

Abstract
Proteins interact with diverse ligands to perform a large number of biological functions, such as gene expression and signal transduction. Accurate identification of these protein–ligand interactions is crucial to the understanding of molecular mechanisms and the development of new drugs. However, traditional biological experiments are time-consuming and expensive. With the development of high-throughput technologies, an increasing amount of protein data is available. In the past decades, many computational methods have been developed to predict protein–ligand interactions. Here, we review a comprehensive set of over 160 protein–ligand interaction predictors, which cover protein–protein, protein−nucleic acid, protein−peptide and protein−other ligands (nucleotide, heme, ion) interactions. We have carried out a comprehensive analysis of the above four types of predictors from several significant perspectives, including their inputs, feature profiles, models, availability, etc. The current methods primarily rely on protein sequences, especially utilizing evolutionary information. The significant improvement in predictions is attributed to deep learning methods. Additionally, sequence-based pretrained models and structure-based approaches are emerging as new trends.
Keywords: protein–ligand interaction, protein–protein interaction, protein–peptide interaction, protein–nucleic acid interaction, protein–other ligands interaction
INTRODUCTION
Proteins are essential participants in most biological processes within cells. In order to carry out cellular functions, proteins interact with a variety of ligands, such as proteins, nucleic acids, peptides, nucleotides, hemes and ions, forming stable complexes. To be specific, protein–protein interactions (PPIs) underlie many cellular processes, such as signal transduction, transport and metabolism [1, 2]. Interactions of proteins and nucleic acids are involved in post-transcriptional modification, gene replication, gene expression and many other biological processes [3–5]. Furthermore, peptides mediate ~40% of protein interactions that contribute to abnormal cellular behaviors causing various diseases [6]. In addition, protein–other ligands (nucleotide, heme, ion) interactions are also indispensable for biological activities [7]. For example, protein–nucleotide interactions play a crucial role in energy provision [8], interactions of proteins and hemes are significant for circadian rhythm and cell-cycle regulation [9], while protein–ion interactions contribute to protein structural stability [10]. Knowledge of protein–ligand interactions facilitates the annotation of protein functions, the comprehension of cellular processes, understanding the pathogenesis of diseases and developing new therapeutic approaches [11–14]. There are some databases that have compiled information on protein–ligand interactions at protein-, residue- and atom-level. Protein-level databases provide information on whether proteins interact with ligands, such as STRING [15], mentha [16] and BioGRID [17]. Residue-level databases, such as BioLip [18] and DisProt [19], provide residue-level annotations that indicate whether amino acids in the protein sequence interact with ligands. Compared with protein-level databases, residue-level databases provide more detailed annotations about the interactions of proteins, nucleic acids, peptides and other ligands. PDB [20] is the atom-level database that provides a large amount of 3D structural information on protein–ligand interactions. However, identification of these protein–ligand interactions by biological experiments, such as affinity chromatography, nuclear magnetic resonance and site-directed mutagenesis, is time-consuming and relatively expensive. Given the continuous advancement of high-throughput sequencing technologies, biological experiments are unable to match the rapid increase in the number of proteins. For example, the Uniprot [21] database contains over 250 million protein sequences (as of April 2023), while BioLiP contains 882 170 proteins (as of 29 September 2023), PDB contains 1278 757 protein structures, including 210 180 experimental structures and 1068 577 computational structures. STRING (as of 26 July 2023) contains 59 309 604 proteins. Therefore, it is highly urgent and challenging to develop computational methods for predicting protein–ligand interactions.
In the past decades, a large number of computational methods have been developed to predict protein–ligand interactions [22–34]. Generally, the pipeline of these methods begins with inputs, from which protein feature profiles are extracted. Models are employed to identify the interactions between proteins and ligands or to infer the specific interaction residues, as illustrated in Figure 1. First, the inputs for these methods can be categorized into three types: protein sequences [24, 35], protein structures [5, 36] and PPI networks [37, 38]. Protein sequences are typically characterized using amino acid binary encodings, such as one-hot encoding, and now protein language models are employed to generate sequence embeddings. The second common type of inputs is protein structures. Similar to sequence-based predictors, structure-based predictors also extract various protein feature profiles to improve predictions. However, given the difficulty in obtaining protein structure data and the direct influence of structure quality on prediction results, only a few methods focus on protein structures compared with sequences. Methods that utilize PPI networks primarily predict PPIs, but compared with methods employing sequence data, these display certain limitations and lower scalability.
Figure 1.

The pipeline of protein–ligand interaction predictors includes inputs, feature profiles, models and prediction. Inputs mainly include protein sequences, protein structures and PPI networks. Feature profiles panel shows various features including amino acid binary encoding, evolutionary information, embedding features based on the protein language model, etc. The model panel displays the commonly used prediction methods including homology-, machine learning- and DL-based methods. The prediction-level and ligand types are shown in the prediction panel.
Second, according to models, these methods can be grouped into two categories: (i) homology-based methods [39, 40], typically identifying annotated proteins similar to the query proteins and assigning those annotations accordingly. (ii) Learning-based methods [24, 35], applicable even to proteins with few similarities to previously characterized ones. Learning-based methods typically utilize prediction models based on machine learning or deep learning (DL) algorithms, optimizing the model for optimal prediction performance on a training dataset of annotated proteins. The optimized model then infers interactions of proteins outside the training dataset. This study primarily focuses on the research pertaining to learning-based methods.
From the perspective of the prediction level, the computational methods can be classified into two classes: protein-level [41, 42] and residue-level [24, 35]. Protein-level methods predict whether a protein interacts with ligands, whereas residue-level methods determine which specific residues within the protein interact with ligands. Residue-level methods offer more detailed annotations compared with protein-level methods. Furthermore, based on the type of interaction ligands, these methods can be divided into four categories. The first category comprises predictors of PPIs [22, 42], encompassing both protein- and residue-level predictors. Protein-level methods infer interactions by utilizing PPI networks or pairs of proteins, while residue-level methods typically employ sliding windows as inputs to identify interaction residues. The second category involves predictions of protein–nucleic acid interactions [24, 35], including protein–DNA, protein–RNA and general protein–nucleic acid interactions. This category includes both protein- and residue-level methods and primarily takes protein sequences as inputs. The third category involves protein–peptide interactions [43, 44] at residue-level and the remaining interaction ligands are classified into the fourth category, such as nucleotides [8, 45, 46], hemes [9, 47, 48] and ions [49–51]. The classification and temporal distribution of all surveyed predictors are illustrated in Figure 2.
Figure 2.

The distribution of protein–ligand interaction predictors across different ligand types (proteins, nucleic acids, peptides and others) and time periods (every 5 years as a time period). The horizontal axis indicates the number of relevant prediction methods and the vertical axis indicates the time period.
In this study, we review a comprehensive set of over 160 predictors section by section, including protein–protein, protein−nucleic acid, protein−peptide and protein–other ligands (nucleotide, heme, ions) interactions. We discuss and try to offer insightful analysis of their inputs, feature profiles, models, availability, etc. Finally, we give a summary and directions for future research.
PROTEIN–PROTEIN INTERACTION
According to the varying inputs and outputs, we grouped 58 PPI predictors into three categories, as shown in Figure 3: (i) PAIR-pro predictors [38, 42], which identify interactions between a pair of proteins while cannot provide the specific positions of the interaction residues; (ii) PAIR-res predictors [52, 53], which predict whether proteins interact with each other and the positions of the interaction residues and (iii) SINGLE-res predictors [54, 55], inferring interaction residues within a single protein. Among the 58 predictors, there are 24 PAIR-pro predictors, 4 PAIR-res predictors and 30 SINGLE-res predictors.
Figure 3.

PPI predictors are divided into three categories based on inputs and outputs: PAIR-pro, PAIR-res and SINGLE-res. The PAIR-pro predictor can predict whether two proteins interact or not, but cannot provide the specific positions of the interaction residues. However, the PAIR-res predictor can predict whether proteins interact with each other and the positions of the interaction residues. In addition, the SINGLE-res predictor can predict the interaction residues on a single protein.
PAIR-pro predictors
PAIR-pro [37, 38, 41, 42, 56–75] predictors, summarized in Table 1, focus on protein-level interactions and accept two proteins as inputs. The most frequently employed input types for PAIR-pro predictors are protein sequences and protein structures, involving a total of 19 predictors. In addition, a few predictors utilize PPI networks or gene ontology (GO) annotation sets as inputs, such as TransformerGo [42] and the predictor proposed by Kovács et al. [71]. The analysis of the dataset for PAIR-pro reveals that the majority of the data originates from public databases, such as the PDB [20], DIP [76], HPRD [77], HIPPLE [78], HINT [79], STRING [15], SKEMPI [80], BioGRID [81], PrePPI [82] and IntAct [83]. Among these, DIP and HPRD are the most commonly utilized PPI databases. Some manually curated datasets have also been extensively employed by researchers, such as the high-quality dataset containing 5594 interaction pairs compiled by Guo et al. [58].
Table 1.
Summary of PAIR-pro predictors in terms of inputs, feature profiles, models and availability
| Predictors | Inputs | Feature profiles | Models | Year | Availability |
|---|---|---|---|---|---|
| Bock et al. [56] | Seqs | Charge, hydrophobicity, surface tension | SVM | 2001 | × |
| Shen et al. [57] | Seqs | Amino acids classification, CT method | SVM | 2007 | × |
| Guo et al. [58] | Seqs | AC (hydrophobicity, hydrophicility, volumes of side chains of amino acids, polarity, polarizability, solvent-accessible surface area and net charge index of side chains of amino acids) | SVM | 2008 | × |
| Yang et al. [59] | Seqs | Amino acids classification, LDs | KNN | 2010 | × |
| LR_PPI [60] | Seqs | Amino acids classification, CT method | Latent dirichlet allocation-RF | 2010 | √ |
| PCA-EELM [61] | Seqs | CT scores, LD, AC (hydrophobicity, volumes of side chains of amino acids, polarity, polarizability, solvent-accessible surface area and net charge index of side chains, Moran autocorrelation | Principal component analysis, ELM | 2013 | × |
| MCDPPI [62] | Seqs | Multi-scale continuous and discontinuous, LD | SVM | 2014 | × |
| You et al. [63] | Seqs | LD | ELM | 2014 | × |
| PR-LPQ Descriptor [64] | Seqs | Physicochemical property response matrix (hydrophobicity) | RF | 2015 | × |
| DeepPPI [65] | Seqs | Amino acid composition, dipeptide composition, LD, Quasi-Sequence-Order descriptors, Amphiphilic Pseudoamino Acid Composition | DNN | 2017 | × |
| Sun et al. [66] | Seqs | AC (hydrophobicity, hydrophilicity, net charge index of side chains, polarity, polarizability, solvent accessible surface area, volume of side chains), CT method | Stacked autoencoder | 2017 | × |
| DANEOsf [37] | Nets | Evolutionary distance, geometric embedding | Density function, Bayes | 2017 | × |
| DPPI [67] | Seqs | Evolutionary profiles | CNN | 2018 | √ |
| DNN-PPI [68] | Seqs | Amino acids encoding | CNN, LSTM | 2018 | × |
| PIPR [69] | Seqs | Amino acids classification, pre-training the Skip-Gram model | RCNN | 2019 | √ |
| LightGBM-PPI [70] | Seqs | Pseudo amino acid composition, autocorrelation descriptor, CT method, LD | LightGBM | 2019 | √ |
| Kovács et al. [71] | Nets | N/A | L3 | 2019 | √ |
| Sim [72] | Nets | N/A | Random network, L3 | 2020 | √ |
| D-SCRIPT [73] | Seqs | Amino acids classification | LSTM | 2021 | √ |
| DeepTrio [41] | Seqs | One-hot encoding | CNN | 2022 | √ |
| FoldDock [74] | Seqs/Strs | MSA | DNN | 2022 | √ |
| TransformerGO [42] | GO | GO terms | Transformer | 2022 | √ |
| protein2vec [75] | GO | Amino acids encoding, GO terms | LSTM, DNN | 2022 | √ |
| PPISB [38] | Nets | N/A | Mixed membership stochastic blockmodel | 2022 | × |
Note: Seqs, Strs and Nets correspond to protein sequences, protein structures and PPI networks.
In the context of machine learning or DL models, proteins are typically encoded as embeddings to serve as inputs. The analysis of feature profiles employed across all PAIR-pro predictors reveals that each amino acid of a protein is generally characterized using an assortment of amino acid binary encodings (such as one-hot encoding, classification encoding), physicochemical profiles (including hydrophobicity, polarity and electrostatic potential), structural features (such as secondary structure (SS), solvent accessibility and molecular surface curvature) and evolutionary information (including conservation, position-weight matrices and coevolution). Each category captures unique and critical aspects of protein features that are essential for understanding protein interactions. Amino acid binary encodings provide a straightforward and efficient strategy for protein sequence representation, and these encodings are also highly interpretable. PPI can be defined as four interaction modes: electrostatic interaction, hydrophobic interaction, steric interaction and hydrogen bond, so physicochemical profiles are often used to reflect these modes [58]. Besides, the interaction between proteins and ligands is closely related to the spatial structure of the proteins, and structural features reveal the spatial structural information of proteins. For example, the use of solvent accessibility is motivated by the fact that interactions occur on the protein surface [84]. Furthermore, interaction residues are typically conserved across homologous protein sequences [84], thus evolutionary information is used in protein–ligand interaction predictions to quantify the conservation of residues. In addition, a variety of popular methods have been proposed to characterize protein sequences, such as local descriptor (LD) [85, 86] and GO terms [87].
Amino acid binary encodings primarily encompass one-hot encoding and amino acid classification encoding. One-hot encoding, alternatively referred to as one-bit valid encoding, utilizes N-bit status registers to record N states. A protein sequence of length L is encoded into an L*20 binary vector through one-hot encoding. It typically depicts amino acids as a 20-dimensional binary vector, with each bit corresponding to a distinct class of amino acids. For amino acid classification encoding, given the dipoles and volumes of the side chains of amino acids reflect electrostatic and hydrophobic interactions that dominate PPIs, respectively, Shen et al. calculated them correspondingly by the density-functional theory method and molecular modeling approach [57], which resulted in the classification of the 20 types of amino acids into 7 categories.
Expanding on the amino acid classification encoding, Shen et al. introduced the conjoint triad (CT) [57] to capture adjacent amino acid information. CT bears similarity to the 3-mer approach, whereby three sequential amino acids are treated as a single entity, and the frequency of each triad's occurrence forms the profile outputs. Through CT, a protein sequence can be represented within a binary space (V, F) where V denotes the vector space of sequence features, with each bit corresponding to a specific triad type, while F signifies the respective frequency corresponding to V. It notably reduces the feature dimension relative to 3-mer coding, while still retaining local information. Among all PPI predictors, over 20% [57, 60, 61, 66, 70] employ CT for encoding protein sequences.
Concurrently, the physicochemical profiles can substantially improve the accuracy of predictions. Features such as electrostatic and hydrophobic profiles, which have been found to be crucial in PPIs, are utilized in over 37% of PAIR-pro predictors. Other features including hydrophilicity, polarity, polarizability, aromaticity, hydrogen bond acceptance or donation, positive or negative ionizability and metallicity, among others, also prove beneficial for PPI predictions [58, 61, 66]. Among predictors utilizing physicochemical profiles, three representations are predominantly employed. The first is direct property values, which necessitate normalization prior to implementation. The second is binary values, such as polarity and non-polarity. The third is auto covariance (AC) [58], which leverages multiple physicochemical properties to construct a comprehensive feature profile tensor. PCA-EELM [61] and predictors [58, 66] utilize AC with properties, such as hydrophobicity, volumes of amino acid side chains, polarity, polarizability, solvent-accessible surface area, and the net charge index of side chains. The demonstrated efficacy of these predictors lends support to the assertion that AC is beneficial for PPI predictions.
Moreover, the structural features used by over 20% of predictors of PAIR-pro [37, 56, 58, 61, 66] can be categorized into three groups according to their sources: (i) primary structural features, such as sequence length and amino acid composition; (ii) secondary structural features, including the SS; (iii) tertiary structural features, encompassing atom composition, surface tension, accessibility and more. Typically, primary and tertiary structural features can be readily obtained from raw protein sequences and structures. However, secondary structural features are generally collected from predictions based on primary structures or calculations of tertiary structures. Therefore, we discuss two approaches to derive SSs from protein sequences and structures, respectively. PSIPRED [88] is a sequence-based tool for predicting SSs, employing a neural network to filter and convolve the protein evolutionary information generated by PSI-BLAST [89], ultimately outputting a protein SS scoring matrix. Conversely, DSSP [90, 91] calculates the most probable SS distribution by analyzing the protein's tertiary structures.
Proteins that derive from the same ancestral protein typically exhibit similar sequences and are likely to share spatial structures and biological functions [92]. Hence, the unknown structure and function of a protein can be inferred based on other annotated proteins with similar sequences. The alignment of sequences from multiple species facilitates the identification of highly conserved residues. Consequently, protein evolutionary information is beneficial for PPI predictions. Six methods [37, 62, 65, 67, 70, 74] utilize evolutionary information to predict PPIs, and there exists a wide variety of evolutionary information, such as multiple sequence alignment (MSA), sequence profiles, evolutionary distance and more. MSA arranges protein sequences to identify similar regions and is utilized in FoldDock [74], which draws inspiration from AlphaFold [93] and employs MSA to predict PPI complexes [69].
On the other hand, descriptors are employed by >20% of the PPI predictors [59, 61–63, 65, 70], and almost all of these predictors utilize LD. LD is a non-aligned method employed to characterize local protein information. It divides each protein into 10 local regions, with varying lengths and compositions. In addition, proteins contain a lot of functional domains, which can be represented by the identifiers of GO terms, and then they can be encoded as embeddings and served as inputs.
In summary, amino acid binary encodings stand out as the most representative and easily accessible. Electrostatic and hydrophobic interactions, which are dominant in PPIs [57], are the most popular profiles among all physicochemical profiles. The application of structural features depends on the inputs. For instance, spatial structural features are more commonly used in structure-based predictors. Since evolutionary information is related to structures and functions of proteins, it is also often used to improve the predictions of PPIs.
Among all PAIR-pro predictors, we note that most of them rely on either machine learning or DL models. In the early stages, there was a tendency toward using machine learning models. For instance, there are four predictors using support vector machines (SVM), two predictors utilizing random forests (RF), two predictors utilizing extreme learning machines (ELM) and several others using techniques like K-nearest neighbors (KNN). However, DL models have become more and more popular in recent years. From 2018, there were only three methods that used machine learning, compared with eight predictors that used DL. Moreover, the complexity of prediction models is incrementally increasing. For example, DPPI [67] harnesses convolutional neural networks (CNN), DNN-PPI [68] engages both CNN and long short-term memory (LSTM) and TransformerGO [42] applies the Transformer model to predict PPIs. Compared with machine learning models, these DL models achieve more accurate predictions. Furthermore, we find that the area under the ROC curve (AUC) has risen as the predominant assessment criterion currently. On the other hand, to evaluate the binary predictions, precision emerges as one of the most frequently employed assessment criteria.
Furthermore, regarding availability, 11 out of the 24 methods provide available source code (10 methods) or web servers (3 methods). Most predictors provide source code rather than web servers. LR_PPI [60], D-SCRIPT [73] and DeepTrio [41] are the only three predictors that provide currently running web servers. Details of availability are shown in Supplementary Table S1. For the latest several predictors, we evaluated their performance separately based on their inputs. For predictors with GO as inputs, TransformerGO improves the performance on average with 5% across all subsets compared with protein2vec [75]. For predictors with PPI networks as inputs, Kovács et al. proposed the L3-based predictor [71], while Sim combined a new link prediction approach with L3 and proved that Sim is always superior to L3 [72]. For sequence-based predictors, we assessed the performance of DeepTrio [41] and D-SCRIPT [73] on the virus–human interaction dataset [41, 94]. DeepTrio attains an AUC value of 79.9% [41] while D-SCRIPT attains that of 66.5% (Supplementary Table S2).
PAIR-res predictors
Compared with PAIR-pro predictors, PAIR-res predictors focus on the interaction residues between protein pairs, which are summarized in Table 2. There are four PAIR-res predictors: BIPSPI [52], Plnet [95], BIPSPI+ [53] and the predictor proposed by Liu et al. [96]. Similar to PAIR-pro predictors, PAIR-res predictors accept protein pairs as inputs. The difference is that these predictors only accept protein sequences or structures as inputs, instead of PPI networks and GO terms that PAIR-pro predictors would use. In particular, BIPSPI+ extends its inputs beyond just pairs of protein sequences or structures. It also supports a combination of a protein sequence and a protein structure as inputs, with different inputs determining unique prediction patterns within BIPSPI+. Our investigation into the dataset shows PAIR-res predictors typically source interaction proteins and residues from PDB and four predictors utilize the protein–protein Docking Benchmark version 5 (DBv5) dataset [52, 97]. The DBv5 comprises 230 non-redundant protein complexes, all of which have bound and unbound structures. Each complex boasts a resolution better than 3.25 Å, and each sequence exceeds 30 amino acids in length.
Table 2.
Summary of PAIR-res predictors in terms of inputs, feature profiles, models and availability
| Predictor | Inputs | Feature profiles | Models | Year | Availability |
|---|---|---|---|---|---|
| BIPSPI [52] | Seqs/Strs | One-hot encoding, PSSM, PSFM, MSA conservation, sequence length, solvent accessibility, SS | XGBoost | 2018 | √ |
| Liu et al. [96] | Seqs | Amino acids encoding, affinities | GNN | 2020 | × |
| Plnet [95] | Strs | Geometry, electrostatics, hydrophobicity | Geometric DL | 2022 | √ |
| BIPSPI+ [53] | Seqs/Strs/Seqs+Strs | One-hot encoding, PSSM, PSFM, MSA conservation, sequence length, solvent accessibility, SS | XGBoost | 2022 | √ |
Note: Seqs and Strs correspond to protein sequences and protein structures.
PAIR-res predictors extract features such as electrostatics, hydrophobicity, solvent accessibility, SS, position-specific scoring matrix (PSSM), etc. based on sequence or structural information. One-hot encoding and PSSM are the two most frequently utilized profiles. PSSM captures evolutionary information and contains crucial information about amino acid occurrence frequency and variation. It is generated through PSI-BLAST [89] that searches in corresponding databases such as Non-Redundant Protein Sequence Database (NR), NRDB90 [98], Swiss-Prot [99], etc. For a protein sequence of length L, the PSSM of the protein is a matrix with a shape of L × 20.
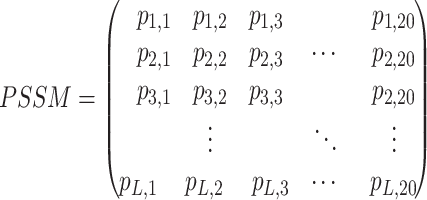 |
(1) |
where  represents the likelihood of the presence of the j amino acid at the i-th position in the protein sequence.
represents the likelihood of the presence of the j amino acid at the i-th position in the protein sequence.
In addition, we note that both machine learning and DL models are prevalent in predicting interaction residues. For instance, BIPSPI utilizes XGBoost, while Plnet and the predictor proposed by Liu et al. leverage graph neural networks (GNN) and geometric DL, respectively.
Furthermore, we examine the availability of source code or web servers of these four methods (Supplementary Table S1). BIPSPI and BIPSPI+ offer web servers and source code, both of which are still working well. Besides, Plnet provides source code. We collected the result of available predictors on DBv5 dataset. As shown in Supplementary Table S2, BIPSPI+ outperforms BIPSPI and Plnet (AUC of 82.3% [52] and 75.3% [95], respectively), achieving the best AUC of 84.8% [53].
SINGLE-res predictors
Unlike PAIR-pro and PAIR-res predictors, SINGLE-res predictors [22, 54, 55, 100–125] accept a single protein as input and infer whether residues in protein sequences interact with other proteins. Table 3 summarizes the main information of SINGLE-res predictors and shows that 19 predictors utilize protein sequences as inputs, while 11 predictors accept protein structures. With the advancement of high-throughput sequencing technologies, protein sequence data are increasing rapidly. Consequently, a large number of sequence-based predictors have been proposed over the past 15 years. Currently, structure-based predictors are also proliferating, which thanks to the advancements in protein structure determination techniques and the emergence of high-quality predictors of protein structures such as AlphaFold [93]. The datasets of SINGLE-res predictors are divided into two classes: structure-annotated proteins and disorder-annotated proteins. Structure-annotated proteins are usually derived from complexes within PDB, while disorder-annotated proteins originate from DisProt [19].
Table 3.
Summary of SINGLE-res predictors in terms of inputs, feature profiles, models and availability
| Predictor | Inputs | Feature profiles | Models | Year | Availability |
|---|---|---|---|---|---|
| Fariselli et al. [100] | Strs | HSSP | ANN | 2002 | × |
| Ofran et al. [101] | Strs | HSSP | ANN | 2003 | × |
| ODA [102] | Strs | Solvent-accessible surface | Surface analysis | 2005 | × |
| Burgoyne et al. [103] | Strs | Hydrophobicity, desolvation, electrostatics, conservative profiles | Surfaces clefts analysis | 2006 | × |
| SPPIDER [104] | Strs | Hydrophobicity, number of contacts, PSSM, amino acid frequencies, entropies, charge, size of side chain, hydrophobicity, the level of surface exposure, the number and distances between surface, the difference between the predicted and observed in an unbound structure surface exposure of an amino acid residue | ML | 2007 | √ |
| ISIS [105] | Seqs | Evolutionary profiles, solvent accessibility, SS | ANN | 2007 | × |
| ANCHOR [106] | Seqs | Amino acids encoding, disorder regions | Pairwise energy estimation | 2009 | √ |
| Raf–Ras [107] | Seqs | SS, average hydrophobicity, accessible surface area, average depth index, average protrusion index, minimal protrusion index, maximal protrusion index, maximal depth index | RF | 2009 | × |
| PSIVER [108] | Seqs | PSSM, relative solvent accessibility | Naïve Bayes classifier | 2010 | √ |
| SPRINGS [109] | Seqs | PSSM, hydropathy, relative solvent accessibility | ANN | 2014 | × |
| LORIS [110] | Seqs | PSSM, hydropathy, relative solvent accessibility | L1-regularized LR | 2014 | × |
| CRF_PPI [111] | Seqs | PSSM, averaged cumulative hydropathy, relative solvent accessibility | RF | 2015 | √ |
| DC-RF-RUS-RF [112] | Seqs | PSSM, averaged cumulative hydropathy, relative solvent accessibility | RF | 2016 | √ |
| SSWRF [113] | Seqs | PSSM, averaged cumulative hydropathy, averaged cumulative relative solvent accessibility | RF, SVM | 2016 | √ |
| RF_PPI [114] | Seqs | Sequence entropy, sequence specificity score, sequence length and HSP length, backbone dynamics, accessibility, SS | RF | 2017 | √ |
| DeepSite [115] | Strs | Hydrophobic, aromatic, hydrogen bond acceptor or donor, positive or negative ionizable and metallic | CNN | 2017 | √ |
| EL-SMURF [116] | Seqs | PSSM-SPF, RER | RF | 2019 | √ |
| SCRIBER [117] | Seqs | Putative protein-binding intrinsically disordered regions, SS, aliphaticity, aromaticity, acidity and size, relative solvent accessibility, evolutionary conservation, relative amino acid propensity | LR | 2019 | √ |
| SASNet [118] | Strs | Atom encoding | CNN | 2019 | √ |
| DLPred [119] | Seqs | PSSM, physical properties (a steric parameter, polarizability, volume, hydrophobicity, isoelectric point, helix probability, sheet probability), hydrophobicity scales, physicochemical characteristics (the number of atoms, electrostatic charges and potential hydrogen bonds), PKx, 3D-1D scores, conservation score, one-hot encoding | LSTM | 2019 | × |
| Deng et al. [120] | Seqs | Residue space sequence, sequence information entropy, relative entropy, residue sequence weight,and residue conservative fraction | XGBoost | 2020 | × |
| DeepPPISP [22] | Seqs | PSSM,SS, sequence | CNN | 2020 | √ |
| ProNA2020 [54] | Seqs | SS, predicted relative solvent accessibility, bio-physical properties of amino acids | Homology, SVM, ProtVec, ANN | 2020 | √ |
| MaSIF-site [121] | Strs | Solvent excluded surface, shape index, distance-dependent curvature, hydropathy index, continuum electrostatics, the location of free electrons and proton donors | CNN | 2020 | √ |
| GraphPPIS [1, 122] | Strs | PSSM, HMM, DSSP | GNN | 2021 | √ |
| DELPHI [55] | Seqs | High-scoring segment pair, 3-mer amino acid embedding, position information, PSSM, evolutionary conservation, putative relative solvent accessibility, relative amino acid propensity, putative protein-binding disorder, hydropathy index, physicochemical characteristics, physical properties, PKx | CNN, RNN | 2021 | √ |
| EGRET [122] | Strs | Distance and relative orientation between the residues, protein language model | Transfer learning | 2022 | √ |
| ScanNet [123] | Strs | Amino acid encoding, SS, accessible surface area, surface convexity and evolutionary conservation | DL | 2022 | √ |
| ProB-Site [124] | Seqs | SS, HMM, PSSM | CNN | 2022 | √ |
| hybridPBRpred [125] | Seqs | SCRIBER [117], DisoRDPbind [126] | Fusion | 2022 | √ |
Note: Seqs and Strs correspond to protein sequences and protein structures.
Similar to PAIR-pro and PAIR-res predictors, SINGLE-res predictors also utilize amino acid binary encodings, physicochemical profiles, structural features and evolutionary information. Furthermore, SINGLE-res predictors employ more extensive protein feature profiles to predict PPI residues. For example, DELPHI [55] uses 3-mer amino acid encoding to characterize proteins. Besides, some predictors [54, 55, 103, 104, 107, 111–113, 115, 117, 119] incorporate a broader range of physicochemical profiles, such as desolvation energy, aliphaticity, aromaticity, acidity and so on [126]. In summary, >66% of predictors use physicochemical profiles to improve predictions. For evolutionary information, the predictors proposed by Fariselli et al. [100] and Ofran et al. [101] utilize features from the homology-derived structures of proteins (HSSP) database [127]. The HSSP database integrates information from one-dimensional sequences and three-dimensional protein structures and aligns each protein with known 3D structures in the PDB with all its probable sequence homologs. HSSP is valuable for analyzing residue conservation in structures and for studying protein evolution and folding. The Hidden Markov Model (HMM) is another evolutionary information widely used in predicting PPI residues, as seen in GraphPPIS [1] and ProB-Site [124]. For a protein sequence of length L, the HMM matrix is shaped as L × 30, where each residue is represented by a 30-dimensional feature vector. HMM matrix is typically normalized using the following formula:
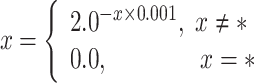 |
(2) |
In contrast to previous works using PSSM, EL-SMURF [116] adopts a different approach. It utilizes the fusion of sequence profile feature in PSSM (PSSM-SPF) and residue evolution rate (RER) to extract features of neighboring residues with a sliding window. It is worth noting that EGRET [122] generates protein embeddings using the self-supervised protein language model ProtTrans [128]. In fact, feature profiles generated by protein language models have some advantages over traditional ones. First, protein language models enable faster feature generation compared with some traditional evolutionary features like PSSM and HMM, which require extensive time for matching. Second, protein language models can generate features for all target proteins, while traditional feature generation tools like PSI-BLAST sometimes fail to match rare proteins and generate feature profiles, which is noticeable when searching small databases like Swiss-Prot. Similar to PAIR-pro and PAIR-res, most SINGLE-res predictors utilize machine learning models at an early stage. With the development of protein databases and DL technology, DL-based predictors are increasingly available. In the past 5 years, >64% of predictors are based on DL, and their model architectures are becoming more and more complex.
Interestingly, cross-prediction rate (CPR) [117] is used to evaluate cross-predictions of other types of interaction residues into protein interaction residues, quantifying the extent to which the model confuses different types of interaction residues. Based on CPR, the area under cross-prediction curve (AUCPC) is also introduced, where the cross-prediction curve is a relation of CPR against recall. Besides, given the imbalance in the dataset, SCRIBER [117] also quantifies the area under the low false positive rate ROC curve (AULC) and normalizes it by dividing the AULC value of the target predictor by the AULC value of the random predictor. Furthermore, we assessed the availability of SINGLE-res predictors (Supplementary Table S1). Out of 30 predictors, 20 predictors are now available to users. Four methods offer user-friendly web platforms, nine provide source code and seven provide both web servers and source code. Besides, on Test_60 dataset [124], we assessed the performance of available predictors published in 2020 and beyond, including DeepPPISP [22], ProNA2020 [54], MaSIF-site [121], GraphPPIS [122], DELPHI [55], ScanNet [123], ProB-Site [124] and hybridPBRpred [125]. As shown in Supplementary Table S2, ProB-Site achieves the best performance with an AUC of 84.4% [124].
PROTEIN–NUCLEIC ACID INTERACTION
We survey 77 protein–nucleic acid interaction predictors and classify them into two categories: (i) protein-level [129, 130] and (ii) residue-level [24, 35], as illustrated in Figure 4. In contrast to PPI predictors, the majority of protein-level predictors for protein–nucleic acid interactions primarily focus on determining whether proteins interact with DNA or RNA, without providing annotations of the specific interaction partners. Therefore, our investigation into protein-level predictors mainly centers around the latest methods proposed since 2018.
Figure 4.

The distribution of protein–nucleic acid interaction predictors at different prediction levels (protein-level, residue-level, both) and for different ligand types (DNA, RNA, both).
A total of 14 protein-level predictors for protein–nucleic acid interactions are investigated, comprising 7 protein–DNA interaction predictors [129–135], 6 protein–RNA interaction predictors [136–141] and 1 predictor for protein–nucleic acid interactions [142], as illustrated in Figure 5. We conduct a comprehensive analysis of the protein feature profiles and availability.
Figure 5.

Predictors of protein–nucleic acid interactions at the protein-level. Protein–RNA interaction predictors are in the left circle, protein–DNA interaction predictors are in the right circle and protein–nucleic acid interaction predictors are in the middle cross section.
PSSM is the most commonly used feature profile among the available options. Notably, iDRBP_MMC [129] and DeepDRBP-2L [130] only employ PSSM as input features for predictions. IDRBP-PPCT [142] combines PSSM with the position-specific frequency matrix (PSFM). PSFM which is also derived from PSI-BLAST, contains protein evolutionary information, indicating the occurrence frequency of residues at specific positions in the protein sequence. Unlike PSSM, PSFM does not account for the mutation probability of residues. Studies have indicated that interaction residues of proteins tend to be evolutionarily conserved [84, 143], which is why evolutionarily conserved features like PSSM are widely used in predicting protein–nucleic acid interactions. Additionally, various PSSM-based feature profiles are employed in protein–nucleic acid interactions. For instance, DBP-DeepCNN [134] reduces PSSM complexity and generates global patterns, using discrete wavelet transform (DWT) for de-noising to create a new protein feature profile, R-PSSM-DWT. PlDBPred [135] considers 10 different PSSM-based feature profiles, including PSSMBLOCK, AADP-PSSM, PSSM-DWT and so on. On the other hand, RBP-TSTL [141] also uses the protein language model to generate embeddings. For availability, four out of seven protein–DNA interaction predictors, five out of six protein–RNA interaction predictors and the predictor for protein–nucleic acid interactions are available to users (Supplementary Table S1).
Additionally, we have summarized 63 methods for predicting protein–nucleic acid interactions at residue-level, which are shown in Table 4 [144]. This comprehensive summary considers various aspects such as ligands, inputs, feature profiles, models and availability. For ligands, we observe that 24 methods are exclusively designed for predicting protein–DNA interaction residues [39, 132, 145–166], while 26 methods focus solely on predicting protein–RNA interaction residues [24, 36, 40, 167–189]. Furthermore, 13 methods are capable of identifying both protein–DNA and protein–RNA interaction residues [3–5, 35, 54, 126, 190–196]. It is worth noting that BindN [190] is the earliest method to provide simultaneous predictions for both types of residues, which was published in 2006. Interestingly, among the eight methods proposed since 2021, five of them offer predictions for both DNA-interaction and RNA-interaction residues. Furthermore, the predictor proposed by Wang et al. [157] predicts DNA-type-specific interaction residues such as single-stranded DNA and double-stranded DNA. DNAgenie [166] takes into account A-DNA (common double-stranded DNA subtype), B-DNA (the most abundant double-stranded DNA conformation) and single-stranded DNA. It is an innovative research trend to predict multiple interaction residues and fine-grained specific interaction residues.
Table 4.
Summary of residue-level protein–nucleic acid interaction predictors in terms of ligands, inputs, feature profiles, models and availability
| Ligands | Predictor | Inputs | Feature profiles | Models | Year | Availability | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| AAE | EVO | SS | RSA | PC | ||||||
| DNA | DBS-pred [145] | Seqs | × | × | × | √ | × | ML (NN) | 2004 | × |
| DBS-PSSM [146] | Seqs | × | √ | × | × | × | ML (NN) | 2005 | × | |
| DNABindR [147] | Seqs | √ | × | × | × | × | ML (Naïve Bayes) | 2006 | × | |
| Ho et al. [148] | Seqs | × | √ | × | × | × | ML (SVM) | 2007 | × | |
| DP-Bind [149] | Seqs | √ | √ | × | × | × | ML (SVM, KLP, PLR) | 2007 | √ | |
| DISIS [150] | Seqs | × | √ | √ | √ | × | ML (SVM, NN) | 2007 | × | |
| BindN-RF [151] | Seqs | × | √ | × | × | √ | ML (RF) | 2009 | × | |
| DBindR [152] | Seqs | √ | √ | √ | × | √ | ML (RF) | 2009 | × | |
| DBD-Threader [39] | Seqs | × | × | × | × | × | Homology | 2009 | × | |
| DNABR [153] | Seqs | √ | √ | × | × | √ | ML (RF) | 2012 | × | |
| Dey et al. [154] | Seqs | × | √ | × | × | √ | ML (SVM) | 2012 | × | |
| DNABind [155] | Seqs/Strs | × | √ | √ | √ | √ | ML (SVM), Homology | 2013 | √ | |
| SPOT-Seq (DNA) [156] | Seqs | × | × | × | × | × | Homology | 2014 | × | |
| Wang et al. [157] | Seqs | × | √ | √ | × | √ | ML (SVM) | 2014 | × | |
| PDNAsite [158] | Seqs | √ | √ | √ | √ | √ | ML (LSA, SVM, ensemble learning) | 2016 | × | |
| Local-DPP [159] | Seqs | × | √ | × | × | × | ML (RF) | 2017 | × | |
| TargetDNA [160] | Seqs | × | √ | × | √ | × | ML (SVM) | 2017 | √ | |
| StackDPPred [132] | Seqs | × | √ | × | × | × | ML(Stacking) | 2019 | √ | |
| DNAPred [161] | Seqs | × | √ | √ | √ | × | ML (SVM, AdaBoost) | 2019 | √ | |
| iProDNA-CapsNet [162] | Seqs | × | √ | × | × | × | DL (capsule neural network) | 2019 | √ | |
| EL_LSTM [163] | Seqs | √ | √ | √ | √ | √ | DL (LSTM, bagging, ensemble learning) | 2020 | × | |
| TargetDBP [164] | Seqs | × | √ | × | √ | × | ML(SVM) | 2020 | √ | |
| funDNApred [165] | Seqs | × | √ | × | √ | × | ML(FCM) | 2020 | × | |
| DNAgenie [166] | Seqs | × | √ | √ | √ | √ | ML | 2021 | √ | |
| RNA | Jeong et al. [167] | Seqs | √ | × | √ | × | × | ML (NN) | 2004 | × |
| Jeong et al. [168] | Seqs | × | √ | × | × | × | ML (NN) | 2006 | × | |
| RNABindR [169] | Seqs | √ | × | × | × | × | ML (Naïve Bayes) | 2007 | × | |
| PRINTR [170] | Seqs | × | √ | √ | √ | × | ML (SVM) | 2008 | × | |
| RISP [171] | Seqs | × | √ | × | × | × | ML (SVM) | 2008 | × | |
| Pprint [172] | Seqs | × | √ | × | × | × | ML (SVM) | 2008 | × | |
| RNAProB [173] | Seqs | × | √ | × | × | × | ML (SVM) | 2008 | × | |
| PiRaNhA [174] | Seqs | × | √ | × | √ | √ | ML (SVM) | 2010 | × | |
| ProteRNA [175] | Seqs | × | √ | √ | × | × | ML (SVM) | 2010 | × | |
| RBRpred [176] | Seqs | √ | √ | √ | √ | × | ML (SVM) | 2010 | × | |
| PRNA [177] | Seqs | × | √ | √ | √ | √ | ML (RF) | 2010 | × | |
| OPRA [178] | Strs | × | × | × | √ | × | Computational | 2010 | × | |
| Wang et al. [179] | Seqs | × | √ | × | √ | √ | ML (SVM) | 2011 | × | |
| PRBR [180] | Seqs | × | √ | √ | × | √ | ML (RF) | 2011 | × | |
| SPOT-Seq [40] | Seqs | × | × | × | × | × | Homology | 2011 | × | |
| Choi et al. [181] | Seqs | × | × | × | √ | √ | ML (SVM) | 2011 | × | |
| RNABindRPlus [182] | Seqs | × | √ | × | × | × | ML (SVM, LR), Homology | 2014 | × | |
| DR_bind1 [183] | Seqs | × | √ | × | √ | √ | Computational | 2014 | √ | |
| aaRNA [184] | Seqs/Strs | √ | √ | √ | √ | √ | ML (NN) | 2014 | × | |
| Ren et al. [185] | Strs | × | √ | × | × | × | ML (ensemble learning) | 2015 | × | |
| PRIdictor [186] | Seqs | × | × | × | √ | √ | ML (SVM) | 2016 | × | |
| RNAProSite [187] | Strs | × | √ | √ | √ | √ | ML (RF) | 2016 | × | |
| PredRBR [188] | Strs | × | √ | √ | √ | √ | ML (GTB) | 2017 | × | |
| RPI-Bind [189] | Strs | × | √ | × | √ | √ | ML (RF, SVM, NN) | 2017 | × | |
| PST-PRNA [36] | Strs | × | √ | √ | √ | √ | DL | 2022 | √ | |
| HybridRNAbind [24] | Seqs | √ | √ | √ | √ | √ | ML(RF) | 2023 | √ | |
| DNA and RNA | BindN [190] | Seqs | × | × | × | × | √ | ML (SVM) | 2006 | × |
| BindN+ [191] | Seqs | × | √ | × | × | √ | ML (SVM) | 2010 | × | |
| NAPS [192] | Seqs | √ | √ | × | × | √ | ML (C4.5) | 2010 | × | |
| SNBRFinder [193] | Seqs | × | √ | √ | √ | √ | ML (SVM), Homology | 2015 | × | |
| DisoRDPbind [126] | Seqs | √ | × | √ | × | √ | ML (LR) | 2015 | √ | |
| DRNApred [4] | Seqs | √ | √ | √ | √ | √ | ML (LR) | 2017 | √ | |
| NucBind [194] | Seqs | × | √ | √ | × | × | ML(SVM), Homology | 2019 | √ | |
| ProNA2020 [54] | Seqs | × | × | √ | √ | √ | ML (SVM, ANN, ProtVec), Homology | 2020 | √ | |
| NCBRPred [195] | Seqs | × | √ | √ | √ | × | DL | 2021 | √ | |
| MTDsite [196] | Seqs | × | √ | × | √ | × | DL | 2021 | √ | |
| GraphBind [5] | Strs | × | √ | √ | × | √ | DL(GNN) | 2021 | √ | |
| DeepDISOBind [35] | Seqs | √ | × | √ | × | × | DL | 2022 | √ | |
| iDRNA-ITF [3] | Seqs | × | √ | √ | × | √ | DL | 2022 | √ | |
Note: Seqs and Strs correspond to protein sequences and protein structures.
AAE, PC, RSA, SS and EVO correspond to amino acid binary encodings, physicochemical profiles, relative solvent accessibility, SS and evolutionary information.
Based on the inputs, we divide the predictors into sequence-based [24] and structure-based [5] predictors. The inputs of 54 predictors are protein sequences, while only 7 predictors infer interaction residues based on protein structures. Furthermore, two predictors [155, 184] simultaneously support both protein sequences and protein structures. The scarcity of structure-based predictors can be attributed to the limited quantity of high-quality protein structure data in prior years. In recent years, the development of protein structure determination techniques and predictors of protein structures such as AlphaFold has provided structure information for a wider range of proteins and led to the development of structure-based predictors.
Then, we find that the datasets of protein–nucleic acid interaction residues are mainly from PDB and BioLip and are generally unbalanced, with positive samples less than negative samples. A recent representative dataset is the one organized by GraphBind [5], which is collected from the BioLip database. Due to the small number of interaction residues, GraphBind applies data augmentation on the dataset, uses bl2seq [197] and TM-align [198] to evaluate the sequence identity and structural similarity between proteins, and then clusters them. The annotation of a protein in the same cluster is transferred to the protein with the largest number of residues to increase the number of interaction residues.
According to the investigation, similar to the previous section, we divide all protein feature profiles into the following five categories: (i) amino acid binary encodings; (ii) physicochemical profiles; (iii) relative solvent accessibility; (iv) SS; (v) evolutionary information, as illustrated in Figure 6. We have observed that almost all the predictors utilize at least one kind of the above features. Evolutionary information is the most popular feature profile, which mainly includes PSSM, HMM, other PSSM-based features and so on. Among these evolutionary features, PSSM is the most representative. Half of the predictors apply physicochemical profiles which are the second commonly used profiles. Besides, tools like ASAquick [199], ACCpro [200], and others are capable of generating the relative solvent accessibility, which is typically presented as an L*1 matrix for a protein of length L and used by 30 predictors. SS is used by 44.4% of predictors, which is mainly generated by PSIPRED and DSSP. Finally, 15 predictors utilize amino acid binary encodings, which are the most direct and easiest to obtain. The most commonly used amino acid binary encodings, described in detail in the PPI section, is to represent each residue with a 20-dimensional vector using one-hot encoding.
Figure 6.

The distribution of features profiles used by predictors of residue-level protein–nucleic acid interactions.
Furthermore, we summarize the models used in 63 predictors, as shown in Table 4. The majority of these predictors (50/63) employ machine learning models, whereas six out of eight predictors proposed after 2021 utilize DL models. It can be seen that DL models have become a research trend for predicting protein–nucleic acid interaction residues. It is worth noting that some protein–nucleic acid interaction residue predictors are based on homology, such as DBD-Threader [39], SPOT-Seq [40], SNBRFinder [193], NucBind [194] and so on. Homology-based methods match the target protein with annotated proteins in the library, and transfer the annotation of similar proteins into the target protein. In addition, GraphBind [5] represents protein structure data and feature profiles as graphs and mines them using GNN, which provides new insights for subsequent related studies. In addition, we find that MCC is the most popular comprehensive assessment criterion and AUC is the most commonly used criterion to evaluate the prediction propensities. Besides, DNAgenie [166] and HybridRNAbind [24] utilize over-prediction rate (OPR) and the area under the over-prediction curve (AUOPC) to evaluate whether a predictor tends to predict residues that don’t interact with ligands as nucleic acid interaction residues.
Finally, in terms of availability, 50 of the 63 predictors provide web servers or source code at the time of publication, and 20 are currently available (Supplementary Table S1). Most early-published predictors are not accessible to users. Among the 31 predictors proposed before 2013, only DP-Bind [149] is still working. Luckily, all eight predictors proposed in 2021 and later can be obtained. For DNA interaction residue predictions, we recommend DNAgenie [166] for fine-grained predictions and DeepDISOBind [35] (AUC of 73.6% [35]) for predictions involving intrinsically disordered proteins compared with DisoRDPbind [126] (AUC of 67.1% [35]). Besides, we assessed the performance of ProNA2020 [54], NCBRPred [195], GraphBind [5] and iDRNA-ITF [3] on DNA-129_Test [3, 5]. iDRNA-ITF achieves the highest AUC of 88.3%, followed by GraphBind (85.5%) and NCBRPred (82.3%) [3]. On the other hand, for RNA interaction residue predictions, we evaluated eight predictors on the test dataset of HybridRNAbind [24], including the structure-annotated and the disorder-annotated proteins. HybridRNAbind is the best-preforming predictor with an AUC of 73.8% [24], followed by iDRNA-ITF (70.0%). Details are shown in Supplementary Table S2.
PROTEIN–PEPTIDE INTERACTION
We investigate nine predictors of protein–peptide interactions, as shown in Table 5. Six protein–peptide interaction predictors accept only proteins as inputs and identify interaction residues in proteins without focusing on specific peptide partners. Specifically, three predictors (SPRINT-Seq [23], PepBind [201] and PepBCL [6]) accept protein sequences as inputs and three predictors (SPRINT-Str [202], PEPSITE [203] and PeptiMap [204]) are based on protein structures. In particular, InterPep [44] and PepNN [43] accept both protein and peptide to make residue-level predictions. It is also worth noting that CAMP [205] takes both protein sequences and peptide sequences as inputs, not only to infer interactions between peptides and proteins, but also to identify their interaction residues in peptides. For datasets, we find that most of the datasets mainly come from PEPSITE, SPRINT-Seq and SPRINT-Str. PDB, BioLiP and DrugBank [206] are the three most commonly used databases.
Table 5.
Summary of protein–peptide interaction predictors in terms of inputs, feature profiles, models and availability
| Predictor | Inputs | Feature profiles | Models | Year | Availability |
|---|---|---|---|---|---|
| PEPSITE [203] | Strs | S-PSSMs | Computational algorithms | 2009 | × |
| PeptiMap [204] | Strs | Functional unit classification, receptor classification (CATH, MSA) | Fragment mapping | 2013 | × |
| SPRINT-Seq [23] | Seqs | One-hot encoding, PSSM, accessible surface area, SS, steric parameter, hydrophobicity, volume, polariz-ability, isoelectric point, helix probability, sheet probability | SVM | 2016 | × |
| SPRINT-Str [202] | Strs | PSSM, SS, amino acid encoding, half sphere exposure, flexibility | RF | 2018 | × |
| PepBind [201] | Seqs | Intrinsic disorder-based features, SS, PSSM, HMM, TM-SITE [207], S-SITE [207] | SVM | 2018 | √ |
| InterPep [44] | Protein Strs+ Peptide Seqs |
TM-align (length, quality), amino acid composition distance, SS, surface information, template peptide information, model information | Homology, cluster, RF | 2019 | √ |
| CAMP [205] | Protein Seqs +Peptide Seqs |
Residue-level structural and physicochemical properties, PSSM, intrinsic disorder tendencies | CNN, attention | 2021 | √ |
| PepNN [43] | Protein Seqs/strs +Peptide Seqs |
Graph representation (amino acid one-hot encoding, residue distance, rotation, residue relative position, torsional backbone angle), protein language model | DNN, transfer learning | 2022 | √ |
| PepBCL [6] | Seqs | Protein language model | Contrastive learning | 2022 | √ |
Note: Seqs and Strs correspond to protein sequences and protein structures.
Because peptides and proteins have many similarities in composition and properties, some feature profiles are also widely used by protein–peptide interaction predictors, such as amino acid binary encodings, evolutionary information (PSSM, HMM, MSA), physicochemical profiles (hydrophobicity), structural features (SS, solvent accessibility). At the same time, some predictors use spatial position specific scoring matrices (S-PSSMs) [203] and template peptide information [44]. In particular, PepNN [43] and PepBCL [6] obtain sequence embedding features by using the protein language model instead of the traditional protein features.
These methods differ in their respective models. Similar to the predictors in previous sections, most methods also employ machine learning to simulate protein–peptide interaction residues. However, PEPSITE [203] and PeptiMap [204] introduce novel algorithms for protein–peptide interaction predictions. PEPSITE uses S-PSSM to infer interactions from known protein–peptide complexes, while PeptiMap computes interaction residues based on fragment mapping. Additionally, PepBind [201] presents a new consensus-based method that combines SVMpep [201] with two homology-based predictors, S-SITE and TM-SITE [207]. In addition, we assess the availability of their web servers and source code, as shown in Supplementary Table S1. Regrettably, only two web servers (PepBind and PepBCL) are currently available, and only four predictors (InterPep, CAMP, PepNN and PepBCL) provide source code for local deployment. Finally, we assessed the performance of available predictors (Supplementary Table S2). According to inputs and motivation, these predictors are classified into three groups: (i) PepBind and PepBCL, which take proteins as inputs and infer interaction residues in proteins, (ii) InterPep and PepNN, which use proteins and peptides as inputs and predict interaction residues in proteins, and (iii) CAMP, to make protein-level predictions or to infer interaction residues in peptides. As reported in PepBCL, PepBCL achieves an AUC of 84.1% on TE125 dataset compared with PepBind (AUC of 79.3%) [6]. The result in PepNN shows that the AUC of PepNN is 83.3%, which is 4% higher than InterPep [43].
PROTEIN–OTHER LIGANDS INTERACTION
Proteins also interact with a variety of other ligands, such as nucleotides, heme, ions, etc. We conduct investigations on them and collect 33 protein–other ligands interaction predictors. It is noteworthy that 32 methods are residue-level predictors [5, 7–10, 45–51, 208–227], aiming to predict the ligand-interaction residues in proteins. In contrast, mebipred [25] is the only protein-level predictor, which can identify whether the protein can interact with Ca, Co, Cu, Fe, K, Mg, Mn, Na, Ni and Zn. Three sequence-derived features are used by the multi-layer perceptron-based predictor, including amino acid composition, physicochemical properties and a count of the metal-binding amino acid 5mers [25]. In this section, we focus on residue-level predictors and the main information of them is summarized in Table 6. According to ligands, residue-level protein–other ligands interaction predictors can be mainly divided into three categories: protein-nucleotide (including ATP, ADP, AMP, GTP, GDP, etc.) interaction residue predictors [8, 45, 46], protein-heme interaction residue predictors [9, 47, 48] and protein-ion (Ca, Mg, Mn, etc.) interaction residue predictors [49–51]. As a result, there are 16 predictors for protein–nucleotide interactions, 7 predictors for protein-heme interactions and 17 predictors for protein-ion interactions, as illustrated in Figure 7. It is worth noting that TargetS [7], TargetCom [219], DELIA [225] and GraphBind [5] are capable of providing simultaneous predictions of protein–nucleotide, protein−heme and protein−ion interaction residues. In the following, protein–other ligands interaction residue predictors will be summarized according to the categories.
Table 6.
Summary of protein–others interaction predictors in terms of ligands, inputs, feature profiles, models and availability
| Predictor | Ligands | Inputs | Feature profiles | Models | Year | Availability |
|---|---|---|---|---|---|---|
| CHED [208] | Co, Cu, Fe, Mn, Ni, Zn | Strs | Geometric search | ML (SVM), decision tree | 2008 | × |
| ATPint [209] | ATP | Seqs | PSSM, Hydrophobicity, Beta-Sheet, Polarity, Solvation potential, Residue interface propensities, Net charge, Average accessible surface area | ML(SVM) | 2009 | √ |
| GTPBinder [210] | GTP | Seqs | One-hot, PSSM | ML(SVM) | 2010 | √ |
| FINDSITE-metal [211] | Ca, Co, Cu, Fe, Mg, Mn, Ni, Zn | Strs | Protein structure modeling | ML(SVM), Homology | 2011 | × |
| Firoz et al. [212] | AMP, ADP, ATP, GMP, GDP, GTP | Seqs | PSSM | ML(SVM) | 2011 | × |
| HemeBIND [47] | HEME | Strs | PSSM, relative accessible surface area (RASA), Depth index (DPX), Protrusion index (CX) | ML(SVM) | 2011 | × |
| hemeNet [48] | HEME | Seqs/Strs | Structural analysis of heme proteins: implications for design and prediction. PSSM, RASA, depth index (DPX), protrusion index (CX). | ML(SVM) | 2011 | √ |
| pfinder [213] | Phosphate | Strs | Position with respect to the solvent accessible surface, clefts on the surface of the protein. | Homology | 2011 | × |
| MetalDetector [214] | Ions | Seqs | HMM | ML (SVM, NN) | 2011 | × |
| ATPsite [215] | ATP | Seqs | PSSM, SS, RSA, conservation scores, amino acid (AA) groups, dihedral angle, Terminal indicator, SS segment indicator for helix/ strand/ coil, Collocation of AA pairs | ML(SVM) | 2011 | × |
| NsitePred [45] | ATP, ADP, AMP, GTP, GDP | Seqs | Sequence, SS, RSA, dihedral angles, PSSM, Terminus indicator, SS segment indicators for helix/strand/coil, Residue conservation scores, collocation of significant AA pairs | ML(SVM) | 2012 | √ |
| TargetS [7] | ATP, ADP, AMP, GDP, GTP, Ca, Mn, Mg, Fe, Zn, HEME | Seqs | PSSM, SS, Ligand-Specific Binding Propensity Feature | Ensemble method (SVM, AdaBoost), cluster | 2013 | × |
| TargetATP [216] | ATP | Seqs | PSSM, SS | Ensemble method (SVM, AdaBoost) | 2013 | √ |
| TargetATPsite [217] | ATP | Seqs | PSSM, Sparse representation of evolution image. | Ensemble method (ATP, AdaBoost) | 2013 | × |
| TargetSOS [8] | ATP, ADP, AMP, GTP, GDP | Seqs | PSSM, SS | ML(SVM, Supervised Over-sampling) | 2014 | √ |
| mFASD [218] | Ca, Cu, Fe, Mg, Mn, Zn | Strs | Functional atoms、local chemical environment、distance between two functional atoms、distance between two functional atom sets | Novel computational algorithms | 2015 | × |
| TargetCom [219] | Cu, Fe, Zn, (SO4)2−, (PO4)3−, ATP, FMN, HEME | Seqs | PSSM, SS, RSA, torsion angles, Conservation scores, COFACTOR [232], TM-SITE [207], S-SITE [207] and COACH [207] | Ensemble method (SVM, AdaBoost) | 2016 | × |
| IonCom [220] | Zn, Cu, Fe, Ca, Mg, Mn, Na, K, (NO2)−, (CO3)2−, (SO4)2−, (PO4)3− | Seqs/Strs | PSSM, SS, RSA, backbone torsion angles, position and segment specific conservation scores, ligand-specific binding propensity. COFACTOR [232], TM-SITE [207], S-SITE [207] and COACH [207] | Ensemble method (SVM, AdaBoost) | 2016 | √ |
| TargetNUCs [221] | ATP, ADP, AMP, GTP, GDP | Seqs | PSSM, SS | Ensemble method | 2016 | √ |
| ATPbind [222] | ATP | Strs | PSSM, SS, solvent accessibility, TM-SITE [207], S-SITE [207] | Ensemble method (SVM) | 2018 | √ |
| Wang et al. [223] | Zn, Cu, Fe, Ca, Mg, Mn, Na, K, Co, (NO2)−, (CO3)2−, (SO4)2−, (PO4)3− | Seqs | Component information, position conservation information, hydropathy, polarization charge, SS, relative solvent accessibility | SMO | 2019 | × |
| Liu et al. [224] | (NO2)−, (CO3)2−, (SO4)2−, (PO4)3− | Seqs | The composition information of amino acid, polarization charge, hydrophilic-hydrophobic, SS and relative solvent availability | KNN | 2019 | × |
| SeqD-HBM [9] | HEME | Seqs | Net charge, solvent accessibility | Stepwise validation | 2019 | × |
| DELIA [225] | Ca, Mn, Mg, ATP, HEME | Strs | PSSM, SS, HMM, RSA, S-SITE-based feature, structure-based distance matrix | DL | 2020 | √ |
| PBSP [226] | Phosphate | Strs | AutoDockFR [233], AutoSite [234] | Energy-based、reverse focused docking | 2021 | √ |
| ATPensemble [227] | ATP | Seqs | PSSM, SS, one-hot | Ensemble method (CNN, LightGBM), Homology | 2021 | √ |
| DeepATPseq [46] | ATP | Seqs | PSFM | Ensemble method (CNN. SVM) | 2021 | √ |
| GraphBind [5] | Ca, Mn, Mg, ATP, HEME | Strs | Pseudo-positions, atom mass, B-factor, whether it is a residue side-chain atom, electronic charge, the number of hydrogen atoms bonded to it, whether it is in a ring, and the van der Waals radius of the atom, SS, PSSM, HMM | DL(GNN) | 2021 | √ |
| MetalSiteHunter [50] | Ca, Fe, Mg, Mn, Zn, Na | Strs | 3D voxels, positive_ionizable, hbond_acceptor, occupancies, negative_ionizable and hbond_donor | DL (3D CNN) | 2022 | √ |
| GASS-Metal [51] | Zn, Ca, Mg, Mn, Cu, Fe, Co, Na, K, Cd, Ni | Strs | Residue position, substitution matrix to handle conservative mutations | Homology, genetic algorithms | 2022 | √ |
| MIB2 [49] | Ca, Cu, Mg, Mn, Zn, Cd, Fe, Ni, Hg, Co, Au, Ba, Pb, Pt, Sm, Sr | Seqs/Strs | BLOSUM62 substitution matrix, weighted contact number of each metal ion | (PS)2 | 2022 | √ |
| LMetalSite [10] | Zn, Ca, Mg, Mn | Seqs | Protein language model | DL | 2022 | √ |
Note: Seqs and Strs correspond to protein sequences and protein structures.
Figure 7.

Protein–other ligands interaction predictors are divided into four categories according to ligand types, with the middle predictors providing protein–nucleotide, heme, ion interaction predictions simultaneously.
Protein–nucleotide interaction
Our investigation reveals that almost 15 out of 16 protein–nucleotide interaction predictors involve protein-adenosine triphosphate (ATP) interactions. This prevalence could be attributed to ATP's biological significance, as it serves as the primary energy source in living organisms. A total of 13 predictors utilize protein sequences as inputs, whereas only GraphBind [5], DELIA [225] and ATPbind [43] are based on protein structures. PDB and BioLiP serve as the primary data sources for these predictors. Several high-quality datasets are commonly used, including those from ATPint [209], ATPsite [215] and ATPbind [43]. An analysis of the protein feature profiles shows that PSSM is used most frequently in protein–nucleotide interaction residue predictors. Further investigation into the models reveals ensemble models (8 out of 15) and SVM (10 out of 15) as the dominant choices. We show that the models integrated in ensemble models are different. For instance, ATPensemble [227] combines a deep CNN with the LightGBM algorithm, while DeepATPseq [46] pairs a deep CNN with SVM. Considering the availability, most provide web servers or source code, and now 11 links remain valid, allowing the prediction of 5 kinds of nucleotides: ATP, ADP, AMP, GTP and GDP. It is a great pity that predictions for other nucleotides such as CMP and PCG are currently inaccessible to users. Finally, we collected the protein-ATP interaction prediction performance of GraphBind, DeepATPseq, ATPensemble and DELIA on PATP-TEST dataset [46]. DeepATPseq achieves the best performance, followed by ATPensemble.
Protein–heme interaction
We investigate seven protein–heme interaction residue predictors: HemeBIND [47], hemeNet [48], TargetS [7] and TargetCom [219], SeqD-HBM [9], DELIA [225] and GraphBind [5]. It is noteworthy that HemeBIND, hemeNet and SeqD-HBM focus exclusively on the predictions of heme interaction residues, while the remaining methods predict other ligands as well. Three predictors derive from protein sequences [7, 9, 219], another three from protein structures [5, 47, 225], with hemeNet [48] providing predictions based on both. Notably, despite the limited protein structure data available in 2011, HemeBIND and hemeNet were developed based on protein structures. Besides, protein–heme interaction residue data usually comes from the BioLiP database. In addition, we summarize the protein feature profiles used by these predictors and learn that in addition to the common profiles such as PSSM and SS, there are also some special features used by these predictors. For example, HemeBIND and hemeNet use the Depth index (DPX) [228] and Protrusion index (CX) [229] features. DPX is the distance between the target atom and its nearest solvent, which is generated by the PSAIA tool [230]. The CX feature represents the degree to which the atom protrudes from the surface of the protein and the composition of CX features is similar to that of DPX. In terms of models, the above seven predictors mainly use SVM, ensemble model and DL model. In particular, SeqD-HBM is based on stepwise validation using the knowledge gained from in-depth spectroscopic studies on heme-peptide complexes. Unfortunately, only three of the above seven predictors are still available, and hemeNet is the only currently available method that specifically focuses on predicting heme interaction residues, which was proposed in 2011. For recently published and available predictors, as reported in GraphBind, GraphBind achieves an AUC of 96.2%, higher than DELIA (95.1%) [5].
Protein–ion interaction
An examination of 17 predictors associated with protein–ion interaction residues reveals that most predictors identify interaction residues with Mn, followed by iron ions, Ca, Mg and Zn. It is important to highlight that different predictor offers diverse insights into ion ligands. For instance, GASS-Metal [51] and MIB2 [49] differentiate between Fe2+ and Fe3+ ions, whereas mFASD [218] and MetalSiteHunter [50] do not. MetalDetector [214] opts not to make fine-grained predictions of protein-metal ions and treats all metal ions as a single class. Interestingly, two predictors, pfinder [213] (proposed in 2011) and PBSP [226] (proposed in 2021), exclusively predict phosphates. Moreover, unlike previously mentioned protein–ligand interaction residue predictors and other protein–ion interaction residue predictors that focus on all types of residues, CHED [208] predicts interaction residues of four types: Cys, His, Glu and Asp. Similar to CHED, MetalDetector identifies Cys and His involved in protein–metal interactions. With regard to inputs, the majority of these methods (11 out of 17) are based on protein structures, with six predictors using protein sequences. Additionally, an investigation into the datasets reveals that protein–ion interaction residue datasets are generally unbalanced and mainly derive from the PDB, BioLip and MetalPDB [231] databases. Notably, datasets corresponding to different ions vary substantially in size. For instance, IonCom [220] uses datasets comprising 379 interaction proteins and 1778 interaction residues for Mn, in contrast to 53 interaction proteins and 536 interaction residues for K. Furthermore, given the prevalence of predictors based on protein structures, the protein feature profiles vary significantly [207, 232–234] . For instance, mFASD incorporates four protein feature profiles: functional atoms, local chemical environment, the distance between two functional atoms and the distance between two functional atoms sets, while MetalSiteHunter employs 3D volume cubes (voxels) features. Notably, similar to previous sections, protein language models are also deployed in this field. LMetalSite [10] uses ProtTrans to produce protein embeddings and generate further predictions. In contrast, RBP-TSTL [141] employs the ProtT5-XL model, EGRET and PepBCL [6] use the ProtBert model and LMetalSite leverages the PROTT5-XL-U50 model, with each protein language model varying in terms of the number of parameters and training strategies. An overview of the algorithms and models reveals that they utilize a variety of innovative algorithms and models, such as MIB2 using the (PS)2 algorithm [235], GASS-Metal combining the homology method and the gene algorithm, and mFASD implementing a novel structure-based computational method. Regarding availability, eight predictors remain accessible to users, covering all ion types. Finally, we assessed the performance of protein–Mn interaction predictors, including LMetalSite, GraphBind and DELIA. The result in LMetalSite shows that LMetalSite (AUC = 96.6%) outperforms GraphBind (AUC = 93.0%) and DELIA (AUC = 90.2%) [10].
SUMMARY
Prompted by the necessity to decipher protein–ligand interactions on a large scale, we review a comprehensive set of over 160 predictors. These encompass interactions between proteins and a range of ligands including proteins, nucleic acids, peptides, nucleotides, hemes and ions. We have scrutinized these predictors through several pertinent lenses, including inputs, feature profiles, models and availability, among others.
According to our investigation, most predictors identify interactions using protein sequences. Especially, evolutionary information, which is derived from protein sequences, is the most widely employed feature profile (used by 67% of all predictors) in the past decades and PSSM stands out as the most representative among evolutionary information. Besides, pretrained large models based on sequences are becoming a new trend. Embeddings from pretrained large models are on pair with traditional features in quality and speed, greatly promoting the development of feature profiles. On the other hand, with the advancements in protein structure determination techniques and predictors of protein structures, an increasing number of structure-based predictors have also emerged, leading to a new trend which is the development of large-scale pretrained models based on multimodal protein data.
Compared with other technologies, DL can yield more accurate predictions and presents the opportunity to unearth deeper information embedded within proteins. In addition, more predictors focus on protein–protein and protein–nucleic acid interactions, indicating that macromolecular ligands are more of a concern. Furthermore, the majority of methods prioritize residue-level predictions over protein-level ones, showing a clear interest in more granular predictions from the perspective of proteins. Besides, some predictors further identify subtypes of ligands, indicating a trend toward fine-grained predictions from the perspective of ligand subtypes.
The current methods for predicting protein interactions focus mainly on structural proteins and individual proteins. However, these methods show significant shortcomings in the prediction of interactions involving inherently disorder proteins and protein complexes, both of which hold critical importance in biological processes. Inherently disorder proteins play a crucial role in cell signaling and regulation, while protein complexes are instrumental in various biological processes such as metabolism, DNA repair and signal transduction. Moreover, these predictors often overlook dynamics, that is, proteins exhibit different interaction mechanisms in the dynamic cellular environment. Therefore, research into inherently disorder proteins, protein complexes and protein dynamics is poised to become the frontier of future studies. Further exploration in these areas is expected to reveal more profound mechanisms of cellular biology, thereby advancing new strategies in drug discovery and disease treatment.
Despite a variety of ligands considered by current protein interaction predictors, the development of predictors for different ligands varies considerably, with some ligands still lacking sufficient predictors. It is necessary to develop new models to predict protein–ligand interactions for less studied ligands, such as GMP and K ions, and explore interactions with a broader range of ligands. Moreover, we find that while the availability of protein–ligand interaction prediction methods is a concern, many predictors remain inaccessible to researchers. The accessibility of these predictors is paramount for researchers and biologists, and we suggest that future studies should prioritize providing access to predictors and maintaining this accessibility.
Key Points
We review over 160 methods for predicting protein–ligand interactions, which focus on protein–protein, protein–nucleic acid, protein–peptide and protein–other ligands (nucleotide, heme, ion) interactions.
Our survey covers various types of methods, including protein- and residue-level predictors, as well as structure- and sequence-based predictors.
A comprehensive analysis is conducted from several significant perspectives including inputs, feature profiles, models, availability and so on. Evolutionary information, which is derived from protein sequences, is the most widely employed feature profile and PSSM stands out as the most representative among evolutionary information.
Finally, the challenges and future development directions are presented.
UNCOMMON ABBREVIATIONS
ELM: extreme learning machine.
L3: network paths of length three
KLR: kernel logistic regression
PLR: penalized logistic regression
LSA: latent semantic analysis
FCM: fuzzy cognitive map
GTB: gradient tree boosting
C4.5: a decision tree algorithm
SMO: sequential minimal optimization
(PS)2: an automatic protein structure prediction server
Supplementary Material
Author Biographies
Pengzhen Jia received the BS degree in computer science from Central South University, Changsha, China, in 2022. Currently, he is working toward the PhD degree in computer science and technology at Central South University, Changsha, China. His current research interests include bioinformatics and protein–ligand interactions.
Fuhao Zhang received the BS degree from the Chongqing University of Posts and Telecommunications, China, in 2014 and PhD degrees from Central South University, China, in 2023. He is currently an associate Professor at College of Information Engineering, Northwest A&F University. His main research interests include bioinformatics and deep learning.
Chaojin Wu received the BS degree in computer science from Central South University, Changsha, China, in 2022. Currently, he is working toward the MS degree in computer science and technology at Central South University, Changsha, China. His current research interests include bioinformatics and protein–ligand interactions.
Min Li received the BS degree in communication engineering and the MS and PhD degrees in computer science from Central South University, Changsha, China, in 2001, 2004 and 2008, respectively. She is currently a Professor at the School of Computer Science and Engineering, Central South University. Her main research interests include bioinformatics and system biology.
Contributor Information
Pengzhen Jia, School of Computer Science and Engineering, Central South University, 932 Lushan Road(S), Changsha 410083, China.
Fuhao Zhang, School of Computer Science and Engineering, Central South University, 932 Lushan Road(S), Changsha 410083, China; College of Information Engineering, Northwest A&F University, No. 3 Taicheng Road, Yangling, Shaanxi 712100, China.
Chaojin Wu, School of Computer Science and Engineering, Central South University, 932 Lushan Road(S), Changsha 410083, China.
Min Li, School of Computer Science and Engineering, Central South University, 932 Lushan Road(S), Changsha 410083, China.
FUNDING
This work is supported by the National Natural Science Foundation of China under Grant No. (62225209), the Science and Technology Innovation Program of Hunan Province (2021RC0048).
DATA AVAILABILITY
The protein-ligand interaction datasets used in this manuscript are publicly available, and their specific sources are summarized as follows: The dataset used for evaluating sequenced-based PAIR-pro predictors is the virus-human interaction dataset [41, 94]. The dataset used for evaluating SINGLE-res predictors is the Test_60 dataset [124]. The DNA-129_Test dataseet is from publications [3, 5]. The dataset used to evaluate protein-RNA interaction residue predictors is from the publication [24].
References
- 1. Yuan Q, Chen J, Zhao H, et al. Structure-aware protein–protein interaction site prediction using deep graph convolutional network. Bioinformatics 2021;38(1):125–32. [DOI] [PubMed] [Google Scholar]
- 2. Zhang J, Kurgan L. Review and comparative assessment of sequence-based predictors of protein-binding residues. Brief Bioinform 2018;19(5):821–37. [DOI] [PubMed] [Google Scholar]
- 3. Wang N, Yan K, Zhang J, Liu B. iDRNA-ITF: identifying DNA- and RNA-binding residues in proteins based on induction and transfer framework. Brief Bioinform 2022;23(4):bbac236. [DOI] [PubMed] [Google Scholar]
- 4. Yan J, Kurgan L. DRNApred, fast sequence-based method that accurately predicts and discriminates DNA- and RNA-binding residues. Nucleic Acids Res 2017;45(10):e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Xia Y, Xia CQ, Pan X, Shen HB. GraphBind: protein structural context embedded rules learned by hierarchical graph neural networks for recognizing nucleic-acid-binding residues. Nucleic Acids Res 2021;49(9):e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang R, Jin J, Zou Q, et al. Predicting protein-peptide binding residues via interpretable deep learning. Bioinformatics 2022;38(13):3351–60. [DOI] [PubMed] [Google Scholar]
- 7. Yu DJ, Hu J, Yang J, et al. Designing template-free predictor for targeting protein-ligand binding sites with classifier ensemble and spatial clustering. IEEE/ACM Trans Comput Biol Bioinform 2013;10(4):994–1008. [DOI] [PubMed] [Google Scholar]
- 8. Hu J, He X, Yu DJ, et al. A new supervised over-sampling algorithm with application to protein-nucleotide binding residue prediction. PloS One 2014;9(9):e107676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wißbrock A, Paul George AA, Brewitz HH, et al. The molecular basis of transient heme-protein interactions: analysis, concept and implementation. Biosci Rep 2019;39(1):BSR20181940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuan Q, Chen S, Wang Y, et al. Alignment-free metal ion-binding site prediction from protein sequence through pretrained language model and multi-task learning. Brief Bioinform 2022;23(6):bbac444. [DOI] [PubMed] [Google Scholar]
- 11. Wells JA, Mcclendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 2007;450(7172):1001–9. [DOI] [PubMed] [Google Scholar]
- 12. De Las Rivas J, Fontanillo C. Protein–protein interaction networks: unraveling the wiring of molecular machines within the cell. Brief Funct Genomics 2012;11(6):489–96. [DOI] [PubMed] [Google Scholar]
- 13. Orii N, Ganapathiraju MK. Wiki-pi: a web-server of annotated human protein-protein interactions to aid in discovery of protein function. PloS One 2012;7(11):e49029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kuzmanov U, Emili A. Protein-protein interaction networks: probing disease mechanisms using model systems. Genome Med 2013;5(4):37–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res 2019;47(D1):D607–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Calderone A, Castagnoli L, Cesareni G. Mentha: a resource for browsing integrated protein-interaction networks. Nat Methods 2013;10(8):690–1. [DOI] [PubMed] [Google Scholar]
- 17. Oughtred R, Stark C, Breitkreutz B-J, et al. The BioGRID interaction database: 2019 update. Nucleic Acids Res 2019;47(D1):D529–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang J, Roy A, Zhang Y. BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res 2013;41(Database issue):D1096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quaglia F, Mészáros B, Salladini E, et al. DisProt in 2022: improved quality and accessibility of protein intrinsic disorder annotation. Nucleic Acids Res 2022;50(D1):D480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. wwPDB consortium . Protein data bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 2019;47(D1):D520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Uniprot: the universal protein knowledgebase in 2023. Nucleic Acids Res 2023;51(D1):D523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zeng M, Zhang F, Wu FX, et al. Protein-protein interaction site prediction through combining local and global features with deep neural networks. Bioinformatics 2020;36(4):1114–20. [DOI] [PubMed] [Google Scholar]
- 23. Taherzadeh G, Yang Y, Zhang T, et al. Sequence-based prediction of protein-peptide binding sites using support vector machine. J Comput Chem 2016;37(13):1223–9. [DOI] [PubMed] [Google Scholar]
- 24. Zhang F, Li M, Zhang J, Kurgan L. HybridRNAbind: prediction of RNA interacting residues across structure-annotated and disorder-annotated proteins. Nucleic Acids Res 2023;51(5):e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aptekmann AA, Buongiorno J, Giovannelli D, et al. Mebipred: identifying metal-binding potential in protein sequence. Bioinformatics 2022;38(14):3532–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yuvaraj N, Srihari K, Chandragandhi S, et al. Analysis of protein-ligand interactions of SARS-Cov-2 against selective drug using deep neural networks. Big Data Min Anal 2021;4(2):76–83. [Google Scholar]
- 27. Wu Y, Gao M, Zeng M, et al. BridgeDPI: a novel graph neural network for predicting drug-protein interactions. Bioinformatics 2022;38(9):2571–8. [DOI] [PubMed] [Google Scholar]
- 28. Li M, Lu Z, Wu Y, Li YH. BACPI: a bi-directional attention neural network for compound-protein interaction and binding affinity prediction. Bioinformatics 2022;38(7):1995–2002. [DOI] [PubMed] [Google Scholar]
- 29. Wang K, Zhou R, Tang J, Li M. GraphscoreDTA: optimized graph neural network for protein-ligand binding affinity prediction. Bioinformatics 2023;39(6):btad340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang K, Li M. Fusion-based deep learning architecture for detecting drug-target binding affinity using target and drug sequence and structure. IEEE J Biomed Health Inform 2023;27(12):6112–20. [DOI] [PubMed] [Google Scholar]
- 31. Wang K, Zhou R, Li Y, Li M. DeepDTAF: a deep learning method to predict protein-ligand binding affinity. Brief Bioinform 2021;22(5):bbab072. [DOI] [PubMed] [Google Scholar]
- 32. Lei C, Lu Z, Wang M, Li M. StackCPA: a stacking model for compound-protein binding affinity prediction based on pocket multi-scale features. Comput Biol Med 2023;164:107131. [DOI] [PubMed] [Google Scholar]
- 33. Wang M, Kurgan L, Li M. A comprehensive assessment and comparison of tools for HLA class I peptide-binding prediction. Brief Bioinform 2023;24(3):bbad150. [DOI] [PubMed] [Google Scholar]
- 34. Zhang F, Li M, Zhang J, et al. DeepPRObind: modular deep learner that accurately predicts structure and disorder-annotated protein binding residues. J Mol Biol 2023;435(14):167945. [DOI] [PubMed] [Google Scholar]
- 35. Zhang F, Zhao B, Shi W, et al. DeepDISOBind: accurate prediction of RNA-, DNA- and protein-binding intrinsically disordered residues with deep multi-task learning. Brief Bioinform 2022;23(1):bbab521. [DOI] [PubMed] [Google Scholar]
- 36. Li P, Liu ZP. PST-PRNA: prediction of RNA-binding sites using protein surface topography and deep learning. Bioinformatics 2022;38(8):2162–8. [DOI] [PubMed] [Google Scholar]
- 37. Huang L, Liao L, Wu CH. Evolutionary analysis and interaction prediction for protein-protein interaction network in geometric space. PloS One 2017;12(9):e0183495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang X, Yang W, Yang Y, et al. PPISB: a novel network-based algorithm of predicting protein-protein interactions with mixed membership stochastic blockmodel. IEEE/ACM Trans Comput Biol Bioinform 2023;20(2):1606–12. [DOI] [PubMed] [Google Scholar]
- 39. Gao M, Skolnick J. A threading-based method for the prediction of DNA-binding proteins with application to the human genome. PLoS Comput Biol 2009;5(11):e1000567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhao H, Yang Y, Zhou Y. Highly accurate and high-resolution function prediction of RNA binding proteins by fold recognition and binding affinity prediction. RNA Biol 2011;8(6):988–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hu X, Feng C, Zhou Y, et al. DeepTrio: a ternary prediction system for protein–protein interaction using mask multiple parallel convolutional neural networks. Bioinformatics 2022;38(3):694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ieremie I, Ewing RM, Niranjan M. TransformerGO: predicting protein–protein interactions by modelling the attention between sets of gene ontology terms. Bioinformatics 2022;38(8):2269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abdin O, Nim S, Wen H, Kim PM. PepNN: a deep attention model for the identification of peptide binding sites. Commun Biol 2022;5(1):503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Johansson-Åkhe I, Mirabello C, Wallner B. Predicting protein-peptide interaction sites using distant protein complexes as structural templates. Sci Rep 2019;9(1):4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen K, Mizianty MJ, Kurgan L. Prediction and analysis of nucleotide-binding residues using sequence and sequence-derived structural descriptors. Bioinformatics 2012;28(3):331–41. [DOI] [PubMed] [Google Scholar]
- 46. Hu J, Zheng LL, Bai YS, et al. Accurate prediction of protein-ATP binding residues using position-specific frequency matrix. Anal Biochem 2021;626:114241. [DOI] [PubMed] [Google Scholar]
- 47. Liu R, Hu J. HemeBIND: a novel method for heme binding residue prediction by combining structural and sequence information. BMC Bioinformatics 2011;12:207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Liu R, Hu J. Computational prediction of heme-binding residues by exploiting residue interaction network. PloS One 2011;6(10):e25560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lu CH, Chen CC, Yu CS, et al. MIB2: metal ion-binding site prediction and modeling server. Bioinformatics 2022;38(18):4428–9. [DOI] [PubMed] [Google Scholar]
- 50. Mohamadi A, Cheng T, Jin L, et al. An ensemble 3D deep-learning model to predict protein metal-binding site. Cell Rep Phys Sci 2022;3(9):101046. [Google Scholar]
- 51. Paiva VA, Mendonça MV, Silveira SA, et al. GASS-metal: identifying metal-binding sites on protein structures using genetic algorithms. Brief Bioinform 2022;23(5):bbac178. [DOI] [PubMed] [Google Scholar]
- 52. Sanchez-Garcia R, Sorzano COS, Carazo JM, Segura J. BIPSPI: a method for the prediction of partner-specific protein–protein interfaces. Bioinformatics 2019;35(3):470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sanchez-Garcia R, Macias J, Sorzano C, et al. BIPSPI+: mining type-specific datasets of protein complexes to improve protein binding site prediction. J Mol Biol 2022;434(11):167556. [DOI] [PubMed] [Google Scholar]
- 54. Qiu J, Bernhofer M, Heinzinger M, et al. ProNA2020 predicts protein–DNA, protein–RNA, and protein–protein binding proteins and residues from sequence. J Mol Biol 2020;432(7):2428–43. [DOI] [PubMed] [Google Scholar]
- 55. Li Y, Golding GB, Ilie L. DELPHI: accurate deep ensemble model for protein interaction sites prediction. Bioinformatics 2021;37(7):896–904. [DOI] [PubMed] [Google Scholar]
- 56. Bock JR, Gough DA. Predicting protein–protein interactions from primary structure. Bioinformatics 2001;17(5):455–60. [DOI] [PubMed] [Google Scholar]
- 57. Shen J, Zhang J, Luo X, et al. Predicting protein–protein interactions based only on sequences information. Proc Natl Acad Sci 2007;104(11):4337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guo Y, Yu L, Wen Z, Li M. Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res 2008;36(9):3025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yang L, Xia JF, Gui J. Prediction of protein-protein interactions from protein sequence using local descriptors. Protein Pept Lett 2010;17(9):1085–90. [DOI] [PubMed] [Google Scholar]
- 60. Pan X-Y, Zhang Y-N, Shen H-B. Large-scale prediction of human protein− protein interactions from amino acid sequence based on latent topic features. J Proteome Res 2010;9(10):4992–5001. [DOI] [PubMed] [Google Scholar]
- 61. You Z-H, Lei Y-K, Zhu L, et al. Prediction of protein-protein interactions from amino acid sequences with ensemble extreme learning machines and principal component analysis. BMC Bioinformatics 2013;14 Suppl 8(Suppl 8):S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. You Z-H, Zhu L, Zheng C-H, et al. Prediction of protein-protein interactions from amino acid sequences using a novel multi-scale continuous and discontinuous feature set. BMC Bioinformatics 2014;15 Suppl 15(Suppl 15):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. You ZH, Li S, Gao X, et al. Large-scale protein-protein interactions detection by integrating big biosensing data with computational model. Biomed Res Int 2014;2014:598129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wong L, You Z-H, Li S, et al. Detection of protein-protein interactions from amino acid sequences using a rotation forest model with a novel PR-LPQ descriptor. In: International Conference on Intelligent Computing. Fuzhou, China: Springer, 2015, 713–20.
- 65. Du X, Sun S, Hu C, et al. DeepPPI: boosting prediction of protein–protein interactions with deep neural networks. J Chem Inf Model 2017;57(6):1499–510. [DOI] [PubMed] [Google Scholar]
- 66. Sun T, Zhou B, Lai L, Pei J. Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinformatics 2017;18:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hashemifar S, Neyshabur B, Khan AA, Xu J. Predicting protein–protein interactions through sequence-based deep learning. Bioinformatics 2018;34(17):i802–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li H, Gong XJ, Yu H, Zhou C. Deep neural network based predictions of protein interactions using primary sequences. Molecules 2018;23(8):1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen M, Ju CJ-T, Zhou G, et al. Multifaceted protein–protein interaction prediction based on Siamese residual RCNN. Bioinformatics 2019;35(14):i305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chen C, Zhang Q, Ma Q, Yu B. LightGBM-PPI: predicting protein-protein interactions through LightGBM with multi-information fusion. Chemom Intel Lab Syst 2019;191:54–64. [Google Scholar]
- 71. Kovács IA, Luck K, Spirohn K, et al. Network-based prediction of protein interactions. Nat Commun 2019;10(1):1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen Y, Wang W, Liu J, et al. Protein interface complementarity and gene duplication improve link prediction of protein-protein interaction network. Front Genet 2020;11:291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Sledzieski S, Singh R, Cowen L, Berger B. D-SCRIPT translates genome to phenome with sequence-based, structure-aware, genome-scale predictions of protein-protein interactions. Cell Syst 2021;12(10):969–82.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bryant P, Pozzati G, Elofsson A. Improved prediction of protein-protein interactions using AlphaFold2. Nat Commun 2022;13(1):1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhang J, Zhu M, Qian Y. protein2vec: predicting protein-protein interactions based on LSTM. IEEE/ACM Trans Comput Biol Bioinform 2022;19(3):1257–66. [DOI] [PubMed] [Google Scholar]
- 76. Xenarios I, Rice DW, Salwinski L, et al. DIP: the database of interacting proteins. Nucleic Acids Res 2000;28(1):289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Keshava Prasad TS, Goel R, Kandasamy K, et al. Human protein reference database--2009 update. Nucleic Acids Res 2009;37(Database issue):D767–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Schaefer MH, Fontaine JF, Vinayagam A, et al. HIPPIE: integrating protein interaction networks with experiment based quality scores. PloS One 2012;7(2):e31826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Das J, Yu H. HINT: high-quality protein interactomes and their applications in understanding human disease. BMC Syst Biol 2012;6:92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Moal IH, Fernández-Recio J. SKEMPI: a structural kinetic and energetic database of mutant protein interactions and its use in empirical models. Bioinformatics 2012;28(20):2600–7. [DOI] [PubMed] [Google Scholar]
- 81. Oughtred R, Rust J, Chang C, et al. The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 2021;30(1):187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Petrey D, Zhao H, Trudeau S, et al. PrePPI: a structure informed proteome-wide database of protein-protein interactions. J Mol Biol 2023;435(14):168052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Del Toro N, Shrivastava A, Ragueneau E, et al. The IntAct database: efficient access to fine-grained molecular interaction data. Nucleic Acids Res 2022;50(D1):D648–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Zhang J, Ma Z, Kurgan L. Comprehensive review and empirical analysis of hallmarks of DNA-, RNA- and protein-binding residues in protein chains. Brief Bioinform 2019;20(4):1250–68. [DOI] [PubMed] [Google Scholar]
- 85. Cui J, Han LY, Li H, et al. Computer prediction of allergen proteins from sequence-derived protein structural and physicochemical properties. Mol Immunol 2007;44(4):514–20. [DOI] [PubMed] [Google Scholar]
- 86. Zhang ZH, Koh JL, Zhang GL, et al. AllerTool: a web server for predicting allergenicity and allergic cross-reactivity in proteins. Bioinformatics 2007;23(4):504–6. [DOI] [PubMed] [Google Scholar]
- 87. Gene ontology consortium: going forward. Nucleic Acids Res 2015;43(Database issue):D1049–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Mcguffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics 2000;16(4):404–5. [DOI] [PubMed] [Google Scholar]
- 89. Altschul SF, Madden TL, Schäffer AA, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997;25(17):3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983;22(12):2577–637. [DOI] [PubMed] [Google Scholar]
- 91. Touw WG, Baakman C, Black J, et al. A series of PDB-related databanks for everyday needs. Nucleic Acids Res 2015;43(D1):D364–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jani J, Pappachan A. Protein analysis: from sequence to structure. In: Singh V, Kumar A (eds). Advances in Bioinformatics. Singapore: Springer Singapore, 2021, 59–82. [Google Scholar]
- 93. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596(7873):583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Liu-Wei W, Kafkas Ş, Chen J, et al. DeepViral: prediction of novel virus-host interactions from protein sequences and infectious disease phenotypes. Bioinformatics 2021;37(17):2722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Dai B, Bailey-Kellogg C. Protein interaction interface region prediction by geometric deep learning. Bioinformatics 2021;37(17):2580–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liu Y, Yuan H, Cai L, et al. Deep learning of high-order interactions for protein interface prediction. In: Proceedings of the 26th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining. Virtual Event, CA, USA: ACM, 2020, 679–87.
- 97. Vreven T, Moal IH, Vangone A, et al. Updates to the integrated protein-protein interaction benchmarks: docking benchmark version 5 and affinity benchmark version 2. J Mol Biol 2015;427(19):3031–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Holm L, Sander C. Removing near-neighbour redundancy from large protein sequence collections. Bioinformatics 1998;14(5):423–9. [DOI] [PubMed] [Google Scholar]
- 99. Gattiker A, Michoud K, Rivoire C, et al. Automated annotation of microbial proteomes in SWISS-PROT. Comput Biol Chem 2003;27(1):49–58. [DOI] [PubMed] [Google Scholar]
- 100. Fariselli P, Pazos F, Valencia A, Casadio R. Prediction of protein–protein interaction sites in heterocomplexes with neural networks. Eur J Biochem 2002;269(5):1356–61. [DOI] [PubMed] [Google Scholar]
- 101. Ofran Y, Rost B. Predicted protein–protein interaction sites from local sequence information. FEBS Lett 2003;544(1-3):236–9. [DOI] [PubMed] [Google Scholar]
- 102. Fernandez-Recio J, Totrov M, Skorodumov C, Abagyan R. Optimal docking area: a new method for predicting protein–protein interaction sites. Proteins 2005;58(1):134–43. [DOI] [PubMed] [Google Scholar]
- 103. Burgoyne NJ, Jackson RM. Predicting protein interaction sites: binding hot-spots in protein–protein and protein–ligand interfaces. Bioinformatics 2006;22(11):1335–42. [DOI] [PubMed] [Google Scholar]
- 104. Porollo A, Meller J. Prediction-based fingerprints of protein–protein interactions. Proteins 2007;66(3):630–45. [DOI] [PubMed] [Google Scholar]
- 105. Ofran Y, Rost B. ISIS: interaction sites identified from sequence. Bioinformatics 2007;23(2):e13–6. [DOI] [PubMed] [Google Scholar]
- 106. Meszaros B, Simon I, Dosztanyi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol 2009;5(5):e1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Sikić M, Tomić S, Vlahovicek K. Prediction of protein-protein interaction sites in sequences and 3D structures by random forests. PLoS Comput Biol 2009;5(1):e1000278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Murakami Y, Mizuguchi K. Applying the Naïve Bayes classifier with kernel density estimation to the prediction of protein-protein interaction sites. Bioinformatics 2010;26(15):1841–8. [DOI] [PubMed] [Google Scholar]
- 109. Singh G, Dhole K, Pai PP, et al. SPRINGS: prediction of protein-protein interaction sites using artificial neural networks. PeerJ PrePrints 2014;2:e266v2. [Google Scholar]
- 110. Dhole K, Singh G, Pai PP, Mondal S. Sequence-based prediction of protein-protein interaction sites with L1-logreg classifier. J Theor Biol 2014;348:47–54. [DOI] [PubMed] [Google Scholar]
- 111. Wei ZS, Yang JY, Shen HB, Yu DJ. A cascade random forests algorithm for predicting protein-protein interaction sites. IEEE Trans Nanobioscience 2015;14(7):746–60. [DOI] [PubMed] [Google Scholar]
- 112. Liu GH, Shen HB, Yu DJ. Prediction of protein-protein interaction sites with machine-learning-based data-cleaning and post-filtering procedures. J Membr Biol 2016;249(1-2):141–53. [DOI] [PubMed] [Google Scholar]
- 113. Wei Z-S, Han K, Yang J-Y, et al. Protein–protein interaction sites prediction by ensembling SVM and sample-weighted random forests. Neurocomputing 2016;193:201–12. [Google Scholar]
- 114. Hou Q, De Geest PFG, Vranken WF, et al. Seeing the trees through the forest: sequence-based homo- and heteromeric protein-protein interaction sites prediction using random forest. Bioinformatics 2017;33(10):1479–87. [DOI] [PubMed] [Google Scholar]
- 115. Jiménez J, Doerr S, Martínez-Rosell G, et al. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 2017;33(19):3036–42. [DOI] [PubMed] [Google Scholar]
- 116. Wang X, Yu B, Ma A, et al. Protein-protein interaction sites prediction by ensemble random forests with synthetic minority oversampling technique. Bioinformatics 2019;35(14):2395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Zhang J, Kurgan L. SCRIBER: accurate and partner type-specific prediction of protein-binding residues from proteins sequences. Bioinformatics 2019;35(14):i343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Townshend R, Bedi R, Suriana P, et al. End-to-end learning on 3D protein structure for interface prediction. Adv Neural Inf Process Syst 2019;15642–51. [Google Scholar]
- 119. Zhang B, Li J, Quan L, et al. Sequence-based prediction of protein-protein interaction sites by simplified long short-term memory network. Neurocomputing 2019;357:86–100. [Google Scholar]
- 120. Deng A, Zhang H, Wang W, et al. Developing computational model to predict protein-protein interaction sites based on the XGBoost algorithm. Int J Mol Sci 2020;21(7):2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Gainza P, Sverrisson F, Monti F, et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat Methods 2020;17(2):184–92. [DOI] [PubMed] [Google Scholar]
- 122. Mahbub S, Bayzid MS. EGRET: edge aggregated graph attention networks and transfer learning improve protein-protein interaction site prediction. Brief Bioinform 2022;23(2):bbab578. [DOI] [PubMed] [Google Scholar]
- 123. Tubiana J, Schneidman-Duhovny D, Wolfson HJ. ScanNet: an interpretable geometric deep learning model for structure-based protein binding site prediction. Nat Methods 2022;19(6):730–9. [DOI] [PubMed] [Google Scholar]
- 124. Khan SH, Tayara H, Chong KT. ProB-site: protein binding site prediction using local features. Cells 2022;11(13):2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Zhang J, Ghadermarzi S, Kurgan L. Prediction of protein-binding residues: dichotomy of sequence-based methods developed using structured complexes versus disordered proteins. Bioinformatics 2020;36(18):4729–38. [DOI] [PubMed] [Google Scholar]
- 126. Peng Z, Kurgan L. High-throughput prediction of RNA, DNA and protein binding regions mediated by intrinsic disorder. Nucleic Acids Res 2015;43(18):e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Dodge C, Schneider R, Sander C. The HSSP database of protein structure—sequence alignments and family profiles. Nucleic Acids Res 1998;26(1):313–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Elnaggar A, Heinzinger M, Dallago C, et al. ProtTrans: toward understanding the language of life through self-supervised learning. IEEE Trans Pattern Anal Mach Intell 2022;44(10):7112–27. [DOI] [PubMed] [Google Scholar]
- 129. Zhang J, Chen Q, Liu B. iDRBP_MMC: identifying DNA-binding proteins and RNA-binding proteins based on multi-label learning model and motif-based convolutional neural network. J Mol Biol 2020;432(22):5860–75. [DOI] [PubMed] [Google Scholar]
- 130. Zhang J, Chen Q, Liu B. DeepDRBP-2L: a new genome annotation predictor for identifying DNA-binding proteins and RNA-binding proteins using convolutional neural network and long short-term memory. IEEE/ACM Trans Comput Biol Bioinform 2021;18(4):1451–63. [DOI] [PubMed] [Google Scholar]
- 131. Rahman MS, Shatabda S, Saha S, et al. DPP-PseAAC: a DNA-binding protein prediction model using Chou's general PseAAC. J Theor Biol 2018;452:22–34. [DOI] [PubMed] [Google Scholar]
- 132. Mishra A, Pokhrel P, Hoque MT. StackDPPred: a stacking based prediction of DNA-binding protein from sequence. Bioinformatics 2019;35(3):433–41. [DOI] [PubMed] [Google Scholar]
- 133. Li G, Du X, Li X, et al. Prediction of DNA binding proteins using local features and long-term dependencies with primary sequences based on deep learning. PeerJ 2021;9:e11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Ali F, Kumar H, Patil S, et al. DBP-DeepCNN: prediction of DNA-binding proteins using wavelet-based denoising and deep learning. Chemom Intel Lab Syst 2022;229:104639. [Google Scholar]
- 135. Pradhan UK, Meher PK, Naha S, et al. PlDBPred: a novel computational model for discovery of DNA binding proteins in plants. Brief Bioinform 2023;24(1):bbac483. [DOI] [PubMed] [Google Scholar]
- 136. Zheng J, Zhang X, Zhao X, et al. Deep-RBPPred: predicting RNA binding proteins in the proteome scale based on deep learning. Sci Rep 2018;8(1):15264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Bressin A, Schulte-Sasse R, Figini D, et al. TriPepSVM: de novo prediction of RNA-binding proteins based on short amino acid motifs. Nucleic Acids Res 2019;47(9):4406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Sun X, Jin T, Chen C, et al. RBPro-RF: use Chou’s 5-steps rule to predict RNA-binding proteins via random forest with elastic net. Chemom Intel Lab Syst 2020;197:103919. [Google Scholar]
- 139. Mishra A, Khanal R, Kabir WU, Hoque T. AIRBP: accurate identification of RNA-binding proteins using machine learning techniques. Artif Intell Med 2021;113:102034. [DOI] [PubMed] [Google Scholar]
- 140. Zhang J, Yan K, Chen Q, Liu B. PreRBP-TL: prediction of species-specific RNA-binding proteins based on transfer learning. Bioinformatics 2022;38(8):2135–43. [DOI] [PubMed] [Google Scholar]
- 141. Peng X, Wang X, Guo Y, et al. RBP-TSTL is a two-stage transfer learning framework for genome-scale prediction of RNA-binding proteins. Brief Bioinform 2022;23(4):bbac215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Wang N, Zhang J, Liu B. IDRBP-PPCT: identifying nucleic acid-binding proteins based on position-specific score matrix and position-specific frequency matrix cross transformation. IEEE/ACM Trans Comput Biol Bioinform 2022;19(4):2284–93. [DOI] [PubMed] [Google Scholar]
- 143. Yan J, Friedrich S, Kurgan L. A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues. Brief Bioinform 2016;17(1):88–105. [DOI] [PubMed] [Google Scholar]
- 144. Li M, Zhang F, Kurgan L. Machine learning methods for predicting protein-nucleic acids interactions. In: Kurgan L (ed.), Machine Learning in Bioinformatics of Protein Sequences: Algorithms, Databases and Resources for Modern Protein Bioinformatics. Singapore: World Scientific, 2023, 265–87. [Google Scholar]
- 145. Ahmad S, Gromiha MM, Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics 2004;20(4):477–86.14990443 [Google Scholar]
- 146. Ahmad S, Sarai A. PSSM-based prediction of DNA binding sites in proteins. BMC Bioinformatics 2005;6:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Yan C, Terribilini M, Wu F, et al. Predicting DNA-binding sites of proteins from amino acid sequence. BMC Bioinformatics 2006;7:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Ho SY, Yu FC, Chang CY, Huang HL. Design of accurate predictors for DNA-binding sites in proteins using hybrid SVM-PSSM method. Biosystems 2007;90(1):234–41. [DOI] [PubMed] [Google Scholar]
- 149. Hwang S, Gou Z, Kuznetsov IB. DP-Bind: a web server for sequence-based prediction of DNA-binding residues in DNA-binding proteins. Bioinformatics 2007;23(5):634–6. [DOI] [PubMed] [Google Scholar]
- 150. Ofran Y, Mysore V, Rost B. Prediction of DNA-binding residues from sequence. Bioinformatics 2007;23(13):i347–53. [DOI] [PubMed] [Google Scholar]
- 151. Wang L, Yang MQ, Yang JY. Prediction of DNA-binding residues from protein sequence information using random forests. BMC Genomics 2009;10 Suppl 1(Suppl 1):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Wu J, Liu H, Duan X, et al. Prediction of DNA-binding residues in proteins from amino acid sequences using a random forest model with a hybrid feature. Bioinformatics 2009;25(1):30–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ma X, Guo J, Liu HD, et al. Sequence-based prediction of DNA-binding residues in proteins with conservation and correlation information. IEEE/ACM Trans Comput Biol Bioinform 2012;9(6):1766–75. [DOI] [PubMed] [Google Scholar]
- 154. Dey S, Pal A, Guharoy M, et al. Characterization and prediction of the binding site in DNA-binding proteins: improvement of accuracy by combining residue composition, evolutionary conservation and structural parameters. Nucleic Acids Res 2012;40(15):7150–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Liu R, Hu J. DNABind: a hybrid algorithm for structure-based prediction of DNA-binding residues by combining machine learning- and template-based approaches. Proteins 2013;81(11):1885–99. [DOI] [PubMed] [Google Scholar]
- 156. Zhao H, Wang J, Zhou Y, Yang Y. Predicting DNA-binding proteins and binding residues by complex structure prediction and application to human proteome. PloS One 2014;9(5):e96694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Wang W, Liu J, Xiong Y, et al. Analysis and classification of DNA-binding sites in single-stranded and double-stranded DNA-binding proteins using protein information. IET Syst Biol 2014;8(4):176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Zhou J, Xu R, He Y, et al. PDNAsite: identification of DNA-binding site from protein sequence by incorporating spatial and sequence context. Sci Rep 2016;6:27653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Wei L, Tang J, Zou Q. Local-DPP: an improved DNA-binding protein prediction method by exploring local evolutionary information. Inform Sci 2017;384:135–44. [Google Scholar]
- 160. Hu J, Li Y, Zhang M, et al. Predicting protein-DNA binding residues by weightedly combining sequence-based features and boosting multiple SVMs. IEEE/ACM Trans Comput Biol Bioinform 2017;14(6):1389–98. [DOI] [PubMed] [Google Scholar]
- 161. Zhu YH, Hu J, Song XN, Yu DJ. DNAPred: accurate identification of DNA-binding sites from protein sequence by ensembled hyperplane-distance-based support vector machines. J Chem Inf Model 2019;59(6):3057–71. [DOI] [PubMed] [Google Scholar]
- 162. Nguyen BP, Nguyen QH, Doan-Ngoc GN, et al. iProDNA-CapsNet: identifying protein-DNA binding residues using capsule neural networks. BMC Bioinformatics 2019;20(Suppl 23):634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zhou J, Lu Q, Xu R, et al. EL_LSTM: prediction of DNA-binding residue from protein sequence by combining long short-term memory and ensemble learning. IEEE/ACM Trans Comput Biol Bioinform 2020;17(1):124–35. [DOI] [PubMed] [Google Scholar]
- 164. Hu J, Zhou XG, Zhu YH, et al. TargetDBP: accurate DNA-binding protein prediction via sequence-based multi-view feature learning. IEEE/ACM Trans Comput Biol Bioinform 2020;17(4):1419–29. [DOI] [PubMed] [Google Scholar]
- 165. Amirkhani A, Kolahdoozi M, Wang C, Kurgan LA. Prediction of DNA-binding residues in local segments of protein sequences with fuzzy cognitive maps. IEEE/ACM Trans Comput Biol Bioinform 2020;17(4):1372–82. [DOI] [PubMed] [Google Scholar]
- 166. Zhang J, Ghadermarzi S, Katuwawala A, Kurgan L. DNAgenie: accurate prediction of DNA-type-specific binding residues in protein sequences. Brief Bioinform 2021;22(6):bbab336. [DOI] [PubMed] [Google Scholar]
- 167. Jeong E, Chung IF, Miyano S. A neural network method for identification of RNA-interacting residues in protein. Genome Inform 2004;15(1):105–16. [PubMed] [Google Scholar]
- 168. Jeong E, Miyano S. A weighted profile based method for protein-RNA interacting residue prediction. In: Priami C, Cardelli L, Emmott S (eds.), Transactions on Computational Systems Biology IV. Berlin, Heidelberg: Springer, 2006, 123–39. [Google Scholar]
- 169. Terribilini M, Sander JD, Lee JH, et al. RNABindR: a server for analyzing and predicting RNA-binding sites in proteins. Nucleic Acids Res 2007;35(Web Server issue):W578–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Wang Y, Xue Z, Shen G, Xu J. PRINTR: prediction of RNA binding sites in proteins using SVM and profiles. Amino Acids 2008;35(2):295–302. [DOI] [PubMed] [Google Scholar]
- 171. Tong J, Jiang P, Lu ZH. RISP: a web-based server for prediction of RNA-binding sites in proteins. Comput Methods Programs Biomed 2008;90(2):148–53. [DOI] [PubMed] [Google Scholar]
- 172. Kumar M, Gromiha MM, Raghava GP. Prediction of RNA binding sites in a protein using SVM and PSSM profile. Proteins 2008;71(1):189–94. [DOI] [PubMed] [Google Scholar]
- 173. Cheng CW, Su EC, Hwang JK, et al. Predicting RNA-binding sites of proteins using support vector machines and evolutionary information. BMC Bioinformatics 2008;9 Suppl 12(Suppl 12):S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Murakami Y, Spriggs RV, Nakamura H, Jones S. PiRaNhA: a server for the computational prediction of RNA-binding residues in protein sequences. Nucleic Acids Res 2010;38(Web Server issue):W412–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Huang YF, Chiu LY, Huang CC, Huang CK. Predicting RNA-binding residues from evolutionary information and sequence conservation. BMC Genomics 2010;11 Suppl 4(Suppl 4):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Zhang T, Zhang H, Chen K, et al. Analysis and prediction of RNA-binding residues using sequence, evolutionary conservation, and predicted secondary structure and solvent accessibility. Curr Protein Pept Sci 2010;11(7):609–28. [DOI] [PubMed] [Google Scholar]
- 177. Liu ZP, Wu LY, Wang Y, et al. Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics 2010;26(13):1616–22. [DOI] [PubMed] [Google Scholar]
- 178. Pérez-Cano L, Fernández-Recio J. Optimal protein-RNA area, OPRA: a propensity-based method to identify RNA-binding sites on proteins. Proteins 2010;78(1):25–35. [DOI] [PubMed] [Google Scholar]
- 179. Wang CC, Fang Y, Xiao J, Li M. Identification of RNA-binding sites in proteins by integrating various sequence information. Amino Acids 2011;40(1):239–48. [DOI] [PubMed] [Google Scholar]
- 180. Ma X, Guo J, Wu J, et al. Prediction of RNA-binding residues in proteins from primary sequence using an enriched random forest model with a novel hybrid feature. Proteins 2011;79(4):1230–9. [DOI] [PubMed] [Google Scholar]
- 181. Choi S, Han K. Prediction of RNA-binding amino acids from protein and RNA sequences. BMC Bioinformatics 2011;12 Suppl 13(Suppl 13):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Walia RR, Xue LC, Wilkins K, et al. RNABindRPlus: a predictor that combines machine learning and sequence homology-based methods to improve the reliability of predicted RNA-binding residues in proteins. PloS One 2014;9(5):e97725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Chen YC, Sargsyan K, Wright JD, et al. Identifying RNA-binding residues based on evolutionary conserved structural and energetic features. Nucleic Acids Res 2014;42(3):e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Li S, Yamashita K, Amada KM, Standley DM. Quantifying sequence and structural features of protein-RNA interactions. Nucleic Acids Res 2014;42(15):10086–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Ren H, Shen Y. RNA-binding residues prediction using structural features. BMC Bioinformatics 2015;16:249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Tuvshinjargal N, Lee W, Park B, Han K. PRIdictor: protein-RNA interaction predictor. Biosystems 2016;139:17–22. [DOI] [PubMed] [Google Scholar]
- 187. Sun M, Wang X, Zou C, et al. Accurate prediction of RNA-binding protein residues with two discriminative structural descriptors. BMC Bioinformatics 2016;17(1):231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tang Y, Liu D, Wang Z, et al. A boosting approach for prediction of protein-RNA binding residues. BMC Bioinformatics 2017;18(Suppl 13):465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Luo J, Liu L, Venkateswaran S, et al. RPI-Bind: a structure-based method for accurate identification of RNA-protein binding sites. Sci Rep 2017;7(1):614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wang L, Brown SJ. BindN: a web-based tool for efficient prediction of DNA and RNA binding sites in amino acid sequences. Nucleic Acids Res 2006;34(Web Server issue):W243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Wang L, Huang C, Yang MQ, Yang JY. BindN+ for accurate prediction of DNA and RNA-binding residues from protein sequence features. BMC Syst Biol 2010;4 Suppl 1(Suppl 1):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Carson MB, Langlois R, Lu H. NAPS: a residue-level nucleic acid-binding prediction server. Nucleic Acids Res 2010;38(Web Server issue):W431–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Yang X, Wang J, Sun J, Liu R. SNBRFinder: a sequence-based hybrid algorithm for enhanced prediction of nucleic acid-binding residues. PloS One 2015;10(7):e0133260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Su H, Liu M, Sun S, et al. Improving the prediction of protein-nucleic acids binding residues via multiple sequence profiles and the consensus of complementary methods. Bioinformatics 2019;35(6):930–6. [DOI] [PubMed] [Google Scholar]
- 195. Zhang J, Chen Q, Liu B. NCBRPred: predicting nucleic acid binding residues in proteins based on multilabel learning. Brief Bioinform 2021;22(5):bbaa397. [DOI] [PubMed] [Google Scholar]
- 196. Sun Z, Zheng S, Zhao H, et al. To improve prediction of binding residues with DNA, RNA, carbohydrate, and peptide via multi-task deep neural networks. IEEE/ACM Trans Comput Biol Bioinform 2022;19(6):3735–43. [DOI] [PubMed] [Google Scholar]
- 197. Mcginnis S, Madden TL. BLAST: at the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res 2004;32(Web Server issue):W20–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 2005;33(7):2302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Faraggi E, Zhou Y, Kloczkowski A. Accurate single-sequence prediction of solvent accessible surface area using local and global features. Proteins 2014;82(11):3170–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Magnan CN, Baldi P. SSpro/ACCpro 5: almost perfect prediction of protein secondary structure and relative solvent accessibility using profiles, machine learning and structural similarity. Bioinformatics 2014;30(18):2592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Zhao Z, Peng Z, Yang J. Improving sequence-based prediction of protein-peptide binding residues by introducing intrinsic disorder and a consensus method. J Chem Inf Model 2018;58(7):1459–68. [DOI] [PubMed] [Google Scholar]
- 202. Taherzadeh G, Zhou Y, Liew AW, Yang Y. Structure-based prediction of protein- peptide binding regions using random forest. Bioinformatics 2018;34(3):477–84. [DOI] [PubMed] [Google Scholar]
- 203. Petsalaki E, Stark A, García-Urdiales E, Russell RB. Accurate prediction of peptide binding sites on protein surfaces. PLoS Comput Biol 2009;5(3):e1000335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Lavi A, Ngan CH, Movshovitz-Attias D, et al. Detection of peptide-binding sites on protein surfaces: the first step toward the modeling and targeting of peptide-mediated interactions. Proteins 2013;81(12):2096–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Lei Y, Li S, Liu Z, et al. A deep-learning framework for multi-level peptide-protein interaction prediction. Nat Commun 2021;12(1):5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46(D1):D1074–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Yang J, Roy A, Zhang Y. Protein-ligand binding site recognition using complementary binding-specific substructure comparison and sequence profile alignment. Bioinformatics 2013;29(20):2588–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Babor M, Gerzon S, Raveh B, et al. Prediction of transition metal-binding sites from apo protein structures. Proteins 2008;70(1):208–17. [DOI] [PubMed] [Google Scholar]
- 209. Chauhan JS, Mishra NK, Raghava GP. Identification of ATP binding residues of a protein from its primary sequence. BMC Bioinformatics 2009;10:434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Chauhan JS, Mishra NK, Raghava GP. Prediction of GTP interacting residues, dipeptides and tripeptides in a protein from its evolutionary information. BMC Bioinformatics 2010;11:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Brylinski M, Skolnick J. FINDSITE-metal: integrating evolutionary information and machine learning for structure-based metal-binding site prediction at the proteome level. Proteins 2011;79(3):735–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Firoz A, Malik A, Joplin KH, et al. Residue propensities, discrimination and binding site prediction of adenine and guanine phosphates. BMC Biochem 2011;12:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Parca L, Gherardini PF, Helmer-Citterich M, Ausiello G. Phosphate binding sites identification in protein structures. Nucleic Acids Res 2011;39(4):1231–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Passerini A, Lippi M, Frasconi P. MetalDetector v2.0: predicting the geometry of metal binding sites from protein sequence. Nucleic Acids Res 2011;39(Web Server issue):W288–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Chen K, Mizianty MJ, Kurgan L. ATPsite: sequence-based prediction of ATP-binding residues. Proteome Sci 2011;9 Suppl 1(Suppl 1):S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Yu D-J, Hu J, Tang Z-M, et al. Improving protein-ATP binding residues prediction by boosting SVMs with random under-sampling. Neurocomputing 2013;104:180–90. [Google Scholar]
- 217. Yu DJ, Hu J, Huang Y, et al. TargetATPsite: a template-free method for ATP-binding sites prediction with residue evolution image sparse representation and classifier ensemble. J Comput Chem 2013;34(11):974–85. [DOI] [PubMed] [Google Scholar]
- 218. He W, Liang Z, Teng M, Niu L. mFASD: a structure-based algorithm for discriminating different types of metal-binding sites. Bioinformatics 2015;31(12):1938–44. [DOI] [PubMed] [Google Scholar]
- 219. Hu X, Wang K, Dong Q. Protein ligand-specific binding residue predictions by an ensemble classifier. BMC Bioinformatics 2016;17(1):470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Hu X, Dong Q, Yang J, Zhang Y. Recognizing metal and acid radical ion-binding sites by integrating ab initio modeling with template-based transferals. Bioinformatics 2016;32(21):3260–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Hu J, Li Y, Yan W-X, et al. KNN-based dynamic query-driven sample rescaling strategy for class imbalance learning. Neurocomputing 2016;191:363–73. [Google Scholar]
- 222. Hu J, Li Y, Zhang Y, Yu DJ. ATPbind: accurate protein-ATP binding site prediction by combining sequence-profiling and structure-based comparisons. J Chem Inf Model 2018;58(2):501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Wang S, Hu X, Feng Z, et al. Recognizing ion ligand binding sites by SMO algorithm. BMC Mol Cell Biol 2019;20(Suppl 3):53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Liu L, Hu X, Feng Z, et al. Prediction of acid radical ion binding residues by K-nearest neighbors classifier. BMC Mol Cell Biol 2019;20(Suppl 3):52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Xia CQ, Pan X, Shen HB. Protein-ligand binding residue prediction enhancement through hybrid deep heterogeneous learning of sequence and structure data. Bioinformatics 2020;36(10):3018–27. [DOI] [PubMed] [Google Scholar]
- 226. Lu ZC, Jiang F, Wu YD. Phosphate binding sites prediction in phosphorylation-dependent protein-protein interactions. Bioinformatics 2021;37(24):4712–8. [DOI] [PubMed] [Google Scholar]
- 227. Song J, Liu G, Jiang J, et al. Prediction of protein-ATP binding residues based on ensemble of deep convolutional neural networks and LightGBM algorithm. Int J Mol Sci 2021;22(2):939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Pintar A, Carugo O, Pongor S. DPX: for the analysis of the protein core. Bioinformatics 2003;19(2):313–4. [DOI] [PubMed] [Google Scholar]
- 229. Jones S, Thornton JM. Analysis of protein-protein interaction sites using surface patches. J Mol Biol 1997;272(1):121–32. [DOI] [PubMed] [Google Scholar]
- 230. Mihel J, Sikić M, Tomić S, et al. PSAIA - protein structure and interaction analyzer. BMC Struct Biol 2008;8:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Putignano V, Rosato A, Banci L, Andreini C. MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res 2018;46(D1):D459–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Roy A, Yang J, Zhang Y. COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 2012;40(Web Server issue):W471–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Ravindranath PA, Forli S, Goodsell DS, et al. AutoDockFR: advances in protein-ligand docking with explicitly specified binding site flexibility. PLoS Comput Biol 2015;11(12):e1004586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Ravindranath PA, Sanner MF. AutoSite: an automated approach for pseudo-ligands prediction-from ligand-binding sites identification to predicting key ligand atoms. Bioinformatics 2016;32(20):3142–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Chen CC, Hwang JK, Yang JM. (PS)2-v2: template-based protein structure prediction server. BMC Bioinformatics 2009;10:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The protein-ligand interaction datasets used in this manuscript are publicly available, and their specific sources are summarized as follows: The dataset used for evaluating sequenced-based PAIR-pro predictors is the virus-human interaction dataset [41, 94]. The dataset used for evaluating SINGLE-res predictors is the Test_60 dataset [124]. The DNA-129_Test dataseet is from publications [3, 5]. The dataset used to evaluate protein-RNA interaction residue predictors is from the publication [24].