

Abstract
Riboswitches are conserved functional domains in mRNA that almost exclusively exist in bacteria. They regulate the biosynthesis and transport of amino acids and essential metabolites, such as coenzymes, nucleobases, and their derivatives by specifically binding small molecules. Due to their ability to precisely discriminate between different cognate molecules as well as their common existence in bacteria, riboswitches have become potential antibacterial drug targets that could deliver urgently needed antibiotics with novel mechanisms of action. In this work, we report the recognition mechanisms of four oxidization products (XAN, AZA, UAC, and HPA) generated during purine degradation by an RNA motif termed the NMT1 riboswitch. Specifically, we investigated the physical interactions between the riboswitch and the oxidized metabolites by computing the changes in the free energy on mutating key nucleobases in the ligand binding pocket of the riboswitch. We discovered that the electrostatic interactions are central to ligand discrimination by this riboswitch. The relative binding free energies of mutations further indicated that some of the mutations can also strengthen the binding affinities of the ligands (AZA, UAC, and HPA). These mechanistic details are also potentially relevant in designing novel compounds targeting riboswitches.
Graphical Abstract

Introduction
Riboswitches are structural motifs underlying an ancient mechanism for genetic regulation in response to the cellular concentration of a metabolite or ion. Riboswitches mostly regulate the expression of bacetrial genes involved in the biosynthesis, transport, or metabolism of small molecules, without the involvement of a specialized protein receptor.1,2 These RNA molecules are commonly found in the 5′-untranslated region (UTR) of mRNAs and are known to regulate genetic expression by an allosteric rearrangement due to the binding of small molecules.3,4 Riboswitches are attractive targets for new antibacterial drugs because currently known riboswitches are primarily bacterial and have evolved to bind small molecules with a high affinity and specificity, which lessens the undesired off target interactions with human molecules.5,6 The antibacterial drugs targeting riboswitches can offer new opportunities to fight antimicrobial resistance, a major global health problem.7
A riboswitch (50–250 nucleotides in length) is a cis-acting regulatory element that is comprised of two mutually interacting domains, namely the aptamer domain and the expression platform.8,9 The aptamer domain binds metabolites or ions with a high selectivity and affinity, and is responsive to intracellular ligand concentrations.10 The expression platform ensures the structural transformation in response to the changes in the aptamer domain so as to modulate either binding of ribosome or transcription anti-termination.8,11 Since the discovery of the first riboswitch in 2002,2 nearly 45 distinct classes of riboswitches have been discovered, experimentally validated, and modeled at the atomic resolution in complex with their cognate ligands.12 Those riboswitches whose ligands were straightforward to identify, have largely been associated with metabolic pathways, but whose target ligands have yet to be identified are known as orphan riboswitch candidates.13,14 The orphan riboswitch candidates have resisted experimental validation for many years,14,15 but their study has led to the discovery of important biological processes once their ligands were identified.16 Thus, it is of significance to probe molecular mechanisms involving conformational changes and ligand recognition mechanisms in riboswitches.
One such RNA motif is the NMT1 riboswitch, which was previously identified through a comparative sequence analysis as a structured, noncoding RNA of unknown function.16,17 Very recently, this NMT1 riboswitch has been identified to bind xanthine (XAN) and uric acid (UAC) to regulate genes associated with purine oxidation/degradation in purine metabolism.17 The consensus sequence model of the NMT1 riboswitch derived from phylogenetic analysis suggested two stems adjoined to a large junction and also suggested that all nucleotides in this junction are highly conserved.17,18 The structures of XAN and its congener 8-azaxanthine (AZA) bound to the NMT1 riboswitch have been recently determined (Figure 1A).19 These tertiary structures suggest 2 stems adjoined to a junction, which is consistent with the predicted secondary structure model of this riboswitch.17,19 These structures also revealed a unique RNA architecture adopting a rod-like fold with an intriguingly structured binding pocket that is critically depended on divalent metal ions.19 These crystal structures further suggest that the highly conserved nucleotides in the junction region are brought into proximity via pairing and stacking interactions which shape the binding pocket specific for XAN.19 Specifically, these structures revealed that XAN inserts between a G-U pair (G10-U40) in the major groove (Figure 1B) and is sandwiched between the base triples A39-G35-C11 and A6-U41-A9 (Figure S1).19 Also, a Mg2+ ion is inner-sphere coordinated to XAN and a non-bridging oxygen of a backbone phosphate group. Further, biochemical studies suggest that mutations in the nucleotides involved in shaping the ligand binding pocket of the NMT1 riboswitch significantly reduce the binding affinity of XAN.19
Figure 1: Structural details of the NMT1 riboswitch and ligand molecules studied in our work.

(A) A cartoon representation of the tertiary structure of the NMT1 riboswitch. The different structural motifs (stems: P1, P2a, and P2b; and junctions: J1 and J2) are uniquely colored and labeled. (B) A zoomed-view of the ligand binding pocket of the NMT1 riboswitch. Several key nucleotides and the ligand are shown in sticks. The metal ions (white) and water molecules (red) are shown by spheres. (C) The chemical structure of each of the four ligands (XAN, AZA, UAC, and HPA) studied in our work.
Biochemical studies have shown that XAN and AZA show comparable binding affinities for the NMT1 riboswitch, XAN showed a K d ~ 3.7 μM, while UAC showed an about 7-fold lower binding affinity (K d ~ 25 μM) (Table S1).17 Another biochemical study reported that another compound (HPA) involved in the purine degradation pathway has no or significantly lower binding affinity for the NMT1 riboswitch even though this compound is a close chemical analogue of the riboswitch’s natural effectors (XAN and UAC).19 Further, it has been shown that mutations in the riboswitch significantly affect their dynamics, affinity for ligands, and their regulatory efficiency.20 However, a structural understanding of how the NMT1 riboswitch discriminates between its effector and their chemically related molecules found in the cellular environment and the effect of mutations on the ligand binding affinity remains unknown. Thus, it is of significance to probe the dynamics and the thermodynamic basis of ligand binding to the NMT1 riboswitch for broadly understanding the role of riboswitches as potential antibacterial drug targets and for identifying novel riboswitch-targeting compounds. Here, MD simulations20–22 and the structure-based free energy calculation methods are useful tools to probe the conformational dynamics and the ligand recognition mechanism of RNA influenced by mutations.20,23 These computational methods have been extensively adopted to explore the interaction mechanisms of biomolecules with small organic ligands with sufficiently high accuracy.24–27
In this study, we have used MD simulations and alchemical free energy calculations,20,23 to gain insights into how the NMT1 riboswitch discriminates close chemical analog molecules and the influence of sequence mutation on their recognition mechanisms. Because identifying novel riboswitch-targeting compounds requires a clear understanding of how these RNA molecules discriminate between their effector and chemically related molecules found in the cellular environment. Therefore, we selected four ligands, including two natural ligands of the NMT1 riboswitch (XAN and UAC), and two of their chemically related molecules, AZA and HPA (Figure 1C). The dynamics and conformational changes in the NMT1 riboswitch were investigated via a total of ~30 μs MD simulations. The results of long MD simulations offer additional insights into understanding the links between ligand binding and conformational changes.
Materials and Methods
Molecular Dynamics Setup
We obtained the atomic coordinates of the wild-type (WT) NMT1 riboswitch complexed with XAN and AZA from the protein data bank (PDB IDs: 7ELR and 7ELS, respectively).19 We constructed the initial structures of UAC and HPA bound to the NMT1 riboswitch by using as a template the crystal structure of XAN (PDB ID: 7ELR).19 We used the Avogadro software to generate atomic coordinates for UAC and HPA ligands.28 The structures of the ligands studied in our work are shown in Figure 1C. We generated the atomic coordinates of XAN, AZA, UAC, and HPA complexed with several mutants of the NMT1 riboswitch (A6G, G10A, C11U, G35A, A39G, and U40C) using VMD.29 We then solvated each system with explicit TIP3P30 water molecules in a periodic simulation domain. To neutralize each system, we added Mg2+ and Cl− ions into the minima of the electrostatic potential computed using the meadionize plugin in VMD.29 We further maintained bulk ionic concentration by adding K+ and Cl− ions (150 mM KCl). The consideration of the protonation and the tautomerization states of the ligands are based on previous experimental studies. For example, experimental studies suggest that the pKa values of the ligands XAN and AZA are 7.7 and 4.8, respectively.19,31 These studies also suggest that a significant population of the ligands XAN and AZA is deprotonated at physiological pH, and the ligand binding pocket of the NMT1 riboswitch can tolerate neutral as well as the tautomeric form of the deprotonated XAN and AZA.19 The chemical structure analysis also suggested that the ligand binding pocket of the NMT1 riboswitch fulfils the requirements for binding of both the neutral and anionic forms of XAN and AZA without a clear preference for one or the other form. Moreover, the isothermal titration calorimetry (ITC) experiments have demonstrated that the neutral and anionic forms of these ligands (XAN and AZA) show comparable affinities for the riboswitch.19 Therefore, we considered the neutral forms of these ligands in our simulation setup. The final system sizes are given in Table S2, and the overall simulation setup is shown in Figure S2.
Before conducting all-atom MD simulations, we energy minimized each system via the conjugate gradient minimization algorithm for 2000 steps. During the initial phase (30 ns) of MD equilibration, we restrained (k = 10 kcal/mol.Å2) the terminal nucleotides and the P-atoms in RNA. We removed the restraints for the P-atoms during subsequent MD simulations. We carried out all conventional MD simulations using the CHARMM3632 force-field for RNA and CHARMM General force-field (CGenFF)33 for ligands (XAN, AZA, UAC and HPA). We have also used the CHARMM36 force-field with improved non-bonded parameters (CHARMM36+NBFix) for magnesium cations in the nucleic acid systems.34 Even though a magnesium cation is not covalently bonded with the surrounding water molecules or with the atoms in nucleic acids, these improved nonbonded parameters keep the coordination geometry of magnesium cations intact as long as the simulation time is shorter than the typical exchange time for the first solvation shell, which is estimated to be ~10 μs.35
We performed all the simulations with a 2-fs time-step in the NPT ensemble. We maintained the temperature and pressure at 310 K and 1 bar using the Langevin thermostat and Nosé-Hoover barostat. We used periodic boundary conditions in all simulations and computed the long-range electrostatic interactions using the Particle Mesh Ewald method.36 We truncated van der Waals interactions with a cut-off of 12 Å and smooth switching taking effect at 10 Å.
We subjected each simulation model (WT and mutant complexes) to one μs long MD trajectory and saved configurations every 20 ps. We used the VMD software29 for generating input files. We performed visualization, analysis, and post-processing of the simulation trajectories using VMD,29 CPPTRAJ,37 and Pymol.38 We used NAMDv3.0 for conducting all simulations.39 The structures shown in our work are representative structures from the dominant cluster of each peptide-RNA complex obtained by clustering the trajectories using cpptraj.37 We applied DBSCAN40 clustering algorithm using 25 minimum number of points (minpoints) with a 1 Å distance cutoff (ϵ) between these points.
Alchemical Free Energy Calculations
Using the alchemical free energy simulation method,41 we computed the relative binding free energies ΔΔG for mutations in riboswitch for each of the four riboswitch-ligand complexes. Specifically, we designed a thermodynamic cycle (Figure S3) where the horizontal arms of the cycle correspond to the binding of a ligand to the wild-type (WT) and mutated riboswitch, and the vertical arms correspond to the alchemical transformation of a WT nucleotide into a mutated nucleotide in a given riboswitch-ligand complex. Since the physical binding of a ligand to the riboswitch (horizontal arms in Figure S3) is not the focus of this study, we computed the free energy changes along the unphysical pathways in liganded (left vertical arm in Figure S3, ΔGcomp) and unliganded riboswitch in an aqueous environment (right vertical arm in Figure S3, ΔGfree) as ΔΔG = ΔGcomp - ΔGfree = ΔGbind (ADE) - ΔGbind (GUA).
We used a hybrid energy function (U) to represent a mixture of two endpoint states of a particular vertical arm of the thermodynamic cycle (Figure S3), as applied in previous studies.24–26,42–44 The molecular topology follows the single topology strategy. A coupling parameter λ connects the end states by modifying the electrostatic, van der Waals energy, and bonded terms. A change from λ = 0 to λ = 1 thus transforms GUA into ADE by means of the mapping energy function: U = (1-λ) U(GUA) + λ U(ADE). The free-energy derivative was calculated as ∂G/∂λ = 〈∂U/∂λ〉λ = 〈U(λ = 1) – U(λ = 0)〉λ, where the brackets “〈 〉” represent averaging over an MD trajectory for a particular value of λ. Further, we used 51 equally spaced λ windows distributed over 10.2 ns simulations for the alchemical transformations of G to A, A to G, U to C, and C to U.
The free-energy derivative at each λ was calculated by computing the difference 〈U(λ = 1) – U(λ = 0)〉λ. For each λ window, the MD simulation lasted for 200 ps, and the data from the last 150 ps was used for averaging. We used the numerical integration (with the trapezoidal method) for calculating the free-energy change for each alchemical transformation. We divided the last 150 ps of the trajectory at each λ into two batches, and report the deviation of the free energy computed from both batches as the statistical error associated with the calculated ΔGcomp or ΔGfree. We averaged the total free energy change (ΔGcomp, ΔGfree) over forward and backward simulations, and repeated in triplicate with different initial velocities, yielding a minimum of 126 ns of simulation data per ΔΔG calculation (Tables S3–S7). We have reported the uncertainities in the averaged ΔGcomp and ΔGfree as the standard error of the mean and have calculated the error in the final ΔΔG by propagating the standard error of the mean associated with the averaged ΔGcomp and ΔGfree values. We obtained a convergence of the computed ΔΔG with a reasonable statistical uncertainty (<1 kcal/mol).25,26,42–44 We report free energy calculations with an aggregate time of ~1.89 μs.
Results
Dynamics of Riboswitch-Ligand Complexes
We performed 1 μs long simulations for all four ligand-bound riboswitch complexes in explicit solvent (Table S2). Further, to assess the structural similarities between the X-ray and MD structures, we monitored the root-mean-squared deviation (RMSD) relative to the initial structures (Figure S4). In Figure S4, we show the evolution of RMSD of the atoms C3′, C4′, C5′, O3′, O5′, and P in NMT1 riboswitch when bound to ligands. The RMSD data show that the MD structures of the NMT1 riboswitch bound to ligands are stable during simulations as they do not significantly deviate from their initial structures (<4 Å) (see Supporting Information). Further, root-mean-squared fluctuation (RMSF) data reveal larger average fluctuations for nucleotides located in the P2b motif (nucleotides G23, U24, U25, and A26) and the unpaired nucleotides in the J1 (nucleotides A7 and A8), P2a (nucleotide C15), and J2 (nucleotide A34) structural motifs of the NMT1 riboswitch (Figure S5).
We also performed the interaction energy decomposition analysis to explore the effects of non-covalent interactions on the affinities of the ligands. The interaction energy is the sum of the electrostatic (elec) energy and the van der Waals (vdw) energy. For each MD trajectory from our simulations, we used the NAMD energy plugin in VMD29 to calculate the interaction energies. In these energy calculations, we used the force-field parameters from MD simulations, the non-bonded cut-off used in conducting MD simulations, as well as the Particle Mesh Ewald method36 for computing electrostatic interactions. It should be noted that the non-bonded energy of a given ligand is not its absolute binding affinity, but is an approximate indicator of the binding affinity emerging from non-covalent interactions of the ligands with the atoms in the riboswitch.
In Figure 2A, we show the average interaction energy between all atoms of each ligand and the NMT1 riboswitch. The average interaction energy of ligands XAN, AZA, UAC and HPA with the NMT1 riboswitch is −63.68 ± 5.93 kcal/mol, −53.07 ± 4.95 kcal/mol, −49 ± 5.27 kcal/mol, and −39.67 ± 4.71 kcal/mol, respectively. The magnitude of the average interaction energy suggests that XAN (−63.68 ± 5.93 kcal/mol) has stronger interactions with the NMT1 riboswitch, while HPA (−39.67 ± 4.71 kcal/mol) has weaker interactions. The trend of the average interaction energy of these four ligands with NMT1 riboswitch agrees well with the experimental studies,17,19 which have shown that the ligand XAN has the highest affinity for the NMT1 riboswitch while the ligand HPA has significantly lower binding affinity for the riboswitch. The contribution from the vdw interactions is similar for four ligands (~ −22 kcal/mol). The contribution from electrostatic interactions is stronger than that from the vdw interactions and varies as −41.46 ± 4.83 kcal/mol, −31.01 ± 4.26 kcal/mol, −27.13 ± 4.41 kcal/mol, and −16.72 ± 3.16 kcal/mol for four ligands XAN, AZA, UAC, and HPA, respectively. Thus, the differences in the electrostatic interaction energies suggest that the electrostatic interaction is the primary driving force for the recognition of these ligands by the NMT1 riboswitch.
Figure 2: Non-bonded interaction energy analysis.

(A) The total electrostatic and van der Waals interaction energies of different ligands (XAN, AZA, UAC, and HPA) with the NMT1 riboswitch, and (B) the energy contribution of each nucleotide in the binding pocket of the XAN-NMT1 complex. The standard error of the mean associated with the averaged non-bonded interaction energies are shown as error bars. See also Figure S8.
We also calculated the interaction energy of each nucleotide in the ligand binding pocket with each ligand. As shown in Figure 2B, the ligand XAN produces interactions stronger than 1.0 kcal/mol with six nucleotides, including A6, G10, G35, A39, U40, and U41. Among these nucleotides, the interaction of XAN with G10 is the strongest. This nucleotide contributes −17.91 ± 1.62 kcal/mol toward the overall interaction of XAN with the riboswitch. Structurally, this interaction energy mainly arises from the hydrogen bonding interactions between XAN and G10 (Figure 3A), primarily involving XAN@N1…G10@O6 and XAN@O2…G10@N1 with 75% and 69% occupancy of these interactions, respectively (Figure S6). Further, these interactions are stabilized by two hydrogen bonds G10@N1…U40@O2 and G10@N2…U40@O2 between the nucleotides G10 and U40 with an occupancy of 72% and 98%, respectively. Similar to G10, the nucleotide U40 forms two hydrogen bonds with XAN, including XAN@N3…U40@O4 and XAN@O2…U40@N3 with an occupancy 68% and 80%, respectively. These interactions of U40 separately contribute a non-bonded interaction energy of −14.25 ± 1.47 kcal/mol to the association of XAN with the riboswitch (Figure 2B). The interaction energies of XAN with A6 and G35 are −9.46 ± 0.96 kcal/mol and −14.21 ± 1.29 kcal/mol, respectively, as structurally confirmed by the stacking interactions of XAN with the nucelobases of A6 and G35 (Figure 3A and Figure S7). The ribose sugar of A6 also forms two hydrogen bonding interactions XAN@N7…A6@O2′ and XAN@O6…A6@O2′ with an occupancy of 68% and 63%, respectively (Figure S6). We observed that XAN makes water mediated interactions with the magnesium ions M1 and M2 which are being further stabilized by the phosphate groups in the RNA backbone (Figure 3A).
Figure 3: Structural insights from MD simulations of the ligand-NMT1 complexes.

For each complex, shown is an initial structure (before MD) and an MD-derived structure highlighting the representative conformation based on the dominant conformational cluster from the respective MD simulation. Selected RNA nucleotides and each ligand are represented as sticks. The interaction patterns of the ligands with the key nucleotides in the binding pocket of the NMT1 riboswitch are shown with dotted lines in the same color as the ligands, and the remaining interactions are shown by white dotted lines. The metal ions and water molecules are shown in white and red spheres, respectively. The fractional occupancies of these interactions are shown in Figure S6.
The ligand AZA binds in a similar way as XAN to the riboswitch but differs in the number of hydrogen bonds and their occupancies (Figure 3B). The nucelobases G10 and U40 each forms a hydrogen bond with AZA as AZA@N1…G10@O6 and AZA@O2…U40@N3 with the occupancies of 98% and 88% (Figure S6), which contribute an interaction energy of −12.18 ± 0.89 kcal/mol and −7.78 ± 0.86 kcal/mol (Figure S8), respectively. The nucleobases A6 and G35 stack with the ligand AZA and contribute the interaction energies −7.94 ± 0.99 kcal/mol and −12.35 ± 1.13 kcal/mol, respectively (Figures S7 and S8). The ribose sugar of A6 also forms a hydrogen bond AZA@N7…A6@O2′ with an occupancy of 97% (Figure S6). The ligand AZA also makes water mediated interactions with the magnesium ions. Further, we observed that the ligand UAC makes an interaction network with riboswitch similar to the ligand AZA but it does not make water mediated interactions with the ions M1 and M2 (Figure 3B, C). The absence of interactions with the ions M1 and M2 are being compensated by a water mediated interaction between UAC and G10, and a hydrogen bond UAC@O8…A6@O2′ (Figure 3C). Therefore, the interaction energy of AZA and UAC are similar (AZA: −53.07 ± 4.95 kcal/mol, and UAC: −49.11 ± 5.27 kcal/mol) (Figure 2A).
For the ligand HPA, we observed that its orientation in the binding pocket is different than three other ligands (XAN, AZA, and UAC) (Figure 3). This is primarily because HPA does not have a carbonyl oxygen (O2) unlike the ligands XAN, AZA, and UAC. Therefore, this ligand (HPA) makes only a water mediated interaction with G10 and forms two hydrogen bonds HPA@N3…U40@N3 and HPA@N9…U40@O4 with the nucleobase U40 with an occupancy of 68% and 66%, respectively (Figures 3D and (S6)). The interaction energies of A6 and G35 are −3.19 ± 0.42 kcal/mol and −0.90 ± 0.27 kcal/mol, respectively (Figure S8), which indicates that the stacking interactions of HPA with the nucleobase A6 and G35 are not stable during MD simulations (Figure S7).
Further, we assessed the interaction pattern of base triples formed in ligand binding pocket of the NMT1 riboswitch. MD simulations suggest that the ligands are stacked between base triples A6-U41-A9 and A39-G35-C11 (Figure S9). We observed that base triple A39-G35-C11 has similar interaction pattern in all four ligand-riboswitch complexes where G35 forms three hydrogen bonds with C11 and two hydrogen bonds with A39. These interactions are stable in all four ligand-riboswitch complexes with an occupancy > 70% (Figure S10). In base triple A6-U41-A9, we observed that the A9-U41 interactions in XAN, AZA, and HPA-bound riboswitch complexes is similar to their interactions observed in X-ray structures. The A9-U41 interactions are disrupted in UAC-riboswitch complex (Figure S9). In all four ligand-riboswitch complexes, we observed that A9 establishes a new hydrogen bond with A6 with an occupancy > 60% (Figure S10). The number of hydrogen bonds and their occupancies in these base triples suggest that A39-G35-C11 is more stable compared to A6-U41-A9 (Figure S10).
Effect of Mutations on Ligand Binding
To evaluate the effect of mutations on the binding ability of different ligands to riboswitch, we have utilized a computationally rigorous TI methodology42–44 for computing the binding affinities of the ligands (XAN, AZA, UAC, and HPA) toward the riboswitch in the mutated and wild-type forms. Specifically, we have mutated six nucleobases (A6, G10, G35, A39, U40, and U41C) which are crucial for the recognition of different ligands. These nucleobases have stronger interaction energies with the ligands and contribute significantly toward the overall binding energy of ligands. Our conventional all-atom MD simulations of the WT NMT1 riboswitch bound to various ligands suggest that a network of electrostatic and aromatic stacking interactions are involved in the recognition of these ligands. Also, the interaction energy of each ligand with the NMT1 riboswitch suggests that the differences in electrostatic interactions contribute toward the discriminatory ability of the riboswitch for each ligand (Figure 2A). Thus, one can hypothesize that a purine to pyrimidine or a pyrimidine to purine mutation will disrupt the ligand binding pocket and alter the binding affinity of each ligand. Therefore, we have mutated purine nucelobases (G, A) to purine nucleobases (A, G) and pyrimidine nucleobases (U, C) to pyrimidine nucleobases (C, U) to quantify the binding contributions of electrostatic interactions. We then used an appropriate thermodynamic cycle (Figure S3) to calculate the relative changes in the binding affinity of ligands on mutations in the riboswitch. The computed free energy (ΔG) resulting from the thermodynamic integration calculations are listed in Table S3–S7.
In Figure 4, we show the calculated relative changes in the binding affinities upon mutations in the NMT1 riboswitch. These calculations revealed the following key features: we observed that the mutation G10A in the NMT1 riboswitch imposes the highest energetic penalty of ~6 kcal/mol for XAN and ~4 kcal/mol for AZA, while the same mutation penalizes UAC and HPA each by ~2 kcal/mol (Figure 4). The A6G mutation reduces the binding affinity of XAN by ~3 kcal/mol and the binding affinities of AZA, UAC, and HPA each by ~1 kcal/mol relative to the WT riboswitch (Figure 4). The mutation G35A reduces the binding affinity of XAN (ΔΔG = 2.06 ± 0.40 kcal/mol), AZA (ΔΔG = 2.65 ± 0.32 kcal/mol), UAC (ΔΔG = 1.51 ± 0.31 kcal/mol), and HPA (ΔΔG = 1.16 ± 0.34 kcal/mol, Figure 4) by ~1–3 kcal/mol relative to the WT riboswitch.
Figure 4: Energetics of riboswitch mutations in each ligand-riboswitch complex.
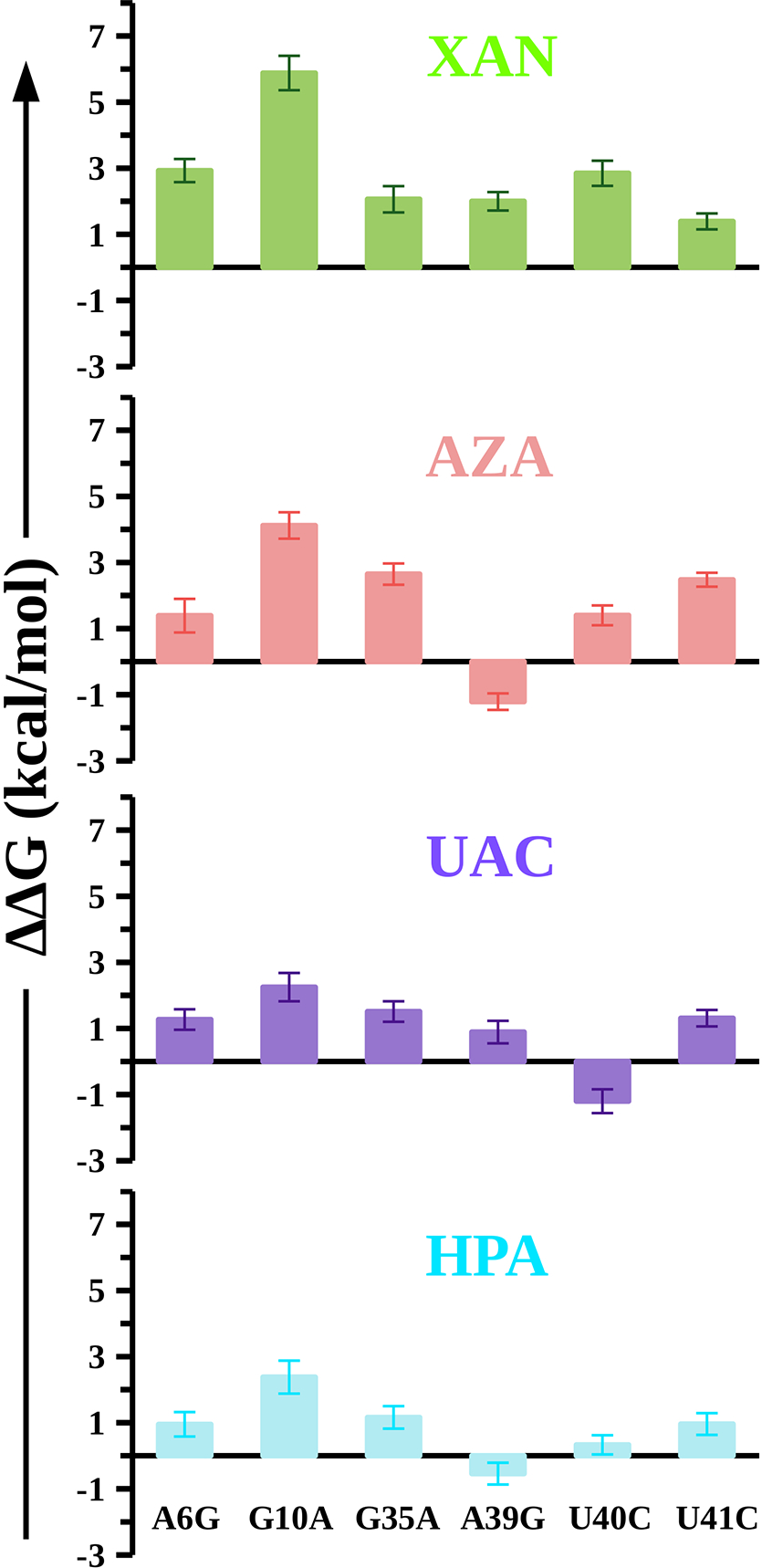
The relative binding free energy (ΔΔG) of mutations in the NMT1 riboswitch when it is bound to each ligand (XAN, AZA, UAC, and HPA). We calculated the error by propagating the standard error of the mean associated with the averaged ΔGcomp and ΔGfree, and the binding free energies are reported in kilocalories per mole (kcal/mol).
The mutation A39G reduces the binding affinity of XAN (ΔΔG = 2.00 ± 0.28 kcal/mol) and UAC (ΔΔG = 0.89 ± 0.34 kcal/mol) by ~1–2 kcal/mol (Figure 4), while the same mutation strengthens the binding affinity of AZA (ΔΔG = −1.21 ± 0.21 kcal/mol) and HPA (ΔΔG = −0.54 ± 0.33 kcal/mol) by ~1 kcal/mol relative to the WT riboswitch. The mutation U40C penalizes the binding of the ligand XAN (ΔΔG = 2.85 ± 0.38 kcal/mol) and AZA (ΔΔG = 1.40 ± 0.30 kcal/mol) by ~1–3 kcal/mol whereas the same mutation strengthens the binding affinity of UAC (ΔΔG = −1.20 ± 0.36 kcal/mol) by ~1 kcal/mol (Figure 4). The same mutation (U40C) does not significantly affect the binding affinity of the ligand HPA (ΔΔG = 0.33 ± 0.29 kcal/mol). The mutation U41C reduces the binding affinities of four ligands XAN (ΔΔG = 1.39 ± 0.24 kcal/mol), AZA (ΔΔG = 2.48 ± 0.21 kcal/mol), UAC (ΔΔG = 1.31 ± 0.25 kcal/mol), and HPA (ΔΔG = 0.96 ± 0.33 kcal/mol) by ~1–2 kcal/mol (Figure 4). Based on these results, we conclude that all six mutations (A6G, G10A, G35A, A39G, U40C, and U41C) in riboswitch significantly reduce the binding affinity of the ligand XAN while the mutation A39G strengthens the binding affinities of the ligands AZA and HPA. Further, the negative sign of calculated ΔΔG on the U40C mutation also suggests that this mutation strengthens the binding affinity of the ligand UAC toward the riboswitch.
Impact of Mutations on the Dynamics of the Riboswitch and Its Interaction Network With the Ligands
We also performed 1 μs long conventional MD simulations for all mutated ligand-bound riboswitch complexes in explicit solvent (Table S2). The RMSD data show that the structures of the mutated riboswitch-ligand complexes are stable during simulations as they do not significantly deviate from their initial structures (<5 Å) (Figure S11). To evaluate the influence of mutations on the residue-level flexibility of the NMT1 riboswitch, we computed the ΔRMSF, the difference of the averaged RMSF per residue of mutated riboswitch and the averaged RMSF per residue of WT riboswitch when bound to the ligands. The ΔRMSF data suggested that the flexibility of the nucleotides is significantly affected by the mutations (Figure S12). The mutations stabilize the nucleotides (C15 and A37) in P2a and nucleotides (A24, A25, and A26) in the P2b structural motif of riboswitch when bound to ligands. Apart from the P2a and P2b structural motifs the flexibility of the remaining structure of the riboswitch is increased by the mutations, especially in the P1 and J1 structural motifs. These ΔRMSF data suggest that even modest sequence alterations in nucleotides have a dramatic impact on the flexibility and the structural stability of the NMT1 riboswitch.
In Figures 5 and 6, we show the representative structures from the dominant cluster of each mutated ligand-riboswitch complexes, which was obtained by a clustering analyses (see methods) of conformations sampled via conventional MD simulations. In Figure 5, we show the structures from the mutated XAN-riboswitch complexes, and in the panels (A) through (F), we show the interaction patterns of key nucleotides with the ligand XAN upon mutations in different nucleotides. We observed that the A6G mutation results in disruption of interactions between the ligand XAN and riboswitch (Figure 5A). This mutation (A6G) also induces the formation of a hydrogen bond between XAN and the nucleobase U41, as XAN@N3…U41@O2 with an occupancy of 98% (Figure S13). The ligand XAN changes its orientation and forms one hydrogen bond with each of the nucleobases A10 and U40, and forms two hydrogen bonds with the nucleobase U41 after the G10A mutation (Figure 5B). For the G35A mutation, the interactions of the ligand XAN are intact with the nucleobases G10 and U40 (Figures 5B and S13) but the interactions of XAN are disrupted upon A39G mutation. This mutation (A39G) also induces the formation of an interaction XAN@N1…U40@O2P with an occupancy of 45% (Figure S13). The mutation U40C and U41C disrupt the interactions of XAN with riboswitch and decrease the occupancy of hydrogen bonds relative to the WT (Figures 5F and S13).
Figure 5: Structural changes in the XAN-NMT1 complexes on mutations.

In panels (A) through (F), we show the interaction pattern of the ligand XAN with the nucleotides in the ligand binding pocket of the riboswitch after A6G, G10A, G35A, A39G, U40C, and U41C mutations, respectively. The fractional occupancies of these interactions are shown in Figure S13. The interactions between XAN and the riboswitch are highlighted by green dotted lines. The ions and water molecules are depicted by white and red spheres, respectively.
Figure 6: Structural changes in the ligand-riboswitch complexes on mutations.

Structural insights from the (A) AZA-NMT1, (B) UAC-NMT1, and (C) HPA-NMT1 complexes upon mutations. The fractional occupancies of the interactions depicted are reported in Figures S18, S23, and S28.
Further, we observed that the stacking of nucleobase A6 with ligand XAN is disrupted upon mutations A6G, A39G, and U40C, and stacking of nucleobase G35 with XAN is disrupted upon mutations A6G, G35A, and A39G (Figure S14). The hydrogen bonds in base triple (A39-G35-C11) remain intact upon mutations (A6G, G10A, A39G, U40C, and U41C) except after mutation G35A, where hydrogen bonds are disrupted between A35-C11 and A35-A39 (Figures S15 and S16). The interactions in base triple A9-A6-C11 are disrupted upon all six mutations in XAN-riboswitch complexes (Figure S15).
In the AZA-riboswitch complex, we observed that the interactions of AZA in ligand binding pocket are disrupted upon mutations (A6G, G10A, G35A, U40C, and U41C) except in the case of A39G mutation (Figures S17 and S18). After the A39G mutation, the ligand AZA forms four hydrogen bonds (Figure 6A) with the nucleobases G10 and U40, as observed in the crystal structure (Figure 1B). The ligand AZA also forms a hydrogen bond with the ribose sugar of A6 and a water-mediated interaction with G39, thereby stabilizing it in the ligand binding pocket of the riboswitch (Figures 6A and S18). The stacking interactions of AZA with the nucleobases A6 and G35 are intact after mutations except on U41C mutation, where stacking of A6 and AZA is disrupted (Figure S19). We observed that the mutation A39G disrupts only one hydrogen bond between G35 and G39, leaving most hydrogen bonds in base triples (A9-A6-C11 and A39-G35-C11) intact (Figures S20 and S21).
In the UAC-riboswitch complex, we observed that the interactions of UAC in the ligand binding pocket are disrupted upon mutations (A6G, G10A, G35A, A39G, and U41C) (Figure S22) except in the case of U40C mutation (Figure 6B). After the U40C mutation, the ligand UAC changes its orientation but it still forms a stable network of interactions with the riboswitch (Figures 6B and S23). The ligand UAC forms two hydrogen bonds with nucleobase C40 and a hydrogen bond with G10 after the U40C mutation (Figure 6B). The nucleobases A6 and G35 make stable stacking interaction with UAC with a >95% occupancy (Figure S24). This mutation (U40C) disrupts hydrogen bonds between A6 and U41 but leaves most hydrogen bonds in base triples (A9-A6-C11 and A39-G35-C11) intact (Figures S25 and S26).
In the HPA-riboswitch complex, we observed that the interactions of HPA in the ligand binding pocket are disrupted upon mutations (A6G, G10A, G35A, U40C, and U41C) (Figure S27) except in the case of A39G mutation (Figure 6C). After the A39G mutation, the ligand HPA forms a stable network of interactions with the riboswitch (Figures 6C, S28, and S29). After this mutation, the interaction network in base triples (A9-A6-C11 and A39-G35-C11) is similar to the interaction pattern in AZA-riboswitch complex after A39G mutation (Figures S20, S30, and S31). The above explained network of hydrogen bonds and stacking interactions between the ligand HPA and the riboswitch upon A39G mutation leads to a stable HPA-riboswitch complex compared to the WT complex.
Discussion
Biochemical studies have established that the ligands XAN and AZA exhibit comparable binding affinities for the NMT1 riboswitch, while the ligand UAC demonstrates approximately 7-fold lower binding affinity.17,19 Interestingly, a compound called HPA, which is part of the purine degradation pathway, either has no binding affinity or significantly lower affinity for the NMT1 riboswitch (Table S1).17,19 Although the experimental structures of the XAN-NMT1 and AZA-NMT1 complexes have provided molecular details of ligand recognition by the NMT1 riboswitch,19 the structural and thermodynamic basis governing the discrimination of natural ligands (XAN and UAC) and their chemical analogues (AZA and HPA) by the NMT1 riboswitch remains unknown. In addition, the structural effects of mutations (in the NMT1 riboswitch) known to alter the ligand binding affinity remain unresolved. We address these questions via MD simulations combined with the alchemical free-energy calculations. Specifically, we report on dynamics of the NMT1-ligand complexes, non-bonded interaction energy between the ligands and the NMT1 riboswitch, and free energy changes on mutations in the riboswitch. We also suggest mutations that can potentially enhance the affinity of the riboswitch toward the ligands (AZA, UAC, and HPA). We also point out that we did not perform free energy calculations for ligand transformations in this study. Our focus is not to examine the actual binding process of the ligands, but instead to gain insights into the interactions between these ligands and the NMT1 riboswitch as well as to understand the impact of mutations in ligand recognition by the riboswitch.
First, we computed the average non-bonded interaction energy of the riboswitch with the ligands (XAN, AZA, UAC, and HPA) to explore the effects of non-covalent interactions on the affinities of ligands. The magnitude of average non-bonded interaction energy suggest that the ligand XAN has a stronger interaction energy followed by the ligands AZA, UAC, and HPA with the riboswitch. This trend of non-bonded interaction energy agrees well with the experimental studies where it has been suggested that XAN has a stronger interaction energy followed by ligands AZA, UAC and, HPA, respectively.17,19 We observed that the contribution from the vdw interaction energy is similar for four ligands (~ −22 kcal/mol, Figure 2A) which is contributed by the stacking interactions of ligands with the nucleotides A6 and G35. These stacking interactions are stable and consistent in all four complexes thereby leading to a similar magnitude of vdw interaction energy. These data suggest that the nucleotides A6 and G35 are critical to the recognition of ligands by the NMT1 riboswitch which is consistent with the experimental observation that altering these nucleotides significantly affects the binding affinities of the ligands.19 The contributions from the electrostatic interactions vary for various ligands XAN (~ −41 kcal/mol), AZA (~ −31 kcal/mol), UAC (~ −27 kcal/mol), and HPA (~ −17 kcal/mol) (Figure 2A). Our simulations suggest that the ligand XAN has a higher number of stable hydrogen bonding interactions with the riboswitch compared to the ligands AZA, UAC, and HPA (Figures 3 and S6), thereby leading to a stronger electrostatic interaction energy for the ligand XAN (Figure 2A).
Further, we calculated the free energy changes for mutations in six key nucleotides (A6, G10, G35, A39, U40, and U41) located in the ligand binding pocket of the NMT1 riboswitch. We selected these six nucleotides on the basis of their non-bonded interaction energy contributions for each ligand (Figure 2B). Our free energy calculations suggest that all six mutations (A6G, G10A, G35A, A39G, U40C, and U41C) in the NMT1 riboswitch reduce the binding affinity of the ligand XAN where the mutation G10A has the highest energetic penalty (~ 6 kcal/mol). Our structural analysis of the XAN-riboswitch complex after G10A mutation suggests that the repulsive interactions between amine group of A10 and the N3 atom of XAN disrupt the interaction network formed by stacking and hydrogen bond interactions (Figure 5B). This disruption of the interaction network in the ligand binding pocket of the riboswitch leads to a reduced binding affinity for the ligand XAN. Our alchemical free energy calculations suggest that the mutation A39G strengthens the binding affinity of the ligands AZA and HPA for the NMT1 riboswitch. The ligands AZA and HPA upon A39G mutation in the riboswitch, form a stable network of hydrogen bonding interactions (Figure 6) thereby leading to an increment in their binding affinities (Figure 4). Further, we observed that the U40C mutation increases the binding affinity of the NMT1 riboswitch for the ligand UAC (Figure 4), which corroborates with the stable hydrogen bonding network of the UAC-NMT1 complex (Figure S23). An experimental study19 suggests that mutating some nucleotides (G10A and U40C) in the ligand binding pocket of the NMT1 riboswitch affects the binding of the ligands (XAN and AZA) although no Kd values for any such mutations were reported (Table S1). Therefore, the calculated relative binding free energies of mutations in the riboswitch cannot be directly compared with the currently available experimental data.
Furthermore, we observed that the structural details on binding of XAN and UAC to the NMT1 riboswitch emerging from our simulations agree well with a previous biochemical study,17 which reported approximately 7-fold lower binding affinity for UAC (K d ~ 25 μM) in comparison to XAN (K d ~ 3.7 μM) (Table S1). Additionally, our observations of decreased overall binding affinity (~2.85 kcal/mol) of XAN on the U40C mutation are consistent with the experimental data reporting no binding,19 while the increased binding affinity of UAC (~−1.20 kcal/mol) on the U40C mutation lacks corresponding experimental evidence for the NMT1-UAC complex. Overall, our study provides structural insights into the binding of the ligands XAN and UAC to the NMT1 riboswitch, corroborating existing experimental findings and shedding light on the impact of riboswitch mutations on ligand recognition. From our results, it is evident that the U40C mutation disrupts the crucial hydrogen bonding interactions necessary for the binding of the ligand XAN. However, this mutation results in the establishment of a stable hydrogen bonding network that increases the binding affinity of UAC. These findings provide an explanation for the computationally observed increment in the UAC binding affinity caused by the U40C mutation in the NMT1 riboswitch. However, further experimental investigations are required to validate our findings involving the effects of mutations on binding of the ligands AZA, UAC, and HPA. We also note that previous studies,20,45–47 employing both computational and experimental approaches, have also demonstrated that mutations in the Guanine riboswitch can induce the formation of new hydrogen bonding interactions with the ligands. The formation of new hydrogen bonding interactions has been shown to contribute significantly to the overall binding affinity of the ligands for the Guanine riboswitch.
The experimental structures of the riboswitch bound to the ligands XAN and AZA suggested that these ligands insert between a wobble base pair (G10-U40) and make a base triple which is sandwiched between the base triples A9-U41-A6 and A39-G35-C11.19 The base triples A9-U41-A6 and A39-G35-C11 form the “floor” and the “roof”, respectively, for the ligand in the binding pocket (Figure S1). Previous studies suggested that base triples play an integral role in ligand recognition by riboswitches and disruption of interactions in base triples leads to a reduction in the binding affinity.48–50 In MD simulations we observed that the hydrogen bonding interactions in these base triples are stable in all four ligand-riboswitch complexes (Figures S9 and S10). The stacking of these base triples further stabilizes the ligand in the binding pocket. Our calculated averaged vdw interaction energy is also similar for all four ligands (~ −22 kcal/mol) but differs in the electrostatic energy (Figure 2A). Therefore, these observations suggest that the differences in the binding affinities of ligands for the NMT1 riboswitch are primarily driven by the electrostatic interactions.
Previously, Sund et al.51 via alchemical calculations have demonstrated that the Adenine riboswitch specifically uses the electrostatic preorganization to discriminate different analogues of Adenine. In our mutational studies, we have observed that upon mutations A39G and U40C in the NMT1 riboswitch, the ligands AZA, UAC, and HPA show higher binding affinities for the mutated riboswitch by forming new hydrogen bonds. Our observations from MD simulations corroborate with the previous computational studies by Hu et al.52 and Chen et al.,20 as they also suggested correlations between the binding affinities of ligands and the role of electrostatic interactions, but we have now quantified these interactions. Therefore, our findings highlight the thermodynamic basis of ligand recognition by the NMT1 riboswitch. While we have not probed the kinetics of ligand binding in this riboswitch, both kinetics and thermodynamics have been suggested to play a role in ligand recognition by other riboswitches.53
Conclusions
In this work, we have reported the structural and thermodynamic basis governing the discrimination of natural ligands (XAN and UAC) and their chemical analogues (AZA and HPA) by the NMT1 riboswitch, and have also investigated the energetics of mutations in the riboswitch that affect its binding to these ligands. We found that the ligands (XAN, AZA, UAC, and HPA) interact with the NMT1 riboswitch through stacking and hydrogen bonding interactions. The base triples (A9-U41-A6 and A39-G35-C11) play an integral role in ligand recognition by forming stacking interaction with the ligand. However, our non-bonded interaction energy data revealed that the energetic contribution of vdw interactions is similar for all four ligands and the energetic contribution of electorstatic interactions varies for these ligands, thereby suggesting that the NMT1 riboswitch uses electrostatic interactions to discriminate between ligands XAN, AZA, UAC, and HPA. These observations also corroborate with previous studies that suggested that riboswitches use electrostatic interactions to discriminate between natural ligands and their chemical anlogues.51,52 Our alchemical free energy calculations demonstrate that the mutations in the NMT1 riboswitch significantly affect the interactions of ligands with several conserved nucleotides (A6, G10, G35, A39, U40, and U41) in the binding pocket. More importantly, the mutations A39G and U40C induce the formation of new hydrogen bonding interactions of AZA, UAC, and HPA with the NMT1 riboswitch which strengthens their binding ability. This observation is further supported by previous studies20,52 which have suggested that the changes in binding affinities of the ligands on mutations in the riboswitch are driven by the loss or gain of the electrostatic interactions. Our mechanistic findings are also potentially relevant in designing novel compounds targeting riboswitches.
Supplementary Material
Acknowledgement
We acknowledge the financial support from the National Science Foundation (CBET-1554558) and the National Institutes of Health (NIH) (R35GM138217). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. We thank computational support through the following resources: Premise, a central shared HPC cluster at UNH supported by the Research Computing Center; and BioMade, a heterogeneous CPU/GPU cluster supported by the NSF EPSCoR award (OIA-1757371).
Footnotes
Supporting Information
Additional data and analyses, steps for system setup and MD simulations, thermodynamic cycle for estimating the relative binding free energy of mutations in the riboswitch, RMSD and RMSF of the ligand-riboswitch complexes, fractional occupancies of all key interactions, structural insights from MD simulations of the ligand-riboswitch complexes
Data and Software Availability
All methodological details for all data generated in this study are available in the Methods section including PDB codes of input files, simulation conditions, error analysis. The input files for conventional MD simulations and TI calculations for mutations in the NMT1-XAN complex are available at github: (https://github.com/amit9869/JCIM_XAN.git). The free energy data averaged over forward and backward transformations for each mutation in complex and solution are also provided (Tables S3–S7). The simulation and visualization software29,38,39 and the force-field for RNA32 used to carry out calculations are openly available. The CHARMM general force-field33 used for the ligands (XAN, AZA, UAC, and HPA) is available in supporting information.
References
- (1).Mironov AS; Gusarov I; Rafikov R; Lopez LE; Shatalin K; Kreneva RA; Perumov DA; Nudler E Sensing small molecules by nascent RNA: a mechanism to control transcription in bacteria. Cell 2002, 111, 747–756. [DOI] [PubMed] [Google Scholar]
- (2).Nahvi A; Sudarsan N; Ebert MS; Zou X; Brown KL; Breaker RR Genetic control by a metabolite binding mRNA. Chem. Biol. 2002, 9, 1043–1049. [DOI] [PubMed] [Google Scholar]
- (3).Winkler W; Nahvi A; Breaker RR Thiamine derivatives bind messenger RNAs directly to regulate bacterial gene expression. Nature 2002, 419, 952–956. [DOI] [PubMed] [Google Scholar]
- (4).Henkin TM Riboswitch RNAs: using RNA to sense cellular metabolism. Genes Dev. 2008, 22, 3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Blount KF; Breaker RR Riboswitches as antibacterial drug targets. Nat. Biotechnol. 2006, 24, 1558–1564. [DOI] [PubMed] [Google Scholar]
- (6).Mulhbacher J; Brouillette E; Allard M; Fortier L-C; Malouin F; Lafontaine DA Novel riboswitch ligand analogs as selective inhibitors of guanine-related metabolic pathways. PLoS pathog. 2010, 6, e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Breaker RR Prospects for riboswitch discovery and analysis. Mol. Cell 2011, 43, 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Serganov A; Nudler E A decade of riboswitches. Cell 2013, 152, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sherwood AV; Henkin TM Riboswitch-mediated gene regulation: novel RNA architectures dictate gene expression responses. Annu. Rev. Microbiol. 2016, 70, 361–374. [DOI] [PubMed] [Google Scholar]
- (10).Garst AD; Edwards AL; Batey RT Riboswitches: structures and mechanisms. Cold Spring Harb. perspect. biol. 2011, 3, a003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Breaker RR Riboswitches and translation control. Cold Spring Harb. perspect. biol. 2018, 10, a032797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McCown PJ; Corbino KA; Stav S; Sherlock ME; Breaker RR Riboswitch diversity and distribution. Rna 2017, 23, 995–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Weinberg Z; Wang JX; Bogue J; Yang J; Corbino K; Moy RH; Breaker RR Comparative genomics reveals 104 candidate structured RNAs from bacteria, archaea, and their metagenomes. Genome Biol. 2010, 11, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Meyer MM; Hammond MC; Salinas Y; Roth A; Sudarsan N; Breaker RR Challenges of ligand identification for riboswitch candidates. RNA Biol. 2011, 8, 5–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Block KF; Hammond MC; Breaker RR Evidence for widespread gene control function by the ydaO riboswitch candidate. J. Bacteriol. 2010, 192, 3983–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sherlock ME; Breaker RR Former orphan riboswitches reveal unexplored areas of bacterial metabolism, signaling, and gene control processes. RNA 2020, 26, 675–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yu D; Breaker RR A bacterial riboswitch class senses xanthine and uric acid to regulate genes associated with purine oxidation. RNA 2020, 26, 960–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Colloc’h N; Hajji ME; Bachet B; l’Hermite G; Schiltz M; Prangé T; Castro B; Mornon J-P Crystal structure of the protein drug urate oxidase-inhibitor complex at 2.05 Å resolution. Nat. Struct. Biol. 1997, 4, 947–952. [DOI] [PubMed] [Google Scholar]
- (19).Xu X; Egger M; Chen H; Bartosik K; Micura R; Ren A Insights into xanthine riboswitch structure and metal ion-mediated ligand recognition. Nucleic Acids Res. 2021, 49, 7139–7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chen J; Wang X; Pang L; Zhang JZ; Zhu T Effect of mutations on binding of ligands to guanine riboswitch probed by free energy perturbation and molecular dynamics simulations. Nucleic Acids Res. 2019, 47, 6618–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lu S; Huang W; Wang Q; Shen Q; Li S; Nussinov R; Zhang J The structural basis of ATP as an allosteric modulator. PLoS Comput. Biol. 2014, 10, e1003831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang J; Shao Q; Cossins BP; Shi J; Chen K; Zhu W Thermodynamics calculation of protein–ligand interactions by QM/MM polarizable charge parameters. J. Biomol. Struct. Dyn. 2016, 34, 163–176. [DOI] [PubMed] [Google Scholar]
- (23).Shirts MR; Mobley DL Biomolecular simulations; Springer, 2013; pp 271–311. [Google Scholar]
- (24).Satpati P; Åqvist J Why base tautomerization does not cause errors in mRNA decoding on the ribosome. Nucleic Acids Res. 2014, 42, 12876–12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kumar A; Basu D; Satpati P Structure-Based Energetics of Stop Codon Recognition by Eukaryotic Release Factor. J. Chem. Inf. Model. 2017, 57, 2321–2328. [DOI] [PubMed] [Google Scholar]
- (26).Kumar A; Vashisth H Role of Mutations in Differential Recognition of Viral RNA Molecules by Peptides. J. Chem. Inf. Model. 2022, 62, 3381–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kumar A; Vashisth H Conformational dynamics and energetics of viral RNA recognition by lab-evolved proteins. Phys. Chem. Chem. Phys. 2021, 23, 24773–24779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hanwell MD; Curtis DE; Lonie DC; Vandermeersch T; Zurek E; Hutchison GR Avogadro: an advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Humphrey W; Dalke A; Schulten K VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (30).Jorgensen WL Free energy calculations: a breakthrough for modeling organic chemistry in solution. Acc. Chem. Res. 1989, 22, 184–189. [Google Scholar]
- (31).Wierzchowski J; Smyk B Excited-state proton transfer in 8-azapurines I: a kinetic analysis of 8-azaxanthine fluorescence. Molecules 2020, 25, 2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Huang J; MacKerell AD Jr CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; MacKerell AD Jr CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yoo J; Aksimentiev A Improved parametrization of Li+, Na+, K+, and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J. Phys. Chem. Lett. 2012, 3, 45–50. [Google Scholar]
- (35).Callahan KM; Casillas-Ituarte NN; Roeselová M; Allen HC; Tobias DJ Solvation of magnesium dication: molecular dynamics simulation and vibrational spectroscopic study of magnesium chloride in aqueous solutions. J. Phys. Chem. A 2010, 114, 5141–5148. [DOI] [PubMed] [Google Scholar]
- (36).Darden T; York D; Pedersen L Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar]
- (37).Roe DR; Cheatham III TE PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (38).DeLano WL Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 2002, 40, 82–92. [Google Scholar]
- (39).Phillips JC; Hardy DJ; Maia JD; Stone JE; Ribeiro J; Singharoy A; Wang Y; Roux B; Aksimentiev A; Luthey-Schulten Z; Kalé L; Schulten K; Chipot C; Tajkhorshid E Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Shao J; Tanner SW; Thompson N; Cheatham TE Clustering molecular dynamics trajectories: 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007, 3, 2312–2334. [DOI] [PubMed] [Google Scholar]
- (41).Zwanzig RW High-temperature equation of state by a perturbation method. II. polar gases. J. Chem. Phys. 1955, 23, 1915–1922. [Google Scholar]
- (42).Kumar A; Satpati P Structure-based thermodynamics of ion selectivity (Mg 2+ versus Ca 2+ and K+ versus Na+) in the active site of the eukaryotic lariat group II intron from algae Pylaiella littoralis. Phys. Chem. Chem. Phys. 2022, 24, 24192–24202. [DOI] [PubMed] [Google Scholar]
- (43).Shukla S; Kumar A; Das D; Satpati P Principle of DNA recognition by sporulation-regulatory protein (Spo0A) in Bacillus subtilis. J. Biomol. Struct. Dyn. 2020, 38, 5186–5194. [DOI] [PubMed] [Google Scholar]
- (44).Kumar A; Åqvist J; Satpati P Principles of tRNAAla Selection by Alanyl–tRNA Synthetase Based on the Critical G3· U70 Base Pair. ACS omega 2019, 4, 15539–15548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Gilbert SD; Love CE; Edwards AL; Batey RT Mutational analysis of the purine riboswitch aptamer domain. Biochem. 2007, 46, 13297–13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Gilbert SD; Reyes FE; Edwards AL; Batey RT Adaptive ligand binding by the purine riboswitch in the recognition of guanine and adenine analogs. Structure 2009, 17, 857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Stoddard CD; Widmann J; Trausch JJ; Marcano-Velázquez JG; Knight R; Batey RT Nucleotides adjacent to the ligand-binding pocket are linked to activity tuning in the purine riboswitch. J. Mol. Biol. 2013, 425, 1596–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Liberman JA; Salim M; Krucinska J; Wedekind JE Structure of a class II preQ1 riboswitch reveals ligand recognition by a new fold. Nat. Chem. Biol. 2013, 9, 353–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Gilbert SD; Rambo RP; Van Tyne D; Batey RT Structure of the SAM-II riboswitch bound to S-adenosylmethionine. Nat. Struct. Mol. Biol. 2008, 15, 177–182. [DOI] [PubMed] [Google Scholar]
- (50).Liberman JA; Suddala KC; Aytenfisu A; Chan D; Belashov IA; Salim M; Mathews DH; Spitale RC; Walter NG; Wedekind JE Structural analysis of a class III preQ1 riboswitch reveals an aptamer distant from a ribosome-binding site regulated by fast dynamics. Proc. Natl. Acad. Sci. 2015, 112, E3485–E3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Sund J; Lind C; Åqvist J Binding site preorganization and ligand discrimination in the purine riboswitch. J. Phys. Chem. B. 2015, 119, 773–782. [DOI] [PubMed] [Google Scholar]
- (52).Hu G; Ma A; Wang J Ligand selectivity mechanism and conformational changes in guanine riboswitch by molecular dynamics simulations and free energy calculations. J. Chem. Inf. Model. 2017, 57, 918–928. [DOI] [PubMed] [Google Scholar]
- (53).Gilbert SD; Stoddard CD; Wise SJ; Batey RT Thermodynamic and kinetic characterization of ligand binding to the purine riboswitch aptamer domain. J. Mol. Biol. 2006, 359, 754–768. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All methodological details for all data generated in this study are available in the Methods section including PDB codes of input files, simulation conditions, error analysis. The input files for conventional MD simulations and TI calculations for mutations in the NMT1-XAN complex are available at github: (https://github.com/amit9869/JCIM_XAN.git). The free energy data averaged over forward and backward transformations for each mutation in complex and solution are also provided (Tables S3–S7). The simulation and visualization software29,38,39 and the force-field for RNA32 used to carry out calculations are openly available. The CHARMM general force-field33 used for the ligands (XAN, AZA, UAC, and HPA) is available in supporting information.