

Significance
The regulation of gene expression corresponds to a key step in a process by which information encoded in the genome is converted into phenotypes. Although the genetic origin of transcription level variation has been extensively analyzed, our knowledge remains very limited regarding the genetic origin of variation in protein abundance at a population level. Here, we generated quantitative proteomes for nearly a thousand natural yeast isolates. By comparison with their transcriptomes, our analyses collectively show that the transcriptome and the proteome are clearly two distinct levels of regulation, governed by distinct genetic bases in natural populations. Taken together, our results highlight the relevance of having access to these two levels of gene expression to better understand the genotype–phenotype relationship.
Keywords: gene regulation, quantitative proteomes, genetic control, pQTL, yeast
Abstract
Gene expression varies between individuals and corresponds to a key step linking genotypes to phenotypes. However, our knowledge regarding the species-wide genetic control of protein abundance, including its dependency on transcript levels, is very limited. Here, we have determined quantitative proteomes of a large population of 942 diverse natural Saccharomyces cerevisiae yeast isolates. We found that mRNA and protein abundances are weakly correlated at the population gene level. While the protein coexpression network recapitulates major biological functions, differential expression patterns reveal proteomic signatures related to specific populations. Comprehensive genetic association analyses highlight that genetic variants associated with variation in protein (pQTL) and transcript (eQTL) levels poorly overlap (3%). Our results demonstrate that transcriptome and proteome are governed by distinct genetic bases, likely explained by protein turnover. It also highlights the importance of integrating these different levels of gene expression to better understand the genotype–phenotype relationship.
Understanding the genetic basis of phenotypic variation in natural populations is one of the main goals of modern biology. Gene expression differs among individuals and is known to be a main determinant of phenotypic variation (1, 2). In humans, the onset and development of numerous diseases have been linked to abnormal regulation of gene expression (3). It is therefore essential to understand how genomic information is expressed through the different layers of gene regulation (i.e., transcriptomes and proteomes). Over the past decades, the development of methods for high-throughput quantification of mRNA and protein abundance has made it possible to explore both the proteome and the transcriptome on a larger scale (4, 5). These approaches facilitated the detection of numerous genetic loci (quantitative trait loci, QTL) affecting either transcript (eQTL) or protein (pQTL) levels (6–11). However, the relationship between transcript and protein levels remains debated and poorly understood at the population level (12).
The transcript-protein correlation provides a first global view of the dependency of the two gene expression layers. Two types of mRNA–protein correlation can be determined, across- and within-gene, reflecting very different dynamics (12–14). The across-gene correlation analysis focuses on the overall correlation of a large set of genes coming from the same sample under a given condition to find out how well the absolute abundances of mRNAs and proteins are correlated. This correlation has been widely investigated in several species, such as humans (15–21), rats and mice (22–25), flies (26), plants (27), or yeast (28–30). Across-gene correlations are consistently high and range from 0.4 to 0.8, suggesting that the absolute number of transcripts and proteins are globally correlated. Therefore, very abundant transcripts generally lead to very abundant proteins and vice versa.
However, the relationship between the transcript and protein abundance at the population level is explored via their variation across samples (e.g., individuals, tissues, or cell lines). Within-gene correlation analysis gives a view on how the protein level of each gene tracks its mRNA level in a population. Different studies have investigated this within-gene correlation in different contexts and organisms, but they often show divergent results. Several surveys of tumors, normal human tissues, as well as pluripotent stem cells have highlighted this discrepancy in estimates with median within-gene correlation coefficients ranging from 0.14 to 0.59 (15, 19, 21, 22, 31–39). Similarly, the overlap of the detected loci influencing mRNA (eQTL) and protein (pQTL) abundance greatly differed across the datasets. It ranges from a very weak overlap of 5.5% in a study on 97 inbred and recombinant mice to nearly 35% in humans (n = 62) and mice (n = 192) (6, 15, 40).
Part of the diverging results might have been driven by technical limitations. For instance, it has been shown that by selecting the most representative peptides in prior proteomic methods, the overall correlation of global transcript and mRNA abundance improves significantly (37, 41). A key difference is also whether the goal of the survey is to correlate absolute number of transcripts and proteins, or relative changes in protein or mRNA levels, which differ between samples. While the absolute number of transcripts and proteins spans several orders of magnitude, the relative expression differences of any individual protein across samples varies within a much narrower range (30, 42). Finally, a main limitation of these studies is that the sample size is much lower than the dimensionality of the problem.
To determine to which extent differences in relative changes in mRNA and protein levels are correlated and the genetic origins of their abundance variation are shared, a large-scale population survey exploring these two facets in a quantitative way was therefore necessary. Here, we took advantage of the 1,011 yeast Saccharomyces cerevisiae population we genome-sequenced and for which we have a species-level understanding of the natural genetic and phenotypic diversity (43). In order to be fully able to compare and analyze in detail the relationship between these two layers of gene regulation, we generated 942 quantitative proteomes in which cells were also cultured in synthetic complete medium supplemented with amino acids using high-throughput mass-spectrometry. We found that protein levels are molecular traits that exhibit considerable variation between individuals and specific signatures related to certain subpopulations. This large available population also makes it possible to generate a detailed map of loci involved in the variation of protein abundance (pQTL) at the species level, via genome-wide association studies (GWAS). Interestingly, local pQTL are less frequent than distant ones (5.7% of the total set of pQTL) but they have a higher impact on their respective traits. Integration of proteomic and transcriptomic datasets acquired under similar conditions allowed comparison of accurate quantification of the mRNA and protein abundance of 629 genes across 889 natural isolates (44). Based on these unique datasets, we clearly demonstrated that the degree of within-gene correlation between protein and mRNA abundance is very low (ρ = 0.165). Consistently, we found that the genetic variants influencing protein and mRNA abundance are very dissimilar. Our study highlights that population-scale proteomes are essential and add a broader dimension to the characterization of the genotype–phenotype relationship when integrated with genomic and transcriptomic information.
Results
Quantitative Proteomes of a Large Collection of Natural Isolates.
We generated a quantitative proteomic dataset for strains of the 1,011 strains collection (43) from cells cultivated in synthetic complete medium with amino acids in order to match the growth medium used for RNA sequencing (44) (Fig. 1A). We had previously acquired a proteome dataset of the 1,011 strains collection, measured with microflow chromatography and SWATH MS (45). For the acquisition of this dataset, we used a proteomic method that allows for an even higher throughput, using analytical flowrate chromatography and Scanning-SWATH MS with a 3 min gradient (46). After cultivation of the yeast isolates in 96-well plates, proteins were extracted, and subjected to reduction, alkylation, and trypsination in a semiautomated workflow using liquid handling robotics (47). Data were recorded using Scanning SWATH acquisition (46) and the raw data was processed using DIA-NN software (version 1.8), which was specifically developed for large-scale proteomic exploration (48). We obtained at first a quantification for 1,048 proteins, corresponding to 4,993 peptides. We applied several quality filters where poor-quality samples were removed from the analysis, and we excluded peptides that were not detected in more than 80% of the samples (Methods). The generated dataset hence encompasses protein abundance quantification for 630 proteins among 942 isolates (Datasets S1 and S2), corresponding to 2,676 peptides (so approximately, four peptides per proteins). This dataset therefore covers the overall genetic diversity of the species and captures the subpopulations that were defined as part of the 1,011 yeast genomes project, including both domesticated and wild clades (43) (SI Appendix, Fig. S1A). We combined the proteomic dataset with transcriptomic data obtained from the 1,011 strains collection (44), which gave access to the quantified expression of both levels for 629 genes across 889 isolates (Fig. 1B and Dataset S1). To be able to properly compare these two datasets, we normalized them with quantile normalization after imputing the missing values using the KNN method (Dataset S3 and SI Appendix, Fig. S1 B and C).
Fig. 1.
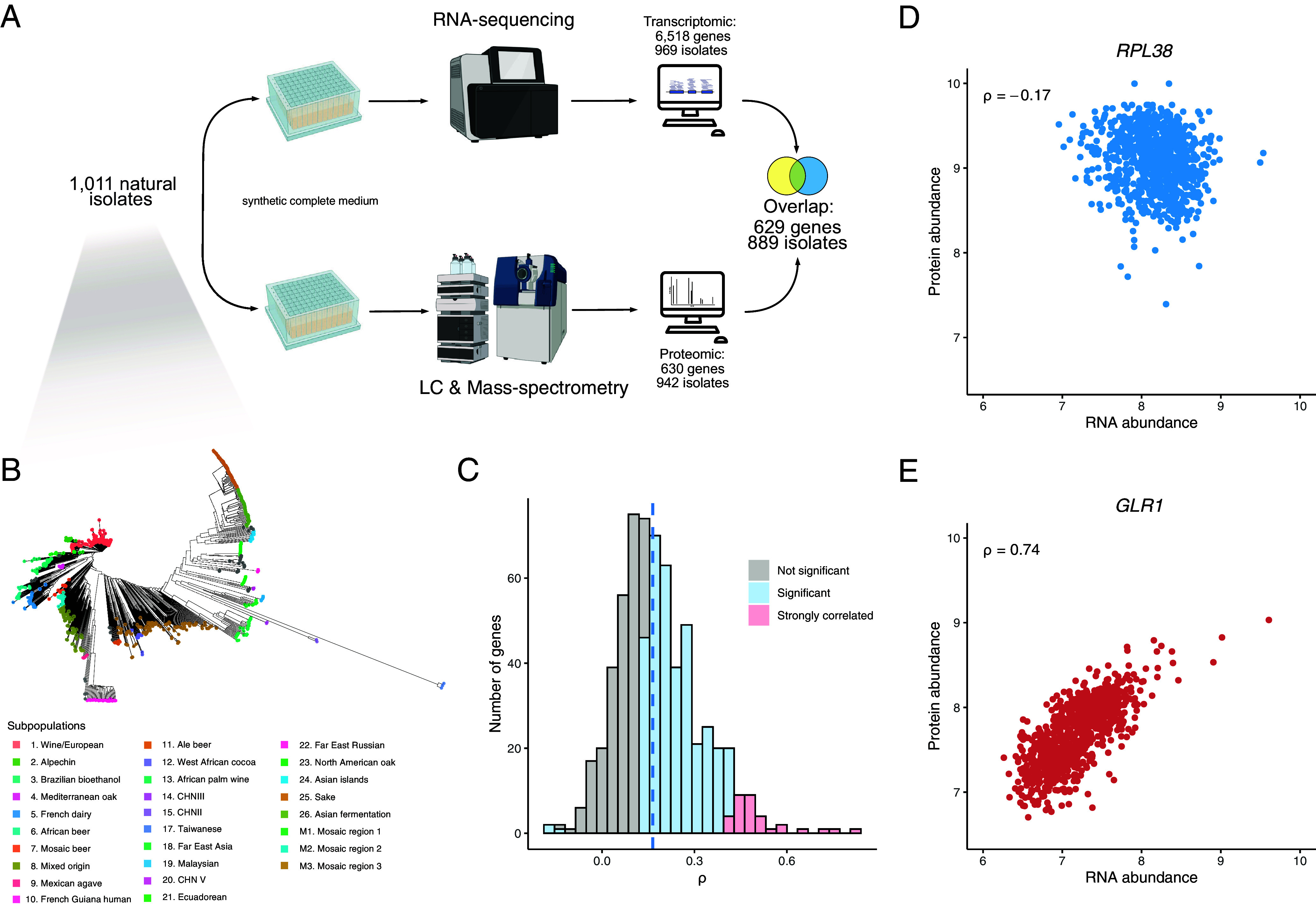
Quantitative proteomes and transcriptomes of a large S. cerevisiae population. (A) The proteomic dataset was generated on isolates grown in synthetic complete (SC) medium with amino acids using a semiautomated sample preparation workflow, and Scanning-SWATH MS (Methods). The overlap between this dataset and the recently generated transcriptomic dataset on the same population in the same condition (44) resulted in 629 protein/transcript abundances across 889 isolates. (B) Phylogenetic trees of the isolates used in this study. Colors correspond to previously defined subpopulations (43). (C) Gene-wise correlation coefficients (Spearman correlation test) between the proteome and the transcriptome. (D and E) mRNA–protein within-gene correlation across isolates for the RPL38 and GLR1 genes (ρ corresponds to the Spearman correlation coefficient with P-values of 4.8 × 10−7 and 2.3 × 10−153, respectively).
To characterize the quantified proteins in our study, we first compared the level of transcription of both the identified and unidentified proteins. Low abundance proteins are less likely to be quantified by proteomics as compared to high abundant proteins (SI Appendix, Fig. S1D). Indeed, 489 out of 629 consistently quantified proteins fall into the 20% highest transcribed genes (n = 1,304). In total, 537 out of 629 quantified proteins were found in the two highest abundance deciles as defined in a recent yeast protein abundance meta-analysis (49) (SI Appendix, Fig. S1E). Overall, proteins related to essential genes and involved in molecular complexes were both significantly enriched in the set of proteins quantified by Scanning SWATH (odd-ratio = 3.5 and 2.2 respectively, Fisher’s exact test, P-values < 2.2 × 10−16) (50–53). Function-wise, we found that metabolism-related genes were overrepresented among the 629 genes included in our study (Dataset S4).
We then investigated the level of variation in protein abundance by calculating the coefficient of variation (CV) for each protein using the nonnormalized dataset. We found an average CV of 31%, varying between 12% and 98% and one high outlier reaching 300% (PDC5, a pyruvate decarboxylase). The precursor-level CVs across quality control samples (15.15%) were much lower than the precursor-level CVs across the natural isolate samples (34.21%), confirming that a biological signal was observed across the isolates (Methods and SI Appendix, Fig. S2A). Moreover, we used a set of seven isolates to perform both technical and biological replicate to ensure the reliability of our proteomic exploration (Methods). The median CV values across all samples, across the biological replicates per strain, as well as within technical replicates were 38.4%, 28.9%, and 18.4%, respectively (SI Appendix, Fig. S2B), clearly demonstrating higher true biological differences between different isolates than technical variance due to either differences in growth, sample processing, or variability of mass spectrometric performance. Gene set enrichment analyses (GSEA) were performed using the CVs and significant enrichment of genes related to amino acid metabolism, respiration, or pyruvate metabolism was found for proteins with a high CV, indicating that they vary the most (Dataset S5). By contrast, proteins with a low CV were significantly related to genes involved in tRNA aminoacylation or protein degradation.
Transcript and Protein Abundances Are Weakly Correlated at the Gene Level across Isolates.
As proteomes and transcriptomes were obtained using the same growth media, our dataset allowed us to characterize the different types of correlation between mRNA and protein abundance across a natural population. We first determined the across-gene correlation, i.e., the concordance between protein and transcript abundance for each isolate, and found a high correlation (median ρ = 0.53, interquartile range of 0.06, SI Appendix, Fig. S3), which is consistent with what was previously described (15–21, 24, 26, 27). We next computed the correlation between the protein and mRNA normalized abundance for each gene across the 889 natural isolates (Fig. 1 C–E and Dataset S6). While the across-gene correlation levels were in line with previous explorations, we found an overall low within-gene correlation level (median ρ = 0.165, interquartile range of 0.17). This value is much lower than the one determined with smaller samples in mice (approximately 0.25) (6, 40) and in human healthy tissues (0.35 and 0.46) (19, 33), but it is in line with what was found in human lymphoblastoid cell lines (0.14) (15). For a total of 385 out of the 629 quantified proteins, the level is significantly correlated with RNA level (Bonferroni corrected P-value < 0.05). Out of these 385 proteins, only 3 show a negative correlation: Rps13, Asc1, and Rpl38 (Fig. 1D), all ribosomal-related proteins. This observation is consistent with previous surveys pointing out that some ribosome-related proteins are negatively correlated with their cognate transcripts (12, 19). But overall, this correlated set of 385 proteins/transcripts is significantly enriched of genes related to several metabolism pathways (Dataset S7). Moreover, the most strongly correlated set of proteins/transcripts (n = 33) show functional enrichment of genes related to mitochondrial respiration (Dataset S8) (Methods). Interestingly, it points out that this specific pathway has similar gene regulation at both levels. Finally, we observed that four genes with very high mRNA–protein correlation were located outside the main correlation index distribution (Fig. 1C). These genes all have correlation coefficients greater than 0.6: SFA1 (alcohol dehydrogenase), HBN1 (unknown function), GLR1 (glutathione oxidoreductase, Fig. 1E), and YLR179C (unknown function). Such a high correlation clearly points to common regulatory mechanisms and genetic bases underlying the two levels of variation, as we have seen below.
Gene Expression Is More Constrained at the Proteome Level.
By combining these proteomic and transcriptomic datasets, we are in a position to simultaneously explore and compare the variation of these two gene expression layers at the population level. We therefore computed the absolute Log2(fold change) value (i.e., |Log2(FC)|) for each gene in each pair of isolates and found that this value is 32% lower on average for the proteome (Fig. 2A), suggesting that protein abundance is less variable and more constrained than mRNA abundance. Furthermore, a higher correlation was observed between proteomes (ρ = 0.92) compared to transcriptomes (ρ = 0.83) (Fig. 2B). Finally, the variance observed for each gene was lower for the proteomic data (SI Appendix, Fig. S4A) and the Euclidean distances between each isolate were smaller when computed with the protein abundance dataset (SI Appendix, Fig. S4B). Overall, these observations reflect and highlight the presence of a global post-transcriptional buffering of the transcriptome variations.
Fig. 2.
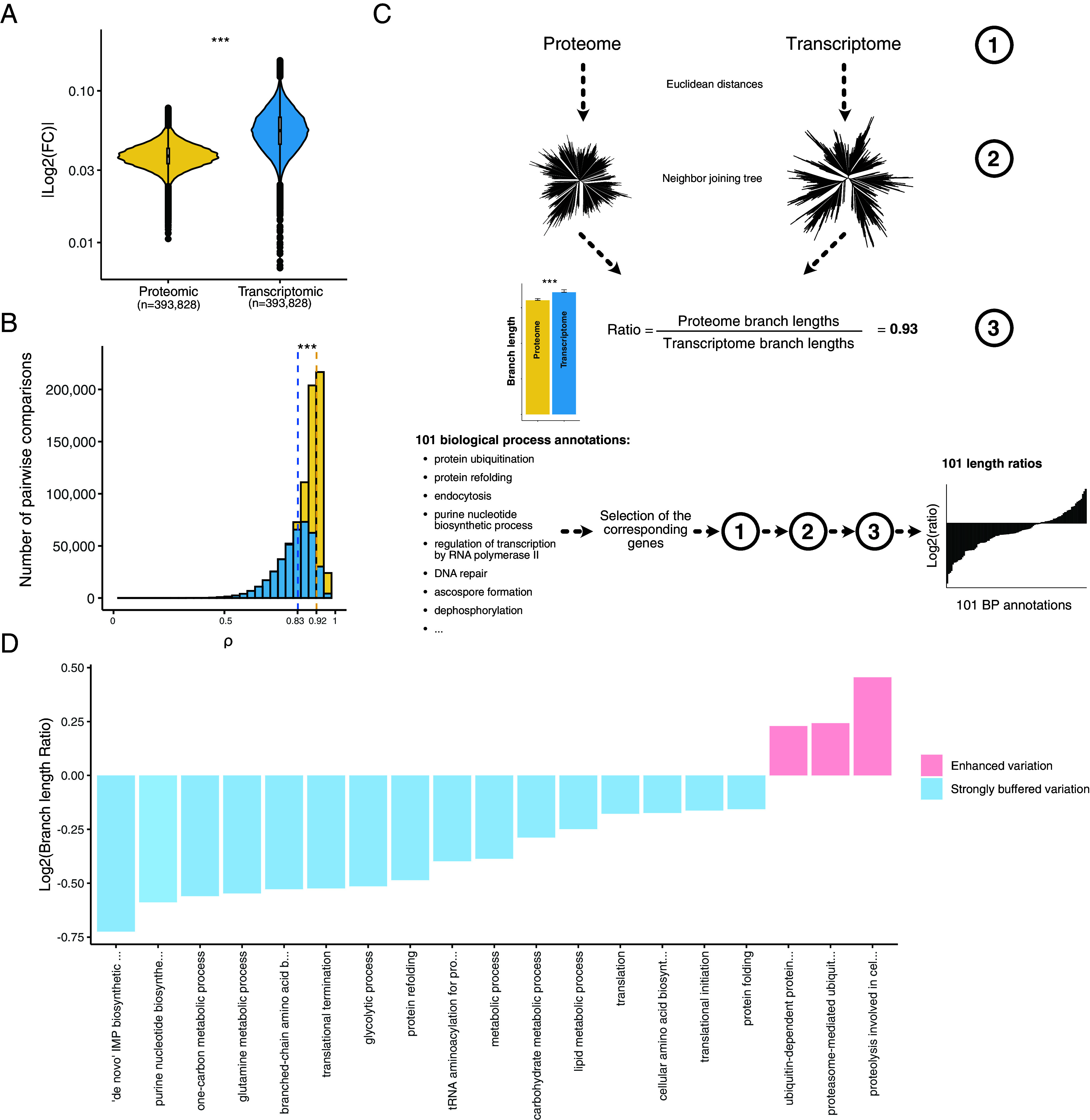
Detection and functional description of the post-transcriptional buffering. (A) Median |log2(fold changes)| computed in each isolate pairwise comparison using both proteomic and transcriptomic data (*** = Wilcoxon test, P-value < 2.2 × 10−16) (Methods). (B) Correlation coefficients from the isolate pairwise comparisons using both protein and transcript abundance (*** = Wilcoxon test, P-value < 2.2 × 10−16). The dotted lines correspond to the median correlation index for the proteomic (yellow) and transcriptomic (blue) data. (C) Cellular functions that are preferentially affected by post-transcriptional buffering. Briefly, using either the proteome and the transcriptome abundances (1) we constructed expression-based neighbor-joining trees (2) and compared the total sum of the branch lengths. We computed a ratio (3) defined by the proteome total branch lengths divided by the transcriptome total branch lengths. Using all the genes, this ratio was equal to 0.93 (overall, the expression evolution is more constrained at the proteome level). We performed the same procedure using subsets of genes corresponding to 101 biological process annotations. The biological processes displaying a ratio lower than 0.93 and a significant difference in terms of branch lengths (Methods) were considered as strongly buffered. The biological processes displaying a ratio higher than 1 and a significant difference in terms of branch lengths had an enhanced abundance variation at the proteome level. (D) Biological processes detected as strongly buffered or with an enhanced variation using the procedure detailed in (C).
Despite recurrent observations (45, 54–58), the post-transcriptional buffering phenomenon remains largely functionally uncharacterized and poorly understood. We sought to better understand this phenomenon at the genetic level by examining the cellular functions that tended to be most affected by post-transcriptional buffering. Briefly, we constructed neighbor-joining trees using the proteome or transcriptome Euclidean distances between each isolate (Fig. 2C and Methods) (57). Total branch length was used as a measure of expression variation and evolution at the species level. We then calculated the ratio between the lengths of the proteome and transcriptome tree branches to quantify the strength of the post-transcriptional buffering phenomenon. The lengths of branches from the proteome-based tree were shorter than those from the transcriptome-based tree, resulting in a length ratio of 0.93 (Fig. 2C and SI Appendix, Fig. S4C). This observation is consistent with the differences in Euclidean distances observed previously (Fig. S4B). We then applied the same procedure to 101 sets of genes, representing central biological processes obtained from a reduced list of gene ontology (GO) annotations (Dataset S9). We found that a total of 16 sets display a ratio lower than 0.93 and a significant difference between the proteome and transcriptome branch lengths, meaning that these sets are strongly affected by the phenomenon of post-transcriptional buffering (Fig. 2D and Dataset S10). Interestingly, 6 out of the 16 sets include genes with functions related to protein production and maturation (Fig. 2D), highlighting that the evolution of the cellular machinery involved in protein production and maturation is highly constrained. The other set of genes are related to several metabolism processes and detected as strongly buffered, despite being highly variable in the proteomic data (Dataset S5). This observation could be due to the fact that metabolism-related genes are among the genes with the greatest variation in mRNA abundance at the species level (44). This variation is largely attenuated at the proteome level but remains important, reflecting differences in metabolic preferences within the population. Moreover, we also found three sets with a ratio higher than 0.93 and a significant difference between the proteome and transcriptome trees, which means that the expression variation of these genes is greater at the proteome level (Fig. 2D and Dataset S10). Interestingly, all of them are related to protein catabolism, highlighting a difference in post-transcriptional mechanism for this specific functional category. Taken together, these results provide deeper insights into post-transcriptional buffering as well as its functional impact.
Architecture of the Proteome Landscape.
Using these datasets, we then sought to understand the main determinants shaping the proteome architecture at the population level. The S. cerevisiae yeast species exhibit a clear population structure (59–61), which potentially can impact the proteome landscape (43) (Dataset S1). We performed a principal component analysis (PCA) with the protein abundance data and found that no clear grouping emerged from the subpopulations when plotting together the 6 first principal components (SI Appendix, Fig. S5 A–C). The same results were observed for transcriptomes (SI Appendix, Fig. S5 D–F). To confirm this, we also computed the Euclidean distance across transcript and protein levels between every pair of isolates and used these to construct a neighbor-joining tree (Fig. 3 B and C). We observed that none of the subpopulations present in the genetic-based tree clearly emerged in either the proteome- or transcriptome-based tree (Fig. 3 A–C). We then attempted to further explore the relationship between the population structure and the proteome and transcriptome profiles by comparing the genetic distances between each pair of isolates with the transcriptome or proteome correlation between the corresponding isolate pairs (SI Appendix, Fig. S5 G and H). We observed a very weak anticorrelation for both transcriptome and proteome (−0.091 and −0.11, respectively, both P-values were below 10−15), suggesting that the population structure is only poorly reflected at the transcriptomic and proteomic level. Taken together, these results indicate that population structure has little effect on the transcriptomes and proteomes of S. cerevisiae species. However, we should emphasize that since our proteome survey mainly includes highly abundant proteins that could be detected in the majority of isolates, a larger coverage of the proteome might reveal a greater similarity between population structure and gene expression profiles.
Fig. 3.
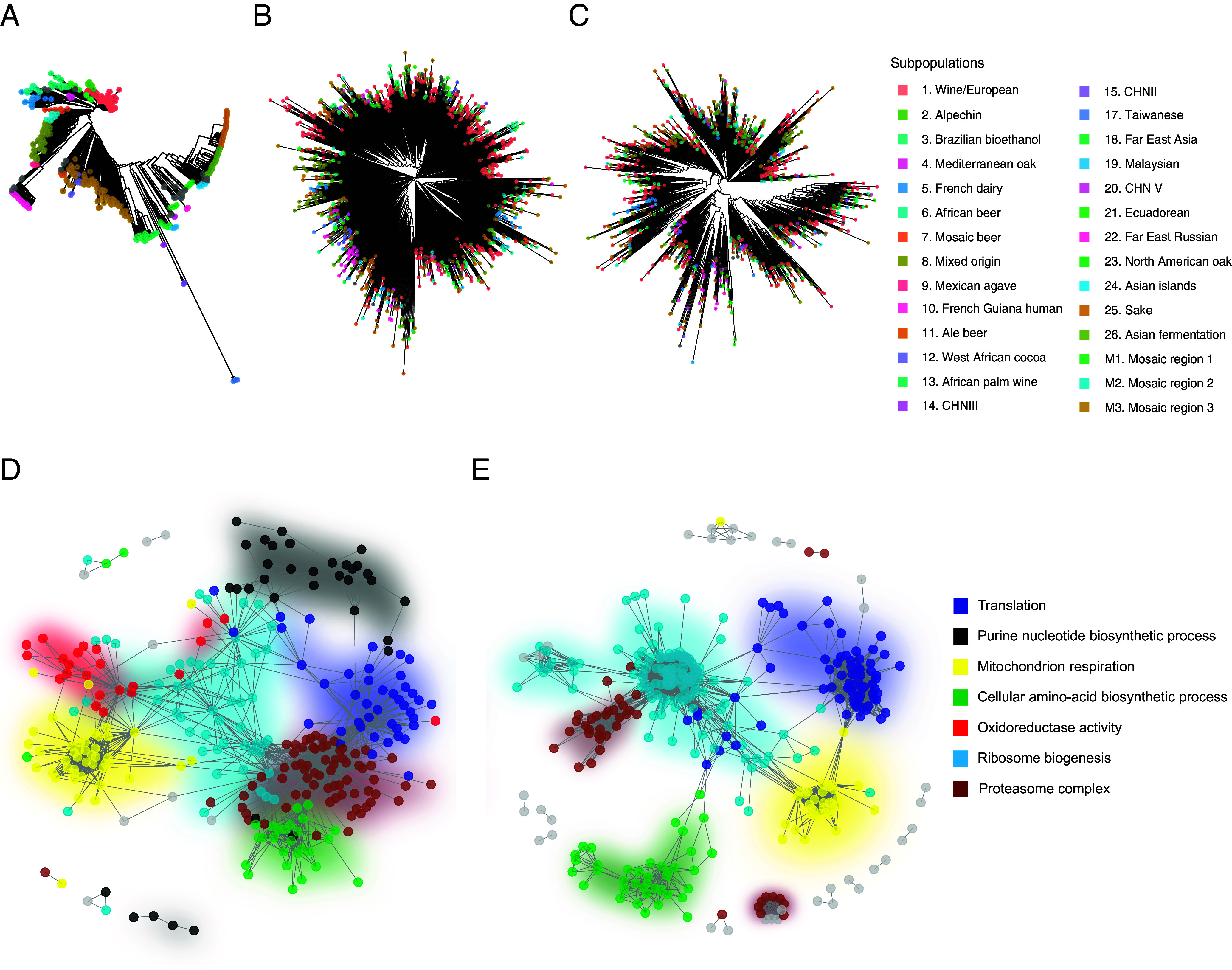
Coexpression network is a major determinant of the proteome organization while the population structure is not. (A–C) Comparison between the phylogenetic tree (A) obtained using the biallelic SNP (as in ref. 43) and the trees obtained from the Euclidean distances based on protein (B) or transcript (C) abundance. Colors correspond to the subpopulations. (D and E) Cellular coexpression network computed with WGCNA using proteomic (D) or transcriptomic (E) data. Colors represent the cellular pathway detected for each coexpression module.
One potential determinant of the proteome organization could be related to coexpression networks that strongly influence the coordination of gene expression or various cellular processes. Using weighted gene co-expression network analysis (WGCNA) (62) on the normalized protein abundance data, we detected seven coexpression modules (Fig. 3D and Dataset S11). Each of these modules corresponds to a specific biological function (Dataset S12 and SI Appendix, Fig. S6) and encompasses between 38 (Cellular amino acid biosynthetic process) and 114 (Ribosome biogenesis) genes. Interestingly, very similar modules were found applying the same procedure on the mRNA normalized data. Five coexpression modules were detected (Fig. 3E and SI Appendix, Fig. S7 and Datasets S13 and S14), and all of them were detected in the seven proteomic modules, suggesting that coexpression patterns recapitulate central cell functions are conserved across the two expression layers (Fig. 3 D and E).
Insight into Subpopulation-Specific Protein Expression.
We also wanted to explore and determine the presence of subpopulation-specific signatures. We therefore sought to identify differential protein expression patterns by comparing each clade to the rest of the population and we detected a total number of 1,129 differentially expressed proteins (DEPs) (corresponding to 465 unique proteins, SI Appendix, Fig. S8 and Dataset S15). An average of 59 DEPs was found per clade, ranging from 218 for the Wine clade to 0 for wild Asian clades represented by a small sample (e.g., CHN, Taiwanese, and Far East Russian) (SI Appendix, Fig. S9A). Several DEPs were adequately related to the ecological origin of the different subpopulations. For example, several subpopulations related to alcoholic fermentation show overexpression of alcohol dehydrogenases, such as ADH4 in Wine and Brazilian bioethanol clades as well as ADH3 in the Sake subpopulation. In the French Dairy subpopulation, we also observed an underexpression of SEC23, a GTPase-activating protein involved in the COPII-related vesicle formation, which could reflect an adaptation to this secretory pathway to the cheese-making environment (63). Overall, these observations suggest that domestication and more generally, ecological constraints are drivers of the proteomic landscape evolution in a natural population. We then performed GSEA based on differential expressed proteins in each subpopulation and found significant enrichments for various biological processes (Fig. 4A and Dataset S16). Many enriched functional categories were associated with respiration-related genes (e.g., “respiratory electron chain transport”). Interestingly, we observed that while most wild clades (8 out of 13) tend to have overexpression of respiration-related proteins, these are underexpressed in domesticated subpopulations (5 out of 7).
Fig. 4.
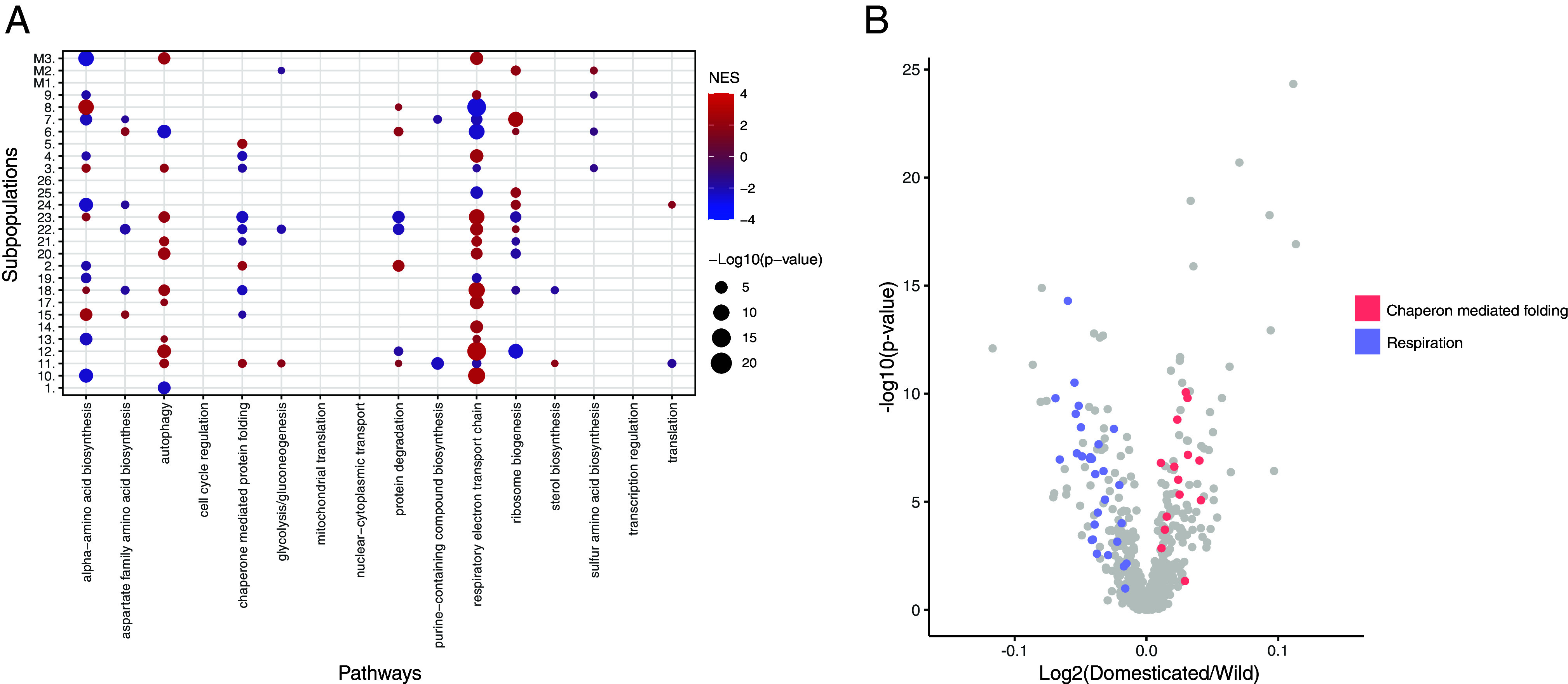
DEPs reveal domestication- and subpopulation-specific metabolic adaptation. (A) GSEA results on the DEPs (using 16 broad functional annotations from ref. 44) of each subpopulation. Colors represent the normalized enrichment score (NES): Red—overexpression, blue—underexpression in subpopulation. (B) Volcano plot of the comparison between wild and domesticated isolates. Colors highlight the genes belonging to two functional annotations related to chaperon-mediated folding and respiration.
We therefore further explored the impact of domestication on the proteome at the population level. Using the same DEP detection method, we assessed the proteome differences between the domesticated and wild isolates (43) and found a total of 133 DEPs (Dataset S17). Among these proteins, other alcohol dehydrogenases such as SFA1 and ADH3 were highly abundant in domesticated isolates. A GSEA performed on this set of DEPs clearly shows an enrichment of underexpressed respiration-related proteins in domesticated clades (Fig. 4B and Dataset S18). Unlike wild isolates, domesticated isolates were selected for fermentation purposes, likely leading to this specific signature. This observation is in line with the previous finding pointing out that the switch from a preference between respiration and fermentation is one of the hallmarks of domestication in yeast (64). In addition, significant enrichment of the functional category “chaperon-mediated protein folding” points to overexpression of this set of proteins in the domesticated isolates (Fig. 4B), which may be an adaptative response to long-term exposure to ethanol, known to induce protein denaturation (65). By performing the same analysis on transcriptomic data (SI Appendix, Fig. S9B and Dataset S17), similar results, showing overexpression of respiration-related genes in domesticated clades, were obtained (Dataset S19).
The Genetic Bases of Protein Abundance at the Population Scale.
To uncover the genetic origins of the proteome variation at the population-scale, we performed genome-wide association studies (GWAS) and considered both SNPs and CNVs that were characterized previously (43). In order to have reliable results and to only capture the impact of genetic variation on protein and transcript abundance, we focused on isolates for which proteomic and transcriptomic data were available, and for which the OD measurements at the time harvest were well correlated (Pearson correlation coefficient > 0.6), resulting in a set of 455 isolates (Dataset S1). In this population, a total of 101,836 SNPs and 631 CNVs were considered, with a minor allele frequency higher than 5%. We performed GWAS using the raw protein abundances of the genes for which we have both levels of expression (i.e., 629 genes). Overall, we detected a total of 528 SNP-pQTL after combining SNP affected by linkage disequilibrium (R2 > 0.6), and 1,009 CNV-pQTL corresponding to 455 and 197 loci and affecting 275 and 44 genes, respectively (Fig. 5A and SI Appendix, Fig. S10A and Datasets S20–S23).
Fig. 5.
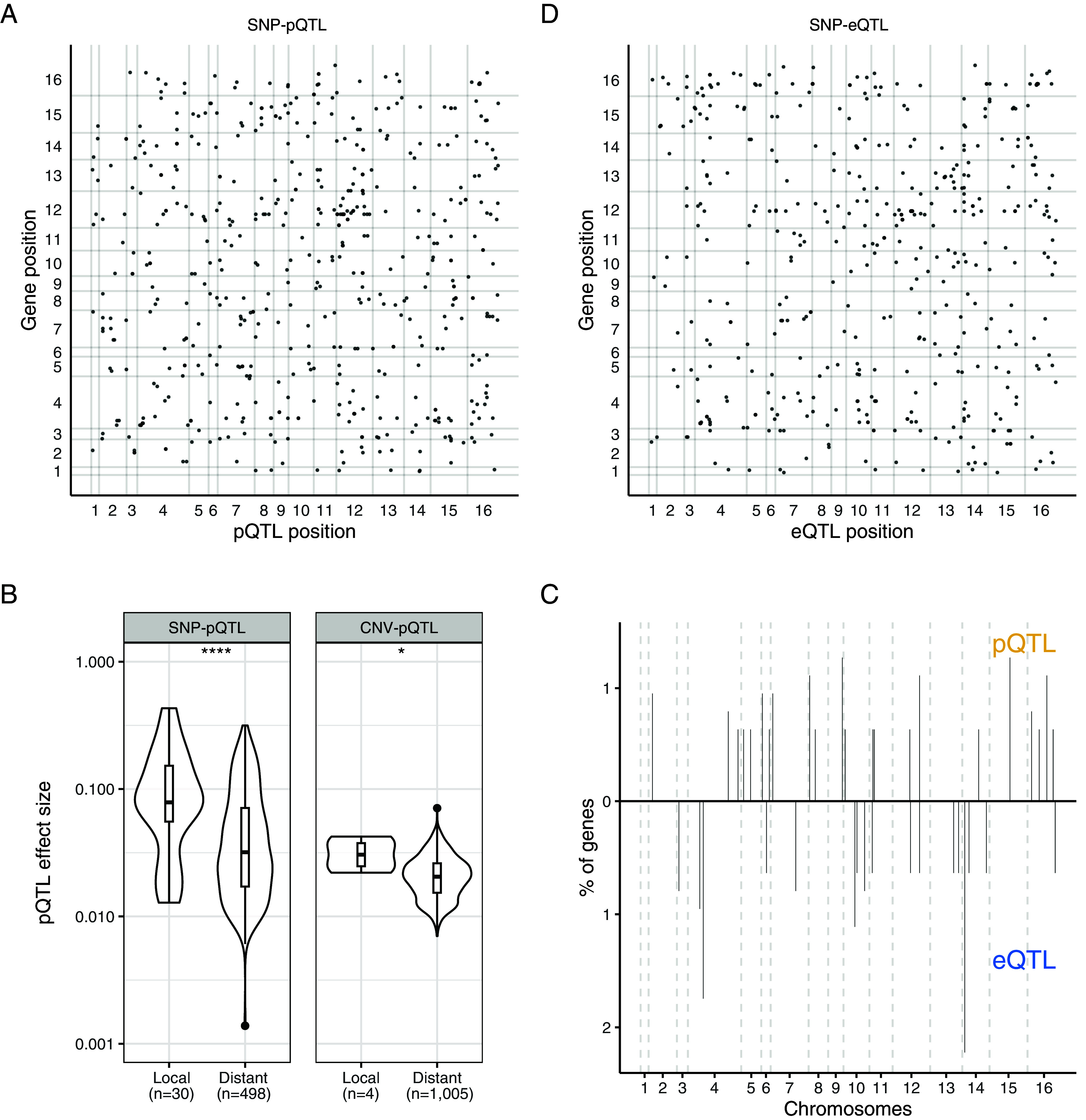
SNP- and CNV-pQTL detection highlights strong differences in the genetic origin of transcript and protein abundance. (A) Map of the SNP-pQTL. The x-axis is the QTL position on the genome and the y-axis the position of the affected gene on the genome. The x and y-axis numbers represent the 16 chromosomes of S. cerevisiae. (B) Effect size difference between the local and distant pQTL for the SNP (P-value = 8.5 × 10−5) and CNV pQTL (P-value = 0.033). (C) Distribution of the SNP-pQTL and SNP-eQTL hotspots along the genome. The y-axis represents the percentage of the 629 genes that are encompassed by each hotspot (defined as a 20 kb window containing four or more distinct SNP). (D) Map of the SNP-eQTL. The x-axis is the QTL position on the genome and the y-axis the position of the affected gene on the genome. The x and y-axis numbers represent the 16 chromosomes of S. cerevisiae.
Among the SNP-pQTL, 5.7% (n = 30) were local-pQTL, showing that regulation of protein abundance is primarily achieved through trans regulation. This fraction is consistent with previous exploration in yeast (66) and lower than what is usually found at the transcriptome level (44, 67). Nonetheless, we observed that the local SNP-pQTL have a higher effect size compared to trans SNP-pQTL (Fig. 5B) and tend to be located near the transcription starting site of the gene (SI Appendix, Fig. S11). We found no strong SNP-pQTL hotspots, suggesting that most of the distant pQTL are evenly distributed throughout the genome (Fig. 5C).
In contrast, CNVs impacting protein abundance had a biased location on chromosomes 1, 3, 8, 9, and 11 (SI Appendix, Fig. S10A). Out of 1,009 CNV-pQTL, a total of 1,000 were located on these chromosomes and affected a gene on their respective chromosome. This observed bias is due to the presence of aneuploidies on these chromosomes in our population (43) and suggests that aneuploidies represent a major source of proteome variation at the population level, even if the effect size of aneuploidy-related CNV-pQTL is not higher than the one from nonaneuploidy CNV-pQTL (SI Appendix, Fig. S12). Only 4 local CNV-pQTL out of 1,009 were detected, and they displayed a significantly higher effect size than the distant CNV-pQTL (Fig. 5B).
We then looked at the extent to which the genetic bases of protein abundance are common with those underlying the abundance of transcripts. We performed GWAS using the transcriptomic dataset and detected 458 SNP-eQTL and 1,143 CNV-eQTL (Fig. 5D and SI Appendix, Fig. S10B and Datasets S20, S21, S24, and S25), which is of the same order of magnitude as the GWAS proteome results, with a significantly higher fraction (Fisher’s Exact Test, P-value = 0.006, odds ratio= 1.94) of local QTL: 10.5% of the total SNP-eQTL are local. Surprisingly, the overlap between the SNP-pQTL and the SNP-eQTL is very low, with only 3% of shared SNP-QTL (n = 15). Interestingly, 12 out of 15 were related to local regulation, meaning that 40% of the local SNP-pQTL (12 out of 30) also impact the cognate transcripts of their target protein. This observation is consistent with previous findings showing that the common regulation between mRNA and protein abundances is mainly related to local regulation (6, 40, 68). Overall, we observed that genes with a strong correlation between transcript and protein abundance tend to have a shared pQTL and eQTL (SI Appendix, Fig. S13). For instance, three out of the four most correlated genes previously mentioned had a shared pQTL and eQTL (SFA1, HBN1, and GLR1). Additionally, we found that the SNP-pQTL distribution across the genome did not match the SNP-eQTL distribution: Only one hotspot was shared across the expression level (Fig. 5C). The reasons for the weak overlap are likely multifactorial, but protein-specific regulation, such as protein degradation, may play a central role. We sought to confirm this by looking at the average protein turnover (45) of the proteins with and without overlapping pQTL and eQTL (SI Appendix, Fig. S14A, see Methods). We found that proteins, for which an overlap between pQTL and eQTL was detected, show a lower turnover rate compared to the other proteins. Consistently, the half-life of proteins with an overlapping SNP-QTL was higher than the rest of the proteome (SI Appendix, Fig. S14B). This observation suggests that protein degradation is probably involved in the large differences observed between the genetic origins of mRNA and protein abundance. However, it is important to emphasize that our results mainly focus on highly abundant proteins. Therefore, we were unable to map a significant portion of the genetic origin of the proteome variation, and we may have missed some overlapping regulations on low-abundance proteins.
In contrast, the overlap between the two sets of CNV-QTL is much higher, as 541 QTLs were shared between the transcriptome and proteome, i.e., approximately 53.6% of the CNV-pQTL. However, these shared CNV-QTLs are all aneuploidy-related CNVs, suggesting that the effect of aneuploidies is persistent through the expression layers (45). None of the nonaneuploidy CNV-QTL (8 CNV-eQTL and 9 CNV-pQTL) were shared. Together, our results highlight that the genetic bases underlying population-level protein abundance are very distinct from those underlying mRNA abundance.
Discussion
Quantifying transcripts and proteins expressed in a large natural population is fundamental for having a better understanding of the genotype–phenotype relationship. In this study, we have quantitatively analyzed the proteome of 942 natural isolates of S. cerevisiae, allowing in-depth exploration of protein abundance and precise characterization of the genetic origins of its variation at the species level.
The S. cerevisiae species is characterized by a complex population structure, with domesticated and wild subpopulations (43). Structured populations are also observed in a large number of other species, such as humans, and their impact on the proteome remains unexplored. In our dataset, the population structure only has a small impact on the proteomic landscape. However, due to the limited proteome coverage, we might miss some specific proteome or transcriptome structure associated with population structure in the less expressed or accessory genes (44). Yet, our observation is consistent with previous results obtained with the transcriptomes of S. cerevisiae isolates (44, 69). In fact, most subpopulations are characterized by specific signatures related to a small set of genes but not to a general pattern.
This dataset allowed us to have better insight into the architecture of the species-wide proteome variation. First, we found that the coexpression network captures main biological functions and is globally conserved across the expression layers. Second, we detected differential protein expression signatures specific to subpopulations, reflecting an adaptation to specific ecological conditions, such as domesticated environments. Similar expression signatures can be also observed using transcriptomic data (44, 70), highlighting that gene expression variation and modulation at both levels is a key mechanism of environmental adaptation.
The species-wide proteomes and transcriptomes obtained in the same condition represent a unique opportunity to compare the gene regulation at both levels. The overall correlation between protein and transcript abundance within each isolate (i.e., the across-gene correlation) appears to be high and this in the whole population, showing again that very abundant transcripts generally lead to very abundant proteins and vice versa (15–21, 24, 26, 27). However, our data allow us to have an accurate estimation of the correlation per gene at the population level and we found that this gene-wise correlation (i.e., the within-gene correlation) is very weak with a median of 0.165, which is lower than most previous estimates based on much smaller human and mice populations (6, 19, 33, 37, 40). Consistent with this result, genome-wide association studies also highlighted that SNPs related to variation in protein (pQTL) and transcript (eQTL) levels poorly overlap (3%), with mostly common local QTL. This high correspondence between local regulation confirms what has been observed in yeast (68): The local regulation of transcript and protein abundance is well conserved across the expression layers. Overall, our results are consistent with one of the first eQTL/pQTL comparisons (66) but unlike other studies, showing a higher overlap (68, 71). However, we should emphasize that we were not able to map the genetic basis of the entire S. cerevisiae proteome and therefore the eQTL/pQTL overlap might be biased and underestimated.
Mechanistically, our results suggest that the regulation of protein degradation has an impact on the variation of the proteome, and therefore on its genetic basis. This observation highlights that when proteins are more affected by specific proteome regulation (in this case, protein degradation), they will exhibit a lower match to the transcriptome. Conversely, proteins with a low turnover rate are more likely to be impacted by variation in transcript abundance. They will therefore likely reflect variation in mRNA abundance.
Although mass spectrometers are highly sensitive, it should be noted the limitation that proteomic methods are biased toward quantification of highly abundant proteins. Indeed, the fraction of the proteome quantified constitutes the vast majority of the total proteomic mass of a cell and is enriched for essential genes as well as in genes most connected in functional networks. Our dataset captures many of the fundamental processes. Yet, results related to low abundant proteins are missed by this approach.
Overall, our study clearly highlights that the dependency between transcript and protein levels is complex, pointing to the importance of post-transcriptional regulation of protein abundance. Proteome and transcriptome are indeed two distinct layers of gene regulation, which need to be further explored to understand the genotype–phenotype relationship. As gene function is ultimately executed by the proteome, while mRNA is the messenger, more proteomic approaches will be needed to create a better understanding of the phenotypic diversity. Our study provides a species-wide insight into the genetics that underlies both proteome and transcriptome diversity in a natural population.
Methods
Cultivation of Library for Proteomics.
The yeast isolate collection was grown on agar containing synthetic complete medium [SC; 6.7 g/L yeast nitrogen base (MP Biomedicals, Cat#114027512-CF), 20 g/L glucose, 2 g/L synthetic complete amino acid mixture (MP Biomedicals, Cat#114400022)]. After 48 h, colonies were inoculated in 200 µL SC liquid medium using a Singer Rotor and incubated at 30 °C overnight without shaking. These precultures were then mixed by pipetting up and down, and diluted 20× by transferring 80 µL per culture to deep-well plates prefilled with 1.55 mL SC liquid medium and one borosilicate glass bead per well. Plates were sealed with a permeable membrane and grown for 8 h at 1,000 rpm, 30 °C to exponential phase. The optical density (Dataset S1) at harvest was measured using an Infinite M Nano (Tecan). Per culture, 1.4 mL of cell suspension was harvested by transferring into a new deep-well plate and subsequent centrifugation (3,220 × g, 5 min, 4 °C). The supernatant was removed by inverting the plates. Cell pellets were immediately cooled on dry ice and stored at −80 °C.
For biological replicate measurements, seven strains (ADE, AEG, ANE, BHH, BPK, CFF, and CNT) were cultivated in quadruplicate as above, but using synthetic minimal medium [6.7 g/L yeast nitrogen base (MP Biomedicals, Cat#114027512-CF), 20 g/L glucose], and using an 11× dilution of the overnight preculture into 1.54 mL total volume of main culture. Samples were harvested as above after 9 h of incubation at 30 °C, 1,000 rpm.
Sample Preparation.
Samples for proteomics were prepared as previously described (42, 45, 47). In brief, samples were processed in 96-well format, with lysis being achieved by bead beating using a Spex Geno/Grinder and 200 µL of lysis buffer (100 mM ammonium bicarbonate, 7 M urea). Samples were reduced and alkylated using DTT (20 µL, 55 mM) and iodoacetamide (20 μL, 120 mM), respectively, diluted with 1 mL 100 mM ammonium bicarbonate, and 500 µL per sample were digested using 2 µg Trypsin/LysC (Promega, Cat#V5072). After 17 h of incubation at 37 °C, 25 µL 20% formic acid was added to the samples, and peptides were purified using solid-phase extraction as described previously (47). Eluted samples were vacuum-dried and subsequently dissolved in 70 µL 0.1% formic acid. An equivoluminal pool of all samples was generated to be used as quality controls (QCs) during MS measurements. The peptide concentration of this pool was determined using a fluorimetric peptide assay kit (Thermo Scientific, Cat#23290). Peptide concentrations per sample were estimated by multiplying the optical density recorded at harvest with the ratio between pool peptide concentration and the median at-harvest optical density.
Samples for biological replicate measurements (ADE, AEG, ANE, BHH, BPK, CFF, and CNT) were processed as above, but using two cycles of bead beating.
LC–MS/MS Measurements.
In brief, peptides were separated on a 3-min high-flow chromatographic gradient and recorded by mass spectrometry using Scanning SWATH (46) using an online coupled 1290 Infinity II LC system (Agilent)—6600+ TripleTOF platform (Sciex). 5 µg of sample were injected onto a reverse-phase HPLC column (Luna®Omega 1.6 µm C18 100A, 30 × 2.1 mm, Phenomenex) and resolved by gradient elution at a flow rate of 800 µL/min and column temperature of 30 °C. Both the order of injection of the sample plates and within the sample plates were randomized, with the pooled sample being injected regularly after 11 samples as a technical control (QC). All solvents were of LC–MS grade. The gradient program used 0.1% formic acid in water (Solvent A) and 0.1% formic acid in acetonitrile (Solvent B) and was as follows: 1% to 40% B in 3 min, increase to 80% B at 1.2 mL over 0.5 min, which was maintained for 0.2 min and followed by equilibration with starting conditions for 1 min. For mass spectrometry analysis, the scanning swath precursor isolation window was 10 m/z; the bin size was set to 1/5th of the window size, the cycle time was 0.7 s, the precursor range 400 m/z to 900 m/z, the fragment range 100 m/z to 1,500 m/z as previously described (46). An IonDrive TurboV source was used with ion source gas 1 (nebulizer gas), ion source gas 2 (heater gas), and curtain gas set to 50 psi, 40 psi, and 25 psi, respectively. The source temperature was set to 450 °C and the ion spray voltage to 5,500 V.
Tryptic digests of biological replicate samples (ADE, AEG, ANE, BHH, BPK, CFF, and CNT) were analyzed in technical quadruplicates by LC–MS/MS using a SCIEX ZenoTOF 7600 mass spectrometer, online coupled to a Waters ACQUITY UPLC M-Class System. Prior to MS analysis, 200 ng sample was chromatographically separated with a 30 min flow 5 µL/min gradient on a Waters HSS T3 column (300 µm × 150 mm, 1.8 µm) heated to 35 °C, where mobile phase A and B are 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. The gradient program included the following separation steps: 1% to 40% B in 19 min, increase for washing to 80% B over 1 min, which was maintained for 0.5 min and followed by equilibration with starting conditions for 6.5 min. For data-independent acquisition, a Zeno SWATH MS/MS acquisition scheme (72) with 85 variable size windows and 11 ms accumulation time was used. Ion source parameters were as follows: Ion source gas 1 and 2 were set as 12 and 60 psi respectively; curtain gas 25, CAD gas 7, and source temperature at 150 °C; Spray voltage was set at 4,500 V.
Data Processing.
The mass spectrometry files were processed following the approach previously described (45). Briefly, an experimental spectral library obtained using the S288c was filtered to reduce the search space to peptides well shared across the strains. This peptide library comprised 4,804 proteins from 47,125 peptides before filtration, and 4,172 proteins from 30,529 peptides after filtration. This library was then used with the software DIA-NN (48) (Version 1.8) and the following parameters: missed cleavages: 0, mass accuracy: 20, mass accuracy MS1: 12, scan windows: 6. The option “MBR” was used to process the data. As the peptides selected were not necessarily present ubiquitously in all the strains, an additional step was required to remove false positives (entries where a peptide is detected in a strain where it should be absent). This represents only ~1% of the total entries of the report.
Samples and entries with insufficient MS2 signal quality (< 1/3 of median MS2 signal) and with entries with Q.Value (>0.01), PG.Q.Value (>0.01), Global.Protein.Q.Value (>0.01), Global.PG.Q.Value (>0.01) were removed. A similar threshold was applied to Lib.PG.Q.Value and Lib.Q.Value to account for the MBR option used. Nonproteotypic precursors were also excluded. Outlier samples were detected based on the total ion chromatograms (TIC) and number of identified precursors per sample (Z-Score > 2.5) and were excluded from further analysis. Precursors were filtered according to their detection rate in the samples, with a threshold set at 80% of detection rate across all the strains, while precursors with a coefficient of variation (CV) above 0.3 in the QC samples were excluded. The CVs of QCs and wild isolates samples were calculated and had a median CV of 15.15% and 34.21%, respectively (SI Appendix, Fig. S2 and Dataset S26).
Overall, our initial quantification encompassed 1,048 proteins (from 5,993 peptides), after filtering peptides that were not detected in 80% of the samples at least, we kept 664 proteins from 2,994 peptides. The filtering of the precursors with a CV higher than 0.3 in the QC sample resulted in the final set of 630 proteins corresponding to 2,676 peptides. Among these 630 proteins, 283 were quantified from two peptides or less. Batch correction was carried out at the precursor level using median batch correction, which consists in bringing the median value of the precursors in the different batches to the same level. Proteins were then quantified from the peptide abundance using the maxLFQ (73) function implemented in the DIA-NN R package. The resulting dataset consists of 630 proteins for 942 strains. We imputed the missing value for further exploration using the KNN imputation method from the impute R package (74).
For biological replicate samples (ADE, AEG, ANE, BHH, BPK, CFF, and CNT), raw data were processed with DIA-NN (version 1.8.1) (48) using the default settings with fragment ion m/z range set from 100 to 1,800, mass accuracy set to 20 ppm at both MS level, scan window set to 7, MBR (match-between-runs) enabled, and quantification strategy set as “Robust LC (high Precision).” A spectral library-free approach using an S. cerevisiae UniProt fasta (UP000000231, downloaded on 27.03.2023) was used for annotation. A filter of 1% FDR on peptide level was set and only proteotypic peptides were considered in the analysis. Four technical repeat injections (one for one biological replicate of strain CNT, one for one biological replicate of strain BHH, and two for one biological replicate of strain BPK) had to be excluded from the variability analysis due to too few proteins being quantified (median number of proteins quantified per sample: 3,763 proteins; cut-off used for exclusion: 3,250 proteins).
Combination of Transcriptomic and Proteomic Data.
Unless specified, all the analysis performed below were conducted using R version 4.1.2. The transcriptomic data were generated previously (44). We used the log2 transcript per million (TPM) data, where the overlap with proteomic data was encompassing 629 genes across 889 isolates, for the genome-wide association studies (see later for the method). For the exploration of gene expression variation, subpopulation-related DEG and gene expression network, we used the variance stabilized data obtained directly from the log2 TPM data. In this case, one gene was removed from the analysis and the reference strain data was not considered, which resulted in an overlap of 628 genes across 888 isolates. To only focus on real expression variation difference between the expression layers, we normalized the proteomic and transcript abundance using quantile normalization. Unless specified, all the analyses described below use the quantile normalized transcriptomic and proteomic data. We recomputed the raw protein abundance coefficient of variation (CV) of each gene by dividing the SD by the mean (using the nonnormalized abundance) and transformed it to a percentage. Based on the CV, we performed a functional exploration by gene set enrichment analysis (GSEA) (75) using the fgsea R package (76) for the gene ontology annotation (77, 78) to detect cellular pathways with a conserved regulation across the population. The within- and across-gene mRNA–protein correlation was performed for each gene or each isolate using a Spearman correlation test. We selected the genes with a mRNA–protein correlation index higher than 0.42 (>95% percentile) and performed gene ontology (GO) enrichment analysis using the biological process (BP) database using the topGO R package (79). For the GO analysis looking at the functional enrichment present in the 630, the gene list reference was the genes encompassed in the transcriptomic data (44). The other GO analyses used the 628 genes as the reference list. All the other GO analyses were performed using the same procedure, unless specified.
Expression Variation Exploration.
We measured the strength of protein and transcript abundance variation using several methods. We computed an absolute transformed Log2(fold change) value (|Log2(FC)|) where in each isolate pairwise comparison (ex: strain A vs. strain B) and for each gene, we performed:
Briefly, the more this value increases, the more different is protein abundance between two isolates for a specific gene. We also computed a pairwise Spearman correlation between the isolates using the normalized proteomic and transcriptomic data. We also gathered the Euclidean distances between the expression profiles of each isolate, as well as the gene expression variance per gene.
We explored the post-transcriptional buffering phenomenon using an approach based on the computation of expression trees (57). First, on both protein and transcript normalized abundances, we constructed a neighbor-joining tree based on the Euclidean distance between each isolate. We computed the total branch length of these two trees and created a ratio of the proteome tree length on the transcriptome tree length. The ratio was equal to 0.93 which is in line with the difference in Euclidean distance between the transcriptome and proteome. We performed 100 bootstrapping tests and used the resulting branch lengths to test the difference between the proteome and the transcriptome tree. We sought to check whether some cellular pathways tended to be more affected by the post-transcriptional buffering phenomenon. To do so, we gathered a reduced biological process GO annotation by computing the similarity between each GO term using the rrvgo R package and the “Resnik” method (80). We discarded terms that are at least 50% overlapping with another term and the terms encompassing no more than five genes, which resulted in a list of 101 terms. For each of these terms, we performed the same tree exploration, but this time with the genes encompassed by each term. We obtained therefore 101 tree length ratios. We selected the terms displaying a ratio lower than 0.93 or higher than 1, and for which the total branch length between the proteome and the transcriptome was significantly different after 10 bootstrapping steps (Bonferroni corrected Wilcoxon test P-value < 0.001).
Transcriptome and Proteome Landscape Exploration.
We sought to check whether the genetic structure of the population had an impact on the transcriptome and proteome structure. We obtained the genetic distances from ref. 43 between pairs of isolates and compared them to the pairwise isolate correlation (Spearman correlation test) obtained with the normalized transcript or protein abundances. We also used both normalized protein and mRNA abundance data to perform PCA using the prcomp function from the stats R package. For the 2 PCA (transcriptomic and proteomic), we plotted the six first principal components (PC) together (PC1-PC2, PC3-PC4, and PC5-PC6) and looked for eventual grouping according to the subpopulation as defined previously (43). We then computed a WGCNA using the WGCNA R package (81) to detect coexpression module in both mRNA and peptide normalized abundance. To do so, we generated a Topological Overlap Matrix (TOM) using the blockwiseModules function. The TOM were calculated based on a signed adjacency matrix with the power of 9 for the mRNA abundance data and 5 for the peptide abundance data. The blockwiseModules automatically detected the coexpression modules by generating a clustering from a dissimilarity matrix (1-TOM) using the following option: detectCutHeight = 0.995; minModuleSize = 30. This resulted in the detection of 5 and 7 transcriptome and proteome modules respectively. We computed an overrepresentation analysis for each coexpression module with the GO terms as annotation and using the mod_ora function from the CEMiTool R package (82) and used the most representative GO terms as the final annotation for each detected module. The two coexpression networks were generated for plotting by computing an adjacency matrix from the TOM (generated previously) and ultimately plotted using the ggnet2 function from the GGally R package.
Transcriptome and Proteome Differentially Expressed Gene Detection.
We used the normalized protein abundance to detect subpopulation-specific (43) differentially expressed proteins (DEPs). The goal was to detect either over- or underexpressed genes by comparing the normalized expression of all the isolates from a subpopulation against the rest of the population using a Wilcoxon test for each gene. The P-value of the test was corrected using a Bonferroni correction with the p.adjust function in R. A gene was considered as differentially expressed if the corrected P-value of the Wilcoxon test was below 0.05. We computed as well a log2 transformed fold change [log2(FC)] value for each gene in each subpopulation using the mean expression of the subpopulation divided by the mean expression of the rest of the population. To further characterize the detected DEPs, we performed a functional exploration using GSEA (with the fgsea function from the fgsea R package) using the log2(FC) value from the DEP exploration as score rankings. In order to have a global view of the pathways that were significantly differentially expressed in each subpopulation, we used the 16 coexpression modules detected and defined previously using the population transcriptome data (44) as biological function annotations for the GSEA. We performed the same procedure but this time comparing the domesticated against the wild isolate (using the clade-wise annotation from ref. 43). This time, the test was performed on both normalized protein and transcript abundances.
Proteome and Transcriptome Genome-Wide Association Studies.
We computed GWAS with a linear mixed model-based method as described previously (43, 44, 83) using FaST-LMM (84). In order to strictly compare the genetic effect on transcript and protein abundance and avoid confounding factors related to different culture phase, we selected a set of 455 out of the 889 isolates for which the harvest OD was correlated (Pearson coefficient > 0.6) across our transcriptomic and proteomic experiments (SI Appendix, Fig. S15) and for which we had both proteomic and transcriptomic measurements. We performed the GWAS using either the transcriptome log2 transformed TPM data or the protein abundance. For each dataset, we performed two separated GWAS, one based on SNP as genotype, and one based on the CNV as genotype. The SNP GWAS was run with total of 101,836 SNP displaying a minor allele frequency (MAF) > 5%. The CNV GWAS was run on a total of 631 CNV (MAF > 5%). We used the SNP matrix for both SNP and CNV GWAS, thus evaluating the kinship between the isolate to account for the population structure. We set a phenotype-specific P-value threshold using 100 permutation tests where the phenotypes were randomly permutated between the isolates. We use the 5% lowest P-value quantile from these permutation tests to define the significance threshold. We finally scaled the significance thresholds of the CNV GWAS to account for the size difference between the SNP and CNV matrices.
Regarding the SNP GWAS, the detected QTL were filtered to avoid false positives detection due to linkage disequilibrium among the SNP as described previously (44). This resulted in the filtration of 78 eQTL and 98 pQTL (out of respectively 536 and 626 QTL). The QTL were considered as “local” QTL when they were located 25 kb around their affected phenotype. We also sought to detect QTL hotspots in both transcriptome and proteome GWAS. We defined a hotspot as a concentration of at least 4 QTL in a 20 kb window.
We compared the protein turnover rate (obtained from ref. 45) of 619 proteins encompassed in our dataset to see whether turnover rate had an impact on the overlap between SNP-eQTL and SNP-pQTL. These data comprise protein degradation rates for 1,836 gene across 55 natural isolates. We computed an average turnover rate per gene and used this value to compare the level of protein degradation of the protein with or without an overlapping QTL.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Dataset S14 (XLSX)
Dataset S15 (XLSX)
Dataset S16 (XLSX)
Dataset S17 (XLSX)
Dataset S18 (XLSX)
Dataset S19 (XLSX)
Dataset S20 (XLSX)
Dataset S21 (XLSX)
Dataset S22 (XLSX)
Dataset S23 (XLSX)
Dataset S24 (XLSX)
Dataset S25 (XLSX)
Dataset S26 (XLSX)
Acknowledgments
We thank Joshua Bloom for insightful discussions and comments on the manuscript. This work was supported by a NIH grant R01 (GM147040-01) and a European Research Council (ERC) Consolidator grant (ERC-CoG 772505) to J.S. This work is also part of Interdisciplinary Thematic Institutes (ITI) Integrative Molecular and Cellular Biology (IMCBio), as part of the ITI 2021-to-2028 program of the University of Strasbourg, CNRS, and Inserm, supported by IdEx Unistra (ANR-10-IDEX-0002). E.M.T. was supported by the PhD Joint Programme CNRS and Weizmann Institute and a fellowship from the medical association la Fondation pour la Recherche Médicale (FDT202204014796). This work was also supported by a European Research Council (ERC) Synergy Grant (ERC-SyG-2020 951475) and a Wellcome Trust Grant (IA 200829/Z/16/Z) to M.R. J.S. is a Fellow of the University of Strasbourg Institute for Advanced Study (USIAS) and a member of the Institut Universitaire de France. P.T. is supported by funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no 101026830.
Author contributions
M.R. and J.S. designed research; E.M.T., J.M., D.L., F.A., M.M., and J.S. performed research; M.R. and J.S. contributed new reagents/analytic tools; E.M.T., P.T., J.M., V.L., A.F., J.H., and J.S. analyzed data; and E.M.T., P.T., J.M., and J.S. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Markus Ralser, Email: ralser359@gmail.com.
Joseph Schacherer, Email: schacherer@unistra.fr.
Data, Materials, and Software Availability
Raw data are available in the PRIDE database (https://www.ebi.ac.uk/pride/) under the project accession name PXD044523 (85). The codes and data are now available on zenodo—https://zenodo.org/records/10567083 (86). In addition, we linked this repository to our laboratory github—https://github.com/HaploTeam/eQTL_pQTL (87).
Supporting Information
References
- 1.Albert F. W., Kruglyak L., The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16, 197–212 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Maurano M. T., et al. , Systematic localization of common disease-associated variation in regulatory DNA. Science 337, 1190–1195 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cookson W., Liang L., Abecasis G., Moffatt M., Lathrop M., Mapping complex disease traits with global gene expression. Nat. Rev. Genet. 10, 184–194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Messner C. B., et al. , Mass spectrometry-based high-throughput proteomics and its role in biomedical studies and systems biology. Proteomics 23, e2200013 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Moyerbrailean G. A., et al. , A high-throughput RNA-seq approach to profile transcriptional responses. Sci. Rep. 5, 14976 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chick J. M., et al. , Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferkingstad E., et al. , Large-scale integration of the plasma proteome with genetics and disease. Nat. Genet. 53, 1712–1721 (2021). [DOI] [PubMed] [Google Scholar]
- 8.Folkersen L., et al. , Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat. Metab. 2, 1135–1148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.The GTEx Consortium, The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.The GTEx Consortium, Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.The GTEx Consortium, Human genomics. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buccitelli C., Selbach M., mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 21, 630–644 (2020), 10.1038/s41576-020-0258-4. [DOI] [PubMed] [Google Scholar]
- 13.Fortelny N., Overall C. M., Pavlidis P., Freue G. V. C., Can we predict protein from mRNA levels? Nature 547, E19–E20 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Liu Y., Beyer A., Aebersold R., On the dependency of cellular protein levels on mRNA abundance. Cell 165, 535–550 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Battle A., et al. , Impact of regulatory variation from RNA to protein. Science 347, 664–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edfors F., et al. , Gene-specific correlation of RNA and protein levels in human cells and tissues. Mol. Syst. Biol. 12, 883 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gautier E.-F., et al. , Comprehensive proteomic analysis of human erythropoiesis. Cell Rep. 16, 1470–1484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salovska B., et al. , Isoform-resolved correlation analysis between mRNA abundance regulation and protein level degradation. Mol. Syst. Biol. 16, e9170 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D., et al. , A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilhelm M., et al. , Mass-spectrometry-based draft of the human proteome. Nature 509, 582–587 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Zhang B., et al. , Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aydin S., et al. , Genetic dissection of the pluripotent proteome through multi-omics data integration. Cell Genom. 3, 100283 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li J. J., Bickel P. J., Biggin M. D., System wide analyses have underestimated protein abundances and the importance of transcription in mammals. PeerJ 2, e270 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moritz C. P., Mühlhaus T., Tenzer S., Schulenborg T., Friauf E., Poor transcript-protein correlation in the brain: Negatively correlating gene products reveal neuronal polarity as a potential cause. J. Neurochem. 149, 582–604 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Schwanhäusser B., et al. , Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Becker K., et al. , Quantifying post-transcriptional regulation in the development of Drosophila melanogaster. Nat. Commun. 9, 4970 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ponnala L., Wang Y., Sun Q., van Wijk K. J., Correlation of mRNA and protein abundance in the developing maize leaf. Plant J. Cell Mol. Biol. 78, 424–440 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Gygi S. P., Rochon Y., Franza B. R., Aebersold R., Correlation between protein and mRNA abundance in yeast. Mol. Cell. Biol. 19, 1720–1730 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ingolia N. T., Ghaemmaghami S., Newman J. R. S., Weissman J. S., Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marguerat S., et al. , Quantitative analysis of fission yeast transcriptomes and proteomes in proliferating and quiescent cells. Cell 151, 671–683 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Archer T. C., et al. , Proteomics, post-translational modifications, and integrative analyses reveal molecular heterogeneity within medulloblastoma subgroups. Cancer Cell 34, 396–410.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang K.-L., et al. , Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat. Commun. 8, 14864 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang L., et al. , A quantitative proteome map of the human body. Cell 183, 269–283.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mertins P., et al. , Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mirauta B. A., et al. , Population-scale proteome variation in human induced pluripotent stem cells. eLife 9, e57390 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mun D.-G., et al. , Proteogenomic characterization of human early-onset gastric cancer. Cancer Cell 35, 111–124.e10 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Upadhya S. R., Ryan C. J., Experimental reproducibility limits the correlation between mRNA and protein abundances in tumor proteomic profiles. Cell Rep. Methods 2, 100288 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasaikar S., et al. , Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities. Cell 177, 1035–1049.e19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang H., et al. , Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghazalpour A., et al. , Comparative analysis of proteome and transcriptome variation in mouse. PLoS Genet. 7, e1001393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alam M. T., et al. , The metabolic background is a global player in Saccharomyces gene expression epistasis. Nat. Microbiol. 1, 1–10 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Messner C. B., et al. , The proteomic landscape of genome-wide genetic perturbations. Cell 186, 2018–2034.e21 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Peter J., et al. , Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556, 339–344 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caudal E., et al. , Pan-transcriptome reveals a large accessory genome contribution to gene expression variation in yeast. bioRxiv [Preprint] (2023). 10.1101/2023.05.17.541122. Accessed 1 June 2023. [DOI] [PMC free article] [PubMed]
- 45.Muenzner J., et al. , The natural diversity of the yeast proteome reveals chromosome-wide dosage compensation in aneuploids. bioRxiv [Preprint] (2022). 10.1101/2022.04.06.487392. Accessed 1 May 2023. [DOI]
- 46.Messner C. B., et al. , Ultra-fast proteomics with scanning SWATH. Nat. Biotechnol. 39, 846–854 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Messner C. B., et al. , Ultra-high-throughput clinical proteomics reveals classifiers of COVID-19 infection. Cell Syst. 11, 11–24.e4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Demichev V., Messner C. B., Vernardis S. I., Lilley K. S., Ralser M., DIA-NN: Neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho B., Baryshnikova A., Brown G. W., Unification of protein abundance datasets yields a quantitative Saccharomyces cerevisiae proteome. Cell Syst. 6, 192–205.e3 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Dowell R. D., et al. , Genotype to phenotype: A complex problem. Science 328, 469–469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giaever G., et al. , Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Pu S., Wong J., Turner B., Cho E., Wodak S. J., Up-to-date catalogues of yeast protein complexes. Nucleic Acids Res. 37, 825–831 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Caudal E., et al. , Loss-of-function mutation survey revealed that genes with background-dependent fitness are rare and functionally related in yeast. Proc. Natl. Acad. Sci. U.S.A. 119, e2204206119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blevins W. R., et al. , Extensive post-transcriptional buffering of gene expression in the response to severe oxidative stress in baker’s yeast. Sci. Rep. 9, 11005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kustatscher G., Grabowski P., Rappsilber J., Pervasive coexpression of spatially proximal genes is buffered at the protein level. Mol. Syst. Biol. 13, 937 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McManus C. J., May G. E., Spealman P., Shteyman A., Ribosome profiling reveals post-transcriptional buffering of divergent gene expression in yeast. Genome Res. 24, 422–430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Z.-Y., et al. , Transcriptome and translatome co-evolution in mammals. Nature 588, 642–647 (2020), 10.1038/s41586-020-2899-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teyssonnière E. M., et al. , Translation variation across genetic backgrounds reveals a post-transcriptional buffering signature in yeast. Nucleic Acids Res. 52, 2434–2445 (2024), 10.1093/nar/gkae030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Donnell S., et al. , Telomere-to-telomere assemblies of 142 strains characterize the genome structural landscape in Saccharomyces cerevisiae. Nat. Genet. 55, 1390–1399 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saada O. A., et al. , Phased polyploid genomes provide deeper insight into the multiple origins of domesticated Saccharomyces cerevisiae beer yeasts. Curr. Biol. 32, 1350–1361.e3 (2022). [DOI] [PubMed] [Google Scholar]
- 61.Schacherer J., Shapiro J. A., Ruderfer D. M., Kruglyak L., Comprehensive polymorphism survey elucidates population structure of Saccharomyces cerevisiae. Nature 458, 342–345 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang B., Horvath S., A general framework for weighted gene co-expression network analysis. Stat. Appl. Genet. Mol. Biol. 4, Article17, 2–40 (2005). [DOI] [PubMed] [Google Scholar]
- 63.Celińska E., Nicaud J.-M., Filamentous fungi-like secretory pathway strayed in a yeast system: Peculiarities of Yarrowia lipolytica secretory pathway underlying its extraordinary performance. Appl. Microbiol. Biotechnol. 103, 39–52 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lahue C., Madden A., Dunn R., Smukowski Heil C., History and domestication of Saccharomyces cerevisiae in bread baking. Front. Genet. 11, 584718 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Auesukaree C., Molecular mechanisms of the yeast adaptive response and tolerance to stresses encountered during ethanol fermentation. J. Biosci. Bioeng. 124, 133–142 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Foss E. J., et al. , Genetic basis of proteome variation in yeast. Nat. Genet. 39, 1369–1375 (2007). [DOI] [PubMed] [Google Scholar]
- 67.Albert F. W., Bloom J. S., Siegel J., Day L., Kruglyak L., Genetics of trans-regulatory variation in gene expression. eLife 7, e35471 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Grossbach J., et al. , The impact of genomic variation on protein phosphorylation states and regulatory networks. Mol. Syst. Biol. 18, e10712 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kita R., Venkataram S., Zhou Y., h.Fraser B., High-resolution mapping of cis-regulatory variation in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 114, E10736–E10744 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hodgins-Davis A., Adomas A. B., Warringer J., Townsend J. P., Abundant gene-by-environment interactions in gene expression reaction norms to copper within Saccharomyces cerevisiae. Genome Biol. Evol. 4, 1061–1079 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Albert F. W., Treusch S., Shockley A. h., Bloom J. S., Kruglyak L., Genetics of single-cell protein abundance variation in large yeast populations. Nature 506, 494–497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Z., et al. , High-throughput proteomics of nanogram-scale samples with Zeno SWATH MS. eLife 11, e83947 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cox J., et al. , Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ*. Mol. Cell. Proteomics 13, 2513–2526 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hastie T., Tibshirani R., Narasimhan B., Chu G., impute: impute: Imputation for microarray data. (2023) 10.18129/B9.bioc.impute, R package version 1.76.0, https://bioconductor.org/packages/impute. [DOI]
- 75.Subramanian A., et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Korotkevich G., et al. , Fast gene set enrichment analysis. bioRxiv [Preprint] (2021). 10.1101/060012. Accessed 15 June 2023. [DOI]
- 77.Ashburner M., et al. , Gene ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gene Ontology Consortium, The gene ontology resource: Enriching a GOld mine. Nucleic Acids Res. 49, D325–D334 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Alexa A., Rahnenfuhrer J., topGO: Enrichment Analysis for Gene Ontology. (2023) 10.18129/B9.bioc.topGO, R package version 2.54.0, https://bioconductor.org/packages/topGO. [DOI]
- 80.Sayols S., rrvgo: a Bioconductor package for interpreting lists of Gene Ontology terms. MicroPublication Biol. (2023). 10.17912/micropub.biology.000811. [DOI] [PMC free article] [PubMed]
- 81.Langfelder P., Horvath S., WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Russo P. S. T., et al. , CEMiTool: A Bioconductor package for performing comprehensive modular co-expression analyses. BMC Bioinform. 19, 56 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tsouris A., Brach G., Friedrich A., Hou J., Schacherer J., Diallel panel reveals a significant impact of low-frequency genetic variants on gene expression variation in yeast. Mol. Syst. Biol. 20, 362–373 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lippert C., et al. , FaST linear mixed models for genome-wide association studies. Nat. Methods 8, 833–835 (2011). [DOI] [PubMed] [Google Scholar]
- 85.Teyssonniere E. M., et al. , Raw data for Species-wide quantitative transcriptomes and proteomes reveal distinct genetic control of gene expression variation in yeast. PRIDE. http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD044523. Deposited 12 August 2023. [DOI] [PMC free article] [PubMed]
- 86.Teyssonniere E. M., et al. , Data and code for Species-wide quantitative transcriptomes and proteomes reveal distinct genetic control of gene expression variation in yeast. Zenodo. 10.5281/zenodo.10567083. Deposited 24 January 2024. [DOI] [PMC free article] [PubMed]
- 87.Teyssonniere E. M., et al. , Data and code for Species-wide quantitative transcriptomes and proteomes reveal distinct genetic control of gene expression variation in yeast. GitHub. https://github.com/HaploTeam/eQTL_pQTL. Deposited 25 January 2024. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Dataset S09 (XLSX)
Dataset S10 (XLSX)
Dataset S11 (XLSX)
Dataset S12 (XLSX)
Dataset S13 (XLSX)
Dataset S14 (XLSX)
Dataset S15 (XLSX)
Dataset S16 (XLSX)
Dataset S17 (XLSX)
Dataset S18 (XLSX)
Dataset S19 (XLSX)
Dataset S20 (XLSX)
Dataset S21 (XLSX)
Dataset S22 (XLSX)
Dataset S23 (XLSX)
Dataset S24 (XLSX)
Dataset S25 (XLSX)
Dataset S26 (XLSX)
Data Availability Statement
Raw data are available in the PRIDE database (https://www.ebi.ac.uk/pride/) under the project accession name PXD044523 (85). The codes and data are now available on zenodo—https://zenodo.org/records/10567083 (86). In addition, we linked this repository to our laboratory github—https://github.com/HaploTeam/eQTL_pQTL (87).