

Abstract
Aging of the world population significantly impacts healthcare globally and specifically the field of transplantation. Together with end-organ dysfunction and prolonged immunosuppression, age increases the frequency of comorbid chronic diseases in transplant candidates and recipients, contributing to inferior outcomes. Although, the frequency of death increases with age, limited use of organs from older deceased donors reflects the concerns about organ durability and inadequate function. Cellular senescence (CS) is a hallmark of aging, which occurs in response to a myriad of cellular stressors, leading to activation of signaling cascades that stably arrest cell-cycle progression to prevent tumorigenesis. In aging and chronic conditions, senescent cells accumulate as the immune system’s ability to clear them wanes, which is causally implicated in the progression of chronic diseases, immune dysfunction, organ damage, decreased regenerative capacity, and aging itself. The intimate interplay between senescent cells, their pro-inflammatory secretome, and immune cells results in a positive feedback loop, propagating chronic sterile inflammation and the spread of CS. Hence, senescent cells in organs from older donors trigger recipient’s alloimmune response resulting in the increased risk of graft loss. Eliminating senescent cells or attenuating their inflammatory phenotype is a novel, potential therapeutic target to improve transplant outcomes and expand utilization of organs from older donors. This review focuses on the current knowledge about the impact of CS on circulating immune cells in the context of organ damage and disease progression, discusses the impact of CS on abdominal solid organs that are commonly transplanted and reviews emerging therapies that target CS.
I. Introduction
The fact that the number of individuals over the age of 65 is more than doubling over the next two decades globally carries significant implications for healthcare and for organ transplantation.1,2 Currently, according to the United Network for Organ Sharing, over 25% of transplant candidates waiting for a heart, lung, liver, or kidney are over 65 years of age. In contrast, only 7% of organs transplanted from deceased donors are in the same age category, primarily due to concerns of inadequate organ function from older donor.3 According to the United States Census Bureau, the projected number of individuals over the age of 65 will surpass the number of individuals under the age of 18 by 2034, which will further exacerbate the problem of aging recipients and donors.4 The risk of organ failure increases with age.5–7 However, multiple analyses of selected older organ recipients demonstrated that advanced age alone is not a contraindication for organ transplantation.8–12 Still the waitlist percentage of older candidates is not reflective of the disease burden in the population as the frequency of medical comorbidities increases exponentially with age, decreasing the likelihood of successful transplant. Frailty and comorbidities reduce the capacity for older recipients to withstand surgical and immunosuppressive stress, contributing to posttransplant organ dysfunction, infections and malignancies that may be related to the immunosenescence of aged organisms. From an organ availability perspective, the mortality increases with age, but organs from older individuals are infrequently used for transplant reflecting concerns about organ durability and function as it is well-established that age contributes to increased immunogenicity and decreased regenerative capacity of transplant organs, thus limiting their clinical use.13,14 Chronological age is the greatest risk factor for most common chronic diseases by orders of magnitude compared to commonly treated risk factors.5 Deciphering what in aging biology mediates that vulnerability to disease and finding ways to therapeutically intervene in that biology has the potential to transform transplantation medicine, extending the age range of donors and recipients.
II. Aging biology
The goal of therapeutically targeting aging biology to extend the period of health in old age is termed Geroscience.15 The pursuit of Geroscience led to identification of nine hallmarks of aging biology by experts in the field.15–17 These include altered intercellular communication, genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence (CS) and stem cell exhaustion. Each hallmark fulfills three distinct criteria: it manifests during normal aging; its aggravation accelerates normal aging; and its amelioration delays normal aging and increases lifespan.16 Recently, the hallmarks of aging were reclassified in a hierarchy with genomic instability, telomeric attrition, epigenetic changes, loss of proteostasis being primary drivers of aging biology, all reflecting mechanisms of accumulation of macromolecular damage in cells.17 The antagonistic hallmarks of aging biology are mechanisms that evolved to combat the primary drivers of aging and include CS, changes in mitochondrial function, and altered nutrient sensing. Finally, the integrative hallmarks of aging comprise systemic responses to chronic activation of the antagonistic hallmarks and include stem cell exhaustion, altered intercellular communication, chronic inflammation, and dysbiosis. At least two of these integrative hallmarks are known to have a direct impact on the success of transplantation: chronic inflammation and stem cell exhaustion.18–20
III. Cellular senescence
CS, as an antagonistic hallmark of aging, is a cellular response to a variety of internal and external stressors, including genotoxic, proteostatic, or organelle stress (Figure 1).21 A signaling cascade is deployed to stably arrest cell-cycle progression. CS protects the organism from pathological conditions such as tumorigenesis.22 A key feature of senescent cells is a robust secretome of growth factors, proteases, and pro-inflammatory cytokines and chemokines.21 This senescence-associated secretory phenotype, or SASP, is involved in tissue remodeling and attraction of immune cells.23 Hence, CS promotes wound healing, which is resolved by immune clearance of senescent cells from the wound site. Senescent cell abundance increases with chronological age in humans.24 CS is also associated with numerous chronic conditions including obesity, metabolic syndrome, diabetes, pulmonary and hepatic fibrosis, osteoporosis, osteoarthritis, steatosis, chronic kidney disease, cardiovascular disease, immune dysfunction, and neurodegenerative diseases such as Alzheimer’s and Parkinson’s.17,25–28 In recent years, CS gained much attention as a target for therapeutic interventions aimed at eliminating senescent cells or attenuating their inflammatory phenotype via senolytics and senomorphics, respectively.29–34
Figure 1. Mechanisms of senescence and characteristics of senescent cells.
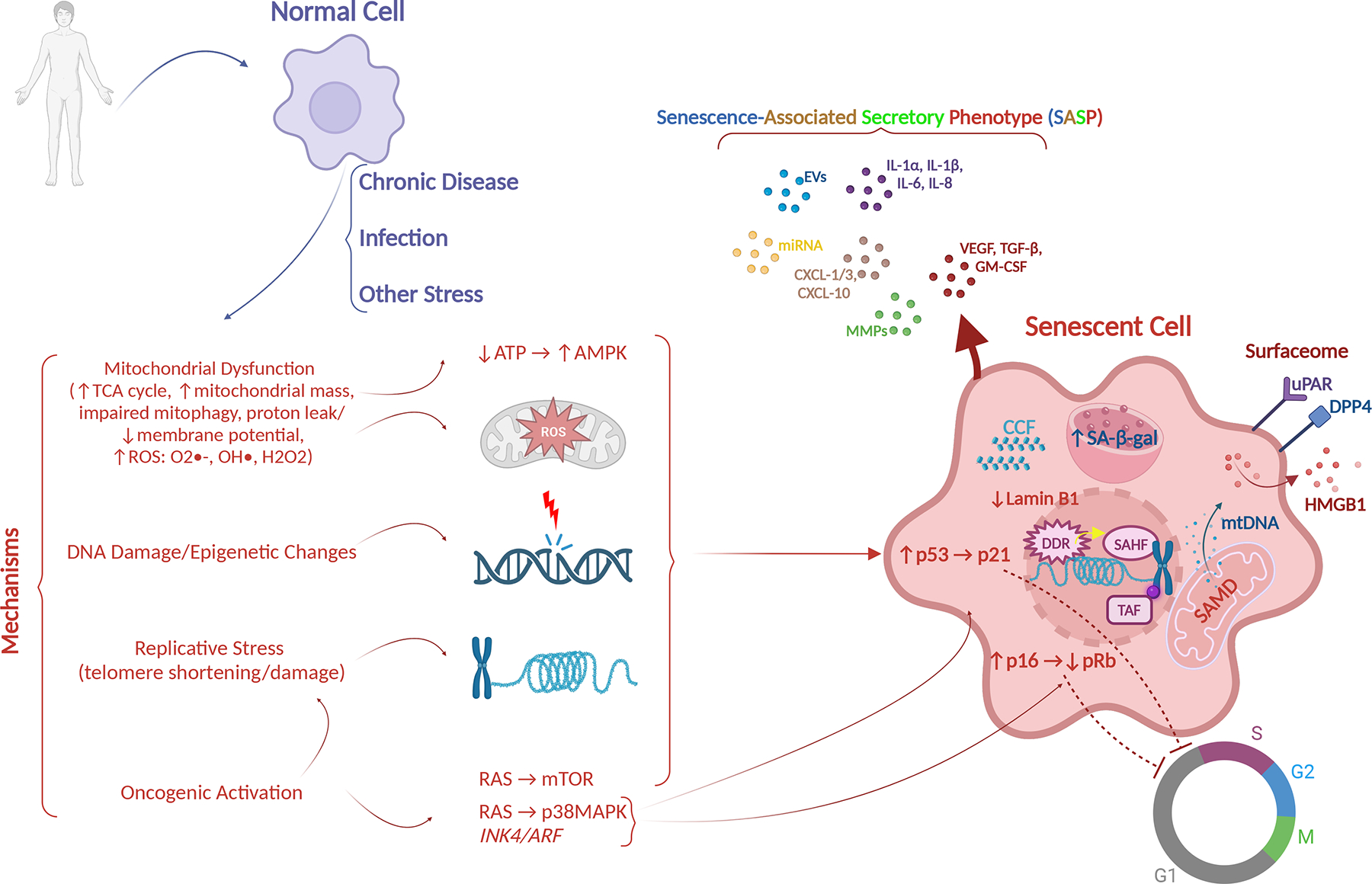
Chronic diseases, infectious agents, trauma, immune dysregulation, metabolic disorders, frailty, and psychosocial stress are associated with increased senescent cells. Senescence arises down stream of signaling events in response to a variety of intracellular stressors, including: (1) mitochondrial dysfunction associated with changes in mitochondrial metabolism, morphology, and increase in ROS; (2) DNA damage and epigenetic changes; (3) replicative stress with telomere shortening and damage; and (4) oncogene activation. These activate the DDR, which converges on p53 and/or p16 activation, which drive cell cycle arrest. Prolonged activation of the DDR induces cellular senescence, characterized by, in addition to the above, SAHF, TAF, SAMD with release of mt-DNA, CCF, metabolic changes including increased lysosomal SA-β-gal activity, expression of new proteins on the cell surface (surfaceome) and a SASP that can spread senescence in a paracrine and endocrine fashion to neighboring cells and organs respectively. AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate; CCF, cytoplasmic chromatin fragments; DDR, DNA damage response; EV, extracellular vescicles; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; MAPK, mitogen-activated protein kinase; miRNA, micro RNA; MMP, matrix metalloproteinase; mtDNA, mitochondrial DNA; mTOR, mammalian target of rapamycin; ROS, reactive oxygen species; SA-β-gal, senescence-associated β-galactosidase; SAHF, senescence-associated heterochromatin foci; SAMD, senescence-associated mitochondrial dysfunction; SASP, senescence-associated secretory phenotype; TAF, telomere-associated foci; TCA, tricarboxylic acid; TGF-β, tumor growth factor beta; VEGF, vascular endothelial growth factor.
A growing body of research demonstrates that the features of senescent cells vary depending on their cell lineage and the type of stress that instigated CS.35 Furthermore, the pattern of senescent cells accumulation varies between organs.16,36,37 Common features of CS include distinct transcriptional changes, upregulation of cell-cycle arrest regulators (p53, p16INK4a and p21CIP1), telomere-associate DNA damage foci, senescence-associated distension of satellites, compromised mitochondrial function, and upregulation of lysosomal senescence-associated β-galactosidase (SA-β-gal) activity.33,38–40 Senescent cells from one organ may affect the entire organism through SASP, which often includes IL-6, IL-8, TNF-α, CCL2, CCL20, and matrix metalloproteases.41 Senescent immune cells appear to be particularly deleterious, perhaps because of their inherent secretory capacity plus the fact that senescent immune cells contribute to immunosenescence, defined as phenotypic changes of immune cell subtypes, increased memory T cells, diminished ability to mount antigen-specific responses, and persistent low-grade inflammation coined as “inflamm-aging”.42–45 With aging and in chronic pathological conditions, there is an intimate interrelationship between senescence of immune and nonimmune cells that results in a positive feedback loop, leading to progressive accumulation of senescent cells that propagates inflammation and immune dysregulation, thereby driving further CS and organ damage.46
In order to appreciate the complexity of CS, it is crucial to understand the role of senescent immune and nonimmune components as they contribute to organ damage in chronic disease and transplantation (Figure 2). Herein, we attempt to define current knowledge about the impact of CS on circulating immune cells in the context of organ damage and disease progression, discuss the impact of CS on abdominal solid organs that are commonly utilized for transplantation (liver, kidneys), and review emerging therapies that target CS with the aim of optimizing and delaying physiological and pathological decline in organ function.
Figure 2. Impact of senescent cells on the organism and target therapies.
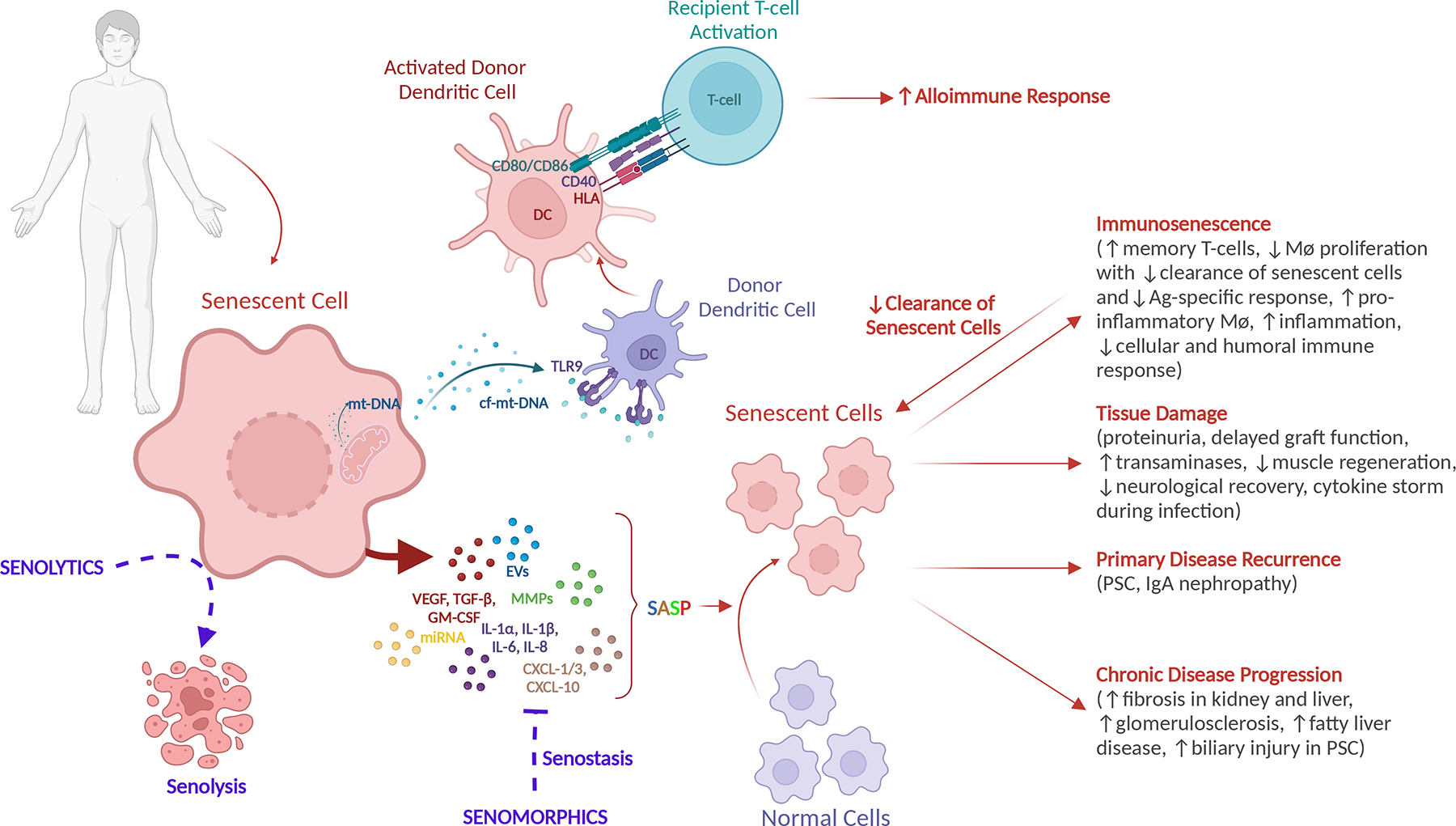
SASP from a variety of senescent cell types can drive healthy, normal cells to senescence resulting in: (1) immunosenescence with accumulation of T-memory cells, decreased in cellular and humoral response, decreased clearance of senescent cells by senescent immune cells, increased inflammation; (2) organ specific damage with renal, hepatic, and endocrine dysfunction, poor neurologic recovery, impaired muscle regeneration, allograft dysfunction; (3) primary disease recurrence (eg, PSC and IgA nephropathy); and (4) chronic disease progression (eg, fibrosis, biliary injury, fat accumulation). In transplantation, ischemia-reperfusion injury and the accumulation of cf-mt-DNA in an older donor causes activation of donor DCs that lead to direct antigen presentation following graft reperfusion and activation of recipient T-cells promoting an alloimmune response and rejection of the transplanted organ. Senotherapies target cellular senescence via senolytics (drugs that target the destruction of senescent cells) and senomorphics (drugs that inhibit the SASP and promote senostasis). cf-mt-DNA, cell-free mitochondrial DNA; DC, dendritic cell; EV, extracellular vescicles; GM-CSF, granulocyte-macrophage colony-stimulating factor; IgA, immunoglobulin A; IL, interleukin; miRNA, micro RNA; MMP, matrix metalloproteinase; mt-DNA, mitochondrial DNA; PSC, primary sclerosing cholangitis; SASP, senescence-associated secretory phenotype; TGF-β, tumor growth factor beta; TLR9, toll-like receptor 9; VEGF, vascular endothelial growth factor.
IMMUNE CELLS
Circulating senescent immune cells contribute to disease progression and organ damage by two distinct mechanisms. First, senescent immune cells drive senescence and damage in solid organs.44 Second, senescent immune cells have altered function, reducing their ability to clear senescent cells.44,47 Healthy immune cells can exacerbate this scenario further. The SASP from senescent cells activates healthy immune cell subpopulations, initiating and propagating an immune response, which can lead to subsequent organ damage.48,49 In the setting of transplantation, the inciting events that contribute to increased systemic CS are multifactorial, including the age of donors and recipients, the chronic disease state of recipients, ischemia reperfusion injury (IRI) at the time of organ implant, alloimmune response to the donor organ, and primary disease recurrence following transplantation.28,47,48,50 This is exacerbated substantially further in allogeneic bone marrow transplantation by pretransplant immunoablative therapy.51,52
I. Preclinical data on senescent immune cells
In mice, deletion of a crucial DNA repair gene, Ercc1, specifically in the hematopoietic system was used to drive primary CS of immune cells.44 This was accomplished using the Vav-iCre promoter to drive Cre recombinase expression throughout the immune compartment. CS arose as a consequence of spontaneous endogenous DNA damage that was no longer repaired in the absence of ERCC1, a subunit of a DNA repair endonuclease. Adult Vav-iCre+/−;Ercc1-/fl mice demonstrate attrition as well as senescence of immune cell subpopulations. Markers of CS (p16Ink4a and p21Cip1 mRNA) and SASP are significantly upregulated in B cellsB220+CD19+, T cellsCD3+, NK cellsCD3-NK1.1+, and macrophagesCD11b+F4/80+.44 In addition, the mice show evidence of premature immunosenescence, including T-cell exhaustion, an increase in memory T-cells (CD44+), upregulation of PD-1 receptor on CD4+ cells, and, as a consequence, impaired cellular and humoral immune responses similar to that of much older wild-type animals.44 Adult Vav-iCre+/−;Ercc1-/fl animals secondarily show an increase in p16Ink4a and p21Cip1 expression, and SA-β-gal activity in parenchymal cells of solid organs, including renal tubule epithelium and hepatocytes. This secondary CS leads to organ damage, e.g., proteinuria, elevated serum transaminases and amylase, and impaired muscle regeneration. Adoptive transfer of splenocytes from the Vav-iCre+/−; Ercc1-/fl mice into young adult recipient animals drives CS in the recipients, confirming the role of senescent immune cells in causing secondary CS.44 In contrast, transplantation of young splenocytes into an aged recipients reduces expression of CS biomarkers in a variety of organs.44 This confirms that senescent immune cells in an older organism lose the capacity to clear senescent cells. Notably, transplantation of splenocytes from old wild-type mice into young recipients is an even more potent driver of CS in trans than immune cells from the transgenic mice.44 These preclinical studies illustrate how aged donor and recipient immune systems could contribute to adverse outcomes in transplantation.
In other preclinical studies instigated by the SARS-CoV2 pandemic, it became clear that senescent cells also modulate the response to pathogen-associated molecular patterns (PAMPs) and infection. When challenged with the classical bacterial PAMP, LPS, senescent cells or old mice harboring senescent cells hyper-respond, secreting elevated levels of pro-inflammatory cytokines and chemokines such as IL1α, IL1β, IL6, IL10, and CCL2, relative to nonsenescent cells or young mice, respectively.53 The same occurs upon challenge with a viral coat protein or upon viral infection of naïve, aged mice, including in the liver, lung, and kidney. This contributes to cytokine storm, multi-organ failure, and mortality in old mice.53 Pharmacologic or genetic clearance of senescent cells in old mice improves mortality by ~50%.53,54 Conversely, SARS-CoV-2 infection drives senescence.55 These studies illustrate how CS in aged recipients or donated organs might negatively impact responses to environmental pathogens. What remains unclear is whether immunosuppression would exacerbate or attenuate this hyperinflammatory, tissue damaging state or potentially accelerate CS.
II. Human studies on senescent immune cells
Similar to the observations in murine aging, human humoral and adaptive immune responses evolve with aging and chronic disease. In the SARS-CoV-2 study cited above, COVID-19 patients show increased CS in their airway mucosa, and increased serum SASP. That SASP drives macrophage and platelet activation, complement lysis, senescence of endothelial cells, and neutrophil extracellular trap formation.55 An aged microenvironment in the lungs drives transcriptional changes in alveolar macrophages, which prevents their proliferation and a robust response to viral infections.56 In chronic kidney disease (CKD), urea levels are correlated with impaired endocytosis and the maturation of monocytes and dendritic cells, resulting in decreased antigen-presentation and maturation of antigen-specific T cells.57,58 Patients on long-term hemodialysis exhibit evidence of premature aging as reflected by a higher percentage of senescent mononuclear cells with significant telomere attrition, a CD14high/CD16low phenotype, a pro-inflammatory cytokine overproduction and upregulation of p53.59 In turn, the pro-inflammatory cytokines of SASP promote chronic inflammation and induce CS via paracrine and endocrine mechanisms, promoting tissue damage and potentially tumorigenesis.60,61 Interestingly, peripheral blood cells from renal transplant recipients demonstrate significantly greater telomere attrition 12 months following transplantation relative to patients on hemodialysis for 12 months or healthy controls.62 The shortening of telomeres is especially pronounced in patients treated with mycophenolate mofetil. In another study, older kidney transplant recipients had decreased frequency of naïve CD4+ and CD8+ T cells, an increased frequency of terminally differentiated senescent NK cells, and increased T cells expressing the immunosenescence marker (KLRG1), which was shown to correlate with increased posttransplant infections and impaired immunity to cancer.63,64 Although, the etiologies of CS differ in the above studies, the accumulation of senescent immune cells in pretransplant and transplant patients contribute to premature aging of organ recipients and the development of age-related diseases. This is a critical area for future research as it is well-documented that the main causes of death among solid organ transplant recipients are infections and age-related chronic conditions such as malignancies and cardiovascular disease.65,66
III. Immune system response to CS signals
There is also substantial data on the role of nonsenescent immune cells responding to senescent cells, leading to propagation of CS and loss of tissue homeostasis. In a murine model of cardiac allotransplantation, senescent cells in old but not young donors are a critical driver of organ immunogenicity, activation of dendritic cells in recipient animals, and immunologic graft loss.48 Clearance of senescent cells from aging hearts prior to transplant dampened age-specific immune response and prolonged survival of allografts.48 The authors identified cell-free mitochondrial DNA (cf-mt-DNA) as a potent driver of CS in this model. They also demonstrated that cf-mt-DNA is increased in the serum of older human deceased donors compared to young and activates human dendritic cells ex vivo.48
In diabetic kidney disease, M1 pro-inflammatory macrophages accumulate in the glomeruli and induce CS in glomerular endothelia cells by increasing the abundance of reactive oxygen species in humans and a preclinical disease model.67 Here, macrophage plasticity should be considered. In the acute injury phase, M2 alternative macrophages are activated by the Th2 type cytokines IL-4 and IL-13, or by M2 phagocytosis of apoptotic cells. This contributes to healing, suppression of inflammation, reduction of immune infiltration, and reduced fibrosis.68 However, senescent cells that are associated with chronic inflammation or aging can inhibit the transition from M1 to M2 via their SASP, thereby propagating tissue injury. For example, following IRI, the intrarenal microenvironment of aged mice inhibits the M1 to M2 transition, leading to the accumulation of M1 and fibrosis.69
In acute liver injury, the SASP factor, CCL2, from senescent murine hepatocytes activates tissue-resident macrophages leading to TGFβ-mediated senescence transfer to nonsenescent hepatocytes and tissue damage.70 Inhibition of this pathway reduces secondary senescence and enhances liver regeneration. Notably, CCL2 was identified as the SASP factor that most consistently reports biological (rather than chronological) age in mice and is increased in frail humans.71 On the other hand, SASP potentiates CD4+T-cell-mediated clearance of senescent premalignant hepatocytes with assistance of tissue-resident monocytes/macrophages.72
Although while young and healthy, CS maintains body homeostasis by promoting regeneration and cancer surveillance, aging and chronic conditions such as obesity, metabolic disorders, cardiovascular disease, CKD and transplantation, promote accelerated accumulation of senescent cells. Senescent cells, whether immune or otherwise, drive chronic sterile inflammation and immunosenescence, which contributes to failure to clear senescent cells. This, in turn, promotes tissue damage and further immune dysfunction. Better understanding of precisely how CS perturbs immune function and promotes inflammation would be valuable for designing novel therapies to improve transplant outcomes.
LIVER
The complex network of hepatic functions includes metabolism, detoxification, coagulation, and immune response. This requires multiple cell types that interact in a spatiotemporal manner to ensure normal function of the organ and the homeostasis of the entire organism.73 Recent single-cell RNA-sequencing efforts provided a detailed human liver atlas that describes over a dozen major types of liver cells.74 Of those, five subtypes, hepatocytes, hepatic stellate cells (HSCs), Kupffer cells, liver sinusoidal endothelial cells (LSECs) and cholangiocytes comprise the majority of hepatic parenchyma with hepatocytes contributing to 70% and HSCs, Kupffer cells, LSECs, cholangiocytes contributing to approximately 30% of the liver.73,75
I. Senescent hepatocytes
To date, the majority of liver aging biology research has focused on hepatocytes.37 CS can be driven by telomere attrition due to replication and cell division or by other types of stress including infection, toxins, oxidative stress, genotoxic entities, ischemia reperfusion, or oncogenic stress.76 Hepatocytes and cholangiocytes do not undergo replicative senescence in humans despite the impressive regenerative capacity of the liver.77,78 This is in part due to a subset of hepatocytes that express telomerase, enabling telomere lengthening and extended replicative capacity.79 In contrast, with healthy aging, the number of polyploid hepatocytes accumulate with time, which limits their replicative capacity, thus defining those cells as senescent.80
In the setting of pathologic conditions, CS provides protection by blocking proliferation of damaged cells early in the disease. However, as disease progresses, there is an accumulation of senescent hepatocytes characterized by increased expression of cell cycle inhibitors p53/p21CIP1 and p16INK4a, larger nuclear area, γH2AX (a marker of DNA damage) and SASP factors that promote the conversion of nonsenescent cells to senescent phenotype in a paracrine manner.28,41,81 In contrast to healthy liver, CS, loss of telomerase expression and loss of regenerative capacity are characteristics of progression from fibrotic disease to cirrhosis.82 Gain of function mutations at the promoter of TERT reintroduces telomerase expression and enables the development of hepatocellular carcinoma in cirrhosis.82
II. Senescent cholangiocytes
As with senescent hepatocytes in pathological conditions, senescent cholangiocytes are characterized by p21WAF1 and p16INK4a overexpression, telomere-shortening, secretion of SASP factors as well as increased SA-β-gal activity. Senescent cholangiocytes are increased in the biliary systems of patients with primary sclerosing cholangitis (PSC) and primary biliary cholangitis.83 Senolysis reduces fibrosis in a murine model of PSC. The SASP of senescent cholangiocytes spreads senescence to bystander cholangiocytes and activates the innate and adaptive immune responses that subsequently leads to the autoimmune response and destruction of the biliary system.84,85 Further, as senescence spreads in the injured ducts, it impairs the removal and replacement of senescent cholangiocytes exacerbating inflammation and loss of bile ducts.85 As in other pathologies, senescent cholangiocytes, characterized by overexpression of p21WAF1/CIP1, are increased in the setting of rejection following liver transplantation and decreased in response to treatment of rejection.86
III. Senescence of other liver cell types
In contrast to hepatocytes and cholangiocytes, HSCs and Kupffer cells demonstrate evidence of replicative senescence as their telomeres undergo shortening in vivo and in vitro.21,78,87 However, the key role of senescence in relation to HSCs involves the regulation of extracellular matrix (ECM) deposition. During liver injury, HSCs acquire a myofibroblastic phenotype and increased secretion of ECM that subsequently leads to fibrosis.88 The induction of senescence in activated HSCs limits their fibrogenic response in the setting of tissue damage by decreasing ECM production and promoting clearance of activated/senescent HSCs by immune cells.89 In the absence of injury, HSCs undergo a milder form of activation characterized by proliferation stimulated by the HSC-related growth factor (PDGFRβ), accumulation of intracellular lipid droplets, an increase in expression of alpha smooth muscle actin (αSMA), collagen 1α1, collagen 1α2, and p-moesin as well as alteration in expression of matrix metalloproteinases and their inhibitors that affect deposition and content of the ECM.90 Interestingly, HSCs from aged livers become hyperresponsive in the setting of injury with overproduction of ECM components leading to architectural distortion, elevated hepatic vascular tone, and in turn exacerbation of liver injury and fibrosis, which may reflect a role of CS.91 These findings are relevant not only for aging patients with primary liver disease but also in the setting of liver transplantation of organs from older donors, as well as the recovery of older live donors following partial liver donation.
LSECs provide a critical contact point between portal circulation and the liver serving the function of bidirectional transfer of metabolites, endocytosis, immunotolerance, and maintenance of hepatic microenvironment.92 Senescent LSECs have elevated p16INK4a expression, evidence of mitochondrial stress, and increased pro-inflammatory markers. This phenotype becomes more prevalent in aging and diseased liver leading to defenestration and reduction of porosity in the sinusoidal endothelium, thus preventing exchange of plasma and plasma proteins.93,94 Decreased sinusoidal permeability triggers a compensatory upregulation of scavenger receptors on the LSECs and promotes uptake of toxic substances, which in turn further promotes senescent phenotypes.94 Ultimately, this leads to increased oxidative stress in liver sinusoids and extrahepatic vasculature as well as LSEC thickening and fibrosis.95,96
Although, Kupffer cells represent the largest population of tissue-resident macrophages in the body, the impact of senescence on these cells is understudied.97 Based on the study of macrophages in other tissues, it is known that aging leads to a reduction in autophagy and phagocytosis, secretion of pro-inflammatory mediators, and alteration of cell morphology.98 As discussed in a prior section, Kupffer cells also act in the role of responders to the aging microenvironment and other senescent cell subpopulations. CS of nonimmune cells contributes to this pro-inflammatory microenvironment, which induces Kupffer cell polarization promoting an M1 proinflammatory phenotype implicated in a number of pathologic liver conditions including fibrosis.98,99
KIDNEY
The complex network of renal functions includes electrolyte, fluid, and acid-base balance, filtration of toxins and waste, hormone production, and regulation of hemodynamics. These diverse functions necessitate a large number of highly specialized cells that include over 16 epithelial subtypes in addition to a significant number of endothelial, immune, and interstitial cell types.100 In aging and pathophysiologic conditions, senescence impacts diverse renal cell subpopulations and compartments with the accumulation of senescent cells as a common feature of the aging kidney and CKD.101–106 This is not surprising as both aging and CKD have similar clinical and pathological presentation. From a clinical standpoint, both manifest with proteinuria, reduced glomerular filtration rate, disruption of electrolyte homeostasis, and fluid retention. Pathohistological findings of the aging kidney and CKD include podocyte loss/dysfunction, glomerular basement membrane thickening, glomerulosclerosis, decline in nephron number/size, tubular atrophy, fibrosis, inflammation, and loss of vascular compartment.107 Eventually both processes render renal vulnerability to toxic or hemodynamic stress and limited regenerative capacity.
I. Senescent renal tubule epithelial cells
The tubular epithelium is the major source of senescent cells in CKD.108 Chronic oxidative, toxic, and metabolic stress induce CS in proximal tubular epithelium. Prolonged injury leads to persistent SASP, inflammation, and the secretion of profibrotic factors.107 For example, indoxyl sulfate, a byproduct of metabolism, induces CS in proximal tubules by upregulating reactive oxygen species and the p53 pathway, suppressing cell proliferation and increasing TGF-β1 and α-SMA, which eventually leads to fibrosis.109 Aging- and uremia-associated oxidative stress leads to cytotoxic DNA damage.110,111 In acute kidney injury, CS of tubular epithelial cells is induced via components of the innate immune system, the Toll-like and interleukin 1 receptors, and leads to persistent inflammation, damage to renal tubules, and fibrosis.112 This is relevant in many aspects of transplantation including delayed graft function, rejection, and infection.
Although proximal tubular epithelium is thought to be the main site of senescent cells, different stressors target diverse cell populations, which impacts the localization and type of CS in the kidney. For example, p16INK4a is significantly upregulated in glomerular and interstitial cell nuclei in patients with glomerular disease compared to normal controls or patients with tubulointerstitial nephritis. The expression of p16INK4a in tubular cytoplasm is higher in kidneys from patients with proteinuria, fibrosis, and atrophy.101 Of note in glomerular disease, podocyte-derived TGF-β1 induces nuclear translocation of p16INK4a in glomerular endothelial cells, leading to endothelial senescence and disease progression.113 Hypertension also contributes to renal CS. In several preclinical models of hypertension, including aldosterone and angiotensin II induced disease, upregulation of SA-β-gal activity and p21Cip1 expression in proximal tubular cells is observed, which contributes to reduced regenerative capacity of tubular endothelium.114,115 Further, in a setting of hypertension, telomere shortening is observed in vascular endothelium.116
II. Senescent glomerular endothelial cells
Other contributors to senescence-associated renal dysfunction are senescent glomerular endothelial cells. Senescent endothelial cells modulate podocyte function through the SASP factor, plasminogen activator inhibitor 1 (PAI-1).117 Inhibition of PAI-1 prevents glomerulosclerosis and podocyte loss in aging animals. In human renal transplantation from elderly donors, PAI-1 expression in senescent endothelium is predictive of poor outcomes.117
Loss of Klotho, an anti-aging factor highly expressed in the kidney, is implicated as a driver of medial vascular calcifications and progression to CKD.118 Klotho-deficient mice demonstrate a progeroid phenotype and tissue-specific age-associated defects.119 Similarly, patients with CKD lack Klotho, further implicating its critical role in renal homeostasis.118 Although, the exact mechanisms are not known, uremic toxins are thought to induce Klotho gene hypermethylation and downregulation of its expression.120 Several studies suggest that Klotho acts systemically to prevent aging phenotypes by decreasing oxidative stress through upregulation of superoxide dismutase, inhibition of endothelial senescence, antifibrotic properties, suppression of the insulin-IGF-1 signaling pathway that in turn is associated with life extension, as well as by systemic regulation of mineral metabolism.111,121 The therapeutic potential of Klotho should be further explored as its systemic delivery has been shown to ameliorate the aging phenotype in klotho mutant mice.122
III. Renal CS and transplantation
In transplantation, there is evidence that CS is correlated with the biological age of the organ and posttransplant function. In the study of human renal allografts, serum creatinine at 6-months posttransplant and subsequent graft dysfunction strongly correlate with donor age and the senescence marker, p16INK4a, on pretransplant graft biopsy.123 Similarly, p16INK4a levels, in combination with chronological donor age, was the best predictor of renal allograft function at 1 year following transplantation. In another study, p16INK4a and p27KIP1 were upregulated in glomeruli, renal tubules, and interstitial cells of kidney allografts with evidence of chronic rejection and in kidneys from older donors when compared to healthy or younger individuals.102
At the time of implant, CS is increased as a result of IRI causing oxidative stress and overexpression of p16INK4a and p21CIP1.124 However, the temporary induction of cell cycle arrest via the p21CIP1 pathway may be advantageous by allowing time for DNA damage repair and has been shown to be beneficial in the setting of IRI, whereas prolonged cell cycle arrest via p16INK4a and senescence contributes to inferior transplant outcomes in animal models.124 In patients with immunoglobulin A nephropathy, markers of CS (increased SA-β-gal activity, p16INK4a and p21CIP1 expression) are significantly elevated in tubular epithelial cells compared to healthy controls, and the increase in these markers correlates with clinical and pathological disease progression.125 These observations are important for management of primary disease and primary disease recurrence following kidney transplantation.
EMERGING THERAPIES
Given the central role of CS in aging and pathogenesis, it is not surprising that CS emerged as a promising new target for therapies, with the coining of a new term “senotherapy”. Senolytics selectively deplete senescent cells, whereas senomorphics suppress markers of senescence or their SASP.126 Senolytics offer some therapeutic advantage as they clear senescent cells, eliminating SASP and require intermittent rather than chronic administration.127 In contrast, senomorphics may prove more advantageous as they do not eliminate acute CS required for wound healing.128
Over a decade ago, the first proof-of-concept animal study demonstrated causal relationship between SC and aging.129 Life-long removal of p16Ink4a-positive senescent cells in the BubR1 progeroid mouse delayed onset of age-related pathologies in these animals and late-life clearance delayed the progression of already acquired age-related disorders.129 This study paved the way for discovery of therapies to target pro-aging and pro-pathologic effects of CS. However, the development of new therapies is hampered by the need to preserve beneficial effects of SC such as tumor suppression and tissue regeneration, avoid potential drug-related toxicities e.g., hyperglycemia, thrombocytopenia, immunosuppression, and the need for tissue-specific targets due to differential tissue vulnerabilities and senescence phenotypes.127,130,131
Transplant candidates and recipients exhibit evidence of premature aging and increased rate of age-related co-morbidities by virtue of suffering from chronic end-stage organ disease and subsequently being exposed to life-long immunosuppression, leading to increased incidence of metabolic and cardiovascular disease, renal dysfunction, infections and cancer.63,132–139 As discussed earlier, the accumulating evidence suggest that CS plays a causal role in the development and progression of these conditions. Therefore, a number of senotherapeutic agents have been tested in the preclinical setting and are slowly introduced into clinical trials to combat age-related diseases (Table 1).
Table 1.
Senotherapy clinical trials with focus on aging, frailty, and age-related conditions.
| Senolytics | |||
|---|---|---|---|
| Drug | Indication | Phase | Trial registration number |
|
Fisetin PI3K/Akt pathway inhibitor |
Frailty | 2 | NCT03675724, NCT03430037, NCT04733534, |
| Healthy participants | 2 | NCT04313634, NCT04313634 | |
|
Luteolin ROS, PGE2, COX2 inhibitor |
Memory in healthy aging | NCT04468854 | |
|
Quercetin PI3K/Akt pathway inhibitor |
Age-related macular degeneration | 2 | NCT05062486 |
|
Dasatinib and quercetin Tyrosine kinase inhibitor and PI3K/Akt pathway |
Alzheimer’s | 1 and 2 | NCT04063124, NCT04785300 |
| 2 | NCT04685590 | ||
| Aging | 1 and 2 | NCT05422885 | |
| 2 | NCT04946383, NCT05838560, NCT04313634 | ||
| Diabetic nephropathy | 2 | NCT02848131 | |
| Frailty | 2 | NCT04733534 | |
| Healthy participants | 2 | NCT04313634 | |
| Liver disease | 1 and 2 | NCT05506488 | |
| Idiopathic pulmonary fibrosis | 1 | NCT02874989 | |
|
UBX1325 Bcl family |
Age-related macular degeneration/diabetic macular edema | 1 | NCT04537884 |
| 2 | NCT05275205, NCT04857996 | ||
|
UBX0101 P53/MDM2 interaction inhibitor |
Osteoarthritis | NA | NCT04349956 |
| 1 | NCT03513016 | ||
| 2 | NCT04229225, NCT04129944 | ||
|
Navitoclax Bcl family |
Liver disease | 1 | NCT02143401 |
|
Catechins Bax/Bcl-2, Nrf2, and PI3K/Akt/mTOR pathways |
Liver disease | 1 | NCT03278925 |
| SASP inhibitor | |||
|
MitoQ Antioxidant, inducer of superoxide dismutase and glutathione peroxidase |
Multiple sclerosis | 1 and 2 | NCT04267926 |
| Aging | 1 | NCT05686967 | |
| Ganoderma lucidum Nrf2, mTOR, and MAPK pathways | Cardiometabolic syndrome | 2 | NCT05079529 |
| Parkinson disease | 3 | NCT03594656 | |
|
Equol Nrf2/ARE pathway |
Cognition in healthy volunteers | 2 | NCT05741060 |
|
Melatonin Keap1/Nrf2/ARE pathway and SIRT1 |
Healthy | NA | NCT05419466 |
| 4 | NCT04831398 | ||
| Aging | NA | NCT03954899 | |
| Stroke | NA | NCT03843008 | |
| 3 | NCT05857046 | ||
| 4 | NCT05247125 | ||
| Multiple sclerosis | 1 | NCT03498131 | |
| 1 and 2 | NCT03540485 | ||
| Encephalopathy | 1 | NCT02621944 | |
| Wound healing | 2 | NCT05122130 | |
| Coronary artery disease | NA | NCT05552586 | |
| Obesity | NA | NCT05759429 | |
| Cardiovascular disease | NA | NCT04631341 | |
| 2 | NCT05257291 | ||
| Renal disease | 3 | NCT05084196, NCT05570526 | |
| Osteoarthritis | NA | NCT04795336 | |
| Osteopenia, osteoporosis | 1 | NCT04233112 | |
| 4 | NCT04864509 | ||
|
Alpha-ketoglutarate Intermediate of tricarboxylic acid (TCA) cycle, imitates caloric restriction |
Healthy aging | 2 | NCT05706389 |
|
Rapamycin mTOR signaling pathway |
Ovarian aging | 2 | NCT05836025 |
| Tuberous sclerosis | 3 | NCT05534672 | |
| Muscle aging | NA | NCT05414292 | |
| Otolaryngologic disease | 2 | NCT05342519 | |
| Amyotrophic lateral sclerosis | 2 | NCT03359538 | |
| Aging, longevity | 1 | NCT04994561, NCT04742777, NCT04608448 | |
| 2 | NCT04488601, NCT02874924 | ||
| Age-related macular degeneration | 2 | NCT00766337 | |
| Alzheimer’s disease | 2 | NCT04629495 | |
| Insulin sensitivity | NA | NCT05233722 | |
| Cardiac function in aging frail adults | 1 | NCT04996719, NCT01649960 | |
| Sarcopenia | 1 | NCT00891696 | |
| Pompe disease | 1 | NCT02240407 | |
|
Sirolimus mTOR signaling pathway |
Aging | 2 | NCT05237687 |
|
Metformin AMPK and NF-κB pathways |
Aging, longevity | 1 | NCT04994561, NCT02308228, NCT03996538, NCT03713801, NCT03107884, NCT04994561, NCT03072485 |
| 3 | NCT04264897, NCT03309007, NCT04221750, NCT03861767 | ||
| 4 | NCT02432287, NCT02745886 | ||
| Age-related macular degeneration | 2 | NCT02684578 | |
| Frailty | 2 | NCT03451006 | |
| Cognitive impairment | 2 | NCT00620191 | |
| 4 | NCT02409238 | ||
| Dementia | 2 | NCT01965756 | |
| Neuromuscular disease | 3 | NCT05532813, NCT01995032 | |
| Visual impairment | 2 | NCT03490019 | |
| Depression | 4 | NCT00834652 | |
| Healthy | 1 | NCT03772964 | |
|
Resveratrol SIRT1/NF-κB pathway |
Friedreich ataxia | 2 | NCT03933163 |
| Peripheral artery disease | 3 | NCT03743636 | |
| Amyotrophic lateral sclerosis | 2 | NCT04654689 | |
| Age-related macular degeneration | 2 | NCT05062486 | |
|
Curcumin
AMPK, sirtuin, PI3K/Akt, NF-κB, and Nrf2 pathways |
Healthy | NA | NCT05414838, NCT05775237 |
| 1 | NCT05334043 | ||
| Alzheimer’s disease | 1 and 2 | NCT05774704 | |
| Amyotrophic lateral sclerosis | 2 | NCT04654689 | |
| Kidney disease | NA | NCT04413266 | |
| 2 | NCT03223883, NCT05714670 | ||
| 3 | NCT05613361, NCT05627843 | ||
| Chronic conditions in kidney transplant recipients | NA | NCT03935958 | |
| Diabetes mellitus | 2 | NCT05753436 | |
| 1 and 2 | NCT05407467 | ||
| Wound | NA | NCT05819632 | |
| 1 | NCT05610878 | ||
| Age-related macular degeneration | 2 | NCT05062486 | |
|
Spermidine Autophagy |
Hypertension | 3 | NCT04405388 |
AMPK, AMP-activated protein kinase; MAPK, mitogen-activated protein kinase; mTOR, mammalian target of rapamycin; NA, not applicable; NCT, National Clinical Trial; NF-κB, nuclear factor kappa B; SASP, senescence-associated secretory phenotype.
Applications of senotherapeutics in treatment of metabolic diseases have been widely studied in preclinical models. Administration of ruxolitinim (a Janus kinase 1 or 2 inhibitor) and senolytic cocktail of dasatinib (D) (tyrosine kinase inhibitor) and quercetin (Q) (kinase/antioxidant) to aged animals successfully ameliorates senescent cells, mitigates lipodystrophy, enhances insulin sensitivity and glucose tolerance, lowers circulating inflammatory mediators, prevents migration of monocytes, decreases the number of macrophages in visceral fat, and reduces frailty.140–142 Similarly, in human trials, senescent cells in subcutaneous fat are associated with altered glycemic control and are cleared within 11 days following the administration of the D+Q cocktail in patients with diabetic kidney disease, although the impact of treatment on renal function and long-term glycemic control is not known.143–145 Further, the administration of the senolytic cocktail improved cardiac and renal function of aged animals, and decreased hepatic steatosis in subsequent study, albeit, it failed to prevent progression of steatosis in animals with nonalcoholic liver disease-induced hepatocellular carcinoma.29,30,141,146 In murine models of cardiovascular disease, BCL2/Bcl-xL inhibitor reduces cardiac hypertrophy and fibrosis with compensatory regeneration in cardiomyocytes and rescues the functional decrease in ejection fraction following myocardial infarction, whereas D+Q reduces CS and plaque calcifications in a model of atherosclerosis.147–149
Frailty, loss of muscle mass and strength are common in transplant candidates and recipients, thus it is very intriguing and promising that a BCL2/Bcl-xL inhibitor reduces the loss of body weight, improves strength, and increases muscle regeneration in a rat model of Duchenne muscular dystrophy.150 Similarly D+Q improves muscle function in mice following the irradiation-induced senescence.34 The first-in-human, open-label clinical trial of D+Q in patients with idiopathic pulmonary fibrosis, a cellular senescence-driven disease, demonstrated improvement in physical function and scores in the Short Physical Performance Battery 5 days after 3-week course of D+Q therapy.151,152
Further, it has been demonstrated that frail transplant candidates have lower cognitive scores than nonfrail and have impaired cognitive recovery following transplantation.153 Although, clinical trials on the use of senolytics in neurodegenerative conditions are in progress (NCTO4063124), it is known that overexpression of senescence markers in variable regions of the brain are associated with neurodegenerative diseases in humans.154–156 Clearance of CS in healthy aged mice and neurodegenerative models with BCL2/Bcl-xL inhibitor or D+Q cocktail enhances brain function, reduces neuroinflammation and accumulation of pathologic markers and improves cognitive deficit in those animals.157–159
The impact of senolytics on the immune system is variable. Administration of BCL2/Bcl-xL inhibitor (senolytic and anti-cancer compound) triggers premature cell death of noncancerous cells infected with influenza A virus leading to dysregulation of pro-inflammatory and anti-viral cytokines, which contributes to an inability of the immune system to clear the virus with subsequent mortality of infected animals.160 In contrast, clearance of senescent cells with fisetin (PI3K/AKT pathway inhibitor) or D+Q in acutely infected aged mice with murine-β-coronavirus (related to SARS-CoV-2) reduces CS, inflammation, increases anti-viral response, and reduces animal mortality from 100% to 50%.53 As of now, it is unclear what will be the direct impact of senotherapeutics on transplant recipients, although, it will likely vary based on mechanism of action. For instance, senomorphic-like agent Rapamycin (mTOR inhibitor) extends lifespan in diverse species and is commonly used in transplant to prevent rejection.161–163 Due to its anti-tumorigenic properties, the drug is utilized in cancer patients as well as transplant patients with a history of malignancy. However, its deleterious impact on the immune system contributes to a high rate of infections and poor healing.164
To date, the majority of clinical studies focus on safety and tolerability of senotherapy in patients and only few report on efficacy.165,166 However, senotherapy provides an opportunity to rejuvenate organs from older donors that are currently underutilized and have a potential to eliminate the gap that exists between organ demand and supply.3 Although, the majority of deaths occur among older individuals, the reluctance of the transplant community to use organs from older donors reflects the concerns that aging is associated with decline in organ function and durability. Moreover, experimental and clinical models demonstrate the increased immunogenicity and graft loss of organs from older donors.48,167,168 Indeed, the transplantation of senescent cells into healthy mice drives frailty, which raises a concern of transferring senescence with organs from older donors during transplantation.142
APPLICATION OF EMERGING THERAPIES TO TRANSPLANTATION
Several experimental strategies using senotherapy have been deployed to optimize organs from older donors for transplantation in preclinical models. Pretreatment of older animals with D+Q cocktail reduces systemic levels of cf-mt-DNA and senescent cells in murine kidneys pre-IRI and decreases circulating levels of pro-inflammatory T-cells after IRI.48 Further, pretreatment of older donors prior to cardiac procurement prolongs cardiac allograft survival in mice particularly in the presence of clinically relevant immunosuppression protocol with the co-stimulatory CTLA4-Ig blockade that dampens T-cell allo-response.48 In both cases, senescent cells are key sources of cf-mt-DNA which is released in the setting of IRI promoting activation of dendritic cells with subsequent initiation of Th1/Th17 alloimmune response. Herein, clearance of senescent cells results in dampening of the immune response and reduces injury of the organ.48 In adult mice, p21Cip1, p16Ink4a, and p19Arf senescence markers are upregulated in multiple cell types when the regenerative capacity of the liver decreases.169 The pretreatment of adult animals with an BCL2/Bcl-xL inhibitor prior to partial hepatectomy reduces p21Cip1 expression, reduces levels of transaminases in the blood, decreases histological damage and increases liver regeneration by day 7 following partial hepatectomy.169 These findings are of clinical relevance for donors and recipients during living donor liver transplantation where the sizes of liver remnant and graft are critical to the recovery of both patients and in the setting of split-liver deceased donor transplantation.
Overall, there are a myriad of opportunities to exploit senotherapies in transplantation from stabilizing potential recipient while waiting for an organ, optimizing recipients following transplantation, improving the quality of organs by pretreatment of older donors with senotherapeutics prior to the donation, improving organ regeneration in older living donors and recipients, and optimizing marginal allografts (liver, kidney, heart, lung) on ex vivo perfusion devices with senotherapeutic treatment prior to transplantation thus prolonging ex vivo and in vivo lifespan of donated organs (Figure 3).31,170
Figure 3. Senotherapies in transplantation.

Clearance of senescent cells and inhibition of senescence-associated secretory phenotype may facilitate reduction in systemic and organ-specific inflammation thus improve metabolism and function of native and transplanted organs, decrease immunogenicity, reverse pathologies in ex vivo perfused organs (eg, steatosis or fibrosis), promote organ regeneration, decrease frailty-associated morbidity in transplant candidates and recipients, decrease primary disease recurrence in transplanted organs, decrease allograft rejection and extend the age of donors and recipients.
In summary, CS of immune and nonimmune compartments play a major role in chronic disease and aging. Manipulating CS to improve outcomes in transplant candidates and recipients by optimizing their overall medical condition and quality of transplanted organs is an emerging field of research. A number of clinical trials on applications of senotherapeutics for chronic conditions are ongoing (Table S1). However, specific use of these therapies in the setting of transplantation and rejuvenation of donor organs need to be explored. D+Q cocktail and BCL2/Bcl-xL inhibitors seem to be promising as therapeutic agents. However, the timing and length of administration, target measurements for success/endpoint of therapies as well as safety profile using senotherapeutics in patients on multi-drug regimens remain to be determined.
Supplementary Material
Acknowledgments
We would like to thank Ms. Terri B. Krause for her assistance in preparation of the manuscript.
Funding:
V.A.K. is supported by NIA K08 AG068374. L.J.N. is supported by NIH/NIA R01 AG063543, P01 AG062413, and U54 AG076041.
Abbreviations Page
- αSMA
alpha smooth muscle actin
- BCL2/Bcl-xL
B-cell lymphoma 2/B-cell lymphoma-extra large
- CCF
cytoplasmic chromatin fragments
- CCL2
chemokines C–C motif chemokine ligand 2
- CCL20
chemokines C–C motif chemokine ligand 20
- cf-mt-DNA
cell-free mitochondrial DNA
- CKD
chronic kidney disease
- CS
cellular senescence
- D
dasatinib
- DC
dendritic cells
- DDR
DNA damage response
- DNA
deoxyribonucleic acid
- ECM
extracellular matrix
- Ercc1
excision repair cross-complementation 1
- EV
extracellular vescicles
- HSC
hepatic stellate cells
- IRI
ischemia reperfusion injury
- IL-6
interleukin 6
- IL-8
interleukin 8
- KLRG1
killer cell lectin-like receptor G1
- LSECs
liver sinusoidal endothelial cells
- M1
pro-inflammatory macrophages
- M2
alternative macrophages
- MMPs
matrix metalloproteinases
- mt-DNA
mitochondrial DNA
- mRNA
messenger ribonucleic acid
- mTOR
mammalian target of rapamycin
- NK
natural killer
- PAI-1
plasminogen activator inhibitor 1
- PD-1
programmed cell death 1
- PSC
primary sclerosing cholangitis
- ROS
reactive oxygen species
- SA-β-gal
senescence-associated β-galactosidase
- SAHF
senescence-associated heterochromatin foci
- SAMD
senescence-associated mitochondrial dysfunction
- SASP
senescence-associated secretory phenotype
- TAF
telomere-associated foci
- TERT
telomerase reverse transcriptase
- TGFβ
tumor growth factor beta
- TNF-α
tumor necrosis factor alpha
- Q
quercetin
Footnotes
Disclosure: The authors declare no conflicts of interest.
References
- 1.World Health Organization. Ageing and health. Updated October 1, 2022. Available at https://www.who.int/news-room/fact-sheets/detail/ageing-and-health. Accessed May 15, 2023.
- 2.Durand F, Levitsky J, Cauchy F, et al. Age and liver transplantation. J Hepatol. 2019;70(4):745–758. [DOI] [PubMed] [Google Scholar]
- 3.Organ Procurement & Transplantation Network. National data. Available at https://optn.transplant.hrsa.gov/data/view-data-reports/national-data/#. Accessed May 15, 2023. [Google Scholar]
- 4.United States Census Bureau. Older people projected to outnumber children for first time in U.S. history. March 13, 2018. Available at https://www.census.gov/newsroom/press-releases/2018/cb18-41-population-projections.html. Accessed May 15, 2023.
- 5.Tian YE, Cropley V, Maier AB, et al. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat Med. 2023;29:1221–1231. [DOI] [PubMed] [Google Scholar]
- 6.United States Renal Data System. 2022. USRDS annual data report. National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases Available at https://usrds-adr.niddk.nih.gov/2022. Accessed May 15, 2023. [Google Scholar]
- 7.National Center for Health Statistics. Leading causes of death and number of deaths, by age: United States, 1980 and 2019. Centers for Disease Control and Prevention. Available at https://www.cdc.gov/nchs/hus/data-finder.htm?&subject=Chronic%20liver%20disease%20and%20cirrhosis. Accessed May 15, 2023. [Google Scholar]
- 8.Cuadrado-Payán E, Montagud-Marrahi E, Casals-Urquiza J, et al. Outcomes in older kidney recipients from older donors: a propensity score analysis. Front Nephrol. 2022;2:1034182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mehta J, Ndubueze O, Tatum D, et al. Kidney transplant outcomes in recipients over the age of 70. Cureus. 2023;15:e34021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwong AJ, Devuni D, Wang C, et al. ; Re-Evaluating Age Limits in Transplantation (REALT) Consortium. Outcomes of liver transplantation among older recipients with nonalcoholic steatohepatitis in a large multicenter US cohort: the Re-Evaluating Age Limits in Transplantation Consortium. Liver Transpl. 2020;26(11):1492–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Okumura K, Lee JS, Dhand A, et al. Trends and outcomes of liver transplantation among older recipients in the United States. World J Transplant. 2022;12(8):259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibulesky L, Leca N, Bakthavatsalam R, et al. Intention-to-treat analysis of patients aged 70 years and older awaiting kidney transplantation in post-kidney allocation system era. Transplantation. IN PRESS. doi: 10.1097/TP.0000000000004677 [DOI] [PubMed] [Google Scholar]
- 13.Colvin MM, Smith CA, Tullius SG, et al. Aging and the immune response to organ transplantation. J Clin Invest. 2017;127(7):2523–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sibulesky L, Leca N, Limaye AP, et al. Survival benefit in older patients transplanted with viremic hepatitis C positive kidneys when compared with high KDPI kidneys. Transplantation. 2022;106(11):2217–2223. [DOI] [PubMed] [Google Scholar]
- 15.Kennedy BK, Berger SL, Brunet A, et al. Geroscience: linking aging to chronic disease. Cell. 2014;159(4):709–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.López-Otín C, Blasco MA, Partridge L, et al. The hallmarks of aging. Cell. 2013;153(6):1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.López-Otín C, Blasco MA, Partridge L, et al. Hallmarks of aging: an expanding universe. Cell. 2023;186(2):243–278. [DOI] [PubMed] [Google Scholar]
- 18.Yang D, De Haan G. Inflammation and aging of hematopoietic stem cells in their niche. Cells. 2021;10(8):1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salminen A Clinical perspectives on the age-related increase of immunosuppressive activity. J Mol Med (Berl). 2022;100(5):697–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cai Y, Wang S, Qu J, et al. Rejuvenation of tissue stem cells by intrinsic and extrinsic factors. Stem Cells Transl Med. 2022;11(3):231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang L, Pitcher LE, Yousefzadeh MJ, et al. Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest. 2022;132(15):e158450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer. 2003;3(5):339–349. [DOI] [PubMed] [Google Scholar]
- 23.Dodig S, Čepelak I, Pavić I. Hallmarks of senescence and aging. Biochem Med (Zagreb). 2019;29(3):030501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Sanoff HK, Cho H, et al. Expression of p16(INK4a) in peripheral blood T-cells is a biomarker of human aging. Aging Cell. 2009;8(4):439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chaib S, Tchkonia T, Kirkland JL. Obesity, senescence, and senolytics. Handb Exp Pharmacol. 2021;274:165–180. [DOI] [PubMed] [Google Scholar]
- 26.Schafer MJ, Miller JD, LeBrasseur NK. Cellular senescence: implications for metabolic disease. Mol Cell Endocrinol. 2017;455:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salminen A Increased immunosuppression impairs tissue homeostasis with aging and age-related diseases. J Mol Med (Berl). 2021;99(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu P, Tang Q, Chen M, et al. Hepatocellular senescence: immunosurveillance and future senescence-induced therapy in hepatocellular carcinoma. Front Oncol. 2020;10:589908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogrodnik M, Miwa S, Tchkonia T, et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun. 2017;8(1):15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogrodnik M, Salmonowicz H, Gladyshev VN. Integrating cellular senescence with the concept of damage accumulation in aging: relevance for clearance of senescent cells. Aging Cell. 2019;18(1):e12841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matsunaga T, Iske J, Schroeter A, et al. The potential of Senolytics in transplantation. Mech Ageing Dev. 2021;200:111582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iske J, Matsunaga T, Zhou H, et al. Donor and recipient age-mismatches: the potential of transferring senescence. Front Immunol. 2021;12:671479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robbins PD, Jurk D, Khosla S, et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu Rev Pharmacol Toxicol. 2021;61(1):779–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Y, Tchkonia T, Pirtskhalava T, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14(4):644–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Consortium SenNet. NIH SenNet Consortium to map senescent cells throughout the human lifespan to understand physiological health. Nat Aging. 2022;2(12):1090–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang C, Jurk D, Maddick M, et al. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009;8(3):311–323. [DOI] [PubMed] [Google Scholar]
- 37.Hunt NJ, Kang SW, Lockwood GP, et al. Hallmarks of aging in the liver. Comput Struct Biotechnol J. 2019;17:1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021;288(1):56–80. [DOI] [PubMed] [Google Scholar]
- 39.Rossiello F, Jurk D, Passos JF, et al. Telomere dysfunction in ageing and age-related diseases. Nat Cell Biol. 2022;24(2):135–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Swanson EC, Rapkin LM, Bazett-Jones DP, et al. Unfolding the story of chromatin organization in senescent cells. Nucleus. 2015;6(4):254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huda N, Liu G, Hong H, et al. Hepatic senescence, the good and the bad. World J Gastroenterol. 2019;25(34):5069–5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee KA, Flores RR, Hwa Jang I. Immune senescence, immunosenescence and aging. Front Aging. 2022;3:900028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Budamagunta V, Foster TC, Zhou D. Cellular senescence in lymphoid organs and immunosenescence. Aging (Albany NY). 2021;13(15):19920–19941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yousefzadeh MJ, Flores RR, Zhu Y, et al. An aged immune system drives senescence and ageing of solid organs. Nature. 2021;594(7861):100–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iske J, Dedeilia A, Xiao Y, et al. The impact of T-cell aging on alloimmunity and inflammaging. Transplantation. IN PRESS. doi: 10.1097/TP.0000000000004715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schroth J, Thiemermann C, Henson SM. Senescence and the aging immune system as major drivers of chronic kidney disease. Front Cell Dev Biol. 2020;8:564461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yousefzadeh MJ, Zhao J, Bukata C, et al. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell. 2020;19(3):e13094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iske J, Seyda M, Heinbokel T, et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat Commun. 2020;11(1):4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narasimhan A, Flores RR, Camell CD, et al. Cellular senescence in obesity and associated complications: a new therapeutic target. Curr Diab Rep. 2022;22(11):537–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wissler Gerdes EO, Zhu Y, Weigand BM, et al. Cellular senescence in aging and age-related diseases: implications for neurodegenerative diseases. In: Söderbom G, Esterline R, Oscarsson J, et al, eds. International Review of Neurobiology. Academic Press; 2020:203–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim JH, Brown SL, Gordon MN. Radiation-induced senescence: therapeutic opportunities. Radiat Oncol. 2023;18(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Prasanna PG, Citrin DE, Hildesheim J, et al. Therapy-induced senescence: opportunities to improve anticancer therapy. J Natl Cancer Inst. 2021;113(10):1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Camell CD, Yousefzadeh MJ, Zhu Y, et al. Senolytics reduce coronavirus-related mortality in old mice. Science. 2021;373(6552):eabe4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pastor-Fernández A, Bertos AR, Sierra-Ramírez A, et al. Treatment with the senolytics dasatinib/quercetin reduces SARS-CoV-2-related mortality in mice. Aging Cell. 2023;22(3):e13771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee S, Yu Y, Trimpert J, et al. Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature. 2021;599(7884):283–289. [DOI] [PubMed] [Google Scholar]
- 56.McQuattie-Pimentel AC, Ren Z, Joshi N, et al. The lung microenvironment shapes a dysfunctional response of alveolar macrophages in aging. J Clin Invest. 2021;131(4):e140299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim WH, Kireta S, Leedham E, et al. Uremia impairs monocyte and monocyte-derived dendritic cell function in hemodialysis patients. Kidney Int. 2007;72(9):1138–1148. [DOI] [PubMed] [Google Scholar]
- 58.Verkade MA, van Druningen CJ, Vaessen LMB, et al. Functional impairment of monocyte-derived dendritic cells in patients with severe chronic kidney disease. Nephrol Dial Transplant. 2007;22(1):128–138. [DOI] [PubMed] [Google Scholar]
- 59.Ramírez R, Carracedo J, Soriano S, et al. Stress-induced premature senescence in mononuclear cells from patients on long-term hemodialysis. Am J Kidney Dis. 2005;45(2):353–359. [DOI] [PubMed] [Google Scholar]
- 60.Ortiz-Montero P, Londoño-Vallejo A, Vernot JP. Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal. 2017;15(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rea IM, Gibson DS, McGilligan V, et al. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9:586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Luttropp K, Nordfors L, McGuinness D, et al. Increased telomere attrition after renal transplantation-impact of antimetabolite therapy. Transplant Direct. 2016;2(12):e116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaenman JM, Rossetti M, Sidwell T, et al. Increased T cell immunosenescence and accelerated maturation phenotypes in older kidney transplant recipients. Hum Immunol. 2018;79(9):659–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nie Y, Liu D, Yang W, et al. Increased expression of TIGIT and KLRG1 correlates with impaired CD56bright NK cell immunity in HPV16-related cervical intraepithelial neoplasia. Virol J. 2022;19(1):68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kirchner V, Gillingham K, Serrano O, et al. Long-term outcomes in 831 kidney transplant recipients with 20 years of graft function. Integr J Med Sci. 2021;8. [Google Scholar]
- 66.Watt KDS, Pedersen RA, Kremers WK, et al. Evolution of causes and risk factors for mortality post-liver transplant: results of the NIDDK long-term follow-up study. Am J Transplant. 2010;10(6):1420–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yu S, Cheng Y, Li B, et al. M1 macrophages accelerate renal glomerular endothelial cell senescence through reactive oxygen species accumulation in streptozotocin-induced diabetic mice. Int Immunopharmacol. 2020;81:106294. [DOI] [PubMed] [Google Scholar]
- 68.Campbell RA, Docherty MH, Ferenbach DA, et al. The role of ageing and parenchymal senescence on macrophage function and fibrosis. Front Immunol. 2021;12:700790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kim MG, Yang J, Ko YS, et al. Impact of aging on transition of acute kidney injury to chronic kidney disease. Sci Rep. 2019;9(1):18445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bird TG, Müller M, Boulter L, et al. TGFβ inhibition restores a regenerative response in acute liver injury by suppressing paracrine senescence. Sci Transl Med. 2018;10(454):eaan1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yousefzadeh MJ, Schafer MJ, Noren Hooten N, et al. Circulating levels of monocyte chemoattractant protein-1 as a potential measure of biological age in mice and frailty in humans. Aging Cell. 2018;17(2):e12706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479(7374):547–551. [DOI] [PubMed] [Google Scholar]
- 73.Ding C, Li Y, Guo F, et al. A cell-type-resolved liver proteome. Mol Cell Proteomics. 2016;15(10):3190–3202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aizarani N, Saviano A, Sagar, et al. A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature. 2019;572(7768):199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Glaser SS, Gaudio E, Miller T, et al. Cholangiocyte proliferation and liver fibrosis. Expert Rev Mol Med. 2009;11:e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Allaire M, Gilgenkrantz H. The aged liver: beyond cellular senescence. Clin Res Hepatol Gastroenterol. 2020;44(1):6–11. [DOI] [PubMed] [Google Scholar]
- 77.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345(6274):458–460. [DOI] [PubMed] [Google Scholar]
- 78.Verma S, Tachtatzis P, Penrhyn-Lowe S, et al. Sustained telomere length in hepatocytes and cholangiocytes with increasing age in normal liver. Hepatology. 2012;56(4):1510–1520. [DOI] [PubMed] [Google Scholar]
- 79.Lin S, Nascimento EM, Gajera CR, et al. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature. 2018;556(7700):244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilkinson PD, Delgado ER, Alencastro F, et al. The polyploid state restricts hepatocyte proliferation and liver regeneration in mice. Hepatology. 2019;69(3):1242–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu L, Yannam GR, Nishikawa T, et al. The microenvironment in hepatocyte regeneration and function in rats with advanced cirrhosis. Hepatology. 2012;55(5):1529–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nault JC, Ningarhari M, Rebouissou S, et al. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol. 2019;16(9):544–558. [DOI] [PubMed] [Google Scholar]
- 83.Moncsek A, Al-Suraih MS, Trussoni CE, et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/− ) mice. Hepatology. 2018;67(1):247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meng L, Quezada M, Levine P, et al. Functional role of cellular senescence in biliary injury. Am J Pathol. 2015;185(3):602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nakanuma Y, Sasaki M, Harada K. Autophagy and senescence in fibrosing cholangiopathies. J Hepatol. 2015;62(4):934–945. [DOI] [PubMed] [Google Scholar]
- 86.Lunz JG, Contrucci S, Ruppert K, et al. Replicative senescence of biliary epithelial cells precedes bile duct loss in chronic liver allograft rejection. Am J Pathol. 2001;158(4):1379–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang M, Serna-Salas S, Damba T, et al. Hepatic stellate cell senescence in liver fibrosis: characteristics, mechanisms and perspectives. Mech Ageing Dev. 2021;199:111572. [DOI] [PubMed] [Google Scholar]
- 88.Guo M Cellular senescence and liver disease: mechanisms and therapeutic strategies. Biomed Pharmacother. 2017;96:1527–1537. [DOI] [PubMed] [Google Scholar]
- 89.Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134(4):657–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Maeso-Díaz R, Ortega-Ribera M, Fernández-Iglesias A, et al. Effects of aging on liver microcirculatory function and sinusoidal phenotype. Aging Cell. 2018;17(6):e12829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Maeso-Díaz R, Ortega-Ribera M, Lafoz E, et al. Aging influences hepatic microvascular biology and liver fibrosis in advanced chronic liver disease. Aging Dis. 2019;10(4):684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gracia-Sancho J, Caparrós E, Fernández-Iglesias A, et al. Role of liver sinusoidal endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol. 2021;18(6):411–431. [DOI] [PubMed] [Google Scholar]
- 93.Mitchell SJ, Huizer-Pajkos A, Cogger VC, et al. Age-related pseudocapillarization of the liver sinusoidal endothelium impairs the hepatic clearance of acetaminophen in rats. J Gerontol A Biol Sci Med Sci. 2011;66A(4):400–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grosse L, Bulavin DV. LSEC model of aging. Aging. 2020;12(11):11152–11160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Oteiza A, Li R, McCuskey RS, et al. Effects of oxidized low-density lipoproteins on the hepatic microvasculature. Am J Physiol Gastrointest Liver Physiol. 2011;301(4):G684–G693. [DOI] [PubMed] [Google Scholar]
- 96.Ito Y, Sørensen KK, Bethea NW, et al. Age-related changes in the hepatic microcirculation in mice. Exp Gerontol. 2007;42(8):789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gregory SH, Wing EJ. Neutrophil-Kupffer cell interaction: a critical component of host defenses to systemic bacterial infections. J Leukoc Biol. 2002;72(2):239–248. [PubMed] [Google Scholar]
- 98.Stahl EC, Haschak MJ, Popovic B, et al. Macrophages in the aging liver and age-related liver disease. Front Immunol. 2018;9:2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alzaid F, Lagadec F, Albuquerque M, et al. IRF5 governs liver macrophage activation that promotes hepatic fibrosis in mice and humans. JCI Insight. 2016;1(20):e88689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Balzer MS, Rohacs T, Susztak K. How many cell types are in the kidney and what do they do? Annu Rev Physiol. 2022;84(1):507–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sis B, Tasanarong A, Khoshjou F, et al. Accelerated expression of senescence associated cell cycle inhibitor p16INK4A in kidneys with glomerular disease. Kidney Int. 2007;71(3):218–226. [DOI] [PubMed] [Google Scholar]
- 102.Chkhotua AB, Gabusi E, Altimari A, et al. Increased expression of p16(INK4a) and p27(Kip1) cyclin-dependent kinase inhibitor genes in aging human kidney and chronic allograft nephropathy. Am J Kidney Dis. 2003;41(6):1303–1313. [DOI] [PubMed] [Google Scholar]
- 103.Park JY, Schutzer WE, Lindsley JN, et al. p21 is decreased in polycystic kidney disease and leads to increased epithelial cell cycle progression: roscovitine augments p21 levels. BMC Nephrol. 2007;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Melk A, Schmidt BM, Vongwiwatana A, et al. Increased expression of senescence-associated cell cycle inhibitor p16INK4a in deteriorating renal transplants and diseased native kidney. Am J Transplant. 2005;5(6):1375–1382. [DOI] [PubMed] [Google Scholar]
- 105.Docherty MH, Baird DP, Hughes J, et al. Cellular senescence and senotherapies in the kidney: current evidence and future directions. Front Pharmacol. 2020;11:755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Docherty MH, O’Sullivan ED, Bonventre JV, et al. Cellular senescence in the kidney. J Am Soc Nephrol. 2019;30(5):726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Xu J, Zhou L, Liu Y. Cellular senescence in kidney fibrosis: pathologic significance and therapeutic strategies. Front Pharmacol. 2020;11:601325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Valentijn FA, Falke LL, Nguyen TQ, et al. Cellular senescence in the aging and diseased kidney. J Cell Commun Signal. 2018;12(1):69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shimizu H, Bolati D, Adijiang A, et al. NF-κB plays an important role in indoxyl sulfate-induced cellular senescence, fibrotic gene expression, and inhibition of proliferation in proximal tubular cells. Am J Physiol Cell Physiol. 2011;301(5):C1201–C1212. [DOI] [PubMed] [Google Scholar]
- 110.Niedernhofer LJ, Garinis GA, Raams A, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444(7122):1038–1043. [DOI] [PubMed] [Google Scholar]
- 111.Stenvinkel P, Luttropp K, McGuinness D, et al. expression is associated with vascular progeria in chronic kidney disease. Aging (Albany NY). 2017;9(2):494–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jin H, Zhang Y, Ding Q, et al. Epithelial innate immunity mediates tubular cell senescence after kidney injury. JCI Insight. 2019;4(2):e125490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ueda S, Tominaga T, Ochi A, et al. TGF-β1 is involved in senescence-related pathways in glomerular endothelial cells via p16 translocation and p21 induction. Sci Rep. 2021;11(1):21643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.McCarthy CG, Wenceslau CF, Webb RC, et al. Novel contributors and mechanisms of cellular senescence in hypertension-associated premature vascular aging. Am J Hypertens. 2019;32(8):709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fan YY, Kohno M, Hitomi H, et al. Aldosterone/mineralocorticoid receptor stimulation induces cellular senescence in the kidney. Endocrinology. 2011;152(2):680–688. [DOI] [PubMed] [Google Scholar]
- 116.Chang E, Harley CB. Telomere length and replicative aging in human vascular tissues. Proc Natl Acad Sci U S A. 1995;92(24):11190–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cohen C, Le Goff O, Soysouvanh F, et al. Glomerular endothelial cell senescence drives age-related kidney disease through PAI-1. EMBO Mol Med. 2021;13(11):e14146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shroff R, Shanahan CM. Klotho: an elixir of youth for the vasculature? J Am Soc Nephrol. 2011;22(1):5–7. [DOI] [PubMed] [Google Scholar]
- 119.Kuro-o M, Matsumura Y, Aizawa H, et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390(6655):45–51. [DOI] [PubMed] [Google Scholar]
- 120.Sun CY, Chang SC, Wu MS. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012;81(7):640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Stenvinkel P, Larsson TE. Chronic kidney disease: a clinical model of premature aging. Am J Kidney Dis. 2013;62(2):339–351. [DOI] [PubMed] [Google Scholar]
- 122.Chen TH, Kuro-O M, Chen CH, et al. The secreted Klotho protein restores phosphate retention and suppresses accelerated aging in Klotho mutant mice. Eur J Pharmacol. 2013;698(1–3):67–73. [DOI] [PubMed] [Google Scholar]
- 123.McGlynn LM, Stevenson K, Lamb K, et al. Cellular senescence in pretransplant renal biopsies predicts postoperative organ function. Aging Cell. 2009;8(1):45–51. [DOI] [PubMed] [Google Scholar]
- 124.van Willigenburg H, de Keizer PLJ, de Bruin RWF. Cellular senescence as a therapeutic target to improve renal transplantation outcome. Pharmacol Res. 2018;130:322–330. [DOI] [PubMed] [Google Scholar]
- 125.Liu J, Yang JR, He YN, et al. Accelerated senescence of renal tubular epithelial cells is associated with disease progression of patients with immunoglobulin A (IgA) nephropathy. Transl Res. 2012;159(6):454–463. [DOI] [PubMed] [Google Scholar]
- 126.Niedernhofer LJ, Robbins PD. Senotherapeutics for healthy ageing. Nat Rev Drug Discov. 2018;17(5):377. [DOI] [PubMed] [Google Scholar]
- 127.Childs BG, Gluscevic M, Baker DJ, et al. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017;16(10):718–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang L, Pitcher LE, Prahalad V, et al. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J. 2023;290(5):1362–1383. [DOI] [PubMed] [Google Scholar]
- 129.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kezic A, Popovic L, Lalic K. mTOR inhibitor therapy and metabolic consequences: where do we stand? Oxid Med Cell Longev. 2018;2018:2640342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rudin CM, Hann CL, Garon EB, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18(11):3163–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pérez-Sáez MJ, Gutiérrez-Dalmau Á, Moreso F, et al. Frailty and kidney transplant candidates. Nefrologia (Engl Ed). 2021;41(3):237–243. [DOI] [PubMed] [Google Scholar]
- 133.Lai JC, Segev DL, McCulloch CE, et al. Physical frailty after liver transplantation. Am J Transplant. 2018;18(8):1986–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lai JC, Sonnenday CJ, Tapper EB, et al. Frailty in liver transplantation: an expert opinion statement from the American Society of Transplantation Liver and Intestinal Community of Practice. Am J Transplant. 2019;19(7):1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lai JC, Ganger DR, Volk ML, et al. Association of frailty and sex with wait list mortality in liver transplant candidates in the multicenter Functional Assessment in Liver Transplantation (FrAILT) Study. JAMA Surg. 2021;156(3):256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gill JS. Cardiovascular disease in transplant recipients: current and future treatment strategies. Clin J Am Soc Nephrol. 2008;3 Suppl 2(Suppl 2):S29–S37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cowan J, Bennett A, Fergusson N, et al. Incidence rate of post-kidney transplant infection: a retrospective cohort study examining infection rates at a large Canadian multicenter tertiary-care facility. Can J Kidney Health Dis. 2018;5:2054358118799692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Romero FA, Razonable RR. Infections in liver transplant recipients. World J Hepatol. 2011;3(4):83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Park B, Yoon J, Choi D, et al. De novo cancer incidence after kidney and liver transplantation: results from a nationwide population based data. Sci Rep. 2019;9(1):17202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xu M, Palmer AK, Ding H, et al. Targeting senescent cells enhances adipogenesis and metabolic function in old age. Elife. 2015;4:e12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Palmer AK, Xu M, Zhu Y, et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell. 2019;18(3):e12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Xu M, Pirtskhalava T, Farr JN, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018;24(8):1246–1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rouault C, Marcelin G, Adriouch S, et al. Senescence-associated β-galactosidase in subcutaneous adipose tissue associates with altered glycaemic status and truncal fat in severe obesity. Diabetologia. 2021;64(1):240–254. [DOI] [PubMed] [Google Scholar]
- 144.Hickson LJ, Langhi Prata LGP, Bobart SA, et al. Corrigendum to ‘Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease’ EBioMedicine 47 (2019) 446–456. EBioMedicine. 2020;52:102595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hickson LJ, Langhi Prata LGP, Bobart SA, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Raffaele M, Kovacovicova K, Frohlich J, et al. Mild exacerbation of obesity- and age-dependent liver disease progression by senolytic cocktail dasatinib + quercetin. Cell Commun Signal. 2021;19(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Walaszczyk A, Dookun E, Redgrave R, et al. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell. 2019;18(3):e12945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Dookun E, Walaszczyk A, Redgrave R, et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell. 2020;19(10):e13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Roos CM, Zhang B, Palmer AK, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016;15(5):973–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sugihara H, Teramoto N, Nakamura K, et al. Cellular senescence-mediated exacerbation of Duchenne muscular dystrophy. Sci Rep. 2020;10(1):16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Nambiar A, Kellogg D, Justice J, et al. Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. EBioMedicine. 2023;90:104481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Justice JN, Nambiar AM, Tchkonia T, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chu NM, Gross AL, Shaffer AA, et al. Frailty and changes in cognitive function after kidney transplantation. J Am Soc Nephrol. 2019;30(2):336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ellison-Hughes GM. First evidence that senolytics are effective at decreasing senescent cells in humans. EBioMedicine. 2020;56:102473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tan FC, Hutchison ER, Eitan E, et al. Are there roles for brain cell senescence in aging and neurodegenerative disorders? Biogerontology. 2014;15(6):643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Orr M, Gonzales M, Garbarino V, et al. Senolytic therapy to modulate the progression of Alzheimer’s Disease (SToMP-AD) - outcomes from the first clinical trial of senolytic therapy for Alzheimer’s disease. Res Sq. IN PRESS. doi: 10.21203/rs.3.rs-2809973/v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ogrodnik M, Evans SA, Fielder E, et al. Whole-body senescent cell clearance alleviates age-related brain inflammation and cognitive impairment in mice. Aging Cell. 2021;20(2):e13296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Zhang P, Kishimoto Y, Grammatikakis I, et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat Neurosci. 2019;22(5):719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Raffaele M, Kovacovicova K, Bonomini F, et al. Senescence-like phenotype in post-mitotic cells of mice entering middle age. Aging (Albany NY). 2020;12(14):13979–13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kakkola L, Denisova OV, Tynell J, et al. Anticancer compound ABT-263 accelerates apoptosis in virus-infected cells and imbalances cytokine production and lowers survival rates of infected mice. Cell Death Dis. 2013;4(7):e742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Laberge RM, Sun Y, Orjalo AV, et al. Author correction: mTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2021;23(5):564–565. [DOI] [PubMed] [Google Scholar]
- 162.Laberge RM, Sun Y, Orjalo AV, et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol. 2015;17(8):1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Angeletti A, Cantarelli C, Riella LV, et al. T-cell exhaustion in organ transplantation. Transplantation. 2022;106(3):489–499. [DOI] [PubMed] [Google Scholar]
- 164.Eiden AM, Zhang S, Gary JM, et al. Molecular pathways: increased susceptibility to infection is a complication of mTOR inhibitor use in cancer therapy. Clin Cancer Res. 2016;22(2):277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Raffaele M, Vinciguerra M. The costs and benefits of senotherapeutics for human health. Lancet Healthy Longev. 2022;3(1):e67–e77. [DOI] [PubMed] [Google Scholar]
- 166.Kim EC, Kim JR. Senotherapeutics: emerging strategy for healthy aging and age-related disease. BMB Rep. 2019;52(1):47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Oberhuber R, Heinbokel T, Cetina Biefer HR, et al. CD11c+ dendritic cells accelerate the rejection of older cardiac transplants via interleukin-17A. Circulation. 2015;132(2):122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Fijter JW, Mallat MJK, Doxiadis IIN, et al. Increased immunogenicity and cause of graft loss of old donor kidneys. J Am Soc Nephrol. 2001;12(7):1538–1546. [DOI] [PubMed] [Google Scholar]
- 169.Ritschka B, Knauer-Meyer T, Gonçalves DS, et al. The senotherapeutic drug ABT-737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev. 2020;34(7–8):489–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zhou H, Tullius SG. Targeting cellular senescence in organ transplantation. Transplantation. 2023;107(7):1413–1415. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.