

Abstract
Single-cell genetic heterogeneity is ubiquitous in microbial populations and an important aspect of microbial biology; however, we lack a broadly applicable and accessible method to study this heterogeneity in microbial populations. Here, we show a simple, robust and generalizable method for high-throughput single-cell sequencing of target genetic loci in diverse microbes using simple droplet microfluidics devices (droplet targeted amplicon sequencing; DoTA-seq). DoTA-seq serves as a platform to perform diverse assays for single-cell genetic analysis of microbial populations. Using DoTA-seq, we demonstrate the ability to simultaneously track the prevalence and taxonomic associations of >10 antibiotic-resistance genes and plasmids within human and mouse gut microbial communities. This workflow is a powerful and accessible platform for high-throughput single-cell sequencing of diverse microbial populations.
Single-cell heterogeneity is ubiquitous in nature and single-cell sequencing is a powerful tool for understanding populations of heterogeneous cells. In bacteria, single-cell genomic heterogeneity plays key roles in evolution1, antimicrobial resistance2, host-colonization3 and pathogenesis4. In these systems, mechanisms of genomic variation such as mutation, phase variation, gene deletion, gene duplication and horizontal gene transfer are frequently observed; however, it is difficult to study this variation at the single-cell level without robust methods for single-cell sequencing. Studying microbes at the single-cell level can provide insights into their individual functions within complex multispecies communities where they naturally reside.
Single-cell genetic heterogeneity is typically observed through colony plating, where single colonies represent populations usually derived from single cells5; however, colony plating fails to represent unculturable taxa, which can play key roles in microbial community functions6,7. In addition, rare variants cannot be reliably detected using this method due to limits in the number of colonies that can be reasonably picked and sequenced. Finally, mechanisms driving heterogeneity that operate on timescales similar to colony growth (for example phase variation) cannot be observed using colony plating methods8.
Despite high demand, methods for single-cell sequencing of microbes are difficult to implement and typically require species-specific protocol optimization9-11. This deficit is due, in part, to the substantial challenges associated with sequencing single microbial cells. Bacteria are small and generate low yields of genetic material per cell. In addition, the diverse makeup of their cell membranes, cell walls and other protective features require different lysis or permeabilization conditions specific to the individual microbial species. These challenges result in single-cell sequencing methods that require complex workflows and/or are not easily generalizable to diverse microbial communities9-11.
Droplet microfluidics, a platform technology where reactions are miniaturized in picoliter-scale droplets and created and manipulated using microfluidic channels, is a promising platform for microbial single-cell sequencing11,12; however, the droplet microfluidics-based methods developed thus far employ advanced microfluidics modules (such as droplet reinjection and merging) that are prone to failure and require specialized expertise, making it unsuitable for typical academic laboratories with moderate budgets and no microfluidics expertise. Thus, deployment of these methods to the broader scientific community has been very limited. As such, there is a pressing need for an accessible and generalizable method for profiling genetic heterogeneity within microbial populations at the single-cell level.
We developed a robust and generalizable droplet microfluidics workflow for quantitative single-cell-targeted genetic profiling of microbes (DoTA-seq). DoTA-seq leverages picoliter-sized hydrogels generated using simple droplet makers to isolate and lyse single microbial cells. Next, a one-step targeted multiplex PCR reaction simultaneously amplifies target DNA and attaches unique DNA barcodes to each cell. Compared to previously published droplet sequencing workflows for single-cell untargeted sequencing11,12, the targeted nature of DoTA-seq enables high capture rates for loci of interest. Therefore, DoTA-seq is suitable for single-cell sequencing assays where the loci of interest are known. DoTA-seq leverages the advantages of ultrahigh-throughput droplet microfluidics without requiring advanced microfluidics modules. Instead, our method relies on simple modules such as microfluidic droplet makers and gel bead re-injectors. Furthermore, due to its simplicity, this workflow is theoretically amenable to be adapted into a completely microfluidics-free workflow in the future13,14. Hence, it has the potential to be adopted widely by the biology research community.
Using DoTA-seq, we demonstrate multiple different single-cell sequencing assays on diverse types of microbial populations, highlighting the broad applicability of the method. We track the taxonomically resolved shifts in the prevalence (the fraction of the population harboring the gene) of 12 antibiotic-resistance genes (ARGs) within a 25-member human gut microbial community exposed to increasing concentration of antibiotics. We profile the taxonomic associations of ARGs and plasmids in a mouse and human fecal sample, respectively. Finally, we quantify the diverse and genetically distinct subpopulations generated by phase variation in the prevalent human gut symbiont Bacteroides fragilis. These highlighted assays are efficiently performed using DoTA-seq and represent just a few of many possible future applications of this workflow.
Results
The DoTA-seq workflow
Droplet microfluidics has been applied to single-cell sequencing methodology with great success11,12,15,16; however, a major drawback of droplet microfluidics is the microfluidic complexity of multistep workflows. In particular, these methods involving a combination of droplet reinjection, splitting and merger steps, which have a have a high failure rate and are difficult to implement without extensive droplet microfluidics expertise11,17,18. As a result, compromises in efficiency are typically made to achieve cell lysis and molecular reactions simultaneously in a one-pot reaction19. In microbial communities, the diversity of bacterial cell walls precludes a one-pot reaction that efficiently lyses all species and performs the desired molecular reactions.
In DoTA-seq, a microfluidic droplet maker is used to encapsulate single bacterial cells into droplets containing acrylamide and a reversible crosslinker. The individual cells are crosslinked into a polyacrylamide matrix, trapping individual cells inside droplet gels (Fig. 1a). Crosslinked polyacrylamide is heat resistant, enabling a wide repertoire of potential lysis conditions to achieve efficient lysis of diverse microbes. The polyacrylamide gel matrix contains pores on the scale of 10–100 nm (ref. 20), allowing detergents and enzymes to diffuse through, lysing the cells and removing cellular material while entrapping genomic and plasmid DNA (Fig. 1b). At limiting dilutions, approximately one in ten droplet gels will contain a cell according to a Poisson distribution (Supplementary Note 1). The droplet maker operates at ~10-kHz frequencies, encapsulating hundreds of thousands of single cells within a few minutes.
Fig. 1 ∣. DoTA-seq profiles single-cell genetic loci in Gram-negative and -positive bacteria.

a, Overview of the DoTA-seq workflow. b, Fluorescent and brightfield image overlay showing lysed of B. subtilis cells inside hydrogels. SYBR Green staining reveals the trapped genomic DNA inside the hydrogels, whereas the lysed cell is no longer visible in brightfield. Scale bar, 20 μm. For more images of the complete lysis workflow, see Extended Data Fig. 1. c, Hydrogels are re-encapsulated with a DoTA-seq PCR mix with unique barcode oligonucleotides at a limiting dilution. Microscopy image shows a representative image of the re-encapsulated gels at the outlet of the microfluidic device. Scale bar, 20 μm. Oligo, oligonucleotide. d, Schematic of the barcoding multiplex PCR reaction inside droplets. Seq, sequencing. e, Grouped bar plot by gene shows the fraction of cells with the given gene detected in B. subtilis (green bars, ~2,000 cells) or E. coli (red bars, ~10,000) cells classified by 16S rRNA sequencing. The numbers on the bars represent the mean number of cells detected. Data points represent independent replicates. O/N, overnight cultures. f, A mock community is generated by combining cultures of eight different bacteria in specific proportions. Relative abundance of the community is determined by DoTA-seq and plotted next to the theoretical values. C. hiranosis, Clostridium hiranonis; L. lactis, Lactococcus lactis.
Following lysis of cells inside the droplet gels, we use a second droplet maker to re-encapsulate the droplet gels together with PCR reagents and random DNA barcode oligonucleotides (synthesized as a 15-bp random sequence flanked by constant sequences) into a larger droplet (Fig. 1c). The barcode oligonucleotides are introduced at a limiting dilution such that most droplets contain one droplet gel and one or zero barcodes according to a Poisson distribution. At this Poisson loading ratio, approximately one in ten droplets contain one barcode and approximately 0.5% of droplets contain more than one barcode (Supplementary Note 1). A droplet that contains more than one barcode can yield a single cell that is counted more than once but these events are low in frequency and occur randomly. Therefore, they do not substantially impact the results. In addition, throughput can be augmented with some loss in quantitative resolution by increasing the Poisson loading ratio of the barcodes (fewer droplets contain zero barcodes, whereas a higher fraction of droplets contains multiple barcodes).
In droplets that contain a lysed cell and a barcode, multiplex PCR simultaneously amplifies both the unique barcode and the target loci, splicing them together into an amplicon sequencing library for Illumina platforms (Fig. 1d). In droplets without a lysed cell or barcode, complete amplicons are not formed and are thus not sequenced. In the data analysis, the sequence of the barcodes labels the amplicons that derive from the same droplet and thus the same cell. In rare instances, multiple different cells could be associated with the same barcode sequence. These cases can often be filtered out based on the presence of conflicting marker sequences (for example 16S rRNA gene) representing two different cells (Supplementary Note 2).
The microfluidic droplet makers used in DoTA-seq are simple in design and easy to use and fabricate. Similar devices can be purchased from multiple commercial sources. All reagents for this workflow are widely available off-the-shelf. In our laboratory, this workflow is regularly performed for five samples at a time for ~10,000 cells per sample pooled into one MiSeq run, which required <8 h of hands-on time starting from cell suspension to sequencing.
DoTA-seq can reliably sequence diverse mixtures of bacteria
To evaluate the ability of DoTA-seq to sequence target loci in both Gram-negative and Gram-positive bacteria, we generated a mixture of freshly grown Escherichia coli MG1655 and Bacillus subtilis PY79 cells as a control community. The E. coli strain contained a 4-kb plasmid harboring red fluorescent protein (RFP) and a pSC101* origin and the B. subtilis strain harbored a chromosomally integrated green fluorescent protein (GFP)21. We designed a DoTA-seq assay to sequence RFP on the plasmid, GFP on the chromosome and the endogenous chromosomal genes dnaG and dnaPolIIIfor E. coli and B. subtilis. In addition, we sequenced the 16S rRNA V3–V5 region for both species (Fig. 1e).
Using DoTA-seq, we sequenced ~10,000 cells per run in technical duplicates. To determine whether the targeted genes were faithfully linked to the correct species at the single-cell level, we first grouped reads with the same barcode to represent reads from a single cell. We used the sequence of the 16S rRNA gene to classify the species of each cell. Next, we grouped all cells of the same species and counted the proportion of cells of each species that contained reads mapping to each gene (gene prevalence). For both E. coli and B. subtilis cells, the expected genes were observed in ~90% of the sequenced cells, showing that DoTA-seq can efficiently capture target genes within single cells (Fig. 1e). At the same time, false positives (genes not belonging to the assigned species) were detected in ~0.2% of cells for the chromosomal genes (dnaG, dnaPolIII and GFP) and ~1% for RFP on the plasmid. The ~4-kb plasmid was orders of magnitude smaller in length than genomic DNA. Therefore, the plasmid has a higher chance of diffusing out of its cognate hydrogel into neighboring gels during the washing, lysis and storage steps. As such, loci on small plasmids will have higher false positive rates than chromosomal loci.
To investigate the accuracy of DoTA-seq on a diverse mixture of bacteria, we generated a mock microbial community consisting of widely varying abundances of diverse Gram-positive and Gram-negative bacteria. In addition, we included one E. coli strain that harbored a plasmid with the rfp gene (~1%) and another E. coli strain that did not harbor this plasmid (~99%) to evaluate the ability of DoTA-seq to detect genetic differences in a single species. We designed DoTA-seq primers targeting rfp, one essential gene for each of the species, and the 16S rRNA gene using our automated primer design pipeline (Methods). Using DoTA-seq, we profiled this mock community in technical duplicates (one sample split into two runs of ~20,000 cells). Most species were well within twofold of the expected relative abundance with low technical variation between the replicates (Fig. 1f). The targeted essential genes were detected in ~70–90% of the cells for each species, indicating a high capture rate for these genes (Extended Data Table 1). These results demonstrated that DoTA-seq can profile diverse bacteria at the single-cell level within multi-species microbial communities.
The deviation from 100% in gene prevalence (~70–90%) of essential genes that are expected to be present in every cell indicates that some genes are more efficiently captured than others. Differences in primer amplification efficiency can contribute to this variation. To mitigate this effect, the primer concentration can be increased or alternative primers can be designed to be higher efficiency. Alternatively, as the primer capture efficiency for each locus is consistent for different samples analyzed using the same primers, we can compare relative changes in gene prevalence across different treatments or samples. A more detailed discussion of primer amplification efficiencies in DoTA-seq and mitigating strategies can be found in Supplementary Note 3.
DoTA-seq reveals gene dynamics within complex microbial communities
Bacterial genomes are highly plastic, able to lose and gain genes in response to changing environmental stressors22,23. These gene dynamics are critical to the emergence of antibiotic-resistant pathogens24. High-throughput single-cell sequencing enables the investigation of these dynamics. To demonstrate how DoTA-seq can be used to track ARGs within complex microbial communities, we generated a synthetic microbial community composed of 25 diverse and prevalent members of human gut microbiota that have been extensively characterized25 (Supplementary Table 1). We identified 12 ARGs within the publicly available genomes of these isolates and designed a DoTA-seq assay to track them by targeting each ARG as well as the 16S rRNA for taxonomic identification. To establish a baseline of the prevalence of these ARGs in this community, we used DoTA-seq to sequence ~3,000 cells per run in technical duplicates (Fig. 2b). The technical variation was small and negatively correlated (Spearman rho = −0.57, P < 6.5 × 10−10) with the number of sequenced cells for each species (Extended Data Fig. 2). This implies that increasing the total number of sequenced cells can reduce the technical variability observed in DoTA-seq, particularly in low-abundance populations.
Fig. 2 ∣. DoTA-seq enables tracking of gene-species dynamics in a complex human gut microbial community.

a, Heat map of ARG–species associations in a 25-member synthetic gut microbial community for a representative replicate. For each species (x axis) and ARG (y axis), the proportion of cells containing the gene (gene prevalence) is shown. The opacity of the background for each box is proportional to the prevalence value. Computationally predicted ARGs based on genome sequence are outlined in blue. ARGs that were not found in the species’ genome sequence but observed using DoTA-seq as well as independently confirmed are outlined in green boxes. ARGs that were not found in a given species’ genome sequence are represented by green text. Species include BA, Bifidobacterium adolescentis; ER, Eubacterium rectale; FP, Faecalibacterium prausnitzii; AC, Anaerostipes caccae; CC, Coprococcus comes; RI, Roseburia intestinalis; DP, Desulfovibrio piger; BH, Blautia hydrogenotrophica; CA, Collinsella aerofaciens; PC, Prevotella copri; DL, Dorea longicatena; CG, Clostridium asparagiforme; BF, Bacteroides fragilis; EL, Eggerthella lenta; CH, Clostridium hiranonis; BO, Bacteroides ovatus; BT, Bacteroides thetaiotaomicron; BU, Bacteroides uniformis; BV, Bacteroides vulgatus; BC, Bacteroides caccae; BY, Bacteroides cellulosilyticus; PJ, Parabacteroides johnsonii; DF Dorea formicigenerans; BL, Bifidobacterium longum subsp. infantis; BP, Bifidobacterium pseudocatenulatum (Supplementary Table 1). b, Bar plot of the average gene prevalence for ARGs for each ARG–species combination that displayed greater than or equal to 10% prevalence. Data points represent values from technical replicates (n = 2). c, Schematic of the experiment for tracking changes in ARGs in response to antibiotics. MLS, erythromycin and lincomycin. d, Relative abundance of species at each measurement time determined by DoTA-seq. The lines corresponding to species that were not detected after passage 1 or did not contain ARGs are semi-transparent. Data points represent values of technical replicates (n = 2). e, Prevalence of ARG–species associations at different passages. Species that were not detected after passage 1 and/or did not contain targeted ARGs are excluded from this graph. Lines correspond to the mean and data points represent technical replicates (n = 2).
DoTA-seq revealed the expected ARG–taxa associations based on the genome sequences (Fig. 2a,b). One exception was the reported existence of the rifampicin resistant allele of the rpoB gene in Bifidobacterium longum (BL) even though this gene was not found in the BL genome sequence; however, we confirmed the presence of this gene in purified BL genomic DNA by PCR, suggesting that the discrepancy was due to an incomplete genome sequence in the public database (Extended Data Fig. 3). Most ARGs were observed at >70% gene prevalence (tetO in Anaerostipes caccae (AC), Dorea longicatena (DL) or cepA in Bacteroides fragilis (BF), for example). Notably, some ARGs were observed at <50% prevalence (ermQ or tetW for example). As these genes are not known to be essential, these results could stem from two possibilities (1) the capture efficiency of the primers for these genes were poor and variable or (2) the bacteria displayed population heterogeneity with respect to the presence/absence of these genes. Considering the first scenario, we analyzed the ratio between the sequencing coverage for target genes and the 16S in each cell. This quantity provides insight into the relative capture efficiencies for each target gene (Supplementary Note 3). This analysis suggests that genes of low prevalence (ermQ and tetW) did not result from poor capture efficiencies (Extended Data Fig. 4). Therefore, we considered the second scenario; if the ARGs were present in a subset of the bacterial populations, then gene prevalence could change in response to antibiotic stress. This hypothesis assumes that primer capture efficiencies are constant across different treatments.
To test this hypothesis, we cultured this community in the presence of erythromycin and lincomycin at successively higher concentrations and measured ARG prevalence with DoTA-seq (Fig. 2c). The addition of erythromycin and lincomycin was expected to confer a fitness advantage to cells that harbored the ermB, ermQ or mef(en2) gene. Our results showed that the growth of most species was inhibited in the presence of the antibiotics (Fig. 2d). Of the species that grew, the prevalence of several ARGs varied drastically across different antibiotic concentrations (Fig. 2e). For example, in Clostridium hiranonis (CH), the prevalence of ermQ increased. In Parabacteroides johnsonii (PJ), the frequencies of mef(en2) and tetQ decreased. In AC, the prevalence of tetO initially decreased, then moderately increased with an increase in the concentration of antibiotics. As the same primer sets were used in all these DoTA-seq runs, our results suggest that the changes in gene prevalence reflect the changes in fraction of cells that contained the gene. Therefore, our results suggest that multiple species established population heterogeneity during outgrowth from a single colony and the fraction of the population harboring the given gene can shift in response to antibiotic stress.
To determine whether the observed variation in gene prevalence reflects temporal changes in the genetic composition of the population, we performed single-cell digital PCR on the communities and colony PCR on streaked out colonies. These experiments were used to independently assess the prevalence of mef(en2), tetQ and cepA genes in the Gram-negative species PJ and BF from the samples described above. Consistent with DoTA-seq, single-cell digital PCR showed that the gene prevalence of mef(en2) and tetQ was ~0.8 at time point 0 and ~0.6 at time point 2 in PJ. In addition, the gene prevalence of cepA was ~0.7 at time point 0 and ~0.2 at time point 2 in BF (Extended Data Fig. 5a-c). Using selective plating of BF colonies, we further confirmed the decreasing trend of cepA prevalence in BF by PCR genotyping of colonies derived from passage 1 (7 of 14 colonies positive for cepA) and passage 2 (0 of 6 colonies positive for cepA) (Extended Data Fig. 5d). In sum, these results are consistent with the trends observed in DoTA-seq and suggest the fraction of the population harboring specific ARGs can change in response to environmental stress.
DoTA-seq reveals gene–taxa relationships in natural microbiomes
To evaluate the capabilities of DoTA-seq on natural microbiome samples, we used DoTA-seq to analyze mouse and human gut microbiome samples. Due to the constraints of microfluidic droplet making, microbial cells were extracted first to remove large debris that may clog the device. Using low-speed centrifugation26, we separated the large debris from the cell suspension in the supernatant (Fig. 3a). Metagenomic shotgun sequencing of pre- and post-extraction samples showed a highly concordant community composition. This implies that the DoTA-seq cell extraction procedure does not substantially alter the community composition (Pearson R = 0.997; Extended Data Fig. 6).
Fig. 3 ∣. DoTA-seq elucidates ARG and plasmid–taxa associations in fecal microbial communities.

a, Schematic of cell extraction procedures for DoTA-seq of natural samples. b, Comparison between 16S rRNA gene profiling and DoTA-seq relative abundance of the major taxa (>0.01% relative abundance) in the mouse fecal community. c, Bar plot of the gene prevalence of ARGs in the mouse fecal sample. The bar represents the average of two technical replicates. d, Bar plot of the gene prevalence of plasmid replicons in the human fecal sample. The bar represents the average of two technical replicates plus one additional technical replicate that sequenced ~37,000 cells. The circular markers represent individual replicates. The red outlines represent replicon–host associations that are also found in a database of human gut MAGs.
To identify DoTA-seq targets in these communities, we first assembled metagenomic shotgun sequencing reads into contigs and searched for ARGs and plasmid replication genes in the mouse and human fecal sample, respectively. Using our automated pipeline (Methods), we designed DoTA-seq primers targeting these genes as well as the 16S rRNA for taxonomic identification. We used a primer set identifying ARG–taxa and plasmid–taxa associations to sequence the microbial cells extracted from both mouse and human fecal samples in technical replicates.
In the mouse fecal sample, DoTA-seq detected all major taxa (≥0.5% relative abundance) and most taxa as low as 0.01% identified by standard 16S rRNA gene sequencing (Fig. 3b). In general, abundant taxa identified by 16S rRNA gene sequencing were also abundant in DoTA-seq results, but the relative abundance of individual taxa exhibited some differences. We would expect some differences in taxonomic abundance due to notable differences between the 16S rRNA gene sequencing and DoTA-seq workflows. For example, there are differences in lysis procedures and sample washing steps. Further, DoTA-seq counts single cells of a given taxonomic identity, whereas the relative abundance based on traditional 16S rRNA gene sequencing can be influenced by the number of 16S rRNA copies, which varies across different taxa27. The primary application of DoTA-seq is not to determine taxonomic relative abundance but to link genes of interest to the taxonomic identity of single cells.
In the commercial human fecal sample there was a systematic under-representation of many (mostly Gram-negative) species in the single-cell sequencing results (Extended Data Fig. 7), which is due to storage in the proprietary preservation buffer that prematurely lysed mostly Gram-negative bacteria (Extended Data Fig. 8). Thus, for single-cell sequencing, samples should be preserved using methods that do not induce cell lysis. All other samples used in this paper were flash frozen and stored at ~80 °C.
Analysis of the ARG–taxa and plasmid–taxa associations in mouse and human fecal samples, respectively, revealed ARGs and plasmids with both broad and narrow host ranges (Fig. 3c,d). For example, in the mouse sample, the ARGs tetQ and tetO were prevalent in multiple taxa (Fig. 3c), whereas ErmB and the Van(D,HD,SD) genes were found only in Ruminococcaceae and Lachnospiraceae at very low prevalence, respectively (Supplementary Table 2). In the human sample, most plasmid replicons were associated with a single family-level taxa, but plasmid replicon 1351 was associated with both Desulfovibrionaceae and Bifidobacteriaceae, which are hosts from two different phyla (Proteobacteria and Actinobacteria), suggesting a broad host range for this replicon (Fig. 3d). To independently confirm these replicon–taxa associations, we performed a BLAST search of each plasmid replicon sequence to a database of metagenome assembled genomes (MAG) from the human microbiome. Except for Marinfilaceae-rep-1702, all other major observed replicon–taxa associations (prevalence ≥5%) were found in this database of MAGs, corroborating the DoTA-seq results (Fig. 3d and Supplementary Table 3). Together, these results demonstrate that DoTA-seq can track the taxonomic association of diverse genes of interest within complex natural microbiomes.
DoTA-seq enables quantification of bacterial subpopulations generated through genetic sequence variation
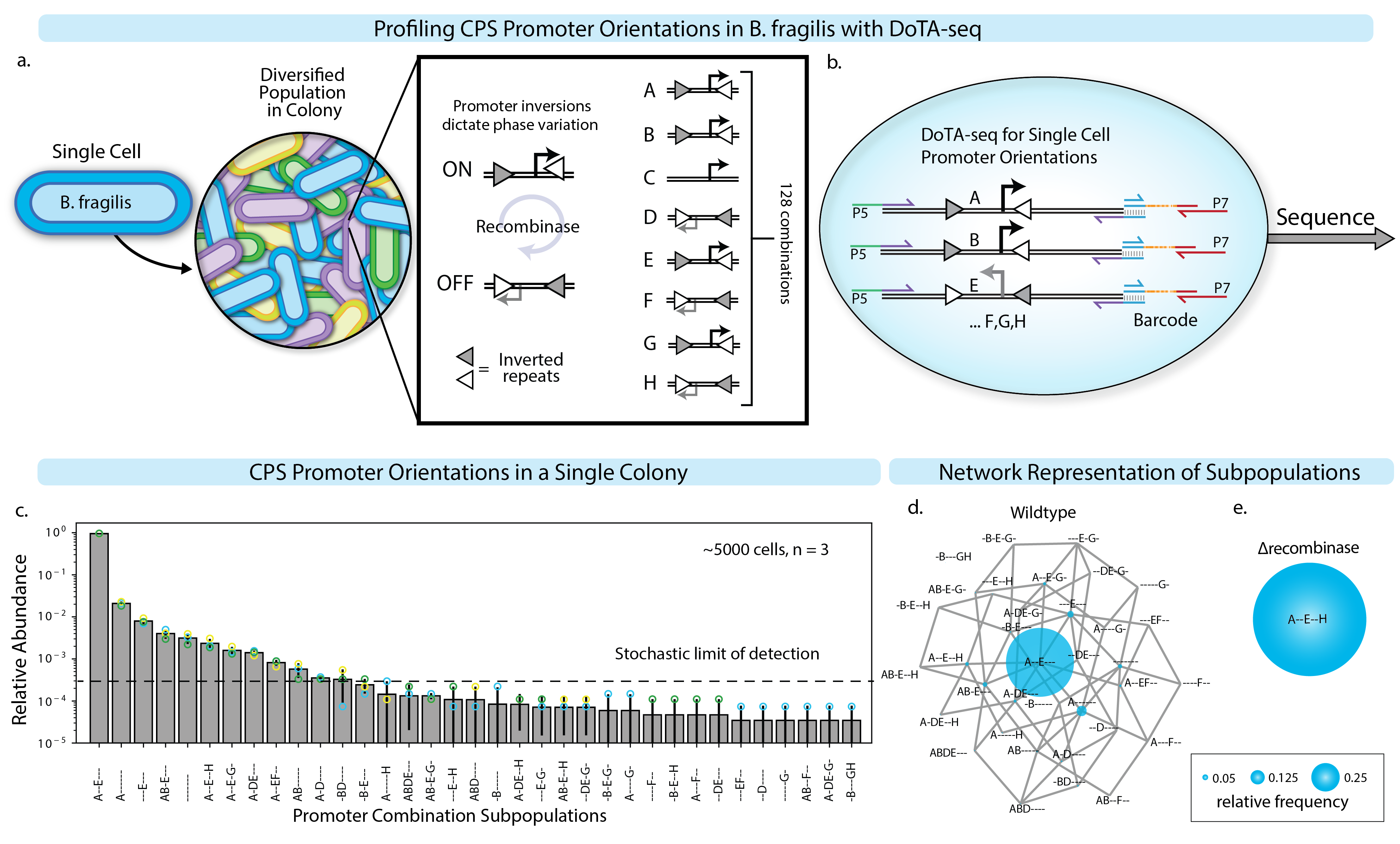
Beyond the presence/absence of genes, DoTA-seq can also be used to profile genetic rearrangements or mutations within the genomes of single cells. The modifications to this workflow of DoTA-seq yields data that is more quantitative compared to the binary (gene presence/absence) measurements of the previously demonstrated applications. In this case, the nucleotide sequence informs the state of each cell and variations in primer capture efficiencies do not bias measurements of the single-cell states under most circumstances. Using this workflow, we profiled the single-cell combinatorial promoter inversion states of the B. fragilis capsular polysaccharide synthesis (CPS) operons28. The promoters of seven operons are regulated by an endogenous recombinase (mpi), which inverts the promoter sequence of each operon, toggling the promoter between an ON and OFF state (Extended Data Fig. 9a). We use the DoTA-seq amplicon sequence of the promoter region to deduce the combinatorial promoter ON/OFF states for each cell (Extended Data Fig. 9b). Sequencing a single B. fragilis colony on an agar plate revealed widespread population heterogeneity (Extended Data Fig. 9c,d). As we show in a separate report8, computational models can be fit to these quantitative data to generate biological insights. In sum, DoTA-seq can be used to determine the presence/absence and nucleotide sequence of target genes within single cells, enabling a wide range of potential applications.
Discussion
Single-cell genetic heterogeneity underlies numerous important phenomena in the microbial world, affecting evolutionary dynamics and microbiome functions1-4,29. DoTA-seq is an accessible and generalizable platform for ultrahigh-throughput single-cell genetic profiling of microbes. As such, the DoTA-seq workflow as presented here should be tuned according to experimental goals. Encapsulating barcode oligonucleotides at limiting dilution results in capture of ~10% of cells injected into the device (Supplementary Note 1). If a higher cell capture efficiency is desired, barcoded hydrogels30 could be used instead of barcode oligonucleotides. Hydrogels pack closely within the microfluidic device to enable delivery of a unique barcode to every droplet (instead of the ~1 in 10 for limiting dilution), potentially yielding close to complete cell capture. Alternatively, barcode oligonucleotides could be added at a higher encapsulation rate, such that on average, multiple unique barcodes are included per droplet and very few droplets contain no barcodes at all. In this case, most cells would be represented by multiple unique barcodes according to the Poisson distribution. Downstream data analyses would have to take this additional layer into account by fitting a Poisson model to the barcode count data to infer the underlying single-cell frequencies.
Designing multiplex primers that efficiently amplify the sequences of interest, while minimizing off-target annealing, is a crucial step in DoTA-seq. The multiplex PCR primers used in DoTA-seq should not produce large primer dimers (>300 bp) when used at the relevant concentrations. Our automated primer generating pipeline generally produces primer sets that satisfy these requirements. In addition, each target loci should be amplified at similar efficiencies, which can be achieved by adjusting the relative primer concentrations (Supplementary Note 3). Although we have not explored the limits for the number of simultaneous targets possible in DoTA-seq, we have generated multiple 10–12-plex DoTA-seq target assays that required little to no additional optimization using our automated pipeline.
DoTA-seq has a wide range of potential applications beyond those explored here. For example, DoTA-seq could be used to identify genes/pathways in taxa of clinical significance within patient microbiomes. DoTA-seq could be used to track the real-time evolution of microbial populations by designing primers targeting mutational hotspots determined from shotgun sequencing31,32. The nucleotide sequence of these amplicons can then be used to accurately reconstruct phylogenetic lineages18. Beyond single cells, DoTA-seq could be used to profile the composition of communities assembled in droplets33. In addition, the DoTA-seq approach could be modified to enable other types of single-cell measurements that are beyond DNA sequencing. For example, we could perform single-cell profiling of phenotypes such as protein expression or cell surface markers via DoTA-seq on cells pre-labeled with DNA barcode-conjugated antibodies34.
The use of microfluidics devices for droplet making presents a barrier to large-scale sample parallelization; however, both droplet-making steps in DoTA-seq can theoretically be replaced with microfluidics-free methods of droplet generation13,14. Thus, we anticipate future versions of this workflow that do not require microfluidics at all, which would substantially boost the throughput and parallelizability of DoTA-seq.
Methods
Oligonucleotides and primers
The oligonucleotides used were purchased from Integrated DNA Technologies as standard single stranded DNA oligonucleotides for universal sequences, an oligonucleotide pool for the ARG primers and as Ultramers for the B. fragilis promoter primers. Ultramers were used to ensure higher synthesis yields of long primers. We found that best performance is achieved with individually ordered and validated primers and carefully combined manually to equal concentrations, rather than using the oligonucleotide pool synthesis methods. Primer sequence and other details can be found in Supplementary Tables 5 and 7-11.
Culturing the 25-member synthetic human gut microbial community
One glycerol stock of the 25-member synthetic community prepared as described above was resuspended in 2 ml YBHI (Supplementary Note 6) in an anaerobic chamber (Coy) with an atmosphere of 2.5 ± 0.5% H2, 15 ± 1% CO2 and balance N2 and incubated at 37 °C for 1 h to recover. After 1 h, the culture was split into two 1-ml tubes for experimental replicates. YBHI was added to each tube to a total of 5 ml with 0.1 μg ml−1l of erythromycin and 0.25 μg ml−1 of lincomycin. After 48 h of incubation at 37°C, 200 μl cells were taken out and added to 200 μl 50% glycerol, labeled ‘time point 1’ and frozen at −80 °C. Then, 1 ml of the culture was added to 4 ml fresh YBHI with 2 μg ml−1 erythromycin and 50 μg ml−1 lincomycin and incubated at 37 °C anaerobically. After an additional 96 h, 200 μl cells were taken out and added to 200 μl 50% glycerol, labeled ‘passage 2’ and frozen at −80 °C.
Microfluidics fabrication
The master mold for the microfluidic devices was fabricated using soft lithography35 in a negative-pressure cleanroom. Thin layers of SU-8 3025 photoresist (MicroChem) were applied to 3-inch silicon wafers (University Wafers) using a spin-coating process to achieve layers of 20 μm for droplet generator 1 and 30 μm for droplet generator 2, respectively. Microfluidic feature patterns were then transferred to the SU-8 layers using a photolithography mask (CAD/Art Services) and a 365 nm collimated LED (Thorlabs M405L4-C1) at 120 mW for 1 min and 45 s. Following exposure, the mold was soft-baked at 95 °C for 5 min before developing the patterned SU-8 in propylene glycol methyl ether acetate for 2 min. The developed master mold was hard-baked at 200 °C for 2 min to complete curing of the SU-8.
Microfluidic devices were cast from the negative mold using polydimethylsiloxane (PDMS) (Sylgard-184) at a 1:11 crosslinker to elastomer ratio and cured at 65 °C for at least 12 h. The devices were then cut from the master mold with a scalpel (Feather) and holes for the inlets and outlets were cut using a 0.75-mm biopsy punch (World Precision Instruments). To close the open microfluidic channels, a glass slide was bonded to the bottom of the PDMS devices. Chemical bonding between the PDMS and glass was achieved on contact following plasma activation with a plasma cleaner (Harrick Plasma). The completed microfluidic channels were treated with Aquapel Glass Treatment (Aquapel, 47100) to render the surfaces hydrophobic.
DoTA-seq workflow
A cell suspension was stained with 1× SYBR Green (Invitrogen) and counted using a hemacytometer (Fisher Scientific, 0267151B) under a Ti-E Eclipse inverted microscope (Nikon) to obtain the cell concentration. This concentration was used to calculate the volume of cells to add to obtain the appropriate concentration for loading into the droplets (2.5 × 107 cells per ml for lambda of 0.1). A polyacrylamide gel solution was prepared as follows: 100 μl acrylamide monomer (Sigma-Aldrich) in water (25% w/v), 15 μl bis-acryloyl cystamine (Santacruz Biotech) in methanol (Sigma-Aldrich) (5% w/v), 10 μl ammonium persulfate (Sigma-Aldrich) (10% w/v), 75 μl PBS and the appropriate number of cells to obtain an encapsulation ratio of 0.1 in 4 pl droplets. This solution was injected into a microfluidic drop maker (device 1) along with BioRad droplet-generation oil for Evagreen (BioRad, 1864005) with 0.5% v/v TEMED (Sigma) dissolved in the oil as a catalyst. Droplet generation was carried out at 1,000 μl h−1 for oil and 600 μl h−1 for the aqueous solutions. Collected droplets were incubated at 37 °C for 10 min to allow gel polymerization. The polymerized gels were removed from the oil as follows: first the oil was drained using a pipette and 1 ml acetone was added, then removed, then 1 ml isopropanol was added, then removed, then 1 ml PBS wash buffer was added to resuspend the gels in an aqueous buffer. The gels were then subject to three more washes in PBS wash buffer.
Cell lysis in the gels was carried out by adding 2× lysozyme buffer (20 mM Tris-HCl, pH 8.0; 10 mM EDTA; NaCl 100 mM and 1% Triton X-100) with 20 mg ml−1 lysozyme (Sigma L6876) to equal volume of gels and incubating at 37 °C for 30 min. The gels were then washed three times in 1 ml PBS +10 mM EDTA, then added to equal volume of 2× proteinase K lysis buffer (Tris, pH 8.0, 20 mM EDTA, 100 mM NaCl and 1% SDS) with 200 μg ml−1 proteinase K and incubated at 55 °C for 30 min. The gels were finally washed four times in 1 ml gel storage buffer (10 mM HEPES, pH 7.5, Tween-20 2%, NaCl 100 mM and EDTA 20 mM) and stored at 4 °C until ready for use in barcoding.
Barcoding of the gels was carried out by first washing the gels four times in 1 ml pre-injection buffer (10 mM HEPES, pH 7.5, NaCl 25 mM, EDTA 0.1 mM and 2% Tween-20) achieving at least 1,000× dilution. The PCR mix consisted of 1× BioRad ddPCR probes mix without dUTPs (BioRad 1863024), 400 nM of P7, 40 nM of Barrev-V3, 400 nM of P5_I5_x, where x represents the unique I5 index used for multiplexing libraries, 20 nM (E. coli/B. subtilis and B. fragilis primers) or 5 nM (ARG primers) of each oligonucleotide in the targeted primer set, 0.015 pM barcode oligonucleotide (freshly diluted from 500 pM stock), 2.5 mM dithiothreitol (from single-use aliquots) to a total of volume of 25 μl. The gel and PCR mix were injected into microfluidic droplet maker device 2 (Supplementary Materials) with BioRad Evagreen droplet oil for encapsulation of gels with PCR mix using 200 μl h−1 for the gel and PCR mix and 900 μl h−1 for the oil. Droplets were collected into a PCR tube (Fisher Scientific, 14222292) until the PCR mix ran out (resulting in ~300 productive droplets per μl PCR mix). In the collected emulsion, excess oil was drained using a pipette and thermocycled as follows: 95 °C 5min, 20 cycles of 95 °C 30 s, 72 °C 10 s, 60 °C 5 min, 72 °C 30 s, then 20 cycles of 95 °C 30 s, 72 °C 10 s, 60 °C 90 s, 72 °C 30 s, then 72 °C 10 min. All steps except for the first and last used a 1 °C s−1 ramp rate. For detailed video instructions on droplet making with gels, please refer to the video article by Demaree et al.36.
After PCR, the coalesced droplets were removed using a pipette, and the emulsion was broken on ice by adding 20 μl of 500 mM EDTA and 20 μl perfluoro-octanol (Sigma, 370533), then the sample was vortexed followed by spin-pulse centrifugation. The aqueous phase was transferred to another tube by pipette, then 20 μl 1 M TCEP (UBP Bio, P1021-100) was added to completely de-crosslink the gels and the resulting solution was vortexed for 10 s to completely dissolve the gels. The whole mixture was cleaned using a Zymo cleanup and concentrator kit (Zymo Research, D4013), then subjected to size selection using SPRI-select beads (Beckman Coulter, B23317) using 0.7× volume of beads. A further round of size selection was performed in 100 mM NaOH, 10% ethanol and 1.4× volume of beads to increase the purity of the library. For the most up-to-date and detailed DoTA-seq protocol, please refer to https://doi.org/10.17504/protocols.io.81wgbxx3ylpk/v1.
Sequencing DoTA-seq libraries
The resultant libraries were quantified using qPCR (NEB E7630S) and sequenced on a MiSeq (Illumina) using a V3 150 cycles kit using custom read 1 and I7 primers. Up to five libraries were pooled per run.
DoTA-seq of mixture culture of E. coli and B. subtilis
E. coli and B. subtilis strains (Supplementary Table 4) were grown overnight separately in LB broth (DOT Scientific) with 34 μg ml−1 chloramphenicol, and 5 μg ml−1 chloramphenicol, 1 μg ml−1 erythromycin and 25 μg ml−1 lincomycin for E. coli and B. subtilis, respectively. Then, 100 μl of the respective overnight cultures were taken, mixed, then washed in 1 ml PBS (Crystalgen) + 0.1% Tween-20 (Sigma-Aldrich) twice before resuspending in 100 μl PBS. The cell suspension was then used as input for DoTA-seq using the E. coli and B. subtilis control primer sets (Supplementary Table 5).
DoTA-seq of mock microbial community
Pure cultures of bacterial isolates were grown in overnight in liquid culture from single colonies according to conditions outlined in Supplementary Table 6. Cells were stained by incubation with 10× SYBR Green and counted with a hemocytometer using CF555 fluorescence to estimate cell concentrations. Aliquots were made in 25% glycerol and flash frozen in liquid nitrogen and stored at −80 °C. A mock community was made by combining different volumes of glycerol stocks according to desired relative abundances and stock cell concentrations (Supplementary Table 6). This mixture was then aliquoted again and flash frozen in liquid nitrogen and stored at −80 °C. For DoTA-seq, each aliquot was thawed on ice, washed in PBS +1% Pluronic F68 (Thermo Fisher, 24040032) and resuspended in 200 μl PBS +1% Pluronic F68. The appropriate number of cells was used in DoTA-seq with mock community primers (Supplementary Table 7) following the DoTA-seq V3 protocol, which can be found at https://doi.org/10.17504/protocols.io.n92ldzox7v5b/v1.
DoTA-seq of the 25-member synthetic community
The 25-member synthetic community (Supplementary Table 1) was prepared as described in Clark R.L. et al.25. Briefly, 25 species were cultured individually in an anaerobic chamber, then mixed to approximate equal proportions based on absorbance at 600 nm (OD600) using a Tecan F200 Plate Reader, with 200 μl in a 96-well microplate. Note that not all monocultures grew to sufficient OD600 to allow equal representation in the final community. The mixture of species was combined with 50% glycerol (Research Products International) and 400-μl aliquots were stored at −80 °C. For each DoTA-seq experiment, a new glycerol stock was thawed, 200 μl cells were spun down and washed twice in 1 ml PBS + 0.1% Tween-20, before resuspension in 100 μl PBS + 0.1% Tween-20. The cell suspension was used as an input for DoTA-seq using the 25-member community primer sets (Supplementary Table 8).
DoTA-seq of the mouse fecal microbial community
Fresh fecal pellets were collected from specific-pathogen-free BALB/c mice and resuspended in 800 μl PBS + 0.1% Pluronic F68 (Thermo Fisher, 24040032) + 25% glycerol with up to five stainless steel 3.2-mm beads (Biospec, 11079132ss). The fecal matter was resuspended by vigorous shaking and vortexing, then the large debris were sedimented by centrifugation at 4 °C at 35g for 20 min. The supernatant containing the cells was extracted, mixed, then aliquoted and flash frozen in liquid nitrogen and stored at −80 °C. For DoTA-seq, an aliquot was thawed on ice, then washed twice with PBS + 1% Pluronic F68, then counted on a hemocytometer as described above. The appropriate number of cells was added to the DoTA-seq workflow as described above and DoTA-seq was performed using primers specified in Supplementary Table 9.
DoTA-seq of the ZymoBIOMICS human fecal reference community
The ZymoBIOMICS human fecal reference sample (Zymo, D6323) was thawed at room temperature and 100 μl was taken and centrifuged at 35g for 20 min to separate cells from large fecal particles. The cells containing supernatant were transferred to a new tube and washed twice with PBS +1% Pluronic F68 and resuspended in 100 μl PBS +1% Pluronic F68. Next, 100 μl overnight cultures of Pseudomonas putida KT2440 and Staphylococcus epidermidis ATCC 12228 grown in LB and BHI broth (Sigma, 53286) respectively were washed twice in PBS +1% Pluronic F68 then resuspended in 100 μl PBS +1% Pluronic F68. We did not analyze the spike-in cells in this study. All cells were stained with 10× SYBR Green and counted on a hemocytometer to estimate cell concentrations. Fecal community and spike-in cells were combined at predetermined ratios and prepared for sequencing with the fecal sample DoTA-seq primer mix (Supplementary Table 10) using the DoTA-seq V3 protocol available on protocols.io at https://doi.org/10.17504/protocols.io.n92ldzox7v5b/v1. For the large-scale sequencing experiment that captured >37,000 cells, the same protocol was followed except no spike-in cells were added and 250 μl of the PCR master mix was used.
Sequencing the capsule polysaccharide promoters in B. fragilis using DoTA-seq
B. fragilis DSM 2151 was streaked onto Petri dishes containing Bacteroides minimal medium with 1.5% w/v agar (Supplementary Note 5). The plate was incubated in the anaerobic chamber at 37 °C for 48 h. After incubation, colonies were randomly picked using a pipette tip and resuspended into PBS + 0.1% Tween-20. The cell suspension was used as an input for step 2 of DoTA-seq using the B. fragilis CPS promoter primer sets, with a slight modification of the protocol. As the population was composed entirely of Gram-negative bacteria that did not require multistep lysis procedures, we simplified the procedure by skipping the lysis step and directly encapsulating single cells with PCR mix. Cells were directly mixed with the PCR mix, proceeding in the second step of DoTA-seq. The PCR mix used was NEB Q5 Ultra Mix (M0544S), and the thermocycling protocol was as follows: 98 °C 2 min, 40 cycles of 98 °C 30 s and 65 °C 5 min, then 72 °C 10 min. The ramp rate was 2 °C s−1. Primers used for these experiments are found in Supplementary Table 11.
DoTA-seq data analysis for synthetic communities
The raw sequencing reads are obtained from the MiSeq. Reads 1 and 2 represent the targeted amplicons. The first index read represents the unique cell barcode, whereas the second index read was used to multiplex libraries from different experiments. Demultiplexing of different libraries (index 2) was performed by MiSeq software. Supplementary Fig. 1 provides a flowchart of the analysis pipeline. Cell barcode demultiplexing and quality control were performed using a custom Python script (R4-parser.ipynb). A typical library will yield ~90% reads after filtering (see Supplementary Table 12 for the sequencing performance of typical DoTA-seq libraries). The filtered reads were mapped to custom built reference databases containing B. fragilis CPS promoter sequences, 16S rRNA sequences and ARGs37 available on GitHub (Code Availability) using Bowtie2 v.2.3.4.1 (ref. 38) using ‘–very-sensitive’ presets. A typical library will yield ~90% mapped reads. The mapped reads were analyzed using custom Python scripts as follows: the mapped reads are organized into read groups consisting of reads with the same unique cell barcode representing amplicons from the same droplet. Read groups with too few reads are removed. For each amplicon target for each read group, a minimum of 1% of the reads of the barcode group, or five reads, whichever is higher, is required to be present to count as a true ‘hit’ for that target. This step removes background crosstalk between the barcodes in the library. When sequencing microbial communities containing different species, 16S rDNA amplicons within each read group are used to taxonomically identify the bacteria represented by the reads within the read group. Read groups with multiple distinct 16S target matches are discarded. When sequencing B. fragilis CPS operons, only read groups containing amplicons for less than all seven targeted amplicons or containing amplicons indicating conflicting promoter orientations are discarded. All scripts are available on GitHub (Code Availability).
DoTA-seq data analysis for natural microbial communities
Using DoTA-seq on communities of unknown composition presents unique data analysis opportunities and challenges. The ability to target multiple loci in a single cell presents the opportunity to simultaneously target multiple marker genes with short read sequencing to obtain more accurate taxonomic classifications; however, traditional pipelines for analysis of microbial taxonomic marker genes do not readily accommodate the single-cell barcoded data format. Marker genes originating from the same droplet should represent a single species. Leveraging this knowledge, we can first group marker gene reads by similarity for each droplet and build a consensus sequence for each group to correct for sequencing/PCR errors. We can then perform taxonomic classification for each droplet by using each consensus sequence for each droplet. We identify multiencapsulated droplets as those that contain representative marker genes that represent distinct taxa. Thus far, we have implemented a proof-of-principle version of this analysis workflow for our natural microbial community datasets.
First, reads were filtered for barcode quality score using a standard DoTA-seq script (R4-parser.ipynb). Then, sequences were converted from ‘fastq’ to ‘fasta’ format with a minimum quality of Q20 using Seqtk (https://github.com/lh3/seqtk). Next, potential chimeras were filtered using USEARCH v.11.0.667_i86linux32 using ‘silva_132_97_16S.fna’ from the ‘SlLVA_132_QllME_release’39 as the reference using ‘sensitive’ mode. Then operational taxanomic units (OTUs) for all chimera-filtered sequences were generated using MMseqs2 v13.45111 (ref. 40) with the settings of ‘–min-seq-id 0.97 -c 0.95–cov-mode 1’. Then, the OTU representative sequences were searched against ‘silva_132_99_16S.fna’ from the ‘SILVA_132_QIIME_release’39 using BLASTN with the settings of ‘-evalue 0.001 -perc_identity 90 -max_target_seqs 1’. The final taxonomy hits were filtered by the criterion of sequence identity ≥95% and query coverage ≥90%. Target genes (non-taxonomic markers) were identified using a database containing those genes and aligned using Bowtie2 as described in the standard DoTA-seq analysis workflow. The results of taxonomic classification of 16S OTU BLAST and target gene alignment with Bowtie2 are combined into one dataset and analyzed in a Jupyter notebook (Unknown-sample-analysis.ipynb). Supplementary Fig. 1 provides a flowchart depicting this pipeline.
Generating DoTA-seq target primers
For defined microbial communities, target genes were first identified from the genome sequences of the constituent strains. In particular, 100 candidate primers were generated from each gene using Primer3 (ref. 41) (http://primer3.org). The thermodynamic ΔG of primer self and heterodimerization of each candidate primer for all target primer genes were calculated using ntthal, which is a submodule of Primer3. A simulated annealing algorithm was then used to select primers for each gene that minimizes the potential of primer dimers. All steps were automated in Jupyter notebooks, which are available at GitHub (Code Availability). For the fecal-derived microbial community, the raw reads obtained from metagenomic shotgun sequencing (see above) were first assembled using Megahit v.1.2.9 (ref. 42) using default options. The resulting contigs are searched using BLAST against the replicon database from Mob-suite43 and the nucleotide database from CARD37 for human and mouse samples, respectively. We filtered the results to matches with more than 500 bp in length and the top 10% identity. For the plasmid replicon primer set, the replicon Inc18 generated large primer dimers in initial PCR tests. As this primer set was not crucial for demonstration purposes, we simply removed this gene target from the list as opposed to testing different Inc18 primers.
Single-cell digital PCR
Cells derived from glycerol stocks of the synthetic human gut community exposed to antibiotics were thawed on ice, then 100 μl cells were washed with 1 ml PBS + 0.1% Tween-20, then resuspended in 30 μl PBS. Then, 70 μl 100% ethanol (Koptec) was added to fix the cells for 10 min at room temperature. After fixation, the cells were washed and then resuspended in 50 μl pre-injection buffer. A PCR mix was prepared using 10 μl BioRad digital PCR mix for probes, 1 μl 20× Primetime probe assay (IDT) for species-specific rpoB gene, 1 μl 20× Primetime probe assay for the ARGs cepA for BF and tetQ and mef(en2) for PJ, 4 μl pre-injection buffer and 4 μl washed cells in pre-injection buffer. Then, 30 μl BioRad droplet-generation oil was added and the solution was vortexed at the highest setting for 30 s with a BioRad BR-2000 vortexer. The resulting emulsion was thermocycled for PCR as follows: 95 °C 10 min and 40 cycles of 94 °C 30 s and 60 °C 1 min with a ramp rate of 2 °C s−1. Then, 10 μl resulting emulsion was loaded into a countess cell-counting chamber (Invitrogen) and imaged with a Nikon Eclipse Ti epifluorescence microscope using 4X objective with a X-cite120 LED light source with 470/525 nm filter and 560/630 nm filters for SYBR Green (FAM for rpoB) and CF555 (HEX for ARGs) channels, respectively. For each sample, at least four images were counted in ImageJ to obtain the ratio of FAM/HEX-positive droplets (probes used for single-cell digital PCR), using a custom macro script (Code Availability) when more than 30 positive (HEX or FAM positive) droplets were present and by manual inspection when fewer than 50 positive droplets were present. Primers for single-cell digital PCR are found in Supplementary Table 13.
Colony PCR for B. fragilis and cepA
Cells derived from glycerol stocks from the synthetic human gut community antibiotic experiment were streaked onto Bacteroides minimal medium agar plates and incubated for 48 h in the anaerobic chamber for outgrowth of colonies. Individual colonies were picked using a pipette tip and inoculated into YBHI growth medium for growth overnight (~16 h) in the anaerobic chamber. Individual cultures were then spun down and resuspended in 1 ml TE buffer. A PCR mix consisting of 5 μl ssoAdvanced Probes mix (BioRad), 0.5 μl each of the 20× Primetime assay for B. fragilis rpoB and for cepA, 4 μl H2O and 1 μl cell suspension. The mix was thermocycled in a BioRad CFX-connect real-time PCR system as follows: 95 °C 3 min, 40 cycles of 95 °C 15 s and 60 °C 45 s with fluorescence detection at 60 °C. The samples with amplification detected in the HEX and FAM channels were determined to have originated from BF colonies and contained the cepA gene. B. fragilis was chosen for this experiment because a selective growth medium (Bacteroides minimal medium; Supplementary Note 5) was available to isolate colonies from the community that were likely to be B. fragilis.
Fluorescence microscopy of droplets and gels and counting of particles and droplets
Droplets (in oil) or gels (in aqueous buffer) were stained with SYBR Green by mixing with equal volumes of SYBR Green-saturated BioRad Oil or dissolving SYBR Green to 10× concentration, respectively. Next, 10 μl SYBR Green-stained droplets or gels were pipetted into a disposable cell-counting chamber slide (C10228) and imaged on a Nikon Eclipse Ti epifluorescence microscope using a 4X objective with a X-cite120 LED light source with 470/525 nm filter and 560/630 nm filters for SYBR Green and CF555 channels, respectively. The resulting raw images were analyzed using ImageJ (FIJI v.1.53f51) using the Find Maxima function on the fluorescence and brightfield images to automatically count the numbers of fluorescent particles and droplets, respectively. In empty droplets or gels, the Find Maxima algorithm failed to identify fluorescent particles, as expected.
Identifying plasmid replicon sequences in MAGs
The website https://opendata.lifebit.ai/table/SGB contains 154,723 microbial genomes assembled from 9,428 samples of the human microbiome from diverse geographic locations, body sites, diseases and lifestyles44. We downloaded all MAGs from the database, performed BLASTN search for all target replicon sequences with settings ‘-evalue 0.001 -perc_identity 0.9 -max_target_seqs 1’ and filtered BLASTN results with an identity cutoff of 90% and replicon coverage cutoff of 70%. Within the filtered results, we used GTDB-Tk45 v.1.8 to extract taxonomic information for all selected MAGs that contained hits to the replicon sequences.
Extended Data
Extended Data Fig. 1 ∣. Tracking cell lysis efficiencies using Cellbrite-Fix 555 and SYBR Green staining in >1000 droplets or gels in each step of the lysis protocol.

Cellbrite (CF555) stains cell membrane and cell wall components while SYBR Green stains DNA, allowing us to identify whether a cell is intact (CF555 and SYBR staining) or lysed (only SYBR staining). (a) Representative fluorescence microscopy overlay of Green Channel (SYBR staining), Red Channel (CF555 staining), and Brightfield (Droplets/Gels) as a water-in-oil emulsion, as gels before lysis (top two images) and as gels after lysis (bottom image). Arrows denote representative droplets containing cells. Scale bars represent 100 μm. (b) The percentage of droplets containing fluorescent particles (# of fluorescent particles divided by the total # of droplets or gels) by SYBR or CF555 staining for each condition. Before lysis, CF555 and SYBR fluorescent particles are approximately equal in abundance. Following lysis, CF555 particles are substantially reduced, indicating lysis of the cells. n represents total number of droplets analyzed for each condition. The discrepancy between % encapsulated cells in pre-lysis emulsion and pre-lysis gel is due to the approximate nature of the droplet counting algorithm. Since the droplet counting algorithms are imperfect, some droplets do not get counted and some get counted multiple times. Therefore, the % of stained cells are only approximate estimates. (c) The percentage of droplets containing unlysed cells at each step. Unlysed cells are defined as particles that are fluorescent in both CF555 and SYBR Green channels. Data shown are result of one independent experiment.
Extended Data Fig. 2 ∣. Coefficient of variation for technical replicates is negatively correlated with number of cells sequenced.

Scatter plot of the average number of cells for that species versus the coefficient of variation (CV) of gene prevalence for two technical replicates for every gene-species pair. CV is calculated as the standard deviation divided by the mean. The negative Spearman correlation (Rho) suggests that a moderate fraction of the variance found between technical replicates can be attributed to stochasticity due to small numbers of cells. P-value is based off comparison to a two-sided t-test null distribution.
Extended Data Fig. 3 ∣. Qualitative check of target gene amplification balance.

Target amplification balance for genes in synthetic community is plotted as a histogram on a log y-scale as a qualitative check of relative amplification efficiency. The X axis represents the fraction of the reads of each barcode group that represents the 16S rRNA gene (ie. barcodes in the middle of the x axis have half their reads map to 16S gene and half map to the target gene, barcodes on the far right contain mostly reads that map to the target gene and not the 16S gene). The distribution of ratios for low prevalence genes (ErmQ, tetW, highlighted in red) are biased towards target genes meaning that the target capture efficiency is high for those targets despite the observed low prevalence. Only cells that contain both 16S reads and at least one additional target gene are included.
Extended Data Fig. 4 ∣. BL rpoB gene is detected via PCR on genomic DNA extract.

Agarose gel showing PCR amplification of the Bifidobacterium specific rpoB gene on the genomic DNA extracted from pure cultures of B. adolescentis (BA), B. psuedocatenulatum (BP), B. longum (BL), or the 25-member synthetic human gut community (Comm). NTC represents no template control. MW molecular weight marker. Gel represents data from one independent experiment.
Extended Data Fig. 5 ∣. Independent confirmation of DoTA-seq results in Fig. 2 by colony and single-cell PCR genotyping.

Droplet PCR genotyping for (a) B. fragilis (BF) and the cepA gene, (b) P.johnsonii (PJ) and the mef(en2) gene, or (c) PJ and the tetQ gene. Cells were fixed and subjected to digital PCR amplification for species marker genes and ARGs in an emulsion PCR (see Methods). Blue bars show the proportion of droplets that showed amplification for the species marker gene as well as the ARG, green bars show the proportion of droplets that show amplification for the species marker gene (rpoB) but not the ARG. Time 0 and time 2 represents timepoints 0 and 2 from the synthetic community antibiotic experiment, respectively. n represents the number of species marker gene positive droplets that were counted in total for each condition. (d) Colony PCR genotyping of B. fragilis (BF) colonies of cells originating from the synthetic human gut community experiment exposed to antibiotics. Colonies from glycerol stocks of samples taken at timepoints 1 and 2 of the synthetic community antibiotics experiment shown in Fig. 3 are genotyped by qPCR for the BF specific rpoB gene and the antibiotic resistance gene cepA. The proportion of cepA positive colonies are shown in red, and proportion of cepA negative colonies are shown in blue. n represents the number of colonies that were positively identified as BF by successful amplification of the BF specific rpoB gene. In total, 24 colonies from timepoint 1 and 32 colonies from timepoint 2 were genotyped (many colonies were not BF). Data are result of one independent experiment.
Extended Data Fig. 6 ∣. Cell suspensions extracted from fecal material highly resembles source material.

Relative abundance of taxa (family level) as profiled by metagenomic sequencing for whole mouse fecal material (Y axis) and cells extracted from mouse fecal material using the gentle centrifugation method (X axis).
Extended Data Fig. 7 ∣. Single-cell sequencing from ZymoBIOMICS preserved human fecal microbiome results in under-representation of gram negative taxa.

Comparison of relative abundance of the major taxa (>0.1% abundance by 16S profiling) between standard 16S profiling and DoTA-seq of the ZymoBIOMICS human fecal microbiome standard. Since this sample was preserved in DNA/RNA shield which prematurely lyses many types of cells (see Supplementary Fig. 9), many taxa are missing or under-represented in DoTA-seq compared to 16S profiling.
Extended Data Fig. 8 ∣. ZymoBIOMICS samples contain premature lysis and under-representation of gram-negative bacteria in single-cell sequencing.

This chart shows DoTA-seq relative abundance of a Zymobiomics microbial community standard, which is a commercial mock microbial community mix preserved in DNA/RNA shield. Comparing the relative abundances with and without washing the cells prior to droplet making, we found that Gram-negative cells (in red) are almost undetected after 2X wash. This suggests that gram-negative cells were already lysed in the buffer and washing removed the cell debris and free-floating genomic DNA, leaving only the gram-positive cells intact for single-cell sequencing.
Extended Data Fig. 9 ∣. DoTA-seq elucidates diverse genetic subpopulations in B. fragilis generated via promoter inversion.

(a) Schematic of the potential diversity generated by B. fragilis CPS operons. There are a total of 8 CPS operons referred to as A-H, 7 of which contain promoters flanked by inverted repeats (triangles). These promoters switch ON and OFF through recombination at the inverted repeats driven by an endogenous recombinase (mpi). (b) Schematic of DoTA-seq reaction with primers designed to flank all 7 invertible promoters. Primers are represented by half-headed arrows. Gray vertical lines represent region of complementarity between amplicons and barcodes. P5 and P7 represent the Illumina sequencing adaptor sequences. (c) Bar plot of the relative frequencies of unique CPS promoter states in a single B. fragilis colony. Promoter states are represented by a 7-letter code, where the letters (A-H) denote that a given promoter is turned ON, and ‘-‘ denote the given promoter is turned OFF. Data points represent technical replicates. Error bars represent 1 s.d. from the mean of technical replicates (n = 3). Bar height represents the mean. Since a subset of combinatorial promoter states were rare in the population and not observed in all technical replicates, we computed the stochastic limit of detection (Supplementary Note 4). (d) An undirected graph network representation of the CPS promoter state subpopulations in (c). Nodes represent CPS promoter combinatorial states where diameter is proportional to relative frequency, and edges connect nodes that are one promoter flip away from each other. (e) Network representation of the measured combinatorial promoter states where the recombinase (mpi) responsible for promoter inversions was deleted. In this strain, the entire population is locked in a single state (A–E-H). The diameter of the node and edges are the same as (d).
Extended Data Table 1 ∣.
Gene prevalence for mock community in Fig. 1f
| Replicate 1 | Gene (Prevaence in species) | |||||||
|---|---|---|---|---|---|---|---|---|
| Species |
B. fragilis-
rpoB |
B. subtilis-
rpoB |
C. hiranonis-
rpoB |
E. coli-
rpoB |
L. lactis-
dnaG |
P. putida-
dnaG |
RFP |
S. epidermidis-
dnaG |
| B. fragilis | 77% | 0% | 1% | 0% | 0% | 0% | 0% | 0% |
| B. subtilis | 0% | 85% | 0% | 0% | 0% | 0% | 0% | 0% |
| C. hiranonis | 0% | 0% | 73% | 0% | 0% | 0% | 0% | 0% |
| E. coli | 0% | 0% | 0% | 90% | 0% | 0% | 1% | 0% |
| L. lactis | 0% | 0% | 0% | 0% | 77% | 0% | 0% | 0% |
| P. putida | 0% | 0% | 0% | 0% | 0% | 100% | 0% | 0% |
| S. epidermidis | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 82% |
| Replicate 2 | Gene (Prevaence in species) | |||||||
| Species |
B. fragilis-
rpoB |
B. subtilis-
rpoB |
C. hiranonis-
rpoB |
E. coli-
rpoB |
L. lactis-
dnaG |
P. putida-
dnaG |
RFP |
S. epidermidis-
dnaG |
| B. fragilis | 79% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| B. subtilis | 0% | 88% | 0% | 0% | 0% | 0% | 0% | 0% |
| C. hiranonis | 0% | 0% | 74% | 0% | 0% | 0% | 0% | 0% |
| E. coli | 0% | 0% | 0% | 91% | 0% | 0% | 1% | 0% |
| L. lactis | 0% | 0% | 0% | 0% | 76% | 0% | 0% | 1% |
| P. putida | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 0% |
| S. epidermidis | 0% | 0% | 0% | 0% | 0% | 0% | 0% | 80% |
Each row represents a different species. Each column represents a species-specific gene. Gene prevalences were computed by dividing the number of cells of a given species harboring a given gene divided by the total number of cells of that species.
Supplementary Material
{kind=link}
Acknowledgements
We thank Y.-Y. Cheng of UW Madison for providing the E. coli and B. subtilis strains and helpful discussions. We thank R. Clark for assistance with the synthetic human gut community. We are also grateful to L. Comstock, University of Chicago for providing the B. fragilis recombinase deletion strain and B. Pfleger of UW Madison for providing the P. putida KT2440 strain. We thank L. Brinkman of the UW Madison animal resources and compliance for providing mouse fecal samples. This research was supported by the National Institutes of Allergy and Infectious Diseases under grant no. R21 AI156438 and R21 AI159980 for O.S.V., National Institute of General Medical Sciences under grant no. R01 GM038660 for R.L. and R35 GM124774 for O.S.V., Army Research Office under grant no. W911NF-19-1-0269, U.S. Department of Agriculture Hatch Award WIS05004 for R.L. and the Burroughs Wellcome Fund through the Careers Award at the Scientific Interfaces for F.L. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41592-023-02157-7.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Competing interests
F.L. and O.S.V. have filed a related US non-provisional patent application entitled ‘Methods for isolating and barcoding nucleic acid’ (Application 18/311,010). The remaining authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41592-023-02157-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41592-023-02157-7.
Data availability
The database of ARGs (CARD database) used to design DoTA-seq primers can be accessed at card.mcmaster.ca. The database of plasmid replicon genes used to design DoTA-seq primers can be accessed as part of MOB-suite at https://github.com/phac-nml/mob-suite. The MAGs used to corroborate plasmid–taxa relationships can be accessed at https://opendata.lifebit.ai/table/SGB. The raw reads and 16S profiling abundance data of the ZymoBIOMICS human fecal community were taken from https://www.fecalreferencedb.com/.
For all DoTA-seq runs, processed data containing barcodes and their mapped associated reads are available from Zenodo at https://doi.org/10.5281/zenodo.6537689.
Raw sequencing reads are available from Zenodo at https://doi.org/10.5281/zenodo.10380035. Source data are provided with this paper.
Code availability
All code used in the analysis of DoTA-seq sequencing data are available on Zenodo at https://doi.org/10.5281/zenodo.10380035. Up-to-date versions of the scripts and code may be found on GitHub at https://github.com/lanfreem/DoTA-seq-Paper.
References
- 1.Jayaraman R. Phase variation and adaptation in bacteria: a ‘Red Queen’s Race’. Curr. Sci 100, 1163–1171 (2011). [Google Scholar]
- 2.Sulaiman JE & Lam H Proteomic investigation of tolerant Escherichia coli populations from cyclic antibiotic treatment. J. Proteome Res 19, 900–913 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Porter NT, Canales P, Peterson DA & Martens EC A subset of polysaccharide capsules in the human symbiont Bacteroides thetaiotaomicron promote increased competitive fitness in the mouse gut. Cell Host Microbe 22, 494–506 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jonsson A-B, Ilver D, Falk P, Pepose J & Normark S Sequence changes in the pilus subunit lead to tropism variation of Neisseria gonorrhoeae to human tissue. Mol. Microbiol 13, 403–416 (1994). [DOI] [PubMed] [Google Scholar]
- 5.Li J. et al. Epigenetic switch driven by DNA inversions dictates phase variation in Streptococcus pneumoniae. PLoS Pathog. 12, e1005762 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcy Y. et al. Dissecting biological ‘dark matter’ with single-cell genetic analysis of rare and uncultivated {TM7} microbes from the human mouth. Proc. Natl Acad. Sci. USA 104, 11889–11894 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rinke C. et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Lan F. et al. Single-cell analysis of multiple invertible promoters reveals differential inversion rates as a strong determinant of bacterial population heterogeneity. Sci. Adv 9, eadg5476 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blattman SB, Jiang W, Oikonomou P & Tavazoie S Prokaryotic single-cell RNA sequencing by in situ combinatorial indexing. Nat. Microbiol 5, 1192–1201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuchina A. et al. Microbial single-cell RNA sequencing by split-pool barcoding. Science 371, eaba5257 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lan F, Demaree B, Ahmed N & Abate AR Single-cell genome sequencing at ultra-high-throughput with microfluidic droplet barcoding. Nat. Biotechnol 35, 640–646 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng W. et al. High-throughput, single-microbe genomics with strain resolution, applied to a human gut microbiome. Science 376, eabm1483 (2022). [DOI] [PubMed] [Google Scholar]
- 13.Hatori MN, Kim SC & Abate AR Particle-templated emulsification for microfluidics-free digital biology. Anal. Chem 90, 9813–9820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clark IC et al. Microfluidics-free single-cell genomics with templated emulsification. Nat. Biotechnol 10.1038/s41587-023-01685-z (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macosko EZ et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein AM et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eastburn DJ, Sciambi A & Abate AR Ultrahigh-throughput mammalian single-cell reverse-transcriptase polymerase chain reaction in microfluidic drops. Anal. Chem 85, 8016–8021 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Lan F, Haliburton JR, Yuan A & Abate AR Droplet barcoding for massively parallel single-molecule deep sequencing. Nat. Commun 7, 11784 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diebold PJ, New FN, Hovan M, Satlin MJ & Brito IL Linking plasmid-based β-lactamases to their bacterial hosts using single-cell fusion PCR. eLife 10, e66834 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmes DL & Stellwagen NC Estimation of polyacrylamide gel pore size from Ferguson plots of linear DNA fragments. II. Comparison of gels with different crosslinker concentrations, added agarose and added linear polyacrylamide. Electrophoresis 12, 612–619 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Cheng Y-Y et al. Efficient plasmid transfer via natural competence in a microbial co-culture. Mol. Syst. Biol 19, e11406 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobrindt U & Hacker J Whole genome plasticity in pathogenic bacteria. Curr. Opin. Microbiol 4, 550–557 (2001). [DOI] [PubMed] [Google Scholar]
- 23.Bonham KS, Wolfe BE & Dutton RJ Extensive horizontal gene transfer in cheese-associated bacteria. eLife 6, e22144 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Murray CJ et al. Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark RL et al. Design of synthetic human gut microbiome assembly and butyrate production. Nat. Commun 12, 3254 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van der Waaij LA, Mesander G, Limburg PC & van der Waaij D Direct flow cytometry of anaerobic bacteria in human feces. Cytometry 16, 270–279 (1994). [DOI] [PubMed] [Google Scholar]
- 27.Louca S, Doebeli M & Parfrey LW Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6, 41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krinos CM et al. Extensive surface diversity of a commensal microorganism by multiple DNA inversions. Nature 414, 555–558 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Hoskisson PA & Smith MCM Hypervariation and phase variation in the bacteriophage ‘resistome’. Curr. Opin. Microbiol 10, 396–400 (2007). [DOI] [PubMed] [Google Scholar]
- 30.Wang Y. et al. Dissolvable polyacrylamide beads for high-throughput droplet DNA barcoding. Adv. Sci 1903463, 1903463 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lourenço M. et al. A mutational hotspot and strong selection contribute to the order of mutations selected for during Escherichia coli adaptation to the gut. PLoS Genet. 12, e1006420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao S. et al. Adaptive evolution within gut microbiomes of healthy people. Cell Host Microbe 25, 656–667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu RH et al. Microbial interaction network inference in microfluidic droplets. Cell Syst. 9, 229–242 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agasti SS, Liong M, Peterson VM, Lee H & Weissleder R Photocleavable DNA barcode–antibody conjugates allow sensitive and multiplexed protein analysis in single cells. J. Am. Chem. Soc 134, 18499–18502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duffy DC, McDonald JC, Schueller OJA & Whitesides GM Rapid prototyping of microfluidic systems in poly(dimethylsiloxane). Anal. Chem 70, 4974–4984 (1998). [DOI] [PubMed] [Google Scholar]
- 36.Demaree B, Weisgerber D, Lan F & Abate AR An ultrahigh-throughput microfluidic platform for single-cell genome sequencing. J. Vis. Exp 2018, 57598 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alcock BP et al. CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quast C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Steinegger M & Söding J MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol 35, 1026–1028 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Untergasser A. et al. Primer3-new capabilities and interfaces. Nucleic Acids Res. 40, e115 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li D, Liu C-M, Luo R, Sadakane K & Lam T-W MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Robertson J & Nash JHE MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genomics 4, e000206 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasolli E. et al. Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176, 649–662 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaumeil P-A, Mussig AJ, Hugenholtz P & Parks DH GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36, 1925–1927 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The database of ARGs (CARD database) used to design DoTA-seq primers can be accessed at card.mcmaster.ca. The database of plasmid replicon genes used to design DoTA-seq primers can be accessed as part of MOB-suite at https://github.com/phac-nml/mob-suite. The MAGs used to corroborate plasmid–taxa relationships can be accessed at https://opendata.lifebit.ai/table/SGB. The raw reads and 16S profiling abundance data of the ZymoBIOMICS human fecal community were taken from https://www.fecalreferencedb.com/.
For all DoTA-seq runs, processed data containing barcodes and their mapped associated reads are available from Zenodo at https://doi.org/10.5281/zenodo.6537689.
Raw sequencing reads are available from Zenodo at https://doi.org/10.5281/zenodo.10380035. Source data are provided with this paper.
All code used in the analysis of DoTA-seq sequencing data are available on Zenodo at https://doi.org/10.5281/zenodo.10380035. Up-to-date versions of the scripts and code may be found on GitHub at https://github.com/lanfreem/DoTA-seq-Paper.