

Abstract
Mutations in the PKLR gene lead to pyruvate kinase (PK) deficiency, causing chronic hemolytic anemia secondary to reduced red cell energy, which is crucial for maintenance of the red cell membrane and function. Heterogeneous clinical manifestations can result in significant morbidity and reduced health-related quality of life. Treatment options have historically been limited to supportive care, including red cell transfusions and splenectomy. Current disease-modifying treatment considerations include an oral allosteric PK activator, mitapivat, which was recently approved for adults with PK deficiency, and gene therapy, which is currently undergoing clinical trials. Studies evaluating the role of PK activators in other congenital hemolytic anemias are ongoing. The long-term effect of treatment with disease-modifying therapy in PK deficiency will require continued evaluation.
Recent advances in pyruvate kinase deficiency
Pyruvate kinase deficiency (see Glossary), a congenital hemolytic anemia caused by a glycolytic pathway defect, was first described in the 1960s. Over the past decade, through registry studies, our understanding of the clinical and genetic heterogeneity, symptoms, and potential complications has expanded. Despite this progress, diagnosing and managing patients with PK deficiency remains challenging because of difficulties in the diagnostic evaluation and heterogeneity of clinical manifestations. Supportive care with blood transfusions, splenectomy, and iron chelation have historically been the main management strategies. A recently approved PK activator, mitapivat, offers an innovative disease-directed approach that may considerably improve the disease burden and quality of life of many patients with PK deficiency. The safety and efficacy of mitapivat in adults with PK deficiency have been demonstrated with successful Phase 2 and Phase 3 clinical trials, and pediatric trials are underway. In addition, as a disease due to a single gene defect, gene therapy is a promising disease-modifying and potentially curative treatment. In the modern era of disease-targeted therapies, recognizing the symptoms and diagnosing patients with PK deficiency is even more critical. This review highlights the recent advances in the diagnosis and natural history of PK deficiency, as well as guidance for monitoring and supportive care, newly approved targeted treatments, and treatments in development.
Diagnosis, symptoms, monitoring, and supportive treatment approaches
Pathophysiology
Mature red blood cells lack a nucleus and mitochondria, thus relying on glycolysis for adenosine triphosphate (ATP) production in order to maintain structure and function. Pyruvate kinase is a key glycolytic enzyme and catalyzes the conversion of phosphoenolpyruvate to pyruvate with the production of ATP (Figure 1). PK deficiency causes red cell dehydration and disturbs the red cell membrane, leading to ineffective erythropoiesis and hemolysis. Reticulocytes, the youngest circulating red blood cells, require higher ATP levels than mature red cells and therefore are the most susceptible to PK deficiency, particularly in the splenic environment [1–3]. As PK is the terminal enzyme in glycolysis, the proximal glycolytic intermediates such as 2,3-biphosphoglycerate (2,3-BPG) are often increased [4,5]. This causes a right shift in the hemoglobin–oxygen dissociation curve, leading to increased tissue oxygenation which can result in better anemia tolerance in some patients [5,6].
Figure 1. Glycolytic pathway.
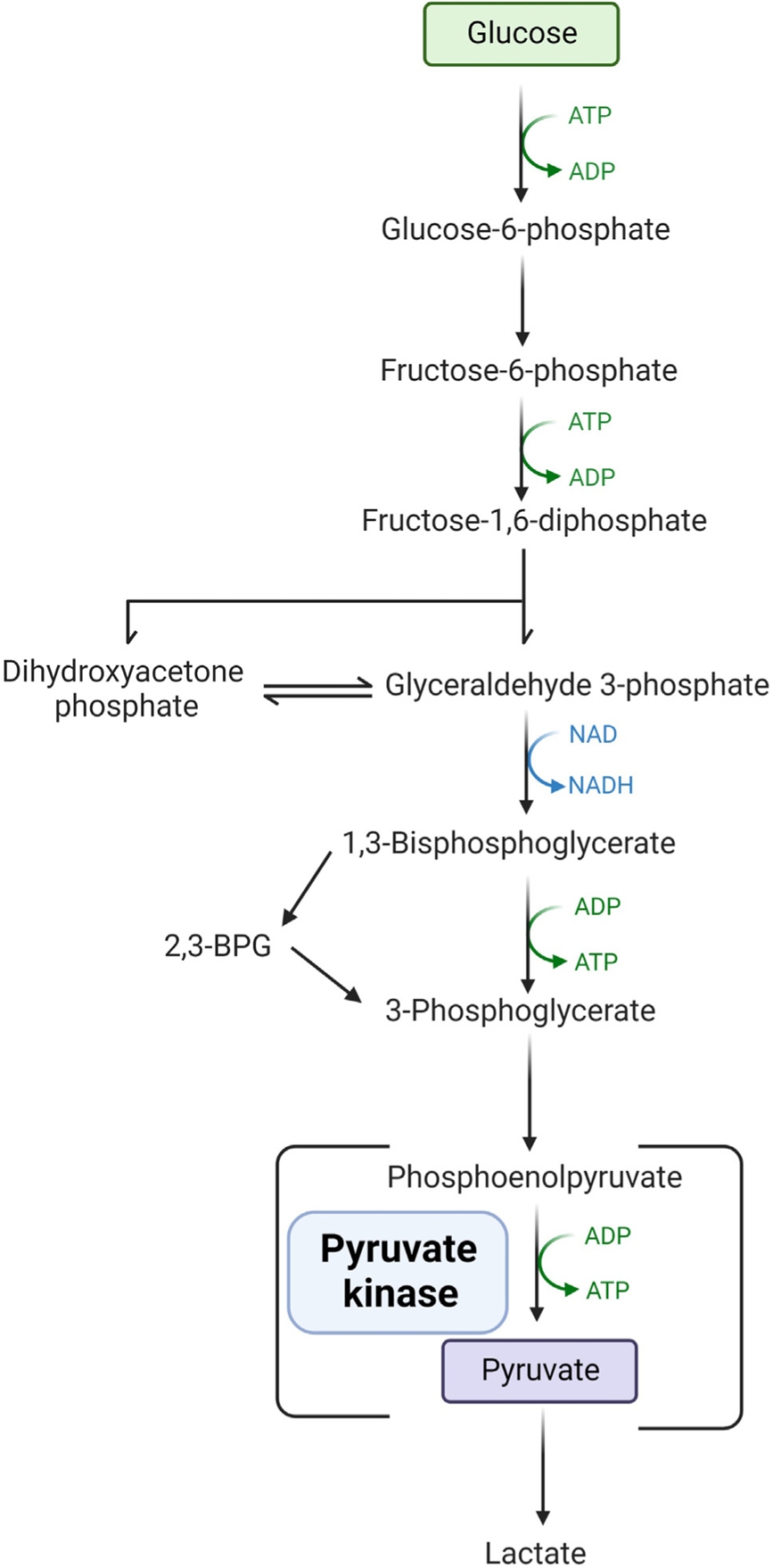
Mature red blood cells lack a nucleus and mitochondria and rely on glycolysis for the production of adenosine triphosphate (ATP). Pyruvate kinase is an enzyme in the terminal part of the pathway converting phosphoenolpyruvate to pyruvate with the production of ATP. With a deficiency of pyruvate kinase, the production of proximal byproducts, such as 2–3-biphosphoglycerate (2,3-BPG), is increased. Abbreviations: ADP: adenosine diphosphate; NAD: nicotinamide adenine dinucleotide; NADPH: nicotinamide adenine dinucleotide phosphate.
The PKLR gene, located on chromosome 1q21, encodes the liver and red cell PK isoenzymes. PK deficiency is autosomal recessive, caused by pathogenic compound heterozygous or homozygous mutations in PKLR. Pathogenic mutations in PKLR affect the PK enzyme by altering its affinity for phosphenolpyruvate (its substrate) or fructose 1,6-bisphosphate (its allosteric activator). PKLR variants can also result in decreased protein stability or altered stability of PK homotetramers [7,8].
Epidemiology
PK deficiency is genetically heterogeneous, with over 300 pathogenic PKLR variants having been described. The variants include single-nucleotide substitutions, as well as intronic and exonic deletions and insertions. The most common mutations include Arg510Gln, which is found in Northern Europe and the USA, followed by Arg486Trp in the Southern European population. A particularly high frequency exists among the Pennsylvania Amish (Arg479His) and Romany communities (1149 base pair deletions resulting in the loss of Exon 11) [9–11]. Most patients have at least one missense PKLR mutation. Highly symptomatic clinical phenotypes are generally associated with mutations that cause premature stop codons, frameshifts, or large deletions; however, there is relatively little predictive value between genotype and phenotype and the severity of the clinical course [12].
The estimated prevalence of PK deficiency is 1 to 8 per 1,000,000 [13–15]. PK deficiency has a worldwide distribution, with a possibly higher frequency in malaria-endemic regions and in isolated populations because of a founder effect. A large number of patients remain undiagnosed for various reasons including difficulties in accessing diagnostic testing, misdiagnosis, and mild disease [16–18].
Molecular and biochemical diagnosis
Testing for PK deficiency should be considered in patients with chronic hemolysis when more common causes have been excluded, including autoimmune hemolytic anemia, red cell membranopathies, and hemoglobinopathies (Figure 2). Diagnosing PK deficiency requires high clinical suspicion, given the widely variable clinical phenotype and nonspecific red cell morphology.
Figure 2. Diagnostic algorithm for the work-up of suspected congenital hemolytic anemia.
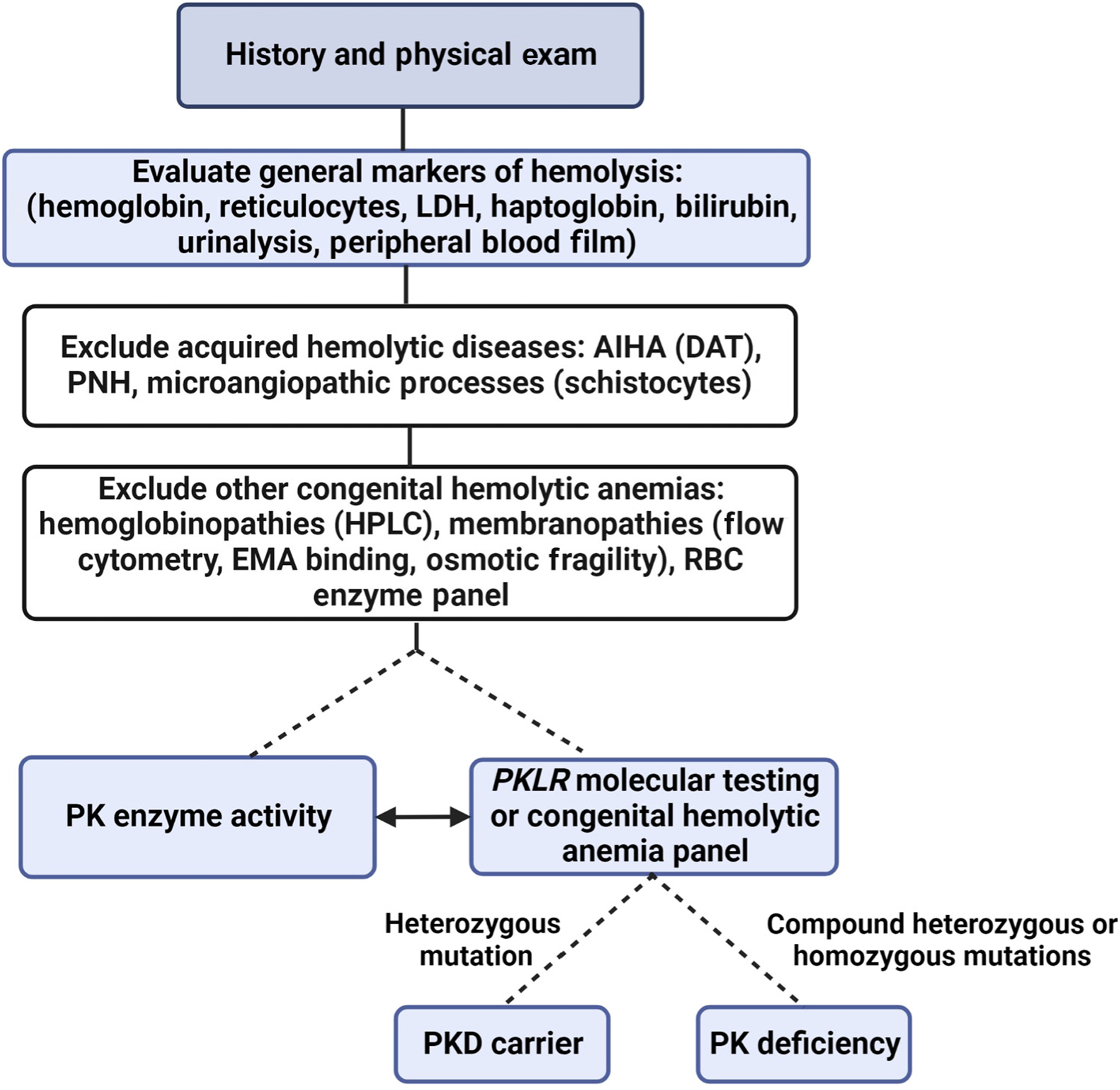
This algorithm shows an approach to the evaluation of a suspected congenital hemolytic anemia. In general, a thorough history and physical exam should be completed. Laboratory testing should evaluate for evidence of hemolysis and include an evaluation of the red cells’ morphology on the peripheral blood film. Acquired causes of hemolytic diseases should be excluded, and a full differential diagnosis for congenital hemolytic anemias should be considered. Pyruvate kinase deficiency should be suspected as a cause of congenital hemolytic anemia after more common causes are excluded, and then testing should include pyruvate kinase activity levels and PKLR genetic testing. Abbreviations: LDH: lactate dehydrogenase; AIHA: autoimmune hemolytic anemia; DAT: direct antiglobulin test; PNH: paroxysmal nocturnal hemoglobinuria; HPLC: high-performance liquid chromatography; EMA: eosin-5′-maleimide; PK: pyruvate kinase; PKD: pyruvate kinase deficiency.
Given the potential limitations of both PK enzyme activity and PKLR genetic testing, the diagnosis of PK deficiency is made through a combination of both tests, if available. Low PK enzyme activity can be found in both carriers and disease states. Falsely normal PK activity can be seen in transfused patients as a result of contamination with healthy red cells or leukocytes, increased reticulocytes, or nonphysiologic substrate concentrations [19,20]. In the presence of reticulocytosis, comparing PK activity with the activity of other age-dependent red cell enzymes, such as hexokinase, increases the sensitivity of PK enzyme testing [21,22]. Somewhat counterintuitively, the measured PK enzyme activity does not correlate with the severity of anemia or hemolysis [1,23,24].
Genetic testing for congenital hemolytic anemias is increasingly available, including through commercial laboratories using next-generation sequencing [25]. Up to 20% of individuals may have newly described PKLR variants of uncertain significance; in these cases, low PK activity can provide additional support for the diagnosis [3,26,27]. Additionally, up to 10% of individuals with reduced PK enzyme activity and a clinical presentation consistent with PK deficiency may have falsely normal genotyping in one or more PKLR alleles if only exon sequencing is performed, due to a missed deep intronic mutation or large deletion [28].
Heterogeneous spectrum of clinical and laboratory manifestations
Laboratory and clinical manifestations of PK deficiency are variable and are secondary to the symptoms and complications of chronic hemolysis (Figure 3).
Figure 3. Symptoms and complications of pyruvate kinase deficiency in adults.
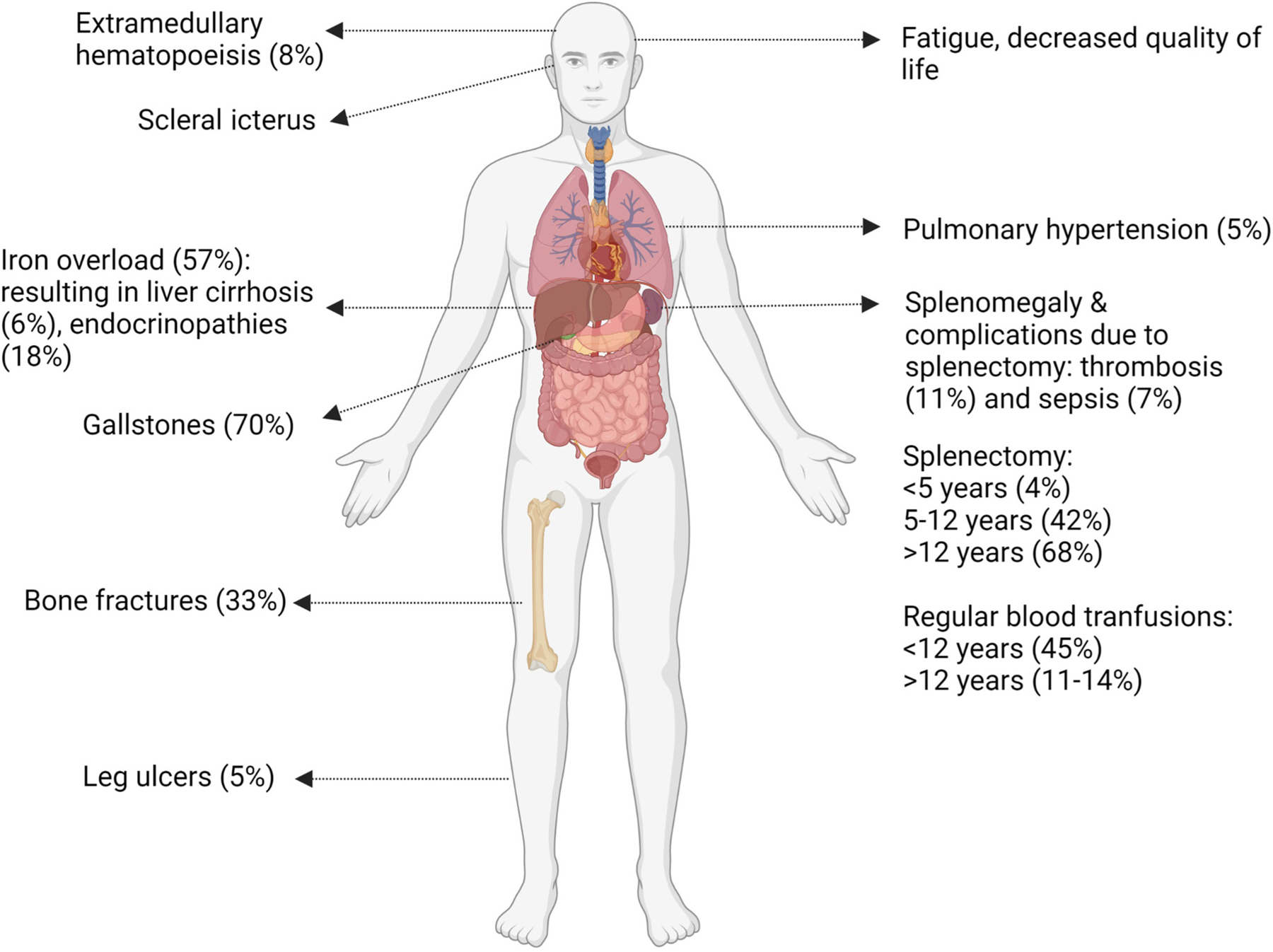
Pyruvate kinase deficiency is clinically heterogeneous, with both common and rare complications arising from both the disease and its historic treatment with blood transfusions and consideration of splenectomy.
Newborns may present with wide-ranging manifestations from unrecognized mild compensated anemia to neonatal hyperbilirubinemia and anemia requiring exchange transfusions to fulminant liver failure [13,23]. Severe manifestations can occur in utero, resulting in restricted growth, hydrops fetalis, prematurity, and/or fetal demise [29,30].
Infants and children generally have jaundice and scleral icterus, splenomegaly, and anemia, with symptoms that may include poor feeding and growth, low energy levels, and irritability. Older children and adolescents can have poor concentration, fatigue, or shortness of breath during activity. Acute exacerbations of anemia may occur during episodes of illness, including infections. Most young children will require regular or intermittent blood transfusions [13]. Severe anemia, iron overload states, persistent jaundice, and genotypes with the presence of two drastic PKLR mutations are associated with a lower quality of life [31]. Reported manifestations in adults include jaundice, pallor, fatigue, poor concentration, and shortness of breath. Pregnancy can increase hemolysis and result in a transient need for transfusions in patients who are not otherwise regularly transfused [2,13,32,33]. The clinical variability is evident in adults, with some individuals experiencing few symptoms of anemia and others, having a significant impact on their everyday quality of life [34]. In general, increased transfusion needs and complications of PK deficiency, such as iron overload requiring chelation or pulmonary hypertension, are associated with a lower quality of life [31]. With advancing age, despite stable laboratory findings, symptoms can increase in the presence of additional comorbidities.
Supportive treatment strategies and associated complications
Transfusions
Initiating red blood cell transfusion therapy depends on growth in children, daily symptoms that impact quality of life, complications, and, to a lesser extent, the nadir hemoglobin. The need for transfusions decreases with age, likely because of decreased hemolytic episodes from viral infections and the timing of splenectomy [13]. Approximately half of children less than 5 years old are regularly transfused at an average interval of 5 weeks. In one large cohort, regular transfusions were required in 30% of children aged 5–12 years and 14% aged 12–18 years [2,13]. Postsplenectomy transfusions are typically required only during intermittent hemolytic episodes, although a subset who undergo splenectomy will continue to require regular transfusions. Chronic anemia may impact quality of life, leading to the reinitiation of regular transfusions in some adults. Regular transfusions are associated with iron overload, the risk of infection, need for intravenous access, and a risk of allosensitization, as well as personal and financial costs. For these reasons, individuals may choose not to be transfused, despite having symptoms of anemia.
Splenectomy
In individuals with PK deficiency who are frequently transfused or have poor quality of life, splenectomy is a supportive treatment typically considered in childhood [35]. Splenectomy only partially alleviates anemia and is not effective for all patients [36–38]. After a splenectomy, most patients have improvements in their hemoglobin and transfusion burden, with a paradoxical increase in the reticulocyte count. Hemolysis persists after splenectomy; therefore, many complications are not resolved by a splenectomy. Splenectomy is typically avoided before the age of 5 years to reduce the risk of postsplenectomy sepsis. In one large cohort, by 12 years of age, nearly half (40%) had undergone a splenectomy and, by the age of 18 years, 70% had done so [13]. Those with less severe hemolysis with a higher presplenectomy hemoglobin and lower indirect bilirubin level are the most likely to benefit from splenectomy [13,34]. In addition to infection, there is a substantially increased lifetime risk of thrombosis postsplenectomy. After a splenectomy, patients must adhere to guidelines regarding vaccines, prophylactic antibiotics, and thromboprophylaxis. Many individuals with PK deficiency still undergo splenectomy in early childhood. As access to newly approved treatments for adults expands and trials are completed for disease-directed therapies in children, fewer individuals with PK deficiency may undergo splenectomy in the future.
Complications
Complications are caused by chronic hemolysis and/or a result of supportive treatment. Iron overload can occur both in transfused and non-transfused patients. In non-transfused patients, increased intestinal iron absorption occurs because of chronic hemolysis and ineffective erythropoiesis [13,39,40]. Iron overload can cause significant morbidity, including chronic liver disease and cirrhosis, bone fractures, cardiac dysfunction, heart failure, and endocrine dysfunction. The toxic effects of circulating free iron can be seen after 10 to 14 red cell transfusions. Regular monitoring allows for adequate treatment via iron chelation [41]. Monitoring of regular ferritin levels is recommended, with annual magnetic resonance imaging (MRI) surveillance (Table 1).
Table 1.
Clinical monitoring in patients with pyruvate kinase deficiency
| Complication | Recommended test(s) | Frequency of monitoring (age <18 years) | Frequency of monitoring (Age ≥18 years) |
|---|---|---|---|
| Hemolytic anemia | Complete blood count, reticulocyte count, bilirubin | Annually or more frequently based on hemolytic parameters and transfusions | Annually or more frequently based on hemolytic parameters and transfusions |
| Iron overload | MRI for liver iron concentration | If regular transfusions: MRI after first 10–14 transfusions, then annually If no regular transfusions: complete first MRI when the patient is able to complete an unsedated study; follow up patients annually if >5 mg/g or every 5 years if <5 mg/g |
If regular transfusions: annually If no regular transfusions: annually if >5 mg/g or every 5 years if <5 mg/g |
| Serum ferritin and transferrin saturation | If regular transfusions: every 3–6 months If no regular transfusions: annually On chelation: every 1 −3 months |
If regular transfusions: every 3–6 months If no regular transfusions: annually On chelation: every 1 −3 months |
|
| Cholestasis | Abdominal ultrasound | Age 2 years then every 2–3 years or until cholecystectomy | Every 2–3 years or until cholecystectomy |
| Osteopenia | DEXA scan, vitamin D levels | First at age 16–18 years and then annually if low | Annually if low |
| Endocrinopathies | Thyroid hormone, sex hormones, fructosamine | - | If regular transfusions (or significant iron overload): annually |
| Pulmonary hypertension, cardiac complications | Echocardiogram | - | Consider after 30 years, before pregnancy, or any time if concerns arise |
| Viral infections | Viral hepatitis serology | If regular transfusions: annually | If regular transfusions: annually |
Pigmented gallstones secondary to hemolysis and bilirubin accumulation occurs in nearly half of patients [13,42,43]. Splenectomy does not decrease this risk; therefore, cholecystectomy is often considered at the time of a splenectomy. Anemia may worsen in the setting of parvovirus infections, resulting in aplastic crisis. Pulmonary hypertension is a rare manifestation that significantly impacts quality of life when it occurs [31].
Patients with PK deficiency have a high rate of reduced bone density, with an earlier onset than in the general population. Reduced bone density may begin in adolescence or early adulthood, and most older adults with PK deficiency have osteopenia or osteoporosis [44]. Leg ulcers and extramedullary hematopoiesis are rare but potentially morbid complications in adults [45–47]. The impact of PK deficiency on quality of life can result in mental health issues; clinicians should monitor for these symptoms and provide psychological support. Monitoring guidance for individuals with PK deficiency is included in Table 1.
Targeted therapeutic strategies in PK deficiency
Mitapivat for treating PK deficiency
Mitapivat (AG-348, Agios Pharmaceuticals, Figure 4) is an oral small molecule that allosterically activates erythrocyte PK. Mitapivat binds to a separate allosteric site from fructose 1,6-bisphosphate on the red cell PK tetramer. Two Phase 3 clinical trials have demonstrated the safety and efficacy of mitapivat for adults with PK deficiency, leading to recent FDA and European Medicines Agency approval [48]. Although additional PK activators are currently in clinical development for the treatment of congenital hemolytic anemias, mitapivat is the only PK activator evaluated to date for PK deficiency. Mitapivat has excellent oral bioavailability with or without food, with a steady state reached after 1 week with dosing of every 12 h. Mitapivat is eliminated via hepatic metabolism by cytochrome P450 enzymes and is a mild to moderate aromatase inhibitor [49]. Clinical studies of mitapivat have not demonstrated meaningful endocrinologic effects to date [50,51]. Ex vivo human studies have demonstrated both increased wild-type and variant PKR enzyme activity after exposure to mitapivat [8].
Figure 4. Mechanism of mitapivat.

Mitapivat, an oral allosteric pyruvate kinase activator, stabilizes and increases the activity of pyruvate kinase, thereby leading to increased intracellular red cell adenosine triphosphate (ATP), resulting in an improvement in anemia and patient-reported outcomes and a reduction in hemolysis. Abbreviation: ADP, adenosine diphosphate.
Two Phase 1 randomized, placebo-controlled, double-blind studies in healthy volunteers assessed the pharmacokinetics, pharmacodynamics, and safety of mitapivat [49]. In a multiple ascending dose study with six healthy volunteer cohorts randomized to receive a placebo or mitapivat, the maximum increase in ATP by day 14 was 60% and the maximum decrease in 2,3-BPG was 47%. There were no serious treatment-emergent adverse events. Treatment-emergent adverse events were more common at high doses of mitapivat (≥700 mg) including headaches, nausea, and/or vomiting. Pharmacodynamic studies and the safety profile supported mitapivat trials in adults with PK deficiency.
The open-label randomized Phase 2 DRIVE-PK trial evaluated the safety and efficacy of mitapivat in adults with PK deficiency who were not receiving regular transfusions, defined as less than four transfusion episodes in the prior 12 months (NCT02476916) [5]. Patients (n = 52) were randomized to 50 mg or 300 mg of mitapivat twice daily for a 24-week period. The primary endpoint of this study was a safety assessment. The secondary objectives were characterizing the pharmacokinetic and pharmacodynamic profile and demonstrating its clinical efficacy, as measured by hemoglobin and hemolysis markers. The therapy was well-tolerated, with the most common adverse events being transient mild headaches, insomnia, and nausea. One individual experienced acute hemolysis when mitapivat was discontinued without tapering. Other rare adverse events included reduced bone mineral density, elevated transaminases, and increased triglycerides. Of the 52 patients, 26 had an increase in hemoglobin of ≥1 g/dl from the baseline, with a mean maximum increase of 3.4 g/dl (range: 1.1–5.8 g/dl). The increase in hemoglobin was rapid (a median duration of 10 days), with a durable response seen with continued dosing. A genotype–response relationship was observed, such that most patients with at least one non-R479H missense mutation had a hemoglobin response, whereas patients with two non-missense mutations or homozygous R479H mutations had poor or no responses [50].
The ACTIVATE trial was a Phase 3 placebo-controlled trial, in which 80 adults with PK deficiency who were not receiving regular transfusions were randomized to receive a placebo or mitapivat twice daily, with potential dose escalation (from 5 mg to 50 mg twice daily) for 24 weeks (NCT03548220) [51]. The inclusion criteria required two PKLR mutations, including at least one non-R479H missense mutation. The primary endpoint was a sustained hemoglobin increase of ≥1.5 g/dl at two or more assessments at weeks 16, 20, or 24 of the trial, a higher response threshold than in the DRIVE-PK study. Secondary endpoints included a change from the baseline in the hemoglobin level, markers of hemolysis and hematopoiesis, and patient-reported outcome measures. The therapy was tolerated well, with similar overall adverse event (AE) rates between the mitapivat and placebo arms, and most AEs being minor and transient. The most common AEs observed with mitapivat, nausea and headache, were both more common in patients receiving the placebo [52]. Mitapivat significantly increased the hemoglobin level, decreased hemolysis, and improved patient-reported outcomes. In this trial, 40% of the patients (16/40) receiving mitapivat met the primary endpoint versus 0% in the placebo arm. Data from an open-label extension study suggested additional benefits, including decreases in ineffective erythropoiesis and iron overload, and stable bone health [53,54].
A Phase 3 single-arm, open-label trial, ACTIVATE-T, evaluated the efficacy and safety of mitapivat in adults with PK deficiency who were regularly transfused (≥6 transfusions in the prior 12 months, NCT03559699) [55]. Eligible patients began with a 16-week dose escalation period followed by a 24-week fixed dose period. The primary endpoint was a reduction in the burden of transfusions, defined as a 33% reduction in transfusion requirements. Secondary endpoints included the proportion of transfusion-free responders and the annualized number of red cell units transfused. Twenty out of 27 enrolled patients completed the study. The primary endpoint was met, with 37% experiencing a reduction in their transfusion burden and 22% achieving a transfusion-free response. Mitapivat was also tolerated well in this trial, with no major adverse events leading to treatment discontinuation.
With the recent FDA and EMA approvals of mitapivat, the first approved therapy for adults with PK deficiency, a treatment trial of mitapivat should be considered in all symptomatic adults with PK deficiency (Figure 5, Key figure). The approved dose ranges from 5 mg to 50 mg twice per day. Given the short onset of action, the response can be assessed over the first few months of treatment and is generally clear within a few weeks of titration to the highest dose of 50 mg twice daily. In those with a hemoglobin response, ongoing use of the drug is required for a durable effect, and abrupt discontinuation should be avoided because of the risk of hemolysis. Trials are currently ongoing in regularly transfused and non-regularly transfused children with PK deficiency (NCT05144256, NCT05175105).
PK activators in other hematologic diseases
PK activators increase the production of ATP which could alleviate the increased metabolic oxidative stress seen in other congenital hemolytic anemias and help maintain the red blood cell membrane and function. Early-phase clinical trials of PK activators, mitapivat and etavopivat, in adults with thalassemia and sickle cell disease have demonstrated an increase in hemoglobin and a reduction in the markers of hemolysis, and a significant decrease in the specific sickling point in the sickle cell population [56–60]. Phase 3 clinical trials of PK activators are ongoing or planned in these patient populations (NCT04770779, NCT04770753, NCT05031780, NCT04987489, and NCT04624659). The role of PK activators in the treatment of red cell disorders may be even broader, as preclinical studies have suggested the potential efficacy of PK activators in additional rare congenital hemolytic anemias, including hereditary spherocytosis [61]. A Phase 2 trial evaluating mitapivat in adults with hereditary spherocytosis and hereditary dyserythropoietic anemia is ongoing. A potent next-generation PK activator, AG-946, has demonstrated safety in a Phase 1 trial of healthy volunteers [62] and is currently being evaluated in sickle cell disease and low-grade myelodysplastic syndromes (NCT04536792 NCT05490446) [63,64].
Hematopoietic stem cell transplantation
Allogeneic hematopoietic stem cell transplantation (HSCT) is a curative treatment option for PK deficiency. However, no clearly described indications for a transplant in this patient population exist. Although HSCT is a curative option, the few published patient cohorts revealed a high rate of morbidity and mortality associated with transplants compared with current standard supportive care [65,66]. A global cohort study described 16 patients with PK deficiency who received transplants in Europe and Asia [67]. In this cohort, the median age at transplantation was 6.5 years, with median follow-up of 2.3 years. The results showed Grade 3–4 graft-versushost disease (GVHD) in a large group of patients (44%), with 5/16 patients (31%) dying from the complications of GVHD. The 2-year cumulative survival was 74%. Various factors such as donor types, conditioning regimens, GVHD and infection prophylaxis affected the prognosis of transplantation. Higher survival was noted in patients receiving transplants prior to 10 years of age, suggesting that HSCT is best considered early in life in the most severely afflicted patients (Figure 5). However, the emergence of PK activators and the ongoing development of gene therapy has further dampened enthusiasm for HSCT in light of its considerable risks.
Gene therapy
Gene therapy has been successful in diseases caused by monogenic defects and other erythrocyte disorders such as thalassemia and sickle cell disease [68]. Both mouse and dog PK-deficient models have shown that gene therapy for PK deficiency can be efficacious; a mouse model demonstrated full correction of the PK deficiency genotype when >25% of genetically corrected cells were transplanted [69–71]. An open-label Phase 1 clinical trial is currently enrolling to evaluate the safety of a hematopoietic cell-based gene therapy for patients with PK deficiency using the Lentivirus-based RP-L301 gene therapy product containing autologous genetically modified CD34+ hematopoietic stem cells with corrected PKLR (NCT04105166). Secondary outcome measures include the genetic correction of cells, transfusion requirements, and reductions in hemolysis and anemia. The interim results of two adults who have undergone gene therapy reported the normalization of hemoglobin, improved hemolysis markers and quality of life, and no transfusions required at 24 months of follow-up. To date, there have been no serious adverse events [72].
Key figure
Algorithm for treatment considerations in pyruvate kinase deficiency
Figure 5.
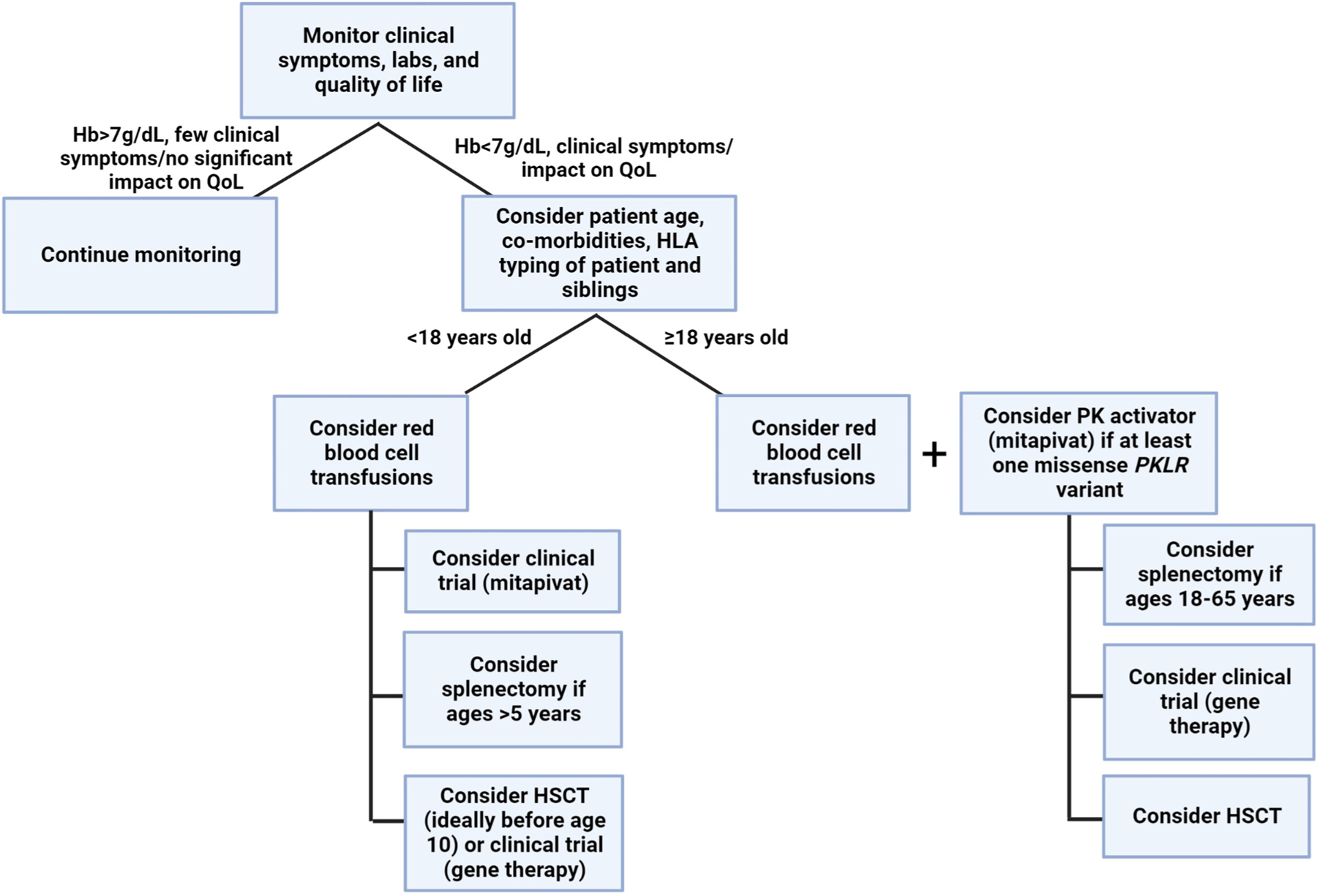
This algorithm shows an approach to the management of pyruvate kinase (PK) deficiency. Clinical signs and symptoms, in addition to hemoglobin levels, should be considered when deciding to initiate supportive care and/or disease-directed therapy. In patients with signs or symptoms of anemia that impact their quality of life, various treatment options should be considered. Symptomatic patients <18 years old should initiate red blood cell transfusions and then consider clinical trials (mitapivat or gene therapy), splenectomy, and/or a hematopoietic stem cell transplant (HSCT). In symptomatic adults ≥18 years, a treatment trial with a PK activator should be initiated, along with consideration of red cell transfusions. Additional options such as HSCT, clinical trials (gene therapy), and splenectomy can be considered if there is no response to a PK activator.
Concluding remarks
PK deficiency is a rare congenital hemolytic anemia caused by homozygous or compound heterozygous mutations in the PKLR gene, leading to decreased production of ATP in the red cells, causing membrane defects and dysfunction. Clinically, patients have chronic hemolysis causing manifestations with variable severity, including jaundice, gallstones, iron overload, extramedullary hematopoiesis, and decreased patient-reported quality of life (see Clinician’s corner). Providers must have a high index of suspicion for this disease and send samples for confirmatory diagnostic testing of PK enzyme activity and genetic sequencing, since lab findings, peripheral blood morphology, and clinical manifestations can be nonspecific.
Clinician’s corner.
PK deficiency is a clinically heterogeneous congenital hemolytic anemia with a wide spectrum of manifestations, including pallor, fatigue, and jaundice, and complications, including iron overload, gallstones, osteopenia, and pulmonary hypertension.
The diagnosis of PK deficiency should be suspected in patients with hemolytic anemia once more common causes, such as autoimmune hemolytic anemia, hemoglobinopathies, and membranopathies, have been excluded. The diagnosis should be confirmed through both the enzyme activity of red blood cells and PKLR genetic testing (targeted gene testing or congenital hemolytic anemia gene panel).
Supportive care with blood transfusions, splenectomy, and iron chelation have historically been the main management strategies and are also associated with the potential for complications.
Complications in patients with PK deficiency can be associated with significant morbidity, including impacts on their daily quality of life and mental health, which requires regular monitoring and management.
Disease-targeted therapy with a recently approved PK activator, mitapivat, offers an innovative disease-directed approach that, in addition to other therapies undergoing trials (gene therapy), may transform the manifestations of the disease and the quality of life of many patients with PK deficiency.
Historically, treatment has focused on supportive care with transfusions, splenectomy, and iron chelation. Recently, treatment has been transformed by disease-directed therapy, including the PK activator, mitapivat, which is now approved for adults with PK deficiency, with studies underway in children. Gene therapy is a promising potentially curative treatment option currently under development, and HSCT is a currently available curative treatment option associated with high morbidity and mortality. PK activation and gene therapy have promise in revolutionizing care for patients by decreasing the rate of both the disease and the treatment-related complications.
Future research should focus on long-term follow-up of disease-directed therapies and the impact on patients’ health and quality of life (see Outstanding questions). These therapies may have lifelong benefits when initiated in children, who could potentially avoid transfusion therapy and splenectomy. The role of PK activators in other congenital hemolytic anemias should be explored. There may be unexpected implications of disease-directed therapies which will require close monitoring in the real-world setting. As treatment options for PK deficiency are both expanded and refined, consensus clinical guidelines for providers should be developed to ensure the widespread adoption of evidence-based care for this patient population.
Outstanding questions.
Will mitapivat allow children with PK deficiency to avoid blood transfusions and splenectomy and their associated complications, as has been demonstrated in adults?
If disease-targeted therapies, including mitapivat and gene therapy, are safe and have short-term efficacy in children, will they have life-long efficacy and transform the natural history of the disease in terms of lifelong symptoms and complications?
Will PK activators be efficacious in raising hemoglobin levels, reducing transfusions, and improving the patients’ quality of life in other congenital and acquired hemolytic anemias?
Which individuals with PK deficiency should consider specific treatments, including hematopoietic stem cell transplant, mitapivat (or other PK activators), splenectomy, and/or transfusions? When is the optimal time to initiate such treatments?
What guidelines should providers follow with regard to monitoring and managing patients with PK deficiency?
Highlights.
Pyruvate kinase (PK) deficiency is a genetically and clinically heterogeneous congenital hemolytic anemia which manifests with a wide range of symptoms and complications.
Given the clinical heterogeneity, clinicians should have a high suspicion of PK deficiency in patients with congenital hemolytic anemias, and both red cell enzyme and genetic testing should be pursued.
Complications in PK deficiency can be associated with significant morbidity and mortality in transfused and non-transfused patients, including iron overload, osteopenia, and impact on quality of life and mental health, which require regular monitoring and management.
A definitive diagnosis of PK deficiency has become critical with the availability of disease-modifying therapy with mitapivat, an oral PK activator which increases hemoglobin and decreases hemolysis in many patients, as well as the potential future curative option of gene therapy.
Acknowledgments
Hanny Al-Samkari receives support from the National Institutes of Health/National Heart, Lung, and Blood Institute (1K23HL159313) and is the recipient of the American Society of Hematology Scholar Award.
Glossary
- Adenosine triphosphate (ATP)
the source of cellular energy
- 2,3-Biphosphoglycerate (2,3-BPG), also 2,3-diphosphoglycerate (2,3-DBG)
a glycolytic metabolic intermediate and regulator of hemoglobin–oxygen affinity and assists with off-loading oxygen into tissues
- Hemolytic anemia
decreased red cell survival (less than 120 days) leading to premature red cell death
- Missense mutation
gene variants that cause an amino acid change
- Phosphoenolpyruvate
glycolytic intermediate
- PKLR
the gene encoding pyruvate kinase that produces the pyruvate kinase protein of liver and red blood cell
- Pyruvate kinase
the key enzyme in an energy-generating step in the glycolytic pathway that leads to the conversion of phosphoenolpyruvate to pyruvate
- Pyruvate kinase activator
a drug that stabilizes pyruvate kinase protein and increases its activity (for example, mitapivat and etavopivat)
- Pyruvate kinase deficiency
a decrease in the activity or half-life of pyruvate kinase protein, leading to a deficiency in energy in red blood cells
- Reticulocyte
the youngest circulating red blood cells, 1–2 days after emerging from the bone marrow
Footnotes
Declaration of interests
H.A-S. has received research funding from Agios, Sobi, Amgen, and Vaderis and is a consultant for Agios, Sobi, Forma, Rigel, Argenx, Moderna, and Novartis. R.F.G. has received research funding from Agios, Sobi, and Novartis and is a consultant for Agios and Sanofi. The remaining authors have no interests to declare.
References
- 1.Bianchi P et al. (2020) Genotype–phenotype correlation and molecular heterogeneity in pyruvate kinase deficiency. Am. J. Hematol 95, 472–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Grace RF and Barcellini W (2020) Management of pyruvate kinase deficiency in children and adults. Blood 136, 1241–1249 [DOI] [PubMed] [Google Scholar]
- 3.Grace RF et al. (2015) Erythrocyte pyruvate kinase deficiency: 2015 status report. Am. J. Hematol 90, 825–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kedar PS et al. (2006) Red cell pyruvate kinase deficiency in neonatal jaundice cases in India. Indian J. Pediatr 73, 985–988 [DOI] [PubMed] [Google Scholar]
- 5.Lakomek M et al. (1994) Erythrocyte pyruvate kinase deficiency. The influence of physiologically important metabolites on the function of normal and defective enzymes. Enzyme Protein 48, 149–163 [PubMed] [Google Scholar]
- 6.Oski FA et al. (1971) The role of the left-shifted or right-shifted oxygen-hemoglobin equilibrium curve. Ann. Intern. Med 74, 44–46 [DOI] [PubMed] [Google Scholar]
- 7.Wang C et al. (2001) Human erythrocyte pyruvate kinase: characterization of the recombinant enzyme and a mutant form (R510Q) causing nonspherocytic hemolytic anemia. Blood 98, 3113–3120 [DOI] [PubMed] [Google Scholar]
- 8.Kung C et al. (2017) AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood 130, 1347–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canu G et al. (2016) Red blood cell PK deficiency: an update of PK-LR gene mutation database. Blood Cells Mol. Dis 57, 100–109 [DOI] [PubMed] [Google Scholar]
- 10.Viprakasit V et al. (2014) Mutations in Kruppel-like factor 1 cause transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Blood 123, 1586–1595 [DOI] [PubMed] [Google Scholar]
- 11.Zanella A et al. (2001) Molecular characterization of the PK-LR gene in sixteen pyruvate kinase-deficient patients. Br. J. Haematol 113, 43–48 [DOI] [PubMed] [Google Scholar]
- 12.Zanella A et al. (2007) Pyruvate kinase deficiency: the genotype-phenotype association. Blood Rev. 21, 217–231 [DOI] [PubMed] [Google Scholar]
- 13.Grace RF et al. (2018) Clinical spectrum of pyruvate kinase deficiency: data from the Pyruvate Kinase Deficiency Natural History Study. Blood 131, 2183–2192 [DOI] [PubMed] [Google Scholar]
- 14.Beutler E and Gelbart T (2000) Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Blood 95, 3585–3588 [PubMed] [Google Scholar]
- 15.Carey PJ et al. (2000) Prevalence of pyruvate kinase deficiency in northern European population in the north of England. Northern Region Haematologists Group. Blood 96, 4005–4006 [PubMed] [Google Scholar]
- 16.Fung RH et al. (1969) Screening of pyruvate kinase deficiency and G6PD deficiency in Chinese newborn in Hong Kong. Arch. Dis. Child 44, 373–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bowman HS et al. (1965) Pyruvate kinase deficient hemolytic anemia in an Amish isolate. Am. J. Hum. Genet. 17, 1–8 [PMC free article] [PubMed] [Google Scholar]
- 18.Rider NL et al. (2011) Erythrocyte pyruvate kinase deficiency in an old-order Amish cohort: longitudinal risk and disease management. Am. J. Hematol 86, 827–834 [DOI] [PubMed] [Google Scholar]
- 19.Lenzner C et al. (1994) Mutations in the pyruvate kinase L gene in patients with hereditary hemolytic anemia. Blood 83, 2817–2822 [PubMed] [Google Scholar]
- 20.Beutler E et al. (1987) Elevated pyruvate kinase activity in patients with hemolytic anemia due to red cell pyruvate kinase “deficiency”. Am. J. Med 83, 899–904 [DOI] [PubMed] [Google Scholar]
- 21.Titapiwatanakun R et al. (2008) Relative red blood cell enzyme levels as a clue to the diagnosis of pyruvate kinase deficiency. Pediatr. Blood Cancer 51, 819–821 [DOI] [PubMed] [Google Scholar]
- 22.Al-Samkari H et al. (2021) The pyruvate kinase (PK) to hexokinase enzyme activity ratio and erythrocyte PK protein level in the diagnosis and phenotype of PK deficiency. Br. J. Haematol 192, 1092–1096 [DOI] [PubMed] [Google Scholar]
- 23.Zanella A et al. (2005) Red cell pyruvate kinase deficiency: molecular and clinical aspects. Br. J. Haematol 130, 11–25 [DOI] [PubMed] [Google Scholar]
- 24.Pekrun A et al. (1995) Diagnosis of pyruvate kinase deficiency in the presence of an elevated reticulocyte count. Dtsch. Med. Wochenschr 120, 1620–1624 [DOI] [PubMed] [Google Scholar]
- 25.Baronciani L and Beutler E (1993) Analysis of pyruvate kinase-deficiency mutations that produce nonspherocytic hemolytic anemia. Proc. Natl. Acad. Sci. U. S. A. 90, 4324–4327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bianchi P et al. (2019) Addressing the diagnostic gaps in pyruvate kinase deficiency: consensus recommendations on the diagnosis of pyruvate kinase deficiency. Am. J. Hematol 94, 149–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaouani M et al. (2017) Molecular basis of pyruvate kinase deficiency among Tunisians: description of new mutations affecting coding and noncoding regions in the PKLR gene. Int. J. Lab. Hematol 39, 223–231 [DOI] [PubMed] [Google Scholar]
- 28.Baronciani L and Beutler E (1994) Prenatal diagnosis of pyruvate kinase deficiency. Blood 84, 2354–2356 [PubMed] [Google Scholar]
- 29.Gilsanz F et al. (1993) Fetal anaemia due to pyruvate kinase deficiency. Arch. Dis. Child 69, 523–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ferreira P et al. (2000) Hydrops fetalis associated with erythrocyte pyruvate kinase deficiency. Eur. J. Pediatr 159, 481–482 [DOI] [PubMed] [Google Scholar]
- 31.Al-Samkari H et al. (2022) Health-related quality of life and fatigue in children and adults with pyruvate kinase deficiency. Blood Adv. 6, 1844–1853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dolan LM et al. (2002) Pyruvate kinase deficiency in pregnancy complicated by iron overload. BJOG 109, 844–846 [DOI] [PubMed] [Google Scholar]
- 33.Amankwah KS et al. (1980) Hemolytic anemia and pyruvate kinase deficiency in pregnancy. Obstet. Gynecol 55, 42S–44S [DOI] [PubMed] [Google Scholar]
- 34.Al-Samkari H et al. (2020) The variable manifestations of disease in pyruvate kinase deficiency and their management. Haematologica 105, 2229–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iolascon A et al. (2017) Recommendations regarding splenectomy in hereditary hemolytic anemias. Haematologica 102, 1304–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leblond PF et al. (1978) Erythrocyte populations in pyruvate kinase deficiency anaemia following splenectomy. II. Cell deformability. Br. J. Haematol 39, 63–70 [DOI] [PubMed] [Google Scholar]
- 37.Tanaka KR and Paglia DE (1971) Pyruvate kinase deficiency. Semin. Hematol 8, 367–396 [PubMed] [Google Scholar]
- 38.Nathan DG et al. (1968) Life-span and organ sequestration of the red cells in pyruvate kinase deficiency. N. Engl. J. Med 278, 73–81 [DOI] [PubMed] [Google Scholar]
- 39.Marshall SR et al. (2003) The dangers of iron overload in pyruvate kinase deficiency. Br. J. Haematol 120, 1090–1091 [DOI] [PubMed] [Google Scholar]
- 40.Chonat S et al. (2021) Pyruvate kinase deficiency in children. Pediatr. Blood Cancer 68, e29148. [DOI] [PubMed] [Google Scholar]
- 41.van Beers EJ et al. (2019) Prevalence and management of iron overload in pyruvate kinase deficiency: report from the Pyruvate Kinase Deficiency Natural History Study. Haematologica 104, e51–e53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Al-Samkari H et al. (2020) Characterization of the severe phenotype of pyruvate kinase deficiency. Am. J. Hematol E281–E285 [DOI] [PubMed] [Google Scholar]
- 43.Boscoe AN et al. (2021) Comorbidities and complications in adults with pyruvate kinase deficiency. Eur. J. Haematol 106, 484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Samkari H et al. (2020) Early-onset osteopenia and osteoporosis in patients with pyruvate kinase deficiency. Blood 136, 30–32 [Google Scholar]
- 45.Plensa E et al. (2005) Paravertebral extramedullary hematopoiesis due to pyruvate kinase deficiency. Haematologica 90, ECR32. [PubMed] [Google Scholar]
- 46.Rutgers MJ et al. (1979) Spinal cord compression by extramedullary hemopoietic tissue in pyruvate-kinase-deficiency-caused hemolytic anemia. Neurology 29, 510–513 [DOI] [PubMed] [Google Scholar]
- 47.Aizawa S et al. (2003) Ineffective erythropoiesis in the spleen of a patient with pyruvate kinase deficiency. Am. J. Hematol 74, 68–72 [DOI] [PubMed] [Google Scholar]
- 48.Al-Samkari H and van Beers EJ (2021) Mitapivat, a novel pyruvate kinase activator, for the treatment of hereditary hemolytic anemias. Ther. Adv. Hematol 12 20406207211066070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang H et al. (2019) Phase 1 single- and multiple-ascending-dose randomized studies of the safety, pharmacokinetics, and pharmacodynamics of AG-348, a first-in-class allosteric activator of pyruvate kinase R, in healthy volunteers. Clin. Pharmacol. Drug Dev 8, 246–259 [DOI] [PubMed] [Google Scholar]
- 50.Grace RF et al. (2019) Safety and efficacy of mitapivat in pyruvate kinase deficiency. N. Engl. J. Med 381, 933–944 [DOI] [PubMed] [Google Scholar]
- 51.Al-Samkari H et al. (2021) ACTIVATE: a phase 3, randomized, multicenter, double-blind, placebo-controlled study of mitapivat in adults with pyruvate kinase deficiency who are not regularly transfused. In Proceedings of the European Haematology Association (EHA) Virtual Congress, pp. 17, Virtual Congress [Google Scholar]
- 52.Al-Samkari H et al. (2022) Mitapivat versus placebo for pyruvate kinase deficiency. N. Engl. J. Med 386, 1432–1442 [DOI] [PubMed] [Google Scholar]
- 53.Barcellini W et al. (2022) Long-term treatment with oral mitapivat is associated with normalization of hemoglobin levels in patients with pyruvate kinase deficiency. In Proceedings of the European Hematology Association, pp. 1429–1430, Virtual Congress [Google Scholar]
- 54.Kuo KHM et al. (2022) Long-term improvements in patient-reported outcomes in patients with pyruvate kinase deficiency treated with mitapivat. Blood 140, 1223–1225 [Google Scholar]
- 55.Glenthøj A et al. (2021) ACTIVATE-T: a phase 3, open-label, multicenter study of mitapivat in adults with pyruvate kinase deficiency who are regularly transfused. In Proceedings of the European Haematology Association (EHA) Virtual Congress, pp. 17, Virtual Congress [Google Scholar]
- 56.Rab MAE et al. (2021) Decreased activity and stability of pyruvate kinase in sickle cell disease: a novel target for mitapivat therapy. Blood 137, 2997–3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu JZ et al. (2022) A phase 1 dose escalation study of the pyruvate kinase activator mitapivat (AG-348) in sickle cell disease. Blood 140, 2053–2062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Dijk MJ et al. (2022) Safety and efficacy of mitapivat, an oral pyruvate kinase activator, in sickle cell disease: a phase 2, open-label study. Am. J. Hematol 97, E226–E229 [DOI] [PubMed] [Google Scholar]
- 59.Schroeder P et al. (2022) Etavopivat, a pyruvate kinase activator in red blood cells, for the treatment of sickle cell disease. J. Pharmacol. Exp. Ther 380, 210–219 [DOI] [PubMed] [Google Scholar]
- 60.Kuo KHM et al. (2022) Safety and efficacy of mitapivat, an oral pyruvate kinase activator, in adults with non-transfusion dependent alpha-thalassaemia or beta-thalassaemia: an open-label, multicentre, phase 2 study. Lancet 400, 493–501 [DOI] [PubMed] [Google Scholar]
- 61.Mattè A et al. (2020) The pyruvate kinase activator mitapivat ameliorates anemia and prevents iron overload in a mouse model of hereditary spherocytosis. Blood 136, 29 [Google Scholar]
- 62.Dai Gurov X et al. (2022) Results from the single and multiple ascending dose study to assess the safety, tolerability, pharmacokinetics, and pharmacodynamics of AG-946 in healthy volunteers. Blood 140, 5426–5427 [Google Scholar]
- 63.Rab MAE et al. (2021) Pharmacodynamic effects of AG-946, a highly potent next-generation activator of pyruvate kinase, in ex vivo treatment of red blood cells from sickle cell disease patients. Blood 138, 2029 [Google Scholar]
- 64.Al-Samkari H et al. (2022) A Phase 2a/2b multicenter study of AG-946 in patients with anemia due to lower-risk myelodysplastic syndromes. Blood 140, 4076–4078 [Google Scholar]
- 65.Kim M et al. (2016) Hemolytic anemia with null PKLR mutations identified using whole exome sequencing and cured by hematopoietic stem cell transplantation combined with splenectomy. Bone Marrow Transplant. 51, 1605–1608 [DOI] [PubMed] [Google Scholar]
- 66.Tanphaichitr VS et al. (2000) Successful bone marrow transplantation in a child with red blood cell pyruvate kinase deficiency. Bone Marrow Transplant. 26, 689–690 [DOI] [PubMed] [Google Scholar]
- 67.van Straaten S et al. (2018) Worldwide study of hematopoietic allogeneic stem cell transplantation in pyruvate kinase deficiency. Haematologica 103, e82–e86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cavazzana-Calvo M et al. (2010) Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 467, 318–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meza NW et al. (2009) Rescue of pyruvate kinase deficiency in mice by gene therapy using the human isoenzyme. Mol. Ther 17, 2000–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Richard RE et al. (2004) Modulating erythrocyte chimerism in a mouse model of pyruvate kinase deficiency. Blood 103, 4432–4439 [DOI] [PubMed] [Google Scholar]
- 71.Tani K et al. (1994) Retrovirus-mediated gene transfer of human pyruvate kinase (PK) cDNA into murine hematopoietic cells: implications for gene therapy of human PK deficiency. Blood 83, 2305–2310 [PubMed] [Google Scholar]
- 72.Shah A (2022) Lentiviral-mediated gene therapy for adults and children with severe pyruvate kinase deficiency: results from a global Phase 1 study. Blood 140, 4902–4903 [Google Scholar]