

Summary
Tissue clearing is an essential prerequisite for 3D volumetric imaging of larger tissues or organs. Here, we present a detailed protocol for optical, aqueous-based clearing of adult murine tissues using EZ Clear. We describe steps to ensure successful perfusion and fixation of organs from the adult mouse and supply guidelines for optimal lipid removal, refractive index matching, and tissue clearing. Finally, we provide imaging parameters for visualizing both exogenous perfused fluorescent dyes and endogenous fluorescence reporters in the adult mouse.
For complete details on the use and execution of this protocol, please refer to Hsu et al.1
Subject areas: Developmental biology, microscopy, Model Organisms, Neuroscience
Graphical abstract
Highlights
-
•
Whole-mount clearing and refractive index matching for adult mouse tissues using EZ Clear
-
•
Instructions for reliable removal of lipids via solvent exchange
-
•
Rapid and robust tissue clearing compatible with lightsheet imaging
-
•
Technique applicable to multiple mouse tissue types
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Tissue clearing is an essential prerequisite for 3D volumetric imaging of larger tissues or organs. Here, we present a detailed protocol for optical, aqueous-based clearing of adult murine tissues using EZ Clear. We describe steps to ensure successful perfusion and fixation of organs from the adult mouse and supply guidelines for optimal lipid removal, refractive index matching, and tissue clearing. Finally, we provide imaging parameters for visualizing both exogenous perfused fluorescent dyes and endogenous fluorescence reporters in the adult mouse.
Before you begin
Before beginning this EZ clearing protocol, multiple parameters must be optimized, including tissue dissection, fixation, and sample mounting for imaging. For example, many adult mouse tissues require 8–12 h of fixation for preserving tissue architecture and fluorescent signal. However, the murine eye requires a brief 1-h fixation period. Time in fixative, and the reagent(s) used for fixation, must be balanced against the wavelength(s) to be imaged, and any autofluorescence created by use of the fixative. Moreover, how mounting will impact refractive index matching and possible image acquisition of the region of interest must be considered, as how a tissue is immobilized may affect how the lightsheet illuminates the sample, as well as the tissue depth that the lightsheet must pass through when imaging. For example, will the tissue be adhered directly to a sample holder via superglue, or embedded in agarose and depressed from a syringe to the imaging chamber? After optimizing these conditions, all reagents should be freshly prepared just prior to beginning this protocol. Ensure that the refractive index of all relevant solutions (i.e., EZ View) is confirmed using a refractometer and that all solutions containing Nycodenz are at room temperature to prevent components from precipitating out of the solution. The protocol below describes the specific steps, sample preparation conditions, and collection parameters for imaging a whole mouse brain, as well as multiple other mouse tissues, using lightsheet fluorescence microscopy, as originally reported in Hsu et al., Elife, 2022.1
Institutional permissions
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animal research was conducted according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) (AN-6680 and AN-8238) at Baylor College of Medicine and the ACUC at the University of Virginia School of Medicine (4446-06-23).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, peptides, and recombinant proteins | ||
| Sodium phosphate, dibasic, 98%+, extra pure, anhydrous. Na2HPO4 (HNa2O4P). FW = 141.96 | Sigma | CAS # 7558-79-4 # 567545 |
| Sodium phosphate, monobasic NaH2PO4·H2O. FW = 119.98 |
Sigma | CAS # 7558-80-7 #S0751 |
| Phosphate-buffered saline (PBS), 10X, pH 7.4. 0.119 M Phosphates, 1.37 M NaCl, 0.027 M KCl |
Fisher | # BP399-20 |
| Tetrahydrofuran (THF), anhydrous, 250 ppm BHT added C3H8O. FW = 72.11 |
Sigma-Aldrich | CAS # 109-99-9 # 186562 |
| Urea (NH2CONH2). FW = 60.06 | Sigma | CAS # 57-13-6 #U5378 |
| Sodium azide; hydrazoic acid sodium salt (NaN3) ≥99.5%. FW = 65.01 | Sigma | CAS # 26628-22-8 #S2002 |
| Nycodenz iohexol, C19H26I3N3O9. FW = 821 | Serumwerk Bernburg | CAS # 66108-05-0 (ProGen cat # 18003) |
| Paraformaldehyde (PFA), (HO(CH2O)nH). FW = 30.03 (as a monomer) | Sigma | CAS # 30525-89-4 #P6148 |
| Sodium hydroxide (NaOH). FW = 40 | Alfa Aesar | CAS # 1310-73-2 #13455 |
| Hydrochloric acid (HCl). FW = 36.46 | Sigma | CAS # 7647-01-0 #H1758 |
| Triethylamine | Sigma | CAS # 121-44-8 #471283 |
| Agarose (standard melting point) | VWR Life Science | CAS # 9012-36-6 #0710 |
| ddH2O | Milli-Q quality | |
| Isoflurane | Covetrus | #029405 |
| Lycopersicon Esculentum (Tomato) Lectin (LEL, TL), DyLight 488 | Vector Laboratories | #DL-1174-1 |
| Lycopersicon Esculentum (Tomato) Lectin (LEL, TL), DyLight 649 | Vector Laboratories | #DL-1178-1 |
| Evans blue | Millipore-Sigma | Cat#: E2119 |
| Other | ||
| Dissection tools (scissors and forceps) | Any | |
| pH meter | Any | |
| WHEATON liquid scintillation vial with attached cap (20 mL) | Millipore-Sigma | #DWK986546 |
| 5 mL disposable plastic syringe(s) | Any | |
| 10 mL disposable plastic syringe(s) | Any | |
| Insulin syringe(s) | BD | #328438 |
| 22G needle(s) | McKesson | #16-N22105 |
| 27G needle(s) | BD | #305109 |
| Loctite Super Glue Ultra Gel Control | Henkel Corp. | #1801751 en-US from any vendor |
| The Translucence specimen holder (magnetic sample holder) | Translucence Biosystems | (https://www.translucencebio.com/mesoscale-imaging-system) |
| Refractometer | Atago | PAL-RI 3850 from any vendor |
| VWR vacuum filtration systems, 0.45 μm pore size | VWR | #76010-408 |
| Fisherbrand syringe filters, 0.45 μm pore size | Fisher Scientific | #09-720-4 |
| Epredia Peel-A-Way disposable embedding molds (22 mm by 22 mm by 20 mm) | Fisher Scientific | #22-19 |
| Lightsheet fluorescence microscope sample holder | Instrument specific | |
| Belly dancer shaker | Stovall Life Science | USBdbo from any vendor |
| NE-300 Just Infusion syringe pump | New Era Pump Systems | #300 |
| Imaris file converter | Oxford Instruments | Version:10.0.1 |
| Imaris Stitcher | Oxford Instruments | Version:10.0.0 |
| Imaris | Oxford Instruments | Version:10.0.1 |
Materials and equipment
CRITICAL: Tetrahydrofuran (THF) is highly flammable. THF is toxic and all steps involving THF should be performed in a fume hood. Avoid inhalation and contact with skin or eyes. This polar solvent will degrade plastic and should only be stored in glass containers.
1 M NaH2PO4 (monobasic) stock solution (1 L).
-
•
Add 120 g of NaH2PO4·H2O to 800 mL ddH2O and mix with a stir bar on a magnetic stirrer until the solids dissolve.
-
•
Raise the solution (i.e., quantum satis or q.s.) to a final volume of 1 L with ddH2O.
-
•
Filter sterilize with a vacuum filtration system (<0.45 μm) and store at 4°C for up to 6 months.
1 M Na2HPO4 (dibasic) stock solution (1 L).
-
•
Add 142 g of Na2HPO4 (HNa2O4P) to 800 mL ddH2O and mix with a stir bar on a magnetic stirrer until the solids dissolve.
-
•
Q.s. to a final volume of 1 L with ddH2O.
-
•
Filter sterilize with a vacuum filtration system (<0.45 μm) and store at 4°C for up to 6 months.
0.1 M Phosphate Buffer (PB) (1 L)
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M NaH2PO4 (monobasic) | 0.0774 M | 22.6 mL |
| 1 M Na2HPO4 (dibasic) | 0.0226 M | 77.4 mL |
| ddH2O | N/A | Add to 1 L |
-
•
Mix 22.6 mL of 1 M NaH2PO4 and 77.4 mL of 1 M Na2HPO4 in 800 mL of ddH2O using a stir bar and a magnetic stirrer.
-
•
Q.s. to a total volume of 1 L with ddH2O.
-
•
Filter sterilize with a vacuum filtration system (<0.45 μm) and store at 4°C for up to 1 month.
Alternative for 0.1 M Phosphate Buffer from powders (PB) (1 L)
| Reagent | Final concentration | Amount |
|---|---|---|
| ddH2O | N/A | 900 mL |
| NaH2PO4 (monobasic) | 0.0258 M | 3.1 g |
| Na2HPO4 (dibasic) | 0.0767 M | 10.9 g |
| ddH2O | N/A | Add to 1 L |
-
•
Mix 3.1 g of NaH2PO4 and 10.9 g of Na2HPO4 in 900 mL of ddH2O.
-
•
Add additional ddH2O to a total volume of 1 L.
-
•
pH should be 7.4 (if made correctly).
-
•
Filter sterilize (<0.45 μm) and store at 4°C for up to 1 month.
0.02 M Phosphate Buffer (PB) (500 mL).
-
•
Add 100 mL of 0.1 M PB to 350 mL ddH2O and mix using a stir bar and magnetic stirrer.
-
•
Q.s. to a total volume of 500 mL.
-
•
Filter sterilize (<0.45 μm) and store at 4°C for up to 1 month.
1X PBS (100 mL).
-
•
Add 100 mL of 10X PBS to 850 mL of ddH2O and mix using a stir bar and magnetic stirrer.
-
•
Q.s. to a total volume of 500 mL.
-
•
Filter sterilize (<0.45 μm) and store at 20°C–25°C for up to 1 month.
10 N Sodium hydroxide (NaOH) solution (1 L).
-
•
Add 400 g of NaOH pellets to 800 mL of ddH2O and mix using a stir bar and magnetic stirrer.
-
•
Q.s. to a total volume of 1 L using ddH2O.
-
•
Store at 20°C–25°C for up to 2 months.
4% paraformaldehyde (PFA) (500 mL)
| Reagent | Final concentration | Amount |
|---|---|---|
| ddH2O | N/A | 450 mL |
| PFA | 4% | 20 g |
| 10 N NaOH | N/A | A few drops to dissolve PFA powder |
| HCl (36.5%–38%) | N/A | A few drops needed to adjust pH to 7.2 |
| 10X PBS | 1X PBS | 50 mL |
-
•
Remove a container of paraformaldehyde powder stored at 4°C and allow the reagent to equilibrate to room temperature for 20–30 min. This step is necessary to prevent condensation from depositing water into the powder.
-
•
Set a hotplate with stirrer functionality to 60°C in a chemical hood with adequate exhaust and then add 425 mL of ddH2O to a 1000 mL glass beaker. Place a clean, autoclaved stir bar in the beaker. Place the beaker on the stir plate and then set the stirrer to 300 RPM.
-
•
Working in a fume hood, while wearing appropriate PPE (i.e., mask, eye protection, lab coat, and gloves), carefully weigh 20 g of paraformaldehyde powder into a plastic weigh boat, then add the paraformaldehyde to the 425 mL of ddH2O in the glass beaker on the stirring hotplate. Mix well using the magnetic stirrer.
-
•
Using a disposable glass transfer pipette, slowly add 10 N NaOH, dropwise, to the stirring paraformaldehyde solution. After a few drops, the solution will begin to clear. Adding too much 10 N NaOH will increase the amount of HCl needed to adjust the final pH to 7.2, so add as little 10 N NaOH as possible to clear the solution.
-
•
After the solution has cleared, transfer the beaker from the hotplate to stirrer at room temperature (to allow the mixture to start to cool to room temperature) and add 50 mL of 10X PBS. Mix well using the magnetic stirrer.
-
•
Adjust the pH of the solution to 7.2 by adding HCl, dropwise, using a glass transfer pipette.
-
•
Adjust the final volume of the solution to 500 mL using ddH2O.
-
•
Filter sterilize (<0.45 μm).
-
•
Aliquot 4% PFA to 50 mL or 15 mL plastic conical tubes.
-
•
Store at −80°C for up to three months.
Note: When making the aliquots for freezing back, do not fill the entire conical tube, as the liquid will expand during freezing and push the lid off.
Note: Ensure that 4% paraformaldehyde (or whatever fixative is used) is either freshly made, or thawed from a frozen stock, immediately prior to tissue fixation.
Alternative: You may add 20 g of room temperature PFA powder to 450 mL of 1X PBS (made by diluting a commercial 10X PBS stock, such as from Thermo Fisher Scientific (Cat # 70011044) in ddH2O) and then adjust the final volume of the solution to 500 mL using 1X PBS.
Lipid removal solution
-
•
Prepare 50%, 70%, 80% and 100% (v/v) tetrahydrofuran (THF): ddH2O solutions by mixing the appropriate volume of tetrahydrofuran with ddH2O in a glass beaker, being sure to do this in a well-ventilated chemical fume hood. Prepare the solution fresh before each experiment.
-
•
Use a clean spatula to stir the solution and make sure it is thoroughly mixed.
-
•
Store at 20°C–25°C for no longer than 1 day.
Note: THF is a polar solvent and it will dissolve a wide range of polar and non-polar compounds, and these dissolved compounds will make THF-containing solutions cloudy and interfere with downstream imaging. Thus, all THF solutions should be prepared and stored only in glass beakers and glass scintillation vials. Avoid making THF-containing solutions in plasticware such as petri dishes, multi-well sample plates, or polypropylene 15 mL or 50 mL conical tubes.
Note: The recipes for THF solutions can be scaled up or down, but the container they are stored in must be as small as possible to prevent oxidation of the solution.
EZ View Clearing Solution (125 mL)
| Reagent | Final concentration | Amount |
|---|---|---|
| 0.02 M Phosphate buffer | 0.0056 M | 35 mL |
| Urea (NH2CONH2) | 7 M | 52.5 g |
| Sodium azide (NaN3) | 0.025% | 31.25 mg |
| Nycodenz iohexol (C19H26I3N3O9) | 80% | 100 g |
-
•
Place a stir bar in a 250 mL glass beaker.
-
•
Pour 35 mL of 0.02 M PB into the beaker.
-
•
Weigh out 52.5 g of urea in a plastic weigh boat and then add this to the 35 mL PB in the glass beaker. Add a magnetic stir bar to the mixture, then place the beaker on a magnetic stirrer.
-
•
Carefully weigh out 31.25 mg of sodium azide and then add this to the mixture. Stir to mix.
-
•
Gently heat the mixture to 37°C and stir (150–250 RPM) until the urea and sodium azide dissolve completely. This should not take more than 15 min.
Note: Urea dissolving in water is an exothermic reaction, meaning the solution absorbs heat from its surroundings as the reaction proceeds. Gently heating up the solution will accelerate the process and prevent the urea from crystalizing in the solution. However, do not heat the solution above 37°C.
-
•
Weigh out 100 g of Nycodenz iohexol in a plastic weigh boat.
-
•
Gradually add Nycodenz iohexol to the PB/Urea/sodium azide solution, 10 g at a time, every 1–2 min, while stirring (at 150–250 RPM), until all 100 g of Nycodenz iohexol have been added. This should take approximately 10–15 min.
-
•
When adding Nycodenz powder to the liquid mixture, prevent clumping and aggregation by turning the stirrer to a setting that promotes vigorous mixing (e.g., > 300 RPM).
-
•
Cover the glass beaker in aluminum foil and allow the solution stir at 37°C (over 300 RPM, or enough to cause vortex stirring). Without vigorous stirring the upper layer will crystalize and the Nycodenz will not fully dissolve. Stirring for 8–12 h is recommended, with aluminum foil covering the top to prevent evaporation. The solution should be clear the following morning.
-
•
Pour the solution into a graduated cylinder and adjust the final volume to 125 mL with additional 0.02 M PB.
-
•
Pour the mixture back into the beaker and continue to stir until the mixture is completely clear (or only has a few particles) and has gone into solution.
-
•
Filter the solution with a 0.45 μm vacuum filtration system.
-
•
Measure the refractive index with a refractometer. The RI should be between 1.512 and 1.519.
-
•
Store the EZ View Clearing Solution at 20°C–25°C for up to 2 months.
Note: This recipe can be scaled up or down to accommodate any sample size or volume.
Note: If a precipitate is seen, the solution can be stored at 32°C–37°C for up to 2 months.
1% agarose
-
•
Add 1 g of agarose powder to 99 mL of ddH2O in a glass beaker or flask.
-
•
Heat the mixture in a microwave with a power setting of “10” for 30 s increments until it boils to dissolve the agarose powder.
-
•
Once the mixture has cooled to approximately 60°C, filter it to remove any debris that may scatter light during imaging. We typically use a 0.45 μm syringe filter system and store the filtered solution in a 50 mL conical tube. Seal the lid on the conical tube and place the tube in a 42°C water bath until ready to mount the sample to prevent the solution from crystalizing.
Note: Depending on the age and manufacturer, different microwaves may require different settings in terms of power or time for boiling the agarose. These parameters must be empirically determined, so be attentive during the first attempts to boil the solution to ensure the mixture does not boil over. Proceeding in 30 second intervals, with power set to 70%, is a conservative approach to achieve boiling if you are unsure of the power of your microwave.
Step-by-step method details
Perfusion and tissue harvest
Timing: Approximately 38 min to 1 h
Perfusion of an adult mouse requires roughly 3 min for anesthesia, 1 min for retroorbital injection, 5 min for circulation of the dye, then 24 min for transcardiac perfusion, and between 5‒25 min to dissect and remove the adult mouse brain. However, these times are approximations and the time required for successful perfusion and tissue harvest vary greatly depending on the volume of fixatives and lectin that are perfused, as well as the dissection skills of the individual.
Whole animal perfusion with fluorescent dye-labeled tomato lectin (Lycopersicon esculentum agglutinin), which binds to poly-N-acetyl lactosamine-type oligosaccharide moieties displayed on the surface of vascular endothelial cells in mice,2,3,4 requires two steps: 1) retro-orbital injection with a small volume of lectin to label the capillary vessels and the venous circulation, and 2) transcardiac perfusion with a larger volume of dye to label large diameter vessels and the arterial circulation (see Figures 1A and 1B).
-
1.Retro-orbital injection of fluorescent lectin.
-
a.Prepare one insulin syringe by filling with 50 μL of the desired contrast agent (e.g., lectin-649).
-
b.Transfer an adult mouse from its home cage to a secure anesthesia induction chamber (approximately 1 L in volume for an adult mouse).
-
c.Turn on the oxygen flow meter to a flow rate of 1–2 L/min.
-
d.Begin the flow of isoflurane, gradually increasing from 0.5% to a final concentration of 4% for induction of anesthesia.
-
e.Once the animal has lost its righting reflex, and its breathing has become deeper and slower, open the induction chamber and transfer the rodent (supporting its entire body and not just grabbing it by the tail) to a tight-fitting nose cone (see Figure 1C).
-
f.Adjust the isoflurane to 2%–3% to maintain anesthesia.
-
g.After waiting approximately 3 min, confirm the plane of anesthesia by absence of the toe pinch reflex.
-
h.Place the mouse on its side and secure the animal using the thumb and middle finger of your non-dominant hand, taking care not to press the neck and/or trachea.
-
i.Apply lubricant to the animal’s eye to prevent it from dehydrating (see Figure 1D).
-
j.Using the index finger and thumb of your non-dominant hand, draw back the skin above and below the orbital socket so that the eye slightly protrudes (see Figure 1E).
-
k.Insert the needle of the syringe prefilled with lectin, at an angle of approximately 45°, and pull slightly on the plunger to suction a visible amount of blood into the syringe.Note: This visual check ensures that the needle is inserted properly.
-
l.Inject the entire contents (50 μL) into the retro-bulbar sinus vein.Note: Wait about 3–5 seconds after expelling the contents of the syringe before proceeding to the next step.
-
m.Gently remove the needle, taking care to not injure the eye and the eyelid.
-
n.Examine the injection site for swelling or trauma.
-
o.If no adverse effects are observed, return the mouse to a clean cage and closely monitor the rodent to ensure full recovery from anesthesia to an alert an ambulatory state.
-
p.Allow the lectin to circulate for a minimum of 5 min before proceeding to the next step.Note: Signs of an unsuccessful retroorbital injection include the dye (or blood) coming out of the eye socket. In this case, prepare and inject the contralateral eye.Note: If rodents are maintained under anesthesia for longer than 5 min, consider providing thermal support (e.g. thermal discs under the cage, etc.) to avoid inducing hypothermia.
-
a.
-
2.Transcardiac perfusion.
-
a.Fill an insulin syringe with the same fluorescent dye used for the retroorbital perfusions (e.g., lectin-649).Note: We use approximately 100 μL to ensure adequate labeling of large diameter vessels (i.e., the middle cerebral artery in the brain, etc.).
-
b.Fill one 10 mL syringe with 10 mL of 20°C–25°C 1X PBS.
-
c.Fill a second 10 mL syringe with 10 mL of ice-cold (4°C) 4% PFA.
-
d.Transfer the adult mouse from its home cage to a secure anesthesia induction chamber (approximately 1 L in volume for an adult mouse).
-
i.Turn on the oxygen flow meter to a flow rate of 1–2 L/min.
-
ii.Begin the flow of isoflurane, gradually increasing from 0.5% to a final concentration of 4% for induction of anesthesia.
-
iii.Once the animal has lost its righting reflex, and breathing has become deeper and slower, open the induction chamber and transfer the rodent (supporting its entire body and not just grabbing it by the tail) to a tight-fitting nose cone (see Figure 1C).
-
iv.Adjust the isoflurane to 2%–3% to maintain anesthesia.
-
v.After waiting approximately 3 min, confirm the plane of anesthesia by absence of the toe pinch reflex.
-
i.
-
e.Transfer the mouse to a Styrofoam board and secure it, abdomen/ventral side toward the experimenter, using either 22G needles or pins through the forelimb and hindlimb paws (see Figure 1F).
-
f.Spray the chest with 70% ethanol, wetting down the animal’s fur to avoid contaminating the surgical field.
-
g.Using sharp dissection scissors, cut through the dermis and peritoneum at the bottom of the chest (see Figures 1F and 1G).
-
h.Continue cutting anteriorly toward the diaphragm and continuing to cut through the rib cage, exposing the lungs and the heart (see Figure 1H).
-
i.Using your non-dominant hand, move the rib cage to gain clear access to the heart.Note: If desired, clamp the bottom of the rib cage with a hemostat, and then set the hemostat away from you to reflect the rib cage and keep the field clear.
-
j.Once the heart is accessible, insert the needle of a syringe preloaded with fluorescent lectin into the left ventricle (see Figure 1I).CRITICAL: Insert the needle at a shallow angle, straight or parallel to the interventricular septum (or the midline of the heart), to avoid puncturing the interventricular septum and inserting the syringe into the right ventricle (which will lead to perfusion of the lungs and inadequate labeling of the large diameter arteries of the brain and other tissues).
-
k.Slowly inject all of the contrast agent.
-
l.Wait for 1 min, leaving the needle inserted in the left ventricle and the heart is still pumping, to ensure circulation of the fluorescent dye.
-
m.Withdraw the needle.
-
n.Next, using dissection scissors, cut the apex of the right atrium.CRITICAL: This step is essential for adequate exsanguination, as it allows blood to exit the circulation.
-
o.Gently pierce the left ventricular wall of the heart, as in Step 12, with a 27G needle affixed to a syringe preloaded with 10 mL of 1X PBS connected to an injection pump.CRITICAL: Take care not to pierce the interventricular septum.
-
p.Holding the needle in place, turn on the syringe pump at a rate of 4 mL/min and perfuse 10 mL of 1X PBS through the animal.Note: This removes all blood (which causes autofluorescence) and excess lectin.
-
q.Turn off the flow pump.
-
r.Leaving the needle in place, gently remove the now empty syringe by twisting it from the Luer lock.
-
s.Carefully (do not disturb the needle inserted into the heart) connect another syringe loaded with freshly prepared, ice-cold, 4% PFA.
-
t.Turn on the pump, and perfuse the mouse with approximately 10 mL of 4% PFA at a flow rate of approximately 4 mL/min.CRITICAL: Delivery of insufficient or small volumes of fixative with result in poor tissue fixation and will negatively impact downstream imaging.Essential: A successful perfusion is essential for optimal sample preparation and subsequent tissue clearing. Signs of a successful transcardiac perfusion include the liver blanching, as blood is replaced with PBS and heme is removed. It should turn color from a deep red to a pale pink. The tail curling and whole-body tremors or twitching (due to fixation of nerves and muscle) are also signs of a successful perfusion.
-
a.
-
3.
Alternative: Retro-orbital and transcardiac injection of Evans Blue.
Evans blue dye is an affordable alternative to expensive fluorescent dyes and tracers, as it robustly labels the endothelium following intravenous administration.3 When Evans blue, a non-cell permeable dye, binds to albumin, it undergoes a conformational shift that produces fluorescence in the far-red spectrum (excitation at 620 nm, emission at 680 nm).5,6,7 We have previously shown that Evans blue dye labeling of the vasculature is compatible with EZ Clear, and it has an excellent signal to noise ratio in the far-red spectrum that is ideal for lightsheet imaging.1-
a.Fill a 31G needle and insulin syringe with approximately 100 μL of Evans Blue solution.Note: This Evans blue solution is 2% Evans blue (w/v) in 0.9% NaCl (w/v) in sterile Milli-Q water, then filter sterilized (0.45 μm), as in Honeycutt and O’Brien, 2021.7
-
b.Transfer the adult mouse from its home cage to a secure anesthesia induction chamber (approximately 1 L in volume for an adult mouse).
-
i.Turn on the oxygen flow meter to a flow rate of 1–2 L/min.
-
ii.Begin the flow of isoflurane, gradually increasing from 0.5% to a final concentration of 4% for induction of anesthesia.
-
iii.Once the animal has lost its righting reflex, and its breathing has become deeper and slower, open the induction chamber and transfer the rodent (supporting its entire body and not just grabbing it by the tail) to a tight-fitting nose cone (see Figure 1C).
-
iv.Adjust the isoflurane to 2%–3% to maintain anesthesia.
-
v.After waiting approximately 3 min, confirm the plane of anesthesia by absence of the toe pinch reflex.
-
i.
-
c.Place the mouse on its side and secure the animal using the thumb and middle finger of your non-dominant hand, taking care not to press the neck and/or trachea.
-
d.Apply lubricant to the animal’s eye to prevent it from dehydrating (see Figure 1D).
-
e.Using the index finger and thumb of your non-dominant hand, draw back the skin above and below the orbital socket so that the eye slightly protrudes (see Figure 1E).
-
f.Insert the needle of the prefilled syringe, at an angle of approximately 45°.
-
g.Pull slightly on the plunger to suction a visible amount of blood into the syringe.Note: This visual check ensures that the needle is inserted properly.
-
h.Inject the entire contents (50 μL) into the retro-bulbar sinus vein.Note: Wait about 3–5 seconds after expelling the contents of the syringe before proceeding to the next step.
-
i.Gently remove the needle, taking care to not injure the eye and the eyelid.
-
j.Examine the injection site for swelling or trauma.
-
k.If no adverse effects are observed, return the mouse to its home cage, allowing the Evans blue to circulate for a minimum of 5 min before proceeding to the next step.Note: Signs of an unsuccessful retroorbital injection include the dye (or blood) coming out of the eye socket. In this case, prepare and inject the contralateral eye.
-
l.Repeat Step 9 to anesthetize the animal.
-
m.Immobilize the animal on a Styrofoam board, as described in Step 10.
-
n.Expose the heart, as described in Step 11.
-
o.Using a 31G insulin syringe, inject 100–150 μL of Evans blue solution through the left ventricle, slowly over 1 min.Note: Similar to lectin, injection of Evans blue is done by hand and does not require the use of injection pumps.
-
p.Leave the needle in place for an additional minute to prevent the Evans blue dye from leaking out of the heart.
-
q.Remove the needle and allow the dye to circulate until the hindlimbs, tail, and nose turn blue, or approximately 5 min.CRITICAL: Do not perfuse with 1X PBS at this time. This would flush the Evans blue dye, which is bound to serum albumin, from the circulation and no fluorescence would be retained.
-
r.Humanely euthanize the animal (while it is still under anesthesia) by a secondary method, such as cervical dislocation.Note: Unlike the lectin perfusion method, where bilateral thoracotomy and collapsing the lungs serves as the primary method and exsanguination and fixation serves as the secondary method, for Evans blue perfusion a secondary method is required to ensure the animal is humanely euthanized.
-
s.Dissect and remove the tissue of interest and immerse it in ice-cold 1X PBS.CRITICAL: After removing the tissue of interest from the animal, we strongly recommend confirming the success of the perfusion using an epifluorescence microscope (see Figures 1J and 1K). If appropriate anatomical landmarks are not visibly labeled, for instance if the Circle of Willis or basilar artery are not labeled following lectin perfusion, then the experiment should be stopped.Note: A detailed protocol for brain dissection can be found in Meyerhoff J et al., J. Vis Experiments, 2021.8 When harvesting a mouse brain, ensure that the dissection is performed carefully to avoid damage/loss to the olfactory bulb region or disruption of any surface tissue(s) of the organ of interest, such as the pial vessels on the surface of the brain, the basilar artery near the brain stem, or damage to vessels in the Circle of Willis when removing the leptomeninges.
-
a.
Figure 1.

Workflow for retro-orbital and transcardiac perfusions
A schematic of retro-orbital (A) and transcardiac (B) perfusion. The nose cone should completely cover the nose and it takes about 3 min until the mouse is anesthetized (C) An eye lubricant is applied to prevent the injected eye from being dehydrated (D) and a desired contrast agent should be injected at an angle of approximately 45° and the needle should be removed without causing any disturbance in the surrounding tissues (E) The red line in panel F indicates an insertion line to cut open the chest and to expose the heart by puncturing diaphragm and cutting through the rib cage (G and H) Insert the pre-loaded syringe into the left ventricle without puncturing into the right ventricle (I) A successful perfusion can be evaluated by examining how evenly the fluorescent signal is distributed across the mid cerebral arteries (MCA) in the left and right hemispheres from a dorsal view (J) and the labeling of the circle of Willis (C.o.W.) and basilar artery (B.A.) from a ventral view (K) ob = olfactory bulb, crb = cerebellum. (Scale bar for J and K = 1000 μm).
Post fixation/clearing preparation
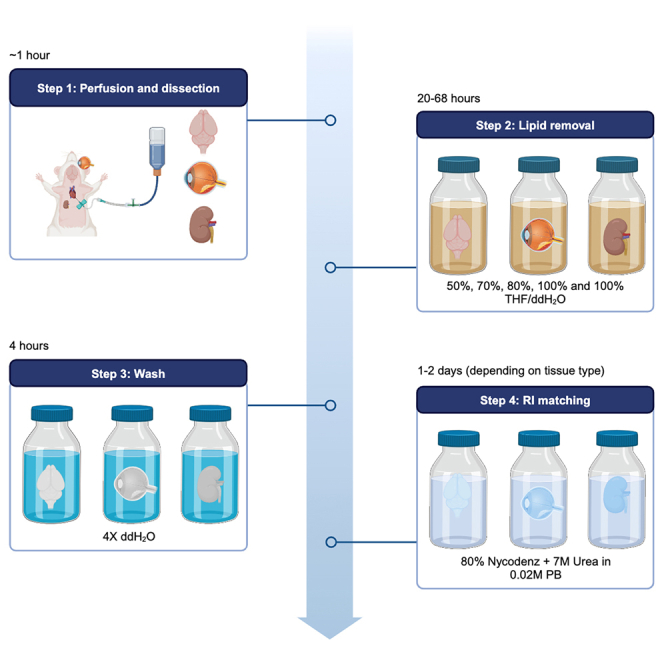
This step ensures adequate preservation of the tissue of interest and integrity of the tissue will be maintained throughout the clearing process. An overview of the entirety of the EZ Clear protocol is shown in Figure 2.
-
4.
Fix the tissue in 4% PFA.
Note: For the adult mouse brain, incubation for 24 h at 4°C is adequate.
Note: For this protocol, we use a setting of “4” on a Belly Dancer orbital shaker to promote solution exchange and washing with gentle agitation.
-
5.
Wash the tissue 3 times in 1X PBS, for 30 min each wash (a total of 90 min washing), at 20°C–25°C with gentle agitation on an orbital shaker.
-
6.
Store the sample at 4°C, shielded from light, in 0.05% sodium azide in 1X PBS to prevent bacterial growth and contamination.
Pause point: This is a stopping point.
Figure 2.

Logic flow of the EZ Clear protocol
Delipidation times may vary depending on the age, genotype and/or disease state. Since lightsheet imaging is time-consuming, the first quality check should be verifying optical transparency using the grid paper and transmitted phase microscopy. Once optical transparency is achieved, the ultimate quality control is determined by lightsheet imaging. We recommend additional delipidation after re-equilibrating in 1X PBS for 8–12 h to improve the lightsheet imaging quality.
Lipid removal
This step removes lipids via solvent exchange, which is required for the future tissue clearing steps.
-
7.
Submerge the sample in a volume of 50% tetrahydrofuran (THF) solution such that the sample is submerged/completely covered.
Note: For an adult mouse brain, place the sample in a 25 mL scintillation vial and then fill the vessel with 50% THF, leaving as little head space as possible to prevent oxidation of the sample.
Note: Be sure to use a glass vessel due to the strong polar solvent properties of THF.
-
8.
Place the sample on an orbital shaker in a vented chemical fume hood.
-
9.
Incubate, with gentle agitation, for 20 h at 20°C–25°C.
Note: Delipidation may take longer if the tissue of interest is lipid rich. If the tissue of interest remains opaque after this initial 20-hour step and refractive index (RI) matching in EZ View solution, re-equilibrate the tissue in 1X PBS for 8–12 h and gradually increase the THF concentration and incubation time for additional delipidation. For serial delipidation, we suggest washing in 50% THF: ddH2O, then 70%, 80%, and 100% THF (2X), for 12 h each wash. This serial delipidation can be stopped at any point and the sample moved to RI matching solution to determine the optimal delipidation duration and THF concentration.
-
10.
Gently aspirate the THF solution, using a glass transfer pipette, being careful not to disturb the sample. Discard the used THF solution in an approved chemical waste container.
-
11.
Wash the delipidated tissue 4 times in 20 mL of milli-Q ddH2O at 20°C–25°C, for 1 h each wash, to remove any remaining THF (a total of 4 h for washing).
-
12.
After the final wash, remove as much ddH2O as possible, being careful not to disturb the sample.
-
13.
Leave the vial open in a vented chemical fume hood for an additional 10 min to promote further evaporation of any residual THF solution.
-
14.
Proceed to mount the sample, or store the delipidated brain in 0.05% sodium azide in 1X PBS at 4°C until ready to proceed.
Note: During the delipidation process, increasing the pH of the THF solution may decrease autofluorescence.9 Depending on the volume of the THF solution, an appropriate amount of triethylamine can be added to raise the pH of the solution to around 9 (note that triethylamine cannot be added to 100% THF).
Tissue clearing with EZ View solution
This refractive index (RI) matching step is essential, as the delipidated tissue will still refract photons if the RI of the tissue does not match that of the imaging media. If the sample will be mounted in agarose for imaging, we recommend clearing the tissue of interest before embedding in agarose, as the agarose slows solvent exchange.
Note: This is the final step if the sample will not be mounted in agarose.
Note: Delipidation and RI matching times vary depending on tissue type (specifically their lipid and mineral composition) and size. We recommend taking a phase image each day that the sample is in RI matching solution to determine when the tissue has been rendered transparent. For this analysis, place the sample on top of paper printed with a measured grid on it. The sample is considered “clear” when the tissue of interest is transparent enough to view the underlying grid lines. However, the degree of clearing is ultimately determined by whether the lightsheet can penetrate through the entire tissue and yield crisp, robust fluorescent signal. All of the mouse organs tested in this protocol became transparent in EZ View solution after one or two days, provided they were appropriately delipidated (Figures 3 and 4). Delipidation and RI matching times for various young and aged adult mouse organs are presented in Table 1.
-
15.
Place the delipidated tissue in a 50 mL conical tube and cover the sample with 20 mL of EZ View solution.
-
16.
Shake gently at room temperature for approximately one or two days, shielded from light.
Note: The EZ View solution changes the refractive index to 1.50 (or greater) and renders the tissue transparent. If more water is introduced into the EZ View solution, it will dilute the concentration of Nycodenz/Urea and decrease the RI of the overall system. The lower the RI, the less transparent the sample will be. Thus, minimize the amount of water being introduced when immersing samples in EZ View. The RI of the solution can be measured after incubating for 8–12 h. If the RI falls below 1.49, replace the solution with fresh EZ View, which will accelerate the clearing process. If the RI has fallen to 1.46, or below, the tissue will not become transparent, no matter how long the sample remains in EZ View. In this case, the sample should be immersed in newly prepared EZ View solution with the correct RI. In all cases, ensure the sample is totally submerged in EZ View and does not dry out during these washes and incubation steps.
Note: At this point, the organs are cleared.
Figure 3.

Phase images of pre-delipidation, post-delipidation and RI-matched organs (after 20 h of delipidation) from 2 month old mice
Eye (A), heart (D), lungs (E), testis (F) and brain (G) are fully cleared after 20 h of delipidation). Liver (B) and kidney (C) are partially cleared. Scale bar = 5 mm.
Figure 4.

Phase images of pre-delipidation, post-delipidation and RI-matched organs (after 20 h and 68 h of delipidation) from 5 month old mice
Eye (A), lungs (E), ovary (G) and brain (H) are fully cleared. Liver (B), heart (D), Kidney (C) and spleen (F) are partially cleared. Additional delipidation improved the clearing of these tissues in EZ View solution. A-F and H, scale bar = 5 mm; in G, scale bar = 2.5 mm.
Table 1.
Summary of delipidation and tissue clearing times employed for individual organs from young and aged adult mice
| Organ | Delipidation time | Tissue clearing time |
|---|---|---|
| 2 month old mice | ||
| Eye | 20 h | 1 day |
| Liver | 20 h | 1 day |
| Kidney | 20 h | 1 day |
| Heart | 20 h | 1 day |
| Lungs | 20 h | 1 day |
| Testes | 20 h | 1 day |
| Brain | 20 h | 1 day |
| 5 months old | ||
| Eye | 20 h | 1 day |
| Liver | 68 h | 1 day |
| Kidney | 68 h | 1 day |
| Heart | 68 h | 1 day |
| Lungs | 20 h | 1 day |
| Spleen | 68 h | 1 day |
| Ovary | 20 h | 1 day |
| Brain | 20 h | 2 days |
Mounting the sample in agarose for imaging
This step details how to assemble a sample holder and mount the cleared tissue, either with agarose or without agarose, for subsequent imaging.
Note: The following mounting protocol is optimized for both the Zeiss Z1 and Z7 lightsheet fluorescence microscopes. A different mounting strategy may need to be employed depending on the available lightsheet fluorescence microscope (LSFM).
-
17.
Prepare a 1% agarose mixture in ddH2O and microwave the mixture for approximately 2 min (until it boils) to dissolve the agarose.
-
18.
After the agarose dissolves, immediately filter sterilize the solution using a vacuum filter (0.45 μm pore size).
Note: This step removes any particles or debris that can refract light and interfere with imaging.
-
19.Add enough 1% agarose solution to a 50 mL conical tube to adequately submerge the tissue (e.g., a mouse brain) (about 5 mL).
-
a.Cap and store this tube in water bath set to 42°C, as this will maintain standard gelling agarose as a liquid.
-
b.Wait until the temperature of the agarose solution equilibrates to that of the water bath to prevent damaging the sample during mounting, as submerging samples in boiling agarose may deform and damage the tissue.
-
a.
-
20.Container Assembly:
-
a.Using a razor blade or scalpel, remove the far end portion of the syringe where a needle would attach.
-
b.Ensure the open end of the syringe is smoothed out to an even surface, as otherwise it may rip a sample if it contacts the edge of the syringe (Figure 5A1).
-
c.Stand the syringe upright with the cut end facing up (a conical tube rack can be used to hold the syringe in position),
-
a.
-
21.Fill the syringe with approximately 3 mL of 1% liquid agarose.
-
a.Wait until the surface of the agarose begins to solidify, then gently place the brain in the syringe. This will help maintain the sample in the middle of the syringe, rather than sinking to the bottom.
-
b.At this point, the brain should be immersed in agarose (add more liquid agarose if necessary).
-
c.Orient the brain such that the cerebellum is toward the plunger and the olfactory bulbs face the open end of the syringe (Figure 5A2).
-
a.
Note: As the agarose solidifies, if needed, carefully reposition the brain into the middle of the syringe by tilting the syringe and nudging the brain into the desired orientation using a paperclip, forceps, or another tool. This will ensure that when the sample is pressed out the syringe, it does not contact with the walls of the syringe and tissue is not torn or deformed.
-
22.
Once the agarose has begun to solidify, gently insert the sample holder by placing it into the open end of the syringe.
Note: Make sure the tissue sample does not sink down to the bottom of the syringe and that the holder does not directly contact the tissue. More agarose can be layered on top of the sample to ensure proper positioning of the sample (Figure 5A3).
-
23.
Leave the sample at 4°C for at least 15 min, or wait for about 1 h in room temperature.
-
24.
Push the agarose-sample complex out of the syringe.
-
25.
Trim excess agarose to limit the amount of material required for RI matching and the diffraction of the lightsheet during imaging (Figure 5A4).
Note: For smaller samples (such as embryos), place the sample in a 10 cm tissue culture dish and then flood the entire dish with 42°C 1% liquid (low-melting point/LMP) agarose solution until the sample is submerged, then aspirate the sample and liquid into the 5 mL syringe (with the distal end of the syringe cut off). Invert the syringe (stand it upwards) once the agarose starts to solidify. An unfolded paperclip or spatula can be used to orient/align the sample (keeping it in the middle, away from the plastic sides of the syringe) once it is in the cylinder. However, if the agarose has not begun to gel, the sample will likely sink to the bottom of the syringe and come to rest on the plunger (which will prohibit successful mounting). Additionally, depending on the sample mounting kit available to the user, and whether full organ imaging is required, agarose embedding may be skipped in favor of simply gluing the sample directly to a sample holder using either super glue (Loctite Super Glue Ultra Gel Control) or a UV-solidified resin (Figure 5B1).
Note: If difficulty is encountered slicing off the top of the syringe, securely grasp a razor blade using forceps and heat it over a Bunsen burner, then use the hot razor blade to evenly slice through a 5 mL plastic syringe. Use caution working with an open flame and be aware that whatever surface is used for cutting may be damaged by melting plastic, or scratched by the razor blade.
Note: Steps 40–48 can be directly reapplied for a sample mounting holder with a magnetic attachment to a lightsheet instrument. The magnetic sample holders in Figures 5C and 5D are provided by Zeiss with the purchase of a Z7 microscope.
Alternative: Agarose embed cleared samples in a cryomold.
-
26.
Add pre-heated agarose into the cryomold, filling it until about 1 cm of empty space remains the top of the mold (see Figure 5D1).
-
27.
Transfer the mold to 4°C for about 15 min until the agarose is completely solidified.
-
28.
Place the tissue of interest on top of the pre-solidified agarose pedestal in the cryomold (see Figure 5D2).
-
29.
Fill the remaining 1 cm empty space with 42°C 1% agarose (see Figure 5D3).
-
30.
Transfer the agarose-tissue complex to 4°C to solidify.
-
31.
Using a razor blade, cut the bottom two corners of the mold and gently separate the mold from the agarose-tissue sample (see Figure 5D4).
-
32.
Trim the agarose as closely to the sample as possible (see Figure 5D5).
Note: Cut the mold vertically to ensure that the light from the laser is not diffracted by an uneven surface.
-
33.
Mount the trimmed sample onto the magnetic sample holder with super glue (Figure 5D6).
Figure 5.

Workflow for mounting cleared tissue samples for lightsheet imaging
Samples can be mounted using agarose (A, C and D) or without agarose (B) for the Zeiss Lightsheet Z1 and Z7 systems with either a sample claw on the Z7 LSFM system (A-B) or a mounting screw on the Z1 and Z7 LSFM systems (C-D). For methods A and C, the tissue of interest and the mounting holder will be mounted in the solidifying agarose subsequently. For method B, the sample will be directly attached by a super glue. For method D, pre-heated agarose will be poured into a cryo-mold until 1 cm space remains at the top. The sample will be placed in the cryomold, chilled at 4°C, then removed from the cryomold and as much agarose as possible trimmed away to allow the sample to fit into the sample holder and decrease the amount of agarose the laser beam must pass through.
Equilibrating the agarose in EZ View
This step matches the refractive index (RI) of the agarose embedded sample to that of the imaging media (e.g., EZ View solution). When the agarose is initially embedded, it appears opaque. In EZ View solution the agarose should gradually be rendered transparent. Light sources and the detection lens of the Zeiss Lightsheet Z1 and Z7 are adjusted to match the refractive index of the imaging media.
-
34.
Place the embedded tissue of interest in a 50 mL conical tube.
-
35.
Fill the conical tube with 20–25 mL of 20°C–25°C EZ View solution, completely submerging the entire agarose-sample complex.
-
36.
Protect the sample from light by covering it in aluminum foil, or placing it in a box.
-
37.
Wrapping the edges of the 50 mL conical tube in parafilm to prevent evaporation.
-
38.
Gently rock the sample, which should be standing upright, at 20°C–25°C until the sample becomes transparent.
Note: If crystals form in the EZ View solution, it is likely due to the temperature of the media. If this happens, heat the mixture to 37°C, and vortex if necessary, until the urea goes back into solution prior to adding this solution to the sample.
Once the tissue is clear, proceed to imaging (e.g., lightsheet fluorescence microscopy).
For guidance using the Z1 or Z7 system, we refer readers to a detailed, step-by-step protocol for image acquisition using lightsheet fluorescent microscopy.10
Expected outcomes
There are two expected outcomes. The first is that the tissue of interest is rendered optically transparent enough (e.g., “cleared”) such that the bottom of the grid is visible by eye (Figures 3 and 4). The second expected outcome is that the use of lightsheet imaging will result in clear visualization of the perfused fluorescent dye(s). As shown herein (see Figures 3 and 4), optimizing the duration of delipidation (through serial washes with increasing concentrations of THF), yields dramatic improvements in the signal to noise ratio and generates superior lightsheet images (see Figures 6 and 7). Moreover, this standard EZ Clear protocol is compatible with transgenic fluorescent reporters, and fluorescence signal should be observed in the expected anatomical location (e.g., Thy1::GFP will label neurons in the brain, as in Figure 8).
Note: Lightsheet imaging will ultimately determine whether clearing is successful for the tissue of interest. Even though the tissue may look optically “cleared” after the RI matching by phase microscopy and transmitted light, further optimization of the delipidation period may be required if inadequate level of signal is observed during fluorescent imaging.
Note: A detailed protocol on how to operate the Zeiss Z7 and acquire 3D images can be found in the protocol paper by Yu et al.10 However, details regarding imaging parameters on the various LSFM platforms used to acquire data in this manuscript are listed in Table 2.
Figure 6.

20 h of delipidation is sufficient to clear most young adult mouse tissues
Lightsheet images of the eye (A), kidney (B), heart (C) testes (D) and brain (E) from 2 month old mice perfused with far-red fluorescent lectin to label the vascular endothelium after 20 h of delipidation (using 50% THF) and RI matching in EZ View. Numbers below correspond to white boxed in regions in the above images. Successful clearing and lightsheet imaging should yield images where smaller blood vessels are clearly resolved, as shown in the bottom panels of the figure. Scale bars in A1, B1 = 400 μm; C1 = 500 μm; A2 = 140 μm; B2, C2 = 200 μm; D1 = 400 μm; E1 = 1500 μm; D2 = 100 μm; E2 = 500 μm.
Figure 7.

Increasing delipidation time from 20 h to 68 h dramatically improves tissue clearing and imaging in aged adult mouse tissues
Lightsheet images of kidney (A and B), heart (C and D), ovary (E), eye (F) and spleen (G) from 5 month old mice following perfusion with far red fluorescent lectin to label blood vessels. Kidney and heart samples were delipidated for 20 h (A and C) and then imaged following RI matching. These exact same tissues were then subjected to an additional 48 h of delipidation prior to a second round of RI matching and imaging (B and D). The kidney sample delipidated for 20 h (A) does not show any discernable vasculature, indicating that this incubation time is not adequate for tissue clearing of 5 month old kidney. After an additional 48 h of delipidation, the vasculature of the 5 month old kidney (B) is evident. The heart after 20 h of delipidation (C) shows clear signal in the vasculature, although some areas lack crisp signal. However, an additional 48 h of delipidation adds detail previously undetected with the 20 h delipidation and improves the signal to noise ratio. 20 h of delipidation is adequate for the ovary (E) and eye (F), whereas the spleen requires an additional 48 h of delipidation to observe optimal signal. Scale bars in A1–D1 = 500 μm; A2–E1 = 200 μm; F1 = 300 μm; G1 = 700 μm; E2 = 70 μm; F2 = 100 μm; G2 = 200 μm.
Figure 8.

EZ Clear preserves fluorescence signal from endogenous fluorescent reporter transgenes
(A) Whole-mount lightsheet image, dorsal view, of the brain from a Tg(Thy1::GFP) (MGI# 3766828)11 adult mouse. Neurons should be identifiable and background signal should be minimal upon successful clearing.
(B‒D) Magnified images show individually labeled neurons through the brain.
(E) GFP positive cells are pseudocolored according to their depth in the imaging plane. This brain was imaged by AxL Cleared Tissue LightSheet (AxL CTLS) from Intelligent Imaging Innovations.
Scale bar in A = 1500 μm; B = 100 μm; C = 100 μm; D = 300 μm; E = 50 μm.
Table 2.
Detailed lightsheet imaging acquisition parameters utilized for individual organs from young and aged adult mice
| Organs | Laser (nm) | Emission Filter (nm) |
Zoom | Laser power | Exposure time (ms) | Lightsheet Thickness (μm) |
Overlap (%) | Raw file Size (Gb) |
Recon File size (Gb) |
|---|---|---|---|---|---|---|---|---|---|
| 2 month old mice perfused with 649-nm tomato lectin | |||||||||
| Eye | 638 | 660 | 0.7 | 14% | 119.85 | 7 | 15 | 28.3 | 10.4 |
| Kidney | 638 | 660 | 0.7 | 14% | 119.85 | 7 | 15 | 66.6 | 39.6 |
| Heart | 638 | 660 | 0.7 | 10% | 119.85 | 7 | 15 | 123 | 53.5 |
| Testis | 638 | 660 | 0.7 | 18% | 149.81 | 7 | 15 | 43.3 | 18.1 |
| #Brain | 640 | 655 | 3.25 | 200 mW | 300 | 10 | 15 | 201.6 | 54.8 |
| 5 month old | |||||||||
| Eye | 638 | 660 | 0.7 | 18 | 249.67 | 5.25 | 15 | 20.6 | 7.4 |
| Kidney | 638 | 660 | 0.7 | 14 | 119.85 | 7 | 15 | 83.6 | 39.9 |
| Heart | 638 | 660 | 0.7 | 10 | 119.85 | 7 | 15 | 95.9 | 39.9 |
| Spleen | 638 | 660 | 0.7 | 14 | 119.85 | 7 | 15 | 102 | 45.7 |
| Ovary | 638 | 660 | 1.0 | 18 | 249.67 | 6 | 15 | 33.6 | 6.0 |
| Brain | 488 | 498 | 0.7 | 7.5 | 99.87 | 4.88 | 10 | 371 | 124 |
∗For all images, the lens magnification is 5X.
∗For all images, “Online Dual Side Fusion” with “Mean”, “Pivot Scan” and “Bi-directional” were checked.
#Imaged by AxL Cleared Tissue LightSheet (AxL CTLS) from Intelligent Imaging Innovations. The rest of the samples were imaged by Zeiss Z7.
Quantification and statistical analysis
-
•
The original Zeiss imaging file was converted using Imaris File Converter (Oxford Instrument, Version:10.0.1). Views were then stitched together using Imaris Stitcher (Oxford Instrument, Version: 10.0.0) with the “Automatic Align” function and aligned manually. Images and a supplementary video were captured by Imaris (Oxford Instrument, Version: 10.0.1).
Limitations
This protocol has been optimized for soft tissue containing no mineralized components. We do not know how, or if, decalcification is needed to clear mineralized tissues such as bone, or whether this would affect the EZ Clear protocol and downstream imaging.
Troubleshooting
Problem 1
Urea or Nycodenz addition leads to precipitate formation. (related to step making EZ View).
Potential solution
-
•
Agarose added too quickly. Add the urea and Nycodenz Iohexol slowly, in 10 g increments, waiting for the material to dissolve before any subsequent additions, and raise the plate temperature to approximately 37°C.
-
•
Stirring, covered to prevent evaporation, for 8–12 h is recommended.
-
•
Properly prepared EZ View solution should be clear with minimal remaining solutes after the filtration step (Figure 9).
Figure 9.

An image of properly prepared EZ View solution
Note that the final, filtered solution should be transparent and free from crystals or debris.
Problem 2
Incorrect sample mounting in agarose. (related to mounting the sample in agarose for imaging).
Potential solution
-
•
Sample added too late. If the tissue is placed in the syringe after the agarose has gelled, the tissue will not sink into the agarose. Add the sample while agarose is still in liquid phase.
-
•
Sample added too early (and sinks to the bottom of the syringe). If the sample is added to liquid agarose before the agarose reaches equilibrium at 42°C, the tissue will be damaged due to the high temperature of the agarose and it will sink to the bottom of the syringe, making proper alignment during mounting quite difficult, if not impossible. Add sample later.
-
•
At higher temperatures, agarose will have reduced viscosity and increased fluidity. If the temperature is too low, the agarose becomes too viscous. To test the viscosity of the solution prior to adding the sample, dispense a few drops of the EZ View solution into a tube containing the agarose and if the drops sink slowly, then the temperature may be correct for addition of the sample.
-
•
Alternatively, the top of the agarose can be prodded with an instrument (e.g., forceps or a paperclip or micropipette tip) to gauge the viscosity and state of the agarose (liquid vs. gel) and whether it is an appropriate time to add the sample.
Problem 3
Incorrect needle placement during retro-orbital injection. (related to Step 5).
Potential solution
-
•
The injection angle is not correct. Depending on the angle, the eye may be damaged, or the contrast agent may not enter the circulation and instead will diffuse and pool around the eye. Re-adjust to a more perpendicular angle in the approach to the eye.
-
•
Incorrect needle placement. Before injecting the fluorescent dye, make sure to draw blood into the syringe. If no blood is aspirated, the needle is incorrectly inserted and needs to be repositioned. We recommend practicing by injecting Evans blue dye into the retroorbital sinus, as delivery can be easily visualized by blue dye circulating throughout the nose, eye, and toes of the animal, enabling assessment of a successful injection.
Problem 4
Poor perfusion of larger diameter vessels. (related to Step 12).
Potential solution
-
•
This likely stems from poor transcardiac perfusion. Do not penetrate the interventricular septum and deliver the contrast agent into the right ventricle (and lungs). This problem may occur during perfusion with the 27G needle for delivery of 1X PBS or 4% PFA (by comparison, the insulin syringe used for contrast agent delivery is quite short). For the transcardiac perfusion the needle should be inserted shallowly, just through the apex of the left ventricle, at an angle parallel to the longitudinal axis of the heart.
-
•
Consider practicing the perfusions by injecting Evans blue dye, as one can determine whether a successful perfusion has occurred directly by visualizing distribution of the blue dye. Similarly, following delivery of PBS and fixative the signs of a successful perfusion, such as blanching of the liver, as well as muscle twitching and tail curling, respectively, should be evident.
-
•
Some experimentalists may to hold the needle in place using a “third hand” adjustable stand with alligator clips and a magnifying glass (as often utilized in soldering applications), as this can prevent the needle from slipping deeper into the heart during the procedure.
Problem 5
Inability to image entire tissue of interest using the Zeiss Z1 or Z7. (related to Expected Outcomes).
Potential solution
-
•
While mounting the tissue, make sure it is placed directly underneath the sample holder, and that the agarose is still warm enough so that the tissue can be manipulated to ensure it is positioned within the center of the syringe (and thus the agarose cylinder when it is expelled from the syringe). If the tissue is not centered, re-equilibrate the tissue in agarose in 1X PBS for 8–12 h and carefully dissect it from the agarose and remount it again.
-
•
If the tissue is too large for the imaging chamber, consider using another lightsheet microscope imaging platform.
Problem 6
Low fluorescence signal. (related to Expected Outcomes).
Potential solution
-
•
If fluorescent lectin cannot be visualized, it may be due to an unsuccessful perfusion. For perfusion tips, please see Problems 3 and 4 potential solutions.
-
•
Examine the level of the fluorescent signal in the mouse brain, or tissue of interest, by taking epifluorescence images with the appropriate filters. For example, if the rodent was perfused with 488-lectin, then visualize the tissue using a 488 nm light source. Example epifluorescence images of a perfused adult mouse brain are shown in Figures 1J and 1K. If an adequate fluorescence intensity and an even distribution of fluorescence signal is not observed at this starting stage, then robust 3D lightsheet images will not be acquired. Revisit Problems 3 and 4.
-
•
If lightsheet imaging fails to show a robust signal from an endogenous fluorescent reporters, such as the Thy1:GFP transgene, re-genotype the tissue or parental animal and ensure the genotype is correct.
-
•
If the endogenous reporter activity is dim or not observable by epifluorescence imaging, consider obtaining a positive control line, such as Thy1:GFP or Tie2:GFP, both available as live mice from Jackson labs (stain #s 007788 and 003658, respectively). These should yield robust signal using epifluoresensce imaging and provide useful positive controls for the fixation, delipidation, RI matching, and imaging portions of the protocol.
-
•
If a robust 3D lightsheet image is not obtained, even after a confirmation of the successful perfusion by epifluorescence imaging, the tissue is likely under-delipidated. Please follow the sample preparation procedures described in Figure 4.
Resource availability
Lead contact
Joshua D. Wythe (jwythe@virginia.edu).
Technical contact
Chih-Wei Hsu (loganh@bcm.edu).
Materials availability
Not applicable.
Data and code availability
Not applicable.
Acknowledgments
The authors thank Ms. Emily Alley for proofreading the final manuscript, and we are grateful to all members of the Dickinson and Wythe laboratories, past and present, for contributing their expertise and valuable discussions to optimize this methodology. The authors would also like to thank Dr. Edward Lachica from Intelligent Imaging Innovations (3i) for his assistance in imaging samples using the AxL CTLS system. This work was supported by the Canadian Institutes of Health Research (PJT-155922 to J.D.W.), National Institutes of Health (R01HD099026 and R01HL146745 to M.E.D. and J.D.W.; R01HL159159 and UC2AR082200 to J.D.W.), and institutional funds from the University of Virginia School of Medicine and generous financial support from the University of Virginia Comprehensive Cancer Center (J.D.W.).
Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health through the NIH HEAL Initiative (https://heal.nih.gov/) under award number UC2AR082200 to J.D.W. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The RE-JOIN consortium consists of Taeyoung (Ted) Ahn, Armen Akopian, Kyle Allen, Alejandro Almarza, Benjamin Arenkiel, Maryam Aslam, Basak Ayaz, Yangjin Bae, Bruna Balbino de Paula, Mario Danilo Boada, Jacqueline Boccanfuso, Jyl Boline, Dawen Cai, Dellina Lane Carpio, Robert Caudle, Racel Cela, Yong Chen, Rui Chen, Brian Constantinescu, Yenisel Cruz-Almeida, Christopher Donnelly, Zelong Dou, Joshua Emrick, Malin Ernberg, Spencer Fullam, Janak Gaire, Akash Gandhi, Terese Geraghty, Benjamin Goolsby, Stacey Greene, Nele Haelterman, Zhiguang Huo, Michael Iadarola, Shingo Ishihara, Sudhish Jayachandran, Zixue Jin, Alisa Johnson, Frank Ko, Priya Kulkarni, Brendan Lee, Yona Levites, Carolina Leynes, Jun Li, Martin Lotz, Lindsey Macpherson, Tristan Maerz, Camilla Majano, Anne-Marie Malfait, Maryann Martone, Simon Mears, Bella Mehta, Emilie Miley, Rachel Miller, Richard Miller, Michael Newton, Alia Obeidat, Soo Oh, Merissa Olmer, Dana Orange, Miguel Otero, Kevin Otto, Folly Patterson, Daniel Perez, Sienna Perry, Theodore Price, Russell Ray, Dongjun Ren, Alexus Roberts, Elizabeth Ronan, Oscar Ruiz, Shad Smith, Mairobys Soccorro Gonzalez, Kaitlin Southern, Joshua Stover, Michael Strinden, Hannah Swahn, Evelyne Tantry, Cristal Villalba Silva, Airam Vivanco Estella, Robin Vroman, Joost Wagenaar, Lai Wang, Kim Worley, Joshua Wythe, Jiansen Yan, Julia Younis, and Yi Zou.
Author contributions
T.A. designed and performed the experiments, collected and analyzed the data, made the figures, wrote the first draft with J.D.W., and reviewed the manuscript. G.E.L. and J.Y. reviewed and edited the first draft. M.E.D. supervised C.-W.H., reviewed the manuscript, and secured funding. C.-W.H. reviewed and edited the manuscript, troubleshot data acquisition methods, and provided experimental guidance to J.Y. and T.A. J.D.W. conceived the study, designed the experiments, obtained funding, wrote the first draft with T.A., and edited all subsequent drafts.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Chih-Wei Hsu, Email: loganh@bcm.edu.
Joshua D. Wythe, Email: jwythe@virginia.edu.
References
- 1.Hsu C.W., Cerda J., 3rd, Kirk J.M., Turner W.D., Rasmussen T.L., Flores Suarez C.P., Dickinson M.E., Wythe J.D. EZ Clear for simple, rapid, and robust mouse whole organ clearing. Elife. 2022;11 doi: 10.7554/eLife.77419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawashima H., Sueyoshi S., Li H., Yamamoto K., Osawa T. Carbohydrate binding specificities of several poly-N-acetyllactosamine-binding lectins. Glycoconj. J. 1990;7:323–334. doi: 10.1007/BF01073376. [DOI] [PubMed] [Google Scholar]
- 3.Robertson R.T., Levine S.T., Haynes S.M., Gutierrez P., Baratta J.L., Tan Z., Longmuir K.J. Use of labeled tomato lectin for imaging vasculature structures. Histochem. Cell Biol. 2015;143:225–234. doi: 10.1007/s00418-014-1301-3. [DOI] [PubMed] [Google Scholar]
- 4.Porter G.A., Palade G.E., Milici A.J. Differential binding of the lectins Griffonia simplicifolia I and Lycopersicon esculentum to microvascular endothelium: organ-specific localization and partial glycoprotein characterization. Eur. J. Cell Biol. 1990;51:85–95. [PubMed] [Google Scholar]
- 5.Namykin A.A., Khorovodov A.P., Semyachkina-Glushkovskaya O.V., Tuchin V.V., Fedosov I.V. Photoinduced Enhancement of Evans Blue Dye Fluorescence in Water Solution of Albumin. Opt. Spectrosc. 2019;126:554–559. doi: 10.1134/S0030400X19050217. [DOI] [Google Scholar]
- 6.Saria A., Lundberg J.M. Evans blue fluorescence: quantitative and morphological evaluation of vascular permeability in animal tissues. J. Neurosci. Methods. 1983;8:41–49. doi: 10.1016/0165-0270(83)90050-X. [DOI] [PubMed] [Google Scholar]
- 7.Honeycutt S.E., O'Brien L.L. Injection of Evans blue dye to fluorescently label and image intact vasculature. Biotechniques. 2021;70:181–185. doi: 10.2144/btn-2020-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyerhoff J., Muhie S., Chakraborty N., Naidu L., Sowe B., Hammamieh R., Jett M., Gautam A. Microdissection of Mouse Brain into Functionally and Anatomically Different Regions. J. Vis. Exp. 2021;10:3791–61941. doi: 10.3791/61941. [DOI] [PubMed] [Google Scholar]
- 9.Kosmidis S., Negrean A., Dranovsky A., Losonczy A., Kandel E.R. A fast, aqueous, reversible three-day tissue clearing method for adult and embryonic mouse brain and whole body. Cell Rep. Methods. 2021;1 doi: 10.1016/j.crmeth.2021.100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu H., Li Q., Sandoval A., Jr., Gibbs H.C., English A., Dunn T., Moth J., Elahi H., Chen B. Pipeline for fluorescent imaging and volumetric analysis of neurons in cleared mouse spinal cords. STAR Protoc. 2022;3 doi: 10.1016/j.xpro.2022.101759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feng G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., Sanes J.R. Imaging Neuronal Subsets in Transgenic Mice Expressing Multiple Spectral Variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.