

Abstract
Background
Menopausal hormone therapy (MHT), a common treatment to relieve symptoms of menopause, is associated with a lower risk of colorectal cancer (CRC). To inform CRC risk prediction and MHT risk-benefit assessment, we aimed to evaluate the joint association of a polygenic risk score (PRS) for CRC and MHT on CRC risk.
Methods
We used data from 28,486 postmenopausal women (11,519 cases and 16,967 controls) of European descent. A PRS based on 141 CRC-associated genetic variants was modeled as a categorical variable in quartiles. Multiplicative interaction between PRS and MHT use was evaluated using logistic regression. Additive interaction was measured using the relative excess risk due to interaction (RERI). 30-year cumulative risks of CRC for 50-year-old women according to MHT use and PRS were calculated.
Results
The reduction in odds ratios by MHT use was larger in women within the highest quartile of PRS compared to that in women within the lowest quartile of PRS (p-value = 2.7 × 10−8). At the highest quartile of PRS, the 30-year CRC risk was statistically significantly lower for women taking any MHT than for women not taking any MHT, 3.7% (3.3%–4.0%) vs 6.1% (5.7%–6.5%) (difference 2.4%, P-value = 1.83 × 10−14); these differences were also statistically significant but smaller in magnitude in the lowest PRS quartile, 1.6% (1.4%–1.8%) vs 2.2% (1.9%–2.4%) (difference 0.6%, P-value = 1.01 × 10−3), indicating 4 times greater reduction in absolute risk associated with any MHT use in the highest compared to the lowest quartile of genetic CRC risk.
Conclusions
MHT use has a greater impact on the reduction of CRC risk for women at higher genetic risk. These findings have implications for the development of risk prediction models for CRC and potentially for the consideration of genetic information in the risk-benefit assessment of MHT use.
Subject terms: Cancer epidemiology, Colorectal cancer, Cancer genetics
Introduction
Colorectal cancer (CRC) is a commonly diagnosed malignancy that ranks third and second in terms of incidence and mortality, respectively, in the world [1]. Genome-wide association studies (GWAS) have identified a large number of genetic risk variants for CRC [2–4]. Aggregating genetic risk variants into a polygenic risk score (PRS) yields a continuous and quantitative measure of the estimated genetic predisposition to a certain disease at the individual level, which could be used to evaluate the impact of particular treatments or lifestyle modifications in individuals with high genetic risk [5].
Menopausal hormone therapy (MHT) is a common and effective treatment for relieving common symptoms of menopause for postmenopausal women, with a rapidly growing multibillion USD global market size [6]. Since the introduction of MHT use in the 1960s, it has been met with very high popularity until the publication of the Women’s Health Initiative (WHI) in 2002, which warned of serious health risks of MHT particularly in relation to breast cancer and cardiovascular disease, resulting in a dramatic decline in MHT use [7, 8]. In the following years, the use of MHT has gradually increased and is expected to further increase as some clinicians have raised the awareness of benefits of MHT potentially outweighing risks for some women’s health based on the women’s individual risk profile [9]. Currently, weighing the benefits and risks for personalized MHT treatment decisions does not take into account of genetic risk; however, it is expected that this would change in the future.
Since the first associations between MHT and CRC were made in the 1980s [10, 11], MHT use has been consistently shown to be associated with a reduced risk of CRC. A meta-analysis including 20 studies reported that both ever-use of estrogen-only MHT (RR: 0.79, 95% CI: 0.69–0.91) and ever-use of combined estrogen-progestogen MHT (RR: 0.74, 95% CI: 0.68–0.81) were associated with a reduced CRC risk [12]. Randomized controlled trial data from the Women’s Health Initiative indicated a lower risk of CRC among women taking estrogen plus progestin and no difference in CRC risk among users of estrogen-only, compared to placebo [13, 14].
Most studies of biological mechanisms have suggested that the protective cellular effect of MHT on CRC is likely to be mediated through nuclear estrogen receptors (i.e., ERα, ERβ) and progesterone receptor, which may involve increasing DNA repair, selectively activating proapoptotic signaling, inhibiting expression of oncogenes, regulating cell cycle progression, changing the miRNA pool and DNA methylation [15]. Nevertheless, these underlying etiologic mechanisms are not fully understood. Further insight into potential biological pathways could be gained by investigating genetic modifiers of CRC risk associated with MHT use. Through a genome-wide association study of gene-environment interaction, we previously identified genetic variants (GRIN2B, DCBLD1) that modified CRC risk associated with MHT use, offering new insights into pathways of CRC carcinogenesis and potential mechanisms involved [16].
CRC is a complex disease resulting from both genetic predisposition and environmental factors [17]. However, it is not yet known whether a genetic risk profile modifies the effect of MHT on CRC risk, i.e., whether there is an interaction between PRS and MHT. For a disease trait, interaction can be commonly described in two ways: multiplicative and additive. Multiplicative interaction focuses on the comparison of relative risk of an exposure (e.g. MHT) for one subgroup compared to another (e.g. high vs. low PRS). Analysis of multiplicative interaction can be performed directly using logistic regression and is typically considered the relevant scale for informing biological etiology. Additive interaction implies the difference in absolute risk due to exposure between one subgroup and another, and can improve the ability to identify relevant subgroups who may benefit the most from public health intervention, which is often neglected in epidemiologic studies. Finding an additive interaction can help guide public health campaigns aimed at identifying sub-populations in whom a specific intervention can lead to the greatest reduction in numbers of new cases, for example, women with high genetic susceptibility may have a greater benefit of reducing CRC risk with MHT use. Given that different information can be gained from studying different types of interactions, it is recommended to present both additive and multiplicative interaction in practice [18]. We therefore aimed to evaluate the joint associations of MHT and a PRS of 141 single nucleotide polymorphisms (SNPs) identified by previous GWAS with CRC risk and to assess both multiplicative and additive measures of interaction [2–4, 19–37]. Additionally, absolute risks were estimated for informing CRC prevention.
Methods
Study participants
We included studies from North America, Australia, and Europe participating in the multi-centered Colon Cancer Family Registry (CCFR), the Colorectal Transdisciplinary Study (CORECT), and the Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO), all with GWAS data available, as previously described [4, 38, 39]. Study details and descriptions can be found in the supplementary section.
Cases were identified as incident invasive colorectal cancer cases and confirmed by medical records, pathological reports, or death certificate information. For cohort studies, nested case-control sets were assembled via risk-set sampling, while population-based controls were used for case-control studies. Controls were matched with cases on age and enrollment date, where applicable.
All studies were approved by their respective Institutional Review Boards, and all study participants provided informed consent.
Exposure assessment
Information on demographics and environmental risk factors was collected by interview and/or structured questionnaire. We carried out a multi-step data-harmonization procedure at the GECCO coordinating center (Fred Hutchinson Cancer Research Center) as described previously [40–42].
Postmenopausal status was defined by using: (I) menopausal status derived from studies, if available; or (II) self-reported menopausal status, if study-derived was not available; or (III) age >55, if neither study-derived nor self-report were available. MHT use was considered using three variables, i.e., any MHT use, estrogen-only use, and combined estrogen-progestogen use at or until the reference date (date of diagnosis for cases, date of interview for controls). Estrogen-only use and combined estrogen-progestogen use were defined to be mutually exclusive, such that for example, combined estrogen-progestogen use excludes the use of estrogen-only or any other MHT at or until the reference time. Non-users of any MHT at or until the reference time were used as the reference group for all three MHT variables. For nested case-control studies from cohorts, the information on MHT use was collected at the enrollment date which was used as reference date. For case-control studies, the information collected on MHT use and duration for cases typically referred to use until diagnosis year or one to two years before diagnosis, depending on the individual studies; controls in case-control studies were similarly requested to provide information about MHT use until the time of recruitment/interview or the past 1–2 years to be consistent with assessment in cases (Supplementary Table 1).
Genotyping, quality control, and imputation
Details on genotyping, imputation, and quality control have been reported previously [2]. In brief, genotyped SNPs were excluded on the basis of call rate (<98%) or evidence of departure from Hardy-Weinberg equilibrium (HWE) in controls (P < 1 × 10−4). For all studies, all autosomal SNPs were imputed to the Haplotype Reference Consortium r1.1 (2016) reference panel via the Michigan Imputation Server [43] and converted into a binary format for data management and analyses using R package BinaryDosage [44]. Imputed common SNPs were restricted based on a pooled MAF ≥ 1% and imputation accuracy (R2 > 0.8). All analyses were restricted to samples clustering with the Utah residents of Northern and Western European ancestry from the CEU population in principal component analysis.
Derivation of polygenic risk score
The PRS was built based on 141 risk variants identified in previous GWAS of CRC risk (Supplementary Table 2) [2–4, 19–37]. The variant-specific weights were determined by the log-odds ratios estimated from prior studies. PRS was calculated by summing the product of the weight and the number of risk alleles for each risk variant across 141 identified genetic risk variants for all study participants. For the known variants identified by GECCO, CCFR, and CORECT studies, the estimates adjusted for winner’s curse [45] (i.e., a statistical effect resulting in the exaggeration of SNP-trait association estimates in the discovery study compared to their true association) were used. We employed quartiles of PRS (PRS.Q) as a categorical variable, using the lowest quartile as the reference group.
Statistical analysis
Statistical analyses were conducted centrally on individual-level data. Logistic regression models were used to assess the association of PRS and MHT with CRC risk by odds ratios (ORs) and 95% confidence intervals (CIs) adjusted for age at the reference time, BMI, study, and the first three principal components to account for potential population substructure. P-values for trend in risks associated with quartiles of PRS were estimated by including the ordinal PRS.Q variable as a continuous variable in the regression models and testing coefficients using the Wald test. Heterogeneity P-values were calculated using Cochran’s Q statistics in study-specific meta-analyses [46].
We assessed multiplicative interaction effects of PRS and MHT variables by taking the products of PRS.Q and MHT variables in logistic regression models and obtained P-values using the likelihood ratio test. We assessed additive interaction effects by the relative excess risk due to interaction (RERI), i.e., departure of the joint effect of PRS and MHT variables from the sum of effect estimates for the two variables, and estimated the variance of RERI by the Delta method [47].
We also calculated the 30-year cumulative risk (%) of CRC for 50-year-old women according to MHT use and PRS to estimate the probability of developing CRC over a 30-year interval from age 50 to 80 years [48]. Specifically, we estimated the age-specific relative risks and attributable risks by three subgroups (≤60 years, 61–70 years, and >70 years) and combined these estimates with CRC incidence rates obtained from the SEER Research Data, 13 Registries, Nov 2019 Sub (1992–2017) [49] for White women only to obtain the baseline age-specific CRC hazard rates. We calculated the absolute risk for any given risk profile of MHT use and PRS, accounting for competing risks from non-CRC mortality rates which were obtained from the National Center for Health Statistics (https://seer.cancer.gov/mortality/). The 95% confidence intervals for absolute risk estimates were calculated based on 100 bootstrap samples.
All analyses were performed using SAS, version 9.4 (SAS Institute Inc, Cary, NC), and R, version 2.15.3 (R Foundation for Statistical Computing, Vienna, Austria) software. A two-sided P-value < 0.05 was considered statistically significant.
Results
Study population
The study sample for analysis comprised 28,486 post-menopausal women (11,519 cases and 16,967 controls) with genotype data and information on the use of any MHT, of which 10,027 women (35.2%) indicated the use of any MHT. A total of 7637 women provided information on the use of estrogen-only and 6887 women on combined estrogen-progestogen use. Among these women, 2156 (28.2%) used estrogen-only and 1509 (21.9%) used combined estrogen-progestogen. Detailed descriptive characteristics of the cases and controls are shown in Supplementary Table 3.
Association of MHT or PRS with CRC risk
MHT use was associated with a reduced CRC risk in our pooled analyses. Compared to non-users, the OR for CRC was 0.71 (95% CI: 0.64–0.78, Supplementary Fig. 1) for women using any MHT, 0.65 (95% CI: 0.53–0.79, Supplementary Fig. 2) for women using estrogen-only, and 0.73 (95% CI: 0.59–0.90, Supplementary Fig. 3) for women using combined estrogen-progestogen. The risk reduction of CRC associated with MHT use is consistent in both cohort and case-control studies (Supplementary Figs. 1–3). The risk for CRC increased with higher quartiles of PRS compared to the lowest quartile [ORs, for PRS.Q2: 1.49 (1.43–1.55); PRS.Q3: 1.92 (1.84–2.00); PRS.Q4: 2.87 (2.76–2.99)].
Joint associations of MHT and PRS with CRC risk
There was a pattern of higher CRC risk with higher quartiles of PRS for both users and non-users of MHT, with a significant linear trend across quartiles of PRS (No MHT use, P for trend = 0.015; MHT use, P for trend = 0.002) (Fig. 1). The increased risks of CRC associated with PRS seemed to be similar in non-users and users of MHT within the same PRS.Q, e.g., for the highest vs lowest PRS quartile, ORs were 2.82 (2.57, 3.09) in non-users and 2.43 (2.15, 2.76) in users of any MHT (Table 1). Similar patterns were also observed for the use of estrogen-only (Table 1, Supplementary Fig. 4) and combined estrogen-progestogen (Table 1, Supplementary Fig. 5).
Fig. 1. Effects of polygenic risk score and menopausal hormone therapy on colorectal cancer risk.

PRS.Q the quartiles of polygenic risk score, OR odds ratio, 95%CI 95% confidence interval, CRC colorectal cancer, MHT menopausal hormone therapy, RERI the relative excess risk due to interaction. The regression model was adjusted for age, BMI, study center, and the first three principal components.
Table 1.
Associations of all MHT variables with CRC risk stratified by quartiles of PRS.
| Quartiles of PRS [OR (95%CI)] | |||||||
|---|---|---|---|---|---|---|---|
| PRS.Q1 | PRS.Q2 | PRS.Q3 | PRS.Q4 | Q2 within strata of MHT | Q3 within strata of MHT | Q4 within strata of MHT | |
| No MHT | 1 (ref.) | 1.42 (1.28, 1.56) | 1.94 (1.77, 2.14) | 2.82 (2.57, 3.09) | 1.42 (1.28, 1.56) | 1.94 (1.77, 2.14) | 2.82 (2.57, 3.09) |
| Any MHT | 0.75 (0.66, 0.85) | 1.09 (0.97, 1.22) | 1.40 (1.25, 1.57) | 1.83 (1.64, 2.04) | 1.45 (1.27, 1.65) | 1.87 (1.64, 2.12) | 2.43 (2.15, 2.76) |
| Within strata of PRS.Q | 0.75 (0.66, 0.85) | 0.77 (0.69, 0.86) | 0.72 (0.65, 0.80) | 0.65 (0.59, 0.72) | P for multiplicative interaction = 0.103 | ||
| RERI | −0.08 (−0.25, 0.09) | −0.29 (−0.50, −0.09) | −0.74a (−1.00, −0.48) | ||||
| No MHT | 1 (ref.) | 1.35 (1.14, 1.60) | 1.84 (1.57, 2.17) | 2.35 (2.00, 2.75) | 1.35 (1.14, 1.60) | 1.84 (1.57, 2.17) | 2.35 (2.00, 2.75) |
| E-only | 0.78 (0.62, 0.98) | 0.91 (0.73, 1.13) | 1.20 (0.97, 1.47) | 1.37 (1.12, 1.68) | 1.16 (0.89, 1.52) | 1.53 (1.19, 1.99) | 1.76 (1.37, 2.26) |
| Within strata of PRS.Q | 0.78 (0.62, 0.98) | 0.67 (0.54, 0.83) | 0.65 (0.53, 0.79) | 0.58 (0.48, 0.71) | P for multiplicative interaction = 0.302 | ||
| RERI | −0.22 (−0.53, 0.09) | −0.43 (−0.79, −0.06) | −0.76 (−1.17, −0.34) | ||||
| No MHT | 1 (ref.) | 1.37 (1.16, 1.62) | 1.86 (1.58, 2.19) | 2.35 (2.00, 2.76) | 1.37 (1.16, 1.62) | 1.86 (1.58, 2.19) | 2.35 (2.00, 2.76) |
| E + P | 0.77 (0.59, 1.01) | 1.02 (0.79, 1.31) | 1.37 (1.08, 1.73) | 1.59 (1.27, 2.00) | 1.32 (0.96, 1.83) | 1.77 (1.30, 2.42) | 2.07 (1.53, 2.80) |
| Within strata of PRS.Q | 0.77 (0.59, 1.01) | 0.74 (0.58, 0.95) | 0.73 (0.59, 0.92) | 0.68 (0.55, 0.84) | P for multiplicative interaction = 0.886 | ||
| RERI | −0.12 (−0.48, 0.23) | −0.27 (−0.68, 0.15) | −0.53 (−1.00, −0.07) | ||||
The regression models were adjusted for age, BMI, study center, and the first three principal components. Statistically significant values are indicated in bold.
The cells of columns “PRS.Q” intersecting rows “Any MHT” or “No MHT” present the ORs of CRC for MHT users or non-users with different PRS.Q using non-users with PRS.Q1 as reference. ORs of CRC for women with different PRS.Q to those with PRS.Q1 stratified by the use of MHT are presented in the right three columns “Q within strata of MHT”. ORs of CRC for MHT users to non-users stratified by PRS.Q are presented in the row “Within strata of PRS.Q”.
CRC colorectal cancer, PRS.Q the quartiles of polygenic risk score, OR odds ratio, 95%CI 95% confidence interval, MHT menopausal hormone therapy, E-only estrogen-only therapy, E + P combined estrogen-progestogen therapy, RERI the relative excess risk due to interaction.
aRERI = −0.74, which equals to OR11-OR10-OR01 + 1 = 1.83–2.82–0.75 + 1, means that the protective effect of MHT use is stronger in the highest quartile of PRS with 74% relative risk reduction more compared to that in the lowest quartile of PRS.
The reduction in odds ratio by MHT use was however stronger in women within the highest quartile of PRS [OR = 0.65 (0.59–0.72)] than that in women within the lowest quartile of PRS [0.75 (0.66–0.85)]. Similar patterns were found for joint associations of MHT types (estrogen-only, combined estrogen-progesterone) and PRS. For all three MHT variables, there was no significant multiplicative interaction with PRS (all P-values > 0.05). However, we observed statistically significant additive interactions consistently across all three MHT variables for the highest quartile of genetic risk [RERI: −0.74 (−1.00, −0.48), P-value = 2.7 ×10−8 for any MHT use; RERI: −0.76 (−1.17, −0.34), P-value = 3.8 ×10−4 for estrogen-only use, and RERI: −0.53 (−1.00, −0.07), P-value = 0.025 for combined estrogen-progestogen use] when compared to the risk excess reductions due to MHT use in those at the lowest quartile of PRS (Table 1, Fig. 1, Supplementary Figs. 4 and 5). In other words, the joint effect of MHT use and high genetic susceptibility on CRC risk differed significantly from that expected from the sum of the individual effects.
We have further analyzed the joint association of MHT and PRS with colorectal cancer risk stratified by tumor anatomical sites (colon, rectum, proximal colon, and distal colon), and observed to some extent statistically significant additive interaction between PRS and all three MHT variables for the different tumor sites (Supplementary Tables 4–7). The magnitudes of RERI for quartiles of PRS across all three MHT variables were more pronounced for risk of distal colon (e.g., −0.35 to −1.12 for any MHT use) than proximal colon (e.g., −0.05 to −0.51 for any MHT use).
Absolute risk estimates for CRC by MHT and PRS
The projected 30-year cumulative risks of CRC for 50-year-old women who used any MHT were consistently lower than those for non-users across quartiles of PRS. The difference in 30-year cumulative risk between users of any MHT and non-users increased with higher quartiles of PRS, implying a greater risk reduction effect of MHT for women at higher genetic risk. At the highest quartile of PRS, the 30-year CRC risk was statistically significantly lower for women taking any MHT than for women not taking any MHT, 3.7% (3.3%–4.0%) vs 6.1% (5.7%–6.5%) (difference 2.4%, P-value = 1.83 ×10−14); these differences were also statistically significant but smaller in magnitude in the lowest PRS quartile, 1.6% (1.4%–1.8%) vs 2.2% (1.9%–2.4%) (difference 0.6%, P-value = 1.01 ×10−3) (Table 2, Fig. 2). The reduction in absolute risk associated with any MHT use was thus 4 times greater in the highest versus lowest quartile of genetic risk (2.4% vs 0.6%, Fig. 2). Similar patterns for 30-year cumulative risks of CRC for 50-year-old women according to quartiles of PRS were also found for estrogen-only use and combined estrogen-progestogen use, respectively (Table 2, Supplementary Figs. 6 and 7).
Table 2.
30-year cumulative risk estimates (%) of CRC for 50-year-old women by use of all MHT variables and quartiles of PRS.
| 30-year absolute risk, % (95% CI) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ca/Co, n | PRS.Q1 | Ca/Co, n | PRS.Q2 | Ca/Co, n | PRS.Q3 | Ca/Co, n | PRS.Q4 | |
| No MHT | 1114/2693 | 2.16 (1.94, 2.39) | 1584/2744 | 3.15 (2.86, 3.43) | 2037/2676 | 4.13 (3.82, 4.43) | 2929/2680 | 6.06 (5.66, 6.46) |
| Any MHT | 580/1544 | 1.59 (1.36, 1.82) | 832/1541 | 2.20 (1.93, 2.47) | 1021/1470 | 2.83 (2.51, 3.14) | 1422/1614 | 3.66 (3.29, 4.03) |
| Diff | 0.57 | 0.95 | 1.30 | 2.40 | ||||
| P value | 1.01 × 10−3 | 3.65 × 10−5 | 3.27 × 10−8 | 1.83 × 10−14 | ||||
| No MHT | 447/628 | 2.45 (2.10, 2.80) | 630/661 | 3.04 (2.70, 3.38) | 776/593 | 4.23 (3.81, 4.65) | 1088/658 | 5.32 (4.83, 5.81) |
| E-Only | 157/279 | 1.88 (1.44, 2.32) | 199/317 | 1.99 (1.55, 2.44) | 253/285 | 2.95 (2.28, 3.62) | 322/344 | 3.13 (2.45, 3.80) |
| Diff | 0.57 | 1.05 | 1.28 | 2.19 | ||||
| P value | 7.93 × 10−2 | 2.85 × 10−4 | 6.39 × 10−3 | 7.26 × 10−6 | ||||
| No MHT | 438/618 | 2.34 (2.03, 2.64) | 622/649 | 2.82 (2.45, 3.18) | 760/583 | 4.06 (3.60, 4.53) | 1064/644 | 4.97 (4.54, 5.40) |
| E + P | 100/188 | 1.89 (0.97, 2.81) | 155/207 | 2.22 (1.26, 3.17) | 187/193 | 2.76 (2.00, 3.52) | 256/223 | 4.11 (2.73, 5.49) |
| Diff | 0.45 | 0.60 | 1.30 | 0.86 | ||||
| P value | 0.44 | 0.35 | 4.56 × 10−3 | 0.32 | ||||
CRC colorectal cancer, Ca/Co number of cases patients and controls individuals, PRS.Q the quartiles of polygenic risk score, 95% CI 95% confidence interval, MHT menopausal hormone therapy, E-only estrogen-only therapy, E + P combined estrogen-progestogen therapy, Diff the estimated difference of absolute risks between MHT users and non-users, P an alpha level of 0.05 (two-sided) was considered to be statistically significant for comparing the absolute risks of MHT users to those of non-users.
Fig. 2. The 30-year cumulative risk estimates (%) of CRC for 50-year-old women, according to any MHT use and quartiles of PRS.
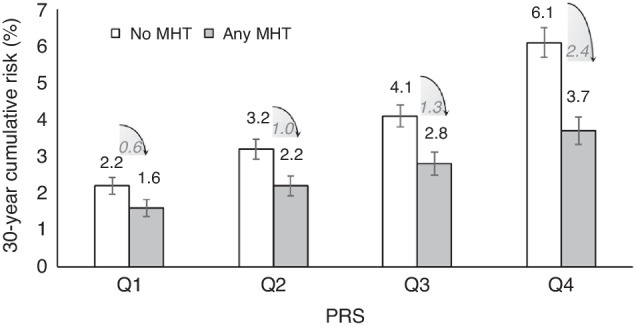
CRC colorectal cancer, PRS.Q the quartiles of polygenic risk score, MHT menopausal hormone therapy.
After stratifying by tumor anatomical sites, we further observed that the reduction in absolute risk associated with MHT use for women with higher genetic risk compared to the lowest quartile of PRS was somewhat greater for distal colon cancer (e.g., 0.22%, 0.30%, and 0.76% for any MHT use) than that for proximal colon cancer (e.g., 0.17%, 0.31%, and 0.62% for any MHT use) (Supplementary Tables 8–11).
Discussion
Based on a large sample size derived from international colorectal cancer consortia, we observed statistically significant modification of MHT associated CRC risk by genetic risk for this disease, which was evidenced by substantial additive interactions between PRS and MHT variables on CRC risk. As such, the reduction in 30-year absolute risk of developing CRC as a result of MHT use was more apparent among 50-year-old women with higher genetic risk profiles, showing that the genetically predetermined increased risk of CRC could be offset to some extent by the use of MHT.
Several previous studies have reported potential association of some genetic modifiers and MHT with CRC risk [40, 50–52]. However, to our knowledge, studies have not investigated associations of aggregated genetic susceptibility with MHT for CRC risk. Although some previous studies reported the joint association of PRS and environmental factors, including diet, lifestyle, and behavior factors, with the risk of CRC [53–61], these studies did not address potential interactions between PRS and MHT. Considering the high use, known risks of use, and the big market value of MHT globally, our study provides new insight on the association between MHT and CRC risk in people with different genetic susceptibilities.
Herein, our study found that MHT has a strong impact on reducing the risk of CRC which may differ by genetic factors, i.e., with increasing genetic susceptibility, women using MHT had a greater reduction in CRC risk compared to non-users. Nevertheless, based on these findings alone we do not simply advocate the use of MHT as a chemoprevention intervention in those with high genetic risk for CRC because of its potential adverse consequences with long-term use of the increased risk of stroke [7] or breast cancer [8]. Instead, our study points to a potential future consideration of genetic risk in evaluating the risk-benefit assessment of MHT use. We do acknowledge that MHT remains widely used and as such, under the model of personalized medicine, it may be possible to use the genetic risk for CRC as input into decisions for or against MHT use when an individual woman is considering using MHT for other reasons such as menopausal symptoms or osteoporosis treatment.
We additionally found some differences in the joint associations of MHT and PRS with CRC risk according to anatomical site of the tumor. When additionally considering women’s PRS, MHT use appeared to have a slightly stronger protective effect across PRS on cancer occurring in the distal colon compared to the proximal colon, and correspondingly slightly stronger protective benefits with increasing PRS in terms of absolute risks. Prior studies indicated that MHT was associated with a stronger reduced cancer risk for the distal colon rather than the proximal colon, without consideration of PRS [62, 63]. The underlying mechanism remains uncertain but may be related to tumor heterogeneity in carcinogenic processes in different sites of the large bowel with different embryonic origins, somatic mutation profile, and microbiomes [64–68]. Further studies are needed to validate the observed tumor site differences and to determine the reasons why the association between MHT and CRC risk is attenuated for the proximal colon.
We investigated the association of PRS and MHT with CRC risk in postmenopausal women by using the largest number of 141 GWAS-identified genetic variants of CRC risk, resulting in a more comprehensive genetic score than any previous study, and with the largest sample size to date. We performed the assessment of both multiplicative and additive interaction, which may provide insight into the mechanisms of disease [18]. It is worth noting that for the gene-environment interaction studies focusing on single SNPs, there is little or no difference between additive and multiplicative interaction due to weak SNP effect size, as commonly observed [69]. However, when PRS is used to capture overall genetic susceptibility, the difference between multiplicative interaction and additive interaction (RERI) may be substantial. In our study, we found statistically significant additive interactions but not significant multiplicative interactions for MHT use. This observation indicates the importance of assessing interaction on both additive and multiplicative scales, where an additive interaction from a public health perspective is a desirable scale for risk stratification because it identifies sub-populations in whom a specific intervention can prevent the largest number of cancer occurrences; taking both genetic and MHT factors into account could be meaningful for making improved predictions for CRC risk as suggested by the results of our study [70].
To our knowledge, this is the first study to report on the joint association and interaction of CRC-related PRS and MHT variables with CRC risk, as well as with tumor site-specific risk, using both multiplicative and additive interaction. Our study also has several potential limitations. First, MHT information in some studies was self-reported; therefore, it may lead to recall bias (in the retrospective studies) or misclassification (in the prospective studies). However, previous studies have found a high validity for self-reported MHT use when compared with population-based prescription databases [71] and a high concordance between self-reported MHT use and physicians’ reports [72]. Second, because some studies asked only about current MHT use at the reference time rather than ever-use of MHT until the reference time, the status of MHT use might be misclassified, which would be likely to result in an underestimation of the strength of association. Third, the SNPs used for PRS as well as the study samples are population specific for postmenopausal women of European ancestries, thus generalization of results to populations of other racial and ethnic groups needs to be further evaluated.
In conclusion, the joint associations of genetic risk as measured by the PRS and the use of MHT with CRC risk show departures from the additive model. MHT use has a stronger impact on the risk reduction of CRC for women at higher genetic risk. These findings will inform the development of risk-prediction models for CRC in the future. They may lead to the consideration of genetic information as an additional factor in the risk-benefit assessment regarding MHT use in both the public health and clinical practice settings.
Supplementary information
Acknowledgements
We thank Prof. Dr. Cornelia M. Ulrich (Huntsman Cancer Institute; Department of Population Health Sciences, University of Utah, Salt Lake City, Utah, USA) for providing a further perspective to polish our manuscript. CCFR: The Colon CFR graciously thanks the generous contributions of their study participants, dedication of study staff, and the financial support from the U.S. National Cancer Institute, without which this important registry would not exist. The authors would like to thank the study participants and staff of the Seattle Colon Cancer Family Registry and the Hormones and Colon Cancer study (CORE Studies). CLUE II: We thank the participants of Clue II and appreciate the continued efforts of the staff at the Johns Hopkins George W. Comstock Center for Public Health Research and Prevention in the conduct of the Clue II Cohort Study. Cancer data was provided by the Maryland Cancer Registry, Center for Cancer Prevention and Control, Maryland Department of Health, with funding from the State of Maryland and the Maryland Cigarette Restitution Fund. The collection and availability of cancer registry data is also supported by the Cooperative Agreement NU58DP006333, funded by the Centers for Disease Control and Prevention. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the Centers for Disease Control and Prevention or the Department of Health and Human Services. CPS-II: The authors express sincere appreciation to all Cancer Prevention Study-II participants, and to each member of the study and biospecimen management group. The authors would like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention’s National Program of Cancer Registries, and cancer registries supported by the National Cancer Institute’s Surveillance Epidemiology and End Results Program. The authors assume full responsibility for all analyses and interpretation of results. The views expressed here are those of the authors and do not necessarily represent the American Cancer Society or the American Cancer Society – Cancer Action Network. DACHS: We thank all participants and cooperating clinicians, and everyone who provided excellent technical assistance. EPIC: Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization. Harvard cohorts: The study protocol was approved by the institutional review boards of the Brigham and Women’s Hospital and Harvard T.H. Chan School of Public Health, and those of participating registries as required. We acknowledge Channing Division of Network Medicine, Department of Medicine, Brigham and Women’s Hospital as home of the NHS. The authors would like to acknowledge the contribution to this study from central cancer registries supported through the Centers for Disease Control and Prevention’s National Program of Cancer Registries (NPCR) and/or the National Cancer Institute’s Surveillance, Epidemiology, and End Results (SEER) Program. Central registries may also be supported by state agencies, universities, and cancer centers. Participating central cancer registries include the following: Alabama, Alaska, Arizona, Arkansas, California, Colorado, Connecticut, Delaware, Florida, Georgia, Hawaii, Idaho, Indiana, Iowa, Kentucky, Louisiana, Massachusetts, Maine, Maryland, Michigan, Mississippi, Montana, Nebraska, Nevada, New Hampshire, New Jersey, New Mexico, New York, North Carolina, North Dakota, Ohio, Oklahoma, Oregon, Pennsylvania, Puerto Rico, Rhode Island, Seattle SEER Registry, South Carolina, Tennessee, Texas, Utah, Virginia, West Virginia, Wyoming. The authors assume full responsibility for analyses and interpretation of these data. Kentucky: We would like to acknowledge the staff at the Kentucky Cancer Registry. LCCS: We acknowledge the contributions of Jennifer Barrett, Robin Waxman, Gillian Smith and Emma Northwood in conducting this study. NCCCS I & II: We would like to thank the study participants, and the NC Colorectal Cancer Study staff. PLCO: The authors thank the PLCO Cancer Screening Trial screening center investigators and the staff from Information Management Services Inc and Westat Inc. Most importantly, we thank the study participants for their contributions that made this study possible. Cancer incidence data have been provided by the District of Columbia Cancer Registry, Georgia Cancer Registry, Hawaii Cancer Registry, Minnesota Cancer Surveillance System, Missouri Cancer Registry, Nevada Central Cancer Registry, Pennsylvania Cancer Registry, Texas Cancer Registry, Virginia Cancer Registry, and Wisconsin Cancer Reporting System. All are supported in part by funds from the Center for Disease Control and Prevention, National Program for Central Registries, local states or by the National Cancer Institute, Surveillance, Epidemiology, and End Results program. The results reported here and the conclusions derived are the sole responsibility of the authors. WHI: The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf.
Author contributions
Writing- original draft: YT. Analysis- statistical methods: YT, YL. Supervising: UP, WJG, LH, JCC. All authors reviewed, edited and approved the final manuscript. JCC and LH are the guarantors and as the corresponding authors attest that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted. YL and LH confirm that they had full access to all the data in the study. JCC and LH confirm that they had final responsibility for the decision to submit for publication.
Funding
Genetics and Epidemiology of Colorectal Cancer Consortium (GECCO): National Cancer Institute, National Institutes of Health, U.S. Department of Health and Human Services (U01 CA137088, R01 CA059045, U01 CA164930, R01201407). Genotyping/ services were provided by the Center for Inherited Disease Research (CIDR) contract number HHSN268201200008I. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA015704. Scientific Computing Infrastructure at Fred Hutch funded by ORIP grant S10OD028685. CLUE II: National Cancer Institute (U01 CA086308, Early Detection Research Network; P30 CA006973), National Institute on Aging (U01 AG018033), and the American Institute for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US government. The Colon Cancer Family Registry (CCFR, www.coloncfr.org) is supported in part by funding from the National Cancer Institute (NCI), National Institutes of Health (NIH) (award U01 CA167551). Support for case ascertainment was provided in part from the Surveillance, Epidemiology, and End Results (SEER) Program and the following U.S. state cancer registries: AZ, CO, MN, NC, NH; and by the Victoria Cancer Registry (Australia) and Ontario Cancer Registry (Canada). The CCFR Set-1 (Illumina 1 M/1M-Duo) scan was supported by NIH awards U01 CA122839 and R01 CA143237 (to GC). The CCFR Set-3 (Affymetrix Axiom CORECT Set array) was supported by NIH award U19 CA148107 and R01 CA081488 (to SBG). The CCFR Set-4 (Illumina OncoArray 600 K SNP array) was supported by NIH award U19 CA148107 (to SBG) and by the Center for Inherited Disease Research (CIDR), which is funded by the NIH to the Johns Hopkins University, contract number HHSN268201200008I. Additional funding for the OFCCR/ARCTIC was through award GL201-043 from the Ontario Research Fund (to BWZ), award 112746 from the Canadian Institutes of Health Research (to TJH), through a Cancer Risk Evaluation (CaRE) Program grant from the Canadian Cancer Society (to SG), and through generous support from the Ontario Ministry of Research and Innovation. The SFCCR Illumina HumanCytoSNP array was supported in part through NCI/NIH awards U01/U24 CA074794 and R01 CA076366 (to PAN). The content of this manuscript does not necessarily reflect the views or policies of the NCI, NIH or any of the collaborating centers in the Colon Cancer Family Registry (CCFR), nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government, any cancer registry, or the CCFR. COLO2&3: National Institutes of Health (R01 CA060987). Colorectal Cancer Transdisciplinary (CORECT) Study: National Institutes of Health (NCI/NIH), U.S. Department of Health and Human Services (grant numbers U19 CA148107, R01 CA081488, P30 CA014089, R01 CA197350; P01 CA196569; R01 CA201407; R01 CA242218), National Institutes of Environmental Health Sciences, National Institutes of Health (grant number T32 ES013678) and a generous gift from Daniel and Maryann Fong. CPS-II: The American Cancer Society funds the creation, maintenance, and updating of the Cancer Prevention Study-II (CPS-II) cohort. The study protocol was approved by the institutional review boards of Emory University, and those of participating registries as required. CRCGEN: Colorectal Cancer Genetics & Genomics, Spanish study was supported by Instituto de Salud Carlos III, co-funded by FEDER funds –a way to build Europe– (grants PI14-613 and PI09-1286), Agency for Management of University and Research Grants (AGAUR) of the Catalan Government (grant 2017SGR723), Junta de Castilla y León (grant LE22A10-2), the Spanish Association Against Cancer (AECC) Scientific Foundation grant GCTRA18022MORE and the Consortium for Biomedical Research in Epidemiology and Public Health (CIBERESP), action Genrisk. Sample collection of this work was supported by the Xarxa de Bancs de Tumors de Catalunya sponsored by Pla Director d’Oncología de Catalunya (XBTC), Plataforma Biobancos PT13/0010/0013 and ICOBIOBANC, sponsored by the Catalan Institute of Oncology. We thank CERCA Programme, Generalitat de Catalunya for institutional support. DACHS: German Research Council (BR 1704/6-1, BR 1704/6-3, BR 1704/6-4, CH 117/1-1, HO 5117/2-1, HE 5998/2-1, KL 2354/3-1, RO 2270/8-1 and BR 1704/17-1), the Interdisciplinary Research Program of the National Center for Tumor Diseases (NCT), Germany, and the German Federal Ministry of Education and Research (01KH0404, 01ER0814, 01ER0815, 01ER1505A and 01ER1505B). DALS: National Institutes of Health (R01 CA048998 to M. L. Slattery). EPIC: The coordination of EPIC is financially supported by International Agency for Research on Cancer (IARC) and also by the Department of Epidemiology and Biostatistics, School of Public Health, Imperial College London which has additional infrastructure support provided by the NIHR Imperial Biomedical Research Centre (BRC). The national cohorts are supported by: Danish Cancer Society (Denmark); Ligue Contre le Cancer, Institut Gustave Roussy, Mutuelle Générale de l’Education Nationale, Institut National de la Santé et de la Recherche Médicale (INSERM) (France); German Cancer Aid, German Cancer Research Center (DKFZ), German Institute of Human Nutrition Potsdam- Rehbruecke (DIfE), Federal Ministry of Education and Research (BMBF) (Germany); Associazione Italiana per la Ricerca sul Cancro-AIRC-Italy, Compagnia di SanPaolo and National Research Council (Italy); Dutch Ministry of Public Health, Welfare and Sports (VWS), Netherlands Cancer Registry (NKR), LK Research Funds, Dutch Prevention Funds, Dutch ZON (Zorg Onderzoek Nederland), World Cancer Research Fund (WCRF), Statistics Netherlands (The Netherlands); Health Research Fund (FIS) - Instituto de Salud Carlos III (ISCIII), Regional Governments of Andalucía, Asturias, Basque Country, Murcia and Navarra, and the Catalan Institute of Oncology - ICO (Spain); Swedish Cancer Society, Swedish Research Council, Region Skåne and Region Västerbotten (Sweden); Cancer Research UK (14136 to EPIC-Norfolk; C8221/A29017 to EPIC-Oxford), Medical Research Council (1000143 to EPIC-Norfolk; MR/M012190/1 to EPIC-Oxford) (United Kingdom). ESTHER_VERDI: This work was supported by grants from the Baden-Württemberg Ministry of Science, Research and Arts and the German Cancer Aid. Harvard cohort (NHS): National Institutes of Health (R01 CA137178, P01 CA087969, UM1 CA186107, K24 DK098311, R01 CA151993, and R35 CA197735). Kentucky: Clinical Investigator Award from Damon Runyon Cancer Research Foundation (CI-8); NCI R01CA136726. LCCS: The Leeds Colorectal Cancer Study was funded by the Food Standards Agency and Cancer Research UK Programme Award (C588/A19167). MCCS: The cohort recruitment was funded by VicHealth and Cancer Council Victoria. The MCCS was further supported by Australian NHMRC grants 509348, 209057, 251553 and 504711 and by infrastructure provided by Cancer Council Victoria. Cases and their vital status were ascertained through the Victorian Cancer Registry (VCR) and the Australian Institute of Health and Welfare (AIHW), including the National Death Index and the Australian Cancer Database. MEC: National Institutes of Health (R37 CA054281, P01 CA033619, and R01 CA063464). MECC: This work was supported by the National Institutes of Health, U.S. Department of Health and Human Services (R01 CA081488, R01 CA197350, U19 CA148107, R01 CA242218, and a generous gift from Daniel and Maryann Fong. NCCCS I & II: National Institutes of Health (R01 CA066635 and P30 DK034987). NFCCR: This work was supported by an Interdisciplinary Health Research Team award from the Canadian Institutes of Health Research (CRT 43821); the National Institutes of Health, U.S. Department of Health and Human Services (U01 CA074783); and National Cancer Institute of Canada grants (18223 and 18226). The authors wish to acknowledge the contribution of Alexandre Belisle and the genotyping team of the McGill University and Génome Québec Innovation Centre, Montréal, Canada, for genotyping the Sequenom panel in the NFCCR samples. Funding was provided to Michael O. Woods by the Canadian Cancer Society Research Institute. PLCO: Intramural Research Program of the Division of Cancer Epidemiology and Genetics and supported by contracts from the Division of Cancer Prevention, National Cancer Institute, NIH, DHHS. Funding was provided by National Institutes of Health (NIH), Genes, Environment and Health Initiative (GEI) Z01 CP 010200, NIH U01 HG004446, and NIH GEI U01 HG 004438. REACH: National Cancer Institute (grant P01 CA074184 to J.D.P. and P.A.N., grants R01 CA097325, R03 CA153323, and K05 CA152715 to P.A.N., and the National Center for Advancing Translational Sciences at the National Institutes of Health (grant KL2 TR000421 to A.N.B.-H.) SMC_COSM: This work is supported by the Swedish Research Council /Infrastructure grant, the Swedish Cancer Foundation, and the Karolinska Institute´s Distinguished Professor Award to Alicja Wolk. UK Biobank: This research has been conducted using the UK Biobank Resource under Application Number 8614. VITAL: National Institutes of Health (K05 CA154337). WHI: The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C. Yu Tian has been supported by the National Natural Science Foundation of China (82204127), and Capital Medical University Startup (3100/12100208). Funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication. Open Access funding enabled and organized by Projekt DEAL.
Data availability
The genotype data as well as limited phenotype data have been deposited at dbGaP under accession numbers phs001415.v1.p1, phs001315.v1.p1, phs001078.v1.p1, phs001499.v1.p1, phs001903.v1.p1, phs001856.v1.p1, phs001045.v1.p1, and phs001499.v1.p1. Further information is available from the corresponding authors upon request.
Code availability
The code, as a part of an international consortium, which was used to generate results of this article, can be accessed on reasonable request to the corresponding authors.
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All studies in consortia were approved by their respective Institutional Review Boards, and all study participants provided informed consent.
Consent for publication
The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, a worldwide license to the Publishers and its licensees in perpetuity, in all forms, formats and media (whether known now or created in the future), to i) publish, reproduce, distribute, display and store the Contribution, ii) translate the Contribution into other languages, create adaptations, reprints, include within collections and create summaries, extracts and/or, abstracts of the Contribution, iii) create any other derivative work(s) based on the Contribution, iv) to exploit all subsidiary rights in the Contribution, v) the inclusion of electronic links from the Contribution to third party material where-ever it may be located; and, vi) license any third party to do any or all of the above.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Yu Tian, Yi Lin.
These authors jointly supervised this work: Ulrike Peters, W. James Gauderman, Li Hsu, Jenny Chang-Claude.
Contributor Information
Li Hsu, Email: lih@fredhutch.org.
Jenny Chang-Claude, Email: j.chang-claude@dkfz.de.
Supplementary information
The online version contains supplementary material available at 10.1038/s41416-024-02638-2.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71:209–49. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Huyghe JR, Bien SA, Harrison TA, Kang HM, Chen S, Schmit SL, et al. Discovery of common and rare genetic risk variants for colorectal cancer. Nat Genet. 2019;51:76–87. doi: 10.1038/s41588-018-0286-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu Y, Kweon SS, Cai Q, Tanikawa C, Shu XO, Jia WH, et al. Identification of Novel Loci and New Risk Variant in Known Loci for Colorectal Cancer Risk in East Asians. Cancer Epidemiol Biomark Prev. 2020;29:477–86. doi: 10.1158/1055-9965.EPI-19-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmit SL, Edlund CK, Schumacher FR, Gong J, Harrison TA, Huyghe JR, et al. Novel Common Genetic Susceptibility Loci for Colorectal Cancer. J Natl Cancer Inst. 2019;111:146–57. doi: 10.1093/jnci/djy099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crouch DJM, Bodmer WF. Polygenic inheritance, GWAS, polygenic risk scores, and the search for functional variants. Proc Natl Acad Sci. 2020;117:18924. doi: 10.1073/pnas.2005634117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hormone Replacement Therapy Market Size, Share & COVID-19 Impact Analysis, By Therapy Type (Estrogen and Combinations Replacement Therapy, Growth Hormone Replacement Therapy, Thyroid Hormone Replacement Therapy, and Testosterone Hormone Replacement Therapy), By Indication (Menopause, Hypothyroidism, Male Hypogonadism, and Growth Hormone Deficiency), By Route of Administration (Oral, Transdermal, and Parenteral), By Distribution Channel (Hospital Pharmacies, Retails Pharmacies & Stores, and Online Pharmacies), and Regional Forecast, 2021–2028: Fortune Business Insights; 2022. Available from: https://www.fortunebusinessinsights.com/hormone-replacement-therapy-hrt-market-102543.
- 7.Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 8.Beral V. Breast cancer and hormone-replacement therapy in the Million Women Study. Lancet. 2003;362:419–27. doi: 10.1016/S0140-6736(03)14596-5. [DOI] [PubMed] [Google Scholar]
- 9.Mehta J, Kling JM, Manson JE. Risks, Benefits, and Treatment Modalities of Menopausal Hormone Therapy: Current Concepts. Front Endocrinol. 2021;12:564781. doi: 10.3389/fendo.2021.564781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McMichael AJ, Potter JD. Reproduction, endogenous and exogenous sex hormones, and colon cancer: a review and hypothesis. J Natl Cancer Inst. 1980;65:1201–7. [PubMed] [Google Scholar]
- 11.Potter JD, McMichael AJ. Large bowel cancer in women in relation to reproductive and hormonal factors: a case-control study. J Natl Cancer Inst. 1983;71:703–9. [PubMed] [Google Scholar]
- 12.Lin KJ, Cheung WY, Lai JY, Giovannucci EL. The effect of estrogen vs. combined estrogen-progestogen therapy on the risk of colorectal cancer. Int J Cancer. 2012;130:419–30. doi: 10.1002/ijc.26026. [DOI] [PubMed] [Google Scholar]
- 13.Chlebowski RT, Wactawski-Wende J, Ritenbaugh C, Hubbell FA, Ascensao J, Rodabough RJ, et al. Estrogen plus progestin and colorectal cancer in postmenopausal women. N Engl J Med. 2004;350:991–1004. doi: 10.1056/NEJMoa032071. [DOI] [PubMed] [Google Scholar]
- 14.Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SA, Black H, et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: the Women’s Health Initiative randomized controlled trial. JAMA. 2004;291:1701–12. doi: 10.1001/jama.291.14.1701. [DOI] [PubMed] [Google Scholar]
- 15.Das PK, Saha J, Pillai S, Lam AK, Gopalan V, Islam F. Implications of estrogen and its receptors in colorectal carcinoma. Cancer Med. 2023;12:4367–79. doi: 10.1002/cam4.5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian Y, Kim AE, Bien SA, Lin Y, Qu C, Harrison TA, et al. Genome-Wide Interaction Analysis of Genetic Variants With Menopausal Hormone Therapy for Colorectal Cancer Risk. J Natl Cancer Inst. 2022;114:1135–48. doi: 10.1093/jnci/djac094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brenner H, Kloor M, Pox CP. Colorectal cancer. Lancet. 2014;383:1490–502. doi: 10.1016/S0140-6736(13)61649-9. [DOI] [PubMed] [Google Scholar]
- 18.VanderWeele TJ, Knol MJ. A Tutorial on Interaction. Epidemiologic. Methods. 2014;3:33–72. [Google Scholar]
- 19.Al-Tassan NA, Whiffin N, Hosking FJ, Palles C, Farrington SM, Dobbins SE, et al. A new GWAS and meta-analysis with 1000Genomes imputation identifies novel risk variants for colorectal cancer. Sci Rep. 2015;5:10442. doi: 10.1038/srep10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Whiffin N, Hosking FJ, Farrington SM, Palles C, Dobbins SE, Zgaga L, et al. Identification of susceptibility loci for colorectal cancer in a genome-wide meta-analysis. Hum Mol Genet. 2014;23:4729–37. doi: 10.1093/hmg/ddu177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Meta-analysis of three genome-wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 2010;42:973–7. doi: 10.1038/ng.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law PJ, Timofeeva M, Fernandez-Rozadilla C, Broderick P, Studd J, Fernandez-Tajes J, et al. Association analyses identify 31 new risk loci for colorectal cancer susceptibility. Nat Commun. 2019;10:2154. doi: 10.1038/s41467-019-09775-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orlando G, Law PJ, Palin K, Tuupanen S, Gylfe A, Hänninen UA, et al. Variation at 2q35 (PNKD and TMBIM1) influences colorectal cancer risk and identifies a pleiotropic effect with inflammatory bowel disease. Hum Mol Genet. 2016;25:2349–59. doi: 10.1093/hmg/ddw087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schumacher FR, Schmit SL, Jiao S, Edlund CK, Wang H, Zhang B, et al. Genome-wide association study of colorectal cancer identifies six new susceptibility loci. Nat Commun. 2015;6:7138. doi: 10.1038/ncomms8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niell BL, Long JC, Rennert G, Gruber SB. Genetic anthropology of the colorectal cancer-susceptibility allele APC I1307K: evidence of genetic drift within the Ashkenazim. Am J Hum Genet. 2003;73:1250–60. doi: 10.1086/379926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia WH, Zhang B, Matsuo K, Shin A, Xiang YB, Jee SH, et al. Genome-wide association analyses in East Asians identify new susceptibility loci for colorectal cancer. Nat Genet. 2013;45:191–6. doi: 10.1038/ng.2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dunlop MG, Dobbins SE, Farrington SM, Jones AM, Palles C, Whiffin N, et al. Common variation near CDKN1A, POLD3 and SHROOM2 influences colorectal cancer risk. Nat Genet. 2012;44:770–6. doi: 10.1038/ng.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng C, Matsuda K, Jia WH, Chang J, Kweon SS, Xiang YB, et al. Identification of Susceptibility Loci and Genes for Colorectal Cancer Risk. Gastroenterology. 2016;150:1633–45. doi: 10.1053/j.gastro.2016.02.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tomlinson IP, Webb E, Carvajal-Carmona L, Broderick P, Howarth K, Pittman AM, et al. A genome-wide association study identifies colorectal cancer susceptibility loci on chromosomes 10p14 and 8q23.3. Nat Genet. 2008;40:623–30. doi: 10.1038/ng.111. [DOI] [PubMed] [Google Scholar]
- 30.Zhang B, Jia WH, Matsuda K, Kweon SS, Matsuo K, Xiang YB, et al. Large-scale genetic study in East Asians identifies six new loci associated with colorectal cancer risk. Nat Genet. 2014;46:533–42. doi: 10.1038/ng.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Burnett T, Kono S, Haiman CA, Iwasaki M, Wilkens LR, et al. Trans-ethnic genome-wide association study of colorectal cancer identifies a new susceptibility locus in VTI1A. Nat Commun. 2014;5:4613. doi: 10.1038/ncomms5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Broderick P, Carvajal-Carmona L, Pittman AM, Webb E, Howarth K, Rowan A, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet. 2007;39:1315–7. doi: 10.1038/ng.2007.18. [DOI] [PubMed] [Google Scholar]
- 33.Tenesa A, Farrington SM, Prendergast JG, Porteous ME, Walker M, Haq N, et al. Genome-wide association scan identifies a colorectal cancer susceptibility locus on 11q23 and replicates risk loci at 8q24 and 18q21. Nat Genet. 2008;40:631–7. doi: 10.1038/ng.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang M, Gu D, Du M, Xu Z, Zhang S, Zhu L, et al. Common genetic variation in ETV6 is associated with colorectal cancer susceptibility. Nat Commun. 2016;7:11478. doi: 10.1038/ncomms11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tomlinson IP, Carvajal-Carmona LG, Dobbins SE, Tenesa A, Jones AM, Howarth K, et al. Multiple common susceptibility variants near BMP pathway loci GREM1, BMP4, and BMP2 explain part of the missing heritability of colorectal cancer. PLoS Genet. 2011;7:e1002105. doi: 10.1371/journal.pgen.1002105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomlinson I, Webb E, Carvajal-Carmona L, Broderick P, Kemp Z, Spain S, et al. A genome-wide association scan of tag SNPs identifies a susceptibility variant for colorectal cancer at 8q24.21. Nat Genet. 2007;39:984–8. doi: 10.1038/ng2085. [DOI] [PubMed] [Google Scholar]
- 37.Houlston RS, Webb E, Broderick P, Pittman AM, Di Bernardo MC, Lubbe S, et al. Meta-analysis of genome-wide association data identifies four new susceptibility loci for colorectal cancer. Nat Genet. 2008;40:1426–35. doi: 10.1038/ng.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hutter CM, Chang-Claude J, Slattery ML, Pflugeisen BM, Lin Y, Duggan D, et al. Characterization of gene-environment interactions for colorectal cancer susceptibility loci. Cancer Res. 2012;72:2036–44. doi: 10.1158/0008-5472.CAN-11-4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peters U, Jiao S, Schumacher FR, Hutter CM, Aragaki AK, Baron JA, et al. Identification of Genetic Susceptibility Loci for Colorectal Tumors in a Genome-Wide Meta-analysis. Gastroenterology. 2013;144:799–807.e24. doi: 10.1053/j.gastro.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garcia-Albeniz X, Rudolph A, Hutter C, White E, Lin Y, Rosse SA, et al. CYP24A1 variant modifies the association between use of oestrogen plus progestogen therapy and colorectal cancer risk. Br J Cancer. 2016;114:221–9. doi: 10.1038/bjc.2015.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gong J, Hutter CM, Newcomb PA, Ulrich CM, Bien SA, Campbell PT, et al. Genome-Wide Interaction Analyses between Genetic Variants and Alcohol Consumption and Smoking for Risk of Colorectal Cancer. PLoS Genet. 2016;12:e1006296. doi: 10.1371/journal.pgen.1006296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jeon J, Du M, Schoen RE, Hoffmeister M, Newcomb PA, Berndt SI, et al. Determining Risk of Colorectal Cancer and Starting Age of Screening Based on Lifestyle, Environmental, and Genetic Factors. Gastroenterology. 2018;154:2152–64.e19. doi: 10.1053/j.gastro.2018.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–7. doi: 10.1038/ng.3656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morrison J. BinaryDosage: Creates, Merges, and Reads Binary Dosage Files. R package version 1.0.0. 2020. https://CRAN.R-project.org/package=BinaryDosage.
- 45.Zollner S, Pritchard JK. Overcoming the winner’s curse: estimating penetrance parameters from case-control data. Am J Hum Genet. 2007;80:605–15. doi: 10.1086/512821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cochran WG. The Combination of Estimates from Different Experiments. Biometrics. 1954;10:101–29. doi: 10.2307/3001666. [DOI] [Google Scholar]
- 47.Hosmer DW, Lemeshow S. Confidence interval estimation of interaction. Epidemiology. 1992;3:452–6. doi: 10.1097/00001648-199209000-00012. [DOI] [PubMed] [Google Scholar]
- 48.Pfeiffer RM, Petracci E. Variance computations for functional of absolute risk estimates. Stat Probab Lett. 2011;81:807–12. doi: 10.1016/j.spl.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.National Cancer Institute, DCCPS, Surveillance Research Program. Surveillance, Epidemiology, and End Results (SEER) program, SEER*Stat database: incidence–SEER 13 regs research data, sub (1992–2017), based on the November 2019 submission. 2020. www.seer.cancer.gov. Accessed November 7, 2020).
- 50.Lin JH, Manson JE, Kraft P, Cochrane BB, Gunter MJ, Chlebowski RT, et al. Estrogen and progesterone-related gene variants and colorectal cancer risk in women. BMC Med Genet. 2011;12:78. doi: 10.1186/1471-2350-12-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rudolph A, Sainz J, Hein R, Hoffmeister M, Frank B, Försti A, et al. Modification of menopausal hormone therapy-associated colorectal cancer risk by polymorphisms in sex steroid signaling, metabolism and transport related genes. Endocr Relat cancer. 2011;18:371–84. doi: 10.1530/ERC-11-0057. [DOI] [PubMed] [Google Scholar]
- 52.Slattery ML, Lundgreen A, Herrick JS, Kadlubar S, Caan BJ, Potter JD, et al. Variation in the CYP19A1 gene and risk of colon and rectal cancer. Cancer causes & control. CCC. 2011;22:955–63. doi: 10.1007/s10552-011-9768-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carr PR, Weigl K, Edelmann D, Jansen L, Chang-Claude J, Brenner H, et al. Estimation of Absolute Risk of Colorectal Cancer Based on Healthy Lifestyle, Genetic Risk, and Colonoscopy Status in a Population-Based Study. Gastroenterology. 2020;159:129–38.e9. doi: 10.1053/j.gastro.2020.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang X, O’Connell K, Jeon J, Song M, Hunter D, Hoffmeister M, et al. Combined effect of modifiable and non-modifiable risk factors for colorectal cancer risk in a pooled analysis of 11 population-based studies. BMJ Open Gastroenterol. 2019;6:e000339. doi: 10.1136/bmjgast-2019-000339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cho YA, Lee J, Oh JH, Chang HJ, Sohn DK, Shin A, et al. Genetic Risk Score, Combined Lifestyle Factors and Risk of Colorectal Cancer. Cancer Res Treat. 2019;51:1033–40. doi: 10.4143/crt.2018.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi J, Jia G, Wen W, Shu XO, Zheng W. Healthy lifestyles, genetic modifiers, and colorectal cancer risk: a prospective cohort study in the UK Biobank. Am J Clin Nutr. 2021;113:810–20. doi: 10.1093/ajcn/nqaa404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen X, Guo F, Chang-Claude J, Hoffmeister M, Brenner H. Physical activity, polygenic risk score, and colorectal cancer risk. Cancer Med. 2023;12:4655–66. [DOI] [PMC free article] [PubMed]
- 58.Chen X, Li H, Guo F, Hoffmeister M, Brenner H. Alcohol consumption, polygenic risk score, and early- and late-onset colorectal cancer risk. EClinicalMedicine. 2022;49:101460. doi: 10.1016/j.eclinm.2022.101460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen X, Hoffmeister M, Brenner H. Red and Processed Meat Intake, Polygenic Risk Score, and Colorectal Cancer Risk. Nutrients. 2022;14:1077. [DOI] [PMC free article] [PubMed]
- 60.Chen X, Guo F, Hoffmeister M, Chang-Claude J, Brenner H. Non-steroidal anti-inflammatory drugs, polygenic risk score and colorectal cancer risk. Aliment Pharm Ther. 2021;54:167–75. doi: 10.1111/apt.16438. [DOI] [PubMed] [Google Scholar]
- 61.Chen X, Jansen L, Guo F, Hoffmeister M, Chang-Claude J, Brenner H. Smoking, Genetic Predisposition, and Colorectal Cancer Risk. Clin Transl Gastroenterol. 2021;12:e00317. doi: 10.14309/ctg.0000000000000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Labadie JD, Harrison TA, Banbury B, Amtay EL, Bernd S, Brenner H, et al. Postmenopausal Hormone Therapy and Colorectal Cancer Risk by Molecularly Defined Subtypes and Tumor Location. JNCI Cancer Spectr. 2020;4:pkaa042. doi: 10.1093/jncics/pkaa042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Limsui D, Vierkant RA, Tillmans LS, Wang AH, Weisenberger DJ, Laird PW, et al. Postmenopausal hormone therapy and colorectal cancer risk by molecularly defined subtypes among older women. Gut. 2012;61:1299–305. doi: 10.1136/gutjnl-2011-300719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee GH, Malietzis G, Askari A, Bernardo D, Al-Hassi HO, Clark SK. Is right-sided colon cancer different to left-sided colorectal cancer? - a systematic review. Eur J Surg Oncol. 2015;41:300–8. doi: 10.1016/j.ejso.2014.11.001. [DOI] [PubMed] [Google Scholar]
- 65.Merlano MC, Granetto C, Fea E, Ricci V, Garrone O. Heterogeneity of colon cancer: from bench to bedside. ESMO Open. 2017;2:e000218. doi: 10.1136/esmoopen-2017-000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Macfarlane GT, Macfarlane LE. Acquisition, evolution and maintenance of the normal gut microbiota. Dig Dis. 2009;27:90–8. doi: 10.1159/000268127. [DOI] [PubMed] [Google Scholar]
- 67.Huyghe JR, Harrison TA, Bien SA, Hampel H, Figueiredo JC, Schmit SL, et al. Genetic architectures of proximal and distal colorectal cancer are partly distinct. Gut. 2021;70:1325–34. doi: 10.1136/gutjnl-2020-321534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang B, Shrubsole MJ, Li G, Cai Q, Edwards T, Smalley WE, et al. Association of genetic variants for colorectal cancer differs by subtypes of polyps in the colorectum. Carcinogenesis. 2012;33:2417–23. doi: 10.1093/carcin/bgs308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gauderman WJ, Mukherjee B, Aschard H, Hsu L, Lewinger JP, Patel CJ, et al. Update on the State of the Science for Analytical Methods for Gene-Environment Interactions. Am J Epidemiol. 2017;186:762–70. doi: 10.1093/aje/kwx228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blot WJ, Day NE. Synergism and interaction: are they equivalent? Am J Epidemiol. 1979;110:99–100. doi: 10.1093/oxfordjournals.aje.a112793. [DOI] [PubMed] [Google Scholar]
- 71.Løkkegaard EL, Johnsen SP, Heitmann BL, Stahlberg C, Pedersen AT, Obel EB, et al. The validity of self-reported use of hormone replacement therapy among Danish nurses. Acta Obstet Gynecol Scand. 2004;83:476–81. doi: 10.1111/j.0001-6349.2004.00376.x. [DOI] [PubMed] [Google Scholar]
- 72.Kropp S, Terboven T, Hedicke J, Mutschelknauss E, Slanger T, Braendle W, et al. Good agreement between physician and self-reported hormone therapy data in a case-control study. J Clin Epidemiol. 2007;60:1280–7. doi: 10.1016/j.jclinepi.2007.02.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genotype data as well as limited phenotype data have been deposited at dbGaP under accession numbers phs001415.v1.p1, phs001315.v1.p1, phs001078.v1.p1, phs001499.v1.p1, phs001903.v1.p1, phs001856.v1.p1, phs001045.v1.p1, and phs001499.v1.p1. Further information is available from the corresponding authors upon request.
The code, as a part of an international consortium, which was used to generate results of this article, can be accessed on reasonable request to the corresponding authors.