjunction with the Adam optimizer. Subsequently, the model was trained on an independent validation set, with the BCE function serving as the loss function. We use 5-fold stratified cross-validation to select the best protein sequence encoding method. In 5-fold cross-validation, the original dataset is evenly divided into five subsets. The model is then iteratively trained on four of these subsets and validated on the remaining subset. This process is repeated five times, and the average of the validation results from the five iterations is calculated to evaluate the performance of the model and reduce the risk of overfitting. In addition, we compare our approach with the other seven benchmark methods, including four baseline models and three existing acetylation site prediction tools. They are Random Forest (RF) [39], Support Vector Machine (SVM) [40] Convolutional Neural Network (CNN) [41] Long Short-Term Memory (LSTM) [42], GPS-PAIL [11], Musitedeep [31] and Deep-PLA [43]. In addition, we also fine-tuned the ProtT5 model, employing it as a baseline for comparison.
Model evaluation and criteria
In this study, non-histone protein sequence with acetylated site are considered as positives samples and non-acetylated site are considered as negatives samples. In the prediction process, acetylated site correctly identified are termed as true positives (TP), while non-acetylated site correctly identified are known as true negatives (TN). Situations where negative sequences are incorrectly classified as positive are labeled as false positives (FP), and cases where positive sequences are wrongly classified as negative are referred to as false negatives (FN). All performance metrics, unless stated otherwise, are averaged and reported. The performance was assessed with four metrics, including accuracy, sensitivity, precision and MCC (Matthew’s correlation coefficient), with a decision probability threshold set to 0.5. Additionally, the area under the ROC curve and the area under the precision-recall (PRC) curve were also used as performance indicators. Equations (6)–(9) describe these evaluation criteria:
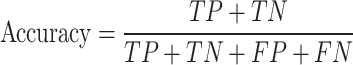 |
(6) |
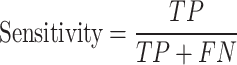 |
(7) |
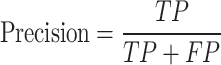 |
(8) |
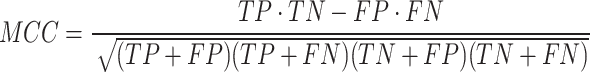 |
(9) |
RESULTS
Window size selection
Numerous studies have focused only on employing a fixed local sliding window in modeling process. Yet, it is important to note that diverse sliding windows may yield varied prediction results. Window size optimization can notably assist in feature selection and enhance prediction performance [44]. For that reason, an equal number of residues on both side of the site of interest (K) is taken as input to capture local sequence information for K. To determine the optimal window size, we conducted experiments across a range of window sizes, which are 11, 15, 21, 25, 31, 35, 41, 45, 51, 55 and 61 (Figure 4). In Figure 4, the mean MCC for different window sizes is shown. We can observe that, as the length of the protein sequence is increased, MCC value reaches the maximum value (0.4359) when the window size is 25. In general, we expect that the longer the sequence is, the more semantic and contextual information it contains. However, it has been declining in the range of 25–61. We presume that this may be because the amino acid pattern that determines the acetylation of central K is at the proximal end of K, and a sequence that is over long will dilute this information. Hence, 25 was selected as the optimized window size value for acetylation residue for subsequent analysis.
Figure 4.

MCC value of TranPTM on the training dataset for sliding window size ranging from 11 to 61.
Performance evaluation of TransPTM
In this section, we first discuss the prediction performance of TransPTM using 5-fold cross-validation on the training set. The ROC curves and PRC curves are plotted in Figure 5, which presents the performance comparison of one-hot encoding and ProtT5 embedding. On 5-fold cross-validation, the average AUC and AUPRC values of our model with one-hot encoding method are 0.74 and 0.45, respectively, while our model with ProtT5 embedding shows average AUC and AUPRC values of 0.83 and 0.64, respectively. We then evaluated and compared the prediction performance of two encoding schemes with independent non-histone acetylation site testing dataset. The prediction results of our TransPTM model with one-hot encoding and ProtT5 embedding are also evaluated in terms of AUC and AUPRC. As shown in Figure 6, ROC curves and PRC curves of the independent testing set have been plotted. Our deep learning predictor with ProtT5 embedding has an accuracy of 0.88, AUC of 0.83 and AUPRC of 0.51. In contrast, our model with one-hot encoding scheme shows an accuracy of 0.86, AUC of 0.75 and AUPRC of 0.35. The results of TransPTM using the ProtT5 embedding method outperform those using the one-hot encoding method for the dataset. We speculate that the superior performance observed can be attributed to the pLMs embedding’s ability to capture contextual dependencies within raw protein sequences, which is crucial for PTM site prediction. On the other hand, one-hot encoding only represents individual amino acid information at each position and does not capture sequence-level dependencies. Therefore, we use TransPTM with ProtT5 embedding for the state-of-the-arts performance comparison.
Figure 5.

Performance of TransPTM (one-hot) and TransPTM (ProtT5) using 5-fold cross-validation on the training dataset. (A) ROCs of TransPTM (one-hot), (B) Precision-recall curves of TransPTM (one-hot), (C) ROCs of TransPTM (ProtT5), (D) Precision-recall curves of TransPTM (ProtT5).
Figure 6.

Performance of TransPTM (one-hot) and TransPTM (ProtT5) on the independent testing dataset. (A) ROC curves, (B) Precision-recall curves.
Comparison analysis can provide insights into the strengths and weaknesses of different methods and guide our future research directions. Therefore, we further employed seven different classifiers including two baseline machine learning models (RF and SVM), two baseline deep learning models (CNN and LSTM) and three existing acetylation site prediction tools (GPS-PAIL [11], MusiteDeep [31] and Deep-PLA [43]) to compare the prediction performance of our model. In addition, we also fine-tuned the ProtT5 model, employing it as a baseline for comparison. The optimized hyper-parameters, as well as the run time for these classifiers, are detailed in Table S1.
Among them, MusiteDeep and Deep-PLA apply state-of-the-art deep learning algorithms, whereas GPS-PAIL implements a traditional machine learning algorithm. Table 2 lists the outputs from all nine methods on the independent non-histone aectylation site testing datasets, including the accuracy, f1 scores, MCC values, AUC and AUPRC. The AUC, AUPRC and MCC values of our method on non-histone acetylation site-independent testing set were significantly higher than those of the state-of-the-art methods. TransPTM secured the accuracy, precision, sensitivity, f1 score, MCC value, AUC and AUPRC of 0.88, 0.61, 0.43, 0.51, 0.45, 0.83 and 0.51, respectively. The outstanding results indicate that TransPTM is a stable predictor with excellent performance, making it the most effective at non-histone acetylation site prediction, as evidenced by the independent testing dataset.
Table 2.
Performance comparison of TransPTM with baseline and state-of-the-art models on independent testing set for non-histone acetylation site prediction
| Classifiers | Accuracy | F1-score | MCC | AUC | AUPRC |
|---|---|---|---|---|---|
| RF | 0.87 | 0.47 | 0.40 | 0.68 | 0.31 |
| SVM | 0.87 | 0.42 | 0.35 | 0.65 | 0.27 |
| CNN | 0.85 | 0.49 | 0.41 | 0.72 | 0.31 |
| LSTM | 0.85 | 0.48 | 0.40 | 0.78 | 0.44 |
| GPS-PAIL | 0.65 | 0.32 | 0.19 | 0.64 | 0.44 |
| MusiteDeep | 0.77 | 0.40 | 0.30 | 0.69 | 0.47 |
| Deep-PLA | 0.61 | 0.29 | 0.14 | 0.60 | 0.42 |
| ProtT5 (fine-tuned) | 0.74 | 0.41 | 0.31 | 0.71 | 0.24 |
| TransPTM | 0.88 | 0.51 | 0.45 | 0.83 | 0.51 |
We assert that there are two distinctive factors behind the observation that TransPTM method improves prediction performance over other methods. The first one is that we represent the embedded protein sequence with graph instead of using sequence information alone. Moreover, we introduce the TransformerConv layer, which uses convolution to extract local features and implement transformer modules to model long-range dependencies during the embedding process. It combines the advantages of both transformer model and CNN network, capturing not only single amino acid information but also long-range relationships between amino acids.
Analysis of amino acids distribution
We conducted statistical analysis on the distribution features of amino acids positions on sequences, comparing the positive and negative subsets. Using the Two Sample Logo web server [23], we found measurable differences between the two groups (t-test: P <0.05), as illustrated in Figure 7A. Amino acids differ in color based on their side chain charge properties, i.e. blue and red mean the positively and negatively charged, respectively. Other colors means neutral amino acids. In general, noticeable distinctions are evident between the acetylated sequence (shown in the upper panel) and the non-acetylated sequence (depicted in the lower panel). Positively charged amino acids, represented in blue, are enriched in the acetylated samples, while the negatively charged amino acids, marked in red, show a higher concentration in the non-acetylated samples. While neutral amino acids (represented by other colors) are evenly distributed on both sides. For all samples, no matter they belong to which side, upstream (position -12 to -1) of sequences tend to having more residues distributed than downstream (position 1 to 12). In particular, lysine (K) are significantly enriched on the upstream positive side of sequence, especially on positions -1, -2, -3, -4, -9 and -10. Moreover, electrically neutral residues leucine(L) mainly distributed in the non-acetylated. The clear discernment of position-specific differences is crucial for the development of reliable tools for PTMs site identification.
Figure 7.

Visualization of amino acids distribution and attention map of training dataset. (A) Two sample logo of positive data (upper panel) and negative data (lower panel). (B) Attention map learned by TransPTM, generated using the best model weights from the fully trained TransPTM, showcasing the learned attention distribution on the training data. (C) and (D) Attention maps of original positive and negative data, processed through the first convolutional layer of the TransPTM model, representing the original, unmodified attention distributions of the data.
Moreover, amino acid residue proportion analysis, Two Sample Logo analysis and MSA analysis have also been implemented to compare the contexts of the acetylated lysine (K) sites on histone and non-histone protein sequences (Figure S1). The results indicate that the amino acid composition near the non-histone proteins acetylation sites is more diverse, more irregular and less conserved than amino acid composition near histone acetylation sites.
Attention interpretation
The self-attention components within the TransformerConv layers empower our model, TransPTM, to assess the influence of amino acids interactions at different positions. Unlike the common perception of a neural network being an opaque ‘black-box’ model, the advantages of our model lie in the self-attention component which provides algorithmic transparency [45]. To demonstrate the interpretability of our model, we applied sequences of 25 amino acids in length, which corresponds to the window size we previously selected. As previously stated, the attention mechanism are applied to produce attention weights. These weights illustrate the emphasis on amino acid feature vectors across different positions. Figure 7B shows the residue–residue attention score of the interactions between amino acids which are located at different positions, where the amino acids with high attention scores are marked in red, and the amino acids with low attention scores are marked in blue. In particular, the amino acids strongly focus on the 13 positions of the protein sequence, where the central lysine (K) is located, while those located at other positions receive comparatively less attention. The areas highlighted by the amino acid on both sides of lysine (K) are relatively balanced, but attention scores in left half area are slightly higher than the right half area. Moreover, positions -1, -2, -4, -9, -10, -11 and -12 show strong attention on central amino acid K. This information shows a consistent trend with the amino acid distribution analysis (Figure 7A). To investigate how residues transfer their attention to those particular positions, Figure 7C and D are introduced as the original attention map of positive samples and negative samples in the training dataset. Since the original input data were not completely trained, amino acids on the protein sequence show only few attention on the central K. These results suggest that the transformer module in our model has reinforced the attention on the amino acids at crucial positions.
TransPTM mechanism visualization
To distinguish the abstract features generated by our TransPTM model from the original protein descriptors, we employed t-SNE dimensionality reduction for visualization. This method maps high-dimensional features into a 2D space and normalizes the values within a range of [-1, 1] [24]. Specifically, we first choose the output of TransformerConv layers to see if this module can map semantically similar vectors to adjacent spaces. Then, we choose the penultimate layer output of MLP module as the output of the TransPTM to see if our model can transform the original data vectors into separable classes. Figure 8 presents the visualization of both the extracted features from our model and the original features of the non-histone acetylation site data. In particular, Figure 8A and B represent the t-SNE visualization of one-hot encoded and ProtT5 embedded 25 amino acid length training data, respectively. Furthermore, we visualized the sample distributions from the outputs of the TransformerConv layer and the penultimate MLP layer of our TransPTM model using training dataset (Figure 8C and D). We can observe that the input data (Figure 8A and B) are jumbled, and the positive and negative samples are intertwined. However, the positive and negative samples were clearly separated after the transformer convolution operation (Figure 8C). Compared with the TransformerConv layer, the spatial distribution of samples from the output of the MLP hidden layer remains consistent (Figure 8D). The comparison results indicate that the original encoded data can be converted into a distinguishable representation through transformer module of TransPTM, which is helpful in further classification by the MLP module.
Figure 8.

t-SNE visualization of original data and extracted features. Red represents the acetylated site, and blue represents the non-acetylated site. (A) Original data encoded by one-hot method, (B) original data embedded by ProtT5 pLM, (C) data embedded by TransformerConv layers, (D) data classified by TransPTM last layer.
The original features were unable to clearly differentiate between positive and negative samples. However, after using the transformer convolution to extract features, a rough separation was achieved, which demonstrates the necessity and effectiveness of the transformer module.
DISCUSSION
Protein acetylation is one of the most common PTMs occurring in various cellular compartments and is important for cellular mechanism investigation. Although a large number of existing machine learning-based acetylation site identification tools have been published, to the best of our knowledge, there is not any benchmark dataset and predictor established for non-histone acetylation site prediction. Therefore, we have completed two unprecedented works to solve these problems.
First, we construct a non-histone acetylation site benchmark dataset, called NHAC, which includes 11 subsets according to the sequence length ranging from 11 to 61 amino acids. There are currently 886 positive samples and 4707 negative samples for each sequence length. In the future data curation, we will implement routine maintenance to ensure that our dataset remains up-to-date and relevant. We will periodically survey relevant review and experimental papers to update our dataset with the latest acetylated non-histone protein sequences. To capture publications not included in review papers, we will conduct regular searches in databases such as PubMed [46] for the most recent non-histone acetylation studies. In parallel, we will also seek collaborations with other researchers and institutions to incorporate wet experimental data. Additionally, we are planning to incorporate longer acetylated non-histone protein sequences, including full-length protein sequences, to enhance the sequence diversity of NHAC.
Secondly, we propose a transformer-based computational model, TransPTM, for non-histone acetylation site identification. Our model employs a pre-trained pLM ProtT5 to construct the site’s feature space. Then, the embedded protein sequence data are fed into a GNN, which combines three TransformerConv layers for feature extraction and an MLP for classification. The 5-fold cross-validation on training dataset and comparison experiments on independent testing datasets indicate that TransPTM has the highest performance for non-histone acetylation site prediction. The strong performance in non-histone acetylation site prediction helps our comprehension of its molecular mechanism and provides a theoretical basis for developing drug targets for diseases. Moreover, the establishment of the benchmark datasets fills the gap in non-histone acetylation site datasets and is beneficial to future researchers to do performance evaluation.
TransPTM has shown promising performance in non-histone acetylation site prediction. However, there are still future works to be completed. Looking ahead, we will further improve the model in the following aspects. First, the prediction performance of TransPTM can be enhanced using extensive structural data due to the success of AlphaFold2 [47]. Furthermore, given the limited quantity of known non-histone acetylation site, in our future work, we plan to incorporate additional techniques such as SMOTE [48] to handle and impute imbalanced datasets.
Key Points
A transformer-based deep learning model is proposed for non-histone acetylation site prediction, which achieves the best performance among the state-of-the-art models.
The establishment of NHAC, a benchmark dataset specifically designed for non-histone acetylation site prediction, enriches the landscape of lysine acetylation site databases.
Pre-trained protein language model ProtT5 is employed for generating contextualized representations of protein sequences by capturing dependencies between amino acid residues.
The self-attention mechanism in TransPTM can reveal key residue positions for non-histone protein acetylation, thereby demonstrating the interpretability of our model.
Supplementary Material
Author Biographies
Lingkuan Meng has been pursuing his PhD degree from the Department of Computer Science, City University of Hong Kong. His research interests include computational proteomics prediction, medicinal chemistry, and drug discovery.
Xingjian Chen has obtained his PhD degree from the Department of Computer Science, City University of Hong Kong. He is now a postdoctoral research fellow at Massachusetts General Hospital and Harvard Medical School. His research interests include computational omics and spatial transcriptomics.
Ke Cheng has obtained his PhD degree from the Department of Chemistry, City University of Hong Kong. His research interest focus on chemical proteomics, theranostics, and nanomaterials.
Nanjun Chen has been pursuing his PhD degree from the Department of Computer Science, City University of Hong Kong. His research interest is bioinformatics algorithm development.
Zetian Zheng has been pursuing his PhD degree from the Department of Computer Science, City University of Hong Kong. His research interests include pharmacogenomics, cancer research, and computational biology.
Fuzhou Wang has been pursuing his PhD degree from the Department of Computer Science, City University of Hong Kong. His research interests reside at the intersection of regulatory genomics and machine learning.
Hongyan Sun is a Professor with the Department of Chemistry, City University of Hong Kong. She has published more than 150 research papers. Her research interests include chemical biology, organic chemistry, and biochemistry.
Ka-Chun Wong is an Associate Professor with the Department of Computer Science, City University of Hong Kong. He leads the East Asian Bioinformatics and Computational Biology laboratory and conducts highimpact computing research.
Contributor Information
Lingkuan Meng, Department of Computer Science, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong.
Xingjian Chen, Cutaneous Biology Research Center, Massachusetts General Hospital, Harvard Medical School, MA 02138, United States.
Ke Cheng, Department of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong.
Nanjun Chen, Department of Computer Science, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong.
Zetian Zheng, Department of Computer Science, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong.
Fuzhou Wang, Department of Computer Science, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong.
Hongyan Sun, Department of Chemistry, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong.
Ka-Chun Wong, Department of Computer Science, City University of Hong Kong, Tat Chee Avenue, Kowloon, Hong Kong; Shenzhen Research Institute, City University of Hong Kong, Shenzhen, China.
FUNDING
This research was substantially sponsored by the research project (Grant No. 32170654 and Grant No. 32000464) supported by the National Natural Science Foundation of China and was substantially supported by the Shenzhen Research Institute, City University of Hong Kong. The work described in this paper was substantially supported by the grant from the Research Grants Council of the Hong Kong Special Administrative Region [CityU 11203723]. The work described in this paper was partially supported by the grants from City University of Hong Kong (2021SIRG036, CityU 9667265, CityU 11203221) and Innovation and Technology Commission (ITB/FBL/9037/22/S).
DATA AVAILABILITY
TransPTM source code and the dataset we constructed are available at https://www.github.com/TransPTM/TransPTM.
AUTHOR CONTRIBUTIONS STATEMENT
Lingkuan Meng: Writing, Conceptualization, Methodology, Visualization. Xingjian Chen: Methodology. Ke Cheng: Methodology. Nanjun Chen: Methodology. Zetian Zheng: Methodology. Fuzhou Wang: Methodology. Hongyan Sun: Writing—review & editing, Supervision. Ka-Chun Wong: Writing—review & editing, Supervision.
References
- 1. Seo J-W, Lee K-J. Post-translational modifications and their biological functions: proteomic analysis and systematic approaches. BMB Rep 2004;37(1):35–44. [DOI] [PubMed] [Google Scholar]
- 2. Krassowski M, Paczkowska M, Cullion K, et al.. Activedriverdb: human disease mutations and genome variation in post-translational modification sites of proteins. Nucleic Acids Res 2018;46(D1):D901–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Keith Keenan E, Zachman DK, Hirschey MD. Discovering the landscape of protein modifications. Mol Cell 2021;81(9):1868–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Walsh CT, Garneau-Tsodikova S, Gatto Jr GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed 2005;44(45):7342–72. [DOI] [PubMed] [Google Scholar]
- 5. Jensen ON. Interpreting the protein language using proteomics. Nat Rev Mol Cell Biol 2006;7(6):391–403. [DOI] [PubMed] [Google Scholar]
- 6. Yang X-J, Seto E. Lysine acetylation: codified crosstalk with other posttranslational modifications. Mol Cell 2008;31(4):449–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meng L, Chan W-S, Huang L, et al.. Mini-review: recent advances in post-translational modification site prediction based on deep learning. Comput Struct Biotechnol J 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
8.
Bannister AJ, Miska EA, Görlich D, Kouzarides T. Acetylation of importin-
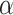 nuclear import factors by cbp/p300. Curr Biol 2000;10(8):467–70.
[DOI] [PubMed] [Google Scholar]
nuclear import factors by cbp/p300. Curr Biol 2000;10(8):467–70.
[DOI] [PubMed] [Google Scholar] - 9. Meng X, Lv Y, Mujahid H, et al.. Proteome-wide lysine acetylation identification in developing rice (oryza sativa) seeds and protein co-modification by acetylation, succinylation, ubiquitination, and phosphorylation. Biochim Biophys Acta-Proteins Proteomics 2018;1866(3):451–63. [DOI] [PubMed] [Google Scholar]
- 10. Watson JD. Molecular Biology of the Gene. Pearson: Always Learning, 2014. [Google Scholar]
- 11. Deng W, Wang C, Zhang Y, et al.. Gps-pail: prediction of lysine acetyltransferase-specific modification sites from protein sequences. Sci Rep 2016;6(1):39787. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
12.
Kalebic N, Sorrentino S, Perlas E, et al..
 tat1 is the major
tat1 is the major  -tubulin acetyltransferase in mice. Nat Commun 2013;4(1):1962.
[DOI] [PubMed] [Google Scholar]
-tubulin acetyltransferase in mice. Nat Commun 2013;4(1):1962.
[DOI] [PubMed] [Google Scholar] - 13. Spange S, Wagner T, Heinzel T, Krämer OH. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol 2009;41(1):185–98. [DOI] [PubMed] [Google Scholar]
- 14. Botrugno OA, Robert T, Vanoli F, et al.. Molecular pathways: old drugs define new pathways: non-histone acetylation at the crossroads of the dna damage response and autophagy. Clin Cancer Res 2012;18(9):2436–42. [DOI] [PubMed] [Google Scholar]
- 15. Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol 2019;20(3):156–74. [DOI] [PubMed] [Google Scholar]
- 16. Grillon JM, Johnson KR, Kotlo K, Danziger RS. Non-histone lysine acetylated proteins in heart failure. Biochimi Biophys Acta Mol Basis Dis 2012;1822(4):607–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Y, Huang H, Zhu M, et al.. Roles of the myst family in the pathogenesis of alzheimer’s disease via histone or non-histone acetylation. Aging Dis 2021;12(1):132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wei G, Roeder RG. Activation of p53 sequence-specific dna binding by acetylation of the p53 c-terminal domain. Cell 1997;90(4):595–606. [DOI] [PubMed] [Google Scholar]
- 19. Meiqi W, Yang Y, Wang H, Yan X. A deep learning method to more accurately recall known lysine acetylation sites. BMC Bioinformatics 2019;20:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Asim MN, Ibrahim MA, Malik MI, et al.. Histone-net: a multi-paradigm computational framework for histone occupancy and modification prediction. Complex Intell Syst 2023;9(1):399–419. [Google Scholar]
- 21. Raffel C, Shazeer N, Roberts A, et al.. Exploring the limits of transfer learning with a unified text-to-text transformer. J Mach Learn Res 2020;21(140):1–67.34305477 [Google Scholar]
- 22. Shi Y, Huang Z, Feng S, et al.. Masked label prediction: unified message passing model for semi-supervised classification. arXiv preprint arXiv:2009.03509. 2021.
- 23. Vacic V, Iakoucheva LM, Radivojac P. Two sample logo: a graphical representation of the differences between two sets of sequence alignments. Bioinformatics 2006;22(12):1536–7. [DOI] [PubMed] [Google Scholar]
- 24. Van der Maaten L, Hinton G. Visualizing data using t-sne. J Mach Learn Res 2008;9(11). [Google Scholar]
- 25. UniProt Consortium . Uniprot: a worldwide hub of protein knowledge. Nucleic Acids Res 2019;47(D1):D506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chou K-C. Prediction of signal peptides using scaled window. Peptides 2001;22(12):1973–9. [DOI] [PubMed] [Google Scholar]
- 27. Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006;22(13):1658–9. [DOI] [PubMed] [Google Scholar]
- 28. Huang Y, Niu B, Gao Y, et al.. Cd-hit suite: a web server for clustering and comparing biological sequences. Bioinformatics 2010;26(5):680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. ElAbd H, Bromberg Y, Hoarfrost A, et al.. Amino acid encoding for deep learning applications. BMC Bioinformatics 2020;21:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. El Allali A, Elhamraoui Z, Daoud R. Machine learning applications in rna modification sites prediction. Comput Struct Biotechnol J 2021;19:5510–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang D, Zeng S, Chunhui X, et al.. Musitedeep: a deep-learning framework for general and kinase-specific phosphorylation site prediction. Bioinformatics 2017;33(24):3909–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Heinzinger M, Littmann M, Sillitoe I, et al.. Contrastive learning on protein embeddings enlightens midnight zone. NAR Genomics Bioinf 2022;4(2):lqac043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mai Ha V, Akbar R, Robert PA, et al.. Linguistically inspired roadmap for building biologically reliable protein language models. Nature. Mach Intell 2023;5(5):485–96. [Google Scholar]
- 34. Teufel F, Armenteros JJA, Johansen AR, et al.. Signalp 6.0 predicts all five types of signal peptides using protein language models. Nat Biotechnol 2022;40(7):1023–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Elnaggar A, Heinzinger M, Dallago C, et al.. Prottrans: towards cracking the language of life’s code through self-supervised deep learning and high performance computing. bioRxiv arXiv:2007.06225, 2020. [Google Scholar]
- 36. Steinegger M, Mirdita M, Söding J. Protein-level assembly increases protein sequence recovery from metagenomic samples manyfold. Nat Methods 2019;16(7):603–6. [DOI] [PubMed] [Google Scholar]
- 37. Suzek BE, Wang Y, Huang H, et al.. Uniref clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 2015;31(6):926–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Asif NA, Sarker Y, Chakrabortty RK, et al.. Graph neural network: a comprehensive review on non-euclidean space. IEEE. Access 2021;9:60588–606. [Google Scholar]
- 39. Ho Tin Kam. Random decision forests. In Proceedings of 3rd international conference on document analysis and recognition, volume 1, pages 278–82. Montreal, QC, Canada: IEEE, 1995. [Google Scholar]
- 40. Cortes C, Vapnik V. Support-vector networks. Mach Learn 1995;20(3):273–97. [Google Scholar]
- 41. Oshea K, Nash R. An introduction to convolutional neural networks. arXiv preprint arXiv:151108458 2015. [Google Scholar]
- 42. Hochreiter S, Schmidhuber J. Long short-term memory. Neural Comput 1997;9(8):1735–80. [DOI] [PubMed] [Google Scholar]
- 43. Kai Y, Zhang Q, Liu Z, et al.. Deep learning based prediction of reversible hat/hdac-specific lysine acetylation. Brief Bioinform 2020;21(5):1798–805. [DOI] [PubMed] [Google Scholar]
- 44. Wuyun Q, Zheng W, Zhang Y, et al.. Improved species-specific lysine acetylation site prediction based on a large variety of features set. PloS One 2016;11(5):e0155370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huang L, Lin J, Liu R, et al.. Coadti: multi-modal co-attention based framework for drug–target interaction annotation. Brief Bioinform 2022;23(6):bbac446. [DOI] [PubMed] [Google Scholar]
- 46. Sayers EW, Bolton EE, Rodney Brister J, et al.. Database resources of the national center for biotechnology information. Nucleic Acids Res 2022;50(D1):D20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jumper J, Evans R, Pritzel A, et al.. Highly accurate protein structure prediction with alphafold. Nature 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chawla NV, Bowyer KW, Hall LO, Kegelmeyer WP. Smote: synthetic minority over-sampling technique. J Artif Intell Res 2002;16:321–57. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TransPTM source code and the dataset we constructed are available at https://www.github.com/TransPTM/TransPTM.