

Abstract
Fragile X Syndrome (FXS) is the most prevalent monogenetic form of intellectual disability and autism. Recently, dysregulation of insulin signaling (IS) and aberrations in mitochondrial function have emerged as robust, evolutionarily conserved components of FXS pathophysiology. However, the mechanisms by which altered IS and mitochondrial dysfunction impact behavior in the context of FXS remain elusive. Here, we show that normalization of IS improves mitochondrial volume and function in flies that lack expression of dfmr1, the Drosophila homolog of the causal gene of FXS in humans. Further, we demonstrate that dysregulation of IS underlies diminished expression of the mitochondrial master regulator PGC-1α/Spargel in dfmr1 mutant flies. These results are behaviorally relevant, as we show that pan-neuronal augmentation of PGC-1α/Spargel improves circadian behavior in dfmr1 mutants. Notably, we also show that modulation of PGC-1α/Spargel expression in wild-type flies phenocopies the dfmr1 mutant circadian defect. Taken together, the results presented herein provide a mechanistic link between mitochondrial function and circadian behavior both in FXS pathogenesis as well as more broadly at the interface between metabolism and behavioral output.
Fragile X Syndrome (FXS) is the most common monogenetic cause of intellectual disability and autism1,2. Affected individuals experience a variety of difficulties that generally preclude their ability to care for themselves and create many challenges for patients and caretakers alike3–8. Currently, there is no cure for FXS. Rather, a better understanding of FXS pathogenesis is necessary to identify effective treatments that improve the capabilities and quality of life of FXS patients.
At the molecular level, FXS is caused by loss of function of the FMR1 gene9–12. The protein product of FMR1, termed FMRP, is an RNA binding protein. While FMRP was initially defined as a translational repressor, advancements in cell-type specific transcriptomic and proteomic approaches have enabled us to learn that FMRP can also promote the expression of many of its targets through multiple modes of gene regulation13–19.
Since the etiology of FXS is well characterized, researchers have been able to generate valuable preclinical disease models3,20,21. With the advent of a wealth of genetic tools, Drosophila melanogaster has emerged as a highly tractable model for the study of FXS22–26. The Drosophila genome encodes a single gene, dfmr1, that is the sole ortholog of the FMR protein family27. The product of dfmr1, termed dFMRP, shares both sequence identity and biochemical properties with those of its mammalian ortholog27. Importantly, flies that harbor loss-of-function mutations in the dfmr1 gene recapitulate many characteristics of FXS. Specifically, dfmr1 mutant flies have neuroanatomical defects as well as deficits in memory, social behavior, and circadian rhythms22–24,28–31.
Studies in preclinical models of FXS have uncovered signaling molecules and pathways that are dysregulated in the disease state. Previously, we reported that several components of the insulin signaling (IS) pathway are elevated in the brains of dfmr1 mutants32. These changes included increased levels of the major insulin-like peptide, elevated phosphoinositide 3-kinase (PI3K) activity, and accumulation of phosphorylated Akt at the plasma membrane32. Aberrant IS appears to be directly linked to behavioral and cognitive function, as genetic manipulations that reduced IS were sufficient to increase circadian rhythmicity and rescue memory deficits in our Drosophila FXS model32. Dysregulation of IS has also been identified as an evolutionarily conserved feature of FXS pathogenesis in murine and patient derived cell models32–40. Moreover, a multitude of genetic and pharmacological approaches that normalize insulin signaling are sufficient to restore behavior and cognition in mammalian FXS models32–39,41. However, the mechanism by which altered IS impinges on behavioral and cognitive function in FXS remains elusive.
To better understand the mechanism by which dysregulation of IS impacts behavioral and cognitive outputs in the dfmr1 mutants, we conducted an unbiased metabolomics analysis. Paradoxically, our metabolic studies revealed reduced levels of carbohydrate and lipid metabolites in the dfmr1 mutants despite increased brain IS; hyperphagia; and normal body size and activity levels42. We also found a robust decrease in the redox ratio of the mitochondrial cofactor nicotinate adenine dinucleotide (NAD+/NADH) and qualitative defects in mitochondrial ultrastructure42. In aggregate, these robust metabolic defects suggest that mitochondrial function is impaired in our Drosophila model of FXS.
The notion that mitochondrial dysfunction underlies behavioral and cognitive impairments in preclinical models of FXS is particularly compelling in light of recent studies that have implicated mitochondrial defects in the pathogenesis of intellectual disability related syndromes and autism43–45. While the central nervous system (CNS) represents 2% of total body weight, it consumes around 20% of inspired oxygen at rest43. This high oxidative demand renders the CNS particularly sensitive to changes in mitochondrial metabolism43. Proper mitochondrial function is especially critical for the establishment of neuronal connectivity, neurogenesis, and synaptic plasticity43,46.
In this study, we characterize alterations of mitochondrial morphology and function in dfmr1 mutant flies and demonstrate that genetic reduction of IS is sufficient to alleviate these mitochondrial defects. Together, these findings suggest that restoration of mitochondrial function is a mechanism by which normalization of IS improves behavior and cognition in a preclinical model of FXS. Moreover, we present evidence that elevated IS underlies diminished expression of the mitochondrial master regulator, PGC-1α, in the heads of dfmr1 mutants. We report that pan-neuronal augmentation of PGC-1α in dfmr1 mutants is sufficient to restore circadian behavior. Beyond their translational relevance for the FXS field, our results indicate a novel role of PGC-1α in the regulation of circadian rhythmicity and thereby provide a mechanism by which metabolism and mitochondrial function inform behavioral output.
Results
Mitochondrial volume and function are diminished in the absence of dFMRP
Prompted by our previous discovery that mitochondria in the indirect flight muscle of dfmr1 mutants appeared to be qualitatively smaller and irregularly shaped, we wanted to quantitatively examine mitochondrial morphology in behaviorally relevant tissues. To do so, we leveraged a genetically encoded chimeric GFP construct (UAS-mitoGFP) that allowed us to label mitochondria in specific subtypes of cells with the binary Gal4/UAS system47. We were particularly interested in visualizing mitochondria in the insulin producing cells (IPCs) of the brain, because we previously found that constitutive expression of the UAS-dfmr1 transgene under the control of a dilp2-Gal4 driver that is specific to the 14 IPCs of the brain was sufficient to restore normal circadian behavior in dfmr1 mutants32. Therefore, we expressed the UAS-mitoGFP transgene in conjunction with the dilp2-Gal4 driver and used confocal microscopy to resolve GFP-labeled mitochondria in the IPC processes that extend ventrally through the brain. We observed that mitochondria in the IPCs of dfmr1 mutants were shorter than controls, with decreased length and aspect ratio relative to iso31Bw- wild-type controls (Fig. 1A–D). Further, the average volume of individual mitochondria and total mitochondrial volume was decreased in the brains of dfmr1 mutants compared to controls (Fig. 1E, F). These findings match recent studies that revealed small mitochondria with decreased aspect ratios in the brains of Fmr1−/y mice during development and synaptic maturation48,49.
Fig. 1 |. Mitochondrial volume and function are compromised in the absence of dFMRP.
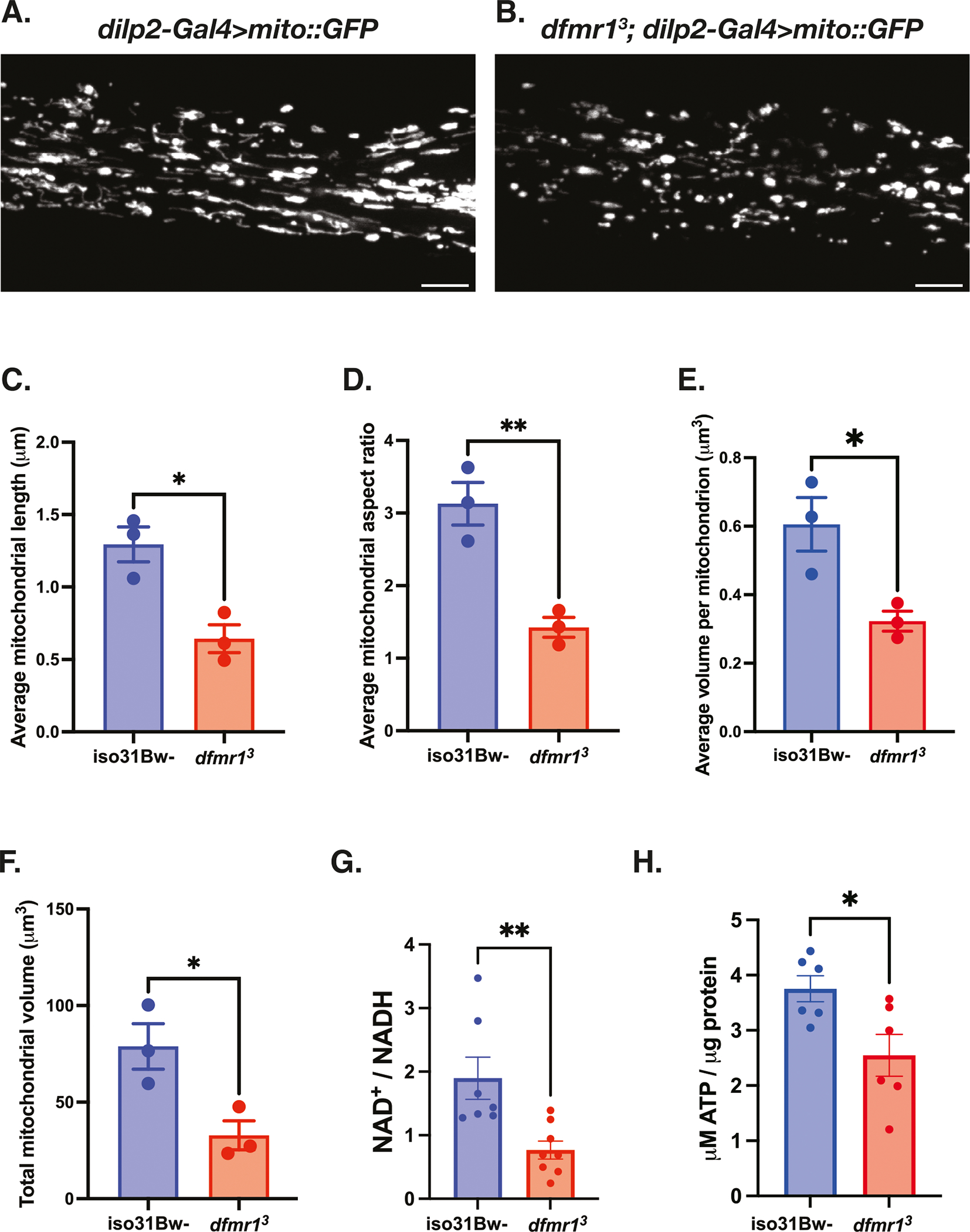
A–F Mitochondria in the insulin producing cells (IPCs) of the brain were labeled by expressing a genetically encoded UAS-mitoGFP construct in conjunction with the dilp2-Gal4 driver. Representative maximum-intensity projections of GFP-labeled mitochondria in the IPC processes of (A) iso31Bw- wild type and (B) dfmr1 mutant flies. Scale bars: 5 μm. Images are oriented with the dorsal side on the left and the ventral side on the right. Quantification of (C) average mitochondrial length, (D) average mitochondrial aspect ratio, (E) average volume per mitochondrion, and (F) average total mitochondrial volume per brain. Sample number (N) per genotype = 3 brains. Unpaired t-tests indicated that the average length, aspect ratio, volume per mitochondrion, and total mitochondrial volume per brain were all significantly reduced in dfmr1 mutants compared to iso31Bw- wild type controls. Values represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01. G Quantification of the NAD+/NADH ratio. Each sample contained 10 fly bodies. Sample number (N) per genotype: iso31Bw- = 7, dfmr1 = 8. An unpaired t-test indicated that the NAD+/NADH ratio was significantly diminished in dfmr1 mutants compared to iso31Bw- wild type controls (p = 0.0059). Values represent mean ± SEM. H Quantification of ATP levels relative to protein content. Each sample contained 5 fly bodies. Sample number (N) per genotype = 6. An unpaired t-test showed that ATP levels were significantly decreased in dfmr1 mutants compared to iso31Bw- wild type controls (p = 0.0224). Values represent mean ± SEM.
Considering that mitochondrial network morphology and bioenergetic capacity are intimately intertwined, we next measured ATP levels in dfmr1 mutants and wild type conspecifics as a direct physiologic readout of mitochondrial function. Consistent with the observed defects in mitochondrial morphology, we found that ATP levels were diminished in the dfmr1 mutant flies compared to wild-type controls (Fig. 1H). Given that the ability of mitochondria to generate ATP is dependent on the redox state of the cofactor NAD+, the diminution of ATP levels that we observed corresponds with our previous discovery42, which we have confirmed herein, that the NAD+/NADH ratio is significantly decreased in dfmr1 mutants (Fig. 1G). Our findings are also consistent with several reports of diminished cytosolic ATP levels in murine models of FXS48–51. Collectively, it appears that the mitochondria of dfmr1 mutant flies are characterized by aberrant mitochondrial network morphology, decreased ATP levels, and a diminished NAD+/NADH ratio.
Genetic reduction of IS ameliorates mitochondrial volume and function in dfmr1 mutants
Once we established that the dfmr1 mutants exhibit several robust hallmarks of mitochondrial dysfunction, we then sought to better understand the contribution of mitochondrial dysfunction to FXS pathophysiology. For these experiments, we introduced one copy of a null allele of the dilp2 gene, which encodes the most abundant Drosophila insulin-like peptide, into the dfmr1 mutant background. We were particularly interested in reduction of dilp2 gene dosage because we have previously demonstrated that this precise genetic manipulation restores behavioral and cognitive function in the dfmr1 mutants32. As an initial indication of mitochondrial health, we conducted TEM experiments on longitudinal sections of isolated thoraces to visualize the impact of genetic reduction of insulin signaling on mitochondrial ultrastructure in dfmr1 mutant flies as well as wild-type controls. We selected the thorax for TEM analysis because this tissue has a distinctive structure of mitochondria aligned along myofibrils to support the high energy demand of flight52. Encouragingly, we observed that in contrast to the small, irregularly shaped mitochondria present in micrographs from dfmr1 mutants, elimination of one copy of the dilp2 gene in dfmr1 mutants restored mitochondrial ultrastructure (Supplementary Fig. 1).
To quantitatively assess the impact of genetic reduction of IS on the mitochondrial network of dfmr1 mutants, we generated dilp2/+, dfmr1 double mutant flies in which mitochondria in the IPCs were labeled with GFP (dilp2-Gal4 > UAS-mitoGFP). Congruent with our TEM findings, we observed that the average length and volume per mitochondrion in the IPCs of dilp2/+, dfmr1 double mutant flies was significantly increased relative to dfmr1 single mutants (Fig. 2A–C). Additionally, the total mitochondrial volume of dilp2/+, dfmr1 double mutants was indistinguishable from that of iso31Bw- control flies (Fig. 2D). Taken together, the results of our TEM and confocal microscopy studies clearly demonstrate that genetic reduction of IS ameliorates brain and peripheral mitochondrial network morphology.
Fig. 2 |. Genetic reduction of IS augments mitochondrial volume and function in dfmr1 mutant flies.
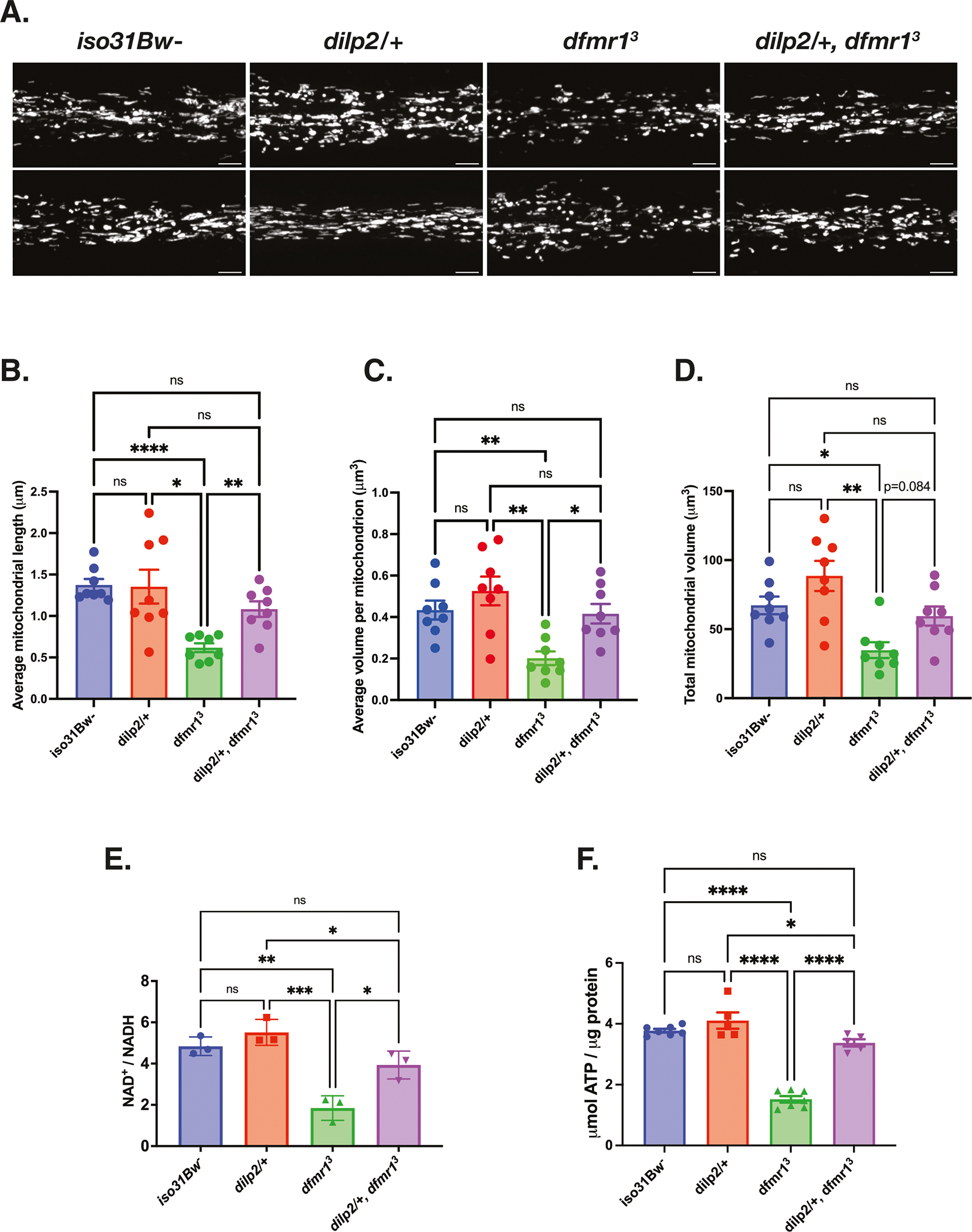
A–D Mitochondria in the insulin producing cells (IPCs) of the brain were labeled by expressing a genetically encoded UAS-mitoGFP construct in conjunction with the dilp2-Gal4 driver. A Two representative maximum-intensity projections of GFP-labeled mitochondria in the IPC processes of iso31Bw- wild type, dilp2/+ heterozygous mutant, dfmr1 homozygous mutant, and dilp2/+, dfmr1 double mutant flies. Scale bars: 5 μm. Images are oriented with the dorsal side on the left and the ventral side on the right. Sample number (N) per genotype = 8 brains. Brown-Forsythe and Welch ANOVA with Dunnet’s T3 multiple comparisons test revealed that genetic reduction of insulin signaling significantly improved (B) average mitochondrial length and (C) average volume per mitochondrion. D Total mitochondrial volume was not significantly increased in dilp2/+, dfmr1 double mutants, but showed a trend towards improvement (p = 0.084). Values represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p < 0.0001. (E) Quantification of the NAD+/NADH ratio.. Each sample contained 10 fly bodies. Sample number (N) per genotype = 3. One-way ANOVAs revealed a significant group effect for the NAD+/NADH ratio (p = 0.0003). Post hoc Tukey tests indicated that while dfmr1 mutant flies had a significantly lower NAD+/NADH ratio than their iso31Bw- and dilp2/+ heterozygous mutant conspecifics, dilp2/+,dfmr1 double mutant flies had a significantly improved NAD+/NADH ratio. Values represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001. F Quantification of ATP levels relative to protein content. Each sample contained 5 fly bodies. Sample number (N) per genotype: iso31Bw- = 6, dilp2/+ = 5, dfmr1 = 6, dilp2/+, dfmr1 = 5. One-way ANOVAs revealed a significant group effect for ATP levels (p < 0.0001). Post hoc Tukey tests indicated that while dfmr1 mutant flies had a significantly lower ATP levels than their iso31Bw- and dilp2/+ mutant conspecifics, dilp2/+,dfmr1 double mutant flies had a significant boost in ATP levels. Values represent mean ± SEM. *p ≤ 0.05, ****p ≤ 0.0001.
Given that normalization of IS substantially improved mitochondrial ultrastructure and network morphology in the dfmr1 mutant flies, we postulated that constitutive elimination of one allele of the dilp2 gene would also ameliorate mitochondrial function in dfmr1 mutants. As predicted, we found that dfmr1 mutants that carried one null allele of the dilp2 gene had a significantly higher whole-body NAD+/NADH ratio and ATP levels than their dfmr1 mutant counterparts (Fig. 2E, F). The rescue of mitochondrial defects by a manipulation that restores circadian rhythmicity and memory in dfmr1 mutant flies suggests that these processes are mechanistically linked. Thus, our results indicate that restoration of mitochondrial function is a potential mechanism by which normalization of IS improves behavior and cognition in the Drosophila model of FXS.
Elevated IS inhibits mitochondrial function by repression of Spargel/ PGC-1α expression in dfmr1 mutants
One intriguing candidate that integrates cellular energy metabolism with mitochondrial biomass and function is PGC-1α expression. Diminished PGC-1α expression is a compelling explanation for the bioenergetic defects in FXS because the PGC-1 family of proteins are transcriptional coactivators that strongly induce mitochondrial biogenesis and function53. Alignment of the Drosophila Spargel (Srl) protein with its three mammalian homologs revealed a high degree of sequence identity, particularly with respect to functional domains53. Similar to its mammalian counterpart, Srl is a transcriptional coactivator that has been shown to modulate mitochondrial biogenesis and energy metabolism53,54. Thus, we can leverage the Drosophila model system to circumvent the functional redundancy present in mammalian models and identify possible roles of PGC-1α/Srl in FXS pathogenesis. Remarkably, flies with loss-of-function mutations in Srl have several phenotypic commonalities with dfmr1 mutants including, similar deficits in energy stores; fragmented mitochondria; locomotor impairment; decreased female fecundity; and shortened lifespan25,42,54–56. The robust phenotypic overlap between dfmr1 and Srl loss-of-function suggests that these genes act in a common pathway to modulate mitochondrial function, behavior, and cognition.
To measure Srl protein levels in the heads of dfmr1 mutant and wild-type flies, we first validated that the mouse monoclonal antibody that specifically recognizes endogenous forms of PGC-1α cross-reacts with the Drosophila Srl protein (Supplementary Fig. 2). As predicted by the convergence of loss-of-function phenotypes and the expected effects of known cellular signaling defects, we observed that Srl protein levels are diminished in the heads of dfmr1 mutants compared to wild-type controls (Fig. 3A, B, Supplementary Fig. 3). This finding is congruent with a previous report that PGC-1α transcript levels are significantly down-regulated in an unbiased transcriptomic analysis of hippocampal neurons in Fmr1−/y mice57.
Fig. 3 |. Diminished Spargel expression in the heads of dfmr1 mutants is restored by normalization of IS.
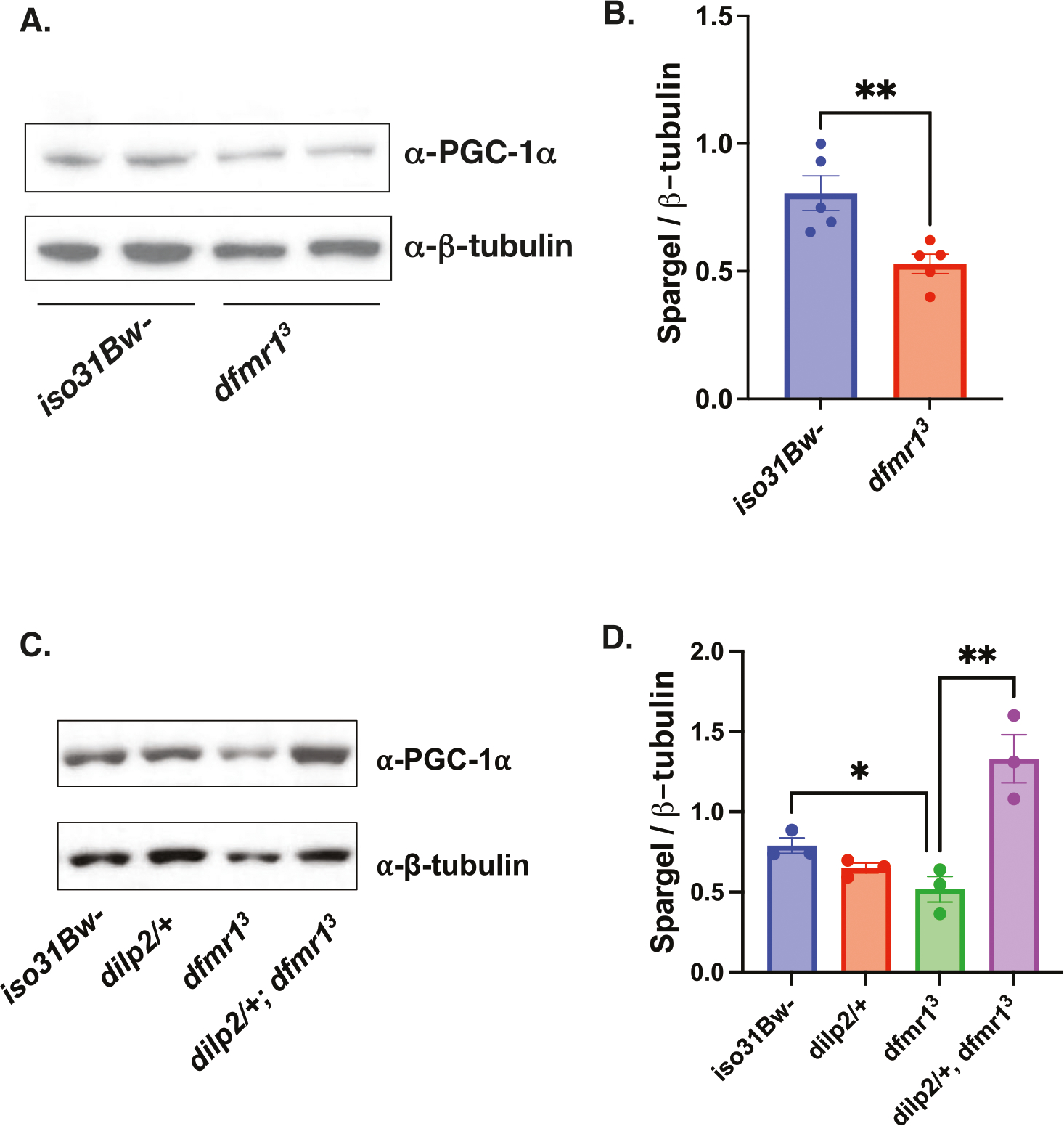
A Western analysis of Spargel expression in extracts from iso31Bw- and dfmr1 mutant fly heads. An antibody to PGC-1α was used to detect Spargel expression (top). β-Tubulin was used as a loading control (bottom). See Supplementary Fig. 3 for full blots. B Quantification of the intensity of Spargel relative to β-Tubulin. An unpaired t-test revealed that Spargel levels are diminished in dfmr1 mutant heads compared to iso31Bw- controls (p = 0.0074). Sample number (N) per genotype = 5. Each sample contained 10 fly heads. Values represent mean ± SEM. **p ≤ 0.01. C Western analysis of Spargel expression in extracts from iso31Bw- wild type, dilp2/+ heterozygous mutant, dfmr1 homozygous mutant, and dilp2/+, dfmr1 double mutant fly heads. See Supplemental Fig. 4 for full blot. D An unpaired t-test indicated that dilp2/+, dfmr1 double mutant flies have higher Spargel expression than dfmr1 single mutants (p = 0.0088). Sample number (N) per genotype = 3. Each sample contained 10 fly heads. Values represent mean ± SEM. **p ≤ 0.01.
To determine whether elevated IS contributes to decreased Srl expression, we measured Srl levels in the heads of dfmr1 mutants that carried a null allele of dilp2. We observed that genetic reduction of IS restored Srl expression in the heads of dfmr1 mutants to wild-type levels (Fig. 3C, D, Supplementary Fig. 4).As anotherindependent manipulation of IS, we acutely administered a highly specific PI3K inhibitor, LY294002, for 5 days post-eclosion and quantified Srl expression in the heads of dfmr1 mutants and wild-type controls. Similar to our findings with genetic reduction of IS, we observed that pharmacologic reduction of IS boosts Srl expression in the heads dfmr1 mutants (Supplementary Fig. 5).
Pan-neuronal augmentation of Spargel expression rescues circadian rhythmicity in the dfmr1 mutants
Next, we queried whether elevation of Srl expression is sufficient to improve behavioral defects in the dfmr1 mutant flies. To genetically boost Srl expression in dfmr1 mutants, we used enhancer promoter (EP) induced overexpression of the Srl locus in the CNS. We obtained flies that contain a UAS element upstream of the endogenous Srl gene, termed SrlEY05931, and made recombinants in the dfmr1 mutant background. The resultant progeny were crossed to dfmr1 mutants that expressed the elav-Gal4 pan-neuronal driver to generate dfmr1 mutant flies that expressed the SrlEY05931 transgene in conjunction with the elav-Gal4 pan-neuronal driver. We then utilized the well-established Drosophila Activity Monitor (DAM) System to record the locomotor activity of these flies in free-running conditions.
Visual inspection of free-running rest:activity rhythms revealed that dfmr1 mutant flies that contained the elav-Gal4 driver in conjunction with the SrlEY05931 transgene had more rhythmic patterns of locomotor activity compared to dfmr1 mutants that contained the elav-Gal4 driver alone (Fig. 4A). To quantify circadian rhythmicity, we calculated fast Fourier transform (FFT) values for flies of each genotype. Conventionally, a fly with an FFT value that is above 0.04 is considered strongly rhythmic. In contrast, flies with FFT values between 0.01 and 0.04 are considered weakly rhythmic and those with FFT values below 0.01 are designated as arrhythmic. Congruent with their actogram appearance, on average, flies that pan-neuronally expressed the SrlEY05931 transgene had higher FFT values than their dfmr1 mutant counterparts that contained the elav-Gal4 driver or SrlEY05931 transgene alone (Fig. 4B). When we stratified the dataset into categories based on the percentage of rhythmic flies, we observed that in contrast to dfmr1 mutants that expressed only the elav-Gal4 driver or SrlEY05931 construct, all dfmr1 mutant flies that contained both the elav-Gal4 driver and the SrlEY05931 construct were strongly rhythmic (Fig. 4D).
Fig. 4 |. Pan-neuronal augmentation of Spargel rescues circadian rhythmicity in dfmr1 mutant flies.
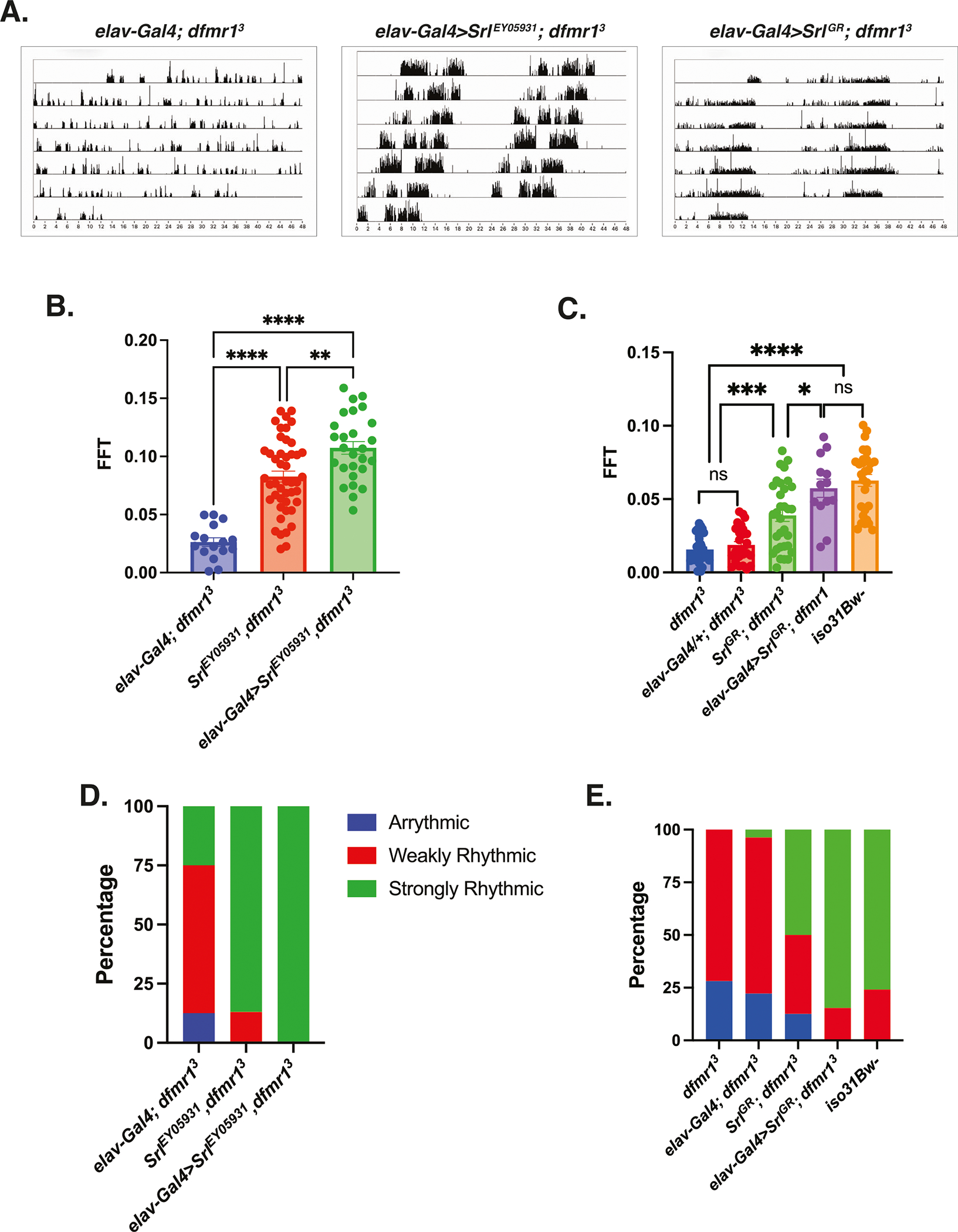
A–E Circadian behavior was evaluated by comparison of actogram appearance, the average fast Fourier transform (FFT) values, and percentage of rhythmic flies to ascertain whether augmentation of Spargel expression improves circadian behavior in dfmr1 mutant flies. Sample number (N) per genotype: (elav-Gal4; dfmr1) = 16–27; (SrlEY05931,dfmr1) = 46, (elav-Gal4>SrlEY05931,dfmr1) = 27, dfmr1 = 32, (SrlGR; dfmr1) = 32, (elav-Gal4>SrlGR; dfmr1) = 13, iso31Bw- = 29. A Representative actograms from flies of the genotypes indicated. In contrast to the free-running rest:activity rhythms of dfmr1 mutants that contain the elav-Gal4 transgene alone, dfmr1 mutants that contain the SrlEY05931 or SrlGR transcript in conjunction with the elav-Gal4 driver display consolidated, rhythmic behavior. B, C The average fast Fourier transform (FFT) was calculated for genetic combinations. One-way ANOVAs revealed a significant group effect for FFT values (p < 0.0001). Post hoc Tukey tests indicated that dfmr1 mutant flies that expressed the (B) SrlEY05931 or (C) SrlGR construct in conjunction with the elav-Gal4 driver had significantly higher FFT values compared to dfmr1 mutant flies that contained either transgene alone. Values represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. D, E The percentages of strongly rhythmic (FFT ≥ 0.04), weakly rhythmic (0.04 > FFT ≥ 0.01), and arrhythmic (FFT < 0.01) flies of each genotype are shown in green, red, and blue, respectively. The percentage of strongly rhythmic flies is increased in dfmr1 mutants that contained the (D) SrlEY05931 or (E) SrlGR construct in conjunction with the elav-Gal4 driver compared to either construct alone.
To independently corroborate our findings, we obtained a UAS-Spargel genomic rescue fragment (SrlGR) to elevate Srl expression in dfmr1 mutants. Similar to what we observed with the SrlEY05931 construct, pan-neuronal expression of SrlGR in dfmr1 mutants also resulted in more consolidated, rest: activity patterns than dfmr1 mutant conspecifics that contained only the elav-Gal4 driver alone (Fig. 4A). Moreover, pan-neuronal expression of SrlGR in dfmr1 mutants increased the average FFT value and percentage of strongly rhythmic flies relative to dfmr1 mutant flies that contained either transgene alone (Fig. 4C, E). The ability of two distinct genetic manipulations that augment Srl expression to ameliorate circadian behavior in dfmr1 mutants strongly suggests that elevation of Srl expression is sufficient to restore circadian rhythmicity. Notably, we did observe a significant increase in circadian rhythmicity for dfmr1 mutant flies that contained the SrlEY05931 or SrlGR transgene alone compared to dfmr1 mutants that contained only the elav-Gal4 driver (Fig. 4). We believe that this is likely due to leaky expression of the UAS constructs whereby Srl expression is moderately increased, albeit not as highly as when the elav-Gal4 driver is present. Therefore, the rescue of circadian rhythmicity by elevation of Srl expression in dfmr1 mutants appears to occur in a dose-dependent manner.
Consistent with the known role of Srl as a modulator of mitochondrial biogenesis, in follow up TEM experiments, we observed that pan-neuronal expression of SrlGR in dfmr1 mutants substantially improved thoracic mitochondrial ultrastructure compared to dfmr1 mutants that contained the elav-Gal4 driver alone (Supplementary Fig. 6). The ability of pan-neuronal expression of SrlGR to restore mitochondrial ultrastructure in the thorax of dfmr1 mutants suggests that this rescue occurs in a cell-non-autonomous manner. Much like our circadian results, the rescue of mitochondrial ultrastructure by augmentation of Srl expression appears to be dose-dependent, as mitochondrial ultrastructure was modestly improved in dfmr1 mutants that caried the SrlGR construct alone. Given that proper mitochondrial ultrastructure is essential to support energy production, we believe that elevation of Srl expression likely ameliorates mitochondrial function in dfmr1 mutants and thereby restores circadian behavior.
Genetic manipulation of Spargel expression phenocopies the dfmr1 mutant circadian defect
As further confirmation that Srl is indeed a major contributor to FXS pathophysiology, particularly with respect to circadian behavior, we next tested whether Srl loss-of-function in a wild-type genetic background phenocopies dfmr1 loss-of-function. For these experiments, we used the daughterless-Gal4 (da-Gal4) driver to ubiquitously express a UAS-SrlRNAi transgene in an otherwise wild-type genetic background. When we assayed circadian behavior, we observed that in contrast to the empty vector control (UAS-CtrlRNAi), ubiquitous knockdown of Srl expression in wild-type flies significantly disrupted free-running locomotor activity rhythms, diminished FFT values, and increased the percentage of flies that were arrhythmic or weakly rhythmic (Fig. 5A, B, E). Considering that pan-neuronal elevation of Srl expression was sufficient to rescue the circadian behavioral defect in dfmr1 mutants, we hypothesized that Srl knockdown exclusively in the CNS would also mimic the dfmr1 mutant phenotype. As expected, we observed that pan-neuronal knockdown of Srl expression in the wild-type genetic background recapitulated the circadian phenotype that we observed with the da-Gal4 driver (Fig. 5A, C, F).
Fig. 5 |. Modulation of Spargel expression in wild-type flies phenocopies the circadian defect observed in dfmr1 mutant flies.
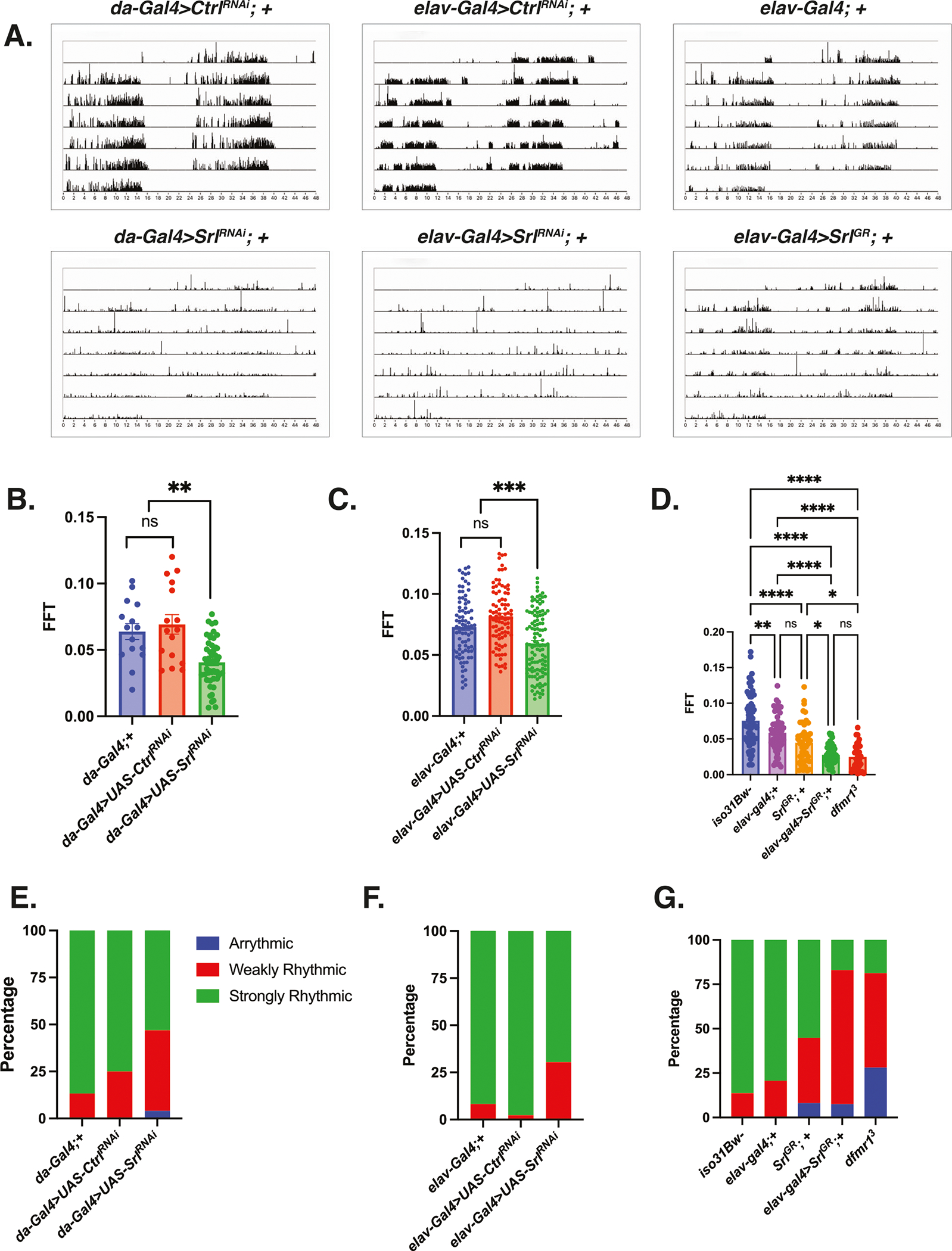
A–G Circadian behavior was evaluated by comparison of actogram appearance, the average fast Fourier transform (FFT) values, and percentage of rhythmic flies for genetic combinations to ascertain whether overexpression or knockdown of Spargel impacts circadian behavior in wild type flies. Sample number (N) per genotype: (da-Gal4;+) = 15, (da-Gal4 > UAS-CtrlRNAi) = 16, (da-Gal4 > UAS-SrlRNAi) = 49, (elav-Gal4;+) = 58–84, (elav-Gal4 > UAS-CtrlRNAi) = 88, (elav-Gal4 > UAS-SrlRNAi) = 115, (SrlGR; +) = 49, (elav-Gal4> SrlGR) = 53, iso31Bw- = 73, dfmr1 = 32. A Representative actograms from flies of the genotypes indicated. While control flies in the top row (da-Gal4 > UAS-CtrlRNAi), (elav-Gal4 > UAS-CtrlRNAi), and (elav-Gal4 > +) display consolidated, rhythmic rest: activity patterns, both Spargel loss-of-function (da-Gal4 or elav-Gal4 > UAS-SrlRNAi) and gain-of-function (elav-Gal4>SrlGR) disrupt rest:activity patterns. B–D The average fast Fourier transform (FFT) was calculated for genetic combinations. One-way ANOVAs revealed a significant group effect for FFT values (p < 0.0001). Post hoc Tukey tests indicated that wild-type flies that expressed a UAS-SrlRNAi construct in conjunction with (B) da-Gal4 or (C) elav-Gal4 had significantly lower FFT values compared to wild-type flies that contained either Gal4 driver alone or a UAS-CtrlRNAi fragment. Similarly, (D) wild-type flies that pan-neuronally expressed a SrlGR over-expression construct (elav-Gal4>SrlGR) had significantly lower FFT values than wild type flies that contained the elav-Gal4 or SrlGR transgene alone. There was no significant difference between the FFT values of the elav-Gal4> SrlGR flies and those of dfmr1 mutants. Values represent mean ± SEM. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001. E–G The percentages of strongly rhythmic (FFT ≥ 0.04), weakly rhythmic (0.04 > FFT ≥ 0.01), and arrhythmic (FFT < 0.01) flies of each genotype are shown in green, red, and blue, respectively. The percentage of weakly rhythmic and arrhythmic flies is increased in genetic combinations that result in (E, F) Spargel loss-of-function (da-Gal4 or elav-Gal4 > UAS-SrlRNAi) and (G) gain-of-function (elav-Gal4> SrlGR) compared to flies that contain each transgene alone.
The ability of Srl loss-of-function to phenocopy the circadian defect that we observe in the dfmr1 mutants prompted us to conduct the inverse experiment to query whether pan-neuronal over-expression of Srl also impacts circadian behavior. Indeed, we observed that targeted overexpression of the SrlGR in the CNS alone results in significantly altered rest:activity rhythms, diminished FFT values, and an increased percentage of weakly rhythmic and arrhythmic flies (Fig. 5A, D, G). While it is counterintuitive that both loss and gain of function manipulations of Srl expression have the same impact on circadian behavior, we postulate that the maintenance of circadian rhythmicity is particularly sensitive to Srl dosage. In this way, both too much and too little Srl expression are detrimental to circadian behavior. Taking the loss- and gain- of function experiments together, it is clear that we have identified a novel role for Srl as a modulator of circadian behavior. This finding is particularly exciting because it positions Srl at the intersection of metabolism, mitochondrial function, and behavior.
Discussion
While the causal gene was first cloned and identified nearly three decades ago, there is still much to be learned about the molecular underpinnings of FXS. Our incomplete understanding of the precise mechanisms that underlie FXS pathogenesis has precluded the identification of effective therapeutic approaches to ameliorate the quality of life of affected individuals. Despite the advancement of several compounds to clinical trials, findings at the bench have had mixed success at the bedside. Rather, it has become evident that the FXS field would benefit from the optimization of preclinical clinical strategies to identify therapeutic candidates and paradigms to predict their clinical efficacy.
One promising route to identify treatments for FXS has been the identification of conserved signaling pathway defects, such as IS, that can be targeted therapeutically26. Our studies in the Drosophila model of FXS pioneered the contribution of dysregulated IS to FXS pathogenesis32. In subsequent follow-up studies, we encountered a metabolic paradox whereby dfmr1 mutant flies have decreased energy stores and are more sensitive to starvation despite elevated IS in the brain and hyperphagia42. Pursuit of an explanation for our discordant metabolic findings lead to our discovery that mitochondria in the dfmr1 mutants have ultrastructural and functional defects42. Studies by other groups further support the notion that mitochondrial dysfunction is a robust, evolutionarily conserved component of FXS pathophysiology49,51,58.
The studies described herein integrate biochemical and physiologic methodologies with behavioral testing to provide a more comprehensive understanding of the contribution of mitochondrial dysfunction to FXS pathogenesis. We demonstrate that mitochondrial volume is diminished in the IPCs of dfmr1 mutants and that ATP levels are decreased in dfmr1 mutants compared to wild-type conspecifics. Strikingly, we report that genetic reduction of IS is sufficient to correct morphologic defects in the mitochondria of dfmr1 mutants as well as augment the NAD+/NADH ratio and ATP levels. As such, our findings indicate that the defect in IS that modulates behavior and cognition in preclinical models of FXS is mechanistically linked to the observed mitochondrial defects. Further, we present evidence that dysregulated IS down-regulates the expression of the mitochondrial master regulator PGC-1α/Srl in the heads of dfmr1 mutants. This result led to the exciting discovery that pan-neuronal augmentation of Srl expression is sufficient to mitigate circadian behavior in dfmr1 mutants. Moreover, genetic manipulation of Srl expression in wild-type flies is sufficient to disrupt circadian behavior. Thus, in addition to implicating Srl in FXS pathogenesis, our findings reveal novel role of Srl in the regulation of circadian behavior. Future studies will be necessary to elucidate the precise mechanisms by which dysregulated IS impinges on PGC-1α expression in the brain. Additionally, it would be interesting to determine the extent to which mitochondrial phenotypes are present in female dfmr1 mutants.
Notably, diminished PGC-1α expression has been reported in other syndromic forms of intellectual disability (ID) and autism, including Down Syndrome and Rett Syndrome43,59. The convergence of multiple distinct genetic forms of ID and autism on decreased PGC-1α expression suggests shared mechanistic underpinnings. Therefore, in a broader sense, further exploration of the precise mechanisms by which PGC-1α expression is compromised in ID and autism will expand our understanding of the contribution of reduced PGC-1α expression to behavioral pathology. Such studies have the potential to uncover novel treatments for FXS and other syndromic forms of ID and autism.
Methods
Fly genetics and husbandry
Fly strains that contain the dfmr13 allele are described in Dockendorff et al. 22. Fly strains that contain the dilp2 mutation were obtained from the Bloomington Stock Center (stock number 30881). The elav-Gal4 and daughterless-Gal4 drivers were derived from the Bloomington Stock Center (stock numbers 8765 and 95282). Flies that contain the UAS-mitoGFP construct were obtained from the Bloomington Stock Center (stock number 8442). The UAS-SrlRNAi and UAS-CtrlRNAi empty vector control lines were obtained from the Vienna Drosophila Resource Center (stock numbers 330271 and 60200). Stocks that contain the SrlEY05931 allele were Bloomington Stock Center (stock number 20009). Stocks that contained the UAS-SpargelGR were obtained from Hugo Stocker. All fly strains were outcrossed to w1118 (iso31Bw-) flies as described in Monyak et al.32. Flies were cultured on a standard cornmeal-molasses medium and maintained in the presence of stringent 12 h light: 12 h dark (LD) cycles at 25 °C.
Mitochondrial morphology in IPCs
We expressed the UAS-mitoGFP reagent described in47 under the control of the dilp2-Gal4 driver to directly visualize mitochondria in the IPCs. Brains were dissected from 4 to 8 days old male flies in 1x PBS and fixed with 4% paraformaldehyde (PFA) in PBS for 20 min at RT. Brains were then washed 3 times for 10 min in PBS-T (PBS + 0.2% Triton-X 100) and mounted on slides in glycerol +2% N-Propyl gallate. Images were acquired with a Perkin Elmer UltraView Vox spinning disk confocal on a Nikon Eclipse Ti Microscope. Experiments were imaged on either a Hamamatsu EMCCD C9100–50 camera or a Hamamatsu CMOS ORCA-Fusion (C11440–20UP). The EMCCD camera was used with Volocity Software [Quorom Technologies/PerkinElmer] and the CMOS camera was used with VisiView (Visitron). Z-stacks encompassing mitoGFP signal were collected at 200-nm step-size. Images were analyzed using ImageJ (NIH). For each brain, a 50 μm long region encompassing the IPCs was cropped for analysis. Mitochondria were manually measured to determine their number and size. Mitochondrial signal was converted to a binary mask using the Pixel classification module of Ilastik, a machine-learning based image segmentation program60. We then used the 3D objects counter function in ImageJ to identify mitochondria and measure the volume per mitochondrion and total mitochondrial volume.
NAD+/NADH quantification
The concentrations of nicotinamide nucleotides were measured using the NAD+/NADH Quantification Colorimetric Kit (Abcam, Waltham, MA) as described in61. Briefly, adult male flies aged 5 to 7 days were collected on dry ice. Fly heads were removed prior to homogenization and the decapitated fly bodies were pooled in groups of 10. The samples were homogenized in 400 μL of the NADH/NAD Extraction Buffer supplied in the kit and the homogenate was centrifuged at 18,407 × g for 5 min at 25 °C to remove debris. The cycling reaction was carried out as per the manufacturer’s instructions for 2 h and the nicotinamide nucleotide concentrations were determined in duplicate. The protein concentration of each sample was measured with the Pierce® BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). The concentration of nicotinamide nucleotide contained in each sample was then normalized to its respective protein content.
ATP measurement
ATP levels were measured using the ATP Determination Kit (Molecular Probes, Eugene, Oregon) as described in62. Adult male flies aged 5 to 7 days were collected on dry ice. Fly heads were removed prior to homogenization and the decapitated fly bodies were pooled in groups of five. The samples were homogenized in 100 μL of ATP homogenization buffer [6 M guanidine HCL, 100 mM Tris (pH7.8), 4 mM EDTA]. An aliquot was boiled at 100 °C for 5 min and centrifuged for 3 min at maximum speed at 4 °C.10 μL of the supernatant was transferred to a 1.5 mL microfuge tube and diluted 1:10 with 90 μL dilution buffer [25 mM Tris (pH 7.8, 100 μM) EDTA]. Subsequently, 10 μL of the diluted supernatant was transferred to another 1.5 ml tube that contained 740 μL of dilution buffer such that the final dilution was 1:750. The diluted homogenate was centrifuged at 20,000 × g for 3 min. To prepare a series of low-concentration ATP standards, the 5 mM ATP stock solution provided with the kit was diluted with ddH2O to reach final concentrations of 0, 0.01, 0.05, 0.1, 0.5, and 1 μM. Next, 10 μL of each standard or sample was transferred in duplicate to a white, opaque 96 well plate. The reaction was started by adding 100 μL of the reaction mixture with a multichannel pipette. Luminescence was measured three times sequentially using a plate reader and the values were averaged. The protein concentration of each sample was measured with the Pierce® BCA Protein Assay Kit (Thermo Scientific, Rockford, IL). The concentration of ATP contained in each sample was then normalized to its respective protein content.
Transmission electron microscopy
Tissues for electron microscopic examination were prepared as described in42. Thoraces were fixed with 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium cacodylate buffer, pH7.4, overnight at 4 °C. After subsequent buffer washes, the samples were post-fixed in 2.0% osmium tetroxide for 1 h at room temperature, and then washed again in buffer followed by dH2O. After dehydration through a graded ethanol series, the tissue was infiltrated and embedded in EMbed-812 (Electron Microscopy Sciences, Fort Washington, PA). Thin sections were stained with lead citrate and examined with a JEOL 1010 electron microscope fitted with a Hamamatsu digital camera and AMT Advantage image capture software.
Western analysis
Adult male flies aged 5–7 days were snap frozen and heads were separated into groups of 10 on dry ice. Protein extracts were prepared from the heads using extraction buffer 20 mM Hepes (pH 7.5), 100 mM KCl, 5% glycerol, 100 μM NA3VO4, 10 mM EDTA, 0.1% Triton X, 1 mM DTT, and (Phosphatase/protease inhibitors) 4X LDS (Invitrogen) and 10X Reducing Agent (Invitrogen) were added before samples were incubated at 70 °C for 10 min to denature and reduce. Samples were separated on a 4–12% Bis-Tris gel (Invitrogen) and transferred to a PVDF membrane (Immobilon-P, Millipore, St. Louis, MO). Enhanced chemiluminescence (SuperSignal West Pico; Thermo Scientific) was used for antibody detection. The following primary antibodies were used: anti-PGC-1α 1:1000 (Millipore, St. Louis, MO), anti-β-tubulin E7 1:20,000 (Developmental Studies Hybridoma Bank, Iowa City, IA). The relative intensity of PGC-1α to β-tubulin was determined using ImageJ (NIH).
Circadian behavior
Circadian analysis was performed as described in Dockendorff et al., 2002. Male flies were collected 0–3 days post eclosion and entrained to a stringent 12 h light: 12 h dark cycle for three days at 25 °C. Flies were then placed in individual tubes containing 5% sucrose, 2% agar, and loaded into monitors (Trikinetics, DAM2 system, Waltham, MA) that were placed in an incubator in constant darkness at 25 °C. The activity of these flies, as indicated by beam breaks, was measured from days 2 to 6. Data were collected in 5 min bins and analyzed with ClockLab software (Actimetrics, Wilmette, IL). Rhythmicity was determined by a FFT analysis.
Statistics
The Prism software package (GraphPad Software, v9.5.1) was used to generate graphs and perform statistical analyses. Unpaired t-tests were used to evaluate pairwise comparisons. Multiple comparisons were investigated using one-way analysis of variance (ANOVA) with post hoc Tukey tests. Multiple comparisons were investigated for datasets with variable standard deviations using Brown-Forsythe and Welch ANOVA with Dunnett’s T3 multiple comparisons test.
Supplementary Material
Acknowledgements
We thank Biao Zuo and Inna Martynyuk at the Electron Microscopy Resource Laboratory at the University of Pennsylvania for assistance with our electron microscopy experiments; Amita Sehgal, PhD at the University of Pennsylvania for use of her circadian facility; and William Haury at the University of Pennsylvania for help with Drosophila husbandry. This work was supported by the National Institutes of Health (MH126257, NS129903) and the Autism Spectrum Program of Excellence at the University of Pennsylvania (https://aspe.med.upenn.edu).
Footnotes
Competing interests
The authors declare no competing interests.
Reprints and permissions information is available at http://www.nature.com/reprints
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s44324-024-00004-7.
Data Availability
The authors declare that the data supporting the findings of this study are available within this manuscript and the accompanying supplementary information files.
References
- 1.Kaufmann WE et al. Autism spectrum disorder in fragile X syndrome: communication, social interaction, and specific behaviors. Am. J. Med, Genet. Part A 129A, 225–234 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Kaufmann WE et al. Autism spectrum disorder in fragile X syndrome: cooccurring conditions and current treatment. Pediatrics 139, S194–s206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardoni B & Mandel J-L Advances in understanding of fragile X pathogenesis and FMRP function, and in identification of X linked mental retardation genes. Curr. Opin. Genet. Dev. 12, 284–293 (2002). [DOI] [PubMed] [Google Scholar]
- 4.de Vries BB et al. The fragile X syndrome. J. Med. Genet. 35, 579–589 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gould EL et al. Melatonin profiles and sleep characteristics in boys with fragile X syndrome: a preliminary study. Am. J. Med. Genet. 95, 307–315 (2000). [PubMed] [Google Scholar]
- 6.Kidd SA et al. Fragile X syndrome: a review of associated medical problems. Pediatrics 134, 995–1005 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Bailey DB, Raspa M & Olmsted MG Using a parent survey to advance knowledge about the nature and consequences of fragile X syndrome. Am. J. Intellect. Dev. Disabil. 115, 447–460 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Hagerman RJ, Rivera SM & Hagerman PJ The fragile X family of disorders: a model for autism and targeted treatments. Curr. Pediatr. Rev. 4, 40–52 (2008). [Google Scholar]
- 9.Verkerk AJMH et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914 (1991). [DOI] [PubMed] [Google Scholar]
- 10.Bell MV et al. Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell 64, 861–866 (1991). [DOI] [PubMed] [Google Scholar]
- 11.Oberlé I et al. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102 (1991). [DOI] [PubMed] [Google Scholar]
- 12.Sutcliffe JS et al. DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1, 397–400 (1992). [DOI] [PubMed] [Google Scholar]
- 13.Richter JD & Zhao X The molecular biology of FMRP: new insights into fragile X syndrome. Nat. Rev. Neurosci. 22, 209–222 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siomi H et al. The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74, 291–298 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Ashley CT et al. FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262, 563–566 (1993). [DOI] [PubMed] [Google Scholar]
- 16.Li Z et al. The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 29, 2276–2283 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Laggerbauer B et al. Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10, 329–338 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Darnell JC et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell 146, 247–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darnell JC & Klann E The translation of translational control by FMRP: therapeutic targets for FXS. Nat. Neurosci. 16, 1530–1536 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bakker CE & Oostra BA Understanding fragile X syndrome: insights from animal models. Cytogenet. Genome Res. 100, 111–123 (2003). [DOI] [PubMed] [Google Scholar]
- 21.Bhogal B & Jongens TA Fragile X syndrome and model organisms: identifying potential routes of therapeutic intervention. Dis. Models Mech. 3, 693–700 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dockendorff TC et al. Drosophila lacking dfmr1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34, 973–984 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Inoue S et al. A role for the Drosophila fragile X-related gene in circadian output. Curr. Biol. 12, 1331–1335 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Morales J et al. Drosophila fragile X protein, DFXR, regulates neuronal morphology and function in the brain. Neuron 34, 961–972 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Zhang YQ et al. Drosophila fragile X-related gene regulates the MAP1B homolog Futsch to control synaptic structure and function. Cell 107, 591–603 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Weisz ED, Monyak RE & Jongens TA Deciphering discord: how drosophila research has enhanced our understanding of the importance of FMRP in different spatial and temporal contexts. Exp. Neurol. 274, 14–24 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Wan L et al. Characterization of dFMR1, a Drosophila melanogaster homolog of the fragile X mental retardation protein. Mol. Cell Biol. 20, 8536–8547 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolduc FV et al. Excess protein synthesis in Drosophila fragile X mutants impairs long-term memory. Nat. Neurosci. 11, 1143–1145 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McBride SM et al. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of fragile X syndrome. Neuron 45, 753–764 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Russo A & DiAntonio A Wnd/DLK is a critical target of FMRP responsible for neurodevelopmental and behavior defects in the drosophila model of fragile X syndrome. Cell Rep. 28, 2581–2593.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tauber JM, Vanlandingham PA & Zhang B Elevated levels of the vesicular monoamine transporter and a novel repetitive behavior in the Drosophila model of fragile X syndrome. PLoS One 6, e27100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monyak RE et al. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol. Psychiatry 22, 1140–1148 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gross C et al. Increased expression of the PI3K enhancer PIKE mediates deficits in synaptic plasticity and behavior in fragile X syndrome. Cell Rep. 11, 727–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gross C et al. Excess phosphoinositide 3-kinase subunit synthesis and activity as a novel therapeutic target in fragile X syndrome. J. Neurosci. 30, 10624–10638 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gross C & Bassell GJ Excess protein synthesis in FXS patient lymphoblastoid cells can be rescued with a p110beta-selective inhibitor. Mol. Med. 18, 336–345 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gross C & Bassell GJ Neuron-specific regulation of class I PI3K catalytic subunits and their dysfunction in brain disorders. Front. Mol. Neurosci. 7, 12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gross C et al. Selective role of the catalytic PI3K subunit p110beta in impaired higher order cognition in fragile X syndrome. Cell Rep. 11, 681–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luhur A et al. Opposing post-transcriptional control of InR by FMRP and LIN-28 adjusts stem cell-based tissue growth. Cell Rep. 21, 2671–2677 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Callan MA et al. Fragile X protein is required for inhibition of insulin signaling and regulates glial-dependent neuroblast reactivation in the developing brain. Brain Res. 1462, 151–161 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Raj N et al. Cell-type-specific profiling of human cellular models of fragile X syndrome reveal PI3K-dependent defects in translation and neurogenesis. Cell Rep. 35, 108991 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monyak RE et al. Insulin signaling misregulation underlies circadian and cognitive deficits in a Drosophila fragile X model. Mol. Psychiatry 90, 1–9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weisz ED et al. Loss of Drosophila FMRP leads to alterations in energy metabolism and mitochondrial function. Hum. Mol. Genet. 27, e560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valenti D et al. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: an overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 46, 202–217 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Wallace DC Bioenergetic origins of complexity and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 1–16 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wallace DC, Fan W & Procaccio V Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. 5, 297–348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mattson MP, Gleichmann M & Cheng A Mitochondria in neuroplasticity and neurological disorders. Neuron 60, 748–766 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rizzuto R et al. Double labelling of subcellular structures with organelle-targeted GFP mutants in vivo. Curr. Biol. 6, 183–188 (1996). [DOI] [PubMed] [Google Scholar]
- 48.Licznerski P et al. ATP Synthase c-Subunit leak causes aberrant cellular metabolism in Fragile X Syndrome. Cell 182, 1170–1185.e9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen M et al. Reduced mitochondrial fusion and Huntingtin levels contribute to impaired dendritic maturation and behavioral deficits in Fmr1-mutant mice. Nat. Neurosci. 22, 386–400 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu B et al. Regulatory discrimination of mRNAs by FMRP controls mouse adult neural stem cell differentiation. Proc. Natl. Acad. Sci. USA 115, E11397–E11405 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.D’Antoni S et al. Aberrant mitochondrial bioenergetics in the cerebral cortex of the Fmr1 knockout mouse model of fragile X syndrome. Biol. Chem. 401, 497–503 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Clark IE, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 441. 1162–1166 (2006). [DOI] [PubMed] [Google Scholar]
- 53.Merzetti EM & Staveley BE spargel, the PGC-1α homologue, in models of Parkinson disease in Drosophila melanogaster. BMC Neurosci. 16, 70 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mukherjee S et al. Emerging functional similarities and divergences between Drosophila Spargel/dPGC-1 and mammalian PGC-1 protein. Front. Genet. 5, 216 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tiefenböck SK et al. The Drosophila PGC-1 homologue Spargel coordinates mitochondrial activity to insulin signalling. Embo J. 29, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ng CH et al. Genetic or pharmacological activation of the Drosophila PGC-1α ortholog spargel rescues the disease phenotypes of genetic models of Parkinson’s disease. Neurobiol. Aging 55, 33–37 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Sawicka K et al. FMRP has a cell-type-specific role in CA1 pyramidal neurons to regulate autism-related transcripts and circadian memory. eLife 8, e46919 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Griffiths KK et al. Inefficient thermogenic mitochondrial respiration due to futile proton leak in a mouse model of fragile X syndrome. FASEB J. 34, 7404–7426 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Valenti D & Vacca RA Brain mitochondrial bioenergetics in genetic neurodevelopmental disorders: focus on down, rett and fragile X syndromes. Int. J. Mol. Sci. 24, 12488 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berg S et al. ilastik: interactive machine learning for (bio)image analysis. Nat. Methods 16, 1226–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 61.Balan V et al. Life span extension and neuronal cell protection by drosophila nicotinamidase. J. Biol. Chem. 283, 27810–27819 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tennessen JM et al. Methods for studying metabolism in Drosophila. Methods 68, 105–115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within this manuscript and the accompanying supplementary information files.