

Abstract
The dynamic state of infection of 11 ducks with the duck hepatitis B virus was investigated. Chronic infections were established in newly hatched ducklings by inoculation with a mixture of wild-type virus and a mutant virus with a partial replication defect. As expected, the wild-type virus was rapidly enriched in the virus population during the spread of infection. Enrichment thereafter was correlated with normal growth of the liver, with the average mutant-to-wild-type ratio stabilizing for at least 2 months beyond the time at which the liver mass stabilized. Using experimentally determined growth rates for the mutant and wild-type viruses, we estimated that after the spread of infection, competition between the two virus strains was limited by the amount of replication required to infect new hepatocytes in the growing livers. The results suggest that, in a chronically infected liver, the selection of variants with a replication rate advantage is inefficient and that the emergence of such variants would depend on induced liver cell turnover, such as that occurring during chronic hepatitis.
Hepadnaviruses cause chronic infections of the liver in a variety of animal species (for reviews, see references 5 and 17). In the absence of inflammation, infection becomes stably established in every susceptible hepatocyte, persisting without causing any apparent cytopathic effect. The noncytopathic state of infection is possible because virus replication is regulated within the infected cell at a level that does not interfere with normal cellular functions. The major mechanism for this regulation is thought to act through copy number control of viral genes in the nucleus (25, 26).
Active viral genes are found as a pool of up to 20 double-stranded covalently closed circular DNA (cccDNA) molecules (14, 20, 26). The pool of cccDNA molecules is established early in the infection. The infecting viral DNA molecule is first converted directly to cccDNA and is transcribed in the nucleus to produce viral mRNAs and proteins (1, 4, 24). The cccDNA is then replicated through transcription of RNA pregenomes, transport of pregenomes to the cytoplasm, and reverse transcription within newly formed viral nucleocapsids to produce double-stranded circular DNA with an open, relaxed conformation (rcDNA) (14, 21, 23). New cccDNA molecules are then formed by the transport of the rcDNA molecules into the nucleus and their conversion to cccDNA (26, 27). By the time that an average of 10 to 20 cccDNA molecules have been formed, sufficient levels of viral envelope proteins, particularly the large envelope protein, preS, have accumulated to direct all rcDNA-containing nucleocapsids into the pathway for enveloped virus assembly and secretion. This process effectively prevents further production of cccDNA as long as sufficient intracellular concentrations of preS protein are maintained in the cell to direct nucleocapsids into the virus assembly pathway (8, 9, 24, 25). The question of the stability of the cccDNA molecules formed during this process is still unresolved (3, 12, 15).
Little is known about the dynamic state of chronic hepadnavirus infections. A high dynamic state would be one in which viral cccDNA molecules are continually being replaced in the liver because of metabolic instability or cell turnover. Conversely, a low dynamic state would be characterized by a high degree of cccDNA stability and a long hepatocellular lifetime, requiring little cccDNA replacement. Because maintenance of the cccDNA pool in each infected cell is necessary for the persistent state of the infection, the dynamics of the infection determines the sensitivity of the persistently infected state of the cell to inhibition of viral DNA synthesis. Inhibition of viral DNA synthesis has been the major strategy for antiviral therapy of chronic hepatitis B infection in humans.
In this report, we describe experiments that elucidate certain aspects of the dynamic state of chronic infection by the duck hepatitis B virus, DHBV. We measured the rate of replacement of a mutant strain of DHBV bearing a partial replication defect with the wild-type DHBV during three phases of infection: (i) the initial spread of infection, (ii) growth of the fully infected liver, and (iii) the fully infected adult liver. We showed that replacement was rapid during the spread of infection but greatly reduced during growth of the liver and initially absent in the adult liver, indicating a low dynamic state of viral competition in the adult liver. Using experimentally determined values for the relative rates of replication of the two viruses, we estimated that the enrichment of wild-type virus could be accounted for by new rounds of replication occurring exclusively in the newly synthesized mass of the growing liver.
MATERIALS AND METHODS
Animals.
One-day-old white Pekin ducklings were obtained from Metzer Farms (Redlands, Calif.). Ducklings testing negative for DHBV infection by dot hybridization were infected by intravenous injection of 0.1 ml of a suspension of virus. Infected birds were housed together, as previously described (28). Ducks were euthanized for necropsy by injection of a sodium pentobarbital solution (200 mg/kg of body weight).
Preparation of virus inocula.
DHBV-16 wild-type virus was obtained from the culture medium of the chicken hepatoma cell line, LMH (7), stably transformed with a plasmid expressing DHBV-16 (16). The DHBV-3 mutant, DR1-13, was obtained from the culture medium of LMH cells transfected with a DR1-13 plasmid expression vector (22). The medium was harvested at day 3 to 8 posttransfection and concentrated 50-fold from the culture fluids by precipitation with 10% polyethylene glycol (PEG 8000), as previously described (24). The virus titer in the concentrated stocks was assayed by selective extraction of DNA from enveloped particles (9) and Southern blot hybridization and compared with standard plasmid DNAs on the same blot. Virus titers were expressed as DHBV genomes. Inocula for injection were prepared by appropriate dilution of the virus stocks in Leibowitz's (L15) medium (Gibco BRL). Procedures used in the analysis of viral DNA replicative intermediates, agarose gel electrophoresis, and blot hybridization were previously published (8).
Analysis of viral DNA in the serum.
The level of viremia was determined by quantitation of viral DNA in the serum by dot hybridization and phosphorimage analysis. Serum (2 μl) was applied directly to nylon membranes, denatured by brief treatment with 0.2 N NaOH, and neutralized with 0.2 M Tris-HCl. DNA was detected on the filter by hybridization with a 32P-labeled riboprobe specific for the minus strand. For analysis of the viral genotype, DNA was extracted directly from 50 μl of serum by digestion with pronase, phenol extraction, and ethanol precipitation (28). The viral DNA was dissolved in 15 μl of Tris-EDTA (TE) (10 mM Tris-HCl [pH = 7.5], 1 mM EDTA), and 5 μl of the sample was then used in a 50-μl PCR.
PCR and sequencing.
Amplification of the serum viral DNA was carried out with a primer set corresponding to nucleotides 2492 to 2516 (biotinylated plus strand), and 2840 to 2818 (minus strand), according the numbering of Mandart et al (13). The standard PCR buffer contained DNA template; 200 μM (each) dATP, dGTP, dCTP, and TTP; 50 mM KCl; 10 mM Tris-HCl, pH 8.3; 1.5 mM MgCl2; 0.02% gelatin; and 38 pmol of each primer in a final volume of 50 μl, with 2.5 units of Taq DNA polymerase (Promega). Amplification was carried out for 35 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 45 s. The biotinylated PCR products (40 μl total) were adsorbed with 20 μl of streptavidin-coated M-280 Dynabeads (DYNAL Corp.) suspended in a solution of 20 mM Tris-HCl [pH 8.0], 2 M NaCl, and 1 mM EDTA and washed two times with 50 μl of TE with the help of a magnetic particle concentrator (DYNAL catalogue no. 120.04). The nonbiotinylated strand was released from the beads by denaturation in 0.1 N NaOH (50 μl), the denaturing solution was removed, and the beads were washed two times with 50 μl of TE. Washed beads with specifically bound biotinylated plus-strand products were used directly in sequencing reactions using a minus-strand primer (nucleotides 2747 to 2729).
Determination of the ratio of wild-type to DR1-13 DNA.
The ratio of wild-type to DR1-13 DNA in the serum samples was determined by the ratio of PCR products derived from the respective templates, using the sequence difference at nucleotide 2547 (C in the plus strand of the wild type and G in DR1-13). This nucleotide difference is responsible for the mutant phenotype of DR1-13 virus (22). Thus, in the sequencing ladder consisting of minus strands, DR1-13 shows a C at position 2547 where the wild-type nucleotide is G. Because the wild-type sequence is compressed in the G lane in this region, we used the intensity of the 2547C band as a measurement of the fraction of DR1-13 sequence in the sample. The intensity of the 2547C band was normalized to the combined intensity of the neighboring upstream C bands at positions 2552 and 2551, since these two positions (as well as all upstream positions) were the same in both viruses. The ratio of 2547C to the sum of 2552C and 2551C was determined for known mixtures of wild-type and DR1-13 cloned DNAs and shown to be linearly proportional to the fraction of DR1-13 DNA in the amplification template (data not shown). Therefore, we used this ratio for each serum virus sample to calculate the fraction of DR1-13 in the sample. The fraction of the wild type (DHBV-16) was taken to be 1 minus the fraction of DR1-13.
Primary hepatocyte cultures.
Primary duck hepatocyte (PDH) cultures were prepared from ducklings approximately 1 week of age by collagenase perfusion of the liver in situ, as previously described (17). Nearly confluent cell layers in 60-mm standard tissue culture dishes were exposed to virus for 24 h beginning 1 day after plating, and the medium was changed daily. Cultures were harvested for DNA extraction at 8 or 9 days postinfection. The cell layers were washed once with a buffered saline solution containing 0.5 mM EDTA and stored at −80°C.
Extraction of cccDNA from PDHs.
Cell layers were lysed by the addition of 0.4 ml of TE containing 0.2% (wt/vol) Nonidet P-40 (NP-40), and cell debris and nuclei were released from the plate by scraping with a rubber policeman. The debris and nuclei were pelleted by microcentrifugation for 1 min, and the pellet was resuspended in 0.2 ml of TE containing 0.2% NP-40. The nuclear suspension was lysed by the addition of 0.2 ml of a solution containing 0.15 N NaOH and 6% sodium dodecyl sulfate. The lysed nuclei were incubated at 37°C for 15 min to allow the cellular DNA to be irreversibly denatured. These conditions, however, did not result in irreversible denaturation of DHBV cccDNA. The alkaline solution was acidified with the addition of 0.1 ml of 3 M acetic acid adjusted to pH 5.0 with KOH, and the potassium-dodecyl sulfate-protein complex, which contained most of the denatured cellular DNA and protein-bound viral DNA, was removed by microfuge centrifugation for 1 min. The supernatant was extracted once with phenol to remove any remaining single-stranded or protein-bound DNA, and the cccDNA fraction was recovered by ethanol precipitation.
Extraction of DNA from enveloped virus particles in culture fluids of PDHs.
Culture fluids from PDHs were clarified of cell debris by low-speed centrifugation. The clarified supernatants were adjusted to 10% vol/vol fetal bovine serum, sodium chloride (1.5 g/45 ml) and PEG 8000 (5 g/45 ml) were added, and the virus was allowed to precipitate overnight at 4°C with gentle stirring. The PEG 8000-precipitable material was recovered by centrifugation at 2,000 × g for 20 min and dissolved in 1 ml of HEPES-buffered saline, 2 mM HEPES [pH 7.45], 0.15 M NaCl) containing 2 mM CaCl2. A portion (0.4 ml) was adjusted to 75 mM with Tris-HCl (pH 8.0), added to pronase (0.5 mg/ml), and incubated at 37°C for 1 h to degrade soluble proteins and nonenveloped viral cores. Magnesium acetate was added to a final concentration of 6 mM, DNase I (type II; Sigma) was added to a concentration of 100 μg/ml, and the samples were digested for 30 min. EDTA (10 mM final concentration) and sodium dodecyl sulfate (0.5% final concentration) were added, and the samples were digested for an additional 30 min. Viral DNA was recovered by phenol extraction and ethanol precipitation. Viral DNA was never recovered from control samples treated with NP-40 to dissociate enveloped virus prior to the addition of pronase (data not shown).
Mathematical treatments. (i) Wild-type enrichment and replication space.
In hepadnavirus infections, the vast majority of viral DNA that is synthesized in the liver is secreted from the hepatocyte and eliminated from the blood and therefore does not participate in further replication. Only a small fraction that is converted to cccDNA for the initiation of new rounds of infection or for maintenance of the cccDNA pool is actually expressed in the liver. Therefore, the genotype of all the virus produced by the infected liver is determined entirely by the cccDNA pool. When two virus strains differing only in their replication rates compete in the liver, the enrichment of one strain over the other in the blood can occur only by changes in the pool of cccDNA, brought about by new cccDNA synthesis. Thus, the enrichment in the blood is a function of the relative rate at which one virus synthesizes new cccDNA molecules from a parental cccDNA (the growth rate), and the number of cycles, or generations, of cccDNA synthesis that has occurred. Each generation of cccDNA synthesis contributes a fixed enrichment of one virus over the other as well as an expansion of the total population of cccDNA molecules.
In these experiments, we calculated the theoretical expansion of a cccDNA population consisting of a mixture of wild-type virus and the slower replicating mutant, DR1-13, from the enrichment of the wild-type strain over the mutant in the blood that we observed. When the growth of both strains follows first-order kinetics, the expansion of each cccDNA population is described by the following expression:
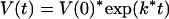 |
1 |
where V(0) is the amount of cccDNA at time zero, V(t) is the amount after time t, and k is the first-order growth-rate constant. The ratio of the wild type (VWT) to DR1-13 (VDR1-13) at time t, divided by their ratio at time zero, here defined as the enrichment (E), is
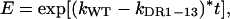 |
2 |
where kWT and kDR1-13 are the first-order growth-rate constants for the wild-type virus and for DR1-13, respectively. This equation can be solved for t, the time required for a particular enrichment, E, to occur.
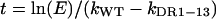 |
3 |
The total expansion of wild-type and DR1-13 cccDNA that is required for enrichment E is given by combining equation 1 for VWT + VDR1-13 and equation 3:
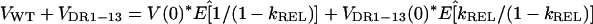 |
4 |
where the relative growth rate, kREL, is defined as kDR1-13/kWT. The relative expansion, S, of the total cccDNA population is
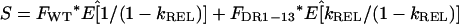 |
5 |
where FWT and FDR1-13 are the fractions of wild-type and DR1-13, respectively, at t = 0. We define S as the fractional increase in “replication space” (see Discussion).
(ii) Averaging.
As seen in equation 3, ln(E) is linearly proportional to the number of generations of growth (growth rate × time) in a mixed population of viruses. In describing the average behavior in the group of birds at each time point, we averaged the log E values to produce the logarithm of a value defined as the geometric mean. The geometric mean represents the enrichment due to the average number of generations of virus growth among various birds.
RESULTS
In previous studies, we have explored how strains of the avian hepadnavirus, DHBV, compete with each other during chronic infection of the liver. We showed that a cytopathic mutant of DHBV is at a severe disadvantage during chronic infection in competition with the noncytopathic wild-type virus (10, 11), and that under certain circumstances, a precore-minus mutant of DHBV is enriched over a wild-type virus (28). In this study, we determined the rate at which one strain of DHBV could be enriched over a slower replicating strain during chronic infection due to a growth-rate advantage. For this purpose, we established mixed infections of ducklings with a low-replication mutant of DHBV in competition with wild-type DHBV.
Replication defect of the DHBV mutant, DR1-13.
The mutant, DR1-13, kindly provided by Dan Loeb (McArdle Laboratory, University of Wisconsin) is partially defective in plus-strand primer translocation, due to a single nucleotide substitution of C→G at position 2547, one nucleotide downstream of DR1. This defect results in enhanced production of in situ-primed linear DNA at the expense of the circular double-stranded DNA, the precursor to cccDNA. Production of functional cccDNA is reduced accordingly. Nevertheless, the replication of DR1-13 is sufficiently rapid to cause chronic experimental infections after inoculation into 3-day-old ducklings. The phenotype of this mutant has been described in detail (22). The DR1-13 mutation does not change the coding of the precore open reading frame.
Emergence of wild-type virus in mixed infections.
Twelve ducklings were inoculated with 2 × 109 viral genomes of a mixture of 1:100 (six birds) or 1:1,000 (six birds) wild-type and DR1-13 viruses from transfected LMH cells. Five of six birds injected with the 1:100 mixture and six of six birds injected with the 1:1,000 mixture developed a peak viremia between 4 and 10 days postinfection and were studied for 72 and 224 days, respectively, the period covering rapid growth to sexual maturity. Each duck was weighed periodically, and blood was obtained for analysis of the genotype of the virus population. Body and organ weights of laboratory-housed Pekin ducks at necropsy, obtained in our laboratory over the last few years, were used to infer a relationship between the total body mass of Pekin ducks at different ages and their liver mass. This relationship is shown in Fig. 1. The average gain in total body mass for the ducks used in this experiment and the calculated average increase in liver mass are shown in Fig. 2.
FIG. 1.

Liver weight as a function of total body weight. Liver and total body weights obtained from the necropsy of 34 white Pekin ducks housed at the University of New Mexico HSC Animal Resource Facility were plotted. The line plotted is a linear regression analysis of the data points, performed with the Microsoft Excel Chart Function tools. The average mature body weight of adult ducks was about 4 kg.
FIG. 2.

Average body weights and calculated liver weights of the infected ducks. The body weights (▵) for 11 ducks with mixed-virus infections were obtained at the indicated times throughout the experiment. The corresponding liver weights (□) were calculated as a fraction of the total body weight using the graph shown in Fig. 1.
All ducks remained viremic, as measured by dot hybridization, throughout the period of study (data not shown). Viral DNA was extracted from each serum sample and analyzed for the genotype of the circulating virus by PCR and direct sequencing. The fraction of wild-type virus in each sample for all ducks is shown in Fig. 3. By day 4 postinfection, wild-type virus was readily detectable in the blood of all birds, and the fraction of wild-type virus was greatly increased over that of the inoculum. After day 4, the fraction of wild-type virus in the blood continued to rise in most birds, but by 40 to 50 days postinfection, this initial rise was usually abated. However, further increases could be seen in five of six birds infected with the 1:1,000 mixture of wild-type and DR1-13 viruses, starting after day 100. The fraction of wild-type virus in individual birds was marked by significant fluctuations that occurred throughout the experiment. These fluctuations were not due to errors inherent in the assay, since repeated assays performed on the same serum sample showed a variance of ±5% of the mean value (Fig. 4).
FIG. 3.

Percent wild type and enrichment for serum virus in 11 ducks with mixed-virus infections. Serum samples from each infected duck were analyzed for the genotype ratio as described in Materials and Methods. The percentage of wild-type virus (□) and the log enrichment of wild-type virus relative to the inoculum (●) is shown for each bird. (A) Five birds infected with 109 virus genomes of a 1:100 mixture of wild-type and DR1-13 viruses. (B) Six birds infected with 109 virus genomes of a 1:1,000 mixture of wild-type and DR1-13 viruses.
FIG. 4.

Reproducibility of the sequencing assay for DR1-13/wild type ratio. Serum (50 μl) from a coinfected duck was divided into six portions, and the total DNA was extracted from each replicate sample and subjected to PCR and direct sequencing. The relevant portions of the G and C lanes are shown for each sample. The intensity of the DR1-13-specific C band at position 2547 (arrow) was measured for each sample and normalized to the combined intensities of the neighboring C bands at positions 2551 and 2552. The normalized value was compared with that obtained for a 1:1 mixture of DR1-13 and wild-type plasmid templates (std) to obtain the proportion of DR1-13, which is indicated as the percentage of DR1-13 below each pair of sample lanes.
To confirm that the DR1-13 genotype as detected in our assay retained its replication defect, we tested the DR1-13 virus present in late serum samples from some of the ducks for its ability to compete with the wild type during outgrowth in a second passage in ducklings. We injected serum samples containing 108 genomes per ml into 2-day-old ducklings and assayed the viremic sera for the presence of the mutant genomes. The results of these assays are shown in Table 1. No DR1-13 virus could be detected in the second-passage serum samples from any of these birds by our assay, indicating that the mutant virus in the serum samples retained a partial replication defect.
TABLE 1.
Results of second passage of serum from mixed infections
| Duck no. | Day of serum collection | Size of inoculum (genomes) | Fraction of DR1-13 in inoculum | No. of ducklings infected | No. of ducks with DR1-13 |
|---|---|---|---|---|---|
| 78 | 78 | 108 | 0.24 | 4 | 0 |
| 84 | 137 | 108 | 0.85 | 4 | 0 |
| 85 | 137 | 108 | 0.84 | 4 | 0 |
| 86 | 137 | 108 | 0.44 | 3 | 0 |
| 87 | 137 | 108 | 0.73 | 3 | 0 |
| 88 | 137 | 108 | 0.78 | 4 | 0 |
| 89 | 137 | 108 | 0.40 | 3 | 0 |
Treatment of data.
In spite of the fluctuations in the fraction of wild-type virus in the blood of individual birds, a general pattern of behavior in the data was apparent. In order to convert the data into a form that quantitatively expressed the extent of wild-type and DR1-13 virus replication in the liver, we calculated the enrichment, E, of wild-type virus at each time point relative to the inoculum. We defined E as the ratio of the wild type to DR1-13 at each time point, divided by the same ratio at a reference time point, i.e., the inoculum at day 0. E is directly proportional to the relative increase in virus titer of the two virus populations according to expansion with first-order kinetics, and therefore log E would be a direct linear function of the number of generations of replication of each virus. Figure 3 shows the values of log E for each bird during the course of the experiment.
The enrichment plots illustrate the true extent of wild-type replacement of DR1-13 during the initial period of spread of infection (about 100-fold enrichment between day 0 and 4), indicating viral growth through multiple generations, with each generation contributing a fixed amount of enrichment. After day 4, when the livers were fully infected, the enrichment of wild-type virus appeared to be highly restricted compared with that occurring during the initial spread of infection.
Fluctuations that occurred in individual birds throughout all phases of enrichment did not occur with any obvious pattern. The lack of a pattern suggested that some factors affecting the ratio of the two viruses in the blood occurred in a manner that was specific for each bird, independently of the actual enrichment or body weight. Since these fluctuations tended to obscure any underlying pattern of enrichment, we attempted to cancel the effects of independent variables by averaging data from the same time points among the entire group of birds. Thus, the mean of the log enrichment would reflect the common pattern of virus growth over time. This value, the log of the geometric mean of the enrichments, is plotted in Fig. 5, beginning at day 4 postinfection with a mean log enrichment of 2.18.
FIG. 5.

Mean log enrichment of wild-type virus and estimated liver growth in all birds. The log enrichments over the inoculum, depicted in Fig. 3, were averaged among all birds at each time point and compared with the mean log estimated liver mass divided by the estimated liver mass at the time of inoculation. The lines represent separate linear regressions of the sequential data from three time intervals, 0 to 45 days postinfection (○), 45 to 101 days postinfection (▵), and 101 to 235 days postinfection (□).
This analysis suggested the presence of three apparent phases of virus growth, as measured by enrichment, after the livers were fully infected at day 4. The first phase corresponded closely with the phase of liver growth, as calculated from the increase in total body mass of each bird, and may have been due to expansion of the virus population into the growing liver. This expansion resulted in an approximately fourfold mean enrichment of wild-type virus. The second phase, during which no enrichment was detected, corresponded to the initial period of stable liver mass, from approximately day 40 to 100 postinfection. In this phase of no wild-type enrichment, the lack of new generations of virus growth may have been due to the lack of production of susceptible cells for virus spread. The third phase of enrichment, resulting in an additional fourfold enrichment of the wild type, also occurred during a time of stable body weight, from which we inferred that liver mass did not increase. However, the livers harvested from four of six ducks in the experiment at 316 days postinfection showed gross abnormalities compared with the livers from five ducks in the experiment that were sacrificed at day 78 postinfection (Table 2). The most striking change in these livers was the replacement of much of the liver parenchyma with amyloid deposits associated with gross changes in liver weight. In general, amyloidosis was not uniform throughout the liver, making it difficult to estimate the actual change in the total mass of hepatocytes. These changes may have been responsible for the third phase of enrichment by providing the opportunity for new generations of virus growth.
TABLE 2.
Characteristics of ducks with mixed infections at necropsy
| Duck no. | Body wt (g) | Liver wt (g) | Histologya |
|---|---|---|---|
| 78 | 3,580 | 92 | M−, A− |
| 79 | 3,420 | 79 | M++, A− |
| 80 | 3,500 | 67 | M+, A− |
| 81 | 3,759 | 87 | M+, A− |
| 83 | 3,527 | 69 | M−, A− |
| 84 | 3,709 | 112 | M−, A− |
| 85 | >4,200 | 180 | M−, A50% (ascites) |
| 86 | 3,395 | 123 | M−, A− |
| 87 | 3,487 | 133 | M−, A 50% (ascites) |
| 88 | 2,900 | 227 | M−, A 70% (ascites) |
| 89 | 3,950 | 268 | M−, A 90% (ascites) |
M, mononuclear cell infiltration; M−, normal (<100 cells/mm2); M+, mild (100 to 250 cells/mm2); M++, moderate (250 to 500 cells/mm2); A, amyloidosis (percentages are estimated hepatocyte replacement); A−, normal.
The lack of wild-type enrichment during a period of stable liver mass, beginning at day 39, suggested that new rounds of virus infection and cccDNA synthesis in the liver were highly restricted, even though virus particles were persistently produced. Lack of new generations of cccDNA synthesis might be due to the absence of production of susceptible cells for infection in the liver and to the resistance of all the existing infected cells to superinfection. If new rounds of virus replication were strictly dependent on the production of susceptible cells in the fully infected livers, the theoretical expansion of the cccDNA population required to produce the observed enrichment should be quantitatively similar to the observed increase in liver mass. This expansion can be estimated if the relative growth rates of the two viruses are known. Therefore, we measured the growth rate of DR1-13 relative to that of wild-type virus.
Relative growth rate of DR1-13.
The growth defect of DR1-13 was measured as the relative rate of secretion of DR1-13 virions per cccDNA molecule in infected primary duck hepatocytes, compared to that of the wild type. These data were obtained using two approaches. In the first approach, PDHs were infected with either wild-type or DR1-13 virus and incubated for 8 days postinfection. The medium was removed from the cells daily and replaced by fresh medium. The amount of virus secreted by the infected cells each day was measured through day 8 postinfection, and the amount of cccDNA for each plate of infected cells was determined at day 8. These data were used to determine the rate of virus production per cccDNA molecule for each virus (Fig. 6).
FIG. 6.

cccDNA and extracellular virus production by DR1-13- or wild-type-infected PDHs. PDH cultures were infected for 24 h with approximately 108 viral genomes of DHBV and incubated with daily medium changes. At the end of 8 days, the cultures were harvested, and cccDNA was extracted. The DNA for enveloped virus particles was extracted from each day's medium. (A) Viral DNAs were mixed with 300 pg of linearized DNA from plasmid pSPDHBV5.1(2X), containing a head-to-tail dimer of the DHBV genome (std), and assayed for viral DNA by agarose gel electrophoresis and blot hybridization. Viral DNA extracted from the 24-h culture medium at the indicated days for wild-type- and DR1-13-infected cells (virus) is seen in the top panel, and cccDNA extracted at 8 days postinfection (ccc) is shown in the bottom panel. The viral DNA yields were calculated by comparison of the viral DNA bands to the internal standard bands by phosphorimage analysis. Samples equivalent in the inoculum (in) of each plate of PDHs were run in each analysis. Each cccDNA sample is that obtained from one 60-mm plate of PDHs, and each viral DNA sample loaded was that obtained from a 16-ml 24-h culture medium (four plates of PDHs). The mean value for the amount of cccDNA per each plate is indicated at the bottom of panel A. (B) DNA from enveloped virus particles in the culture medium over 8 days postinfection is plotted as the cumulative total DNA per milliliter (4 ml per plate). The final slope of the curve indicates the rate of virus release for wild-type- or DR1-13-infected cells. The calculated relative rate of virus release of DR1-13-infected cells, normalized to that of cccDNA, 0.69.
In a second approach, plates were infected by a mixture of both viruses and incubated with daily medium changes. After 8 days, the viral DNA in the medium and the cccDNA in the cells was extracted, and the ratio of the two viruses was determined for each fraction by PCR amplification and direct sequencing (Fig. 7). Both experiments yielded similar results, i.e., the relative rate of synthesis of viral DNA from cccDNA by the DR1-13 mutant was 0.64, and that of the wild-type virus was 0.69. We used an average of these two numbers (0.67) in the following calculations.
FIG. 7.

Enrichment of wild-type virus in coinfected PDHs. Hepatocytes infected with 109 viral genomes of a mixture of wild-type and DR1-13 viruses were incubated with daily medium changes. At 9 days postinfection, the 24-h culture fluids were removed from each plate, and DNA from enveloped virus particles was extracted. The cccDNA was extracted from each corresponding plate of PDHs. The amount of DR1-13 DNA relative to wild-type DNA was determined for each sample by PCR and direct sequencing. The G and C lanes for each cccDNA (C) sample and its corresponding virus (V) sample are shown. The amount of DR1-13 in each sample was determined, and these data were used to determine the relative rate of DR1-13 virus production per cccDNA. The average relative replication rate for seven matched samples is 0.64 ± 0.08.
Calculations of virus expansion.
We considered two models of how new cccDNA synthesis from virus spreading into the growing liver would be reflected in the serum virus produced by this new cccDNA (Fig. 8). In model 1, new infected hepatocytes are derived by division of existing infected hepatocytes, resulting in simple dilution of the intracellular viral DNA forms, including cccDNA. Virus expansion would consist of a doubling of cccDNA in each progeny nucleus, followed by a restoration of intracellular replicative intermediate levels. We further assumed that cccDNA would be produced by direct conversion of the preexisting double-stranded DNA of the parental cell, including both linear and rcDNA. The majority of the progeny cccDNA derived from linear DNA would not be competent for further DNA synthesis, while that derived from rcDNA would be replication competent, so the amount of viral DNA produced by the DR1-13 cccDNA pool would be less than the amount produced by the wild-type cccDNA pool. In a dividing cell infected by both viruses, the enrichment of wild-type virus in the serum would reflect this defect over one generation of viral DNA synthesis from cccDNA; that is, the ratio of DR1-13 to wild-type virus produced by the newly synthesized cccDNA in each progeny cell would be 0.67 of the ratio produced by the original cccDNA in each progeny cell.
FIG. 8.

Two models for expansion of virus in the growing liver. The two models differ in the origin of new liver cells during liver growth or turnover. Model 1: infected liver cells divide, resulting in a reduction of cccDNA copy number per nucleus in the progeny cells. New cccDNA molecules (cccDNA∗) are made directly from preexisting rcDNA and linear DNA in the cytoplasm. The additional virus secreted in the serum is derived from the new cccDNA molecules. Model 2: new uninfected hepatocytes are derived from progenitor cells and are infected de novo by rcDNA- and linear DNA-containing virus in the serum. New cccDNA molecules (cccDNA∗) derived from the infecting virus constitute a new pool of cccDNA analogous to the cccDNA molecules synthesized in model 1. rcDNA and linear DNA is synthesized in the newly infected cells and is used for cccDNA amplification (cccDNA∗). Virus secreted in the serum is derived primarily from the amplified cccDNA pool. The spread of infection as described in model 1 results in the secretion of virus that is one generation removed from the previous virus population. In model 2, virus released in the blood is two generations removed from the previous virus population. The first and second generations of virus from cccDNA are indicated as numbered circles.
In model 2, new hepatocytes in the growing liver arise from a pool of uninfected progenitor cells, and these new hepatocytes are then infected de novo by extracellular virus. In this case, virus secreted in the serum by these newly infected cells would be derived from two generations of double-stranded DNA synthesis from cccDNA: the first generation occurring during amplification of new cccDNA derived from the extracellular virus, and the second generation occurring during virus production from the amplified pool, as indicated in Fig. 8. Thus, the ratio of DR1-13 to wild-type virus produced by the newly infected cells would be (0.67)2, or 0.45 of that produced by the resident infected cells.
Using 0.67 as the relative growth rate of DR1-13 in model 1, or 0.45 as the rate in model 2, we calculated the expansion of cccDNA that would be required to produced the observed enrichments for each time point between day 4 postinfection and day 42, the time of maximum growth of the liver, according to equation 5 in Materials and Methods. Assuming that all virus spread during growth was due to the division of mixed-infection cells (model 1), the virus expansion required for the enrichment observed during growth of the liver was two- to threefold higher than the amount of liver growth observed (Fig. 9A). In contrast, if all virus spread was due to new cycles of infection of susceptible cells (model 2), the amount of virus expansion required was actually less than the amount of liver growth observed (Fig. 9B). Thus, the observed expansion of liver mass was intermediate between the values predicted if either model alone accounted for the entire amount of virus expansion in the liver. The relatively close correspondence in the magnitude and the time of virus expansion predicted by either model to the increase in liver mass lends support to the notion that wild-type enrichment was dependent on the production of new hepatocytes.
FIG. 9.

Relative increase in replication space and liver mass. The mean log increase in replication space and liver mass, normalized to day 4 postinfection, is plotted versus the time postinfection. (A) The calculations assume a relative growth rate for DR1-13 of 0.67 that of wild-type virus (model 1; see text for details). (B) The calculated replication space assumes a relative growth rate of (0.67)2 = 0.45 for DR1-13 (model 2).
DISCUSSION
Sources of variation.
An unanticipated degree of fluctuation in the fractions of wild-type virus in the serum was seen in the serial samples from individual birds, as well between birds during the course of this experiment. As previously mentioned, these fluctuations were not inherent in the assay itself. We concluded that variation in the actual ratio of the two viruses in the blood occurred from week to week. We do not know the source of this variation. Because the wild-type virus was the DHBV-16 strain and the DR1-13 mutant was derived from DHBV-3, it is possible that strain differences, independently of relative growth rates, caused the two populations of virus to be susceptible to the influences of strain-specific factors. For example, small differences in the two viral envelope proteins might cause fluctuations in the ratios of circulating viruses in the blood, independently of the ratio of the two strains in the liver, due to differentials in the antibody response. Other scenarios are also possible. The effects of fluctuations seemed to be successfully subtracted by averaging of the data to produce an enrichment curve that was subject to our simple interpretation. However, our interpretation depends on the assumption that fluctuations in the serum were random with respect to the actual enrichments in the livers.
Relationship between virus growth and enrichment during mixed infection.
The enrichment of virus strains that compete for spread to susceptible cells during an initial infection of the liver is determined by their relative growth rates; i.e., faster replicating viruses are expected to be enriched over slower replicating viruses. Similarly, in a single cell simultaneously coinfected by two virus strains, it is expected that the virus that replicates more rapidly will be enriched over the slower replicating virus, since it would become more highly represented in the amplified pool of cccDNA. Once the pool of cccDNA is established, further enrichment would not occur, since the size of this pool is limited by inhibition of further cccDNA synthesis. These predictions were supported by our previous results showing that in vivo enrichment of wild-type virus over a variant with a partial replication defect occurred rapidly during the spread of infection, but the rate of enrichment was reduced by a factor of 8 to 9 once the liver was fully infected (28).
Because virus replication occurs according to quasi-first-order kinetics, the enrichment of one virus strain over another during growth in the same environment can be calculated if the relative growth rate constants of the two virus strains are known. In addition, the total expansion of virus (in this case, the cccDNA population) that is required for this enrichment can be calculated if the starting fractions of the two virus strains are known. The magnitude of this expansion is related quantitatively to the observed enrichment by equation 5 in Materials and Methods. Using this equation, we calculated the virus expansion during any given period by measuring the wild-type enrichment in a representative sample of the virus population.
In chronic hepadnavirus infections, net virus expansion cannot occur indefinitely. The maximum amount of virus, or viral cccDNA, in the liver is limited by (i) the number of hepatocytes that can be infected and (ii) the maximum number of cccDNA copies per hepatocyte. In a fully infected liver, new cccDNA synthesis is prevented unless uninfected cells are generated by liver growth or cell turnover or unless existing cccDNA molecules are lost and replaced within the cell. Turnover of cells or cccDNA thus provides an opportunity for enrichment of one virus strain over another through competitive growth. In the absence of other selective factors, any enrichment signifies that an effective expansion of virus population has occurred through new generations of cccDNA synthesis, even if there is no net increase in the amount of cccDNA in the liver. The magnitude of this effective expansion can be measured as changes in a property of the liver we define here as replication space.
Replication space.
Replication space is the potential of the liver to accommodate a replicating virus, in the case of hepadnaviruses, cccDNA molecules or their equivalent. More precisely, in an hepadnavirus infection, replication space may be thought of as occurring in discrete units, each of which may or may not be occupied by a cccDNA molecule. In this abstract concept of replication space, only one molecule of cccDNA may occupy each unit of space. In a growing liver, the total number of units of replication space increases, while during liver cell turnover, units of replication space are destroyed and are replaced with new unoccupied units of replication space as the liver regenerates. In effect, synthesis of a new molecule of cccDNA can occur only if a unit of unoccupied space is available. The concept of replication space allows us to analyze quantitatively the production of new generations of virus without knowing whether this new virus is formed by turnover of infected cells or by virus turnover inside the infected cells. The production of replication space implies that a corresponding dynamic state of virus replication, or cccDNA synthesis in the case of hepadnaviruses, exists at some level in the infected tissue.
Replication space could be created in the liver by an increase in liver mass, by hepatocyte turnover, and by cccDNA turnover. However, any replication space created that was not accessible to both viruses would not have been detected as enrichment in our experiments, even though new viral cccDNA synthesis may have occurred. For example, turnover of cccDNA within a hepatocyte infected by a single virus genotype would not allow competition for the new space created if the cell could not be superinfected. Similarly, division of an hepatocyte infected by a single virus genotype would not allow competition from another virus if the progeny cells could not be superinfected. Superinfection of a persistently infected cell has been reported to be inefficient (18); moreover, it is difficult to imagine how exogenous virus particles could compete with endogenous replicative intermediates for the addition of new molecules to the nuclear pool of cccDNA.
Replication space and liver growth.
The observed increase in liver mass during the phase of liver growth was intermediate between the increase in replication space predicted by two different simple models for the origin of new liver cells. This result suggests that normal liver growth may occur by a mixture of mechanisms. In addition, either model can be complicated by the possibility of segregation of the two viruses into two populations of hepatocytes or if cell division altered the stability of the virus in the dividing cells. Little is known about the frequency or stability of dual infection at the cellular level or how persistent infection may be perturbed by cell division.
However, these experiments suggest at least one aspect of the dynamic state of DHBV infections, i.e., persistence of the infection in the quiescent liver is characterized by very little initiation of new rounds of exogenous infection. This implies that a variant occurring in a single cell in a persistently infected liver would be unable to spread throughout the liver because of a replication advantage if replication space were not created in the liver by destruction of cccDNA or cells. If these conclusions also apply to the human liver, the dependence of enrichment on processes that create replication space may explain the long lag period that occurs before resistant mutants of HBV emerge in patients being treated with the antiviral nucleoside analog lamivudine (2, 6, 29). In this case, the enrichment of such mutants can be viewed as a measure of the effectiveness of the antiviral therapy in eliminating cccDNA of the lamivudine-sensitive virus from the liver. More generally, any antiviral measures in which resistant variants can occur would be predicted to result in the emergence of such variants, accelerated in proportion to the effectiveness of the therapy in eliminating cccDNA and creating unoccupied replication space. Our experiments suggest that the availability of replication space is the limiting factor for enrichment of growth variants.
Replication space as a measure of cell turnover.
The apparent low dynamic state of the hepadnaviral infection suggests that most viral variants that emerge in the infected liver do so under conditions of cell destruction, whether or not such destruction directly selects for cells infected by the emerging mutant. Thus, a rapidly growing variant may emerge as a predominant genotype only during periods of acute inflammation and cell destruction, even if cells infected by the variant are as equally susceptible to the inflammatory processes as those infected by the resident virus. This effect can appear as a correlation between exacerbation of liver disease and the emergence of a viral variant and can lead to the erroneous conclusion that a particular variant arising under those conditions is more pathogenic because it grows more rapidly.
Such an effect may account for the late enrichment of the wild-type virus, which is nonpathogenic, in five birds in this study following 2 months of stable ratios of wild-type to mutant virus. Thus, the enrichment of a rapidly growing virus in the liver may be used to measure the extent of cell turnover during liver disease. For example, the geometric mean increase in replication space between day 100 and 224 in our experiment was calculated to be 4- to 25-fold, depending on our assumptions about the mechanism of spread. This increase corresponds to the equivalent of 2 to 4.6 doublings of the liver, respectively, in a period of about 18 weeks. This amount of liver growth would correspond to the regeneration resulting from a hepatocyte half-life of 63 or 27 days, respectively, during this period.
ACKNOWLEDGMENTS
We thank Bai Hua Zhang and C. J. Ramey for excellent technical assistance and W. S. Mason, Fox Chase Cancer Center, for helpful discussions and for critical reading of the manuscript.
This work was supported by a grant from the National Cancer Institute, no. CA42542.
REFERENCES
- 1.Cattaneo R, Will H, Schaller H. Hepatitis B virus transcription in the infected liver. EMBO J. 1984;3:2191–2196. doi: 10.1002/j.1460-2075.1984.tb02113.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chayama K, Suzuki Y, Kobayashi M, Kobayashi M, Tsubota A, Hashimoto M, Miyano Y, Koike H, Kobayashi M, Koida I, Arase Y, Saitoh S, Murashima N, Ikeda K, Kumada H. Emergence and takeover of YMDD motif mutant hepatitis B virus during long-term lamivudine therapy and re-takeover by wild type after cessation of therapy. Hepatology. 1998;27:1711–1716. doi: 10.1002/hep.510270634. [DOI] [PubMed] [Google Scholar]
- 3.Civitico G M, Locarnini S A. The half-life of duck hepatitis B virus supercoiled DNA in congenitally infected primary hepatocyte cultures. Virology. 1994;203:81–89. doi: 10.1006/viro.1994.1457. [DOI] [PubMed] [Google Scholar]
- 4.Enders G H, Ganem D, Varmus H. Mapping the major transcripts of ground squirrel hepatitis virus: the presumptive template for reverse transcriptase is terminally redundant. Cell. 1985;42:297–308. doi: 10.1016/s0092-8674(85)80125-2. [DOI] [PubMed] [Google Scholar]
- 5.Ganem D. Hepadnaviridae and their replication. In: Fields B N, Knipe D M, Howley P M, et al., editors. Fields Virology. 3rd ed. Philadelphia, Pa: Lippincott-Raven Publishers; 1996. pp. 2703–2737. [Google Scholar]
- 6.Genovesi E V, Lamb L, Medina I, Taylor D, Seifer M, Innaimo S, Colonno R J, Standring D N, Clark J M. Efficacy of the carbocyclic 2′-deoxyguanosine nucleoside BMS-200475 in the woodchuck model of hepatitis B virus infection. Antimicrob Agents Chemother. 1998;42:3209–3217. doi: 10.1128/aac.42.12.3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawaguchi T, Nomura K, Hirayama Y, Kitagawa T. Establishment and characterization of a chicken hepatocellular carcinoma cell line LMH. Cancer Res. 1987;47:4460–4464. [PubMed] [Google Scholar]
- 8.Lenhoff R, Summers J. Construction of avian hepadnavirus variants with enhanced replication and cytopathicity in primary hepatocytes. J Virol. 1994;68:5706–5713. doi: 10.1128/jvi.68.9.5706-5713.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenhoff R, Summers J. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J Virol. 1994;68:4565–4571. doi: 10.1128/jvi.68.7.4565-4571.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lenhoff R L, Luscombe C A, Summers J. Acute liver injury following infection with a cytopathic duck hepatitis B virus. Hepatology. 1999;29:563–571. doi: 10.1002/hep.510290236. [DOI] [PubMed] [Google Scholar]
- 11.Lenhoff R L, Luscombe C A, Summers J. Competition in vivo between a cytopathic variant and a wild type duck hepatitis B virus. Virology. 1998;251:85–96. doi: 10.1006/viro.1998.9394. [DOI] [PubMed] [Google Scholar]
- 12.Luscombe C, Pedersen J, Uren E, Locarnini S. Long-term ganciclovir chemotherapy for congenital duck hepatitis B virus infection in vivo: effect on intrahepatic-viral DNA, RNA, and protein expression. Hepatology. 1996;24:766–773. doi: 10.1053/jhep.1996.v24.pm0008855174. [DOI] [PubMed] [Google Scholar]
- 13.Mandart E, Kay A, Galibert F. Nucleotide sequence of a cloned duck hepatitis B virus genome: comparison with woodchuck and human hepatitis B virus sequences. J Virol. 1984;49:782–792. doi: 10.1128/jvi.49.3.782-792.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mason W S, Aldrich C, Summers J, Taylor J M. Asymmetric replication of duck hepatitis B virus DNA in liver cells (free minus strand DNA) Proc Natl Acad Sci USA. 1982;79:3997–4001. doi: 10.1073/pnas.79.13.3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moraleda G, Saputelli J, Aldrich C E, Averett D, Condreay L, Mason W S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J Virol. 1997;71:9392–9399. doi: 10.1128/jvi.71.12.9392-9399.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moraleda G, Wu T T, Jilbert A R, Aldrich C E, Condreay L D, Larsen S H, Tang J C, Colacino J M, Mason W S. Inhibition of duck hepatitis B virus replication by hypericin. Antivir Res. 1993;20:235–247. doi: 10.1016/0166-3542(93)90023-c. [DOI] [PubMed] [Google Scholar]
- 17.Nassal M, Schaller H. Hepatitis B virus replication—an update. J Viral Hepat. 1996;3:217–226. doi: 10.1111/j.1365-2893.1996.tb00047.x. [DOI] [PubMed] [Google Scholar]
- 18.Protzer U, Nassal M, Chiang P W, Kirschfink M, Schaller H. Interferon gene transfer by a hepatitis B virus vector efficiently suppresses wild-type virus infection. Proc Natl Acad Sci USA. 1999;96:10818–10823. doi: 10.1073/pnas.96.19.10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pugh J C, Summers J. Infection and uptake of duck hepatitis B virus by duck hepatocytes maintained in the presence of dimethyl sulfoxide. Virology. 1989;172:564–572. doi: 10.1016/0042-6822(89)90199-2. [DOI] [PubMed] [Google Scholar]
- 20.Ruiz-Opazo N, Chakraborty P R, Shafritz D A. Evidence for supercoiled hepatitis B virus DNA in chimpanzee liver and serum Dane particles: possible implications in persistent HBV infection. Cell. 1982;29:129–136. doi: 10.1016/0092-8674(82)90097-6. [DOI] [PubMed] [Google Scholar]
- 21.Seeger C, Ganem D, Varmus H E. Biochemical and genetic evidence for the hepatitis B virus replication strategy. Science. 1986;232:477–484. doi: 10.1126/science.3961490. [DOI] [PubMed] [Google Scholar]
- 22.Staprans S, Loeb D, Ganem D. Mutations affecting hepadnavirus plus-strand synthesis dissociate primer cleavage from translocation and reveal the origin of linear viral DNA. J Virol. 1991;65:1255–1262. doi: 10.1128/jvi.65.3.1255-1262.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Summers J, Mason W S. Replication of the genome of hepatitis B-like virus by reverse transcription of an RNA intermediate. Cell. 1982;29:403–415. doi: 10.1016/0092-8674(82)90157-x. [DOI] [PubMed] [Google Scholar]
- 24.Summers J, Smith P, Huang M, Yu M. Regulatory and morphogenetic effects of mutations in the envelope proteins of an avian hepadnavirus. J Virol. 1991;65:1310–1317. doi: 10.1128/jvi.65.3.1310-1317.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Summers J, Smith P, Horwich A L. Hepadnaviral envelope proteins regulate amplification of covalently closed circular DNA. J Virol. 1990;64:2819–2824. doi: 10.1128/jvi.64.6.2819-2824.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tuttleman J, Pourcel C, Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- 27.Wu T T, Coates L, Aldrich C E, Summers J, Mason W S. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology. 1990;175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y-Y, Summers J. Enrichment of a precore-minus mutant in mixed infections with duck hepatitis B virus. J Virol. 1999;73:3616–3622. doi: 10.1128/jvi.73.5.3616-3622.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou T, Saputelli J, Aldrich C E, Deslauriers M, Condreay L D, Mason W S. Emergence of drug-resistant populations of woodchuck hepatitis virus in woodchucks treated with the antiviral nucleoside lamivudine. Antimicrob Agents Chemother. 1999;43:1947–1954. doi: 10.1128/aac.43.8.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]