

Alzheimer’s disease (AD) is the most prevalent form of dementia, i.e., progressive memory loss and profound cognitive dysfunction, resulting in a considerable societal burden. At the neuropathological level, the brains of AD patients exhibit amyloid-β (Aβ) plaques, neurofibrillary tangles, and neuroinflammation (Sala Frigerio and De Strooper, 2016). The growing number of individuals affected with AD underscores the pressing need for the development of effective treatments, and a cure remains elusive. The pathogenesis of AD involves intricate molecular and cellular mechanisms that lead to progressive neurodegeneration and cognitive decline. A central tenet of AD pathogenesis is the amyloid cascade hypothesis, which posits that the accumulation of Aβ peptides plays a pivotal role in disease progression. Aβ derives from the amyloid precursor protein (APP) by BACE1 (β-secretase) and γ-secretase cleavages, and aggregates into plaques that eventually disrupt neuronal function. Concurrently, abnormal phosphorylation of the tau protein leads to the formation of neurofibrillary tangles, contributing to neuronal degeneration. Neuroinflammation, oxidative stress, mitochondrial dysfunction, and synaptic impairment further compound the pathology (Sala Frigerio and De Strooper, 2016). The intricate interplay of these phenomena underscores the challenges in treating AD, necessitating innovative therapeutic approaches to halt or slow disease progression effectively. Recently, monoclonal antibody drugs, like Aducanumab, Lecanemab, and Donanemab, have shown the ability to decelerate memory and cognitive decline in phase III clinical trials of early-stage AD (Boxer and Sperling, 2023). Aducanumab is designed to bind Aβ aggregates in both the oligomeric and fibrillar states rather than amyloid monomers, while Lecanemab has been proposed to target so called Aβ protofibrils. Donanemab is directed against N-terminally modified form of Aβ. These clinical trials collectively suggest that the approach to target Aβ represents an effective strategy for treating AD, particularly in its early stages.
While antibody drugs have gained significant attention for their effects in mitigating memory and cognitive deterioration in early-stage AD, several crucial aspects warrant consideration, including (1) Efficacy and side effects: While the antibodies do play a role in slowing the progression of AD, the magnitude of their efficacy may vary, and the benefits need to be weighed against the probability of side effects (Boxer and Sperling, 2023). This underscores the importance of a thorough assessment of both the benefits and potential drawbacks of these drugs. (2) Blood-brain barrier permeability: The development of compounds for AD faces a notable failure rate, with many tested compounds, particularly antibodies, exhibiting poor permeability across the blood-brain barrier. This limitation poses an obstacle in treating central nervous system disorders and emphasizes the need for innovative approaches to enhance drug delivery. (3) Late-stage AD treatment: AD is typically diagnosed in its later stages, characterized by observable cognitive decline and memory impairment. It remains unclear whether drugs effective in early-stage AD exhibit the same efficacy in later stages. This highlights the necessity for treatment strategies that take into account the evolving nature of the disease. (4) Multifactorial nature of AD: Aβ is recognized as a significant contributor to AD, but it is probably not the sole driving force, and factors like tau pathology, neuro-inflammation, and oxidative stress also play integral roles. Thus, there is a growing acknowledgment of the need to develop interventions that comprehensively target the multifactorial aspects of AD.
Molecular chaperones play a crucial role in preserving cellular protein homeostasis (proteostasis). ATP-independent molecular chaperones, often referred to as “holdases”, maintain (partially) unfolded client proteins in a folding-competent state without necessarily refolding them, leaving the task of refolding or degradation to other cellular systems. Experimental data suggest that these chaperones, including small heat shock proteins (sHsps), can prevent or resolve protein aggregation also in neurodegenerative diseases including AD. For instance, Hsp27 (HspB1) impedes the formation of tau fibrils by engaging in weak interactions with early species during the aggregation process, and mitigates the toxicity of Aβ oligomers by sequestering them and transforming them into larger, non-toxic aggregates (Wentink et al., 2019). αB-crystallin (HspB5) binds to both wildtype Aβ42 fibrils and fibrils formed from the Aβ42E22G (Arctic) mutant and subsequently inhibits amyloid fibril formation (Wentink et al., 2019). Further, Nuclebindin-1 (NUCB1), identified as a novel chaperone-like amyloid-binding protein, demonstrates inhibitory effects on the aggregation of islet amyloid polypeptide (IAPP) linked to type 2 diabetes, α-synuclein associated with Parkinson’s disease, transthyretin V30M mutant related to familial amyloid polyneuropathy, and Aβ42 (Bonito-Oliva et al., 2017). In contrast, ATP-dependent molecular chaperones assist substrates in adopting their native conformation (“foldases”) or prepare them for degradation. DNAJB6, a member of the Hsp40 heat shock protein family, demonstrates a high efficiency in inhibiting Aβ42 amyloid formation (Wentink et al., 2019). The extracellular secretion of Hsp70 demonstrates protection against Aβ42-induced toxicity, effectively mitigating neurotoxicity in adult eyes, reducing cell death, preserving the structural integrity of adult neurons, alleviating locomotor dysfunction, and extending lifespan. Additionally, engineered Hsp70 chaperones prove effective in preventing Aβ42-induced memory impairments in a Drosophila model (Wentink et al., 2019). While the majority of information about molecular chaperones centers around sHSPs primarily located intracellularly, the discovery of extracellular proteins with ATP-independent molecular chaperone functions likely reflects the need for chaperoning outside the cell. Clusterin, a major extracellular chaperone (Humphreys et al., 1999), has been demonstrated to inhibit Aβ42 aggregation by disrupting the nucleation process rather than affecting the fibril elongation rate. In rat brains, microinjection of Aβ42 oligomers, pre-incubated for 1 hour with extracellular chaperones such as clusterin or α2-macroglobulin, prevents Aβ42-induced learning and memory impairments, reduces Aβ42-induced gliosis and neuronal degeneration, and suppresses oligomer cytotoxicity. Additionally, an intracellular form of clusterin has been identified as a tau-interacting protein (Foster et al., 2019). Furthermore, intraventricular infusion of clusterin has been shown to ameliorate cognition and reduce pathology in the Tg6799 model of AD (Qi et al., 2018), while peripheral administration of human recombinant clusterin was able to modulate brain Aβ levels in APP23 mice (de Retana et al., 2019). Interestingly, disrupted proteostasis and persistent unfolded protein response activity are identified in AD, and notably, chemical chaperone treatment successfully restores proteostasis in a mouse model of AD, leading to improved cognition and reduced pathology (Hafycz et al., 2023). While these findings underscore the potential of chaperones for the multi-targeted treatment of AD, challenges may exist in their ability to cross the blood-brain barrier. Further, as Aβ accumulates extracellularly in senile plaques and tau accumulates intracellularly in tangles in AD brain, further research is needed to optimize therapeutic strategies for molecular chaperones in AD.
In contrast to classical molecular chaperones, the BRICHOS domain, consisting of approximately 100 amino acid residues, stands out as an anti-amyloid molecular chaperone. BRICHOS is present in proproteins encompassing an amyloid-forming region, in which the BRICHOS domain is thought to prevent the amyloidogenic region from forming aggregates (Leppert et al., 2023). The term “BRICHOS” is derived from the initially identified proproteins containing this domain, namely Bri2, chondromodulin-1, and prosurfactant protein C. Under physiological conditions, BRICHOS supposedly facilitates the proper folding of the amyloid-prone region within the respective proprotein, and is then proteolytically released (Sánchez-Pulido et al., 2002; Chen et al., 2022). Recent research has shown that the recombinant BRICHOS domain is a versatile anti-amyloid molecular chaperone with a broad substrate spectrum, encompassing also “alien” client peptides (i.e., not parts of the BRICHOS proproteins) associated with various human amyloid diseases as well as bacterial functional amyloid-forming peptides and proteins, including AD-relevant Aβ42 and Aβ40, as well as Huntington’s disease-associated huntingtin peptide, Parkinson’s disease-linked α-synuclein, aortic amyloid-forming peptide medin, functional amyloid-forming protein C, and the de novo-designed peptide β17 (Figure 1). For instance, BRICHOS binds to amyloid fibrils of Aβ42 and IAPP, effectively reduce their cellular toxicity and interact with the smallest emerging toxic Aβ oligomers. BRICHOS also binds to amyloid fibrils associated with Huntington’s disease and α-synuclein linked to Parkinson’s disease. Intriguingly, BRICHOS efficiently reduces, and can rescue, Aβ neurotoxicity in mouse hippocampal slice preparations (Leppert et al., 2023). BRICHOS passes the blood-brain barrier in wild-type mice and AD mouse models, allowing intravenous administration (Leppert et al., 2023). The preventive and therapeutic effects of BRICHOS against amyloid fibril formation, toxicity, and AD have undergone rigorous testing (Figure 1; Leppert et al., 2023). In a Drosophila melanogaster fly model expressing Aβ42, brain-expressed BRICHOS gave a reduction in Aβ42 deposition, improved lifespan, enhanced locomotor activity, and rescued pathological eye phenotypes. Co-expression of BRICHOS resulted in a notable shift in intracellular distribution of Aβ42 in the Drosophila mushroom body. In an AD mouse model overexpressing mutant APP and presenilin 1, coexpression of BRICHOS led to reduced Aβ levels and aggregation without impacting APP processing. Mice coexpressing BRICHOS exhibited improved memory and reduced neuroinflammation compared to AβPP/presenilin 1 control animals that overexpressed green fluorescent protein. Recently, in an APP knockin (APPNL-F) AD mouse model, which harbors the Swedish (KM670/671NL) and Beyreuther/Iberian (I716F) mutations and develops Aβ plaque pathology, astrogliosis, and microgliosis starting from an age of about 9–12 months, repeated intravenous BRICHOS treatment from age of 19 months for 2 months, reduced plaque burden and gliosis in the cortex. With a similar administration approach, BRICHOS was applied to a 3-month APPNL-G-F knockin AD mouse model, which in addition carries the Arctic mutation (E693G) mutation and develops AD-like pathology features from an age of about 2–4 months because of the more aggressive fibril formation of Arctic Aβ. The treatment with BRICHOS improved recognition and working memory reduced Aβ plaque deposition, and decreased activation of astrocytes and microglia (Manchanda et al., 2023). Recombinant human BRICHOS has also been administrated to 10 months old APPNL-G-F knockin mice, i.e., at a stage where AD pathology is established since long, which led to reduced Aβ amyloid burden, mitigated astro- and microgliosis, and altered AD-relevant gene expression. Taken together, the anti-amyloid chaperone domain BRICHOS emerges as a promising candidate for multi-targeted treatment of AD, which warrants further investigation through clinical trials.
Figure 1.
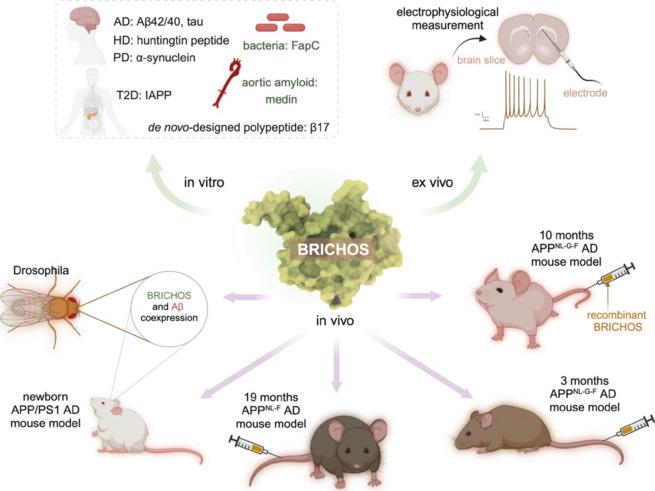
Activities and applications of the anti-amyloid chaperone domain BRICHOS.
The BRICHOS domain has been evaluated extensively, including (1) in vitro regarding activity against amyloid fibril formation of different amyloidogenic peptides/proteins (top left); (2) ex vivo, as the ability to prevent or rescue amyloid-induced neurotoxicity assessed by electrophysiological measurements on mouse brain slice preparations (top right); and (3) in vivo in different animal models, including fruit flies and AD mouse models at various ages (bottom panel). Specifically, the BRICHOS domain interacts with various amyloid-forming peptides and proteins, encompassing AD-relevant Aβ42, Aβ40, and tau (unpublished data), as well as HD-associated huntingtin peptide, PD-linked α-synuclein, aortic amyloid-forming peptide medin, FapC, and the de novo-designed peptide β17. Notably, BRICHOS efficiently reduces and rescues Aβ neurotoxicity in mouse hippocampal slice preparations, specifically through preventing a decline in γ-oscillations. The preventive and therapeutic effects of BRICHOS have further been rigorously examined across diverse animal models. This includes coexpression experiments with Aβ42 in the Drosophila fruit fly and the APP/PS1 AD mouse model from birth, along with intravenous administration of BRICHOS in the APPNL-F knockin AD mouse model from the age of 19 months, while the pathology onset of these AD mice occurs around 15 months. BRICHOS have also been administrated intravenously in the 3-month and 10-month (manuscript submitted) APPNL-G-F knockin AD mouse model, which exhibits AD-like pathology features from an age of approximately 2–4 months. BRICHOS consistently demonstrates its ability to reduce Aβ amyloid burden, mitigate neuroinflammation, and enhance recognition and working memory. These compelling outcomes position BRICHOS as a highly promising candidate for the treatment of AD. The central cartoon represents BRICHOS tertiary structure as predicted by Alphafold (Leppert et al., 2023). The syringe stands for the injection of recombinant BRICHOS proteins through the tail vein. Created with BioRender.com. AD: Alzheimer’s disease; APP: amyloid precursor protein; Aβ: amyloid-β; FapC: functional amyloid-forming protein C; HD: Huntington’s disease; IAPP: islet amyloid polypeptide; PD: Parkinson’s disease; PS1: presenilin 1; T2D: type 2 diabetes.
In summary, the two primary hallmarks of AD are senile plaques and neurofibrillary tangles, stemming from the aggregation of Aβ and tau proteins, respectively. While monoclonal antibody drugs were specifically designed to target Aβ, molecular chaperones, particularly the anti-amyloid chaperone domain BRICHOS, exhibit abilities to interfere with various amyloid-forming peptides and proteins, including Aβ and tau. Notably, BRICHOS has demonstrated superior efficacy in preventing the formation of toxic Aβ oligomers compared to the monoclonal antibody aducanumab and mechainism underlying antibodies and molecular chaperone (-like) proteins discussed in (Abelein and Johansson, 2023). Further research efforts are crucial to optimize approaches for effective AD treatment.
This work was supported by grants from the Alzheimer’s Association Research Grant (to GC), Olle Engkvists Stiftelse (to GC), the Petrus and Augusta Hedlunds Stiftelse (to GC), Åke Wibergs stiftelse (to GC), the Swedish Alzheimer foundation (to GC), the Åhlén Stiftelsens (to GC), Karolinska Institutet Research Foundation Grant (to GC), the Stiftelsen för Gamla Tjänarinnor (to GC), the Stiftelsen Sigurd och Elsa Goljes Minne (to GC), the Loo and Hans Osterman Foundation (to GC), Geriatric Diseases Foundation at Karolinska Institutet (to GC), the Gun and Bertil Stohne’s Foundation (to GC), the Magnus Bergvall Foundation (to GC).
Footnotes
C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- Abelein A, Johansson J. Amyloid inhibition by molecular chaperones can be translated to Alzheimer’s pathology. RSC Med Chem. 2023;14:848–857. doi: 10.1039/d3md00040k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonito-Oliva A, Barbash S, Sakmar TP, Graham WV. Nucleobindin 1 binds to multiple types of pre-fibrillar amyloid and inhibits fibrillization. Sci Rep. 2017;7:42880. doi: 10.1038/srep42880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer AL, Sperling R. Accelerating Alzheimer’s therapeutic development: the past and future of clinical trials. Cell. 2023;186:4757–4772. doi: 10.1016/j.cell.2023.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Andrade-Talavera Y, Zhong X, Hassan S, Biverstål H, Poska H, Abelein A, Leppert A, Kronqvist N, Rising A, Hebert H, Koeck PJB, Fisahn A, Johansson J. Abilities of the BRICHOS domain to prevent neurotoxicity and fibril formation are dependent on a highly conserved Asp residue. RSC Chem Biol. 2022;3:1342–1358. doi: 10.1039/d2cb00187j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Retana SF, Marazuela P, Solé M, Colell G, Bonaterra A, Sánchez-Quesada JL, Montaner J, Maspoch D, Cano-Sarabia M, Hernández-Guillamon M. Peripheral administration of human recombinant ApoJ/clusterin modulates brain beta-amyloid levels in APP23 mice. Alzheimers Res Ther. 2019;11:42. doi: 10.1186/s13195-019-0498-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster EM, Dangla-Valls A, Lovestone S, Ribe EM, Buckley NJ. Clusterin in Alzheimer’s disease: mechanisms, genetics, and lessons from other pathologies. Front Neurosci. 2019;13:164. doi: 10.3389/fnins.2019.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafycz JM, Strus E, Naidoo NN. Early and late chaperone intervention therapy boosts XBP1s and ADAM10, restores proteostasis, and rescues learning in Alzheimer’s disease mice. bioRxiv. 2023 doi: 10.1101/2023.05.23.541973. [Google Scholar]
- Humphreys DT, Carver JA, Easterbrook-Smith SB, Wilson MR. Clusterin has chaperone-like activity similar to that of small heat shock proteins. J Biol Chem. 1999;274:6875–6881. doi: 10.1074/jbc.274.11.6875. [DOI] [PubMed] [Google Scholar]
- Leppert A, Poska H, Landreh M, Abelein A, Chen G, Johansson J. A new kid in the folding funnel: molecular chaperone activities of the BRICHOS domain. Protein Sci. 2023;32:e4645. doi: 10.1002/pro.4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchanda S, Galan-Acosta L, Abelein A, Tambaro S, Chen G, Nilsson P, Johansson J. Intravenous treatment with a molecular chaperone designed against β-amyloid toxicity improves Alzheimer’s disease pathology in mouse models. Mol Ther. 2023;31:487–502. doi: 10.1016/j.ymthe.2022.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi XM, Wang C, Chu XK, Li G, Ma JF. Intraventricular infusion of clusterin ameliorated cognition and pathology in Tg6799 model of Alzheimer’s disease. BMC Neurosci. 2018;19:2. doi: 10.1186/s12868-018-0402-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala Frigerio C, De Strooper B. Alzheimer’s disease mechanisms and emerging roads to novel therapeutics. Annu Rev Neurosci. 2016;39:57–79. doi: 10.1146/annurev-neuro-070815-014015. [DOI] [PubMed] [Google Scholar]
- Sánchez-Pulido L, Devos D, Valencia A. BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci. 2002;27:329–332. doi: 10.1016/s0968-0004(02)02134-5. [DOI] [PubMed] [Google Scholar]
- Wentink A, Nussbaum-Krammer C, Bukau B. Modulation of amyloid states by molecular chaperones. Cold Spring Harb Perspect Biol. 2019;11:a033969. doi: 10.1101/cshperspect.a033969. [DOI] [PMC free article] [PubMed] [Google Scholar]