

Abstract
Carbonyls and alkenes are versatile functional groups, whose reactivities are cornerstones of organic synthesis. The selective combination of two carbonyls to form an alkene – a carbonyl cross-metathesis – would be a valuable tool for their exchange. Yet, this important synthetic challenge remains unsolved. Although alkene/alkene and alkene/carbonyl cross-metathesis reactions are known, there is a lack of analogous methods for deoxygenative cross-coupling of two carbonyls. Here, we report a pair of strategies for the cross-metathesis of unbiased carbonyls, allowing an aldehyde to be chemo- and stereo-selectively combined with another aldehyde or ketone. These mild, catalytic methods are promoted by earth-abundant metal salts and enable rapid access to an unprecedentedly broad range of either Z or E alkenes by two distinct mechanisms – entailing transiently generated: (1) carbenes and ylides (via Fe catalysis) or (2) doubly nucleophilic gem-di-metallics (via Cr catalysis).
Given the synthetic importance of carbonyls and alkenes, mechanistically novel methods for their interchange are highly valued.1 An important example is alkene metathesis, wherein two alkenes are structurally shuffled (Fig. 1a).2 Although some methods enable cross-reactivity, an inherent challenge remains in differentiating the two alkenes to favor cross-selectivity over dimerization. Alkene-carbonyl metathesis provides another path by exchanging an alkene with a carbonyl, and valuable catalytic strategies exploit this inherent chemo-selectivity.3,6–8 Alternatively, the metathesis of carbonyls to form an alkene provides a mechanistically distinct means of differentiating a product alkene from its carbonyl precursors. Deoxygenative carbonyl dimerization has been developed using low-valent titanium (i.e., the McMurry coupling).9–11 Nonetheless, cross-selectivity remains unsolved.4,5 Here, we present two, distinct strategies for cross-metathesis of carbonyls, entailing either: (1) carbene-to-ylide transfer or (2) gem-di-metalation. By these mechanistically divergent methods, an aldehyde may now be chemo- and stereo- selectively combined with another carbonyl to form either a Z or E alkene. This transform, an endothermic reverse of ozonolysis, is enabled by catalytic transformation of electrophilic carbonyls into either zwitterionic ylides or doubly nucleophilic gem-di-metals.
Fig. 1: Carbonyl metathesis strategies are rare and cross-selectivity mechanisms must be developed.
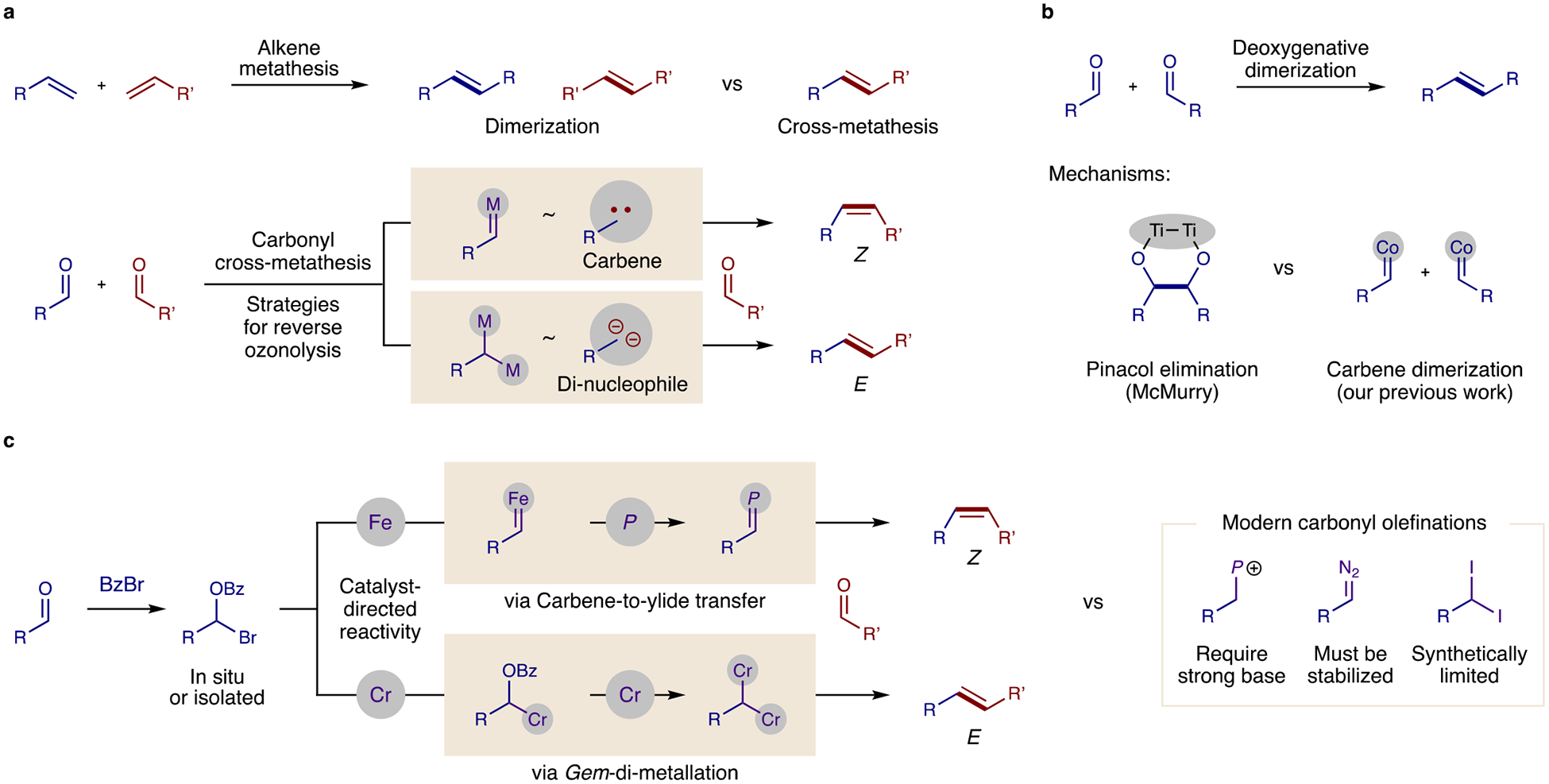
a, Reactivity and selectivity challenges are shown. Cross-selective, alkene metathesis is rare, yet synthetically valuable. Carbonyl cross-metathesis (a reverse ozonolysis of an unsymmetric alkene) is unknown. b, State of the art methods for deoxygenative olefination of carbonyls only yield dimeric products. c, Two complementary strategies to access cross-selective reactivity are presented via either (1) Fe-catalyzed carbene/ylide or (2) Cr-catalyzed gem-di-metal mechanisms. Synthetic advantages of these approaches include mild (base-, diazo-, and dihalo- free) conditions that employ unbiased, enolizable aldehydes and yield robust, divergent (Z or E) stereoselectivity.
The challenge of enabling cross-selectivity in the McMurry coupling is tied to its mechanism (Fig. 1b).9 In it, Ti-mediated reduction of a carbonyl and ketyl radical coupling forms pinacol, which is subsequently deoxygenated as TiO2 to yield an alkene. Since there is no mechanism for carbonyl selectivity in ketyl formation nor for the ensuing pinacol coupling, a statistical mixture of cross-coupling and dimerization products is inherent. The only exceptions are certain carbonyls (e.g., diaryl ketones, glyoxylates), whose double reductions to anion nucleophiles are thermodynamically biased.12,13 Other novel alternatives to the McMurry coupling are similarly limited to differentiation by at least one aryl carbonyl partner.14–17
Towards broader, chemoselective carbonyl activation, we recently developed an in situ acyl halide addition of aldehydes to form ketyl radicals by atom transfer catalysis.18 This enables aldehyde cross-coupling with several other electrophiles.19–23 However, carbonyl olefination also requires a mechanism for oxygen deletion24,25 (e.g. Ti to TiO2, in the McMurry reaction). Thus, we developed a method to convert aldehydes to carbenes, wherein aldehydes are deoxygenated via transient α-benzoyl zinc intermediates (Fig. 1b).26 Notably, we observed stereo-selective carbene dimerization to E-alkenes with a Co catalyst. To reverse polarity of these electrophilic carbenes and combine them with another carbonyl, we included sulfide co-catalysts to access sulfonium ylide reactivity.27 Yet, this strategy affords epoxides rather than alkenes (i.e., only one carbonyl is deoxygenated).26 Instead, to promote carbonyl metathesis, double deoxygenation of two carbonyls is necessary.
To enable synergistic deoxygenation of a second carbonyl, we have now developed two distinct catalytic strategies to access either a Z or E alkene stereoselectively (Fig. 1c). The first approach is inspired by a modified Wittig mechanism.1 In this case, the ensuing deoxygenation is enabled by converting a transient carbene to a phosphonium ylide. This method complements pioneering approaches by Carreira, Lebel, and Aggarwal entailing carbene transfer from diazo reagents,28–30 by now permitting the use of enolizable alkyl aldehydes as carbene precursors. And for comparison with other modern ylide-based approaches, Silvi recently intercepted the Wittig reaction by decarboxylative radical addition of α-stabilized acids to vinyl phosphoniums,31 and Ott developed a P=P reagent to promote a double Wittig of aryl aldehydes.16 Yet, in our proposal, we expected this chemoselective conversion of aldehydes to ylides to now enable catalytic coupling of unbiased, alkyl aldehydes. Moreover, this base-free olefination is expected to be stereoselective – affording Z-alkenes, in contrast with diazo-mediated E-olefinations.28–30
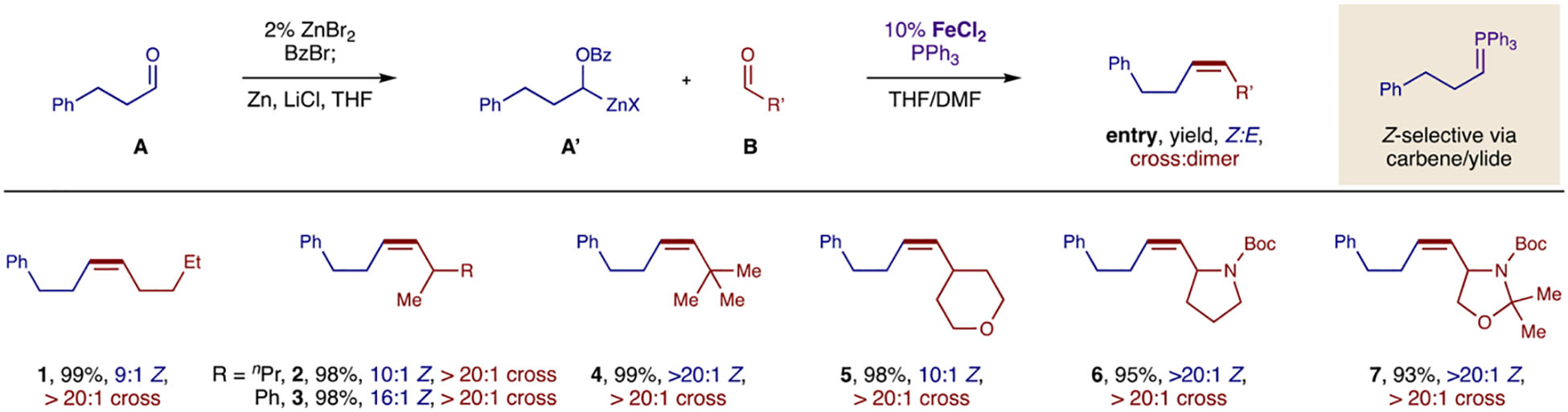
To test our first proposal (Table 1), we converted unbiased, enolizable, alkyl aldehyde A to its stable α-benzyloxy bromide by in situ BzBr addition with a ZnBr2 catalyst. This inter-mediate may be purified by column chromatography or recrystallization, and stored cold indefinitely or may be used directly without purification. Upon its Zn-insertion to form alkyl zinc carbenoid, A’ (mediated by LiCl), an iron catalyst (10% FeCl2), phosphine (Ph3P), and acceptor aldehyde B were added to the mixture and let stir at room temperature. In support of our mechanistic hypothesis, the product alkenes 1–7 are formed with complete cross-selectivity (>20:1 by 1H NMR) since the phosphonium ylide is exclusively generated from aldehyde A, and the ylide does not dimerize, but only reacts with aldehyde B. The high efficiency and cross-selectivity observed also indicates that transfer of the iron carbene to Ph3P outpaces dimerization. To our delight, Z-alkene formation is also highly stereoselective (up to >20:1 with LiCl in THF/DMF) in contrast to diazo variants.28–30 The cross-coupling of two linear n-alkyl aldehydes (1) yields 9:1 Z:E selectivity, and trapping with α-branched (alkyl or aryl) aldehydes affords even higher stereo-selectivity (2, 10:1 Z and 3, 16:1 Z). Further steric hindrance is also well-tolerated (and more stereoselective), as in the case of pivaldehyde (4, >20:1 Z) and heterocyclic aldehydes (5-7, >20:1 Z).
Table 1:
Discovery and synthetic scope of an Fe-catalyzed, carbonyl cross-metathesis (Z-selective).
|
Reaction conditions: Aldehyde A (0.2 mmol, 1 equiv), ZnBr2 (2 mol%), BzBr (1.2 equiv) in CH2Cl2 (0.6 mL) at −10 °C for 2 h; α-OBzBr of A (2 equiv), LiCl (2 equiv), Zn (3 equiv) in THF (1 mL) at r.t. for 8 h; carbenoid A’ (2 equiv), FeCl2 (10 mol%), Ph3P (2 equiv), aldehyde B (1 equiv) in THF/DMF (2 mL/0.5 mL) at r.t. for 12 h. Isolated yields. Cross- and Stereo- selectivity measured by 1H NMR.
THF, tetrahydrofuran; DMF, N,N-dimethylformamide.
We next sought to develop a parallel strategy to access E-alkenes via a distinct deoxygenation mechanism (Fig. 1c). Here, we were inspired by the Takai reaction, wherein gem-dimetals stereoselectively olefinate aldehydes.32,33 In this phosphine-free approach, gem-diiodides are converted to dizinc or dichromium intermediates by insertion of either Zn or Cr reductants.34 Like the reagents developed by Nysted or Tebbe, metal oxide formation drives alkene generation and carbonyl deoxygenation in these cases.1 Yet, advantages of the Cr-based method include high E-selectivity and broad reactivity with either aryl or alkyl aldehydes, including base-sensitive, enolizable ones. Still, the need for unstable gem-diiodides, or related precursors, remains a major synthetic limitation since harsh iodination conditions and the potential for elimination typically limit generality and accessibility.35,36 Motherwell and Boland have each extended this reaction to employ other carbonyl derivatives as the gem-di-metal precursor.37,38 However, neither approach is cross-selective, catalytic, or exhibits the wide synthetic scope of the Takai method. In contrast, we anticipated chemoselective in situ aldehyde preactivation and catalytic stereocontrol could enable a broadly useful carbonyl cross-metathesis.
To examine our proposed E-selective cross-metathesis (Table 2), aldehyde A was converted to its α-OBz bromide and added to aldehyde B and CrCl2 in a 1:1:1 ratio. In this preliminary stoichiometric example, we were pleased to see complete cross-selectivity (>10:1) and stereoselectivity (>20:1) in the formation of alkene 8. Toward a catalytic variant, we were inspired by how Fürstner rendered the Nozaki−Hiyama−Kishi reaction catalytic in Cr by incorporating a Mn reductant and chlorosilane.39 Recent examples have also shown the value of bipyridyl ligands and coordinating solvents to enable reductive catalytic turnover of Cr.40–42 We investigated several ligands and reductants and found the combination of Mn, LiI, Me3SiCl, and 4,4′-di-t-butyl-bipyridyl ligand (dtbbpy) to be most effective. Upon inclusion of 10% dtbbpy in THF, Cr catalyst loading may be reduced to 25% CrCl2. Yet, while product 8 forms efficiently (79%) in this case, cross- (8:1) and stereo- (10:1 E) selectivity are diminished for this dichromium-mediated reaction. Thus, 50% CrCl2 was selected to enable broad, robust efficiency (>90% yield) and selectivity (>10:1 cross; >20:1 E:Z).
Table 2:
Discovery of a Cr-catalyzed cross-metathesis of aldehydes (E-selective).
| Entry | CrCl2 | dtbbpy | Yield 8 | E:Z | Cross |
|---|---|---|---|---|---|
| 1 | 100% | — | 90% | >20:1 | >10:1 |
| 2 | 0% | 10% | 0% | — | — |
| 3 | 25% | 10% | 79% | 10:1 | 8:1 |
| 4 | 50% | 10% | 95% | >20:1 | >10:1 |
| 5 | 100% | 10% | 91% | >20:1 | >10:1 |
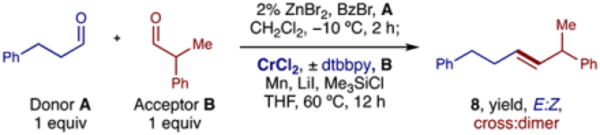
Reaction conditions: Aldehyde A (0.2 mmol, 1 equiv), ZnBr2 (2 mol%), BzBr (1.2 equiv) in CH2Cl2 (0.2 mL) at −10 °C for 2 h; then add to aldehyde B (1 equiv), CrCl2 (X mol%), dtbbpy (Y mol%), Mn (2 equiv), LiI (3 equiv), Me3SiCl (2 equiv) in THF (2.5 mL) at 60 °C for 4–12 h. Isolated yields. Cross- and Stereo- selectivity measured by 1H NMR. THF, tetrahydrofuran; dtbbpy, 4,4′-di-tert-butyl-2,2′-bipyridyl.
To evaluate the synthetic utility of this Cr-catalyzed carbonyl cross-metathesis (Fig. 2a), a diverse range of aldehydes were investigated as the donor (A) and acceptor (B) components. Both were shown to have broad scope and generality. For example, acceptor B may consist of linear (9–12) and branched (13–20) aldehydes, including isotopes of formaldehyde (9), d-acetaldehyde (10), and citronellal (12), as well as medicinally relevant, small rings and heterocycles (14–18). Large sterics are also well-tolerated (19–20). In each of these cases (8, 13–20), high chemo- and stereo- selectivity is observed (>10:1 cross; >20:1 E:Z). Conversely, aryl and heteroaryl aldehydes are also efficient and cross-selective but lack stereoselectivity (21–27). Notably, ketones may also be employed as acceptors (28–34) with their efficiency illustrating the robustness of this dichromium reactivity.
Fig. 2: The Cr-catalyzed carbonyl metathesis has wide scope with robust E- and cross-selectivity.
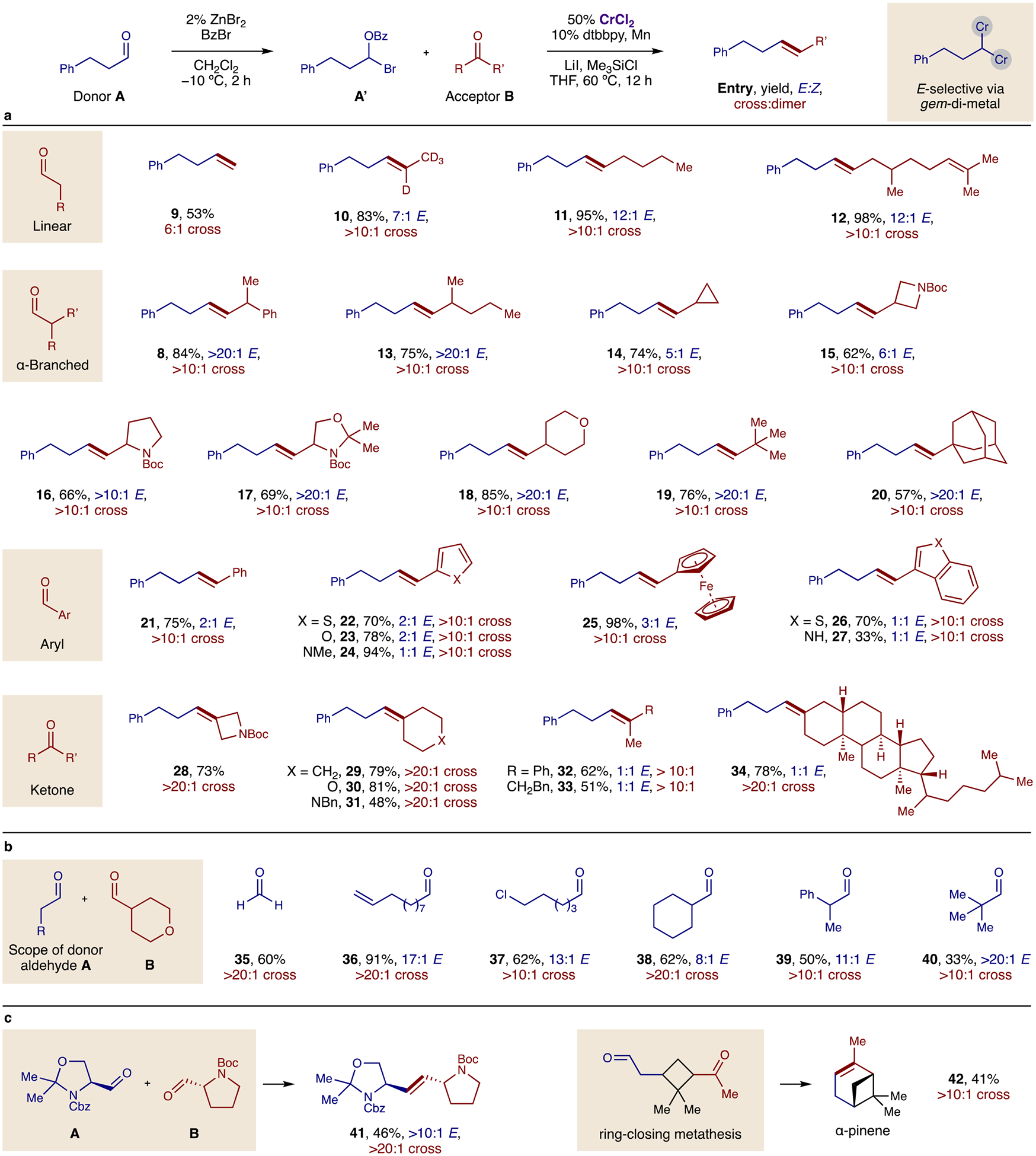
a, Acceptor B may be a linear or branched, alkyl or aryl aldehyde, as well as a ketone. b, Donor A may be a wide range of aldehydes, including the smallest formaldehyde or most hindered, pivaldehyde. c, Complex molecule applications show the utility of this olefination. See Table 2 for conditions.
To probe the mildness and unique tunability of the aldehyde activation, the donor component A of this cross-metathesis was also varied (Fig. 2b). These experiments show a similarly broad scope. For example, formaldehyde (35) may be employed, as well as aldehydes containing reactive functional groups, such as alkenes (36) or halides (37). Moreover, sterically hindered, α-branched and enolizable aldehydes are tolerated (38–40) – overcoming significant limitations of the Wittig reaction.1 Complex aldehydes were also coupled to form a heteroatom-rich alkene (41) and showcase the synthetic utility of this mild, catalytic protocol (Fig. 2c). Finally, ring-closing metathesis was performed on a molecule bearing an aldehyde and ketone to generate α-pinene 42.
To benchmark the unprecedented cross-selectivity of these two new strategies, we evaluated the metathesis of aldehydes using other current state-of-the-art methods. First, our previous Co-catalyzed carbene dimerization method (Fig. 3a) yields a 4:1 mixture of dimers (43 and 44) to cross-coupled alkene 8 since there is no mechanism for cross-selectivity. Surprisingly, the Ti-mediated McMurry coupling does not afford any alkene when two alkyl aldehydes (A and C) are employed (Fig. 3b). Yet, if a more easily reduced aryl aldehyde D is combined with aldehyde A, then a 1:1 mixture of D-D dimerization (45) and A-D cross-coupling (21) is observed. In this case, although there is a method for differentiating Ti-ketyl radical generation (favoring D > A), there is no selectivity mechanism for its cross-coupling.
Fig. 3: Cross-metathesis: selectivity probes and mechanism.
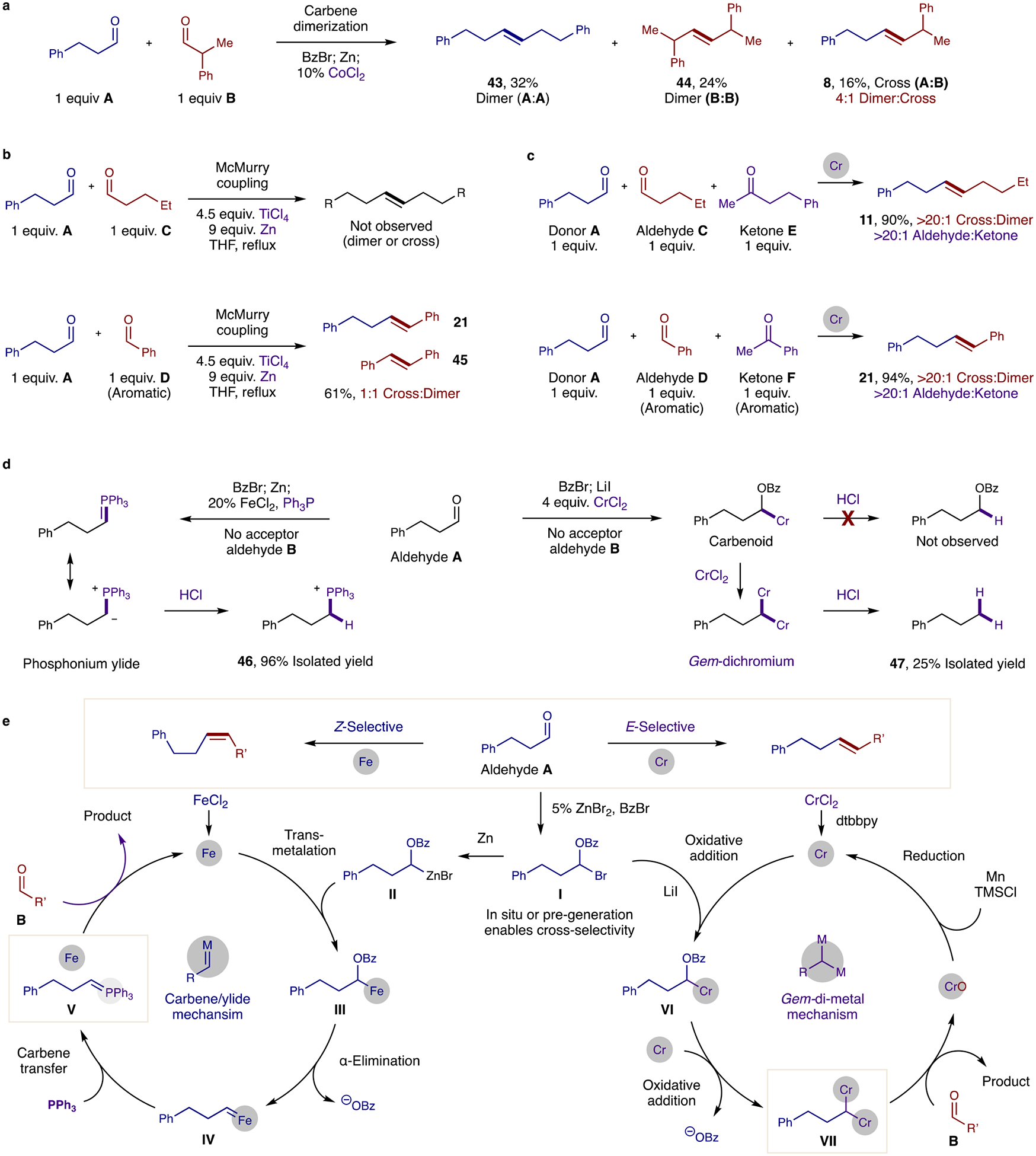
No cross-selectivity is observed via previous methods, including: a, Co-catalyzed carbene dimerization, or b, Ti-mediated McMurry coupling with either alkyl or aryl aldehyde acceptors. c, In contrast, Cr-catalysis affords cross- and chemo- selectivity, even in three-component competitions with aldehyde and ketone acceptors. d, In the absence of an aldehyde acceptor, key proposed intermediates were isolated to support each mechanism, including phosphonium 46 by ylide protonation and doubly reduced 47 by gem-dichromium protodemetalation. e, Proposed mechanisms for each strategy are shown. Cross-selectivity is ensured by transient formation of I from aldehyde A. Z-selectivity (left catalytic cycle) occurs by Zn-insertion of I to II, followed by transmetallation to an FeCl2 catalyst. Upon α-elimination of III to alkyl carbene IV, carbene transfer to PPh3 affords catalyst turnover and ylide V, which enables Z-selective olefination of B. In contrast, E-selectivity (right catalytic cycle) occurs by double Cr-insertions of I to VI to VII. This catalytically generated gem-dichromium arises from two oxidative additions of a L·CrCl2 catalyst to the α-oxy carbon of I, in a net deoxygenation of carbonyl A. Upon E-selective olefination of B by doubly nucleophilic organometal VII, CrO reduction permits catalyst turnover.
Since we have already shown selective formation of these two products (3, 8, 21) with >10:1 cross-selectivity, we next sought to evaluate the rate of reactivity between aldehyde and ketone acceptors in our Cr-catalyzed method (Fig. 3c). In this case, when donor aldehyde A (1 equiv.) is subjected to two acceptors, aldehyde C and ketone E (1 equiv. each), the aldehyde partners are exclusively coupled with >20:1 aldehyde:ketone selectivity and >20:1 cross-selectivity. When aryl aldehyde D and ketone F (1 equiv. each) are employed as acceptors in this three-component competition, the same >20:1 aldehyde:ketone chemoselectivity is observed.
Next, we sought to support our mechanistic hypotheses by isolating key intermediates of each method (Fig. 3d). When aldehyde A is subjected to the Z-selective conditions in the absence of an acceptor aldehyde B, the phosphonium ylide is expected to be present stoichiometrically, even with 20% Fe catalyst. In fact, quenching with HCl yields alkyl phosphonium 46 in >95% isolated yield – validating the intermediacy of an ylide in this Z-selective reaction. In parallel, aldehyde A was subjected to the E-selective reaction, albeit with stoichiometric CrCl2 to isolate a transient organometallic intermediate. If α-OBz-Cr-carbenoid (via only one Cr reduction) predominates, then HCl quenching should yield the reduced benzoate ester. However, this product was not observed. Instead, acidic quenching yields a fully deoxygenated product 47, which results from the doubly reduced gem-dichromium intermediate. Since LiI accelerates this stoichiometric CrCl2 reaction, we expect it also plays a role in activating the α-OBz species for reduction by Cr.
Given these observations, we propose the following two mechanisms (Fig. 3e). For the Fe-catalyzed carbene/ylide strategy (shown on the left), in situ activation of aldehyde A by BzBr (with ZnBr2 catalyst) yields α-OBz bromide I. Next, Zn insertion of I affords carbenoid II. Transmetallation with the Fe catalyst then yields α-OBz organoiron III, which may rapidly α-eliminate the OBz anion to generate Fe-carbene IV. Upon carbene transfer to Ph3P, phosphonium ylide V is formed (as observed by its protonated analog 46), which can chemoselectively combine with aldehyde B to form Z-alkene while recycling the Fe catalyst. Conversely, in the gem-di-metal, Cr-catalyzed mechanism (shown on right), α-OBz bromide I may be directly and chemoselectively reduced by Cr(II) to Cr-carbenoid VI – accelerated by LiI, and in the presence of acceptor aldehyde B. A second Cr-mediated reduction yields the key gem-dichromium VII, which enables deoxgenative olefination of aldehyde B to afford the E-alkene and a chromium oxide. By combination of Mn, LiI, and TMSCl, this CrOn is reduced back to Cr(II) to turn over the Cr catalytic cycle.
In conclusion, two catalytic methods have been developed for the chemo- and stereo- selective cross-metathesis of carbonyls. These deoxygenative strategies enable the reverse of modern ozonolysis reactions43,44 by combining two carbonyls to form an alkene – a valuable addition to the synthetic toolkit.
Methods.
Cr-catalyzed cross-metathesis (E-selective):
To ZnBr2 (5 mol%) in a dry vial, was added CH2Cl2 (0.2 mL) and BzBr (1.2 equiv.). Next, aldehyde A (0.2 mmol, 1 equiv.) was added slowly and stirred at −10 °C for 2 h, then purified by chromatography or recrystallization, or used crude. Separately, to a dry vial of CrCl2 (50 mol%), dtbbpy (10 mol%), Mn (2 equiv.), LiI (3 equiv.), Me3SiCl (2 equiv.), THF (1 mL), and a stir bar, was added aldehyde B (1 equiv.). The aldehyde A mixture was then added with THF (1.5 mL), stirred at 60 °C for 4–12 h, then quenched and purified by chromatography to afford the E-alkene product.
Fe-catalyzed cross-metathesis (Z-selective):
To ZnBr2 (2 mol%) in a dry vial, was added CH2Cl2 (0.6 mL) and BzBr (1.2 equiv.). Aldehyde A (0.2 mmol, 1 equiv.) was added slowly and stirred at −10 °C for 2 h, then purified by chromatography. Next, this α-OBz bromide (2 equiv.) in THF (1 mL) was slowly added to a dry vial of LiCl (2 equiv.) and Zn dust (3 equiv.), then stirred at room temperature for 8 h. The filtrate of this alkyl zinc carbenoid solution was then transferred to FeCl2 (10 mol%), Ph3P (2 equiv.), aldehyde (1 equiv.), THF/DMF (2 mL/0.5 mL), and then stirred at room temperature for 12 h. Purification by chromatography affords the Z-alkene product.
Supplementary Material
Acknowledgements.
We thank the National Institutes of Health (R35 GM119812), National Science Foundation (CAREER 1654656), and Brown Science Foundation (BIA) for funding (to D.A.N.), and we are grateful to Taylor Bednar for independent verification of the robustness of this method.
Footnotes
Competing interests.
The authors declare no competing interests.
Data availability.
The data supporting the findings of this study are included in the Supplementary Information.
References
- 1.Takeda T Modern Carbonyl Olefination - Methods and Applications. Synthesis 2004, 1532–1532 (2004). [Google Scholar]
- 2.Hoveyda AH & Zhugralin AR The remarkable metal-catalysed olefin metathesis reaction. Nature 450, 243–251 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Albright H et al. Carbonyl-Olefin Metathesis. Chem. Rev 121, 9359–9406 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asako S & Ilies L Olefin Synthesis by Deoxygenative Coupling of Carbonyl Compounds: From Stoichiometric to Catalytic. Chem. Lett 49, 1386–1393 (2020). [Google Scholar]
- 5.Bongso A, Roswanda R & Syah YM Recent advances of carbonyl olefination via McMurry coupling reaction. RSC Adv. 12, 15885–15909 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Griffith AK, Vanos CM & Lambert TH Organocatalytic carbonyl-olefin metathesis. J. Am. Chem. Soc 134, 18581–18584 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Ludwig JR, Zimmerman PM, Gianino JB & Schindler CS Iron(III)-catalysed carbonyl-olefin metathesis. Nature 533, 374–379 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Pitzer L, Sandfort F, Strieth-Kalthoff F & Glorius F Carbonyl–Olefin Cross-Metathesis Through a Visible-Light-Induced 1,3-Diol Formation and Fragmentation Sequence. Angew. Chem. Int. Ed 57, 16219–16223 (2018). [DOI] [PubMed] [Google Scholar]
- 9.McMurry JE Carbonyl-Coupling Reactions Using Low-Valent Titanium. Chem. Rev 89, 1513–1524 (1989). [Google Scholar]
- 10.McMurry JE & Fleming MP New Method for the Reductive Coupling of Carbonyls to Olefins. Synthesis of β-Carotene. J. Am. Chem. Soc 96, 4708–4709 (1974). [DOI] [PubMed] [Google Scholar]
- 11.Mukaiyama T, Sato T & Hanna J Reductive Coupling of Carbonyl Compounds to Pinacols and Olefins by Using TiCl4 and Zn. Chem. Lett 2, 1041–1044 (1973). [Google Scholar]
- 12.McMurry JE, Fleming MP, Kees KL & Krepski LR Titanium-Induced Reductive Coupling of Carbonyls to Olefins. J. Org. Chem 43, 3255–3266 (1978). [Google Scholar]
- 13.Mukaiyama T, Sugimura H, Ohno T & Kobayashi S Reductive Cross Coupling Reaction of a Glyoxylate with Carbonyl Compounds. A Facile Synthesis of α,β-Dihydroxycarboxylate Based on a Low Valent Titanium Compound. Chem. Lett 18, 1401–1404 (1989). [Google Scholar]
- 14.Wang H, Dai XJ & Li CJ Aldehydes as alkyl carbanion equivalents for additions to carbonyl compounds. Nat. Chem 9, 374–378 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Wei W et al. Ruthenium(II)-catalyzed olefination: Via carbonyl reductive cross-coupling. Chem. Sci 8, 8193–8197 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Esfandiarfard K, Mai J & Ott S Unsymmetrical E-Alkenes from the Stereoselective Reductive Coupling of Two Aldehydes. J. Am. Chem. Soc 139, 2940–2943 (2017). [DOI] [PubMed] [Google Scholar]
- 17.Wang S, Lokesh N, Hioe J, Gschwind RM & König B Photoinitiated carbonyl-metathesis: Deoxygenative reductive olefination of aromatic aldehydes via photoredox catalysis. Chem. Sci 10, 4580–4587 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang L, Lear JM, Rafferty SM, Fosu SC & Nagib DA Ketyl radical reactivity via atom transfer catalysis. Science 362, 225–229 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang ZP & Fu GC Convergent Catalytic Asymmetric Synthesis of Esters of Chiral Dialkyl Carbinols. J. Am. Chem. Soc 142, 5870–5875 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rafferty SM, Rutherford JE, Zhang L, Wang L & Nagib DA Cross-Selective Aza-Pinacol Coupling via Atom Transfer Catalysis. J. Am. Chem. Soc 143, 5622–5628 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang H-M, Bellotti P, Kim S, Zhang X & Glorius F Catalytic multicomponent reaction involving a ketyl-type radical. Nat. Synth 1, 464–474 (2022). [Google Scholar]
- 22.Huang HM, Bellotti P, Erchinger JE, Paulisch TO & Glorius F Radical Carbonyl Umpolung Arylation via Dual Nickel Catalysis. J. Am. Chem. Soc 144, 1899–1909 (2022). [DOI] [PubMed] [Google Scholar]
- 23.Zhu C, Lee SC, Chen H, Yue H & Rueping M Reductive Cross-Coupling of α-Oxy Halides Enabled by Thermal Catalysis, Photocatalysis, Electrocatalysis, or Mechanochemistry. Angew. Chem. Int. Ed 61, e202204212 (2022). [DOI] [PubMed] [Google Scholar]
- 24.Kennedy SH, Dherange BD, Berger KJ & Levin MD Skeletal editing through direct nitrogen deletion of secondary amines. Nature 593, 223–227 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Dong Z & MacMillan DWC Metallaphotoredox-enabled deoxygenative arylation of alcohols. Nature 598, 451–456 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang L, DeMuynck BM, Paneque AN, Rutherford JE & Nagib DA Carbene reactivity from alkyl and aryl aldehydes. Science 377, 649–654 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aggarwal VK & Winn CL Catalytic, asymmetric sulfur ylide-mediated epoxidation of carbonyl compounds: Scope, selectivity, and applications in synthesis. Acc. Chem. Res 37, 611–620 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Ledford BE & Carreira EM Synthesis of unsaturated esters from aldehydes: An inexpensive, practical alternative to the Horner-Emmons reaction under neutral conditions. Tetrahedron Lett. 38, 8125–8128 (1997). [Google Scholar]
- 29.Lebel H, Paquet V & Proulx C Methylenation of aldehydes: Transition metal catalyzed formation of salt-free phosphorus ylides. Angew. Chem. Int. Ed 40, 2887–2890 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Aggarwal VK, Fulton JR, Sheldon CG & De Vicente J Generation of phosphoranes derived from phosphites. A new class of phosphorus ylides leading to high E selectivity with semi-stabilizing groups in Wittig Olefinations. J. Am. Chem. Soc 125, 6034–6035 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Filippini D & Silvi M Visible light-driven conjunctive olefination. Nat. Chem 14, 66–70 (2022). [DOI] [PubMed] [Google Scholar]
- 32.Takai K, Hotta Y, Oshima K & Nozaki H Effective methods of carbonyl methylenation using CH2I2-Zn-Me3Al and CH2Br2-Zn-TiCl4 system. Tetrahedron Lett. 19, 2417–2420 (1978). [Google Scholar]
- 33.Okazoe T, Takai K & Utimoto K (E)-Selective Olefination of Aldehydes by means of gem-Dichromium Reagents Derived by Reduction of gem-Diiodoalkanes with Chromium(II) Chloride. J. Am. Chem. Soc 109, 951–953 (1987). [Google Scholar]
- 34.Kochi JK & Mocadlo PE Facile Reduction of Alkyl Halides with Chromium(II) Complexes. Alkylchromium Species as Intermediates. J. Am. Chem. Soc 88, 4094–4096 (1966). [Google Scholar]
- 35.Marek I & Normant JF Synthesis and reactivity of sp3-geminated organodimetallics. Chem. Rev 96, 3241–3267 (1996). [DOI] [PubMed] [Google Scholar]
- 36.Nallagonda R, Padala K & Masarwa A gem-Diborylalkanes: recent advances in their preparation, transformation and application. Org. Biomol. Chem 16, 1050–1064 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Banerjee AK, Sulbaran De Carrasco MC, Frydrych-Houge CSV & Motherwell WB Observations on the reductive deoxygenation of aryl and α,β-unsaturated carbonyl compounds with chlorotrimethylsilane and zinc. J. Chem. Soc. Chem. Commun 1803–1805 (1986). doi: 10.1039/C39860001803 [DOI] [Google Scholar]
- 38.Knecht M & Boland W (E)-Selective Alkylidenation of Aldehydes with Reagents derived from α-Acetoxy bromides, Zinc and CrCl3. Synlett 1993, 837–838 (1993). [Google Scholar]
- 39.Fürstner A & Shi N Nozaki-Hiyama-Kishi reactions catalytic in chromium. J. Am. Chem. Soc 118, 12349–12357 (1996). [Google Scholar]
- 40.Zhao K & Knowles RR Contra-Thermodynamic Positional Isomerization of Olefins. J. Am. Chem. Soc 144, 137–144 (2022). [DOI] [PubMed] [Google Scholar]
- 41.Zhang C, Lin Z, Zhu Y & Wang C Chromium-Catalyzed Allylic Defluorinative Ketyl Olefin Coupling. J. Am. Chem. Soc 143, 11602–11610 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Schwarz JL, Schäfers F, Tlahuext-Aca A, Lückemeier L & Glorius F Diastereoselective Allylation of Aldehydes by Dual Photoredox and Chromium Catalysis. J. Am. Chem. Soc 140, 12705–12709 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Ruffoni A, Hampton C, Simonetti M & Leonori D Photoexcited nitroarenes for the oxidative cleavage of alkenes. Nature 610, 81–86 (2022). [DOI] [PubMed] [Google Scholar]
- 44.Wise DE et al. Photoinduced Oxygen Transfer Using Nitroarenes for the Anaerobic Cleavage of Alkenes. J. Am. Chem. Soc 144, 15437–15442 (2022). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are included in the Supplementary Information.