

Abstract
Cell membrane coating has recently emerged as a promising biomimetic approach to engineering nanoparticles (NPs) for targeted drug delivery. However, simple cell membrane coating may not meet the need for efficient drug delivery to the brain. Here, we report a novel molecular engineering strategy to modify the surface of NPs with a cell membrane coating for enhanced brain penetration. By using poly(lactic-co-glycolic) acid (PLGA) NPs as our model, we show that delivery of NPs to the ischemic brain is enhanced through surface coating with the membrane of neural stem cells (NSCs), and the delivery efficiency can be further increased using membrane isolated from NSCs engineered for overexpression of CXCR4. We found that this enhancement is mediated by the chemotactic interaction of CXCR4 with SDF-1, which is enriched in the ischemic microenvironment. We demonstrate that the resulting CXCR4-overexpressing membrane-coated NPs, termed CMNPs, significantly augments the efficacy of glyburide, an anti-edema agent, for stroke treatment. The study suggests a new approach to improving drug delivery to the ischemic brain and establishes a novel formulation of glyburide that can be potentially translated into clinical applications to improve management of human patients with stroke.
Keywords: neural stem cells, nanoparticles, CXCR4, Stroke, glyburide
1. Introduction
Stroke, which accounts for one in every 20 deaths worldwide, represents a major public health problem [1]. Despite tremendous progress in understanding the biology of stroke, effective pharmacological management of this disease remains elusive [2]. One of the major hurdles for translating the biological findings to improving clinical management of stroke has been the inability to overcome the blood brain barrier (BBB), a biological barrier that protects the brain from toxins and infections in physiological conditions but also prevents drug penetration to the brain. In later stages of stroke, the BBB is partially compromised,[3, 4] but remains largely intact within the therapeutic window for most therapeutic treatment. Therefore, the degree of BBB disruption may not allow delivery of pharmacologically significant quantities of drugs for effective treatment [5, 6].
The BBB can be potentially overcome by emerging nanotechnology, as nanoparticles (NPs) can be engineered through various mechanisms, such as chemical conjugation of ligands that target receptors expressed in the BBB, to enhance their brain penetrability [7–10]. Nonetheless, accumulating evidence shows that the majority of existing approaches cannot enhance drug delivery to a degree that is sufficient for stroke treatment [9–11]. New approaches to enabling further enhanced BBB penetrability is in great demand.
In this study, we synthesized and characterized poly(lactic-co-glycolic) acid (PLGA) NPs surface coated with membrane isolated from neural stem cells (NSCs) for targeted drug delivery to the ischemic brain. We found that coating with NSC membrane efficiently enhanced the accumulation of NPs in the ischemic brain and that the delivery efficiency was further enhanced by using membrane isolated from NSCs engineered to overexpress CXCR4. To simplify the nomenclature, we designated the NPs coating with membrane NSCs and CXCR4-overexpressing NSCs as MNPs and CMNPs, respectively. We characterized CMNPs for stroke treatment by delivering glyburide, a promising anti-stroke agent which we recently tested in human patients [6], and found that CMNPs effectively enhanced the therapeutic benefit of glyburide. This study suggests a novel approach to engineering cell membrane coated NPs for targeted drug delivery to the brain.
2. Results
Biomimetic engineering through surface coating with cell membrane has emerged as a promising approach for targeted drug delivery [12, 13]. By hijacking the biological interaction of cells with the host, this approach may enhance the circulation of NPs in the blood and promote the interaction of NPs with specific biological targets [12]. NSCs have been explored for treatment of ischemic stroke in preclinical and early clinical studies [14, 15]. It has been shown that NSCs express various chemokine factors, which enable them to migrate to the injured area [14, 15]. Among all the chemokines, CXCR4 represents the primary factor driving the NSC tropism through interaction with its ligand SDF-1, whose expression level is elevated in the ischemic region [16, 17]. Here, we set to test whether efficient delivery of NPs to the ischemic brain can be achieved through surface coating of membrane of NSCs that are biologically engineered to overexpress CXCR4 (Figure 1).
Figure 1.

Schematic diagram of targeted delivery of NPs to the ischemic brain through surface coating of membrane of CXCR4-overexpressing NSCs. NSCs are engineered through lentiviral transduction to overexpress CXCR4. Cell membrane is extracted and coated on to the surface of PLGA NPs via extrusion. After intravenous administration, the resulting CMNPs selectively accumulate in the ischemic microenvironment through interaction with SDF-1, a ligand of CXCR4 that is preferentially accumulated. CMNPs locally release cargo therapeutics, such as glyburide, and promote stroke repair and recovery.
2.1. CXCR4 overexpression enhances the migration of NSCs to the ischemic brain
Previous studies have shown that NSCs have significant tropism to the ischemic brain [14, 15]. Therefore, we engineered mouse NSCs to express both luciferase and GFP through lentiviral transduction (Figure S1a,b), and administered them (5X104 cells) to the lateral ventricle on the contralateral hemisphere of ischemic brain. Ischemia was induced through middle cerebral artery occlusion (MCAO) surgery. After 48 hours, the mice were subjected to imaging for luciferase signal using in vivo imaging system (IVIS). As expected, we found that NSCs migrated to the ipsilateral hemisphere (Figure 2a). Next, the mice were euthanized, and the brains were isolated and sliced. Ex vivo imaging of the isolated brains and brain slices confirmed that NSCs (green, from GFP) selectively accumulated in the ischemic region (white, from triphenyltetrazolium chloride (TTC) staining) (Figure 2a).
Figure 2.

Overexpression of CXCR4 enhances the tropism of NSCs to the ischemic brain. a) Characterization of tropism of NSCs with and without CXCR4 overexpression to the ischemic brain (n=3). b) Representative images of the expression of SDF-1 in the ischemic or normal brain. c) Flow cytometry analysis of CXCR4 expression in the indicated NSCs. d) Quantification of luciferase expression in the ischemic brain after NSCs transplantation (n=6).
The tropism of NSCs to the ischemic regions is known to mediate through the interaction of chemokine factors on the surface of NSCs and their ligands enriched in the ischemic microenvironment [14, 15]. One of the major driving forces of this interactions is the SDF-1-CXCR4 axis [16–18]. Consisting with previous findings [19], we found that the level of SDF-1 in the ischemic region was significantly increased 24 hours after ischemia-reperfusion (Figure 2b). In contrast, the level of SDF-1 in the contralateral hemisphere remained at a low level. To test if increasing the expression of CXCR4 enhances the tropism of NSCs to the ischemic regions, we engineered NSCs to overexpress CXCR4 through lentiviral transduction. The overexpression of CXCR4 was confirmed by both flow cytometry and immunostaining (Figure 2c, Figure S1b). The resulting CXCR4-overexpressing NSCs, designated as NSCsCXCR4, were engineered to express both luciferase and GFP (Figure S1), and administered to MCAO mice. We found that overexpression of CXCR4 significantly increased the accumulation of NSCs (green GFP signal) in the ischemic region (white TTC staining) by 1.6-fold, based on quantification of GFP signal (Figure 2a, d).
2.2. Synthesis and characterization of membrane coated NPs
The significant tropism of NSCs to the ischemic brain can be attributed to the cell membrane, on which various chemokine factors are anchored. To take advantage of the unique surface property of NSCs, we fabricated PLGA NPs with surface coating of NSC membrane. PLGA NPs were synthesized via the standard emulsion using ethyl acetate (EA) as the solvent. NSC membrane was prepared by gradient centrifugation after freeze-thaw and sonication of the cells, and coated onto the surface of NPs through mechanical extrusion using a 200 nm filter [12]. We found that both MNPs and CMNPs, which were surface coated with membrane of NSCs and membrane of NSCsCXCR4, respectively, were spherical in shape, and, unlike uncoated PLGA NPs, had visible core-shell structures (inserts in Figure 3a–c). Quantification of hydrodynamic diameter by dynamic light scattering (DLS) found that MNPs and CMNPs, which were comparable in size, were slightly larger than uncoated PLGA NPs (Figure 3d), and unlike the uncoated PLGA NPs, both MNPs and CMNPs were stable in size after incubation in serum-containing medium (Figure 3e). Compared to PLGA NPs, cell membrane-coated had a lower Zeta potential (Figure 3f), which is consistent with previous reports [20]. The decrease of Zeta potential likely resulted from the cell membrane coating, which has lower surface charge than uncoated PLGA NPs. Successful coating of cell membrane was further confirmed by Western Blot (Figure 3g) and flow cytometry analysis (Figure 3h). Immunostaining demonstrated that CXCR4 was presented on the surface of membrane-coated NPs and that the amount of CXCR4 on CMNPs is greater than that on MNPs (Figure 3i).
Figure 3.
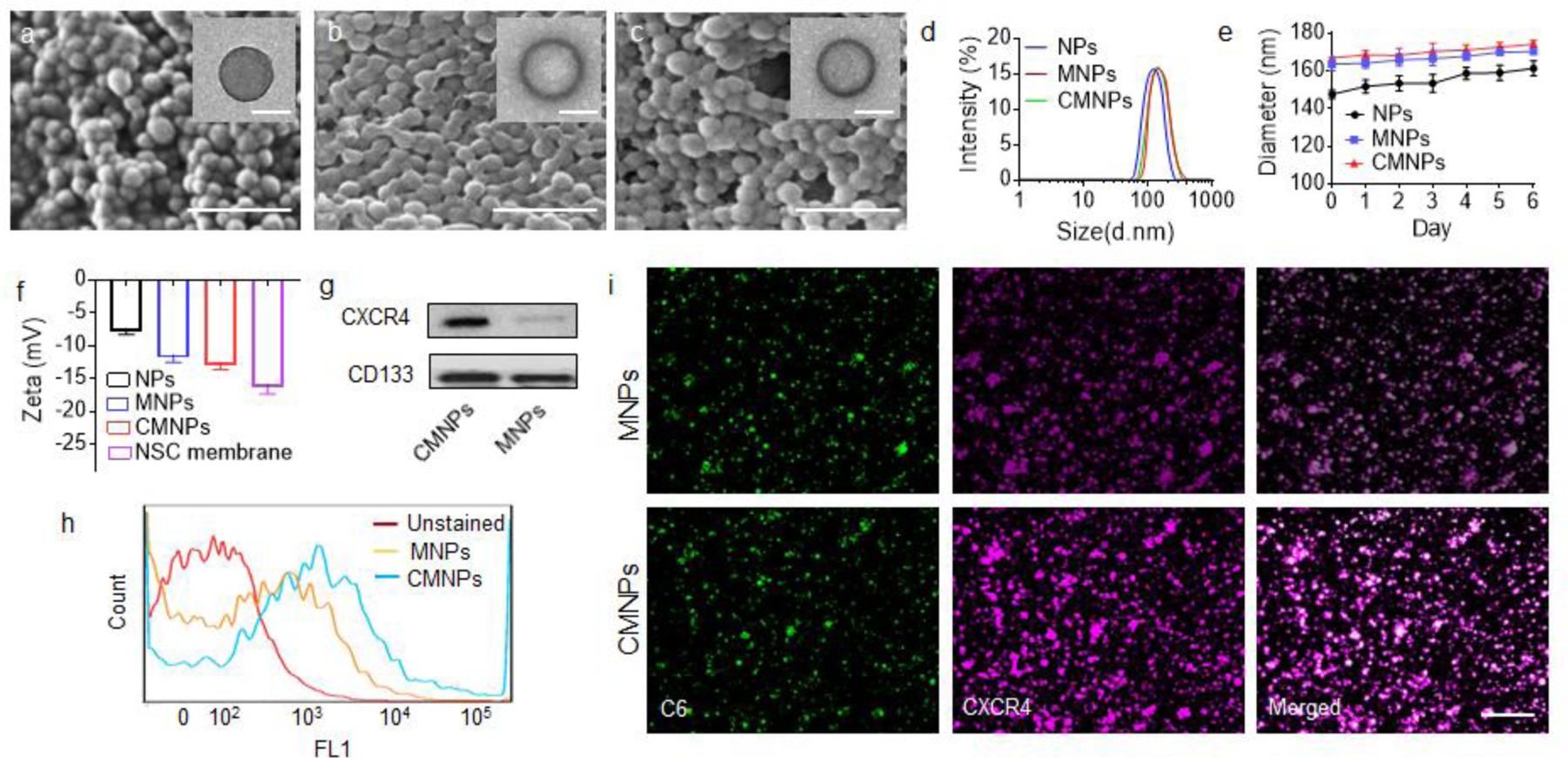
NP synthesis and characterization. a-c) Representative images of PLGA NPs (a), MNPs (b), and CMNPs (c), as captured by SEM (scare bar: 500 nm) and TEM (insert, scare bar: 100 nm). d,e) DLS analysis of the hydrodynamic diameters (d) and the change of the hydrodynamic diameter with time in serum-containing medium of the indicated NPs. f) Zeta potential of cell membrane and the indicated NPs (n=3). g) Western Blot analysis of the expression of CXCR4 and CD133 on the membrane isolated from the indicated NPs. h,i) Flow cytometry analysis (f) and confocal imaging analysis (i) of the presence of CXCR4 on the surface of the indicated NPs. For confocal imaging analysis, the NPs were synthesized with encapsulation of coumarin 6 and stained with CXCR4 antidoby, following with Alexa Fluor 546- conjugated secondary anbitody. Scale bar: 1 µm.
2.3. CXCR4- mediated chemotactic recruitment of CMNPs to the ischemic brain
To evaluate if NSC membrane coating enhanced targeted delivery of NPs to the ischemic brain, membrane coated NPs, including MNPs and CMNPs, and control PLGA NPs, were synthesized with encapsulation of IR780, an infrared fluorescent dye. The resulting NPs were administered to mice after MCAO surgery. After 24 hours, the mice were imaged using IVIS and then euthanized. The brains were harvested, sliced, and subjected to imaging. Results of live imaging and ex vivo imaging of the brains and brain slices suggested that coating with NSC membrane significantly enhanced the delivery of NPs to the ischemic brain after intravenous administration, and overexpression of CXCR4 on the membrane further enhanced the brain targeting efficiency (Figure 4a). Both MNPs and CMNPs, as located by IR780 fluorescence, accumulated in the ischemic regions, as determined by TTC staining, with great specificity. Quantification of IR780 fluorescence revealed that CMNPs accumulated in the ischemic region in efficiency 2.7 and 2.1 times greater than the naked PLGA NPs and MNPs, respectively (Figure 4b).
Figure 4.
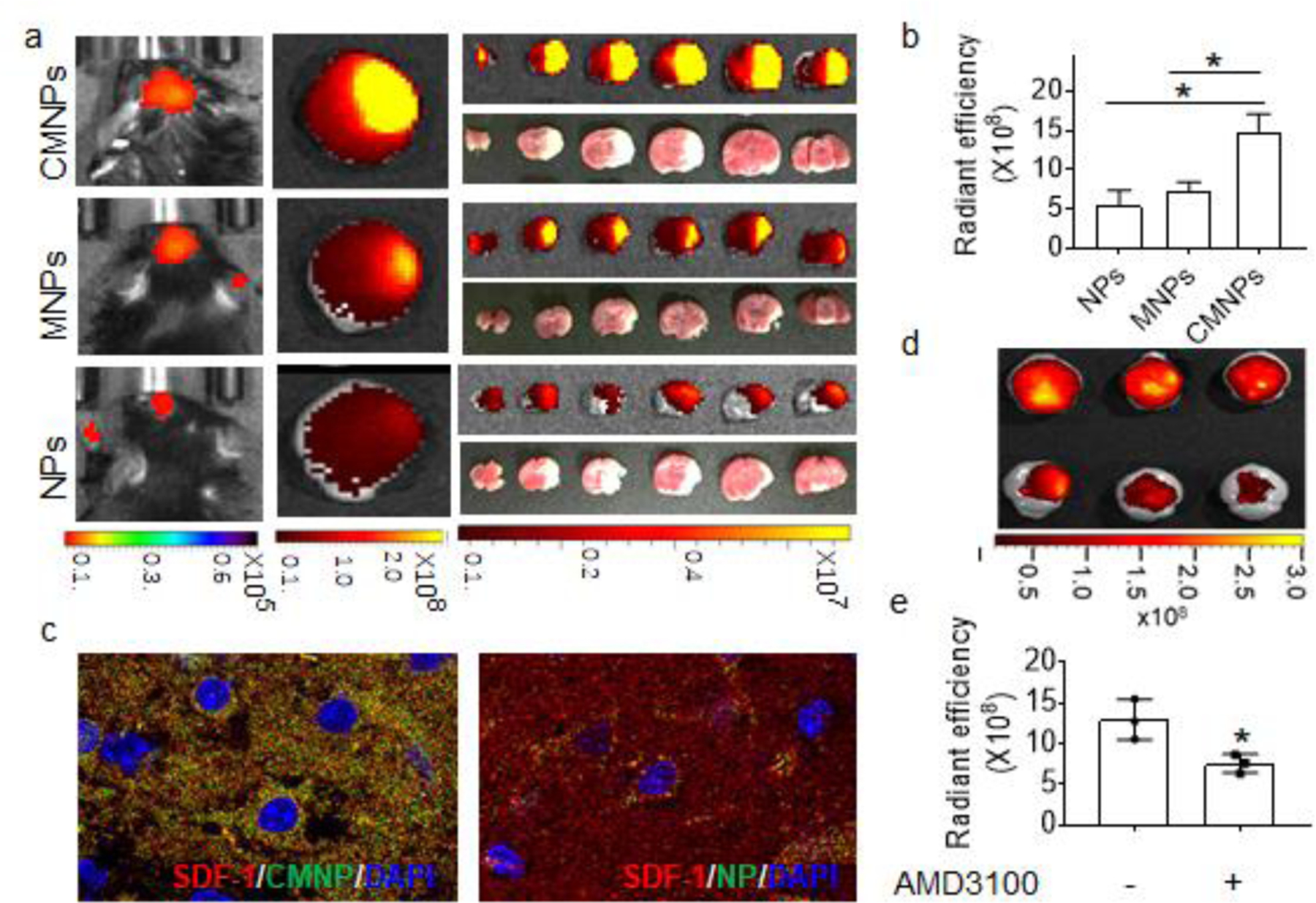
CXCR4- mediated chemotactic recruitment of NPs to the ischemic brain. a) IVIS image of IR-780 loaded NPs in ischemic brain (n=3). b) Semi-quantification of the indicated NPs in the ischemic brain based on IR-780 fluorescence (n=3). c) Representative images of SDF-1 and the indicated NPs distributed in the ischemic area. Red: SDF-1. Green: NPs. Blue: DAPI. d,e) Representative images (d) and semi-quantification (e) of CMNPs in the brain isolated from MCAO mice received intravenous administration of IR780-loaded CMNPs with and without pre-treatment of AMD3100 (n=3).
To determine if the observed significant efficiency of CMNPs was mediated by the interaction of CXCR4 with SDF-1, we examined the distribution of CMNPs and SDF-1 in the ischemic region. CMNPs and control PLGA NPs were synthesized with encapsulation of Coumarin 6 (C6) and administered to mice after MCAO. After 24 hours, the mice were euthanized. The brains were sliced and subjected to immunostaining for SDF-1. Confocal microscopic analysis found that the amount of CMNPs accumulated in the penumbra region was significantly greater than that of uncoated NPs, and that the distribution of CMNPs, as indicated by the green fluorescence signal from C6, overlapped with that SDF-1, as detected by red immunofluorescence (Figure 4c). Next, we assessed if blocking CXCR4 reduced CMNP brain penetration. Stroke-bearing mice were established and received intravenous administration of IR780- loaded CMNPs with or without pre-treatment with AMD3100, a well-characterized antagonist of CXCR4 [21]. After 24 hours, the accumulation of CMNPs in the brain was imaged and quantified based on the fluorescence of IR780. We found that pre-treatment with AMD3100 reduced the accumulation of CMNPs in the ischemic brain by 31% (Figure 4d,e). Taken together, these results suggest that penetration of CMNPs to the ischemic brain is partially mediated through CXCR4- based chemotactic recruitment.
2.4. CMNPs for targeted delivery of glyburide for stroke treatment
We assessed CMNPs for targeted delivery of therapeutic for stroke treatment. Glyburide, a promising anti-stroke agent that we recently tested in human stroke patients in a phase II clinical trial [6], was encapsulated and evaluated in MCAO mice. Glyburide is a potent drug for stroke treatment and is typically given at a dose of 5 µg/kg in mouse studies [22]. Therefore, we encapsulated glyburide with a low drug loading at 5%. Encapsulation of glyburide did not alter the morphology of CMNPs (Figure 5a), which released 22.5 % of glyburide within 48 h (Figure 5b). To characterize the therapeutic benefit, glyburide-loaded CMNPs, designated as Gly-CMNPs, were intravenously administered to MCAO mice at a dose equivalent of 5 µg/kg of glyburide 0, 24, and 48 h after surgery. Mice were monitored for survival and behavior, and were euthanized at day 7. We found that treatment with glyburide-loaded CMNPs significantly improved the survival (Figure 5c, Logrank test p = 0.002), reduced infarct volumes by 58% (Figure 5d, Figure S2), and improved neurological scores (Figure 5e). In contrast, both free glyburide and glyburide-loaded MNPs, or Gly-MNPs, administrated at the same dose failed to provide comparable efficacy, which is consistent with our previous findings [22]. Further analyses by H&E staining of major organs, serum aspartate aminotransferase (AST) assay, and alanine aminotransferase (ALT) assay showed that both CMNPs and Gly-CMNPs did not induce tissue damage nor liver toxicity, suggesting that they are biocompatible for intravenous use (Figure S3).
Figure 5.
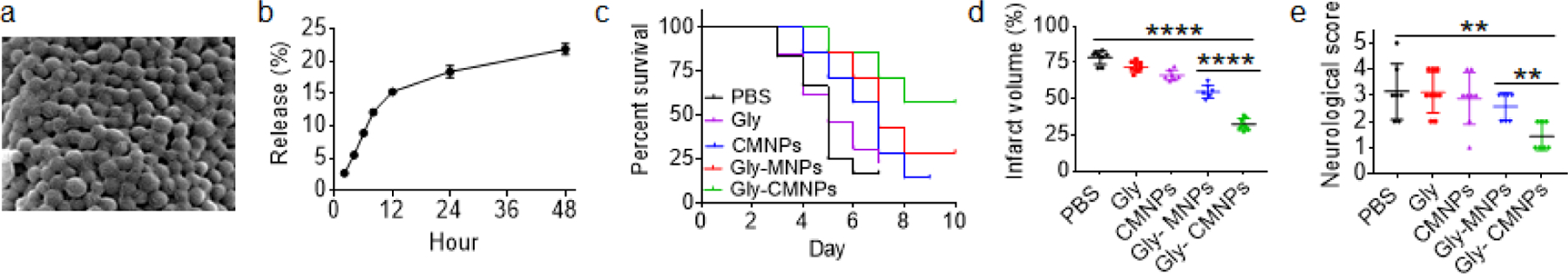
Characterization of glyburide-loaded CMNPs for stroke treatment. a) A representative SEM image of glyburide-loaded CMNP. b) Accumulated release of glyburide from glyburide-loaded CMNPs with time (n=3). c-e) Kaplan-Meier survival analysis (c, n=7–10), infarct volume (d, day 3 after surgery, n=5), and neurological scores (e, day 3 after surgery, n=5) of MCAO mice receiving the indicated treatments.
3. Discussion
Through biomimetic functionalization via surface coating with cell membrane, NPs could be have a wide range of functions, such as biocompatibility for prolonged blood circulation[23], chemotactic ability for disease targeting [24], and immunogenicity for immune stimulation [25]. Further enhancement targeted delivery can be achieved through traditional ligand conjugation approaches using lipid linkers [26, 27]. However, this is not ideal for display of large transmembrane protein receptors or their ligands, such as CXCR4 protein, as there is inadequate space to allow for optimal interactions with their binding partners. To overcome this limitation, we designed cell membrane coated NPs for targeted drug delivery by engineering cell membrane to overexpress CXCR4. We found that this approach significantly enhanced the stroke-targeting delivery of NPs to the ischemic microenvironment, and the enhancement was resulted from the chemotactic interaction between CXCR4 and SDF-1 enriched in the ischemic region (Figure 4).
We assessed the NSC membrane-coated NPs for targeted delivery of glyburide for stroke treatment. Glyburide is a potent inhibitor of ATP-sensitive potassium channels (KATP) subunit sulfonylurea receptor 1 (SUR1) that can effectively reduce stroke-induced cerebella [28]. In the recently completed phase II GAMES-RP clinical trial, we found that systemic delivery of glyburide improved the survival of patients with stroke, but failed to significantly provide favorable clinical outcome [6]. Glyburide is known to be a substrate for several transports and cannot penetrate the BBB in a therapeutic quantity [29, 30]. The observed unmet efficacy could be attributed to its limited ability to penetrate the brain. We demonstrated that glyburide delivered via CMNPs effectively enhanced mouse survival, reduced infarct volumes and improved neurological scores (Figure 5); while, in the same condition, free glyburide showed limited therapeutic benefits.
CMNPs have the potential to be utilized as a platform for drug delivery to brain injuries other than stroke since NSCs are capable of migrating to the injured brain through chemotactic interaction [31, 32]. As an example, we characterized the NPs developed in mice bearing traumatic brain injury (TBI), for which glyburide is known to be effective [33]. We found that, consistent with the findings in the stroke model, SDF-1 is highly expression in the injuried brain (Figure S4a), coating with NSC membrane increased the delivery of NPs to the injured region and overexpression of CXCR4 further enhanced the delivery efficiency (Figure S4b–d), and compared to free glyburide, glyburide-loaded CMNPs demonstrated significantly greater effects in reducing lesion volume (Figure S4f,g).
4. Conclusion
In summary, we demonstrated that the biomimetic approach through surface coating with NSC membrane enhanced the delivery of NPs to the injured brain. We developed a novel approach to modify the surface of NPs with cell membrane coating and showed that this approach further enhances brain targeting delivery. We showed that CMNPs significantly enhanced the efficacy of glyburide for brain injury treatment. Due to their excellent therapeutic benefits, glyburide-loaded CMNPs have the potential to be translated into clinical applications for management of human patients with brain injuries.
5. Experimental Section
Materials:
AMD3100 tetrahydrochloride was purchased from Santa Cruz Biotechnology. All other chemicals were purchased from Sigma-Aldrich unless otherwise noted.
Cell culture and engineering:
NSCs isolated from C57BL/6 mouse were obtained from Cyagen (Santa Clara, USA), and cultured in in DMEM-F12 medium (ThermoFisher) supplemented with B-27 (ThermoFisher), 20ng/ml EGF(Peprotech), 20ng/ml bFGF (Peprotech), and 1% penicillin-streptomycin (Sigma) at 37°C and 5% CO2., CXCR4 cDNA, which was purchased from GE Dharmacon (Catalog # MMM1013-202762695), and DNA for expression of luciferase/GFP, which was amplified from pEGFPLuc (Clontech), were subcloned into pCDH-CMV-MCS-T2A-Puro lentiviral vector (System Biosciences). Production and transduction of artificial lentivirus were performed according to our previous report [34]. NSCs with stable gene overexpression were obtained through antibiotic selection using puromycin at 1ug/ml.
Cell membrane preparation:
Cell membrane was prepared by a hypotonic lysing and freeze-thaw process with minor modification [23, 25]. Briefly, 1×107 NSCs were collected and resuspended in 1ml hypotonic lysing buffer (pH=7.0) containing 10 mM Tris, 10 mM MgCl2 and 1x EDTA-free protease inhibitor, and incubated on ice for 3 hours. Afterwards, the homogenate was subjected to freeze-thaw followed by centrifugation at 7000 g for 10 min at 4 °C. The supernatant was centrifugated at 13000g for 60 min. The resulting precipitate was resuspended in 1ml DNase free/RNase free water (Invitrogen) and stored at −80°C until use.
Synthesis of PLGA nanoparticles, membrane coated nanoparticles:
PLGA NPs were synthesized according to our previously reported procedures [35, 36]. Briefly, 50 mg PLGA polymer (0.67 dL g−1,50:50 ratio; Absorbable Polymers) and cargo agent IR780, C6, or glyburide was dissolved in 1 ml ethyl acetate (EA). Afterward, the solution was added dropwise into a tube with 2 ml 2.5% Polyvinyl alcohol (PVA, MW 30,000–70,000) under vortex and ultrasonicated to form an oil/water solution. The solution was poured into a beaker with 35 ml 0.3% PVA and stirred at 520 rpm for overnight at 4 °C. NPs were collected by centrifugation at 18,000 rpm for 20 min, washed with water twice. To form cell membrane coated PLGA nanoparticles, 0.5 ml PLGA NPs and 0.5 ml cell membrane suspension were mixed in a 1 ml syringe (Hamilton) and extruded through a 200 nm membrane filter (Whatman) in a mechanical extruder for 7 times.
Scanning electron microscopy (SEM):
The morphology of NPs was determined by SEM. Briefly, lyophilized NPs were mounted on a carbon tape and sputter -coated with gold using a sputter with current set at 40 mA (Dynavac Mini Coater, Dynavac, USA). The images were carried out by a LaB electron gun with an accelerating voltage of 3 kV using a Philips XL 30 SEM.
Transmission electron microscope (TEM):
The structure of NSC-membrane coated NPs was determined by TEM. Briefly, a drop of NP suspension was added onto a glow-discharged carbon-coated grid (Electron Microscopy Sciences) and, after 2 minutes, rinsed with 10 drops of distilled water. Three drops of 1% phosphotungstic acid were deposited on the sample followed by a rinse process with 10 drops of distilled water. The grid was dried and imaged by a FEI 200kV sphere microscope.
Dynamic light scattering (DLS):
The hydrodynamic size and zeta potential of NPs were measured by DLS. Briefly, 0.1 mg NPs in 1 ml ddH2O water was added to a transparent cuvette, which was subjected to measurement using a Zetasizer (Malvern).
Mouse models:
Male C57BL/6 mice (Charles River Laboratories, USA), ~20 g each, were given free access to food and water before all experiments. All animal experiments were approved by the Yale University Institutional Animal Care and Utilization Committee. Stroke was established through MCAO surgery according to our previous reports [22, 37, 38]. TBI was induced through a standard controlled cortical impact (CCI) method. Briefly, mice were anesthetized with isoflurane and placed in a stereotaxic frame. A 3-mm craniotomy was made unilaterally at AP - 0.5, ML + 0.5 mm, without damage to underlying dura mater. A 2-mm striker in a Hattaras Precision Cortical Impactor (Hattaras, USA) was used to impact the exposed dura to produce a traumatic lesion. TBI was generated using 1.5 m/sec speed, 1 mm depth, and 155 msec dwell time. After the impact, the wound was stitched, and the mouse was subjected to treatment and evaluation.
Fluorescent imaging:
Mice bearing stroke or TBI were prepared and randomly assigned into experimental groups (n=3). Immediately after surgery, IR780- loaded nanoparticles were administered intravenously through the tail vein. Dose for each group was adjusted according to the fluorescence intensity to ensure that each mouse received the same amount of dye. Twenty-four hours later, mice were sacrificed to isolate the brain and imaged by IVIS imaging system (Xenogen) with excitation wavelength of 745 nm and emission wavelength of 820 nm for IR780- loaded NPs. Fluorescence intensity in each brain was quantified using Living Image 3.0 (Caliper, CA). To correlate the location of NPs and ischemic region, the brains were sliced coronally with 2mm thickness and stained with 2,3,5-triphenyltetrazolium chloride (TTC) by incubating the brain sections in a 2%TTC solution for 20 min at 37 °C. The images of the TTC staining and the IT-780 fluorescence were captured with a camera and IVIS imaging system, respectively.
Determination of the therapeutic benefits:
Mice received MCAO surgery and randomly divided into 4 groups (n = 7), which received treatment of PBS, blank CMNPs, glyburide-loaded CMNPs at a dose equivalent to 5 μg/kg of glyburide, and the same amount of free glyburide, respectively. Mice received three injections, which were given intravenously at 0, 24 and 48 h after surgery. Afterwards, the mice were monitored for survival for 7 days and euthanized if one of the following criteria was met: (1) the mouse’s body weight dropped below 15% of its initial weight, or (2) the mouse became lethargic or sick and unable to feed. For the study to determine the impact of treatments on infarct volume and neurological score, a cohort of mice were prepared (n = 5) and received the same treatments as described above. Three days later, the neurological score of each mouse was assessed by a standard behavioral test,[37, 38] and were scored as follows: (1) normal motor function, (2) flexion of torso and contralateral forelimb when animal was lifted by the tail, (3) hemiparalysis resulting in circling to the contralateral side when held by tail on flat surface, but normal posture at rest, (4) leaning to the contralateral side at rest, and (5) no spontaneous motor activity. Therapeutic evaluations were carried out using an unbiased approach; the reviewer who scored mouse function was unaware of which treatment group each mouse belonged to. After the evaluation, the mice were sacrificed and the brains were excised, sectioned, and stained with TTC. The infarct area in each slice was quantified using ImageJ (NIH). For evaluation of the therapeutic benefit for TBI, mice received CCI surgery and were randomly divided into 4 groups (n = 4), which received the same treatments as described above. 4 days later, the mice were sacrificed and the brains were excised, sectioned, and subjected to Nissl staining. The lesion volumes were calculated using ImageJ.
Fluorescent immunohistochemistry for SDF-1:
Seventy-two hours after surgery, mice were anesthetized and intracardially perfused with PBS followed by 4% paraformaldehyde. Brains were dissected and placed in 4% paraformaldehyde for 2 days and transferred into 30% sucrose. After fixation, the brains were sliced at 30um by cryostat section. The sections were transferred to 24 cell culture plates and treated by 0.1%Triton X-100 and washed for 3 times by PBS. After blocking with 4% BSA for 30 minutes and washed 3 times, the sections were incubated with anti-SDF-1 antibody (1:500) (MAB350-100 R&D) for overnight at 4 °C. The sections were incubated with goat anti-mouse IgG(Santa Cruz Biotechnology #G3108) for 30 min after washed by PBS for 3 times. The samples were mounted and imaged using a confocal microscope (Leica TCS SP8).
Characterization of drug encapsulation and release:
For characterization of drug encapsulation, IR780- or glyburide- loaded nanoparticles were dissolved in DMSO. Free IR780 and glyburide released form the NPs were quantified by a microplate reader and HPLC, respectively. For characterization of drug release, glyburide- loaded NPs (3–5 mg) were suspended in 1.0 ml of phosphate buffer (pH 7.4), and incubated at 37º with gentle shaking (270 rpm). Release of glyburide was monitored at several time intervals. At each sampling time, the NP suspension was centrifuged for 10 min at 18,000 rpm. The supernatant was removed for quantification of glyburide by HPLC and an equivale volume of PBS was replaced for continued monitoring of release.
Characterization of CXCR4 on the surface of cells and NPs:
The presence of CXCR4 on cell membrane and the surface of NPs was verified by Western Blot and flow cytometric analysis. For Western Blot analysis, CMNPs were collected by centrifugation at 18000 rpm, 40 min, 4 °C to remove the uncoated membranes. NSCs or NPs were incubated in RIPA lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing protease inhibitor cocktail (Roche) on ice for 5 min. Then, the lysates were centrifuged at 13,000×g for 5 min at 4 °C, and the supernatant was subjected to BCA protein assay (Thermo Scientific) for the quantification of the total protein. After that, the supernatant was mixed with SDS loading buffer and heated at 100 °C for 5 min. An equivalent 20 μg of protein per sample was loaded into each well of a Tris/glycine SDS-polyacrylamide gelatin in an electrophoresis chamber system (Bio-Rad). The protein was transferred to polyvinylidene fluoride membranes (Cell Signaling), which was then blocked with 5% nonfat milk in TBS-T (Tris-HCl 50 mM, NaCl 150 mM, Tween-80 0.1%) for 1 h at room temperature. The blots were probed by antibody against CXCR4 (#NB100-74396, Novus), overnight at 4 °C and then incubated with the HRP anti-rabbit IgG (invitrogen, 656120) and imaged.
For flow cytometry analysis of NSCs, 1×106 cells were collected, fixed using 4% formaldehyde, and washed by PBS for 3 times. Then, the cells were resuspended in 1ml PBS with anti-CXCR4 (#NB100-74396, Novus) and incubated on ice for 1 h. After washing with PBS for 3 times, the cell suspension was centrifuged, resuspended in 1ml PBS with second antibody goat anti rabbit-IgG (Invitrogen, A11010) and incubated for 30 min. The cells were then collected and analyzed by a FACscalibur flow cytometer (BD Biosciences). For analysis of NPs, 1 mg NPs were resuspended in 1ml PBS and incubated anti-CXCR4 for 1h. The NPs were collected by centrifugation at 18,000 rpm/min for 5 min, washed with PBS for 3 times, resuspended in 1ml PBS with anti-rabbit IgG (Invitrogen, A11010), and incubated in dark for 30min. After washing with PBS for 3 times, the NPs were subjected to flow cytometry analysis.
Statistical analysis:
All data were collected in triplicate and reported as mean and standard deviation. Comparison between the groups were performed using a t-test. Logrank test was used to analyze multiple comparisons by GraphPad Prism 7.0. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 were considered significant.
Supplementary Material
Acknowledgements
J. Ma and S. Zhang contributed equally to this work. This work was supported by NIH grant NS095817 and AHA grant 18TPA34170180.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Junning Ma, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Shenqi Zhang, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Jun Liu, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Fuyao Liu, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Fenyi Du, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Miao Li, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Ann T. Chen, Department of Biomedical Engineering, Yale University, New Haven, CT 06511, USA
Youmei Bao, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Hee Won Suh, Department of Biomedical Engineering, Yale University, New Haven, CT 06511, USA.
Jonathan Avery, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Gang Deng, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Yu Zhou, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Peng Wu, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
Kevin Sheth, Department of Neurology, Yale University, New Haven, CT, 06510, USA.
Haijun Wang, Department of Neurosurgery, The First Affiliated Hospital Sun Yat-Sen University, Guangzhou, Guangdong, 510080, China..
Jiangbing Zhou, Department of Neurosurgery, Yale University, New Haven, CT 06511, USA.
References
- [1].Feigin VL; Forouzanfar MH; Krishnamurthi R; Mensah GA; Connor M; Bennett DA; Moran AE; Sacco RL; Anderson L; Truelsen T; O’Donnell M; Venketasubramanian N; Barker-Collo S; Lawes CMM; Wang WZ; Shinohara Y; Witt E; Ezzati M; Naghavi M; Murray C; Fa GBDIR; Grp GSE, Lancet 2014, 383 (9913), 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Janowitz T; Menon DK, Sci Transl Med 2010, 2 (27), 27rv1. [DOI] [PubMed] [Google Scholar]
- [3].Latour LL; Kang DW; Ezzeddine MA; Chalela JA; Warach S, Annals of neurology 2004, 56 (4), 468–77. [DOI] [PubMed] [Google Scholar]
- [4].Warach S; Latour LL, Stroke; a journal of cerebral circulation 2004, 35 (11 Suppl 1), 2659–61. [DOI] [PubMed] [Google Scholar]
- [5].Kaplan B; Brint S; Tanabe J; Jacewicz M; Wang XJ; Pulsinelli W, Stroke; a journal of cerebral circulation 1991, 22 (8), 1032–9. [DOI] [PubMed] [Google Scholar]
- [6].Sheth KN; Elm JJ; Molyneaux BJ; Hinson H; Beslow LA; Sze GK; Ostwaldt AC; Del Zoppo GJ; Simard JM; Jacobson S; Kimberly WT, Lancet Neurol 2016. [DOI] [PubMed] [Google Scholar]
- [7].Deeken JF; Loscher W, Clinical cancer research : an official journal of the American Association for Cancer Research 2007, 13 (6), 1663–74. [DOI] [PubMed] [Google Scholar]
- [8].Dominguez A; Suarez-Merino B; Goni-de-Cerio F, Journal of nanoscience and nanotechnology 2014, 14 (1), 766–79. [DOI] [PubMed] [Google Scholar]
- [9].Patel T; Zhou J; Piepmeier JM; Saltzman WM, Advanced drug delivery reviews 2012, 64 (7), 701–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou J; Atsina KB; Himes BT; Strohbehn GW; Saltzman WM, Cancer J 2012, 18 (1), 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sarmah D; Saraf J; Kaur H; Pravalika K; Tekade RK; Borah A; Kalia K; Dave KR; Bhattacharya P, Micromachines-Basel 2017, 8 (9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fang RH; Kroll AV; Gao W; Zhang L, Adv Mater 2018, 30 (23), e1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Liang H; Huang K; Su T; Li Z; Hu S; Dinh PU; Wrona EA; Shao C; Qiao L; Vandergriff AC; Hensley MT; Cores J; Allen T; Zhang H; Zeng Q; Xing J; Freytes DO; Shen D; Yu Z; Cheng K, ACS nano 2018, 12 (7), 6536–6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Boese AC; Le QE; Pham D; Hamblin MH; Lee JP, Stem Cell Res Ther 2018, 9 (1), 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sinden JD; Hicks C; Stroemer P; Vishnubhatla I; Corteling R, Stem Cells Dev 2017, 26 (13), 933–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lee SP; Youn SW; Cho HJ; Li L; Kim TY; Yook HS; Chung JW; Hur J; Yoon CH; Park KW; Oh BH; Park YB; Kim HS, Circulation 2006, 114 (2), 150–159. [DOI] [PubMed] [Google Scholar]
- [17].De Falco E; Porcelli D; Torella AR; Straino S; Iachininoto MG; Orlandi A; Truffa S; Biglioli P; Napolitano M; Capogrossi MC; Pesce M, Blood 2004, 104 (12), 3472–3482. [DOI] [PubMed] [Google Scholar]
- [18].Imitola J; Raddassi K; Park KI; Mueller FJ; Nieto M; Teng YD; Frenkel D; Li J; Sidman RL; Walsh CA; Snyder EY; Khoury SJ, Proceedings of the National Academy of Sciences of the United States of America 2004, 101 (52), 18117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hill WD; Hess DC; Martin-Studdard A; Carothers JJ; Zheng JQ; Hale D; Maeda M; Fagan SC; Carroll JE; Conway SJ, J Neuropath Exp Neur 2004, 63 (1), 84–96. [DOI] [PubMed] [Google Scholar]
- [20].Rao L; Meng QF; Huang QQ; Wang ZX; Yu GT; Li A; Ma WJ; Zhang NG; Guo SS; Zhao XZ; Liu K; Yuan YF; Liu W, Adv Funct Mater 2018, 28 (34). [Google Scholar]
- [21].Cashen AF; Nervi B; DiPersio J, Future oncology 2007, 3 (1), 19–27. [DOI] [PubMed] [Google Scholar]
- [22].Guo X; Deng G; Liu J; Zou P; Du F; Liu F; Chen AT; Hu R; Li M; Zhang S; Tang Z; Han L; Liu J; Sheth KN; Chen Q; Gou X; Zhou J, ACS nano 2018, 12 (8), 8723–8732. [DOI] [PubMed] [Google Scholar]
- [23].Hu CM; Zhang L; Aryal S; Cheung C; Fang RH; Zhang L, Proc Natl Acad Sci U S A 2011, 108 (27), 10980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Xuan M; Shao J; Dai L; Li J; He Q, ACS Appl Mater Interfaces 2016, 8 (15), 9610–8. [DOI] [PubMed] [Google Scholar]
- [25].Kroll AV; Fang RH; Jiang Y; Zhou J; Wei X; Yu CL; Gao J; Luk BT; Dehaini D; Gao W; Zhang L, Adv Mater 2017, 29 (47). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fang RH; Hu CM; Chen KN; Luk BT; Carpenter CW; Gao W; Li S; Zhang DE; Lu W; Zhang L, Nanoscale 2013, 5 (19), 8884–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Chai Z; Hu X; Wei X; Zhan C; Lu L; Jiang K; Su B; Ruan H; Ran D; Fang RH; Zhang L; Lu W, Journal of controlled release : official journal of the Controlled Release Society 2017, 264, 102–111. [DOI] [PubMed] [Google Scholar]
- [28].Simard JM; Chen M; Tarasov KV; Bhatta S; Ivanova S; Melnitchenko L; Tsymbalyuk N; West GA; Gerzanich V, Nature medicine 2006, 12 (4), 433–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tournier N; Saba W; Cisternino S; Peyronneau M-A; Damont A; Goutal S; Dubois A; Dolle F; Scherrmann J-M; Valette H; Kuhnast B; Bottlaender M, Aaps J 2013, 15 (4), 1082–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lahmann C; Kramer HB; Ashcroft FM, PloS one 2015, 10 (7), e0134476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ludwig PE; Thankam FG; Patil AA; Chamczuk AJ; Agrawal DK, Neural regeneration research 2018, 13 (1), 7–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xue L; Wang J; Wang W; Yang Z; Hu Z; Hu M; Ding P, Cell Biochem Biophys 2014, 70 (3), 1609–16. [DOI] [PubMed] [Google Scholar]
- [33].Patel AD; Gerzanich V; Geng Z; Simard JM, Journal of neuropathology and experimental neurology 2010, 69 (12), 1177–90. [DOI] [PubMed] [Google Scholar]
- [34].Chen Y; Gou X; Kong DK; Wang X; Wang J; Chen Z; Huang C; Zhou J, Oncotarget 2015, 6 (32), 32575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhou J; Patel TR; Fu M; Bertram JP; Saltzman WM, Biomaterials 2012, 33 (2), 583–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhou J; Patel TR; Sirianni RW; Strohbehn G; Zheng M-Q; Duong N; Schafbauer T; Huttner AJ; Huang Y; Carson RE; Zhang Y; Sullivan DJ Jr.; Piepmeier JM; Saltzman WM, Proc Natl Acad Sci U S A 2013, 110 (29), 11751–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Cai Q; Chen Z; Kong DK; Wang J; Xu Z; Liu B; Chen Q; Zhou J, Neurosci Lett 2015, 597, 127–31. [DOI] [PubMed] [Google Scholar]
- [38].Han L; Cai Q; Tian D; Kong DK; Gou X; Chen Z; Strittmatter SM; Wang Z; Sheth KN; Zhou J, Nanomedicine 2016, 12 (7), 1833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.