

Abstract
Purpose of Review
This study aims to review diagnosis, potential complications, and clinical management in craniofacial fibrous dysplasia.
Recent Findings
Fibrous dysplasia (FD) is a rare mosaic disorder in which normal bone and marrow are replaced with expansile fibro-osseous lesions. Disease presents along a broad spectrum and may be associated with extraskeletal features as part of McCune-Albright syndrome (MAS). The craniofacial skeleton is one of the most commonly impacted areas in FD, and its functional and anatomical complexities create unique challenges for diagnosis and management.
Summary
This review summarizes current approaches to diagnosis and management in FD/MAS, with emphasis on the clinical and therapeutic implications for the craniofacial skeleton.
Keywords: Fibrous dysplasia, Craniofacial fibrous dysplasia, McCune Albright syndrome, Rare bone disease
Introduction
Fibrous dysplasia/McCune-Albright syndrome (FD/MAS) is a rare complex disorder leading to fractures, deformities, pain, and functional impairment [1]. This mosaic disease arises from gain-of-function variants in GNAS, encoding the α-subunit of the Gs G-coupled protein receptor [2], resulting in constitutive receptor activation and inappropriate downstream production of cyclic-AMP. In bone, constitutive Gαs signaling inhibits normal skeletal stem cell differentiation, creating expansile fibro-osseous lesions filled with abnormally proliferating osteoprogenitor cells [3, 4]. FD can affect one bone (monostotic FD) or multiple bones (polyostotic FD), and may occur in association with skin hyperpigmentation and hyperfunctioning endocrinopathies as part of MAS. The extent of the disease depends in part upon the timing and location in which the GNAS variant occurs. Variants that arise early in embryogenesis, prior to gastrulation, can result in involvement of tissues derived from multiple germ layers (such as the skin, bone, and endocrine systems), while later variants may result in more limited disease [1].
The craniofacial skeleton is one of the most commonly impacted areas in FD, and its functional and anatomical complexities create unique challenges for diagnosis and management. The purpose of this review is to provide an updated overview of FD/MAS, with an emphasis on the clinical and therapeutic implications for the craniofacial skeleton.
Clinical Presentation of FD/MAS
FD/MAS has variable clinical presentations that evolve throughout the lifespan. Extraskeletal features typically present in childhood. Hyperpigmented macules are apparent at or shortly after birth, and typically have characteristic features including irregular borders and location respecting the midline of the body (Fig.1 A and B). The majority of girls with MAS develop estrogen-producing ovarian cysts, leading to precocious puberty and vaginal bleeding (Fig.1C). Hyperthyroidism develops in approximately half of patients with MAS, and is associated with characteristic ultrasound abnormalities, including general homogeneity with mixed hyperechoic and hypoechoic features (Fig. 1D). Less common endocrinopathies include growth hormone excess and neonatal hypercortisolism [5]. Many patients with FD/MAS present initially with macules and/or endocrinopathies, and on further evaluation are found to have FD.
Fig. 1.
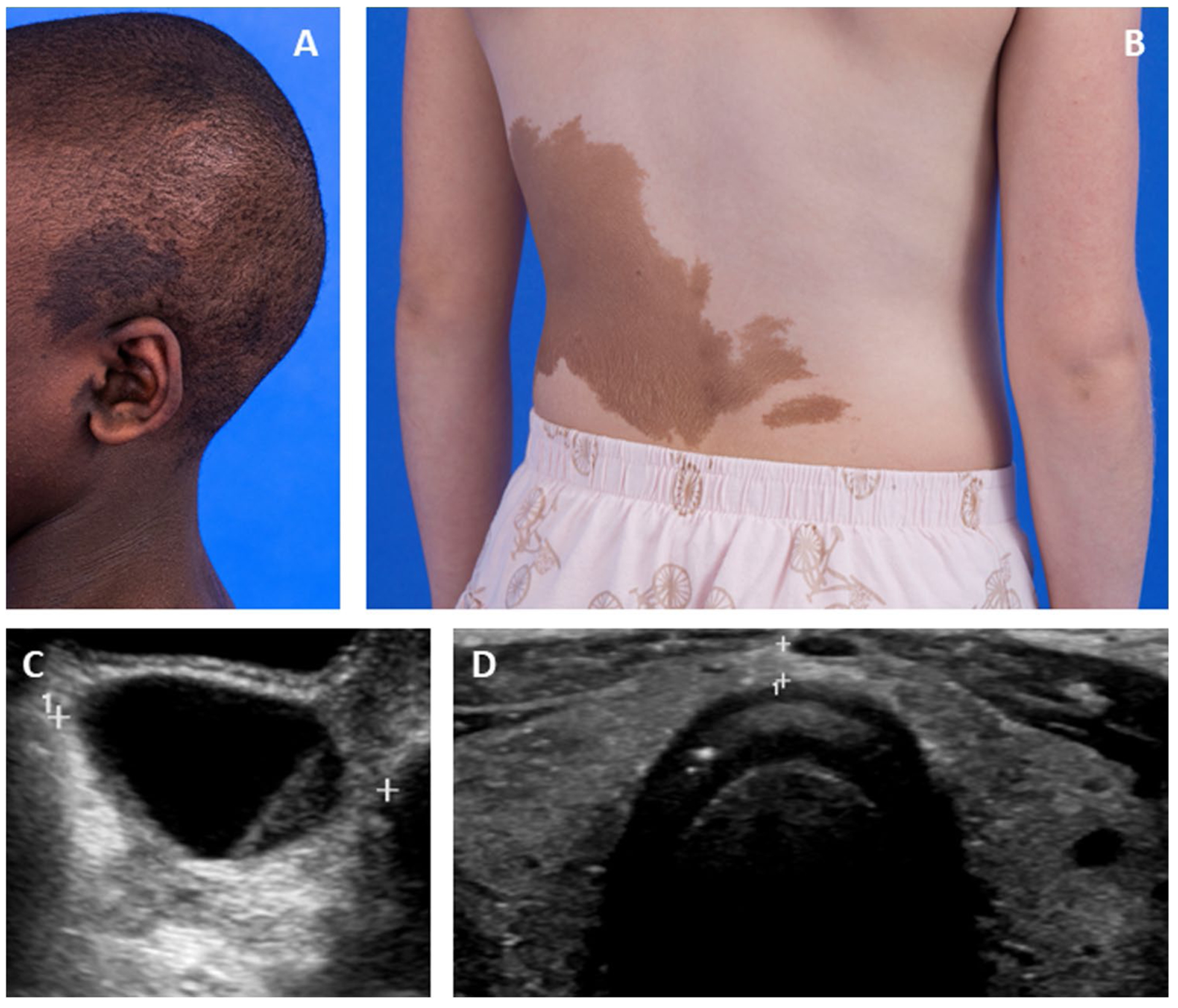
Extraskeletal features of McCune-Albright syndrome. A Photograph showing typical hyperpigmented macules overlying a patient’s skull and ear. B Photograph showing typical hyperpigmented macules on a patient’s trunk. C Pelvic ultrasound image showing a typical ovarian cyst associated with precocious puberty. D Thyroid ultrasound image showing diffuse heterogeneity of both lobes
FD lesions typically become apparent during the first few years of life, expand over childhood, and reach final disease burden in late adolescence [6, 7]. Lesions become less metabolically active in adulthood, potentially due to apoptosis or senescence of variant-bearing osteoprogenitor cells [8]. The location and evolution of the FD lesions, and degree of involvement in each bone, dictate the potential complications. Involvement of the appendicular skeleton can lead to fractures, long bone deformities, and impaired ambulation. Axial skeletal involvement can result in rib fractures and scoliosis, which can rarely be progressive [9].
Clinical sequelae of craniofacial FD arise primarily due to bone expansion, and are dictated by location, extent, and evolution of each lesion. Some lesions may remain asymptomatic and be detected only incidentally, while others may present with varying degrees of facial asymmetry and functional impairment. Specific manifestations of craniofacial FD are discussed in detail in later sections.
Diagnosis of FD
The diagnosis of FD is made using a combination of clinical, radiographic, and (if needed) pathologic and molecular evaluations. The presence of FD and/or any extraskeletal features of MAS should prompt a screening evaluation to identify the extent of disease. This should include a nuclear medicine bone scan (such as technetium-99 or 18F-NaF PET/CT) to identify all skeletal lesions (Fig. 2C), biochemical screening for endocrinopathies, and thyroid and gonadal ultrasounds. Specific screening recommendations are clearly outlined by Javaid MK et al. [10]. A clinical diagnosis of FD can generally be made in the presence of 2 or more typically appearing skeletal lesions, and/or extraskeletal features of MAS. In patients with a single monostotic lesion and no extraskeletal features, biopsy and identification of a GNAS variant are required to establish a diagnosis of FD.
Fig. 2.
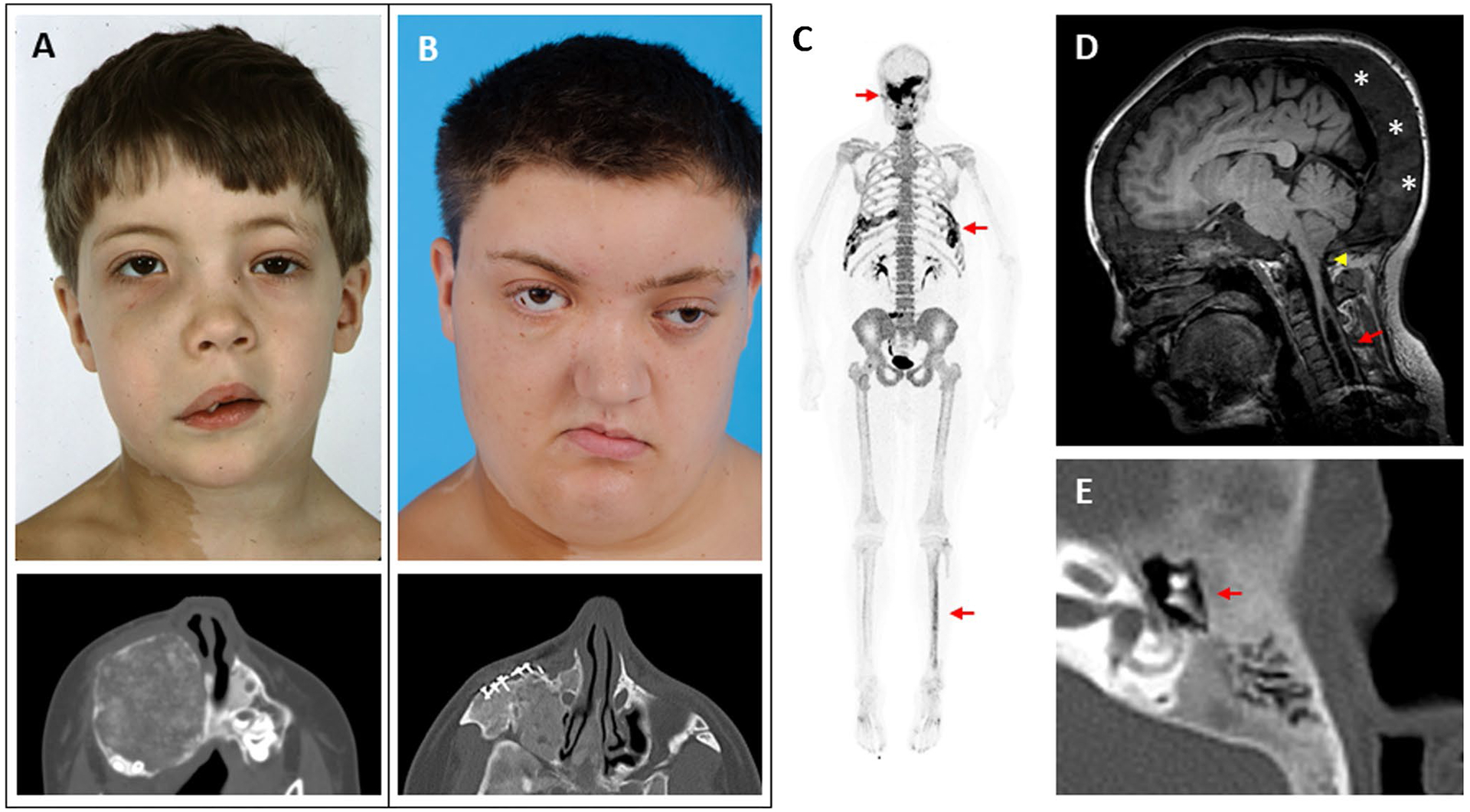
Clinical images of craniofacial fibrous dysplasia (FD). The left-hand panels show images from the same patient at ages 6 (A) and 18 years (B). Note the progression in facial asymmetry, including expansion of the right-sided face and orbital dystopia. The lower panels show axial computed tomography views of his right maxilla, including surgical implants from a reconstructive procedure performed at age 7. C 18F-NaF PET/CT scan performed in this patient at age 25 years. Areas of increased tracer uptake correspond to FD lesions in his skull, ribs, and tibia (red arrows). D Brain MRI from a 17-year-old girl with craniofacial FD and headaches. The cerebellar tonsils extend below the foramen magnum, consistent with a Chiari I malformation (yellow arrowhead). An associated syrinx has developed in the spinal cavity (red arrow). Note the diffuse expansion of the posterior cranium involved with FD (asterisks). E Computed tomography scan of the temporal bone in a 13-year-old patient with mild conductive hearing loss. The etpitympanum is diffusely involved with FD (red arrows), surrounding the ossicular chain
Radiographic features of craniofacial FD evolve throughout the lifespan. Young patients present with homogenous, radio-opaque lesions described as having a “ground glass” appearance on computed tomography (CT) scans, which evolve to a mixed lytic and sclerotic appearance in adulthood (Fig. 2 A, B, and E). Craniofacial FD lesions are not well-demarcated, often crossing bony sutures, but not breaching the bony cortices [11]. A standard non-contrast craniofacial CT imaging with reduced slice thickness of 3 mm is recommended to define the anatomy of individual lesions and to establish the extent of disease. FD does not show consistent enhancement patterns on MRI, making this a less reliable modality for diagnosing FD and monitoring its progression [12]. Radiographic features such as cortical perforation or irregular borders can indicate a secondary process and require further workup to rule-out malignant degeneration.
Histologic evaluation of FD is frequently impacted by sampling error, and overlap in appearance between FD and other benign fibro-osseous lesions. Typical histologic features include a background of fibrous stroma interspersed with abnormally formed woven bone, typically arranged in discontinuous, curvilinear trabeculae (Fig. 3). Typical marrow features are absent, and prominent osteoclasts and cartilaginous foci may be variably present [3].
Fig. 3.
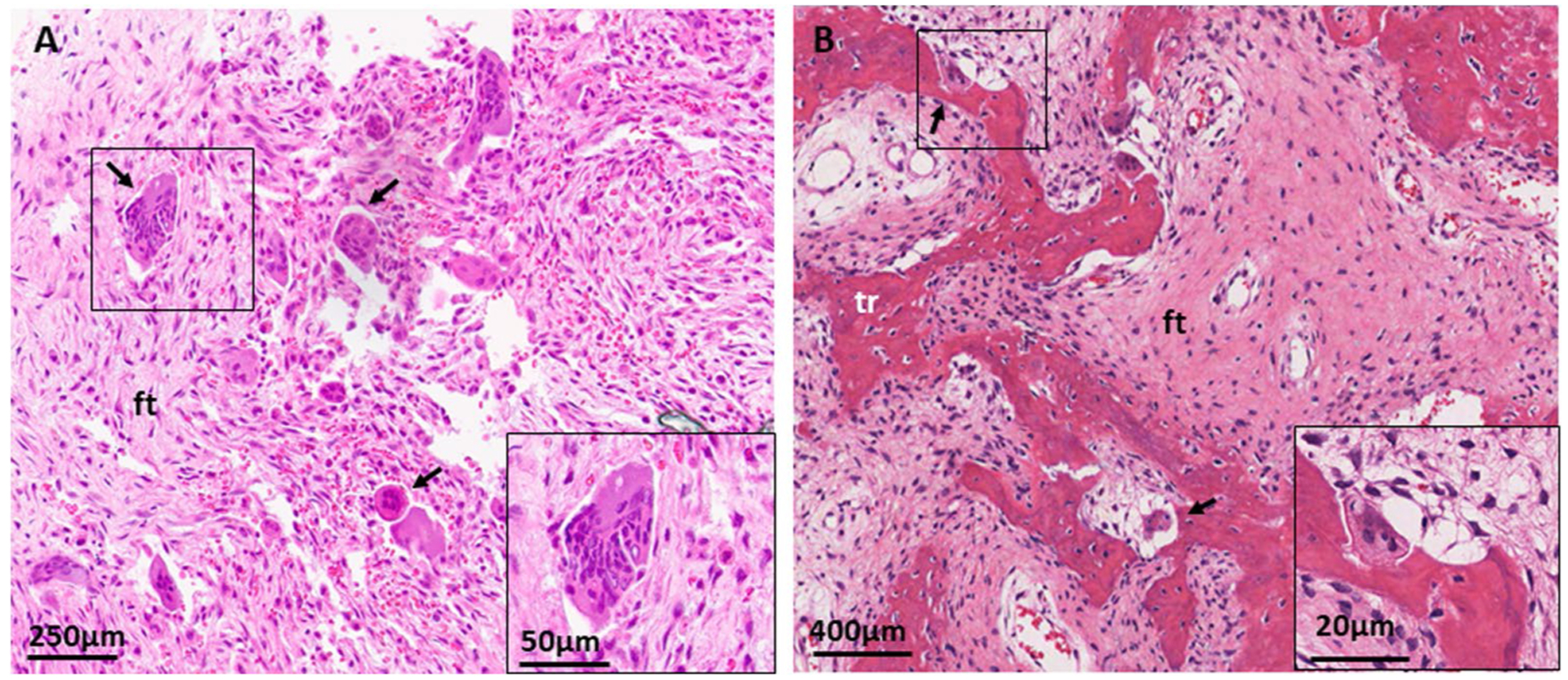
Histologic features of craniofacial fibrous dysplasia. A H&E section from a sphenoid bone shows diffuse fibrous tissue (ft) with prominent, irregularly situated osteoclasts (black arrows). B H&E section from a maxilla shows discontinuous, curvilinear trabeculae (tr) on a background on fibrous tissue (ft). Again noted are prominent osteoclasts (black arrows)
Differential Diagnosis of Craniofacial FD
Craniofacial FD falls under a class of disorders termed benign fibro-osseous lesions, which share similar clinical, radiographic, and histological features. This class encompasses a variety of entities, including ossifying fibromas, cemento-osseous dysplasias, giant cell tumors, and others, many of which require distinct treatment paradigms [13]. FD also shares similar features with low grade osteosarcoma, which can be distinguished using genetic markers such as MDM2 and CDK4 [14].
When lesions present with aggressive features, such as rapid expansion, severe pain, and/or cortical destruction, the differential should be expanded to include malignant processes, aneurysmal bone cysts, and other secondary vascular anomalies. FD lesions carry a small risk of malignant transformation ranging from 0.4 to 4%, most commonly into osteosarcoma [15, 16]. Cerebrovascular abnormalities have been reported in up to 1.9% of patients with craniofacial FD, including aneurysms, arteriovenous malformations, and venous stenosis [17, 18].
Clinical Features of Craniofacial FD
Facial and Sinonasal Involvement
Facial asymmetry is common and often the first clinically evident feature of craniofacial FD. Patients typically present in childhood with a painless, slowly expanding mass, particularly when the maxillary, mandibular, frontal, or zygomatic bones are involved [19, 20] (Fig. 2 A and B). Sinonasal involvement has been reported with variable prevalence, ranging from 36% in a series of primarily monostotic FD patients to 92% in a more severely affected cohort [21, 22]. The most frequent sites of involvement are the sphenoid sinus, followed by the frontal, maxillary, and ethmoid sinuses [21, 22]. While sinus involvement is often asymptomatic, it may also present with rhinorrhea, nasal congestion or obstruction, pain, sinusitis, or headaches.
Ophthalmologic Features
Ophthalmologic complications of craniofacial FD are uncommon but may be highly morbid. Involvement of the orbital bones can lead to proptosis, hypertelorism, diplopia, and other visual disturbances [23, 24]. Skull base involvement with optic canal narrowing is common, but only rarely leads to optic neuropathy [25]. In one large cohort, the primary risk factors associated with vision loss included a history of prophylactic optic nerve decompression surgeries and uncontrolled growth hormone excess [25, 26]. Vision loss may also occur in association with acute or chronic aneurysmal bone cysts [27•, 28, 29]. Optic disc edema has been reported in up to 3% of patients with craniofacial FD [30]. Its precise etiology is unclear; however, it appears to arise more commonly in younger patients, those with structural craniofacial abnormalities (such as Chiari malformations), and in association with leuprolide therapy for precocious puberty [30].
Hearing Loss
The temporal bone is a frequent site of FD involvement, and may be associated with mild to moderate hearing impairment in up to 30% of patients [31, 32]. The etiology is often multifactorial and can involve conductive and/or sensorineural components, depending on FD lesion location and behavior. External auditory canal narrowing may result in conductive hearing loss, as well as cerumen impaction and cholesteotoma. Conductive hearing loss may also result from epitympanum involvement and ossicular crowding [31, 33] (Fig. 2E). Sensorineural hearing loss is associated with internal auditory canal deformity and/or FD invasion of the otic capsule [31].
Oral and Dental Complications
Craniofacial FD involving the mandible and maxilla may result in dental displacement and malocclusion [11, 34]. Teeth may become crowded, abnormally spaced, or rotated, and can demonstrate splaying of roots around the FD lesion [11]. Akintoye et al. further described other dental anomalies including dentin dysplasia, taurodontic pulp chambers, odontomas, and a high caries index [20]. This series also noted no evidence of FD progression following dental procedures such as restorations, extractions, and orthodontic treatment [20].
Skull Base Deformities
FD expansion constricting the posterior cranial fossa may result in cranial constriction and/or settling, leading to the skull base deformities Chiari malformation and/or basilar invagination, respectively [35] (Fig. 2D). MAS-associated hyperthyroidism and hypophosphatemia are associated with an increased risk of skull base deformities, likely due to deleterious effects of these endocrinopathies on FD bone. Skull-base deformities are frequently asymptomatic; however, in rare cases, they may be progressive and require surgical correction to prevent neurologic sequelae.
Clinical Management
Management in craniofacial FD requires a multidisciplinary approach. Screening for and treating MAS endocrinopathies is a critical component; in particular, growth hormone excess has been associated with craniofacial FD expansion [36, 37] and an increased risk of vision and hearing loss [25, 31], as well as an increased risk of post-operative FD regrowth [38]. Early diagnosis and treatment of growth hormone excess can potentially mitigate craniofacial expansion and morbidity [39].
The goal of management in craniofacial FD is focused on maintaining function and aesthetics. Surgery is the mainstay of treatment for facial asymmetry; however, outcomes are frequently suboptimal due to post-operative FD regrowth [38]. Larger reconstruction procedures with placement of surgical implants result in lower rates of recurrence (45%) compared to less aggressive recontouring procedures (82%) [38]; however, they are associated with increased operative and post-operative risks. Sequential procedures may be required to attain facial symmetry. Procedures performed after skeletal maturity (> 16 years of age) are likely associated with decreased recurrence rates [40•]. Observation with close monitoring is often the most effective strategy.
Patients with optic canal involvement should be monitored with ophthalmologic exams at least yearly, and developing visual field defects or otherwise abnormal exams should raise concern for optic neuropathy. Optical coherence tomography is a technique that assesses the thickness of the retinal nerve fiber layer using high-resolution optic nerve cross sections, and can be used to more accurately assesses for developing optic neuropathy as part of a traditional ophthalmologic exam [41, 42]. A recent study by Pan et al. demonstrated that retinal nerve fiber layer thickness accurately identified optic neuropathy in FD patients, and was a better indicator than conventional CT assessment of optic canal stenosis and optic nerve stretch [27]. The majority of patients with FD encasement of the optic nerve remained stable over time without development of optic neuropathy, even in the presence of radiological evidence of optic nerve compression [25]. Surgical intervention should therefore only be considered when there is concern for developing visual impairment. Prophylactic optic nerve decompression increases risk of vision loss and is therefore contraindicated [26, 43].
Patients with craniofacial FD should undergo routine monitoring to track the clinical behavior of lesions over time. Serial imaging with CT and/or MRI should be performed; however, there is a paucity of data to guide intervals between imaging studies. Clinicians should therefore use their judgement, and in general perform imaging as guided by symptom progression. In general, imaging should be performed more frequently in younger patients than adults, because this is the period of time when lesions are most progressive [6, 7]. Imaging review should include assessment for skull base deformities, and patients should be followed with routine neurologic exams and assessed for concerning symptoms, such as headache, neck pain, and unsteadiness [35].
Patients with craniofacial FD can safely undergo routine dental, endodontic, and orthodontic procedures. There is some evidence that craniofacial FD may be associated with a higher caries index [20], and it is important for patients maintain regular dental evaluations. Clinicians may consider recommending additional measures such as topical fluoride and electric toothbrushes to aid with hygiene.
Medical therapies for management of craniofacial FD are lacking. Bisphosphonates are a class of antiresorptive medication that bind hydroxyapatite and inhibit osteoclast activity. Bisphosphonate treatment was initially advocated for FD based on the presence of prominent tissue osteoclastogenesis (Fig. 3); however, there is no convincing evidence of any effects on lesion activity or progression [44–46]. Intravenous bisphosphonates may be helpful for treatment of FD-related bone pain, although their efficacy in treating craniofacial pain is less clear [10]. Osteonecrosis of the jaw has been reported in patients with craniofacial FD treated with bisphosphonates [47]; risk factors appear to be similar to the general population, including high dose, long-term intravenous treatment, preceding invasive oral procedures, and poor dental hygiene [48].
Denosumab is another class of antiresorptive medication which shows promise in the treatment of FD. It is a monoclonal antibody of the IgG2 immunoglobulin isotype to receptor activator of nuclear kappa-B ligand (RANKL), a central regulator of bone resorption. Denosumab acts by mimicking the inhibitory effects of osteoprotegerin, a decoy receptor produced by osteoblasts which prevents interaction of RANKL with its receptor RANK and inhibits osteoclast differentiation. RANKL is highly expressed by FD cells, and serum levels are positively correlated with disease burden [49, 50]. Retrospective series suggest denosumab has beneficial effects on FD lesion activity and expansion [50–52]. However, unlike bisphosphonates, the anti-resorptive effects of denosumab are transient, and post-discontinuation bone turnover frequently rebounds above pre-treatment levels. This results in loss of therapeutic effects, and has been associated with life-threatening hypercalcemia in 2 patients with FD [50, 52]. Long-term treatment with denosumab is also associated with osteonecrosis of the jaw and high bone mass. Ongoing clinical trials in adult (NCT03571191) and pediatric (NCT05419050) patients with FD will provide further information about the clinical use of denosumab in patients with FD. In particular, there is considerable interest about whether denosumab may prevent progression of FD during childhood, which is the period of time during which disease burden is established. However, at present, denosumab treatment should be limited to compassionate use for treatment of severe morbidity, and in clinical trials.
Conclusions
FD/MAS is a complex disease which evolves over the lifespan. The craniofacial skeleton offers unique diagnostic and management challenges, and monitoring and treatment approaches must be individualized according to age, lesion behavior, and symptoms. A multidisciplinary team is required for appropriate diagnostic work up and monitoring to provide optimal patient care. While treatment is currently centered around secondary and tertiary preventative measures, there is a need to develop novel tools and treatments to target lesion progression, particularly in childhood.
Funding
This work was funded by the Intramural Research Program of the NIDCR, National Institutes of Health. NIDCR receives funding from Amgen, Inc, Ultragenyx, Inc, and Kyowa Kirin, Inc for studies related to fibrous dysplasia.
National Institute of Dental and Craniofacial Research,DE-000758,Alison Boyce
Footnotes
Conflict of interest The authors report no additional conflicts of interest.
Data availability
This is a review article which does not contain any unpublished data.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
- 1.Boyce AM, Florenzano P, de Castro LF, Collins MT. Fibrous Dysplasia / McCune-Albright Syndrome. In: Adam MP, Everman DB, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2015. pp. 1993–2023. [Google Scholar]
- 2.Weinstein LS, Liu J, Sakamoto A, Xie T, Chen M. Minireview: GNAS: normal and abnormal functions. Endocrinology. 2004;145(12):5459–64. 10.1210/en.2004-0865. [DOI] [PubMed] [Google Scholar]
- 3.Riminucci M, Liu B, Corsi A, et al. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999;187(2):249–58. . [DOI] [PubMed] [Google Scholar]
- 4.Riminucci M, Saggio I, Robey PG, Bianco P. Fibrous dysplasia as a stem cell disease. J Bone Miner Res. 2006;21(Suppl 2):P125–31. 10.1359/jbmr.06s224. [DOI] [PubMed] [Google Scholar]
- 5.Collins MT, Singer FR, Eugster E. McCune-Albright syndrome and the extraskeletal manifestations of fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S4. 10.1186/1750-1172-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hart ES, Kelly MH, Brillante B, et al. Onset, progression, and plateau of skeletal lesions in fibrous dysplasia and the relationship to functional outcome. J Bone Miner Res. 2007;22(9):1468–74. 10.1359/jbmr.070511. [DOI] [PubMed] [Google Scholar]
- 7.Szymczuk V, Taylor J, Michel Z, Sinaii N, Boyce AM. Skeletal disease acquisition in fibrous dysplasia: natural history and indicators of lesion progression in children. J Bone Miner Res. 2022;37(8):1473–8. [DOI] [PubMed] [Google Scholar]
- 8.Kuznetsov SA, Cherman N, Riminucci M, Collins MT, Robey PG, Bianco P. Age-dependent demise of GNAS-mutated skeletal stem cells and “normalization” of fibrous dysplasia of bone. J Bone Miner Res. 2008;23(11):1731–40. 10.1359/jbmr.080609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leet AI, Magur E, Lee JS, Wientroub S, Robey PG, Collins MT. Fibrous dysplasia in the spine: prevalence of lesions and association with scoliosis. J Bone Joint Surg Am. 2004;86(3):531–7. [PubMed] [Google Scholar]
- 10.Javaid MK, Boyce A, Appelman-Dijkstra N, et al. Best practice management guidelines for fibrous dysplasia/McCune-Albright syndrome: a consensus statement from the FD/MAS international consortium. Orphanet J Rare Dis. 2019;14(1):139. 10.1186/s13023-019-1102-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burke AB, Collins MT, Boyce AM. Fibrous dysplasia of bone: craniofacial and dental implications. Oral Dis. 2017;23(6):697–708. 10.1111/odi.12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kushchayeva YS, Kushchayev SV, Glushko TY, et al. Fibrous dysplasia for radiologists: beyond ground glass bone matrix. Insights Imaging. 2018;9(6):1035–56. 10.1007/s13244-018-0666-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JS, FitzGibbon EJ, Chen YR, et al. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S2. 10.1186/1750-1172-7-s1-s2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dujardin F, Binh MB, Bouvier C, et al. MDM2 and CDK4 immunohistochemistry is a valuable tool in the differential diagnosis of low-grade osteosarcomas and other primary fibro-osseous lesions of the bone. Mod Pathol. 2011;24(5):624–37. 10.1038/modpathol.2010.229. [DOI] [PubMed] [Google Scholar]
- 15.Schwartz DT, Alpert M. The malignant transformation of fibrous dysplasia. Am J Med Sci. 1964;247:1–20. 10.1097/00000441-196401000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994;73(5):1411–24. . [DOI] [PubMed] [Google Scholar]
- 17.Pan KS, de Castro LF, Roszko KL, et al. Successful intravascular treatment of an intraosseous arteriovenous fistula in fibrous dysplasia. Calcif Tissue Int. 2020;107(2):195–200. 10.1007/s00223-020-00712-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song X, Li Z. Co-existing of craniofacial fibrous dysplasia and cerebrovascular diseases: a series of 22 cases and review of the literature. Orphanet J Rare Dis. 2021;16(1):471. 10.1186/s13023-021-02102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sweeney K, Kaban LB. Natural history and progression of craniofacial fibrous dysplasia: a retrospective evaluation of 114 patients from Massachusetts General Hospital. J Oral Maxillofac Surg. 2020;78(11):1966–80. 10.1016/j.joms.2020.05.036. [DOI] [PubMed] [Google Scholar]
- 20.Akintoye SO, Lee JS, Feimster T, et al. Dental characteristics of fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2003;96(3):275–82. 10.1016/s1079-2104(03)00225-7. [DOI] [PubMed] [Google Scholar]
- 21.Kapitanov DKA, Nersesyan M, Vicheva D. Sinonasal fibrous dysplasia: our 10-years experience. J of IMAB. 2019;25(2):2583–8. [Google Scholar]
- 22.DeKlotz TR, Kim HJ, Kelly M, Collins MT. Sinonasal disease in polyostotic fibrous dysplasia and McCune-Albright Syndrome. Laryngoscope. 2013;123(4):823–8. 10.1002/lary.23758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bibby K, McFadzean R. Fibrous dysplasia of the orbit. Br J Ophthalmol. 1994;78(4):266–70. 10.1136/bjo.78.4.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ricalde P, Horswell BB. Craniofacial fibrous dysplasia of the fronto-orbital region: a case series and literature review. J Oral Maxillofac Surg. 2001;59(2):157–67 (discussion 67–8). [DOI] [PubMed] [Google Scholar]
- 25.Cutler CM, Lee JS, Butman JA, et al. Long-term outcome of optic nerve encasement and optic nerve decompression in patients with fibrous dysplasia: risk factors for blindness and safety of observation. Neurosurgery. 2006;59(5):1011–7. 10.1227/01.Neu.0000254440.02736.E3. (discussion 17–8). [DOI] [PubMed] [Google Scholar]
- 26.Lee JS, FitzGibbon E, Butman JA, et al. Normal vision despite narrowing of the optic canal in fibrous dysplasia. N Engl J Med. 2002;347(21):1670–6. 10.1056/NEJMoa020742. [DOI] [PubMed] [Google Scholar]
- 27.•.Pan KS, FitzGibbon EJ, Vitale S, Lee JS, Collins MT, Boyce AM. Utility of optical coherence tomography in the diagnosis and management of optic neuropathy in patients with fibrous dysplasia. J Bone Miner Res. 2020;35(11):2199–210. 10.1002/jbmr.4129. [DOI] [PubMed] [Google Scholar]; This large retrospective series demonstrated the utility of optic coherence tomography in patients with FD, and established age-related normative values.
- 28.Ninomiya H, Ozeki M, Nozawa A, et al. A rare pediatric case of McCune-Albright syndrome with acute visual disturbance: Case report. Medicine (Baltimore). 2022;101(6):e28815. 10.1097/md.0000000000028815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Terkawi AS, Al-Qahtani KH, Baksh E, Soualmi L, Mohamed Ael B, Sabbagh AJ. Fibrous dysplasia and aneurysmal bone cyst of the skull base presenting with blindness: a report of a rare locally aggressive example. Head Neck Oncol. 2011;3:15. 10.1186/1758-3284-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raborn LN, Pan KS, FitzGibbon EJ, Collins MT, Boyce AM. Optic disc edema in fibrous dysplasia/McCune-Albright syndrome: prevalence, etiologies, and clinical implications. Bone. 2021;143:115661. 10.1016/j.bone.2020.115661. [DOI] [PubMed] [Google Scholar]
- 31.Boyce AM, Brewer C, DeKlotz TR, et al. Association of hearing loss and otologic outcomes with fibrous dysplasia. JAMA Otolaryngol Head Neck Surg. 2018;144(2):102–7. 10.1001/jamaoto.2017.2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frisch CD, Carlson ML, Kahue CN, et al. Fibrous dysplasia of the temporal bone: a review of 66 cases. Laryngoscope. 2015;125(6):1438–43. 10.1002/lary.25078. [DOI] [PubMed] [Google Scholar]
- 33.Megerian CA, Sofferman RA, McKenna MJ, Eavey RD, Nadol JB Jr. Fibrous dysplasia of the temporal bone: ten new cases demonstrating the spectrum of otologic sequelae. Am J Otol. 1995;16(4):408–19. [PubMed] [Google Scholar]
- 34.Akintoye SO, Boyce AM, Collins MT. Dental perspectives in fibrous dysplasia and McCune-Albright syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116(3):e149–55. 10.1016/j.oooo.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan KS, Heiss JD, Brown SM, Collins MT, Boyce AM. Chiari I Malformation and basilar invagination in fibrous dysplasia: prevalence, mechanisms, and clinical implications. J Bone Miner Res. 2018;33(11):1990–8. 10.1002/jbmr.3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roszko KL, Collins MT, Boyce AM. Mosaic effects of growth hormone on fibrous dysplasia of bone. N Engl J Med. 2018;379(20):1964–5. 10.1056/NEJMc1808583. [DOI] [PubMed] [Google Scholar]
- 37.Tessaris D, Boyce AM, Zacharin M, et al. Growth hormone-Insulin-like growth factor 1 axis hyperactivity on bone fibrous dysplasia in McCune-Albright syndrome. Clin Endocrinol (Oxf). 2018;89(1):56–64. 10.1111/cen.13722. [DOI] [PubMed] [Google Scholar]
- 38.Boyce AM, Burke A, Cutler Peck C, DuFresne CR, Lee JS, Collins MT. Surgical management of polyostotic craniofacial fibrous dysplasia: long-term outcomes and predictors for post-operative regrowth. Plast Reconstr Surg. 2016;137(6):1833–9. 10.1097/PRS.0000000000002151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyce AM, Glover M, Kelly MH, et al. Optic neuropathy in McCune-Albright syndrome: effects of early diagnosis and treatment of growth hormone excess. J Clin Endocrinol Metab. 2013;98(1):E126–34. 10.1210/jc.2012-2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.•.Park JW, Jung JH, Park SJ, Lim SY. Evaluation of natural growth rate and recommended age for shaving procedure by volumetric analysis of craniofacial fibrous dysplasia. Head Neck. 2020;42(10):2863–71. 10.1002/hed.26337. [DOI] [PubMed] [Google Scholar]; This retrospective series investigated risk factors for post-operative regrowth, and reported that patients with ≥16 had improved surgical outcomes.
- 41.Yu M, Lin C, Weinreb RN, Lai G, Chiu V, Leung CK. Risk of visual field progression in glaucoma patients with progressive retinal nerve fiber layer thinning: a 5-year prospective study. Ophthalmology. 2016;123(6):1201–10. 10.1016/j.ophtha.2016.02.017. [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Martin E, Ara JR, Martin J, et al. Retinal and optic nerve degeneration in patients with multiple sclerosis followed up for 5 years. Ophthalmology. 2017;124(5):688–96. 10.1016/j.ophtha.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 43.Amit M, Collins MT, FitzGibbon EJ, Butman JA, Fliss DM, Gil Z. Surgery versus watchful waiting in patients with craniofacial fibrous dysplasia–a meta-analysis. PLoS One. 2011;6(9):e25179. 10.1371/journal.pone.0025179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boyce AM, Kelly MH, Brillante BA, et al. A randomized, double blind, placebo-controlled trial of alendronate treatment for fibrous dysplasia of bone. J Clin Endocrinol Metab. 2014;99(11):4133–40. 10.1210/jc.2014-1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Florenzano P, Pan KS, Brown SM, et al. Age-related changes and effects of bisphosphonates on bone turnover and disease progression in fibrous dysplasia of bone. J Bone Miner Res. 2019;34(4):653–60. 10.1002/jbmr.3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plotkin H, Rauch F, Zeitlin L, Munns C, Travers R, Glorieux FH. Effect of pamidronate treatment in children with polyostotic fibrous dysplasia of bone. J Clin Endocrinol Metab. 2003;88(10):4569–75. 10.1210/jc.2003-030050. [DOI] [PubMed] [Google Scholar]
- 47.Nadella S, Mupparapu M, Akintoye SO. Risk of developing spontaneous MRONJ in fibrous dysplasia patients treated with bisphosphonates: a systematic review of the literature. Quintessence Int. 2022;53(7):616–23. 10.3290/j.qi.b3082785. [DOI] [PubMed] [Google Scholar]
- 48.Metwally T, Burke A, Tsai JY, Collins MT, Boyce AM. Fibrous dysplasia and medication-related osteonecrosis of the jaw. J Oral Maxillofac Surg. 2016;74(10):1983–99. 10.1016/j.joms.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Castro LF, Burke AB, Wang HD, et al. Activation of RANK/RANKL/OPG pathway is involved in the pathophysiology of fibrous dysplasia and associated with disease burden. J Bone Miner Res. 2019;34(2):290–4. 10.1002/jbmr.3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Boyce AM, Chong WH, Yao J, et al. Denosumab treatment for fibrous dysplasia. J Bone Miner Res. 2012;27(7):1462–70. 10.1002/jbmr.1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Majoor BCJ, Papapoulos SE, Dijkstra PDS, Fiocco M, Hamdy NAT, Appelman-Dijkstra NM. Denosumab in patients with fibrous dysplasia previously treated with bisphosphonates. J Clin Endocrinol Metab. 2019;104(12):6069–78. 10.1210/jc.2018-02543. [DOI] [PubMed] [Google Scholar]
- 52.Raborn LN, Burke AB, Ebb DH, Collins MT, Kaban LB, Boyce AM. Denosumab for craniofacial fibrous dysplasia: duration of efficacy and post-treatment effects. Osteoporos Int. 2021;32(9):1889–93. 10.1007/s00198-021-05895-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This is a review article which does not contain any unpublished data.