

Abstract
Transposable elements (TE) play critical roles in shaping genome evolution. Highly repetitive TE sequences are also a major source of assembly gaps making it difficult to fully understand the impact of these elements on host genomes. The increased capacity of long-read sequencing technologies to span highly repetitive regions promises to provide new insights into patterns of TE activity across diverse taxa. Here we report the generation of highly contiguous reference genomes using PacBio long-read and Omni-C technologies for three species of Passerellidae sparrow. We compared these assemblies to three chromosome-level sparrow assemblies and nine other sparrow assemblies generated using a variety of short- and long-read technologies. All long-read based assemblies were longer (range: 1.12 to 1.41 Gb) than short-read assemblies (0.91 to 1.08 Gb) and assembly length was strongly correlated with the amount of repeat content. Repeat content for Bell's sparrow (31.2% of genome) was the highest level ever reported within the order Passeriformes, which comprises over half of avian diversity. The highest levels of repeat content (79.2% to 93.7%) were found on the W chromosome relative to other regions of the genome. Finally, we show that proliferation of different TE classes varied even among species with similar levels of repeat content. These patterns support a dynamic model of TE expansion and contraction even in a clade where TEs were once thought to be fairly depauperate and static. Our work highlights how the resolution of difficult-to-assemble regions of the genome with new sequencing technologies promises to transform our understanding of avian genome evolution.
Keywords: Passerellidae, transposable elements, genome size, California Conservation Genomics Project, C-value
Significance.
Transposable elements (TEs) are a major driver of genome size evolution, but their activity can be difficult to study given their role in causing genome assembly gaps. We explored TE detectability in relation to genome sequencing technology across 14 sparrow genomes. First, we find sequencing technology to be a major confounding factor in TE detection. Second, in genomes assembled from long-reads we find higher levels of TEs than previously reported in songbirds. Third, the high levels of TEs emerged from unique histories of TE proliferation and deletion across species. These findings highlight how the latest generation of sequencing technologies promise to reveal novel insights into TE dynamics that may have been missed from analyses based on short-read genome assemblies.
Introduction
The dynamics of transposable element (TE) activity within host genomes are a major driver of genome evolution (Ågren and Wright 2011). Transposable elements proliferate throughout the genome either through copy-and-paste mechanisms (Class I elements; e.g. long interspersed nuclear elements [LINEs]) or through cut-and-paste mechanisms (class II elements; e.g. DNA transposons). The mobility of these elements in the genome contributes to structural variation (e.g. indels, inversions), alterations to gene expression, and the evolution of gene regulatory networks (Feschotte 2008; Schrader and Schmitz 2019). Given the genomic disruption potentially caused by TEs, the majority of new TE insertions are likely deleterious and species exhibit a wide range of defense mechanisms to both silence and delete TEs from the genome (Goodier 2016). Nevertheless, co-option of TEs by the host genome has led to the evolution of novel phenotypes (Mi et al. 2000; Cornelis et al. 2017) including color polymorphisms (van't Hof et al. 2016; Kratochwil et al. 2022), increased immunity (Brosh et al. 2022), insecticide resistance (Daborn et al. 2002), and speciation (Serrato-Capuchina and Matute 2018). To date, much of TE biology has focused on model organisms with well-characterized genomic resources. The generation of high-quality genomes for a diversity of nonmodel organisms (Teeling et al. 2018; Feng et al. 2020; Rhie et al. 2021; Lewin et al. 2022) promises to broaden our understanding of how co-evolutionary dynamics between TEs and their host shape genome evolution.
Avian genomes provide an illuminating case of how expanding the diversity of available genome assemblies has altered our understanding of TE dynamics. Among amniotes, birds exhibit the smallest and most constrained genomes. Although contraction of avian genomes likely began prior to the evolution of flight (Organ et al. 2007), the high metabolic demand of flight is the leading hypothesis for continued constraint on avian genome size evolution (Hughes and Hughes 1995; Andrews et al. 2009; Wright et al. 2014). Consistent with a hypothesis of constrained genome evolution, the first avian genomes sequenced revealed low repeat content (<10%), little recent TE activity, and high chromosomal stability (Ellegren 2010). Detailed TE annotation of an increasing diversity of avian genome assemblies has since challenged the early narrative of low repeat content and high stability. First, comparative analyses across 12 avian genomes showed that the apparent stability in avian genome size was actually the product of a more dynamic history of genomic expansions offset by large-scale deletions (Kapusta et al. 2017). Second, extensive variation in the timing and proliferation of TE elements has been discovered across birds (Kapusta and Suh 2017; Suh et al. 2018; Galbraith et al. 2021). This includes the discovery of relatively high repeat content of 20% to 30% in the orders Piciformes (woodpeckers and allies) and Bucerotiformes (hornbills and hoopoes; Zhang et al. 2014; Manthey et al. 2018; Feng et al. 2020). Third, novel TEs have been discovered in avian lineages that derive from horizontal gene transfer from filarial nematodes (Suh et al. 2016). Finally, highly contiguous assemblies have confirmed that previous challenges to assembling the W chromosome were due in part to its role as a refugium for long terminal repeat (LTR) retrotransposons (Peona et al. 2021a, b; Warmuth et al. 2022).
Our understanding of TE dynamics in avian genomes is poised to advance further with the increased use of long-read sequencing technologies (Kapusta and Suh 2017; Rhie et al. 2021). Repetitive regions of the genome, including centromeres, telomeres, and the W chromosome, are a major source of assembly gaps. Consequently, repetitive DNA is thought to make up a large proportion of the 7% to 42% of the genomic DNA missing from short-read genome assemblies relative to flow cytometry or densitometry estimates of genome size (hereafter the C-value; Peona et al. 2018). Indeed, a recent comparison of assembly methods for the paradise crow (Lycocorax pyrrhopterus) showed that gaps in short-read assemblies were primarily caused by LTR retrotransposons and simple repeats (Peona et al. 2021a). Further, a recent comparison of activity levels of the chicken repeat 1 (CR1) retrotransposon across 117 avian genomes found a relationship between assembly contiguity (scaffold N50) and number of full length CR1s identified in individual genomes (Galbraith et al. 2021). This pattern was found across all genomes analyzed and also explained intra-generic variation in CR1 insertions. Detailed TE annotation of highly contiguous genomes will be essential for overcoming the confounding influence of assembly quality on patterns of TE diversity. In particular, studies leveraging highly contiguous genomes to explore TE dynamics across shorter evolutionary time scales are lacking but will be essential for understanding the contributions of these elements to the generation of avian diversity.
To this end, we performed in-depth TE annotations of highly contiguous genomes generated from six closely related sparrow species in the family Passerellidae. Passerellidae sparrows are a diverse clade of oscine Passeriformes, with 132 recognized species that are found throughout the Americas from northern Canada to southern Chile (Winkler et al. 2020). We generated de novo genome assemblies for Bell's sparrow (Artemisiospiza belli), Savannah sparrow (Passerculus sandwichensis), and song sparrow (Melospiza melodia) for this paper as part of the California Conservation Genomics Project (CCGP; Shaffer et al. 2022). We analyze these assemblies alongside three genomes recently sequenced by the Vertebrate Genomes Project (VGP; Rhie et al. 2021) for saltmarsh (Ammospiza caudacutus), Nelson's (Ammospiza nelsoni), and swamp sparrow (Melospiza georgiana), for a study of the genomic basis of tidal marsh adaptation. These new genome assemblies come from six members of the “grassland” sparrow clade (Klicka et al. 2014). True to their name, all species can be generally found in a variety of shrub and grassland habitats across North America. Savannah and song sparrow are the two most ecologically and geographically widespread species occupying a broad range of tundra, alpine, meadow, prairie, marsh, and shrub habitats from Alaska and northern Canada south through Mexico to Guatemala (Arcese et al. 2020; Wheelwright and Rising 2020). Bell's sparrow is found primarily in more arid chaparral and coastal sage habitat from northwestern California south into Baja California, Mexico and east into the southern San Joaquin valley and Mojave desert of southeastern California (Cicero and Koo 2012). Nelson's and swamp sparrow can primarily be found in central to eastern North America, principally in marsh habitats (Herbert and Mowbray 2020; Shriver et al. 2020). Saltmarsh sparrow is exclusively found in tidal marsh habitats of the Atlantic coast, and Nelson's, swamp, song, and Savannah sparrow all include tidal marsh specialist subspecies (Greenberg et al. 2006; Walsh et al. 2019a).
Song and Savannah sparrow are two of the most polytypic North America bird species, with 25 and 17 subspecies described in the song (Patten and Pruett 2009) and Savannah sparrow (Wheelwright and Rising 2020), respectively. In general, subspecific divergence across ecological gradients has long made all six species the focus of geographic variation and speciation studies (Marshall 1948; Aldrich 1984; Rising 2001; Cicero and Johnson 2006; Walsh et al. 2017, 2019b, 2021; Mikles et al. 2020; Clark et al. 2022). Bell's sparrow also forms a narrow hybrid zone with the sagebrush sparrow (Artemisiospiza nevadensis) in Owen's Valley of eastern California (Cicero and Johnson 2007; Cicero and Koo 2012), while Nelson's and saltmarsh sparrow hybridize along the coast of southern Maine (Rising and Avise 1993; Shriver et al. 2005; Walsh et al. 2015). Additionally, studies of these six sparrow species have provided important insights into avian life history and demography (Nice 1937; Johnston 1954; Keller et al. 1994; Keller and Arcese 1998; Marr et al. 2002; Freeman-Gallant et al. 2005; Ruskin et al. 2017a, b; Field et al. 2018), physiology (Poulson 1965; Greenberg et al. 2012; Benham and Cheviron 2020), vocal learning and behavior (Marler and Peters 1977; Searcy and Marler 1981; Williams et al. 2022), and migratory behavior (Moore 1978 ; Able and Able 1996). The generation of highly contiguous reference genomes for these sparrow species with in-depth TE annotations will thus provide a critically important resource for future research in this intensively studied clade.
In addition to the six new sparrow genome assemblies, nine other assemblies were analyzed from across the Passerellidae family. The previous assemblies were produced using a variety of short- and long-read sequencing approaches. Previously sequenced genomes also include short-read assemblies for both the song and saltmarsh sparrows, which allows for intra-specific comparisons to assess the impact of sequencing technology on repeat annotation. We take advantage of the diverse genomic resources available from within this single avian family to ask: (i) what is the impact of sequencing technology and assembly completeness on TE element annotation? And (ii) how do the evolutionary dynamics of TEs vary among closely related sparrow species? Addressing these questions will be important for determining how different sequencing approaches may introduce bias into comparative genomics analyses. In addition, our comparisons provide insights into how analyses based on short-read assemblies may miss important dynamics of avian genome evolution.
Results
Genome Assemblies
The three CCGP assemblies included a low number of 337 scaffolds in the Savannah sparrow and a high number of 1,339 scaffolds in Bell's sparrow; total assembly lengths ranged from 1.15 to 1.40 Gb (Table 1; supplementary figs. S1 to S3, Supplementary Material online). All assembly metrics indicate that the genomes are highly contiguous with contig N50 ranging from 5.98 to 8.31 Mb and scaffold N50 from 17.08 to 25.78 Mb. The largest contig length was over 32.13 Mb and the longest scaffold over 99.81 Mb. Over 95% of the genes in the avian orthologous database were found to be complete and single copy in BUSCO. These metrics indicate that the genomes generated de novo by the CCGP pipeline are in line with overall contiguity and completeness metrics for the three genomes generated by the VGP. Genomes for swamp, saltmarsh, and Nelson's sparrow showed similar contig N50 ranging from 8.25 to 12.04 Mb, but scaffold N50s were approximately 3 × as large (74.25 to 78.44 Mb). The VGP genomes were also assembled into chromosome-level assemblies with 36 to 40 chromosomes identified based on decreasing order of size. BUSCO scores were highly similar between the two sets of genomes ranging from 94.8% in Nelson's sparrow to 95.6% in Bell's sparrow. For the VGP genomes, BUSCO scores exceeded 98% when run using protein mode in the NCBI Eukaryotic Genome Annotation Pipeline (Thibaud-Nissen et al. 2013). Jupiter plots showed CCGP sparrow scaffolds mapping to most chromosomes of the zebra finch genome, with little evidence for inversions or translocations that may be indicative of misassemblies (Fig. 1). Similarly, although contact maps for the primary assemblies of the three CCGP genomes show some level of fragmentation, they also reveal little evidence for inversions or translocations (supplementary fig. S4, Supplementary Material online). Given their greater contiguity, we only describe the primary assemblies here, but the sequences corresponding to both primary and alternate assemblies for each of the CCGP species are available on NCBI (See supplementary table S1, Supplementary Material online and Data availability for details).
Table 1.
Comparison of assembly quality statistics and BUSCO search results among the three CCGP (left three) and three VGP (right three) genomes. BUSCO results for all genomes were obtained using the 8,338 universal single copy genes in birds found in the aves_odb10 database
| Genome metrics | Savannah Sparrow (Passerculus sandwichensis) | Bell's Sparrow (Artemisiospiza belli) | Song Sparrow (Melospiza melodia) | Swamp sparrow (Melospiza georgiana) | Nelson's sparrow (Ammospiza nelsoni) | Saltmarsh sparrow (Ammospiza caudacuta) |
|---|---|---|---|---|---|---|
| Chromosomes | NA | NA | NA | 36 | 37 | 40 |
| # contigs | 676 | 1,539 | 823 | 276 | 292 | 645 |
| Largest contig (bp) | 32,137,824 | 35,931,659 | 59,497,540 | 29,976,604 | 50,792,433 | 43,812,934 |
| Total length (bp) | 1,152,258,190 | 1,401,798,777 | 1,356,272,071 | 1,160,782,308 | 1,180,370,373 | 1,239,216,328 |
| GC (%) | 43.1 | 43.46 | 44.45 | 43.24 | 43.01 | 43.5 |
| N50 | 5,981,027 | 8,253,817 | 8,311,625 | 10,446,106 | 12,036,358 | 8,252,193 |
| N75 | 2,762,208 | 1,578,947 | 3,378,466 | 3,855,203 | 4,591,448 | 3,265,797 |
| L50 | 50 | 45 | 39 | 36 | 27 | 39 |
| # scaffolds | 337 | 1,339 | 501 | 40 | 77 | 282 |
| Largest scaffold (bp) | 124,432,526 | 99,814,828 | 153,992,920 | 155,044,423 | 155,447,619 | 157,152,855 |
| Total length (bp) | 1,152,292,115 | 1,401,818,823 | 1,356,304,709 | 1,162,015,399 | 1,185,463,352 | 1,241,209,685 |
| GC (%) | 43.1 | 43.46 | 44.45 | 43.24 | 43.01 | 43.5 |
| N50 | 18,220,233 | 17,082,054 | 25,784,215 | 74,254,230 | 74,723,840 | 78,443,464 |
| N75 | 6,722,078 | 2,980,250 | 6,297,809 | 23,937,375 | 21,551,278 | 22,481,186 |
| L50 | 17 | 20 | 14 | 6 | 6 | 6 |
| # N's per 100 kbp | 2.94 | 1.43 | 2.38 | 106.12 | 429.62 | 160.60 |
| BUSCO_results (%) | ||||||
| complete | 95.2 | 95.6 | 95.5 | 94.9 | 94.8 | 95.4 |
| complete and single copy | 94.6 | 95.2 | 95.0 | 94.5 | 94.4 | 94.9 |
| complete and duplicate | 0.6 | 0.4 | 0.5 | 0.4 | 0.4 | 0.5 |
| fragmented | 2.1 | 1.5 | 1.6 | 1.8 | 1.9 | 1.8 |
| missing | 2.7 | 2.9 | 2.9 | 3.3 | 3.3 | 2.8 |
Fig. 1.

Jupiter plot comparing higher level synteny and completeness between the zebra finch (Taeniopygia guttata) genome (bTaeGut.4) and each of the three CCGP draft assemblies of Passerellidae sparrow species. Zebra finch chromosomes are on the left in each plot (colored) and sparrow scaffolds are on the right (light gray). Twists represent reversed orientation of scaffolds between assemblies. Song and Bell's sparrow reference genome samples were both from females, whereas the Savannah sparrow reference was from a male. Song and Bell's sparrow illustrations reproduced with the permission of https://birdsoftheworld.org with permission from Lynx Edicions. Savannah sparrow illustration contributed by Jillian Nichol Ditner.
Genome Size Variation in Sparrows
Adjusted genome size estimates from C-values of Passerellidae sparrows varied by 0.5 Gb from 1.13 Gb in Savannah sparrow (Passerculus sandwichensis) to 1.63 Gb in gray-browed brushfinch (Arremon assimilis), with a mean of 1.36 Gb (Fig. 2a). Previous short-read genome assemblies of sparrows varied in length from 0.91 Gb in the grasshopper sparrow to 1.05 Gb in the white-throated sparrow assembly, which were 0.16 to 0.42 Gb smaller than the corresponding C-value estimates of genome size for these species. Assembly lengths for the CCGP and VGP sparrow genomes varied from 1.15 Gb in the Savannah sparrow to 1.40 Gb in Bell's sparrow (Fig. 2a). Recently released long-read assemblies of the white-crowned sparrow (1.12 Gb) and California towhee (1.41 Gb) span a similar range. The length of the assemblies reported here closely approximated the C-value estimates of genome size for the Savannah sparrow (mean 1.18 Gb vs. 1.15 Gb assembly) and the song sparrow (mean 1.40 Gb vs. 1.35 Gb assembly), but was more divergent in the swamp sparrow (1.46 Gb vs. 1.16 Gb assembly). No C-value estimates exist for the other three sparrow species; however, alternate estimates of genome size are available from the kmer profiles analyzed in GenomeScope (supplementary figs. S1 to S3, Supplementary Material online; https://www.genomeark.org/genomeark-all/). These profiles suggest that Nelson's (assembly: 1.18 Gb; GenomeScope: 1.19 Gb) and saltmarsh sparrow (1.24 vs. 1.22) assemblies closely match the expected genome length estimated from the kmer profile. In contrast, all three CCGP sparrow genome assemblies exceed the kmer profile genome size estimates (for example Bell's sparrow assembly: 1.40 Gb vs. GenomeScope estimate: 1.13); whereas, the curated swamp sparrow assembly (1.16 Gb) was considerably shorter than the estimated length from GenomeScope (1.33 Gb). Together these data underscore the high level of completeness of the assemblies generated using long-read approaches, with less than 3% of the genome missing from most species. The swamp sparrow assembly appears to be an exception with 12% to 20% of the genome content potentially missing (depending on GenomeScope or C-value estimate). Purged repeat content may explain some of the missing data from the swamp sparrow assembly. Repeat content in the swamp sparrow was estimated to span 18.26% of the genome using our Passerellidae repeat library in RepeatMasker, while k-mer estimates of repeat content were 30.4% from GenomeScope. Sparrow genome assemblies that used primarily short-read data were inferred to be missing as much as 12% to 30% of the genomic DNA (Fig. 2b).
Fig. 2.
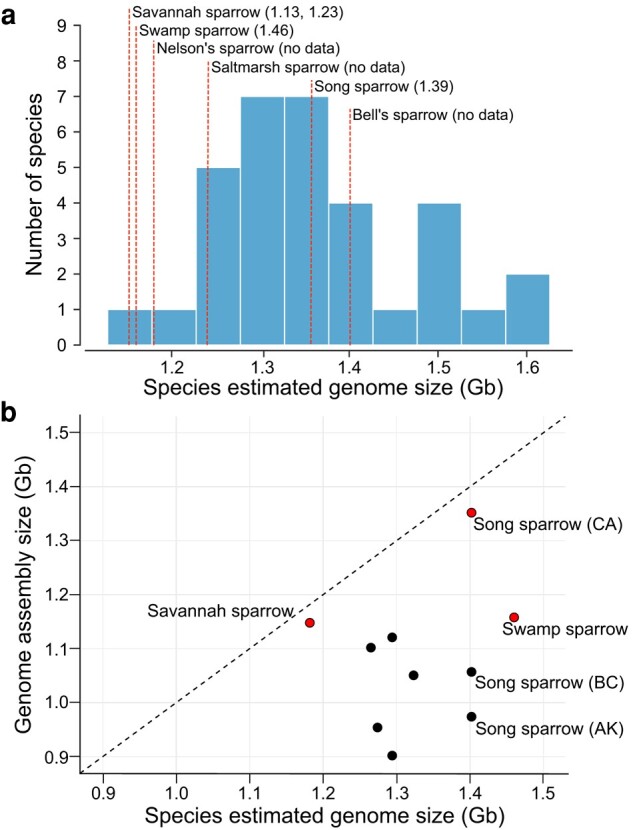
a) Histogram showing variation in the genome sizes for 21 species (33 individuals) of Passerellidae sparrows estimated from Feulgen image analysis densitometry methods. C-values were adjusted based on the assumption that 1 pg = 0.978 Gb (Dolezel et al. 2003). Genome size of the draft assemblies generated through the CCGP and VGP shown as red dashed lines with corrected c-value estimate (Gb) in parentheses. For Passerculus sandwichensis two estimates of genome size were found in the Animal Genome Size Database. b) Comparison of estimated genome size from C-values versus genome assembly size (Gb). Black dashed line indicates the 1-to-1 line indicating equal estimates of genome size from the two metrics. Red dots are newly generated assemblies reported here. Nelson's, saltmarsh, and Bell's sparrow lack independent estimates of C-values and are not shown on this plot. Black dots denote previously published genome assemblies, which all show a shorter assembly length relative to the C-value estimate of genome size.
Passerellidae De Novo Repeat Library
De novo identification of transposable elements in RepeatModeler2 followed by manual curation led to the identification of 514, 704, and 650 TE subfamilies within the Savannah, song, and Bell's sparrows, respectively. Merging of the three sparrow libraries produced a final Passerellidae TE library with 1,272 elements. This includes 361 elements shared by two or more sparrow species and 234, 341, and 336 elements unique to Savannah, song, and Bell's sparrows, respectively. Similar to other avian species, LINE and LTR elements represent the majority of TEs identified in these sparrow species. These include 15 shared LINE subfamilies and 68 shared LTR subfamilies across all three species. Song sparrows had the most unique LINE elements (n = 58), whereas Bell's sparrow had the most unique LTR elements (n = 122). Savannah sparrow had the least number of both elements identified (supplementary fig. S5, Supplementary Material online).
The curated Passerellidae repeat library was used to annotate all 15 sparrow assemblies. Annotation results from RepeatMasker showed that repeat content comprises a considerably higher percentage of the genome in the more contiguous, long-read assemblies (Fig. 3a). Bell's sparrow, song sparrow, and California towhee showed the greatest proportion of repeat content with repeats comprising over 29% of the genome (Table 2; Fig. 3a). The Savannah sparrow exhibited the lowest proportion of repeats (16.5%) among the long-read assemblies, but this still exceeded the 6.5% to 10.3% of the genome covered by repeat content identified in other sparrow species. Indeed, we found contig N50 to be highly predictive of total repeat content discovered in genome assemblies. All eight sparrow assemblies with a contig N50 greater than 1 Mb had significantly more repeat content than assemblies with contig N50 less than 1 Mb (Fig. 3b; t = −6.174; df = 7.8; P = 0.0003). All assemblies with a contig N50 > 1 Mb were assembled using PacBio HiFi long-reads; whereas only 1 of 7 of the assemblies with contig N50 less than 1 Mb included PacBio long-read sequencing in the assembly. Variation in assembly length among species also strongly predicted repeat content (adjusted R2 = 0.95, P-value <<0.0001; Fig. 4c). This pattern was replicated among different assemblies of the song sparrow and saltmarsh sparrow. Repeat content increased from 7.1% (0.978 Gb assembly) to 10.3% (1.06 Gb assembly) to 29.5% (1.36 Gb assembly) as assembly length increased in the three song sparrow assemblies. In the saltmarsh sparrow, repeat content more than doubled from 10.6% in the short-read assembly (1.07 Gb) to 24.2% in the long-read assembly (1.24 Gb). Finally, the amount of repeat content significantly decreased as the percent missing DNA increased for each assembly with missing DNA inferred from the difference between C-value estimate and assembly length (adjusted R2 = 0.57, P-value = 0.007; Fig. 3d).
Fig. 3.

a) Percentage of the genome comprising interspersed repeats, including: retroelements (LINE, SINE, LTR), DNA transposons (DNA), rolling-circles (RC), and unclassified elements (white-crowned sparrow, WCSP; California towhee, CALT; swamp sparrow, SWSP; Nelson's sparrow, NESP; saltmarsh sparrow short-read, SALS_SR; saltmarsh sparrow long-read, SALS_VG; Savannah sparrow, SAVS; Bell's sparrow, BESP; song sparrow, SOSP; white-throated sparrow, WTSP; dark-eyed junco, DEJU; chipping sparrow, CHSP; grasshopper sparrow, GRSP). b) The relationship between contig N50 and genome-wide repeat content. Significantly higher levels of repeat content were discovered in genomes with a contig N50 greater than 1 Mb. All of which were generated with PacBio long-read technology. c) Correlation between percent repeat content identified in each genome and the length of the assembled genome in Gb. d) Correlation between percent repeat content and the amount of DNA inferred to be missing from each of the sparrow assemblies. C-value is assumed to be the more accurate estimate of total genome length. Percent missing DNA from each sparrow assembly is estimated as the difference between the c-value and assembly length.
Table 2.
Percentage of each genome spanned by different classes of repeats. Estimates of each class of repeat region identified within RepeatMasker using the sparrow TE libraries generated de novo with RepeatModeler2
| Element class: | Savannah sparrow (Passerculus sandwichensis) | Bell's sparrow (Artemisiospiza belli) | Song sparrow (Melospiza melodia) | Swamp sparrow (Melospiza georgiana) | Nelson's sparrow (Ammospiza nelsoni) | Saltmarsh sparrow (Ammospiza caudacuta) |
|---|---|---|---|---|---|---|
| LINE | 4.41 | 7.8 | 14.58 | 5.37 | 5.63 | 6.75 |
| SINE | 0.05 | 0.04 | 0.02 | 0.05 | 0.05 | 0.05 |
| LTR | 7.28 | 21.21 | 8.22 | 8.25 | 7.72 | 7.06 |
| DNA transposons | 0.09 | 0.10 | 0.09 | 0.08 | 0.08 | 0.1 |
| Rolling-circles | 0.02 | 0.02 | 0.01 | 0.01 | 0.02 | 0.05 |
| Unclassified | 4.66 | 2.01 | 6.56 | 4.51 | 5.81 | 8.40 |
| Total interspersed repeats: | 16.49 | 31.16 | 29.49 | 18.26 | 19.29 | 22.35 |
| Autosomal chromosomes: | 16.17 | 30.91 | 28.84 | 15.85 | 16.11 | 19.48 |
| Z chromosome: | 20.72 | 23.75 | 25.28 | 22.40 | 26.42 | 26.46 |
| W chromosome: | NA | 82.59 | 91.03 | 93.73 | 79.23 | NA |
| Other repeat regions: | ||||||
| Small RNA | 0.04 | 0.04 | 0.03 | 0.06 | 0.05 | 0.04 |
| Satellites | 0.33 | 0.21 | 0.24 | 0.25 | 0.22 | 0.25 |
| Simple repeats | 1.35 | 1.11 | 1.05 | 1.25 | 1.19 | 1.21 |
| Low complexity | 0.25 | 0.20 | 0.20 | 0.23 | 0.28 | 0.32 |
Fig. 4.

a to b) Comparison of transposable element annotation across three song sparrow (Melospiza melodia) assemblies. a) Total number of transposable elements found in each assembly. b) Total genomic content (Mb) of transposable element content identified in each of the three song sparrow assemblies. MelMel_AK (red) is an assembly from an Alaskan bird sequenced using short-read and Chicago library technology. MelMel_BC (green) is a bird from British Columbia sequenced using Illumina short-read and PacBio SMRT long-read approaches MelMel_CA was generated using Hi-C and PacBio long-read approaches for this paper. c and d) Comparison of TE annotations in two saltmarsh sparrow assemblies (Ammospiza caudacuta). (c) Total number of transposable elements found in each assembly. b) Total genomic content (Mb) of transposable element content identified in each of the three song sparrow assemblies. SALS_SR (Red) was assembled from Illumina short reads and the SALS_VG (blue) assembly was assembled using PacBio long-read and Omni-C approaches with the Vertebrate Genomics Project pipeline.
The comparison among the three song sparrow genome assemblies showed that LINEs and LTRs comprised the greatest number and total base pairs of newly discovered TE sequence (Fig. 4a). We found an additional 8,375 (a ∼5% increase) line elements in the California versus British Columbia song sparrow assemblies. Despite only a small increase in the total number of elements, we find that these LINE elements span an additional 155 Mb of DNA in the California song sparrow genome (Fig. 4b). This discrepancy likely stems both from different elements segregating at different frequencies in each population and LINEs in the California genome being of greater length on average. One of the most abundant LINE elements in the California song sparrow genome was found across all three of the CCGP sparrow genomes, but was missing from the British Columbia song sparrow genome. This element was >6500 bp in length and nearly 4,000 full length copies were found across the California song sparrow genome. Comparisons between a short-read and long-read assembly of the saltmarsh sparrow revealed a similar pattern (Fig. 4c and d). A small increase in the total number of LINE (∼3%) and LTR (∼16%) elements led to respective increases of 43.1 Mb and 43.8 Mb of TE DNA discovered in these genomes. These results further support the inference that missing DNA from previous assemblies corresponds to longer TE elements and may have been a major contributor to gaps.
The prevalence of different TE classes varied across our three genome assemblies. LTRs were the most abundant element within all sparrow genomes except the song sparrow assembly where LINE elements were the most abundant (14.58%; Table 2). Across different chromosomes, the density of repeat content (79.23% to 93.73%) was highest on the W chromosomes. W chromosome repeat content was particularly high in the genus Melospiza with over 90% of the W chromosome spanned by repetitive elements in both the song and swamp sparrow. Z chromosome repeat content tended to be higher than autosomal repeat content for all species except song and Bell's sparrow (Table 2).
Timing of Repeat Proliferation
We extracted an average of 4,663 (range: 3,699 to 4,839) ultra-conserved element (UCE) loci from the 17 reference genomes queried (supplementary table S2, Supplementary Material online). From these loci we constructed a concatenated data matrix of 4,196 UCE loci shared across 95% of samples with a total of 4,815,326 base pairs. The concatenated maximum likelihood tree was well-resolved with all nodes receiving bootstrap support of 100. This topology was used as input into MCMCtree to estimate divergence times among the focal species (see supplementary fig. S6, Supplementary Material online for full phylogeny). This time-calibrated phylogeny indicated that the white-crowned sparrow split from the other sequenced sparrow species at 13.3 Mya (95% HPD: 7.4 to 17.8; Fig. 5), Bell's sparrow diverged from other species in the grassland sparrow clade 7.9 Mya (95% HPD: 4.5 to 10.8), Ammospiza sparrows (Nelson's and saltmarsh) split from Savannah, swamp, and song sparrow 6.8 Mya (95% HPD: 3.8 to 9.3), and Savannah sparrow diverged from the Melospiza sparrows 5.8 Mya (95% HPD: 3.3 to 7.9). Within the context of these divergence times, members of the Passerellidae family show sharply divergent histories of transposable element proliferation (Fig. 5). All members of the grassland sparrow clade show evidence for a spike in LINE element activity in the autosomes ∼25 to 30 Ma. In contrast, the white-crowned sparrow does not show evidence for this spike, but rather shows a normal distribution of LINE element divergence centered at ∼40 to 50 Ma. Although the timing of LINE proliferation in grassland sparrows appears to predate divergence estimates among Passerellidae species (Fig. 5), the contrast with white-crowned sparrow suggests it may have occurred more recently following divergence of these different sparrow lineages. Despite shared evidence for this period of LINE activity, song sparrows have retained more LINE elements from this proliferation (∼14% of the genome) than the other five sparrow species (only 1% to 3% of genomes). Bell's sparrow shows a unique pulse of LTR proliferation approximately 12 Mya and a very recent (<5 Mya) proliferation of both LINE and LTR elements in the autosomes. For species with an assembled W chromosome, all show a steady accumulation of LTR elements on the W chromosome, with endogenous retroviruses (ERVs) being the most prolific and representing up to a maximum of 69.2% in the song sparrow.
Fig. 5.

Transposable element (TE) landscapes for the autosomal, Z, and W chromosomes. Left panel shows time-calibrated UCE phylogeny of seven sparrow species. Branch labels indicate mean estimate of divergence time for each node with purple bars indicating 95% HPD error around that estimate. All nodes in the topology received bootstrap support of 100%. Right panel shows TE landscapes for each species. Percent divergence on the x axis was calculated as the percent Kimura 2-parameter (K2P) distance with CpG sites excluded. The abundance of TEs in each percent divergence bin was normalized as a percentage of the chromosome length on the y axis.
Discussion
Highly Contiguous and Complete Genomes Reveal High Repeat Content
We generated highly contiguous and complete genomes of three sparrow species in the family Passerellidae that we compared with three chromosome-level genomes generated by the Vertebrate Genome Project. Contig N50 for the three newly generated genomes exceeded 92%, and the scaffold N50 exceeded 85% of all avian genomes recently surveyed by Bravo et al. (2021). Assembly length for the six species analyzed here also exceeds assembly lengths for all short-read based assemblies generated to date (1.16 to 1.40 Gb vs. 0.91 to 1.05 Gb). The longer length of these assemblies more closely approximates independent estimates of genome size from Feulgen image analysis densitometry (C-value), with song and Savannah sparrow missing only 2% to 3% of genomic sequence relative to C-value size estimates. Longer assemblies were also associated with greater levels of repeat content. The high percentage of total interspersed repeats discovered in the song sparrow (31.2%), Bell's sparrow (29.5%), and California towhee (30.9%; also see Black et al. 2023) genomes are the highest levels ever reported for Passeriformes and more closely resemble levels of repeat content described in the avian orders Piciformes and Bucerotiformes (Manthey et al. 2018; Feng et al. 2020). Although our finding is a novel result for passerine genome assemblies, reassociation kinetic studies found about 36% of the dark-eyed junco genome to be repetitive DNA (Shields and Straus 1975). Recent long-read assemblies for jays in the passerine family Corvidae also show repeat content in-line with the results presented here (Benham et al. 2023; DeRaad et al. 2023). Further, high levels of repeat content in these sparrows matches predictions that much of the missing genomic data from avian short-read assemblies are likely repetitive DNA (Elliott and Gregory 2015; Kapusta and Suh 2017; Peona et al. 2018). We expect that the generation of additional highly contiguous and complete genomes using third generation sequencing technology will also find higher levels of repeat content in avian genomes than previously appreciated.
Previous sparrow genome assemblies generated using short-read methods were found to be missing ∼12% to 30% of DNA sequence relative to C-values (Fig. 2b). The majority of this missing DNA is likely associated with highly repetitive regions of the genome that caused gaps in prior assemblies. Gaps associated with repeat regions is a well-established phenomenon and recent comparisons among sequencing technologies point to long contiguous reads as essential for spanning these gaps (Rhie et al. 2021). Similarly, we find that sparrow assemblies generated using PacBio long-read sequence data exhibited elevated contig N50 and higher percent repeat content, and that percent repeat content decreased in assemblies inferred to be missing a greater percentage of DNA (Fig. 3). This was also true for intra-specific comparisons of multiple song and saltmarsh sparrow genomes where repeat content increased with assembly length and contiguity. The diversity of transposable elements can vary significantly within and among populations (e.g. Ficedula flycatchers; Suh et al. 2018). Although we compared song sparrow genomes from three different subspecies that could differ in repeat content and genome size, the patterns for song sparrow are consistent with work showing increased TE discovery and abundance in long-read versus short-read assemblies of the same individual (Peona et al. 2021a). Our results are also consistent with prior work linking metrics of genome contiguity with levels of repeat content detected in genome assemblies (Galbraith et al. 2021). Overall, our analyses among closely related sparrow species confirms that sequencing technology appears to be a major confounding factor in comparative research on TE diversity.
Genome Size Evolution in Birds
Transposable elements are widely recognized as an important driver of genome size evolution across Eukaryotes (Kidwell 2002; Elliott and Gregory 2015). Low repeat content and high synteny contributed to an early view that avian genomes were relatively stable and constrained in size as an adaptation for the metabolic demands of flight (Hughes and Hughes 1995; Wright et al. 2014). This perspective has been challenged with evidence of a more dynamic history of avian genome expansions followed by large-scale deletions (Kapusta et al. 2017). Genome size variation in the Passerellidae, as measured by densitometry and cytometry methods, ranges from 1.13 to 1.63 Gb, with the Savannah sparrow possessing the smallest genome of any sparrow measured to date. Differences in assembly length across all sparrow species were entirely related to repeat content (Fig. 3). Further, TE composition differed significantly even in species with similar genome assembly lengths (e.g. Bell's and song sparrow).
Unlike the other sparrow species analyzed, CR1 LINE elements were found to be the most abundant TE class within the song sparrow genome. The TE landscape of the song sparrow genome indicates that the majority of LINE DNA stems from a period of increased activity 25 to 30 million years ago. All six sparrow species in the grassland clade show a spike in LINE activity during this period, but much of the LINE DNA from this period was eliminated in species other than the song sparrow. In contrast, the white-crowned sparrow shows a more “bell-curve” shape of LINE element proliferation with a peak of less than 0.5% at ∼30 to 40 Mya (about 20% divergence from consensus). The white-crowned sparrow pattern more closely resembles TE landscapes observed in other Passerifomes birds such as Ficedula flycatchers (Muscicapidae; Suh et al. 2018) and Estrildidae finches (Boman et al. 2019). These patterns point to a proliferation of LINE elements within the grassland sparrow clade that more likely occurred after divergence from the white-crowned sparrow ∼13.3 Mya. This discrepancy in the timing of activity could reflect the use of a genome wide estimate of mutation rate from Ficedula flycatchers (Smeds et al. 2016) that may be underestimating the true mutation rate for Passerellidae sparrows and/or transposable elements. Indeed, many of the host genome's defense mechanisms against TE proliferation involve DNA-editing enzymes, such as APOBECs, which mutate TE sequences to silence their activity in the genome (Goodier 2016; Knisbacher and Levanon 2016).
LTR elements were the most abundant TE within all sparrow genomes except the song sparrow. Proliferation of these elements has also been more recent, beginning ∼12 million years ago and continuing to the present. Recent proliferation of LTR elements was especially pronounced in Bell's sparrow. Recent proliferation of LTR elements more closely aligns with patterns of TE expansion observed in the zebra finch (Kapusta and Suh 2017) and the Eurasian blackcap (Sylvia atricapilla; Bours et al. 2023). The reasons for recent LTR expansions in songbirds versus other avian lineages (e.g. Chicken; Warren et al. 2017) are not entirely clear. One possibility is that competition for similar genomic insertion sites between LTR and LINE elements could be mediated by host defenses. Recent work in the deer mouse (Peromyscus maniculatus; Gozashti et al. 2023) provided evidence for a cycle initiated by greater host repression of ancient ERV (a type of LTR) that allowed for greater LINE proliferation in the genome. This was followed by the invasion of the deer mouse genome by a novel ERV that was hypothesized to have a greater immunity to host defense mechanisms and greater potential to outcompete LINE elements for insertion sites. A related possibility could reflect the accumulation of LTR elements on the W chromosome, many of which remain transcriptionally active and could seed invasions of the autosomal chromosomes (see below; Peona et al. 2021b). Whether either of these scenarios contributes to the recent expansions of LTR elements in songbird genomes awaits further study; however, the expanding number of avian genomes assembled using long-read sequence data will be essential for understanding the dynamics of TE proliferation and deletion in the evolution of avian genomes.
Differences in genome size and repeat content could be the result of a number of different mechanisms involved in TE silencing, deletion, or expansion in host genomes (Goodier 2016). A wide range of epigenetic mechanisms exist to silence TE activity in plants and animals (reviewed in Slotkin and Martienssen 2007). In birds, methylation of CpG and nonCpG sites in TEs with DNA methyltransferases is the primary mechanism of TE silencing that has been documented (Derks et al. 2016; Kapusta and Suh 2017). Mutating TE sequences is another mechanism hosts deploy to defend against TE proliferation. APOBEC genes induce C-to-U mutations in retrotransposons leading to inactivation and degradation of these elements. The genomes of zebra finch and other bird species exhibit signatures of high APOBEC activity (Knisbacher and Levanon 2016). An important mechanism for the removal of LTR elements from the genome is ectopic recombination. This process deletes most of the element sequence leaving only a single LTR and correlates with recombination rate variation across the genome in birds (Ji and DeWoody 2016). Finally, demographic differences among populations could influence TE dynamics, with TEs predicted to insert and spread more rapidly in populations with a small effective population size (Ne; Lynch and Conery 2003). Demographic analyses of the different sparrow species provide some support for this hypothesis as both Nelson's and saltmarsh sparrows have been inferred to experience historical bottlenecks and lower Ne than other species (Walsh et al. 2019a, b; Walsh et al. 2021). In contrast, the Savannah sparrow has the lowest repeat content and has been inferred to maintain high and constant effective population sizes (Benham and Cheviron 2019), which may be important for combating TE proliferation and maintaining a smaller genome. However, the relationship between Ne and TE proliferation is not necessarily straightforward (Whitney and Garland 2010) and some authors argue that TE expansions may be even more likely in species with large Ne (Ågren and Wright 2011).
Disruption of a host's TE repression mechanisms can lead to TE expansions. Stressful conditions (e.g. thermal stress) can disrupt epigenetic silencing of TEs in the host genome, leading to TE expansions (Capy et al. 2000; Slotkin and Martienssen 2007). Furthermore, co-evolutionary arms races between TEs and the host genomes could lead to divergence in TE repressors among populations or closely related species. Subsequent hybridization among these lineages could allow TEs to escape their repressors and proliferate throughout the genome of hybrids (Bingham et al. 1982; Serrato-Capuchina and Matute 2018). Examples of TE re-activation following hybridization have been documented in both plants (Josefsson et al. 2006) and animals (O'Neill et al. 1998). In hybrid Helianthus sunflower species, proliferation of LTR elements was found to contribute significantly to a 50% increase in the genome size of hybrids relative to parental species (Ungerer et al. 2006). Intriguingly, Bell's sparrow individual used to generate the reference assembly for this project comes from the same subspecies known to hybridize with sagebrush sparrow in a contact zone centered ca. 120 to 150 km. to the northwest of the collecting locality. Whether recent hybridization between Bell's and sagebrush or Nelson's and saltmarsh sparrow lineages led to a TE expansion remains to be determined. However, the dynamic patterns of genome size evolution within sparrows indicates that the Passerellidae are an exciting model for future research on the dynamics of TE evolution.
TE Element Proliferation on Sex Chromosomes
The potential deleterious effects of TE insertions are thought to explain a general trend of TE prevalence in regions of lower recombination rate (Rizzon et al. 2002; Ji and DeWoody 2016; Kent et al. 2017). These patterns are especially pronounced on the Y/W sex chromosomes where a lack of recombination, low gene density, and small effective population sizes are thought to allow for TE accumulation (Charlesworth and Langly 1989; Bachtrog 2003). This high TE abundance is thought to be a major contributing factor to the challenges of sequencing and assembling the W chromosome in birds and Y chromosome in mammals (Tomaszkiewicz et al. 2017). Consistent with these expectations for the nonrecombining W chromosome, we found that repeat content on the W chromosome was dramatically higher across all four female birds sequenced relative to autosomal or Z chromosomes. Interspersed repeats comprised 79.2% and 82.6% of Nelson's and Bell's sparrow W chromosome, respectively, while the song and swamp sparrow (both Melospiza) possessed W chromosomes with over 90% repeat content. Previous reports of repeat content on the W chromosome range from 22% in the emu (Dromaius novaehollandiae; Peona et al. 2021a, b) to over 84% in the hooded crow (e.g. Corvus cornix; Warmuth et al. 2022) and 89% in Steller's jay (e.g. Cyanocitta stelleri; Benham et al. 2023). Similar to other avian W chromosome assemblies, endogenous retroviral elements are the dominant element representing 42.3% of the song to 69.2% of Bell's sparrow W chromosome assembly. Peona et al. (2021b) also showed that a disproportionately large percentage of LTR elements on the avian W chromosome are full length retroviral elements that continue to be actively transcribed. The capacity of active elements to spread from the W to other regions of the genome makes the W chromosome a likely source for the recent activity and abundance of endogenous retroviral elements in Passeriformes (Warren et al. 2010; Zhang et al. 2014; Warmuth et al. 2022). Kapusta and Suh (2017) posited that the abundance of these elements in Passeriformes may have played critical roles in their high levels of diversification. The highly complete genome assemblies generated via third generation sequencing techniques will provide new opportunities to test this hypothesis.
Conclusions
Here we report on the release of three highly contiguous assemblies of sparrows in the family Passerellidae. The combination of long-read and Omni-C technology enabled the generation of nearly complete and highly contiguous assemblies. Analysis of these genomes revealed a previously underappreciated abundance of repetitive elements in the genomes of songbirds and suggests that much of the missing data from other avian assemblies are likely comprised of repeat content. As third generation sequencing technologies become the standard in avian genome assembly, the dynamics of TE element proliferation and genome size evolution across different evolutionary timescales will become better understood. Our results point to the strong role repetitive element proliferation and deletion plays in the dynamics of avian genome size evolution, even among closely related species.
Materials and Methods
CCGP Genome Sampling
We sequenced liver from an adult female Bell's sparrow (Artemisiospiza belli canescens) collected on June 25, 2018 at Hunter Cabin, 1.5 Mi. east of Jackass Spring, Death Valley National Park, Inyo Co., California (36.54758°N, 117.48786°W; elevation: 6860 ft). Blood and liver tissue samples were sequenced from an immature female song sparrow (Melospiza melodia gouldii) captured on September 5, 2020 in oak woodland habitat at Mitsui Ranch, Sonoma Mountain, Sonoma Co., California (38.33131°N; 122.57720°W). Liver tissue for sequencing was collected from an adult male Savannah sparrow (Passerculus sandwichensis alaudinus) on May 20, 2015 in tidal marsh habitat at the San Francisco Bay National Wildlife Refuge, Santa Clara Co., California (37° 26.029′N; 122° 0.996′W: elevation: 4 m). Bell's and song sparrow individuals were collected with approval of California Department of Fish and Wildlife (CDFW permit #: SCP-458), the U.S. Fish and Wildlife Service (USFWS permit #: MB153526), Death Valley National Park (Bell's sparrow only; permit#: DEVA-2015-SCI-0040), and following protocols approved by the University of California, Berkeley IACUC (AUP-2016-04-8665-1). The Savannah sparrow sample was also collected with approval from CDFW (permit #: SCP-012913), USFWS (permit #: MB24360B-0), the San Francisco Bay National Wildlife Refuge (special use permit: 2015-015), and using methods approved by the University of Illinois, Urbana-Champaign IACUC (protocol #: 13418). Voucher specimens were deposited at the Museum of Vertebrate Zoology, Berkeley, CA for Bell's sparrow (https://arctos.database.museum/guid/MVZ:Bird:192114) and song sparrow (https://arctos.database.museum/guid/MVZ:Bird:193390). A specimen voucher of the Savannah sparrow was deposited at the Field Museum of Natural History in Chicago, IL (FMNH:Birds:499929).
DNA Extraction, Library Preparation, and Sequencing for CCGP Genomes
High molecular weight (HMW) genomic DNA (gDNA) for PacBio HiFi library preparation was extracted from 27 and 15 mg of liver tissue from Bell's and Savannah sparrow samples, respectively. Extractions were performed using the Nanobind Tissue Big DNA kit following the manufacturer's instructions (Pacific BioSciences—PacBio, Menlo Park, CA). For song sparrow, HMW gDNA was isolated from whole blood preserved in EDTA. A total of 30 µl of whole blood was added to 2 ml of lysis buffer containing 100 mM NaCl, 10 mM Tris-HCl pH 8.0, 25 mM EDTA, 0.5% (w/v) SDS and 100 µg/ml Proteinase K. Lysis was carried out at room temperature for a few hours until the solution was homogenous. The lysate was treated with 20 µg/ml RNase A at 37 °C for 30 min and cleaned with equal volumes of phenol/chloroform using phase lock gels (Quantabio Cat # 2302830). DNA was precipitated by adding 0.4 × volume of 5 M ammonium acetate and 3 × volume of ice-cold ethanol. The DNA pellet was washed twice with 70% ethanol and resuspended in an elution buffer (10 mM Tris, pH 8.0), and purity was estimated using absorbance ratios (260/280 = 1.81 to 1.84 and 260/230 = 2.29 to 2.40) on a NanoDrop ND-1000 spectrophotometer. The final DNA yield (Bell's: 13 μg; Savannah: 16 μg; song: 150 μg total) was quantified using the Quantus Fluorometer (QuantiFluor ONE dsDNA Dye assay; Promega, Madison, WI). The size distribution of the HMW DNA was estimated using the Femto Pulse system (Agilent, Santa Clara, CA): 62% of the fragments were >140 Kb for Bell's sparrow; 60% of the fragments were >140 Kb for Savannah sparrow; and 85% of the DNA was found in fragments >120 Kb for song sparrow.
The HiFi SMRTbell library was constructed using the SMRTbell Express Template Prep Kit v2.0 following the manufacturer's protocols (Pacific Biosciences—PacBio, Menlo Park, CA; Cat. #100-938-900). HMW gDNA was sheared to a target DNA size distribution between 15 to 20 Kb and concentrated using 0.45 × of AMPure PB beads (PacBio Cat. #100-265-900) for the removal of single-strand overhangs at 37 °C for 15 min. Enzymatic steps of DNA damage repair were performed at 37 °C for 30 min, followed by the end repair and A-tailing steps at 20 °C for 10 min and 65 °C for 30 min. Ligation of overhang adapter v3 was performed at 20 °C for 60 min with subsequent heating to 65 °C for 10 min to inactivate the ligase. Finally, DNA product was nuclease treated at 37 °C for 1 h. To collect fragments greater than 9 Kb, the resulting SMRTbell library was purified and concentrated with 0.45 × Ampure PB beads (PacBio, Cat. #100-265-900) for size selection using the BluePippin system (Sage Science, Beverly, MA; Cat #BLF7510). The 15 to 20 Kb average HiFi SMRTbell library was sequenced at the University of California Davis DNA Technologies Core (Davis, CA) using two 8 M SMRT cells, Sequel II sequencing chemistry 2.0, and 30-hour movies each on a PacBio Sequel II sequencer.
The Omni-C library was prepared using the Dovetail Omni-C Kit (Dovetail Genomics, CA) according to the manufacturer's protocol with slight modifications. First, specimen tissue was ground thoroughly with a mortar and pestle while cooled with liquid nitrogen. Subsequently, chromatin was fixed in place in the nucleus and then passed through 100 μm and 40 μm cell strainers to remove large debris. Fixed chromatin was digested under various conditions of DNase I until a suitable fragment length distribution of DNA molecules was obtained. Chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter containing ends. After proximity ligation, crosslinks were reversed and the DNA purified from proteins. Purified DNA was treated to remove biotin that was not internal to ligated fragments. An NGS library was generated using an NEB Ultra II DNA Library Prep kit (NEB, Ipswich, MA) with an Illumina compatible y-adaptor. Biotin-containing fragments were then captured using streptavidin beads. The post-capture product was split into two replicates prior to PCR enrichment to preserve library complexity with each replicate receiving unique dual indices. The library was sequenced at the Vincent J. Coates Genomics Sequencing Lab (Berkeley, CA) on an Illumina NovaSeq platform (Illumina, San Diego, CA) to generate over 100 million 150 bp paired end reads per species. See supplementary table S3, Supplementary Material online for details on PacBio and Illumina sequencing.
Assembly of CCGP Genomes
We assembled the genome of the three CCGP sparrows following the CCGP assembly pipeline Version 3.0 (see Lin et al. 2022; supplementary table S4, Supplementary Material online). The pipeline takes advantage of long and highly accurate PacBio HiFi reads alongside chromatin capture Omni-C data to produce high-quality and highly contiguous genome assemblies while minimizing manual curation.
In brief, we removed remnant adapter sequences from the PacBio HiFi dataset for all three assemblies using HiFiAdapterFilt (Sim et al. 2022) and obtained the initial dual assembly with the filtered PacBio reads using HiFiasm (Cheng et al. 2022). The dual assembly consists of a primary and alternate assembly: the primary assembly is more complete and consists of longer phased blocks, while the alternate consists of haplotigs (contigs with the same haplotype) in heterozygous regions and is more fragmented. Given the characteristics of the latter, it cannot be considered a complete assembly of its own but rather is a complement of the primary assembly (https://lh3.github.io/2021/04/17/concepts-in-phased-assemblies, https://www.ncbi.nlm.nih.gov/grc/help/definitions/).
Next, we identified sequences corresponding to haplotypic duplications, contig overlaps and repeats on the primary assembly with purge_dups (Guan et al. 2020) and transferred these sequences to the corresponding alternate assembly. We aligned the Omni-C data to both assemblies following the Arima Genomics Mapping Pipeline (https://github.com/ArimaGenomics/mapping_pipeline) and used SALSA to produce scaffolds for the primary assembly (Ghurye et al. 2017, Ghurye et al. 2019).
Omni-C contact maps for the primary assembly were produced by aligning the Omni-C data with BWA-MEM (Li 2013), identifying ligation junctions, and generating Omni-C pairs using pairtools (Open2C et al. 2023). We generated a multiresolution Omni-C matrix with cooler (Abdennur and Mirny 2020) and balanced it with hicExplorer (Ramírez et al. 2018). To visualize and check contact maps for misassemblies, we used HiGlass (Kerpedjiev et al. 2018) and the PretextSuite (https://github.com/wtsi-hpag/PretextView; https://github.com/wtsi-hpag/PretextMap; https://github.com/wtsi-hpag/PretextSnapshot). In detail, if we identified a strong off-diagonal signal in the proximity of a join that was made by the scaffolder, and a lack of signal in the consecutive genomic region, we dissolved it by breaking the scaffolds at the coordinates of the join. After this process, no further manual joins were made. Some of the remaining gaps (joins generated by the scaffolder) were closed using the PacBio HiFi reads and YAGCloser (https://github.com/merlyescalona/yagcloser). We checked for contamination using the BlobToolKit Framework (Challis et al. 2020). Finally, upon submission of the assemblies to NCBI, we trimmed remnants of sequence adaptors and mitochondrial contamination identified during NCBI's own contamination screening.
We assembled the mitochondrial genomes for each of the sparrows from their corresponding PacBio HiFi reads starting from the same mitochondrial sequence of Zonotrichia albicollis (NCBI:NC_053110.1; Feng et al. 2020, B10K Project Consortium) and using the reference-guided pipeline MitoHiFi (Allio et al. 2020; Uliano-Silva et al. 2021). After completion of the nuclear genomes, we searched for matches of the resulting mitochondrial assembly sequence in their nuclear genome assembly using BLAST+ (Camacho et al. 2009), filtering out contigs and scaffolds from the nuclear genome with a sequence identity >99% and a size smaller than the mitochondrial assembly sequence.
Genome Assembly Assessment
We generated k-mer counts from the PacBio HiFi reads using meryl (https://github.com/marbl/meryl). GenomeScope2.0 (Ranallo-Benavidez et al. 2020) was used to estimate genome features including genome size, heterozygosity, and repeat content from the resulting k-mer spectrum. To obtain general contiguity metrics, we ran QUAST (Gurevich et al. 2013). Genome quality and completeness were quantified with BUSCO 5.6.1 (Manni et al. 2021) using Augustus (Stanke et al. 2008), genome mode, and the 8,338 genes in the Aves ortholog database (aves_odb10). Assessment of base level accuracy (QV) and k-mer completeness was performed using the previously generated meryl database and merqury (Rhie et al. 2020). We further estimated genome assembly accuracy via a BUSCO gene set frameshift analysis using the pipeline described in Korlach et al. (2017). We followed the quality metric nomenclature established by Rhie et al. (2021), with the genome quality code x.y.Q.C, where, x = log10[contig NG50]; y = log10[scaffold NG50]; Q = Phred base accuracy QV (quality value); C = % genome represented by the first “n” scaffolds, following a known karyotype of 2n = 74 for P. sandwichensis (Bird Chromosome database—V3.0/2022; Degrandi et al. 2020) and estimated 2n = 80 for both M. melodia and A. belli; this estimation is the median number of chromosomes from closely related species (Genome on a Tree—GoaT; tax_tree(1729112); Challis et al. 2023). Quality metrics for the notation were calculated on the primary assemblies. Finally, we used the JupiterPlot pipeline (https://github.com/JustinChu/JupiterPlot) to visualize higher level synteny between the scaffolds of each sparrow assembly and chromosomes of the zebra finch (Taeniopygia guttata) genome assembly (Warren et al. 2010). Scaffolds representing 80% of each draft assembly (ng = 80) were mapped to zebra finch chromosomes exceeding 1 Mb in length (m = 1,000,000).
VGP Genome Sampling
The three CCGP genomes were compared to three chromosome-level assemblies of closely related sparrows generated by the Vertebrate Genomes Project following protocols outlined in Rhie et al. (2021). These samples were sequenced from blood samples (100 to 200 µl in ethanol) taken from female Nelson's and swamp sparrows and a saltmarsh sparrow that was identified as a female in the field but lacked the W chromosome in the final assembly. Nelson's sparrow sample was collected by Nicole Guido (Saltmarsh Habitat and Avian Research Program) in South Branch Marsh River, Waldo County, Maine (44.5864°N; 68.8591°W) on July 31, 2020. The swamp sparrow sample was collected by Jonathan Clark (University of New Hampshire) in Durham, Rockingham County, New Hampshire (43.14°N; 71.00°W) on July 23, 2020. The saltmarsh sparrow sample was collected by Chris Elphick (University of Connecticut) from Barn Island Wildlife Management Area, New London County, Connecticut (41.338°N; 71.8677°W) on August 19 2020. Sample collection occurred under permits of the Maine Division of Inland Fisheries and Wildlife (#2020-314), Connecticut Department of Energy and Environmental Protection (#0221012b), and New Hampshire Fish and Game, and followed protocols approved under the University of New Hampshire IACUC (#190401). All individuals were released at the capture location immediately after sampling. DNA extracted from these three species was sequenced to 31.6 to 34.6 × coverage of PacBio Sequel II HiFi long reads, 254 to 450 × coverage of Bionano Genomics DLS, and 103 to 112 × coverage for Arima Hi-C v2. Genome assemblies were generated from these data with the VGP standard assembly pipeline version 2.0, which included hifiasm v0.15.4, purge_dups v. 1.2.5, solve v. 3.6.1, salsa v. 2.3. To ensure comparable results, we also ran BUSCO on the VGP genomes using identical parameters to the CCGP genome assessment above and with the aves_odb10 database. Further details on raw data, sequence evaluations and curated assemblies can be found on VGP Genome Ark pages for Nelson's sparrow (https://genomeark.github.io/genomeark-all/Ammospiza_nelsoni.html), saltmarsh sparrow (https://genomeark.github.io/genomeark-all/Ammodramus_caudacutus.html), and swamp sparrow (https://genomeark.github.io/genomeark-all/Melospiza_georgiana.html).
Genome Size Variation Within Sparrows
We estimated the distribution of genome sizes in Passerellidae sparrows with c-value data from 33 individuals of 21 species archived in the Animal Genome Size Database (Gregory 2022). The majority of these C-value estimates were generated using Feulgen image analysis densitometry with original values reported in Andrews et al. (2009) and Wright et al. (2014). This includes all species for which we compared C-values to assembly lengths. C-value estimates of genome size were converted from picograms to base pairs using a conversion of 1 pg = 0.978 Gb (Dolezel et al. 2003). We additionally obtained genome assembly length from nine publicly available assemblies that were sequenced previously for members of the Passerellidae. Six of these were based on Illumina short-read sequence data and include a second song sparrow genome from Alaska (Louha et al. 2020), a short-read genome of the saltmarsh sparrow (Ammospiza caudacuta; Walsh et al. 2019a), plus genomes for the white-throated sparrow (Zonotrichia albicollis; Tuttle et al. 2016), dark-eyed junco (Junco hyemalis; Friis et al. 2022), chipping sparrow (Spizella passerina; Feng et al. 2020), and grasshopper sparrow (Ammodramus savannarum; Carneiro 2021). The remaining three assemblies were generated using PacBio long-read sequence data and include a third song sparrow genome from British Columbia (Feng et al. 2020), a white-crowned sparrow genome (Zonotrichia leucophrys; Wu et al. 2024), and a contig-level assembly of the California towhee (Melozone crissalis; Black et al. 2023). For complete GenBank accession details, sequencing, and assembly methods for these genomes see supplementary table S5, Supplementary Material online. We compared C-value estimates of genome size and genome assembly length for all of the assemblies that included both these estimates of genome size (10 of 15 total).
Repeat Annotation
We performed detailed de novo repeat annotation and manual curation of repeat libraries for Bell's, song, and Savannah sparrow genomes sequenced by the CCGP in the program RepeatModeler2 with the ltrstruct option selected to improve identification of LTR elements (Flynn et al. 2020). Consensus transposable element libraries generated from RepeatModeler2 were then curated manually following protocols and methods of Goubert et al. (2022). First, we removed any redundancy in the de novo repeat libraries using cd-hit-est (Li and Godzik 2006) to cluster any consensus sequences together that were 80 base pairs in length and shared >80% similarity over more than 80% of their length. This corresponds to the 80-80-80 rule of Wicker et al. (2007) frequently used to classify TE elements as a single subfamily. We then prioritized elements for manual curation that were at least 1,000 base pairs in length and had at least 10 blastn hits in the genome assembly. For each consensus sequence prioritized for manual curation, we used blastn (Camacho et al. 2009) to identify other members of each TE subfamily in the genome, and for each blastn hit we added 2,000 bp of flanking sequence to both ends of the sequence. We then aligned the extended sequences using mafft (Katoh and Standley 2013) and removed gaps automatically using T-coffee (Notredame et al. 2000). The multiple sequence alignment produced by mafft was visualized in aliview (Larsson 2014), and the termini of each element were identified based on canonical motifs of different element classes (e.g. 5′ TG and 3′ CA dinucleotides in LTR elements). A consensus sequence of the trimmed multiple sequence alignment was then generated using the cons tool in EMBOSS (Rice et al. 2000). Finally, the program TE-Aid (https://github.com/clemgoub/TE-Aid) was used to confirm structural properties and the presence of open reading frames for the expected proteins characteristic of each class of TE element. This process of blast, extension, and alignment was repeated iteratively for each element until termini were discovered. Following manual curation of TE sequences, we again used cd-hit-est with the same settings as above to cluster sequences belonging to the same subfamily. The final set of manually curated sequences was then compared against a library of avian TE elements downloaded from repbase as well as other recently published TE datasets (e.g. Dromaius novaehollandiae, Peona et al. 2021b) using cd-hit-est and the 80-80-80 rule above as a threshold to classify curated elements as belonging to previously identified TE subfamilies. We assigned the following species-specific prefixes for newly identified repeat elements in each sparrow species: pasSan (Passerculus sandwichensis), melMel (Melospiza melodia), and artBel (Artemisiospiza belli). For new TE subfamilies shared across two or more of the sparrow species we assigned the prefix Passerellidae. For each element, the prefix was followed by the superfamily identity (e.g. LTR/ERV1). For elements where we could not confidently identify the complete consensus sequence we added the suffix .inc.
Curated TE libraries for all three sparrow species were merged into a single Passerellidae repeat library that was then used to annotate transposable element diversity in the genome assemblies of the three CCGP sparrow genomes using RepeatMasker v. 4.1.2 (Smit et al. 2015 ). We additionally used the de novo Passerellidae TE library to annotate the genome assemblies of the three VGP sparrow genomes and the nine previously sequenced sparrow genomes available on Genbank. Secondly, for seven sparrow species with a contig N50 > 1 Mb and at least a scaffold level assembly, we performed separate RepeatMasker runs on the autosomes, Z chromosome, and W chromosome (if present). For the CCGP genomes, scaffolds were assigned to these different chromosomes based on homology with the Zebra Finch chromosomes using Minimap2 (Li 2018). Finally, we assessed temporal patterns of TE activity for these seven sparrow species. For autosomal, Z, and W chromosomes, we used the calcDivergenceFromAlign.pl script in the RepeatMasker package to estimate the Kimura 2-parameter (K2P) distance of each TE element from the consensus sequence. K2P distances were used to generate barplots for the LTR, SINE, LINE, and DNA classes of TE elements found in the genome. For autosomal loci, we additionally used a mutation rate estimate of 2.3 × 10−9 per site per year from another Passerine species (Ficedula albicollis, Smeds et al. 2016) to estimate the approximate timing of repeat activity.
Sparrow Phylogeny Construction
To further contextualize the history of transposable element proliferation across sparrows, we constructed a time-calibrated phylogeny using ultra-conserved elements (UCE) extracted from all available sparrow genomes (14 taxa including 12 species and 3 song sparrow subspecies) as well as the medium ground finch (Geospiza fortis), island canary (Serinus canaria), and zebra finch (Taeniopygia guttata) as outgroup taxa. We used the UCE-5k-probe-set and the phyluce pipeline (Faircloth 2016) to align probes to each genome, extended sequences by 1,000 bp on either flank, aligned sequences using mafft v. 7.49 (Katoh and Standley 2013), and produced a final PHYLIP file of the UCEs. We used RAxML-NG (Stamatakis 2014) on the CIPRES science portal (Miller et al. 2010) to generate a maximum-likelihood phylogeny of the concatenated UCE loci using the GTRCAT model, rapid bootstrapping, and the autoMRE bootstrapping criterion. Secondly, we used MCMCtree to estimate a time-calibrated phylogeny based on the topology generated by RAxML-NG. We used the fossil sparrow Ammodramus hatcheri (Steadman 1981) to calibrate the node between the grasshopper sparrow (Ammodramus savannarum) and all other sparrows. Following Oliveros et al. (2019), we set the calibration time to 12 Mya bounded by a minimum age of 7.5 Mya and a maximum age of 18.6 Mya. We implemented a model of independent rates among branches and drawing from a log-normal distribution. We performed two independent runs of MCMCtree with each run starting from a different random seed. The first 50,000 iterations were removed as burnin before running for another 100 million iterations with a sample taken every 1,000 iterations. We assessed convergence of the two runs using tracer v. 1.6.0 (Rambaut et al. 2018).
Supplementary Material
Acknowledgments
We thank Joshua Ho for assistance with tissue subsampling, and Nicole Guido, Jonathan Clark and Chris Elphick for sample collection. PacBio Sequel II library prep and sequencing was carried out at the DNA Technologies and Expression Analysis Cores at the UC Davis Genome Center, supported by NIH Shared Instrumentation Grant 1S10OD010786-01. Deep sequencing of Omni-C libraries used the Novaseq S4 sequencing platforms at the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 OD018174 Instrumentation Grant. We thank the staff at the UC Davis DNA Technologies and Expression Analysis Cores and the UC Santa Cruz Paleogenomics Laboratory for their diligence and dedication to generating high-quality sequence data. We thank Erich Jarvis and personnel at the Vertebrate Genomes Project for conducting the sequencing and assembly of Nelson's, saltmarsh, and swamp sparrow. Publication made possible in part by support from the Berkeley Research Impact Initiative (BRII) sponsored by the UC Berkeley Library.
Contributor Information
Phred M Benham, Museum of Vertebrate Zoology, University of California Berkeley, Berkeley, CA 94720, USA; Department of Integrative Biology, University of California Berkeley, Berkeley, CA 94720, USA.
Carla Cicero, Museum of Vertebrate Zoology, University of California Berkeley, Berkeley, CA 94720, USA.
Merly Escalona, Department of Biomolecular Engineering, University of California Santa Cruz, Santa Cruz, CA 95064, USA.
Eric Beraut, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA 95064, USA.
Colin Fairbairn, Department of Ecology and Evolutionary Biology, University of California, Santa Cruz, Santa Cruz, CA 95064, USA.
Mohan P A Marimuthu, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California-Davis, Davis, CA 95616, USA.
Oanh Nguyen, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California-Davis, Davis, CA 95616, USA.
Ruta Sahasrabudhe, DNA Technologies and Expression Analysis Core Laboratory, Genome Center, University of California-Davis, Davis, CA 95616, USA.
Benjamin L King, Department of Molecular and Biomedical Sciences, University of Maine, Orono, ME 04469, USA.
W Kelley Thomas, Department of Molecular, Cellular and Biomedical Sciences, University of New Hampshire, Durham, NH 03824, USA.
Adrienne I Kovach, Department of Natural Resources and the Environment, University of New Hampshire, Durham, NH 03824, USA.
Michael W Nachman, Museum of Vertebrate Zoology, University of California Berkeley, Berkeley, CA 94720, USA; Department of Integrative Biology, University of California Berkeley, Berkeley, CA 94720, USA.
Rauri C K Bowie, Museum of Vertebrate Zoology, University of California Berkeley, Berkeley, CA 94720, USA; Department of Integrative Biology, University of California Berkeley, Berkeley, CA 94720, USA.
Supplementary Material
Supplementary material is available at Genome Biology and Evolution online.
Funding
This work was supported by the California Conservation Genomics Project, with funding provided to the University of California by the State of California, State Budget Act of 2019 [UC Award ID RSI-19-690224]. Additional funding was provided by National Science Foundation Grant #1826777.
Data Availability
Data generated for this study are available under NCBI BioProject PRJNA720569. Raw sequencing data for the Savannah sparrow sample FMNH:Bird:499929 (NCBI BioSample SAMN24839580) are deposited in the NCBI Short-Read Archive (SRA) under SRS12336030. Raw sequencing data for the song sparrow sample MVZ:Bird:193390 (NCBI BioSample SAMN24817870, SAMN24817871) are deposited in the NCBI SRA under SRS12452128. Raw sequencing data for Bell's sparrow sample MVZ:Bird:192114 (NCBI BioSample SAMN24224802) are deposited in the NCBI SRA under SRS11988259. See supplementary table S5, Supplementary Material online for the GenBank accession, BioProject, and BioSample numbers associated with the VGP and other genomes analyzed. For the CCGP genomes, assembly scripts and other data for the analyses presented can be found at the following GitHub repository: www.github.com/ccgproject/ccgp_assembly. Transposable element library, UCE sequences, and other supplemental data and code can be found on Dryad: https://doi.org/10.5061/dryad.cjsxksncs.
Literature Cited
- Abdennur N, Mirny LA. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics. 2020:36(1):311–316. 10.1093/bioinformatics/btz540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Able KP, Able MA. The flexible migratory orientation system of the savannah sparrow (Passerculus sandwichensis). J Exp Biol. 1996:199(Pt 1):3–8. 10.1242/jeb.199.1.3. [DOI] [PubMed] [Google Scholar]
- Ågren JA, Wright SI. Co-evolution between transposable elements and their hosts: a major factor in genome size evolution? Chromosome Res. 2011:19(6):777–786. 10.1007/s10577-011-9229-0. [DOI] [PubMed] [Google Scholar]
- Aldrich JW. Ecogeographical variation in size and proportions of song sparrows (Melospiza melodia). Ornithol Monogr. 1984:35:iii–134. 10.2307/40166779. [DOI] [Google Scholar]
- Allio R, Schomaker-Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F. MitoFinder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour. 2020:20(4):892–905. 10.1111/1755-0998.13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews CB, Mackenzie SA, Gregory TR. Genome size and wing parameters in passerine birds. Proc Biol Sci. 2009:276(1654):55–61. 10.1098/rspb.2008.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcese P, Sogge MK, Marr AB, Patten MA. Song sparrow (Melospiza melodia), version 1.0. In: Poole AF, Gill FB, editors. Birds of the world. Ithaca (NY): Cornell Lab of Ornithology; 2020. 10.2173/bow.sonspa.01. [DOI] [Google Scholar]
- Bachtrog D. Accumulation of Spock and Worf, two novel non-LTR retrotransposons, on the neo-Y chromosome of Drosophila miranda. Mol Biol Evol. 2003:20(2):173–181. 10.1093/molbev/msg035. [DOI] [PubMed] [Google Scholar]
- Benham PM, Cheviron ZA. Divergent mitochondrial lineages arose within a large, panmictic population of the Savannah sparrow (Passerculus sandwichensis). Mol Ecol. 2019:28(7):1765–1783. 10.1111/mec.15049. [DOI] [PubMed] [Google Scholar]
- Benham PM, Cheviron ZA. Population history and the selective landscape shape patterns of osmoregulatory trait divergence in tidal marsh Savannah sparrows (Passerculus sandwichensis). Evolution. 2020:74(1):57–72. 10.1111/evo.13886. [DOI] [PubMed] [Google Scholar]
- Benham PM, Cicero C, DeRaad DA, McCormack JE, Wayne RK, Escalona M, Beraut E, Marimuthu MPA, Nguyen O, Nachman MW, et al. A highly contiguous reference genome for the Steller's hay (Cyanocitta stelleri). J Hered. 2023:114(5):549–560. 10.1093/jhered/esad042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingham PM, Kidwell MG, Rubin GM. The molecular basis of P-M hybrid dysgenesis: the role of the P element, a P-strain-specific transposon family. Cell. 1982:29(3):995–1004. 10.1016/0092-8674(82)90463-9. [DOI] [PubMed] [Google Scholar]
- Black A, Yoon J, McCreedy C, Janjua S, Heenkenda E, Mathur S, Ferree E, Fesnock A, Hernandez A, DeWoody A. Conservation genomics of California towhee (Melozone crissalis) in relation to the official list of endangered and threatened wildlife. Authorea. 2023. https://doi.org/10.2254. preprint: not peer reviewed. [Google Scholar]
- Boman J, Frankl-Vilches C, da Silva dos Santos M, de Oliveira EHC, Gahr M, Suh A. The genome of blue-capped cordon-bleu uncovers hidden diversity of LTR retrotransposons in zebra finch. Genes. 2019:10(4):301. 10.3390/genes10040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bours A, Pruisscher P, Bascón-Cardozo K, Odenthal-Hesse L, Liedvogel M. The blackcap (sylvia atricapilla) genome reveals a recent accumulation of LTR retrotransposons. Sci Rep. 2023:13(1):16471. 10.1038/s41598-023-43090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo GA, Schmitt CJ, Edwards SV. What have we learned from the first 500 avian genomes? Ann Rev Ecol Evol Syst. 2021:52:611–639. 10.1146/annurev-ecolsys-012121-085928. [DOI] [Google Scholar]
- Brosh O, Fabian DK, Cogni R, Tolosana I, Day JP, Olivieri F, Merckx M, Akilli N, Szkuta P, Jiggins FM. A novel transposable element-mediated mechanism causes antiviral resistance in Drosophila through truncating the Veneno protein. Proc Natl Acad Sci U S A. 2022:119(29):e2122026119. 10.1073/pnas.2122026119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinformatics. 2009:10(1):1–9. 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capy P, Gasperi G, Biémont C, Bazin C. Stress and transposable elements: co-evolution or useful parasites? Heredity (Edinb). 2000:85(2):101–106. 10.1046/j.1365-2540.2000.00751.x. [DOI] [PubMed] [Google Scholar]
- Carneiro CM. Genomic insight into the demographic history and structure of the grasshopper sparrow (Ammodramus savannarum) [M.sc. thesis]. [Gainesville]: University of Florida; 2021.
- Challis R, Kumar S, Sotero-Caio C, Brown M, Blaxter M. Genomes on a Tree (GoaT): a versatile, scalable search engine for genomic and sequencing project metadata across the eukaryotic tree of life. Wellcome Open Res. 2023:8:24–27. 10.12688/wellcomeopenres.18658.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis R, Richards E, Rajan J, Cochrane G, Blaxter M. BlobToolKit—interactive quality assessment of genome assemblies. G3 (Bethesda). 2020:10(4):1361–1374. 10.1534/g3.119.400908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Langley CH. The population genetics of Drosophila transposable elements. Ann Rev Genet. 1989:23(1):251–287. 10.1146/annurev.ge.23.120189.001343. [DOI] [PubMed] [Google Scholar]
- Cheng H, Jarvis ED, Fedrigo O, Koepfli K-P, Urban L, Gemmell NJ, Li H. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotechnol. 2022:40(9):1332–1335. 10.1038/s41587-022-01261-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicero C, Johnson NK. Diagnosability of subspecies: lessons from Sage Sparrows (Amphispiza belli) for analysis of geographic variation in birds. Auk. 2006:123(1):266–274. 10.1093/auk/123.1.266. [DOI] [Google Scholar]
- Cicero C, Johnson NK. Narrow contact of desert sage sparrows (Amphispiza belli nevadensis and A. B. Canescens) in owens valley, Eastern California: evidence from mitochondrial DNA, morphology, and gis-based niche models. Ornithological Monogr. 2007:63(1):78–95. 10.1642/0078-6594(2007)63[78:NCODSS]2.0.CO;2. [DOI] [Google Scholar]
- Cicero C, Koo MS. The role of niche divergence and phenotypic adaptation in promoting lineage diversification in the Sage Sparrow (Artemisiospiza belli, Aves: Emberizidae). Biol J Linn Soc. 2012:107(2):332–354. 10.1111/j.1095-8312.2012.01942.x. [DOI] [Google Scholar]
- Clark JD, Benham PM, Maldonado JE, Luther DA, Lim HC. Maintenance of local adaptation despite gene flow in a coastal songbird. Evolution. 2022:76(7):1481–1494. 10.1111/evo.14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornelis G, Funk M, Vernochet C, Leal F, Tarazona OA, Meurice G, Heidmann O, Dupressoir A, Miralles A, Ramirez-Pinilla MP, et al. An endogenous retroviral envelope syncytin and its cognate receptor identified in the viviparous placental Mabuya lizard. Proc Natl Acad Sci U S A. 2017:114(51):E10991–E11000. 10.1073/pnas.1714590114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daborn PJ, Yen JL, Bogwitz MR, Le Goff G, Feil E, Jeffers S, Tijet N, Perry T, Heckel D, Batterham P, et al. A single P450 allele associated with insecticide resistance in Drosophila. Science. 2002:297(5590):2253–2256. 10.1126/science.1074170. [DOI] [PubMed] [Google Scholar]
- Degrandi TM, Barcellos S A, Costa A L, Garnero A D, Hass I V, Gunski RJ. Introducing the bird chromosome database: an overview of cytogenetic studies in birds. Cytogenet Genome Res. 2020:160(4):199–205. 10.1159/000507768. [DOI] [PubMed] [Google Scholar]
- DeRaad DA, Escalona M, Benham PM, Marimuthu MPA, Sahasrabudhe RM, Nguyen O, Chumchim N, Beraut E, Fairbairn CW, Seligmann W, et al. De novo assembly of a chromosome-level reference genome for the California Scrub-Jay, Aphelocoma californica. J Hered. 2023:114(6):669–680. 10.1093/jhered/esad047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks MFL, Schachtschneider KM, Madsen O, Schijlen E, Verhoeven KJF, van Oers K. Gene and transposable element methylation in great tit (Parus major) brain and blood. BMC Genomics. 2016:17(1):1–13. 10.1186/s12864-016-2653-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolezel J, Bartos J, Voglmayr H, Greilhuber J. Nuclear DNA content and genome size of trout and human. Cytometry. 2003:51A(2):127–128. 10.1002/cyto.a.10013. [DOI] [PubMed] [Google Scholar]
- Ellegren H. Evolutionary stasis: the stable chromosomes of birds. Trends Ecol Evol. 2010:25(5):283–291. 10.1016/j.tree.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Elliott TA, Gregory TR. What's in a genome? The C-value enigma and the evolution of eukaryotic genome content. Philos Trans R Soc B Biol Sci. 2015:370(1678):20140331. 10.1098/rstb.2014.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth BC. PHYLUCE is a software package for the analysis of conserved genomic loci. Bioinformatics. 2016:32(5):786–788. 10.1093/bioinformatics/btv646. [DOI] [PubMed] [Google Scholar]
- Feng S, Stiller J, Deng Y, Armstrong J, Fang Q, Reeve AH, Xie D, Chen G, Guo C, Faircloth BC, et al. Dense sampling of bird diversity increases power of comparative genomics. Nature. 2020:587(7833):252–257. 10.1038/s41586-020-2873-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008:9(5):397–405. 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field CR, Ruskin KJ, Benvenuti B, Borowske A, Cohen JB, Garey L, Hodgman TP, Kern RA, King E, Kocek AR, et al. Quantifying the importance of geographic replication and representativeness when estimating demographic rates, using a coastal species as a case study. Ecography. 2018:41(6):971–981. 10.1111/ecog.02424. [DOI] [Google Scholar]
- Flynn JM, Hubley R, Goubert C, Rosen J, Clark AG, Feschotte C, Smit AF. RepeatModeler2 for automated genomic discovery of transposable element families. Proc Natl Acad Sci U S A. 2020:117(17):9451–9457. 10.1073/pnas.1921046117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman-Gallant CR, Wheelwright NT, Meiklejohn KE, States SL, Sollecito SV. Little effect of extrapair paternity on the opportunity for sexual selection in Savannah sparrows (Passerculus sandwichensis). Evolution. 2005:59(2):422–430. 10.1111/j.0014-3820.2005.tb01000.x. [DOI] [PubMed] [Google Scholar]
- Friis G, Vizueta J, Ketterson ED, Milá B. A high-quality genome assembly and annotation of the dark-eyed junco Junco hyemalis, a recently diversified songbird. G3 (Bethesda). 2022:12(6):jkac083. 10.1093/g3journal/jkac083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbraith JD, Kortschak RD, Suh A, Adelson DL. Genome stability is in the eye of the beholder: CR1 retrotransposon activity varies significantly across Avian diversity. Genome Biol Evol. 2021:13(12):evab259. 10.1093/gbe/evab259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye J, Pop M, Koren S, Bickhart D, Chin C-S. Scaffolding of long read assemblies using long range contact information. BMC Genomics. 2017:18(1):527. 10.1186/s12864-017-3879-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghurye J, Rhie A, Walenz BP, Schmitt A, Selvaraj S, Pop M, Phillippy AM, Koren S. Integrating Hi-C links with assembly graphs for chromosome-scale assembly. PLoS Comput Biol. 2019:15(8):e1007273. 10.1371/journal.pcbi.1007273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodier JL. Restricting retrotransposons: a review. Mob DNA. 2016:7:16. 10.1186/s13100-016-0070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubert C, Craig RJ, Bilat AF, Peona V, Vogan AA, Protasio AV. A beginner's guide to manual curation of transposable elements. Mob DNA. 2022:13(1):1–19. 10.1186/s13100-021-00259-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozashti L, Feschotte C, Hoekstra HE. Transposable element interactions shape the ecology of the deer mouse genome. Mol Biol Evol. 2023:40(4):1–17. 10.1093/molbev/msad069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg R, Cadena V, Danner RM, Tattersall G. Heat loss may explain bill size differences between birds occupying different habitats. PLoS One. 2012:7(7):e40933. 10.1371/journal.pone.0040933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg R, Maldonado JE, Droege S, McDonald MV. Tidal marshes: a global perspective on the evolution and conservation of their terrestrial vertebrates. BioScience 2006:56(8):675–685.. 10.1641/0006-3568(2006)56[675:TMAGPO]2.0.CO;2. [DOI] [Google Scholar]
- Gregory TR. Animal Genome Size Database. 2022. [accessed 2022 February 7]. http://www.genomesize.com.
- Guan D, McCarthy SA, Wood J, Howe K, Wang Y, Durbin R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 2020:36(9):2896–2898. 10.1093/bioinformatics/btaa025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013:29(8):1072–1075. 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert JA, Mowbray TB. Swamp Sparrow (Melospiza georgiana), version 1.0. In: Rodewald PG, editor. Birds of the world. Ithaca (NY): Cornell Lab of Ornithology; 2020. 10.2173/bow.swaspa.01. [DOI] [Google Scholar]
- Hughes AL, Hughes MK. Small genomes for better flyers. Nature. 1995:377(6548):391. 10.1038/377391a0. [DOI] [PubMed] [Google Scholar]
- Ji Y, DeWoody JA. Genomic landscape of long terminal repeat retrotransposons (LTR-RTs) and solo LTRs as shaped by ectopic recombination in chicken and zebra finch. J Mol Evol. 2016:82(6):251–263. 10.1007/s00239-016-9741-0. [DOI] [PubMed] [Google Scholar]
- Johnston RF. Variation in breeding season and clutch size in song sparrows of the Pacific coast. Condor. 1954:56(5):268–273. 10.2307/1364850. [DOI] [Google Scholar]
- Josefsson C, Dilkes B, Comai L. Parent-dependent loss of gene silencing during interspecies hybridization. Curr Biol. 2006:16(13):1322–1328. 10.1016/j.cub.2006.05.045. [DOI] [PubMed] [Google Scholar]
- Kapusta A, Suh A. Evolution of bird genomes—a transposon's-eye view. Ann N Y Acad Sci. 2017:1389(1):164–185. 10.1111/nyas.13295. [DOI] [PubMed] [Google Scholar]
- Kapusta A, Suh A, Feschotte C. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci U S A. 2017:114(8):E1460–E1469. 10.1073/pnas.1616702114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013:30(4):772–780. 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller LF, Arcese P. No evidence for inbreeding avoidance in a natural population of song sparrows (Melospiza melodia). Am Nat. 1998:152(3):380–392. 10.1086/286176. [DOI] [PubMed] [Google Scholar]
- Keller LF, Arcese P, Smith JNM, Hochachka WM, Stearns SC. Selection against inbred song sparrows during a natural population bottleneck. Nature. 1994:372(6504):356–357. 10.1038/372356a0. [DOI] [PubMed] [Google Scholar]
- Kent TV, Uzunović J, Wright SI. Coevolution between transposable elements and recombination. Philos Trans R Soc B Biol Sci. 2017:372(1736):20160458. 10.1098/rstb.2016.0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerpedjiev P, Abdennur N, Lekschas F, McCallum C, Dinkla K, Strobelt H, Luber JM, Ouellette SB, Azhir A, Kumar N, et al. Higlass: web-based visual exploration and analysis of genome interaction maps. Genome Biol. 2018:19(1):125. 10.1186/s13059-018-1486-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidwell MG. Transposable elements and the evolution of genome size in eukaryotes. Genetica. 2002:115(1):49–63. 10.1023/A:1016072014259. [DOI] [PubMed] [Google Scholar]
- Klicka J, Keith Barker F, Burns KJ, Lanyon SM, Lovette IJ, Chaves JA, Bryson RW. A comprehensive multilocus assessment of sparrow (aves: passerellidae) relationships. Mol Phylogenet Evol. 2014:77(1):177–182. 10.1016/j.ympev.2014.04.025. [DOI] [PubMed] [Google Scholar]
- Knisbacher BA, Levanon EY. DNA editing of LTR retrotransposons reveals the impact of APOBECs on vertebrate genomes. Mol Biol Evol. 2016:33(2):554–567. 10.1093/molbev/msv239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korlach J, Gedman G, Kingan SB, Chin CS, Howard JT, Audet JN, Cantin L, Jarvis ED. De novo PacBio long-read and phased avian genome assemblies correct and add to reference genes generated with intermediate and short reads. GigaScience. 2017:6(10):1–16. 10.1093/gigascience/gix085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratochwil CF, Kautt AF, Nater A, Härer A, Liang Y, Henning F, Meyer A. An intronic transposon insertion associates with a trans-species color polymorphism in Midas cichlid fishes. Nat Commun. 2022:13(1):296. 10.1038/s41467-021-27685-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics. 2014:30(22):3276–3278. 10.1093/bioinformatics/btu531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin HA, Richards S, Lieberman Aiden E, Allende ML, Archibald JM, Bálint M, Barker KB, Baumgartner B, Belov K, Bertorelle G, et al. The Earth BioGenome Project 2020: starting the clock. Proc Natl Acad Sci. 2022:119(4):1–7. 10.1073/pnas.2115635118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM, arXiv: 1303.3997, preprint: not peer reviewed.
- Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018:34(18):3094–3100. 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006:22(13):1658–1659. 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- Lin M, Escalona M, Sahasrabudhe R, Nguyen O, Beraut E, Buchalski MR, Wayne RK. A reference genome assembly of the bobcat, Lynx rufus. J Hered. 2022:113(6):615–623. 10.1093/jhered/esac031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louha, S., Ray, D. A., Winker, K., Glenn, T. C. (2020). A high-quality genome assembly of the North American Song Sparrow, Melospiza melodia. G3.10(4):1159–1166. doi: 10.1534/g3.119.400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS. The origins of genome complexity. Science. 2003:302(5649):1401–1404. 10.1126/science.1089370. [DOI] [PubMed] [Google Scholar]
- Manni M, Berkeley MR, Seppey M, Simão FA, Zdobnov EM. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 2021:38(10):4647–4654. 10.1093/molbev/msab199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthey JD, Moyle RG, Boissinot S. Multiple and independent phases of transposable element amplification in the genomes of piciformes (woodpeckers and allies). Genome Biol Evol. 2018:10(6):1–35. 10.1093/gbe/evy105/5020728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marler P, Peters S. Selective vocal learning in a sparrow. Science. 1977:198(4316):519–521. 10.1126/science.198.4316.519. [DOI] [PubMed] [Google Scholar]
- Marr AB, Keller LF, Arcese P. Heterosis and outbreeding depression in descendants of natural immigrants to an inbred population of song sparrows (Melospiza Melodia). Evolution. 2002:56(1):131–142. 10.1111/j.0014-3820.2002.tb00855.x. [DOI] [PubMed] [Google Scholar]
- Marshall JT. Ecologic races of song sparrows in the San Francisco Bay Region: Part II. Geographic variation. Condor. 1948:50(6):233–256. 10.2307/1364817. [DOI] [Google Scholar]
- Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang X-Y, Edouard P, Howes S, et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000:403(6771):785–789. 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- Mikles CS, Aguillon SM, Chan YL, Arcese P, Benham PM, Lovette IJ, Walsh J. Genomic differentiation and local adaptation on a microgeographic scale in a resident songbird. Mol Ecol. 2020:29(22):4295–4307. 10.1111/mec.15647. [DOI] [PubMed] [Google Scholar]
- Miller MA, Pfeiffer W, Schwartz T. Creating the CIPRES science gateway for inference of large phylogenetic trees. In: 2010 Gateway computing environments workshop (GCE); 2010. p. 1–8. [Google Scholar]
- Moore FR. Sunset and the orientation of a nocturnal migrant bird. Nature. 1978:274(5667):154–156. 10.1038/274154a0. [DOI] [Google Scholar]
- Nice MM. Studies in the life history of the song sparrow. I. A population study of the song sparrow. Trans Linnaean Soc New York. 1937:4:1–247. [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000:302(1):205–217. 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Oliveros CH, Field DJ, Ksepka DT, Barker FK, Aleixo A, Andersen MJ, Alström P, Benz BW, Braun EL, Braun MJ, et al. Earth history and the passerine superradiation. Proc Natl Acad Sci U S A. 2019:116(16):7916–7925. 10.1073/pnas.1813206116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill R, O'Neill M, Graves J. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature. 1998:393(6680):68–72. 10.1038/29985. [DOI] [PubMed] [Google Scholar]
- Open2C, Abdennur N, Fudenberg G, Flyamer IM, Galitsyna AA, Goloborodko A, Imakaev M, Venev SV. Pairtools: from sequencing data to chromosome contacts. bioRxiv. 2023. 10.1101/2023.02.13.528389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Organ CL, Shedlock AM, Meade A, Pagel M, Edwards SV. Origin of avian genome size and structure in non-avian dinosaurs. Nature. 2007:446(7132):180–184. 10.1038/nature05621. [DOI] [PubMed] [Google Scholar]
- Patten MA, Pruett CL. The Song Sparrow, Melospiza melodia, as a ring species: patterns of geographic variation, a revision of subspecies, and implications for speciation. Syst Biodivers. 2009:7(1):33–62. 10.1017/S1477200008002867. [DOI] [Google Scholar]
- Peona V, Blom MPK, Xu L, Burri R, Sullivan S, Bunikis I, Liachko I, Haryoko T, Jønsson KA, Zhou Q, et al. Identifying the causes and consequences of assembly gaps using a multiplatform genome assembly of a bird-of-paradise. Mol Ecol Resour. 2021a:21(1):263–286. 10.1111/1755-0998.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peona V, Palacios-Gimenez OM, Blommaert J, Liu J, Haryoko T, Jønsson KA, Irestedt M, Zhou Q, Jern P, Suh A. The avian W chromosome is a refugium for endogenous retroviruses with likely effects on female-biased mutational load and genetic incompatibilities. Philos Trans R Soc B Biol Sci. 2021b:376(1833):1833. 10.1098/rstb.2020.0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peona V, Weissensteiner MH, Suh A. How complete are “complete” genome assemblies? —an avian perspective. Mol Ecol Resour. 2018:18(6):1188–1195. 10.1111/1755-0998.12933. [DOI] [PubMed] [Google Scholar]
- Poulson TL. Countercurrent multipliers in avian kidneys. Science. 1965:148(3668):389–391. 10.1126/science.148.3668.389. [DOI] [PubMed] [Google Scholar]
- Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA. Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Syst Biol. 2018:67(5):901–904. 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Bhardwaj V, Arrigoni L, Lam KC, Grüning BA, Villaveces J, Habermann B, Akhtar A, Manke T. High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat Commun. 2018:9(1):189. 10.1038/s41467-017-02525-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranallo-Benavidez TR, Jaron KS, Schatz MC. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat Commun. 2020:11(1):1432. 10.1038/s41467-020-14998-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, McCarthy SA, Fedrigo O, Damas J, Formenti G, Koren S, Uliano-Silva M, Chow W, Fungtammasan A, Kim J, et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021:592(7856):737–746. 10.1038/s41586-021-03451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhie A, Walenz BP, Koren S, Phillippy AM. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biol. 2020:21(1):245. 10.1186/s13059-020-02134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P, Longden L, Bleasby A. EMBOSS: the European molecular biology open software suite. Trends Genet. 2000:16(6):276–277. 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
- Rising JD. Geographic variation in size and shape of Savannah Sparrows (Passerculus sandwichensis). Stud Avian Biol. 2001:23:1–65. [Google Scholar]
- Rising D, Avise JC. Application of genealogical-concordance principles to the taxonomy and evolutionary history of the sharp-tailed sparrow (Ammodramus caudacutus). Auk. 1993:110(4):844–856. 10.2307/4088638. [DOI] [Google Scholar]
- Rizzon C, Marais G, Gouy M, Biémont C. Recombination rate and the distribution of transposable elements in the Drosophila melanogaster genome. Genome Res. 2002:12(3):400–407. 10.1101/gr.210802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin KJ, Ettersona MA, Hodgman TP, Borowske A, Cohen JB, Elphick CS, Field CR, Kern RA, King E, Kocek AR, et al. Demographic analysis demonstrates systematic but independent abiotic and biotic stressors across 59% of a global species range. Auk. 2017a:134(4):903–916. 10.1642/AUK-16-230.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruskin KJ, Etterson MA, Hodgman TP, Borowske A, Cohen JB, Elphick CS, Field CR, Kern RA, King E, Kocek AR, et al. Seasonal fecundity is not related to range position across a species’ global range despite a central peak in abundance. Oecologia. 2017b:183(1):291–301. 10.1007/s00442-016-3745-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader L, Schmitz J. The impact of transposable elements in adaptive evolution. Mol Ecol. 2019:28(6):1537–1549. 10.1111/mec.14794. [DOI] [PubMed] [Google Scholar]
- Searcy WA, Marler P. A test for responsiveness to song structure and programming in female sparrows. Science. 1981:213(4510):926–928. 10.1126/science.213.4510.926. [DOI] [PubMed] [Google Scholar]
- Serrato-Capuchina A, Matute DR. The role of transposable elements in speciation. Genes (Basel). 2018:9(5):254. 10.3390/genes9050254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer HB, Toffelmier E, Corbett-Detig RB, Escalona M, Erickson B, Fiedler P, Gold M, Harrigan RJ, Hodges S, Luckau TK, et al. Landscape genomics to enable conservation actions: the California Conservation Genomics Project. J Hered. 2022:113(6):577–588. 10.1093/jhered/esac020. [DOI] [PubMed] [Google Scholar]
- Shields GF, Straus NA. DNA-DNA hybridization studies of birds. Evolution. 1975:29(1):159. 10.2307/2407149. [DOI] [PubMed] [Google Scholar]
- Shriver WG, Gibbs JP, Vickery PD, Gibbs HL, Hodgman TP, Jones PT, Jacques CN. Concordance between morphological and molecular markers in assessing hybridization between sharp-tailed sparrows in New England. Auk. 2005:122(1):94–107.. 10.1642/0004-8038(2005)122[0094:CBMAMM]2.0.CO;2. [DOI] [Google Scholar]
- Shriver WG, Hodgman TP, Hanson AR. Nelson's Sparrow (Ammospiza nelsoni), version 1.0. In: Rodewald PG, editor. Birds of the world. Ithaca (NY): Cornell Lab of Ornithology; 2020. 10.2173/bow.nstspa.01. [DOI] [Google Scholar]
- Sim SB, Corpuz RL, Simmonds TJ, Geib SM. HiFiAdapterFilt, a memory efficient read processing pipeline, prevents occurrence of adapter sequence in PacBio HiFi reads and their negative impacts on genome assembly. BMC Genomics. 2022:23(1):157. 10.1186/s12864-022-08375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007:8(4):272–285. 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- Smeds L, Qvarnstrom A, Ellegren H. Direct estimate of the rate of germline mutation in a bird. Genome Res. 2016:26(9):1211–1218. 10.1101/gr.204669.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. RepeatMasker Open-4.0.2013-2015. 2015.
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014:30(9):1312–1313. 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke M, Diekhans M, Baertsch R, Haussler D. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics. 2008:24(5):637–644. 10.1093/bioinformatics/btn013. [DOI] [PubMed] [Google Scholar]
- Steadman DW. A re-examination of Palaeostruthus Hatcheri (Shufeldt), a late Miocene sparrow from Kansas. J Vertebr Paleontol. 1981:1(2):171–173. 10.1080/02724634.1981.10011889. [DOI] [Google Scholar]
- Suh A, Smeds L, Ellegren H. Abundant recent activity of retrovirus-like retrotransposons within and among flycatcher species implies a rich source of structural variation in songbird genomes. Mol Ecol. 2018:27(1):99–111. 10.1111/mec.14439. [DOI] [PubMed] [Google Scholar]
- Suh A, Witt CC, Menger J, Sadanandan KR, Podsiadlowski L, Gerth M, Weigert A, McGuire JA, Mudge J, Edwards SV, et al. Ancient horizontal transfers of retrotransposons between birds and ancestors of human pathogenic nematodes. Nat Commun. 2016:7(1):11396. 10.1038/ncomms11396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teeling EC, Vernes SC, Dávalos LM, Ray DA, Gilbert MTP, Myers E. Bat biology, genomes, and the Bat1K project: to generate chromosome-level genomes for all living bat species. Annu Rev Anim Biosci. 2018:6(1):23–46. 10.1146/annurev-animal-022516-022811. [DOI] [PubMed] [Google Scholar]
- Thibaud-Nissen F, Souvorov A, Murphy T, DiCuccio M, Kitts P. Eukaryotic genome annotation pipeline. 2013 Nov 14. The NCBI handbook. 2nd ed. Bethesda (MD): National Center for Biotechnology Information (US); 2013. p. 111–130. [Google Scholar]
- Tomaszkiewicz M, Medvedev P, Makova KD. Y and W chromosome assemblies: approaches and discoveries. Trends Genet. 2017:33(4):266–282. 10.1016/j.tig.2017.01.008. [DOI] [PubMed] [Google Scholar]
- Tuttle EM, Bergland AO, Korody ML, Brewer MS, Newhouse DJ, Minx P, Stager M, Betuel A, Cheviron ZA, Warren WC, et al. Divergence and functional degradation of a sex chromosome-like supergene. Curr Biol. 2016:26(3):344–350. 10.1016/j.cub.2015.11.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uliano-Silva M, Ferreira Nunes JG, Krasheninnikova K, McCarthy SA. marcelauliano/MitoHiFi: mitohifi_v2.0 (v2.0). Zenodo. 2021. 10.5281/zenodo.5205678. [DOI]
- Ungerer MC, Strakosh SC, Zhen Y. Genome expansion in three hybrid sunflower species is associated with retrotransposon proliferation. Curr Biol. 2006:16(20):R872–R873. 10.1016/j.cub.2006.09.020. [DOI] [PubMed] [Google Scholar]
- Van’t Hof AE, Campagne P, Rigden DJ, Yung CJ, Lingley J, Quail MA, Hall N, Darby AC, Saccheri IJ. The industrial melanism mutation in British peppered moths is a transposable element. Nature. 2016:534(7605):102–105. 10.1038/nature17951. [DOI] [PubMed] [Google Scholar]
- Walsh J, Benham PM, Deane-Coe PE, Arcese P, Butcher BG, Chan YL, Cheviron ZA, Elphick CS, Kovach AI, Olsen BJ, et al. Genomics of rapid ecological divergence and parallel adaptation in songbirds. Evol Lett. 2019a:3-4(4):324–338. 10.1002/evl3.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J, Clucas G, MacManes M, Thomas WK, Kovach AI. Divergent selection and drift shape the genomes of two avian sister species spanning a saline-freshwater ecotone. Ecol Evol. 2019b:9(23):13477–13494. 10.1002/ece3.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh J, Kovach AI, Benham PM, Clucas GV, Winder GI, Lovette I. Genomic data reveal the biogeographic and demographic history of Ammospiza sparrows in Northeast Tidal Marshes. J Biogeogr. 2021:48(9):2360–2374. 10.1111/jbi.14208. [DOI] [Google Scholar]
- Walsh J, Lovette I, Winder V, Elphick C, Olsen B, Shriver G, Kovach AI. Subspecies delineation amid phenotypic, geographic, and genetic discordance. Mol Ecol. 2017:26(5):1242–1255. 10.1111/mec.14010. [DOI] [PubMed] [Google Scholar]
- Walsh J, Shriver WG, Olsen BJ, O’brien KM, Kovach AI. Relationship of phenotypic variation and genetic admixture in the Saltmarsh-Nelson’s sparrow hybrid zone. Auk. 2015:132(3):704–716. 10.1642/AUK-14-299.1. [DOI] [Google Scholar]
- Warmuth VM, Weissensteiner MH, Wolf JBW. Accumulation and ineffective silencing of transposable elements on an avian W Chromosome. Genome Res. 2022:32(4):671–681. 10.1101/gr.275465.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WC, Clayton DF, Ellegren H, Arnold AP, Hillier LW, Künstner A, Searle S, White S, Vilella AJ, Fairley S, et al. The genome of a songbird. Nature. 2010:464(7289):757–762. 10.1038/nature08819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren WC, Hillier LW, Tomlinson C, Minx P, Kremitzki M, Graves T, Markovic C, Bouk N, Pruitt KD, Thibaud-Nissen F, et al. A new chicken genome assembly provides insight into avian genome structure. G3 (Bethesda). 2017:7(1):109–117. 10.1534/g3.116.035923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelwright NT, Rising JD. Savannah Sparrow (Passerculus sandwichensis), version 1.0. In: Poole AF, editor. Birds of the world. Ithaca (NY): Cornell Lab of Ornithology; 2020. 10.2173/bow.savspa.01. [DOI] [Google Scholar]
- Whitney KD, Garland T. Did genetic drift drive increases in genome complexity? PLoS Genet. 2010:6(8):e1001080. 10.1371/journal.pgen.1001080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, et al. A unified classification system for eukaryotic transposable elements. Nat Rev Genet. 2007:8(12):973–982. 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- Williams H, Scharf A, Ryba AR, Ryan Norris D, Mennill DJ, Newman AEM, Doucet SM, Blackwood JC. Cumulative cultural evolution and mechanisms for cultural selection in wild bird songs. Nat Commun. 2022:13(1):4001. 10.1038/s41467-022-31621-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler DW, Billerman SM, Lovette IJ. New world sparrows (Passerellidae), version 1.0. In: Billerman SM, Keeney BK, Rodewald PG, Schulenberg TS, editors. Birds of the world. Ithaca (NY): Cornell Lab of Ornithology; 2020. 10.2173/bow.passer3.01. [DOI] [Google Scholar]
- Wright NA, Gregory TR, Witt CC. Metabolic “engines” of flight drive genome size reduction in birds. Proc Biol Sci. 2014:281(1779):20132780. 10.1098/rspb.2013.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Z, Miedzinska K, Krause JS, Pérez JH, Wingfield JC, Meddle SL, Smith J. A chromosome-level genome assembly of a free-living white-crowned sparrow (Zonotrichia leucophrys gambelii). Sci Data. 2024:11(1):1–10. 10.1038/s41597-024-02929-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Li C, Li Q, Li B, Larkin DM, Lee C, Storz JF, Antunes A, Greenwold MJ, Meredith RW, et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science. 2014:346(6215):1311–1321. 10.1126/science.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated for this study are available under NCBI BioProject PRJNA720569. Raw sequencing data for the Savannah sparrow sample FMNH:Bird:499929 (NCBI BioSample SAMN24839580) are deposited in the NCBI Short-Read Archive (SRA) under SRS12336030. Raw sequencing data for the song sparrow sample MVZ:Bird:193390 (NCBI BioSample SAMN24817870, SAMN24817871) are deposited in the NCBI SRA under SRS12452128. Raw sequencing data for Bell's sparrow sample MVZ:Bird:192114 (NCBI BioSample SAMN24224802) are deposited in the NCBI SRA under SRS11988259. See supplementary table S5, Supplementary Material online for the GenBank accession, BioProject, and BioSample numbers associated with the VGP and other genomes analyzed. For the CCGP genomes, assembly scripts and other data for the analyses presented can be found at the following GitHub repository: www.github.com/ccgproject/ccgp_assembly. Transposable element library, UCE sequences, and other supplemental data and code can be found on Dryad: https://doi.org/10.5061/dryad.cjsxksncs.