

Abstract
This article delineates the systematic identification, synthesis, and impurity control methods used during the manufacturing process development of tecovirimat, an antiviral drug that treats monkeypox. Critical impurities were synthesized, and their chemical structure was confirmed through NMR analysis, GC, and HPLC mass spectrometry. The results established a thorough approach to identify, address, and control impurities to produce high-quality tecovirimat drug substance in accordance with International Conference on Harmonization (ICH)-compliant standards. This study is the first of its kind to evaluate both process and genotoxic impurities in tecovirimat, demonstrating effective control measures during commercial sample investigations and scaling up to a 60-kg batch size. The findings highlight the importance of critical impurity characterization and control in pharmaceutical development and production to ensure the safety and efficacy of the final product.
Keywords: Tecovirimat, Genotoxic impurities, Process development, International Conference on Harmonization (ICH), Quality control
1. Introduction
Tecovirimat (1), a tetracyclic Active Pharmaceutical Ingredient (API) was discovered by SIGA Technologies as an oral antiviral drug for the treatment of monkeypox. It inhibits a viral protein essential for cellular transmission of the virus, suggesting its inhibitory effect on orthopoxvirus [1]. The U.S. Food and Drug Administration (FDA) granted approval for the commercial use of this antiviral drug in late 2012 [2]. It is an achiral structured API due to a plane of symmetry [3].
The synthesis of tecovirimat involves a crucial Diels–Alder reaction between cycloheptatriene (2) and maleic anhydride (3) (Scheme 1) [4]. The successful construction of this core cyclic hydrocarbons and Diels−Alder adducts has been a significant challenge during the drug's preparation. The initial step in producing the API involves the synthesis of cycloheptatriene (2) through a high-temperature dehydrochlorination–isomerization–ring expansion process [[5], [6], [7], [8]]. Unfortunately, this process yields undesirable toluene as a byproduct, which accounts for approximately 65 % of the final product.
Scheme 1.

Previous Synthetic Route for the Synthesis of Tecovirimat (1) [9].
Several process impurities, along with genotoxic impurities of anhydride derivatives 4, and an undesired isomer impurity 5 at levels of 20 %, have been documented as competitive side reactions, resulting in a low yield (28 %) of the key intermediate 4 [9,10]. A recent patent from SIGA did not provide a yield, but it reported the presence of the undesired isomer impurity 5 at a 7 % level at the end of the reaction [11]. Similarly, Leitich and Sprintschnik have studied this and related reactions. Although they also did not provide a yield, they observed that the undesired isomer impurity 5 ranges from 3 % at 45 °C to 6 % at 110 °C when the reaction was carried out in nitromethane [12].
From the previous study, hydrazide compound 7 was installed through amine condensation to the cyclic anhydride fragment 4 in refluxing EtOH, resulting in the production of tecovirimat with a 51 % yield [9,10]. However, the potential genotoxic effect of the hydrazide compound was not disclosed, and the synergistic reaction between the transferred impurities of cyclic anhydrazide and hydrazide components contributing to the low yields of the final API was not addressed. SIGA further proposed other two alternative approaches to the final API, but these routes suffer from bis-addition impurities, formed at a level of 11.6 % [9]. Subsequently, a cycloaddition with cycloheptatriene (2) was carried out in toluene at 95−110 °C, yielding tecovirimat at a 65 % yield with a 94:6 ratio of desired to undesired isomeric impurities [9].
Delivering a high-quality drug substance requires careful attention to the impurities present in the active pharmaceutical ingredient (API). Therefore, in order to guarantee product quality and, ultimately, patient safety, the identification, quantification, and control methods of these impurities that originate in the manufacturing process become crucial components of drug development [13].
It's worth noting that, in order for drug substances to meet International Conference on Harmonization (ICH) guidelines on impurities, impurities at levels more than 0.10 % or 1.0 mg (whichever is lower) should be identified for drugs with a daily maximum dose of 2 g or less. Any potential impurities that are thought to be unusually potent and have toxic or therapeutic effects at levels below 0.1 % should also be identified [13].
To the best of our knowledge, easy access to these impurities is limited. Moreover, there are only a few reported synthesis routes, primarily in patents, for the synthesis of tecovirimat [3]. Although there are several reports on related intermediates, no synthetic details of impurities have been documented. This study represents the first report in which many critical genotoxic and process impurities of the tecovirimat drug substance have been addressed, as well as their control strategies in the manufacturing process. As a result, this research provides valuable insights for synthetic organic chemists on critical impurities, particularly genotoxic ones, in tecovirimat synthesis, and methods to obtain a pure compound.
2. Results and discussion
During the initial phase of process development of tecovirimat (1), we obtained the API with a 74 % isolated yield by following the experimental and purification procedures outlined in SIGA Technologies' patent method (Scheme 1) [9]. While the laboratory-scale studies demonstrated the effective production of the API, it also generated excessive undesired byproducts that exceed ICH guidelines [13] based on HPLC analysis. Improving the process and controlling the formation of side products became imperative to meet targeted limits. To determine possible impurity structures and content levels at each step of synthesis, we conducted qualitative evaluation through NMR analysis, GC, and HPLC.
A combination of gas chromatography (GC) and mass spectroscopy (MS) was employed to separate and identify the impurities present in intermediates of compound 1. We verified seven general impurities and eleven genotoxic impurities (Table 1), and their structures were confirmed using NMR and mass spectroscopy analysis. The synthetic route to the final compound 1 was disclosed by SIGA Technologies in patent literature [3,9], and our research group successfully produced the API on a 60 kg batch based on our improved method (Scheme 2) [14].
Table: 1.
Structure of related impurities of tecovirimat (1).
| General impurity |
Genotoxic impurity [[15], [16], [17], [18]] |
||
|---|---|---|---|
| Impurity | Structure | Impurity | Structure |
| Compound/Impurity 2 | 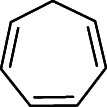 |
Compound/impurity 9 |  |
| Impurity 3 | 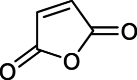 |
Hydrazine hydrate (impurity 6a) | |
| Compound/Impurity 4 | 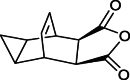 |
Compound/impurity 7 | 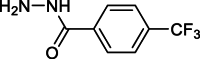 |
| Impurity 5 | 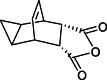 |
Impurity 7b |  |
| Impurity 4a |  |
Impurity 7c |  |
| Impurity 4b | 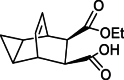 |
Impurity 1a | 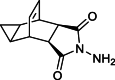 |
| Impurity 4c | 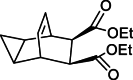 |
Impurity 1d |  |
| Impurity 1b |  |
||
| Impurity 1c | 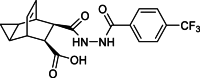 |
||
| Impurity 7a | 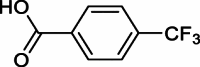 |
||
| Impurity 7d |  |
||
Scheme 2.

Improved industrial Manufacturing Process for Synthesis of Tecovirimat (1)a [14]
aReaction conditions: (i) HCCl3, TEBA (0.05 equiv), 50 % NaOH (aqueous) (4.0 vol), 50–60 °C; 6 h; (ii) 2-pyrrolidone (3.0 vol), 170–180 °C, 4 h; (iii) toluene (1 vol), 80−90 °C, 3 h; (iv) 80 % hydrazine hydrate (1.5 equiv), reflux, 3 h (v) DIEA (0.04 equiv), EtOH (10 vol), reflux, 4 h.
During the synthesis of compound 1, the yields of products 9, 2, 4, and 7 were notably low due to the formation of in-process and genotoxic impurities resulting from degradation, incomplete reactions, or side reactions. The HPLC analysis (figure S11, supporting information) revealed that a number of isomer adduct impurities could originate in the Diels-Alder reaction involving 2 and 3 (Scheme 2) [4,14]. Additionally, potential genotoxic impurities could originate from the hydrazinolysis reaction of 6 to produce 7 [15,16] (Scheme 2), and subsequently from the amine condensation process involving intermediates 4 and 7 (Scheme 2) [9,10,14]. These impurities have been systematically categorized into general/in-process and genotoxic impurities, and are detailed in Table 1.
2.1. Impurity synthesis, identification, and structure verification
To further validate the impurities and improve the purity of compound 1, we purposely synthesized various in-process and genotoxic-related impurities. Firstly, in the Diels-Alder reaction step, the dienophile compound 3 was readily added to the diene 2 in additional toluene to yield compound 4 in a crude form. However, a residual amount of about 0.2 % of compound 9 remained in the starting material 2. Alternatively, the decomposition or rearrangement of the Diels-Alder adducts, based on the stereochemistry of the diene 2 and dienophile 3, could result in the formation of 10 % isomer impurity 5 in the Diels-Alder reaction mixture of compound 4. Impurities 4a and 4b were encountered during the post-treatment of compound 4 with ethanol and water. Furthermore, in the hydrazinolysis step, the ester group in the benzoate moiety of compound 6 could undergo substitution with the hydrazide group, resulting in the formation of a hydrobenzo/benzohydrazide intermediate 7. The hydrazine reaction could potentially yield genotoxic/mutagenic impurities 6a, with the residual reactive methyl ester fragments 6b and 6c remaining in 7 capable of reacting with 6a through hydrazinolysis, leading to the formation of in-process impurities 7b and 7c. Subsequently, 6 could react with residual solvent 80 % hydrazine hydrate, culminating the formation of impurity 7d. Upon the reaction of intermediates 7 and 4 in ethanol and catalytic DIEA, the condensation of benzohydrazide with cycloproa(2)isobenofuran-1,3(3aH)-dione (4) results in the formation of the title compound 1.
Upon the detection of residual cyclic compound 3 in 4, the presence of multiple stereocenters could potentially serve as sites for the formation of isomer impurities that may subsequently be transferred to the final API. Consequently, the cyclic compound 3 remaining in the Diels-Alder product 4 may further reacts with compound 7, potentially resulting in the formation of genotoxic impurity 1d. The formation of the exo and edo fragments of compound 4 can facilitate the formation of endoisomer impurity 1a and 1b. Impurity 1a originates from the reaction between the residual hydrazine solvent 6a and compound 4, while impurity 1b results from the reaction of isomer byproduct 5 and compound 7. Subsequently, ring-opening impurity 1c could be formed from the reaction of 7 and impurity 4a or 4b. Conversely, impurities of benzohydrazide products 7b and 7c undergoes condensation with compound 4, leading to the formation of positional impurities 1e and 1f in a single batch process. However, in subsequent batches, impurities 1e and 1f were effectively controlled in the genotoxic impurity 7c and 7d. As a result, impurities 1e and 1f were not observed in subsequent industrial batch scale production. The formation of impurities should be carefully considered, given that the desired product 1 is achiral due to its plane of symmetry [3]. In order to investigate and validate the potential structures of compounds and related impurities, a substantial amount of related compounds of 1 was required. Therefore, we carried out the synthesis of intermediates 2, 4, 7, 9 and 1.
We meticulously repeated the improved procedure outlined in Scheme 2 [14] to obtain the desired products of all the intermediates, as well as the crude and the final API.
Based on Scheme 3 [12,19,20], compound 8 reacted with 50 % aqueous sodium hydroxide (5 equiv) and chloroform (4.0 vol) catalyzed by a water-soluble phase transfer catalyst, benzyl triethylammonium chloride (0.05 equiv), resulting in an aqueous biphasic system. After extraction with water and re-distillation, compound 9 was isolated as a yellow-to-brown viscous oil in 74 % yield and with a purity of 99.55 % as determined by GC analysis (Fig. S4, supporting information). Compound 2 was further prepared from 9 by reactive distillation in 2-pyrrolidone at 170–180 °C, and isolated by distillation [14]. GC analysis showed that about 0.02 % and 0.3 % of compound 8 and 7,7-dichlorobicyclo[4.1.0]heptane 9, respectively, were identified in compound 2, while approximately 10 % toluene was observed as a byproduct. Additionally, residual solvents including chloroform, benzene, and dichloromethane were identified in product 2 at a 6 % level. In order to remove or reduce the impurities in 2, extraction and re-distillation was attempted. Following re-distillation, compound 8 and residual solvents were completely purged. Successively, the levels of impurity 9 and toluene in compound 2 were reduced to 66 ppm and 8 %, respectively (Fig. S8, supporting information). This purification process culminated in an 82 % yield and a purity of 92.85 % for compound 2 as determined by GC analysis. The structure of compound 2 was confirmed by 1 H and 13C NMR analysis respectively (Figs. S5–S6, supporting information).
Scheme 3.

Preparation of intermediate 2 and its related Impurities.
Compounds 2 and 3 were identified as starting materials for synthesizing compound 4, and subsequently for the synthesis of impurities 5, and 4a-4c, as shown in Scheme 4 [4]. Compound 4 was prepared from 2 by reacting with 1.05 equivalent of maleic anhydride 3 in toluene at 120 °C, resulting in crude anhydride 4. Quantitative results indicated that compound 5 was a major isomer impurity, constituting about 10 % of the crude reaction mixture, aligning with findings reported by SIGA and colleagues [9]. Therefore, compound 5 was prepared from the mother liquor, while ring opening byproduct impurities 4a, 4b and 4c comprising approximately 0.04 %, were observed in product 4. Hydrolysis of product 4 was achieved by reacting with an aqueous solution of sodium hydroxide, which was then neutralized with hydrochloric acid to give ring opening impurities 4a, 4b and 4c. In subsequent industrial batch production, impurity 4a was not detected in compound 4.
Scheme 4.

Formation of Isomer impurities 5, 4a-4c.
In the synthetic route outlined in the patent literature (Scheme 1) [9,10], it is noted that impurities, particularly the undesired isomer impurity 5, may be transferred into the amine condensation reaction at a concentration of about ≤0.03 % in the mother liquid. During this process, they can be transformed into impurity 1b, which is an isomer of the active pharmaceutical ingredient (API), and subsequently into impurity 1c through hydrogenation. The opening ring impurities 5 and 4b were of great concern, as they could significantly impact the quality of intermediate 4 and, consequently, the final API.
We chose a nonpolar solvent in a polar solvent mixture for recrystallization of the crude anhydride 4 since our desired product 4 is a discreetly polar compound. Therefore, an MTBE and ethanol (10:1) solvent mixture was deemed to provide a good balance of solubility and selectivity for purification by recrystallization of crude product 4 and its impurities 4a, 4b, 4c, and 5. This purification approach effectively mitigated the formation of related impurities, notably reducing the undesired isomer impurity 5 and 4b, to levels of 0.018 %, each, with a complete elimination of the ring-opening impurity 4a. As a result, compound 4 was isolated as a crystalline solid with an 82 % yield and 99.85 % purity determined by HPLC analysis (Fig. S11, supporting information).
According to the synthetic route depicted in Scheme 5 [9], the synthesis of compound 7 involved the hydrazinolysis of methyl ester 6 using an 80 % hydrazine hydrate solution in a mixture of methanol and water. Impurity 6a was detected in the crude reaction mixture as residual solvent, while 7b, 7c, and 7d were identified as general impurities of compound 7 and genotoxic impurities in the API. In our improved approach (Scheme 2) [14], it is evident that impurities originating from intermediates of tecovirimat, particularly intermediate 7 can significantly impact the quality of the API. This is a critical concern, particularly considering the potential genotoxicity [15,16,21], which underscores the importance of our efforts to improve the synthesis process.
Scheme 5.

Synthesis of Intermediate 7 and its related Impurities 7a-7d
We confirmed and synthesized the benzohydrazide impurities 7b, 7c, and 7d as outlined in Scheme 5. Impurity 7b and 7c were obtained by the reaction of 6b and 6c with 80 % hydrazine hydrate concurrently. Impurity 7d was produced by the deprotonating methyl-2-trifluorobenzoate (6) with sodium methoxide in methanol, followed by the nucleophilic attack of 4-trifluoromethylbenzoyl hydrazide 7 via hydrazinolysis reaction to afford the target impurity.
Upon detecting impurities 7b and 7c in the crude reaction mixture, we devised purification methods to minimize and or eliminate the related impurities of compound 7. Given the significance of removing these genotoxic impurities, particular attention was directed towards optimizing the crystallization conditions employed in the purification process. Considering the highly-solubility of compound 7 in organic solvents, we hypothesized that the precipitation induced by the addition of an antisolvent such as water could serve as an effective isolation technique. Various polar solvents, including methanol, ethanol, MTBE, and acetone were explored for the crystallization conditions.
The optimal conditions, in line with previous literature, involve utilizing MTBE as the solvent and water as an antisolvent for titrating the crude product [14]. Table 2 presents the impurity removal assessment experiments conducted to determine robust process conditions for the high-purity isolation of product 7. By slowly titrating the reaction mixture with 4 vol of water and subsequently crystallizing in 3 vol of MTBE, the desired product 7 was obtained in a commendable 77 % yield, 99.96 % purity, successfully eliminating impurity 6a and significantly reducing genotoxic impurities 7b and 7c to levels below 21 ppm and 66 ppm, respectively. In an attempt to remove impurities 7a-c, the crude product was titrated with 4 vol of water, effectively removing in-process impurity 7a. Subsequent crystallization of the crude product with 3 vol of MTBE led to a significant reduction in genotoxic impurities 7b and 7c to below 21 ppm and 66 ppm, respectively, while a minimal amount of impurity 7d (0.01 %) remained in product 7 (Table 2, entry 8) (also see Fig. S29, supporting information).
Table: 2.
Optimization of the crystallization solvent for effective removal of impurities of 4-(trifluoromethyl)benzohydrazide (7)a.
| Entry | Impurity content in Crude (%) | Titration bolvent (vol.) | Purification conditions | Yield of 7 (%)b | Purity of 7 (%)c | Impurity content in purified 7 (%)d |
|---|---|---|---|---|---|---|
| 1 | 0.12 | 2 V Water | 0.5 V methanol | 65 | 86.72 % | 0.09 |
| 2 | 0.12 | 4 V water | – | 72 | 91.16 % | 0.10 |
| 3 | 0.15 | 4 V Water | 1 V methanol | 67 | 89.97 % | 0.08 |
| 4 | 0.14 | 4 V Water | 2 V acetone | 73 | 95.30 % | 0.05 |
| 5 | 0.18 | 4V Water | 0.5 V methanol | 77 | 96.97 % | 0.10 |
| 6 | 0.07 | 3 V Water | 1 V MTBE | 79 | 98.68 % | 0.08 |
| 7 | 0.14 | 4 V Water | 2 V MTBE | 82 | 99.05 % | 0.05 |
| 8 | 0.10 | 4 V Water | 3 V MTBE | 87 | 99.96 % | 0.01 |
| 9 | 0.10 | 5 V Water | 5 V MTBE | 80 | 79.34 % | 0.04 |
Reaction conditions: 6 (1.0 eq), hydrazine hydrate as solvent (1.5 eq), reflux.
Calculated yields were given as isolated yields.
Purity determined by HPLC.
Impurity content in 7 determined by HPLC.
In the amine condensation step of synthesizing the API, the improved route incorporates a single-step reaction involving intermediate 4 and equimolar amounts of hydrazide 7 with EtOH in the presence of catalytic diisopropylethylamine, resulting in the successful production of the target API, as shown in Scheme 9 [9,10,14]. The likelihood of residues from compounds 4 and 7 being present in the final API is expected. Considering tecovirimat's starting material, products and derivatives of benzohydrazide, including compound 4, are key focus areas due to their potential genotoxic impurity characteristics [15,16,21]. Moreover, impurities originating from intermediate 4 were carried into the reaction and transformed into new impurities. In light of patient safety concerns, meticulous control over compounds 4 and 7 as potential impurities in the drug substance is imperative. Initially, we considered synthesizing the known impurities in the crude product. The direct synthesis of impurity 1b from the mother liquor of 1 appeared logical. As depicted in Scheme 9, impurity 1a was obtained through the reaction of 4 with 80 % hydrazine hydrate in methanol. In-process impurity 1c resulted from the condensation of compounds 4 and 7 in methanol. Furthermore, the reaction of compound 4 with maleic anhydride 3 resulted in the formation of genotoxic impurity 1d.
Scheme 9.

Synthesis of Impurities 1a-1f.
Our focus then shifted to developing a downstream isolation process to ensure effective removal of genotoxic impurities and establish efficient process control. We noted that the successful removal of impurities 1a-f hinged significantly on the specific crystallization conditions employed. The purification and removal of residues from compounds 4, 7, and their related impurities from the API were accomplished through recrystallization from an ethanol-water mixture (5:1, v/v). HPLC validation was conducted to evaluate compounds 4, 7, and 1a-1c in the API (see Figs. S15, S29, S49-S61, supporting information), with method detection ensuring comprehensive removal of all related impurities. The analysis results presented in Table 3 conclusively demonstrate that the purification process implemented guarantees that tecovirimat, obtained on 60 kg scale with a 96 % yield, 99.96 % purity by HPLC, is completely free from all related impurities.
Table: 3.
HPLC Analysis Report of Impurities in Crude and Purified Samples for API, and the respective limit in accordance with ICH [13,22].
| Sample/Impurity | Testing standards/limits (by ICH guideline) |
Test Results (determined by HPLC) |
|||||
|---|---|---|---|---|---|---|---|
| Crude |
API |
Crude |
Final API |
||||
| Batch 1 | Batch 2 | Batch 1 | Batch 2 | Batch 3 | |||
| Impurity 1b | ≤0.05 % | ≤0.05 % | 0.01 % | N.D | 0.01 % | N.D | N.D |
| Impurity 7a | ≤0.05 % | ≤0.05 % | N.D | N.D | N.D. | N.D | N.D |
| Impurity 9 | ≤66 ppm | ≤66 ppm | N.D | N.D | N.D. | N.D | N.D |
| Impurity 7 | ≤0.05 % | ≤66 ppm | N.D | N.D | N.D | N.D | N.D |
| Impurity 7b | ≤0.05 % | ≤66 ppm | N.D | N.D | N.D | N.D | N.D |
| Impurity 7c | ≤0.05 % | ≤66 ppm | N.D | N.D | 8 ppm | 7 ppm | 6 ppm |
| Impurity 4 | ≤0.50 % | ≤0.50 % | 0.01 % | 0.02 % | N.D | N.D | N.D |
| Impurity 4a | ≤0.50 % | ≤0.50 % | N.D | 0.01 % | N.D | N.D | N.D |
| Residual Solvents | |||||||
| Methanol | ≤0.3 % | ≤0.3 % | N.D | N.D | N.D | N.D | N.D |
| Dichloromethane | ≤0.06 % | ≤0.06 % | 0.004 % | ND | 0.004 % | N.D | N.D |
| Toluene | ≤0.089 % | ≤0.089 % | N.D | N.D | N.D | N.D | N.D |
| Methyl Tert-Butyl Ether | ≤0.5 % | ≤0.5 % | N.D | N.D | N.D | N.D | N.D |
| Ethanol | ≤0.5 % | ≤0.5 % | 0.02 % | 0.02 % | 0.02 % | 0.02 % | 0.03 % |
| Benzene | ≤0.0002 % | ≤0.0002 % | 0.00004 % | N.D | 0.00004 % | N.D | N.D |
| Chloroform | ≤0.006 % | ≤0.006 % | N.D | N.D | N.D | N.D | N.D |
| 2-Pyrrolidone | ≤0.05 % | ≤0.05 % | N.D | N.D | N.D | N.D | N.D |
| Moisture content | 4.2%–5.0 % | 4.2%–5.0 % | 4.9 % | 4.9 % | 4.7 % | 4.9 % | 4.7 % |
| Content (in terms of anhydrous) | 98.0 %–102.0 % | 98.0 %–102.0 % | 100.4 % | 100.4 % | 100.4 % | 99.5 % | 100.0 % |
N.D: Not detected.
Our study unequivocally demonstrates that incremental purification throughout the synthesis process proves to be more effective than deferring purification until the final stage. This finding corroborates with previous reports by Nair [23] and Kim [24], reinforcing the importance of meticulous impurity control at each step of the synthesis process. In contrast to the HPLC analysis when we repeated SIGA's method, the HPLC results showed undesired hydrazine 7 and 7d in the API at 4 % and 0.57 % levels, respectively, with impurities 1a-1d ranging from 0.02 to 1.18 % (Fig. S67, supporting information). This exceeds the ICH guidelines [13] and highlights the necessity of maintaining strict impurity control at each step to mitigate potential accumulation and unexpected reactions, thereby eliminating competitive side products and genotoxic impurities.
Our systematic approach has substantially improved the synthesis of tecovirimat, ensuring product quality and safety. This aligns with our research group's overarching concept of “Control from Root Design,” which emphasizes the design and evaluation of a synthetic route from the beginning of an R&D project to create a safer, more efficient, and cost-effective industrial process.
Safety Precaution! Proper safety measures must be prioritized when handling starting materials and final products during the industrial production of tecovirimat API, particularly when operating at or above a batch size of 60 kg. Failure to implement effective measures to prevent oxidation (through inerting) during feeding may result in a vapor cloud explosion, which may pose harm to personnel. Ensuring the implementation of robust safety procedures, including the installation of appropriate process equipment and technology to guarantee optimal inerting, is crucial in mitigating such risks.
3. Conclusion
The identification, synthesis, and effective control of critical impurities in tecovirimat have been achieved through carefully selected improved isolation conditions and downstream isolation processes. This approach resulted in a significant decrease in the production of the major competitive side products particularly isomer impurity of Diels-Alder adducts intermediate 4 from ≤20 % to <0.05 %, and successful elimination of originating and transferred impurities in the API, particularly the benzohydrazide genotoxic impurities. The comprehensive approach outlined in this study offers valuable insights and guidance for process chemists in the generics industry to meet various requirements of drug regulatory agencies. The success of this research in achieving acceptable control of impurities by ICH guidelines in both small-scale and industrial runs of up to a 60-kg batch size underscores its potential application for the synthesis of tecovirimat API on a multi-ton scale.
4. Experimental section
4.1. General information
If not specifically stated, then all reagents and solvents were purchased from commercial suppliers and used without further purification.
For NMR spectroscopy, the 1H NMR and 13C NMR spectra were applied to characterize the structures of all compounds and related impurities. The spectra were obtained on a Bruker 400 MHz instrument using the residual signal of deuterated solvent (DMSO‑d6) as the internal standard. The chemical shifts (δ) for 1H and 13C are given in parts per million (ppm), with coupling constants reported in Hz, with multiplicities denoted as singlet (s), doublet (d), triplet (t), quartet (q), doublet of doublets (dd), multiplets (m), and so on.
The Electrospray ionization (ESI) mass spectra were measured by a Thermo Fisher FINNIGAN LTQ instrument. For Thin layer chromatography (TLC) analysis, pre-coated plates (silica gel 60 GF-254, 0.25 mm) were employed using UV light as the visualization agent (254 nm). Organic solutions were concentrated under reduced pressure on a Heidolph rotary evaporator. GC samples were analyzed on an Agilent Technologies 6890 GS system by using an HP-5 column.
Analytical High performance liquid chromatography (HPLC) for liquid phase was carried out on an Agilent 1260 HPLC workstation, equipped with a Waters Xbridge-C18 system (4.6 × 150 mm, 3.5 μm). Tecovirimat, related compounds, and impurities were synthesized and characterized following our previous study [14], established literature procedures [9], and known methods, with appropriate modifications.
4.1.1. Synthesis of compound/impurity 9
Compound/impurity 9 was synthesized from 8 following our previously reported procedure [14]. A mix of cyclohexene 8 (30 kg, 365.20 mol, 1 eq), benzyltriethylammonium chloride (4.2 kg, 18.44 mol, 0.05 eq), and chloroform (180 kg, 4 v) was warmed up for 20 min at 45–50 °C. Then, a solution of 50 % sodium hydroxide (73.3 kg, 1830 mol, 5 eq) was added with stirring. The reaction temperature was carefully maintained between 50 and 60 °C for 1.5 h. Then, the mixture was stirred for 6 more hours at 55–60 °C before being cooled to room temperature and mixed with 90 L of water. The organic layer was isolated, concentrated under vacuum at a temperature of 40–50 °C to obtain a crude product, which was subsequently distilled at 3 kPa using a vacuum reactor equipped with a water pump. A clear yellow to brown viscous oil of 7,7-dichlorobicyclo-[4-1.0]heptane 9 was obtained from the 60–90 °C fraction, resulting in a 75 % yield of 45 kg. The purity of the compound was determined to be 99.55 % by GC analysis. 1H NMR (400 MHz, DMSO‑d6) δ 2.45 (d, 1H), 2.26 (m, 1H), 2.03 (m, 1H), 1.69 (m, 2H). 13C NMR (101 MHz, DMSO‑d6) δ 68.2, 25.6, 20.2, 18.8.
4.1.2. Synthesis of compound/impurity 2
Compound/impurity 2 was prepared following similar steps as reported in our previous study [14]. In a reactor with a distillation device was charged 7,7-dichlorobicyclo-[4.1.0]heptane 9 (44.8 kg (271.42 mol) of and 2-pyrrolidone (134 kg (3.0 v). The reaction was initiated by heating the mixture to 170–180 °C, leading to the formation of product 2 over the course of the reaction. Subsequently, the 80–100 °C fraction was collected at atmospheric pressure for a duration of 4 h. After removing the aqueous phase, the organic phase was washed with a saturated sodium bicarbonate aqueous solution to obtain a crude product, which was then re-distilled under 4 kPa pressure. The resulting 20–25 °C fraction yielded product 2 with a weight of 20 kg, 82 % yield, and 92.85 % purity as determined by GC. The identity of the 1,3,5-cycloheptatriene 2 was confirmed via GC-MS analysis. The product's toluene content was 8 %, probably much lower. 1H NMR (400 MHz, DMSO‑d6) δ 6.59 (m, 2H), 6.18 (m, 2H), 5.35 (m, 2H), 2.18 (m, 2H). 13C NMR (101 MHz, DMSO‑d6) δ 130.8, 126.4, 120.5, 27.4.
4.1.3. Synthesis of compound 4 and related impurity (4a-c)
The procedure for synthesizing compound 4 was adapted from our previous work [14]. Maleic anhydride 3 (21.1 kg, 216.52 mol, 1.05 eq) and 1,3,5-cycloheptatriene 2 (19 kg, 206.21 mol, 1.0 eq) were stirred in toluene (200.0 kg) at 80–90 °C for 3–4 h. Upon cooling, the reaction mixture was crystallized with the addition of MTBE (70 kg), and then filtered. The mother liquor was concentrated, and the crude product was purified by prep-HPLC to isolate isomer impurity 5. The resulting filter cake was dried and further recrystallized from a mixture of MTBE: ethanol (10:1) to afford white crystals of compound 4 (32 kg, 99.85 % purity by HPLC) in 82 % yield. Compound 4 1H NMR (400 MHz, DMSO‑d6) δ 5.86 (s, 2H), 3.40 (t, J = 1.9 Hz, 2H), 3.29 (ddq, J = 4.5, 2.8, 1.7 Hz, 2H), 1.16 (s, 2H), 0.29 (s, 1H), 0.04 (s, 1H).13C NMR (101 MHz, DMSO‑d6) δ 173.3, 128.3, 45.7, 40.1, 39.9, 39.7, 39.5, 39.2 (d, J = 21.0 Hz), 38.8, 33.0, 8.9, 8.7, 4.6. HRMS (ESI): m/z [M + H]+ calcd for C11H11O3 191.0703, found: 191.0707; Impurity 5 (98.95 purity by HPLC) 1H NMR (400 MHz, DMSO‑d6) δ 6.06–5.73 (dd, J = 5.0, 3.2 Hz, 2H), 3.29–3.21 (dpt, J = 5.1, 3.2, 1.7 Hz, 2H), 3.20–3.13 (t, J = 1.6 Hz, 2H), 1.05–0.83 (dq, J = 7.9, 2.7 Hz, 2H), 0.24–0.11 (td, J = 7.5, 5.2 Hz, 1H), 0.05 (dt, J = 5.3, 3.4 Hz, 1H). HRMS (ESI): m/z [M + H]+ calcd for C11H11O3 191.0703, found: 191.0710.
4.1.4. Synthesis of impurity 4a
A solution of NaOH (0.64 g, 0.016 mol, 1 eq) was added to compound 4 (3 g, 0.016 mol, 1 eq) and stirred continuously in a mixture of THF and water (4:1) for 2 h at 60 °C. Upon completion of the reaction, the pH of the solution was adjusted to 3 using 1 M hydrochloric acid. Subsequently, the mixture was allowed to stand for 10 min before being extracted with EA. The organic layer was separated and concentrated to yield a solid product, which was further purified by column chromatography on PE/EA 1:1, resulting in the isolation of 2.6 g of impurity 4a in an 85 % yield with a purity of 98.38 % by HPLC. 1H NMR (400 MHz, DMSO‑d6) δ 11.83 (s, 2H), 5.72 (dd, J = 4.9, 3.3 Hz, 2H), 3.02–2.96 (m, 2H), 2.94 (d, J = 1.0 Hz, 2H), 0.95 (ddt, J = 7.3, 5.2, 2.4 Hz, 2H), 0.06 (d, J = 5.1 Hz, 1H), −0.00 (dt, J = 5.1, 3.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 174.3, 127.6, 48.2, 34.2, 9.6, 3.0. 13C NMR (101 MHz, DMSO‑d6) δ 173.7, 173.5, 129.6, 129.3, 46.9, 46.7, 32.5, 31.9, 5.9, 5.7, 2.1, 1.9. HRMS (ESI): m/z [M − H]- calcd for C11H13O4 207.0633, found: 207.0678.
4.1.5. Synthesis of impurity 4b
Subsequently, DMAP (2 g, 0.02 mol, 1 eq) was added to compound 4 (3 g, 0.016 mol, 1 eq) and continuously stirred in ethanol (5 v) for 2 h at 60 °C. After completion, ethanol (2 v), 1 M hydrochloric acid (1 v) and water (5 v) were added to the reaction mixture, followed by standing for 10 min for aqueous layer separation and concentration to afford solid. The solid was further purified by column chromatography on PE:EA 5:1 to afford impurity 4b (3.2 g, 82 % yield). 1H NMR (400 MHz, DMSO‑d6) δ 12.00 (s, 1H), 5.74 (d, J = 18.3 Hz, 2H), 3.91 (dd, J = 10.9, 7.1 Hz, 2H), 3.06–2.96 (m, 4H), 0.11–0.03 (m, 1H), 0.00 (dt, J = 5.2, 3.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 174.1, 172.9, 127.8, 127.4, 59.9, 48.2, 47.8, 34.4, 33.7, 14.4, 9.6, 9.5, 3.0. HRMS (ESI): m/z [M + H]+ calcd for C11H16O4 236.1049, found: 236.1013.
4.1.6. Synthesis of impurity 4c
To impurity 4b (2.5 g, 0.01 mol, 1 eq) and DMF (5 v, 0.06 mol) was added potassium carbonate (4 g, 0.03 mol, 3 eq) and stirred for 10 min at 60 °C. Afterwards, Iodoethane (3 g, 0.02 mol, 2 eq) was added and the reaction mixture was stirred continually for 4 h. After completion, ethyl ether (3 v) and water (4 v) were added for aqueous layer separation and concentrated to obtain the crude solid (2.5 g). The solid was further purified using column chromatography to afford impurity 4c (2 g, 72 % yield). 1H NMR (400 MHz, DMSO‑d6) δ 5.76 (dd, J = 5.0, 3.3 Hz, 2H), 3.94 (dd, J = 9.2, 7.1 Hz, 4H), 3.12–3.07 (m, 2H), 3.03 (q, J = 2.9 Hz, 2H), 1.14 (t, J = 7.1 Hz, 6H), 1.02–0.97 (m, 2H), 0.10 (d, J = 5.3 Hz, 1H), 0.05–0.00 (m, 1H). 13C NMR (101 MHz, DMSO) δ 172.6, 127.6, 60.0, 47.9, 40.6, 40.4, 40.2, 40.0, 39.8, 39.5, 39.3, 34.1, 14.4, 9.4, 3.0. HRMS (ESI): m/z [M + H]+ calcd for C15H20O4 264.1364, 264.1378.
4.1.7. Synthesis of compound/impurity 7
Impurity 7 was the same as compound 7 (4-(trifluoromethyl)benzohydrazide) shown in Scheme 5. It could be synthesized by the process route outlined in previous studies [9,10,14]. The reactor was filled with a mixture of methyl 4-(trifluoromethyl)benzoate 6 (50 kg, 1.0 eq 244.92 mol) and 80 % hydrazine hydrate (impurity 6a) (18.4 kg, 1.5 eq, 367.38 mol). The mixture was stirred at 65–70 °C for an hour, followed by the gradual addition of a mixture of methanol and water (25 kg/200 kg). Subsequently, the mixture was refluxed for 4 h, filtered, and dried. The crude product was then titrated with water (200 kg, 4 v) and crystallized with MTBE (150 kg, 3 v) to give white solid of 4-(trifluoromethyl)benzohydrazide 7 weighing 38 kg, with 77.4 % yield and a purity of 99.96 % as determined by HPLC.1H NMR (400 MHz, DMSO‑d6) δ 10.02 (s, 1H), 8.01 (s, 2H), 7.82 (s, 2H), 4.60 (s, 2H). 13C NMR (101 MHz, DMSO‑d6) δ 164.5, 137.1, 131.3 (d, J = 31.9 Hz), 131.0, 127.8, 125.3, 122.6, 119.8, 39.5. HRMS (ESI): m/z [M + H]+ calcd for C8H8F3N2O 205.0583, found: 205.0601; Impurity 7a 1H NMR (400 MHz, DMSO‑d6) δ 13.48 (s, 1H), 8.13 (d, J = 8.1 Hz, 2H), 7.85 (d, J = 8.2 Hz, 2H). 13C NMR (101 MHz, DMSO) δ 166.6, 135.0, 133.1, 132.7, 130.5, 126.0, 126.0, 125.9. HRMS (ESI): m/z [M − H]- calcd for C8H4F3O2 189.0169, found:189.0184.
4.1.8. Synthesis of impurity 7b
The synthesis route of Impurity 7b is shown in Scheme 6. Impurity 6b (2 g, 0.01 mol, 1.5 eq) was stirred in 80 % hydrazine hydrate (impurity 6a) (2 g, 0.04 mol, 1.5 eq) at 75 °C for 3 h. When reaction completion was observed by TLC, the reaction mixture was evaporated under vacuum, and the solid was crystallized in methanol, which was further recrystallize with MTBE (3 v) to give the desired product Impurity 7b as a white solid (1.7 g, 84 % yield, 97.28 % purity by HPLC). 1H NMR (400 MHz, DMSO‑d6) δ 9.62 (s, 1H), 7.79 (dd, J = 7.8, 1.3 Hz, 1H), 7.76–7.61 (m, 2H), 7.52–7.46 (m, 1H), 13C NMR (101 MHz, DMSO) δ 166.9, 135.6, 132.8, 130.3, 129.3, 126.8, 126.7, 126.7, 126.6, 126.6. HRMS (ESI): m/z [M + H]+ calcd for C8H8F3N2O 205.0583, found: 205.0604.
Scheme 6.

Synthesis of intermediate 7b
4.1.9. Synthesis of impurity 7c
The synthesis route of Impurity 7c is outlined in Scheme 7. A quantity of Impurity 6c (2 g, 0.01 mol, 1eq) was stirred with 80 % hydrazine hydrate (impurity 6a) (2 g, 0.04 mol, 1.5 eq) at 75 °C for 3 h. Upon observing completion of the reaction by TLC, the reaction mixture was subjected to vacuum evaporation, and the resulting solid was crystallized in methanol. Subsequent recrystallization with MTBE (3 v) yielded the desired product Impurity 7c as a white solid (1.58 g, 80 % yield, 99.40 % purity by HPLC). 1H NMR (400 MHz, DMSO‑d6) δ 10.06 (s, 1H), 8.27–8.04 (m, 2H), 7.88 (d, J = 7.8 Hz, 1H), 7.71 (t, J = 7.8 Hz, 1H), 4.60 (s, 2H). 13C NMR (101 MHz, DMSO) δ 164.7, 134.6, 131.4, 130.1, 128.1, 128.0, 124.1, 124.0, 124.0, 124. HRMS (ESI): m/z [M + H]+ calcd for C8H8F3N2O 205.0583 found: 205.0605.
Scheme 7.

Synthesis of intermediate 7c
4.1.10. Synthesis of impurity 7d
The synthesis route of Impurity 7d is shown in Scheme 8. Methyl 4-(trifluoromethyl)benzoate 6 (2 g, 0.01 mol, 1 eq), sodium methoxide (1.1 g, 0.02 mol, 2 eq) and methanol (3 v) were stirred at 70 °C for 3 h. Subsequently, 4-(trifluoromethyl)benzohydrazide 7 (2 g, 0.01 mol, 1 eq) was added, then reaction mixture was continuously stirred overnight at 70 °C. The solvent was evaporated under vacuum, and the crude product was purified by prep-HPLC to isolate impurity 7d (3.2 g, 88 % yield, 100 % purity by HPLC) as a white solid. 1H NMR (400 MHz, DMSO‑d6) δ 10.98–10.84 (s, 2H), 8.23–8.08 (d, J = 8.1 Hz, 4H), 8.00–7.85 (d, J = 8.2 Hz, 4H). 13C NMR (101 MHz, DMSO) δ 165.2, 136.6, 132.4, 132.1, 128.9, 128.3, 126.1, 126.1, 126.0, 125.6. HRMS (ESI): m/z [M + H]+ calcd for C16H11F6N2O2 377.0719, found: 377.0810.
Scheme 8.

Synthesis of intermediate 7d.
4.1.11. Synthesis of tecovirimat (1)
The title compound 1 was synthesized following our reported method [14]. In a reactor, compound 4 (0.87 kg, 167.16 mol, 1.0 eq), compound 7 (35.7 kg, 175.55 mol, 1.05 eq) and DIEA (0.87 kg, 6.69 mol, 0.04 eq) were introduced in 250 kg of ethanol (10 v). The reaction mixture was then refluxed for 4 h. Upon observing completion of the reaction by TLC, the reaction was quenched by the addition of water (64 kg, 2 v). The resulting suspension was cooled to 25 °C, and the precipitated solid was filtered, dried at 50 °C to give a crude product of 65 kg. The mother liquor was concentrated by vacuum to give impurity 1b. The crude product was recrystallized with ethanol: water (300 kg, 5:1 v/v). Tecovirimat was obtained as white solid (60 kg, 96 % yield, 99.96 % purity). Compound 1 1H NMR (400 MHz, DMSO‑d6) δ 11.40 (s, 1H), 8.10 (d, J = 8.3 Hz, 2H), 7.93 (d, J = 8.3 Hz, 2H), 5.90–5.67 (m, 2H), 3.26 (d, J = 27.6 Hz, 4H), 1.18 (d, J = 8.4 Hz, 2H), 0.32–0.19 (m, 1H), 0.07 (q, J = 6.3, 4.2 Hz, 1H). 13C NMR (101 MHz, DMSO‑d6) δ 174.7, 163.5, 163.1, 135.0, 134.6, 132.4, 132.1, 128.6, 127.6, 127.3, 125.8, 125.8, 125.7, 125.7, 125.1, 122.4, 119.7, 43.3, 43.0, 33.0, 32.8, 9.2, 4.1. LRMS (ESI): m/z [M + H]+ calcd for C19H16F3N2O3 376.1 found: 376.4; Impurity 1b (98.1 purity by HPLC). 1H NMR (400 MHz, DMSO‑d6) δ 11.35 (s, 1H), 8.12 (d, J = 8.1 Hz, 2H), 7.96 (t, J = 8.6 Hz, 2H), 5.95 (dd, J = 4.9, 3.3 Hz, 2H), 3.28 (ddd, J = 7.0, 3.4, 1.8 Hz, 2H), 2.97 (d, J = 20.3 Hz, 2H), 1.22–1.06 (m, 1H), 0.20–0.13 (m, 1H). 13C NMR (101 MHz, DMSO) δ 174.4, 171.6, 164.6, 136.9, 132.1, 131.8, 128.8, 128.4, 126.5, 126.2, 125.9, 125.8, 125.8, 48.3, 47.0, 36.6, 33.0, 10.1, 9.4, 2.9. HRMS (ESI): m/z [M + H]+ calcd for C19H14F3N2O3 375.1108, found: 377.1106.
4.1.12. Synthesis of impurity 1a
The synthesis route for Impurity 1a is shown in Scheme 9. Compound 4 (2 g, 0.01 mol) and 80 % hydrazine hydrate (impurity 6a) (1 g, 0.02 mol) were stirred in methanol (20 ml) at reflux for 5 h. Upon confirmation of reaction completion by TLC, the reaction mixture was evaporated under vacuum to give off-white solid as the desired product Impurity 1a (1.63 g, 76 % yield, 99.6 % purity by HPLC). 1H NMR (400 MHz, DMSO‑d6) δ 5.68 (dd, J = 4.9, 3.3 Hz, 2H), 4.88 (s, 2H), 3.20 (dtt, J = 5.1, 3.7, 1.9 Hz, 2H), 2.99 (t, J = 1.8 Hz, 2H), 1.12 (ddt, J = 7.9, 4.0, 2.3 Hz, 2H), 0.23 (td, J = 7.4, 5.4 Hz, 1H), 0.03 (dt, J = 5.4, 3.7 Hz, 1H). 13C NMR (101 MHz, DMSO‑d6) δ 176.0, 127.8, 43.4, 33.2, 9.7, 4.6. HRMS (ESI): m/z [M + H]+ calcd for C11H13N2O2 205.0972, found: 205.0988.
4.1.13. Synthesis of impurity 1c
The method used to synthesize Impurity 1c is shown in Scheme 9. Compound 4 (4.6 g, 0.02 mol) and compound 7 (5 g, 0.02 mol) were stirred in methanol (50 ml) at reflux for 5 h. Upon completion of the reaction, as indicated by TLC, the reaction mixture was quenched with water. It was subsequently filtered and dried, resulting in the formation of a white solid identified as the desired product Impurity 1c (7.3 g, 76.6 % yield, 96.6 % purity by HPLC). 1H NMR (400 MHz, DMSO‑d6) δ 11.40 (s, 1H), 10.58 (d, J = 1.4 Hz, 1H), 9.94 (s, 1H), 8.06 (d, J = 8.1 Hz, 2H), 7.87 (d, J = 8.2 Hz, 2H), 5.91–5.75 (m, 1 zH), 5.59 (d, J = 6.8 Hz, 1H), 3.20–3.15 (m, 1H), 3.13–3.09 (m, 2H), 3.00 (dp, J = 6.2, 1.7 Hz, 2H), 2.89–2.82 (m, 1H), 1.14–0.79 (m, 1H). 13C NMR (101 MHz, DMSO‑d6) δ 174.4, 171.6, 164.6, 136.9, 132.1, 128.8, 128.4, 126.5, 125.9, 48.3, 47.0, 36.6, 10.1, 9.4, 2.9. HRMS (ESI): m/z [M + H]+ calcd for C19H16F3N2O4 393.1068, found: 393.1072.
4.1.14. Synthesis of impurity 1d
The synthesis process for Impurity 1d is shown in Scheme 9. To a solution of Maleic anhydride 3 (2 g, 0.02 mol) and compound 7 (4 g, 0.02 mol) in xylene (40 ml), the mixture was stirred and refluxed for 5 h. Upon observing completion of the reaction by TLC, the solvent was evaporated from the reaction mixture under vacuum, to give an off-white solid. The solid was further purified by column chromatography (PE: EA 10:1) to afford product Impurity 1d (4.7 g, 81 % yield, 99.06 % purity by HPLC). 1H NMR (400 MHz, DMSO‑d6) δ 11.37 (s, 1H), 8.14 (d, J = 8.1 Hz, 2H), 7.96 (d, J = 8.2 Hz, 2H), 7.28 (s, 2H). 13C NMR (101 MHz, DMSO‑d6) δ 168.5, 164.8, 134.4, 129.1, 126.3, 126.2. HRMS (ESI): m/z [M + H]+ calcd for C12H6F3N2O3 283.0336 found: 283.0346.
Funding statement
This work was supported by the Institute of Drug Innovation of the Chinese Academy of Sciences for research on antiviral protease inhibitors (Grant No. CASIMM120234003), the China-Uzbekistan New Drug, Belt and Road Joint Laboratory Construction and Innovative Drug Research for National Key R&D Program of China (Grant No. 2020YFE0205600), and the University of Chinese Academy of Sciences (UCAS) for the UCAS Scholarship for International students.
Data availability statement
The supplementary data associated with this article including copies of 1H NMR, 13C NMR, mass spectra, and additional data for all compounds are available in supporting information file.
CRediT authorship contribution statement
Emmanuel Mintah Bonku: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Hongjian Qin: Supervision, Methodology, Formal analysis, Conceptualization. Abdullajon Odilov: Methodology, Investigation. Safomuddin Abduahadi: Methodology, Investigation. Samuel Desta Guma: Methodology, Investigation. Feipu Yang: Supervision, Conceptualization. Xinglong Xing: Validation, Supervision, Formal analysis, Data curation, Conceptualization. Xukun Wang: Methodology, Investigation, Formal analysis. Jingshan Shen: Validation, Supervision, Resources, Project administration, Funding acquisition.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2024.e29559.
Contributor Information
Xinglong Xing, Email: xinglong.xing@vigonvita.cn.
Jingshan Shen, Email: shenjingshan@simm.ac.cn.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Tecovirimat, SIGA . European Medicines Agency; 2022. https://www.ema.europa.eu/en/medicines/human/EPAR/tecovirimat-siga [Google Scholar]
- 2.2018. https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation 2021.
- 3.Hughes D.L. Review of the patent literature: synthesis and final forms of antiviral drugs tecovirimat and baloxavir marboxil. Org. Process Res. Dev. 2019;23:1298–1307. [Google Scholar]
- 4.Funel J.A.A.S. Industrial applications of the Diels–alder reaction. Angew. Chem., Int. Ed. 2013;52(14):3822–3863. doi: 10.1002/anie.201201636. [DOI] [PubMed] [Google Scholar]
- 5.(a) McNamara O.A., Maguire A.R. The norcaradiene-cycloheptatriene equilibrium. Tetrahedron. 2011;67(1):9–40. [Google Scholar]; (b) Maier G. Das norcaradien‐problem. Angew. Chem. 1967;79(10):446–458. [Google Scholar]; (c) Reisman S.E., Nani R.R., Levin S. Buchner and beyond: arene cyclopropanation as applied to natural product total synthesis. Synlett. 2011;(17):2437–2442. [Google Scholar]; (d) Schwiebert K.E., Stryker J.M. Transition cetal-mediated [3+ 2+ 2] allyl/alkyne cycloaddition reactions. A new reactivity pattern for the synthesis of seven-membered carbocycles. J. Am. Chem. Soc. 1995;117(31):8275–8276. [Google Scholar]; (e) Etkin N., Dzwiniel T.L., Schweibert K.E., Stryker J.M. Cobalt-mediated intermolecular allyl/alkyne [3+ 2+ 2] cycloaddition reactions. A practical metal template for convergent synthesis of functionalized seven-membered rings. J. Am. Chem. Soc. 1998;120(37):9702–9703. [Google Scholar]; (f) Older C.M., McDonald R., Stryker J.M. Unprecedented coordination modes and demetalation pathways for unbridged polyenyl ligands. Ruthenium η, η4-cycloheptadienyl complexes from allyl/alkyne cycloaddition. J. Am. Chem. Soc. 2005;127(41):14202–14203. doi: 10.1021/ja0556023. [DOI] [PubMed] [Google Scholar]; (g) Tsukada N., Sakaihara Y., Inoue Y. Palladium-catalyzed cycloheptatriene formation by [3+ 2+ 2] cocyclization of 2-substituted allylic alcohols and alkynes. Tetrahedron Lett. 2007;48(23):4019–4021. [Google Scholar]; (h) Kamikawa K., Shimizu Y., Matsuzaka H., Uemura M. Stereoselective [3+ 2+ 2] cycloaddition utilizing optically active binuclear Fischer carbene complexes with alkynes. J. Organomet. Chem. 2005;690(24–25):5922–5928. [Google Scholar]
- 6.Winberg W. Synthesis of cycloheptatriene. J. Org. Chem. 1959;24(2):264–265. [Google Scholar]
- 7.Robinson G.C. Pyrolytic conversion of 7, 7-dichloronorcarane to cycloheptatriene. J. Org. Chem. 1964;29(11):3433–3434. [Google Scholar]
- 8.Parham W.S., Throckmorton J.R., Kuncl K., Dodson R.M. Reactions of enol ethers with carbenes. V. Rearrangements of dihalocyclopropanes derived from six-, seven-, and eight-membered cyclic enol Ethers1. J. Am. Chem. Soc. 1965;87(2):321–328. [Google Scholar]
- 9.(a) Jordan R., Bailey T.R., Rippin S.R., Dai D. Compounds, compositions and methods for treatment and prevention of orthopoxvirus infections and associated diseases. U.S. Patent. Sep 10, 2013;8 530,509 B2. [Google Scholar]; (b) Jordan R., Bailey T.R., Rippin S.R., Dai D.U.S. Patent. Aug 12, 2014;8:802. 714 B2. [Google Scholar]; (c) Tyavanagimatt S.R., Stone M.A.C.L., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. Polymorphic forms of ST-246 and methods of preparation. U.S. Patent Appl. 2011/0236434. Sep 29, 2011 [Google Scholar]; (d) Tyavanagimatt S.R., Anderson M.A.C.L.S., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. Polymorphic forms of ST-246 and methods of preparation. U.S. Patent Appl. 2016/0107993 A1. Apr 21, 2016 [Google Scholar]; (e) Tyavanagimatt S.R., Anderson M.A.C.L.S., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. U.S. Patent Appl. 2018/0193308. Jul 12, 2018 [Google Scholar]; (f) Tyavanagimatt S.R., Anderson M.A.C.L.S., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. U.S. Patent Appl. 2018/0311213 A1. Nov 1, 2018 [Google Scholar]; (g) Tyavanagimatt S.R., Anderson M.A.C.L.S., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A.U.S. Aug 14, 2018. Patent 9,744,154 B2, Aug 29, 2017. B2. [Google Scholar]; (h) Tyavanagimatt S.R., Anderson M.A.C.L.S., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. U.S. Patent 10,045,964 B2. Aug 14, 2018 [Google Scholar]; (i) Tyavanagimatt S.R., Stone M.A.C.L., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. Polymorphic forms of ST-246 and methods of preparation. U.S. Patent 9,339,466B2. May 17, 2016 [Google Scholar]; (j) Dong M.-x., Zhang J., Peng S.-q., Lu H., Yun L.-h., Jiang S., Dai Q.-y. Tricyclononene carboxamide derivatives as novel anti-HIV-1 agents. Eur. J. Med. Chem. 2010;45:4096–4103. doi: 10.1016/j.ejmech.2010.05.070. [DOI] [PubMed] [Google Scholar]; (k) Dai Q., Dong M., Hu J. Crystal Thereof and Preparation Method Thereof. Chinese Patent Appl. CN101445478; Jun 3, 2009. Compound ST-246 containing a crystal water. [Google Scholar]; (l) Wang H., Zhang D., Dou Y., Xu L., Guo Y., Sun Y., Bei Z., Yang Y., Dong P., Li L., Yang L. Dec 8, 2010. Preparation Method of ST-246 Stereoisomer, Chinese Patent Appl. CN101906064. [Google Scholar]; (m) Jordan R.B.T.R., Rippin S.R. 2005. Compounds, Compositions and Methods for Treatment and Prevention of Orthopoxvirus Infections and Associated Diseases; p. 56. Patent WO 2004/112718 A3. [Google Scholar]; (n) Dai D. Methods of preparing tecovirimat. U.S. Patent. Dec 18, 2018 10,155,723 B2. [Google Scholar]
- 10.Bailey T.R., Rippin S.R., Opsitnick E., Burns C.J., Pevear D.C., Collett M.S., Rhodes G., Tophan S., Huggins J.W., Baker R.O., et al. N-(3, 3a, 4, 4a, 5, 5a, 6, 6a-octahydro-1, 3-dioxo-4, 6-ethenocycloprop[2]isoindol-2-(1 H)-yl) carboxamides: identification of novel orthopoxvirus egress inhibitors. J. Med. Chem. 2007;50(7):1442–1444. doi: 10.1021/jm061484y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Tyavanagimatt S.R., Stone M.A.C.L., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. Polymorphic forms of ST-246 and methods of preparation. U.S. Patent Appl. 2011/0236434. Sep 29, 2011 [Google Scholar]; (b) Tyavanagimatt S.R., Anderson M.A.C.L.S., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. Polymorphic forms of ST-246 and methods of preparation. U.S. Patent Appl. 2016/0107993 A1. Apr 21, 2016 [Google Scholar]; (c) Tyavanagimatt S.R., Anderson M.A.C.L.S., Weimers W.C., Nelson D., Bolken T.C., Hruby D.E., O'Neill M.H., Sweetapple G., McCloughan K.A. U.S. Patent Appl. 2018/0193308. Jul 12, 2018 [Google Scholar]
- 12.Leitich J.S.G., Sprintschnik G. Verläuft die thermische Cycloaddition von Maleinsäureanhydrid an Cycloheptatrien, 7H Benzocyclohepten und 7H-Benzocyclohepten-7, 7-dicarbonitril nach [4π+ 2π] oder [2π+ 2π+ 2π] Chem. Ber. 1986;119(5):1640–1660. [Google Scholar]
- 13.International conferences on harmonization of technical requirements for registration of pharmaceuticals for human use, ICH harmonized tripartite guideline, impurities in new drug substances. 2006;Q3A(R2) [Google Scholar]
- 14.Bonku E.M., Qin H., Odilov A., Yang F., Xing X., Wang X., Shen J. Efficient large-scale process for tecovirimat via reactive distillation for the preparation of cycloheptatriene. Org. Process Res. Dev. 2023;27(11):1984–1991. [Google Scholar]
- 15.Elder D.P., Snodin D., Teasdale A. Control and analysis of hydrazine, hydrazides and hydrazones—genotoxic impurities in active pharmaceutical ingredients (APIs) and drug products. J. Pharmaceut. Biomed. Anal. 2011;54(5):900–910. doi: 10.1016/j.jpba.2010.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Ma L., Ma Y.-N., Chen Z., Jiang Y. Structural alerts of genotoxic impurities. Chinese Journal of New Drugs. 2014;23:2106–2111. [Google Scholar]
- 17.Benigni R., Bossa C., Tcheremenskaia O., Worth A. 2009. Development of Structural Alerts for the in Vivo Micronucleus Assay in Rodents. [Google Scholar]
- 18.(a) Snodin D.J. Genotoxic impurities: from structural alerts to qualification. Org. Process Res. Dev. 2010;14(4):960–976. [Google Scholar]; (b) Sawatari K., Nakanishi Y., Matsushima T. Relationships between chemical structures and mutagenicity: a preliminary survey for a database of mutagenicity test results of new work place chemicals. Ind. Health. 2001;39(4):341–345. doi: 10.2486/indhealth.39.341. [DOI] [PubMed] [Google Scholar]
- 19.von E Doering W., Hoffmann A.K., Hoffmann A.K. The addition of dichlorocarbene to olefins. J. Am. Chem. Soc. 1954;76(23):6162–6165. [Google Scholar]
- 20.Winberg H. Synthesis of cycloheptatriene. J. Org. Chem. 1959;24(2):264–265. [Google Scholar]
- 21.Bhavana V., Srinivasa Rao Y. A review on analysis of genotoxic impurities. Int. J. Sci. Res. 2018;9 SR20808120645. [Google Scholar]
- 22.(a) ICH Q3B (R2) 2006. Impurities in New Drug Products. [Google Scholar]; (b) ICH Q3C (R6 . 2006. Impurities: Guidelines for Residual Solvents. [Google Scholar]; (c) Ich M.7. Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit the potential carcinogenic risk. Guidance for Industry. 2014 [Google Scholar]
- 23.Nair A.S., Singh A.K., Kumar A., Kumar S., Sukumaran S., Koyiparambath V.P., Mathew B. FDA-approved trifluoromethyl group-containing Drugs: a review of 20 years. Processes. 2022;10(10):2054. [Google Scholar]
- 24.Kim E.J., Kim J.H., Kim M.S., Jeong S.H., Choi D.H. Process analytical technology tools for monitoring pharmaceutical unit operations: a control strategy for continuous process verification. Pharmaceutics. 2021;13(6):919. doi: 10.3390/pharmaceutics13060919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The supplementary data associated with this article including copies of 1H NMR, 13C NMR, mass spectra, and additional data for all compounds are available in supporting information file.