

Abstract
Use of tumor-suppressive microRNAs (miRNAs) as anti-cancer agents is hindered by the lack of effective delivery vehicles, entrapment of the miRNA within endocytic compartments, and rapid degradation of miRNA by nucleases. To address these issues, we developed a miRNA delivery strategy that includes (1) a targeting ligand, (2) an endosomal escape agent, nigericin and (3) a chemically modified miRNA. The delivery ligand, DUPA (2-[3-(1,3-dicarboxy propyl) ureido] pentanedioic acid), was selected based on its specificity for prostate-specific membrane antigen (PSMA), a receptor routinely upregulated in prostate cancer—one of the leading causes of cancer death among men. DUPA was conjugated to the tumor suppressive miRNA, miR-34a (DUPA-miR-34a) based on the ability of miR-34a to inhibit prostate cancer cell proliferation. To mediate endosomal escape, nigericin was incorporated into the complex, resulting in DUPA-nigericin-miR-34a. Both DUPA-miR-34a and DUPA-nigericin-miR-34a specifically bound to, and were taken up by, PSMA-expressing cells in vitro and in vivo. And while both DUPA-miR-34a and DUPA-nigericin-miR-34a downregulated miR-34a target genes, only DUPA-nigericin-miR-34a decreased cell proliferation in vitro and delayed tumor growth in vivo. Tumor growth was further reduced using a fully modified version of miR-34a that has significantly increased stability.
Keywords: MT: Delivery Strategies, microRNA, DUPA, PSMA, ligand, delivery, vehicle-free, prostate cancer, endosomal escape, miR-34
Graphical abstract
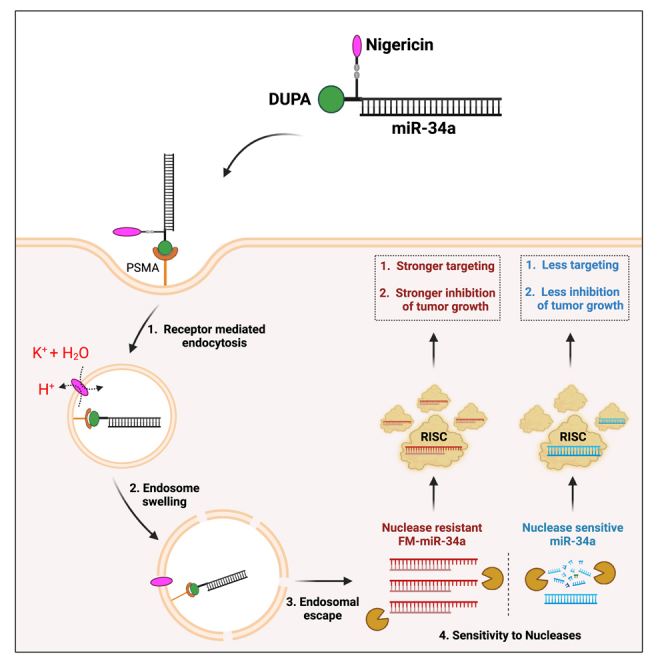
Kasinski and colleagues report on the utility of an endosomal escape agent, together with a targeting ligand, and a chemically modified miRNA that achieves robust cytosolic accumulation of the conjugated and stabilized miRNA leading to an enhanced anti-tumor effect.
Introduction
miRNAs are small non-coding RNAs that regulate gene expression through binding to sites in the 3′UTR of target mRNAs. Through this binding, coupled with recruitment of various proteins including Argonaute, miRNAs affect target gene expression through mRNA destabilization or by causing translation repression.1,2,3,4,5 Because miRNAs bind with imperfect complementarity, a single miRNA has the capacity to regulate hundreds of target mRNAs. This pleotropic role has resulted in miRNAs being referred to as “master regulators of the genome.” Indeed, if the expression of even one of these “master regulators” is altered, various phenotypic consequences can ensue.
While microRNAs (miRNAs) are dysregulated in nearly every disease, in cancer, miRNAs are generally depleted. The depleted miRNAs are typically classified as tumor suppressive miRNAs. Restoring some of these tumor-suppressive miRNAs, including miR-34a or let-7, leads to the inhibition of tumor growth in vivo and abrogates other essential processes, such as invasion, migration, and metastasis.6,7,8,9 Based on these anti-cancer attributes, miR-34a was the first tumor-suppressive miRNA to be investigated as an anti-cancer agent in clinical trials; however, the study was terminated due to severe immune-related effects.8,10 These immune-related events were predicted to be due to excess, minimally modified miRNA along with the liposomal delivery vehicle used to encapsulate the RNA. To overcome the toxicity associated with the vehicle, we previously generated a vehicle-free, ligand-mediated delivery approach. In this approach, miR-34a is directly conjugated to a folate, a high-affinity ligand of the folate receptor.11 The folate receptor is overexpressed in many cancers that originate in the lung, breast, ovary, kidney, colon, and brain.12 Nonetheless, folate receptor expression is sparse in prostate cancer, limiting the utility of folate-miRNA conjugates (FolamiRs) for treating prostate cancer.
In the presence of the correct ligand-receptor pair, ligand-mediated delivery can lead to the robust accumulation of conjugated cargo to the intended cells; however, one of the challenges associated with ligand-mediated delivery is endosomal entrapment of the miRNA conjugates, which limits the bioactivity of the delivered miRNA.13,14 Previously, we generated an intramolecular FolamiR conjugate containing the endosomal escape agent nigericin, which significantly enhanced the cytosolic localization and subsequent activity of the conjugated miRNA.13 Nonetheless, FolamiRs are ineffective in cancers with no or low folate receptor expression, such as prostate cancer. Here we describe the synthesis and advancement of a specific ligand conjugate in combination with nigericin for delivery of miRNAs to prostate cancer.
Prostate cancer is one of the leading causes of death among men.15 Most of the current treatments for prostate cancer, such as androgen deprivation therapy, are associated with resistance.16 Thus, finding new therapeutic approaches is essential for successful prostate cancer treatment. miR-34a represents a promising therapeutic; it inhibits prostate cancer development, survival, and invasiveness by repressing multiple targets, including oncogenic c-Myc and the androgen receptor.17,18,19,20,21 Clinically, miR-34a expression is associated with the overall survival of prostate cancer patients, further supporting its tumor-suppressive role.20 Regardless, delivery and sustained activity of tumor suppressive miRNAs, such as miR-34a, has remained a critical bottleneck in clinical advancement.
Various strategies have been tested for the delivery of small RNAs, including viral-based delivery, encapsulating nanoparticles, aptamers, and most recently small molecules, such as N-acetylglucosamine, folate, and 2-[3-(1,3-dicarboxypropyl) ureido] pentanedioic acid (DUPA).14 DUPA is a glutamate urea synthetic ligand that binds to prostate-specific membrane antigen (PSMA) with high affinity (inhibitory constant = 8 nM) and specificity.14,22,23 PSMA, also known as glutamate carboxypeptidase II, is a surface receptor specifically overexpressed in prostate cancer. When bound by its antibody, PSMA is constitutively internalized, a feature that makes PSMA a good candidate for miRNA delivery.24,25 Importantly, PSMA expression in prostate cancer cells is 100- to 1,000-fold higher in comparison with normal tissues and its expression level increases with cancer aggressiveness, recurrence, and metastasis.23,26,27 While folate has been shown to bind to PSMA,28 the low affinity between folate and PSMA makes our previous FolamiR strategy not suitable for targeting prostate cancer,29,30 which spearheaded our effort to adapt DUPA as a ligand for miRNA delivery to PSMA-overexpressing prostate cancer.
In this study, we developed a vehicle-free strategy to deliver miR-34a to prostate cancer based on three critical components. First, miR-34a was directly conjugated to DUPA to enable specific delivery to PSMA-expressing prostate cancer cells. Second, nigericin—a small molecule ionophore—was incorporated into the therapeutic design to enhance the release of miR-34a from the endosomes. Third, a fully modified and stabilized version of miR-34a was evaluated in comparison with a partially modified version. We show that this strategy results in targeted delivery of miR-34a to prostate cancer cells and that miR-34a delivered using DUPA-nigericin results in a significant delay in tumor growth in vivo, with fully modified miR-34a generating an enhanced effect.
Results
Generation, validation, and evaluation of DUPA-miR-34a conjugation and its binding to PSMA-expressing cells
We first validated the ability of miR-34a to inhibit the proliferation of LNCaP prostate cells and compared its effect to other miRNAs with known tumor suppressive roles in prostate cancer.31,32,33,34 As shown in Figure 1A, transfection of LNCaP cells with miR-34a significantly inhibited cell proliferation in comparison to other tumor-suppressive miRNAs, such as let-7a and let-7c. Next, to achieve targeting specificity, click chemistry was used to link DUPA, a high-affinity PSMA ligand, to miR-34a. To generate DUPA-miR-34a conjugates, the 5′ end of the miR-34a sense strand, which was modified with an azide, was linked to DUPA-dibenzocyclooctyne (DUPA-DBCO) following incubation at a 1:10 M ratio. Subsequently, the guide strand of miR-34a was annealed to the DUPA-miR-34a sense strand, resulting in the formation of a DUPA-miR-34a duplex (Figures 1B and S1). A short spacer containing two phenylalanine residues was incorporated between DUPA and miR-34a to further enhance the stoichiometry of the design and allow efficient binding of DUPA to PSMA35 (see Figure 1B). Conjugation and generation of the DUPA-miR-34a duplex were confirmed using polyacrylamide gel electrophoresis (Figure 1C).
Figure 1.

DUPA-miR-34a conjugation and binding to prostate cancer cells
(A) Effect of transfecting various tumor suppressive miRNAs on proliferation of LNCaP cells, measured by SRB assay. Experiment repeated twice with five technical replicates in each experiment. One representative biological replicate is shown, one-way ANOVA. ∗∗∗∗p < 0.0001, error bars are mean ± SD. (B) Schematic of DUPA-miR-34a synthesis. (C) Gel red-stained polyacrylamide gel indicating successful conjugation of DUPA-miR-34a based on the shift in mobility. (D) Flow cytometry histograms of miR-34a-Atto.647N (with or without DUPA conjugation) treated cells in the presence or absence of the PSMA inhibitor, PMPA. (E and F) Flow cytometry analysis of DUPA binding to PSMA positive cancer cells in a dose-dependent (E) and time-dependent (F) manner using DUPA-AF488.
To evaluate uptake and efficacy of DUPA-miR constructs, LNCaP and 22Rv1 prostate cancer cell lines were selected owing to their elevated PSMA expression (Figure S2A). Using these cell lines and the PSMA negative (−) control cell line, A549, binding of DUPA-miR-34a conjugates to the PSMA receptor was assessed. Cells were exposed to DUPA conjugated to a fluorescent version of miR-34a, miR-34a-Atto.647N. In the absence of a transfection reagent, cells treated with DUPA-miR-34a-Atto.647N responded with a significant increase in the Atto.647N signal. This was true for both PSMA+ cell lines, LNCaP and 22RV1, but not for A549 cells, suggesting that binding of DUPA-miR-34a is PSMA dependent (Figure 1D). The PSMA dependency was further confirmed via co-treatment of cells with DUPA-miR-34a-Atto.647N and the PSMA inhibitor, 2-(phosphonomethyl) pentanedioic acid (PMPA) (Figure 1D). For both PSMA+ cell lines, binding of DUPA-miR-34a-Atto.647N was significantly attenuated in the presence of 10 μM PMPA providing further support that binding is PSMA mediated (Figure 1D). To confirm that gymnosis was not contributing to the uptake of miR-34a, cells were treated with either unconjugated miR-34a-Atto.647N or DUPA-miR-34a-Atto.647N. The data indicate that DUPA is required for binding of DUPA-miR-34a to PSMA-expressing cells, and that binding is not mediated by gymnosis (Figure 1D). Dose- and time-dependent binding was also confirmed using DUPA conjugated to Alexa Fluor 488 (DUPA-AF488) (Figures 1E and 1F).
DUPA-miR-34a uptake by prostate cancer cells is PSMA mediated
While a diagnostic agent only needs to bind the surface of a cell, a therapeutic, such as an miRNA, must also be internalized. Thus, to verify that the DUPA conjugates are internalized by cells following binding to PSMA, LNCaP cells were treated with either miR-34a-Atto.647N or DUPA-miR-34a-Atto.647N, and intracellular accumulation of Atto.647N was evaluated using confocal microscopy. Only the targeted DUPA-miR-34a-Atto.647N, but not miR-34a-Atto.647N, was internalized by LNCaP cells (Figure 2A), which increased in a time-dependent manner (Figure 2B). To further confirm uptake of DUPA-miR-34a, internalized miR-34a was quantified using RT-qPCR. Treatment of LNCaP cells, but not A549 cells, with DUPA-miR-34a resulted in a significant enrichment of miR-34a inside the cell relative to untreated cells (Figure 2C).
Figure 2.

DUPA-miR-34a uptake and internalization by prostate cancer cells is DUPA and PSMA mediated
(A) Representative confocal images of LNCaP cells treated with 100 nM DUPA-miR-34a-Atto.647N or miR-34a-Atto.647N (no DUPA) for 4 h, Scale bar, 10 μM. (B) Representative confocal images of LNCaP cells treated with 100 nM DUPA-miR-34a-Atto.647N (red) at different time points after treatment. Scale bar, 10 μM. (C) Quantification of miR-34a uptake in PSMA positive LNCaP cells using qRT-PCR. LNCaP cells were treated (100 nM) or transfected (+Lipo, 10 nM) with DUPA-miR-34a or DUPA-NC (siLuc2, NC) for 18 h (top). PSMA negative A549 cells treated with DUPA-NC or DUPA-miR-34a, or transfected with a NC miRNA (miR-NC) or with a commercial miR-34a mimic. One representative experiment is depicted from a total of three biological experiments. ∗∗p < 0.01, ∗∗∗∗p < 0.0001, one-way ANOVA. Error bars are mean ± SD. (D) Targeted silencing of miR-34a Renilla sensor after transfection of LNCaP-miR-34a sensor cells with 50 nM DUPA-miR-34a, miR-34a duplex, or respective controls for 24 h. One representative experiment is depicted from a total of three biological experiments. ∗∗∗∗p < 0.0001, one-way ANOVA. Error bars are mean ± SD. (E) Dose-dependent response of LNCaP-miR-34a sensor cells following treatment with DUPA-miR-34a, miR-34a duplex, or respective controls for 96 h (n = 2). ∗∗p < 0.01, one-way ANOVA. Error bars are mean ± SD. (F) DUPA-miR-34a competition assay following treatment of LNCaP-miR-34a sensor cells with DUPA-miR-34a (100 nM, 96 h) in the presence of 10-, 100-, or 500-fold higher concentrations of PMPA (n = 2). ∗∗p < 0.01, one-way ANOVA. Error bars are mean ± SD. (G) Representative confocal images of LNCaP cells treated with 100 nM DUPA-miR-34a-Atto.647 (pink) for the indicated time points. Late endosomes and lysosomes were stained with LysoTracker (green). Nuclei were stained by Hoechst (blue). Insets in the top left corner of DUPA-miR-34a-treated samples represent scatterplot of LysoTracker vs. miR-34a fluorescence intensities. Appearance of intracellular white fluorescence indicates late endosomal/lysosomal localization of DUPA-conjugates. Scale bar, 10 μM. (H) Representative graph showing quantification of miR-34a colocalization with late endosome/lysosomes at the indicated times by Pearson’s correlation coefficient analysis (n = 2). ∗p < 0.00001, two-tailed unpaired t test. Error bars are mean ± SD.
To verify that DUPA conjugation does not impair miR-34a gene targeting activity, DUPA-miR-34a was transfected into LNCaP cells engineered to overexpress a miR-34a Renilla reporter. Transfecting these LNCaP miR-34a sensor cells with either DUPA-miR-34a or the unconjugated duplex resulted in a similar reduction in Renilla (Figure 2D), suggesting that DUPA does not alter miR-34a activity. Using the same sensor cells, activity of the conjugate was evaluated in the absence of an exogenous transfection reagent. In this case, cells were simply treated with various doses of DUPA-miR-34a. While treatment of cells with the unconjugated miR-34a duplex did not alter Renilla activity, treatment with 100 or 200 nM DUPA-miR-34a significantly decreased Renilla expression (Figure 2E). Importantly, the decrease in Renilla was attenuated in the presence of the PSMA antagonist, PMPA, lending additional support that uptake is PSMA mediated (Figure 2F).
Because receptor-mediated endocytosis via PSMA is the most likely mechanism of uptake of DUPA-conjugates, we next tracked the intracellular distribution of DUPA conjugates in relation to the late endosomal compartments. LNCaP cells were imaged using confocal microscopy following treatment with DUPA-miR-34a-Atto.647N (pink) and after incubation with LysoTracker (green), a late endosome/lysosome marker. At both 4 and 8 h after treatment, DUPA-miR-34a-Atto.647N conjugates co-localized with LysoTracker, suggesting sequestration into late endosomes or lysosomes (Figures 2G and 2H), as previously reported.36,37
Intramolecular conjugation of nigericin enhances endosomal escape of miR-34a delivered via DUPA
We previously determined that conjugating a single molecule of nigericin into the FolamiR design enhanced the cytosolic enrichment and activity of folate-conjugated miRNAs through facilitating endosomal escape.13 As a small molecule ionophore, nigericin mediates the exchange of potassium ions (K+) with a proton. Because K+ is osmotically active but the proton is not, water molecules enter the endosome with K+ but do not escape with the proton, leading to endosomal swelling.13,38 Thus, to overcome the proposed endosomal entrapment of DUPA conjugates, we generated an intramolecular conjugate containing nigericin and DUPA (DUPA-nigericin-DBCO) (Figures 3A and S3). We initially tested whether the inclusion of nigericin results in swelling of the endocytic vesicles as a result of its ability to disrupt the osmotic equilibrium within the endosomes. Using LAMP1 as a marker of endosomes, we observed an increase in the size of LAMP1+ vesicles following treatment of LNCaP cells with DUPA-nigericin-DBCO in comparison to DUPA-DBCO treated cells (Figure 3B). Next, DUPA-nigericin-miR-34a conjugates were synthesized by incubating the 5′ end of the azide-modified sense strand of miR-34a with DUPA-nigericin-DBCO at a molar ratio of 1:40. This was followed by annealing of the antisense strand to generate DUPA-nigericin-miR-34a duplexes (Figure 3C). We hypothesized that inclusion of nigericin should increase the abundance of miR-34a in the cytosol of the cells following treatment with DUPA-nigericin-miR-34a. To test this, treated cells were fractionated into a membranous fraction (containing organelles, including endosomes) and a membrane-depleted fraction (cytosolic fraction). Successful fractionation was indicated via enrichment of the endosomal protein LAMP1 and depletion of cytosolic GAPDH in the membrane-containing fraction, while the reverse was observed in the cytosol fraction (Figure 3D). Quantifying miR-34a from each of the fractions highlighted a significant enrichment of miR-34a in the cytosolic fraction when cells were treated with DUPA-nigericin-miR-34a (Figure 3D), suggesting that incorporating nigericin enhances the endocytic release of miR-34a.
Figure 3.

MiR-34a delivery using DUPA-nigericin facilitates cytosolic enrichment and early response
(A) Schematic of DUPA-nigericin miR-34a conjugate. (B) Evaluation of morphological changes in early and late endosomes in LNCaP cells after treatment with DUPA or DUPA-nigericin. LNCaP cells were treated with either DUPA or DUPA-nigericin for 5 h followed by imaging and measuring of late endosomes (LAMP1+ vesicles, white arrows). ∗∗∗∗p ˂ 0.0001, two-tailed unpaired t test. Error bars are mean ± SD. (C) Gel-red-stained polyacrylamide gel indicates successful conjugation of DUPA-nigericin-miR-34a (lane 6), as visualized by the shift in mobility. nig, nigericin. ∗Excess unconjugated miR-34a-5P guide strand. (D) Left, Western blot verifying the separation of cytosol fraction (C) and membrane-containing organelles fraction (M) as determined by LAMP1 and GAPDH. Right, miR-34a quantified using qRT-PCR from the cytosolic fractions obtained from LNCaP cells treated with 100 nM DUPA-miR-34a or DUPA-nigericin-miR-34a for 18 h. The experiment repeated twice with three technical replicates in each experiment. ∗p < 0.05, ∗∗∗p ˂ 0.001, two-tailed unpaired t test. Error bars are mean ± SD. Significance was determined relative to the corresponding NC. nig, nigericin. (E) Targeted silencing of miR-34a Renilla sensor after the treatment of LNCaP-miR-34a sensor cells with 100 nM DUPA-miR-34a, DUPA-nigericin-miR-34a, or respective controls for the indicated time points (n = 3). ∗p < 0.05, ∗∗p < 0.01, one-way ANOVA. Error bars are mean ± SD, significance was determined relative to untreated. nig, nigericin. (F) Renilla luciferase expression following treatment of miR-34a sensor cells with miR-34a duplex, folate (Fol)-nigericin-NC or Fol-nigericin-miR-34a 72 h after treatment. nig, nigericin. Error bars are mean ± SD (G) Cell proliferation of LNCaP cells treated with DUPA-miR-34a or DUPA-nigericin-miR-34a for 96 h. Data were normalized to the untreated. Statistical significance was determined using one-way ANOVA, ∗p < 0.05, ∗∗∗∗p < 0.0001. nig, nigericin; ns, not significant. Error bars are the mean ± SD.
To evaluate the ability of DUPA-miR-34a and DUPA-nigericin-miR-34a conjugates to repress miR-34a targets, LNCaP cells expressing the miR-34a Renilla luciferase sensor were treated with DUPA-nigericin-miR-34a or DUPA-miR-34a. Treatment with DUPA-nigericin-miR-34a resulted in earlier knockdown than when cells were treated with DUPA-miR-34a (Figure 3E). And, while PSMA is known to associate weakly with folate, treating the same LNCaP cells with folate-nigericin-miR-34a had no effect on reporter activity (Figure 3F).29,30 These data lend further support to the use of the high-affinity DUPA ligand over folate for delivery to PSMA-overexpressing tumors. Phenotypically, the proliferation of LNCaP cells was also significantly decreased following treatment of LNCaP with DUPA-nigericin-miR-34a (Figure 3G). Collectively, these data suggest that DUPA drives specific uptake into PSMA-expressing cells and that the incorporation of nigericin likely facilitates an earlier release of miR-34a from the endocytic vesicles and an earlier downregulation of miR-34a targets, which leads to a more significant, although modest effect on proliferation.
Enhanced stability and targeting using a fully modified version of miR-34
The modest effect on proliferation was hypothesized to be attributed to poor stability of the delivered miRNA once it escaped the endosome and encountered intracellular nucleases. To address this possibility, we utilized our recently developed fully modified version of miR-34a (FM-miR-34a). FM-miR-34a contains an alternating pattern of 2′-O-methyl and 2′-fluoro in place of the less stable 2′-OH found in ribose and phosphorothioates in place of select phosphodiester bonds (Figure 4A). The inactive (sense) strand of miR-34a is also shortened to accommodate the enhanced thermostability that occurs due to the modifications that would otherwise reduce unwinding by the endogenous miRNA machinery. Collectively, these modifications stabilize the RNA and decrease immune recognition while maintaining activity.39 Indeed, in an in vitro serum stability assay miR-34a began to degrade within 30 min, while FM-miR-34a was stabilized for at least 8 h (Figure 4B). To determine whether FM-miR-34a maintained its targeting efficacy, target gene repression in LNCaP or 22RV1 cells was evaluated following transfection. Transfection of FM-miR-34a led to a decrease in the miR-34a targets c-Myc, MET, and AR, which was either more pronounced or equal to the effects achieved with the original miR-34a or a commercially available miR-34a mimic (Figures 4C and 4D). Phenotypically, transfection of either miR-34a or FM-miR-34a decreased the invasive ability of LNCaP cells (Figure 4E).
Figure 4.

Chemically stabilized FM-miR-34a has increased stability and activity
(A) miR-34a was modified on all ribose sugars at the 2′ position and select phosphodiester bonds were changed to more stable phosphorothioate bonds to generate a FM-miR-34a. Modification positions are indicated in the schematic. (B) Both miR-34a and FM-miR-34a were incubated in the presence of 50% FBS for the indicated times followed by resolving on glycerol tolerant polyacrylamide gels. Shown is a representative image from at least three independent biological replicates. The top band, which represents the intact miR-34a is quantified in the graph. (C and D) LNCaP (C) or 22RV1 (D) cells were transfected with the indicated concentrations of miR-34a or FM-miR-34a or 50 nM of NC or miR-34a mimic (Mimic). The indicated target genes were evaluated by immunoblot 72 h later. Values below each lane represent band intensity normalized to β-actin and relative to UT (untreated). (E) Representative images (left) and quantification (right) of LNCaP cells that invaded through a matrix over 96 h following transfection with miR-34a or FM-miR-34a for 48 h prior to seeding in transwell chambers (n = 2). ∗ p < 0.05, one-way ANOVA. (F) Targeted silencing of miR-34a Renilla sensor after the transfection of LNCaP miR-34a-sensor-cells with 50 nM DUPA-FM-miR-34a, DUPA-nigericin-FM-miR-34a, or respective controls for 48 h. Data represent two biological replicates with at least six technical replicates each. ∗∗∗∗p < 0.0001, one-way ANOVA. Error bars are mean ± SD, significance was determined relative to untreated. nig, nigericin. (G) miR-34a was quantified using qRT-PCR from the cytosolic fractions obtained from LNCaP cells treated with either 100 nM DUPA-FM-miR-34a, DUPA-nigericin-FM-miR-34a, or their respective controls for 18 h. Experiment repeated twice with three technical replicates in each experiment. ∗∗p < 0.01, one-way ANOVA. Error bars are mean ± SD; nig, nigericin.
To verify that conjugation to the DUPA ligand does not alter FM-miR-34a activity, LNCaP sensor cells were transfected with DUPA-miR-34a or DUPA-FM-miR-34a and Renilla activity was quantified. Both constructs generated a significant decrease in Renilla activity, suggesting that conjugation does not affect FM-miR-34a activity (Figure 4F). To evaluate the effects of nigericin on distribution of FM-miR-34a, cells were treated with DUPA and DUPA-nigericin conjugates and miR-34a levels in the cytosol were evaluated. Similar to DUPA-nigericin-miR-34a (Figure 3D), treatment of cells with DUPA-nigericin-FM-miR-34a generated a significant increase in cytosolic miR-34a over conjugates without nigericin (Figure 4G).
Targeted delivery of miR-34a to tumors using DUPA conjugates
While only a modest effect on proliferation was observed in cells in culture, we hypothesized a greater effect in vivo due to the slower growth kinetics, which would decrease replication-mediated dilution of the miRNA. To test this hypothesis, we first evaluated the distribution of DUPA conjugates in vivo using DUPA conjugated to a near-infrared dye (DUPA-NIR) (see Figure S4 for synthesis and LC-MS validation). NRG mice with preformed LNCaP tumors were injected with a single dose (10 nmol) of DUPA-NIR and NIR signal distribution was visualized at 1, 24, and 48 h after injection. DUPA-NIR specifically labeled LNCaP tumors, but not PSMA-negative A549 tumors (Figures 5A–5D), and the signal was competed away with PMPA (Figures 5A and 5B), validating the specific binding of DUPA-NIR to PSMA-expressing tumors in vivo. The specificity of DUPA-NIR was further validated following ex vivo imaging of excised tumors and whole organs (liver, intestines, spleen, heart, lungs, and kidneys), which again confirmed that DUPA-NIR conjugates were specifically taken up by PSMA(+) tumors with minimal uptake by other organs except kidneys (Figures 5B and 5D), which is expected as murine kidneys express a high amount of PSMA. In both models, DUPA-NIR signal in the tumor was normalized to that in the kidney, which indicated between a 2.5- and 5-fold increase of DUPA-NIR in PSMA-expressing tumors (LNCaP and 22Rv1) in comparison with A549 tumors (Figure 5E). To determine the lowest dose of DUPA-NIR that results in significant tumor uptake, LNCaP tumor-bearing mice were intravenously injected with increasing doses of DUPA-NIR followed by whole animal imaging. The results indicate a significant accumulation of DUPA-NIR within LNCaP tumors with concentrations of 4 nmol or greater (Figure 5F).
Figure 5.

Evaluation of the biodistribution of DUPA conjugates in vivo
(A) Whole animal imaging of animals with LNCaP (PSMA+) xenografts following systemic injection of 10 nmol DUPA-NIR at the indicated time points (n = 3, one representative mouse from each treatment shown). Pretreatment with the PSMA inhibitor, PMPA, reduced accumulation of NIR signal in LNCaP tumors. (B) Gross images of excised LNCaP tumors and whole organs visualized for NIR signal. Data are graphically represented from all animals included in the study (n = 3). ∗p < 0.05, two-tailed unpaired t test. Error bars are mean ± SD. (C) Representative whole animal imaging of mice implanted with 22Rv1 (PSMA+) or A549 (PSMA−) cells following systemic injection of DUPA-NIR. Images were acquired 1, 24, and 48 h after delivery. Red boxes represent the tumors (n = 3, one representative mouse from each treatment shown). (D) Gross images of representative excised 22Rv1 and A549 tumors and organs visualized for NIR signal. NIR signal from tumors/organs is represented graphically from all animals included in the study, (n = 3). Error bars are mean ± SD. (E) Quantification of NIR signal in LNCaP, 22Rv1, or A549 tumors normalized to NIR signal from kidneys from experiments in (B) and (D) (n = 3). ∗∗p < 0.01, one-way ANOVA. Error bars are mean ± SD. (F) Representative whole animal imaging of mice implanted with LNCaP (PSMA+) tumors 1, 24, and 48 h after tail vein injection of the indicated doses of DUPA-NIR (left) and gross images of excised LNCaP tumors and whole organs visualized for NIR signal after the last time points (right) (n = 3).
DUPA-nigericin-miR-34a treatment reduces LNCaP tumor growth
The therapeutic efficacy of DUPA-miR-34a and DUPA-nigericin-miR-34a, conjugated to either miR-34a or FM-miR-34a, was determined using LNCaP tumor-bearing mice. Animals were treated with 4 nmol of the indicated conjugate, or the corresponding negative control (NC) via tail vein injection once every 4 days for a total of five doses (see Figure 6A for a schematic of the treatment plan). Treatment with DUPA-nigericin-miR-34a resulted in a significant reduction in tumor growth in comparison to untreated mice or mice treated with the NC based on caliper measurements (Figure 6B). Importantly, the inclusion of the FM-miR-34a (DUPA-nigericin-FM-miR-34a) generated a further significant decrease. While there was no change in body weight during the course of study for animals administered the unmodified miR-34a conjugates, there was a modest yet significant decrease in body weight in animals administered the FM-miR-34a conjugates that would need to be evaluated more extensively in future studies (Figure 6C).
Figure 6.

DUPA-Nig-FM-miR-34a treatment decrease the tumor burden of LNCaP xenografts
(A) Schematic of the treatment schedule. (B) Tumor volumes of LNCaP tumor following intravenous administration of DUPA-miR-34a, DUPA-nigericin-miR-34a, DUPA-FM-miR-34a, DUPA-nigericin-FM-miR-34a, or respective controls (n = 6 mice per group, 4 nmol, intravenous injection, once every 4 days). Error bars are means ± SEM, ∗p < 0.05, ∗∗∗∗p < 0.0001, #p < 0.05; two-way ANOVA significance determined against respective NC (DUPA-Nig-FM-miR-34a relative to DUPA-Nig-NC; DUPA-miR-34a relative to DUPA-NC) is indicated by asterisks colored relative to the group being compared; significance determined between DUPA-Nig-miR-34a and DUPA-Nig-FM-miR-34a is indicated by #. (C) Body weight measurement throughout the treatment period. Error bars are means ± SEM. ∗∗∗∗p < 0.0001; two-way ANOVA. On day 19, significance was determined against the respective NC treatment group.
Discussion
miRNA dysregulation in cancer has resulted in the interest in two potential miRNA-based therapeutic strategies. The first involves restoring tumor-suppressive miRNAs to inhibit oncogenic signals. The second is based on antagonizing oncogenic miRNAs to inhibit their function, also referred to as anti-miRNA-based therapy. Although these therapeutic strategies have shown some promising results in pre-clinical studies, the clinical application of miRNA replacement therapy has been thwarted. The reasons include the absence of a delivery method that can effectively target the miRNAs to the tumors while preventing delivery to normal tissues,10,40 the inability of the miRNAs to reach the cytosol and be incorporated into the endogenous silencing machinery, due to entrapment in endosomes,13,14,41,42 and the instability of the RNA leading to its rapid degradation and loss of activity. To address these limitations, we have developed a delivery strategy that involves attaching a targeting ligand to a relevant tumor-suppressive miRNA, together with an endosomal escape molecule. We further expanded on this design by developing and evaluating the effects of a fully modified miRNA in comparison with a partially modified version. This simple design eliminates the need for using complex components such as peptides or proteins, decreasing the cost of synthesis and enabling large-scale manufacturing. The ligand chosen was a small molecule, DUPA, which binds PSMA on the surface of prostate cancer cells with nanomolar affinity.14,35 DUPA not only facilitates specific delivery, but its structure is also amenable for the engineering of an endosomal escape agent, nigericin, without affecting its binding to the receptor. We show that DUPA-miR-34a and DUPA-nigericin-miR-34a specifically bind to and are taken up by LNCaP cells in a PSMA-dependent manner. While both DUPA-miR-34a and DUPA-nigericin-miR-34a resulted in a significant decrease of miR-34a reporter in vitro, DUPA-nigericin-miR-34a resulted in an earlier response, likely due to the early release of the miRNA from the endosomes. Phenotypically, a modest decrease in cell proliferation was observed following the treatment of cells in culture with DUPA-nigericin-miR-34a, and effect that was more pronounced in vivo. In vivo, DUPA conjugates bound specifically and rapidly to PSMA-expressing tumors with minimal uptake by normal tissues. Systemic delivery of DUPA-nigericin-miR-34a resulted in a significant delay in tumor growth in comparison with DUPA-miR-34a or the NCs. Importantly, the newly generated FM-miR-34a further enhanced the anti-tumor activity of the conjugate. Indeed, the minimally modified miR-34a used in these studies would be subject to both serum and cellular nucleases, leading to degradation and thus preventing sustained targeting. Modifications to the chemical structure of miR-34a, including changes to the ribose sugar and phosphodiester backbone, significantly increased stability and potency, leading to improved activity and could potentially decrease dosing. Nonetheless, due to some moderate weight loss, further studies will need to be done to gain a better understanding of the long-term safety of the modification pattern used.
Interestingly, in the tumors that were treated with DUPA-nigericin-FM-miR-34a, there seemed to be a significant increase in inflammatory cells in the tumors. Our initial analysis suggests that the increased inflammation in the DUPA-nigericin-FM-miR-34a group was due to neutrophilic infiltrate. Whether the neutrophils are part of an anti-tumor or tissue repair response, or are influencing a pro-tumor or immunosuppressive response, would need to be evaluated.
While the DUPA-nigericin conjugates seem to be better than those that lack nigericin, the overall activity remains modest. There are a variety of advancements that can be considered to enhance these first-generation DUPA-nigericin conjugates. Some of these include (1) increasing the nigericin stoichiometry to enhance additional endosomal escape, and (2) combining this agent with other standard-of-care treatments. Regardless, it is unlikely that miRNA therapeutics will advance as mono-therapeutic agents, especially for heterogeneous diseases such as prostate cancer. Despite the notable impact of DUPA-nigericin-FM-miR-34a on tumor growth in vivo, the tumor continued to grow, underscoring the need for a combinatorial approach. Based on the ability of miR-34a to target multiple genes involved in driving chemotherapeutic resistance, pretreating patients with DUPA-nigericin-FM-miR-34a could sensitize these tumors to standard-of-care agents, such as chemotherapeutics or androgen deprivation therapy, approaches that should be considered in the future. In conclusion, this study expands the applications of miRNA targeted therapeutics to prostate cancer and opens the way to deliver other tumor suppressive miRNAs besides miR-34a, either alone or in combination with other standard of care therapeutics.
Materials and methods
Cell culture
Prostate cancer LNCaP cells, 22Rv1 cells and the lung epithelial A549 cells (CLL-185), all mycoplasma-free as determined using MycoAlert Mycoplasma Detection Kit (Lonza), were grown using RPMI 1640 medium (Hyclone, GE Healthcare Life Sciences) supplemented with 10% fetal bovine serum (FBS) (Sigma), 100 U mL−1 penicillin and streptomycin (Hyclone, GE Healthcare Life Sciences) and the cells were maintained at 37°C in 5% CO2. LNCaP cells were grown on tissue culture dishes after coating with poly-D-lysine (Thermo Fisher Scientific).
Preparation of DUPA-miRNAs and DUPA-nigericin-miRNA conjugates
DUPA-miR-34a and DUPA-nigericin-miR-34a molecules were prepared using click chemistry reaction as previously used to prepare FolamiR molecules.11 In brief, the azide oligo (miR-34a-azide) and DUPA-DBCO or DUPA-nigericin-DBCO were mixed at a 1:10 or 1:40 M ratio in water at room temperature for 10 h followed by cooling to 4°C for 4 h to make DUPA-miR-34a or DUPA-nigericin-miR-34a single strands, respectively. An unconjugated DUPA ligand was cleaned from the reaction using Oligo Clean & Concentrator kit (Zymo Research) per the manufacturer’s instructions. miR-34a-5p (guide strand) was added to the conjugated DUPA-miR-34a or DUPA-nigericin-miR-34a single strands at a (1:1) M ratio in the presence of annealing buffer (10 mM Tris buffer, pH 7 [Sigma], 50 mM NaCl [Sigma], and 1 mM EDTA [Sigma]), followed by incubation at 95°C for 5 min, cooling to room temperature for 1 h, and then was used for cell treatment or otherwise stored at −80°C. Successful conjugation was verified by running 15% polyacrylamide gel followed by gel red staining. For experiments with the NC siLuc2, the following sequences were used: antisense strand, 5′-GGACGAGGACGAGCACUUCUU-3′; sense strand, 5′-GAAGUGCUCGUCCUCGUCCUU-3′.
Flow cytometry, immunofluorescence, and confocal microscopy
To evaluate the surface expression of PSMA, LNCaP and A549 cells were incubated with Alexa 488 anti-PSMA antibody (FOLH1, BioLegend) for 1 h on ice followed by washing with PBS (13684; Cell Signaling) and their fluorescence was measured using LSRFortessa flow cytometer (BD Biosciences) or by visualization using an Olympus IX73 microscope, Olympus DP80 camera, and CellSens 1.11. To evaluate the binding of DUPA-miR-34a-Atto.647N or DUPA-AF488 to PSMA, LNCaP, 22Rv1, and A549 cells were incubated with 100 nM DUPA-AF488 or DUPA-miR-34a-Atto.647N in the absence or presence of 10 μM PMPA at 4°C for 1 h followed by washing with PBS and measuring the fluorescence intensity using flow cytometry. To evaluate the cellular uptake of DUPA-miR-34a conjugates, 50,000 LNCaP cells were seeded on a poly-D-lysine (A3890401, Thermo Fisher Scientific) coated coverslip in a 24-well plate. Cells were treated with 100 nM DUPA-miR-34a-Atto.647 or miR-34a-Atto.647 for 4 h followed by washing with PBS and fixation using 2% paraformaldehyde in PBS for 10 min at room temperature followed by incubation with Hoechst at 1:1,000 dilution in PBS for 30 min followed by washing with PBS. After mounting the coverslips on glass slides using ProLong Glass Antifade Mountant (P36982, Thermo Fisher Scientific), cells were imaged using a Nikon A1R-MP confocal microscope and analyzed using NIS-Elements Software. In a different experiment, LNCaP cells were treated with DUPA-miR-34a-Atto-647 for 4, 8, 12, and 24 h followed by processing and imaging as mentioned above. For endosome colocalization study, LNCaP cells were incubated with 100 nM DUPA-miR-34a-Atto.647 for 4 and 8 h in RPMI complete media. Late endosomes/lysosomes were stained by adding 1 μM LysoTracker Red (L7528, Thermo Fisher Scientific) to the cells 1 h prior to the collection time point. Cells were washed with ice-cold PBS followed by processing and imaging as above. To determine the effect of nigericin on the vesicle’s size, LNCaP cells were treated with 100 nM DUPA-DBCO or DUPA-nigericin-DBCO. After 5 h, cells were washed with PBS, fixed with 4% paraformaldehyde for 15 min at room temperature. After washing with PBS, cells were permeabilized using 0.5% saponin in PBS for 10 min. To maintain permeabilization in the next steps, 0.1% saponin was added to all reagents. After incubation with 0.5% saponin, cells were incubated in blocking buffer (1% bovine serum albumin in PBS) for 30 min. Next, cells were stained with LAMP1 antibody (Cell Signaling, 9091S) overnight at 4°C, washed with PBS, and incubated with anti-rabbit secondary antibody conjugated to Alexa Fluor 488 (Thermo Fisher Scientific, A11034) and nuclear stain, Hoechst, in PBS containing 0.1% saponin. Cells were washed with PBS and processed as above for imaging using confocal microscopy. To determine the size of the vesicles, z-stacks were opened in Nikon NIS-Elements Analysis software in their native format. To identify LAMP1-positive late endosomes, a region of interest was drawn manually or using the automated tool to trace the perimeter of the cell. Vesicles that clearly represent a puncta or a hollow puncta morphology were included in the analysis. Using the distance measurement tool (measure > distance measurement …), vesicle diameter was measured along the longest dimension. A minimum of 10 vesicles per cell were measured and a total of approximately 30 vesicles were measured per treatment. The data were exported to Excel and plotted using GraphPad Prism (version 9.5.1).
qRT-PCR
To evaluate the internalization of DUPA-miR-34a, 200,000 LNCaP or A549 cells per well were seeded in 12-well plates. The next day, cells were treated with 100 nM DUPA-NC or DUPA-miR-34a in 5% FBS containing RPMI media for 18 h. Cells transfected with 10 nM DUPA-miR-34a using lipofectamine RNAiMAX (Life Technologies) served as a positive control. After washing the cells with PBS, cells were incubated with 100 μg/mL RNase A for 5 min at 37°C to degrade any surface-bounded DUPA-miR-34a followed by extensive washing with PBS to remove any leftover RNase. Total RNA was isolated using the miRneasy Kit (217004, Qiagen) according to the manufacturer’s instruction. After DNase I digestion (79254, Qiagen) to remove genomic DNA, RNA integrity was evaluated by running 1.5% agarose gel and RNA concentration was quantified using NanoDrop 2000 spectrophotometer. We used 1 μg total RNA to make cDNA using an miScript Reverse Transcriptase kit (218161, Qiagen) using HiFlex buffer per manufacturer’s instructions. Real-time PCR was performed using miScript SYBR Green PCR Kit (Qiagen) with the following primers: MiR-34a-5p (MiScript primer assay; Qiagen) and RNU6B (non-target RNA, MiScript primer assay; Qiagen). Data were then analyzed using the 2−ΔΔCt method and expressed as fold change.
DUPA-miR and DUPA-nigericin-miR activity in vitro
To evaluate miR-34a activity, miR-34a sensor plasmid was generated by inserting miR-34a target sequence into the 3′UTR of Renilla luciferase in the vector (psiCHECK, Promega). To determine DUPA-miR-34a or DUPA-nigericin-miR-34a activity, sensor cells were seeded in a 96-well plate and treated with 100 nM DUPA-miR-34a, DUPA-NC, DUPA-nigericin-miR-34a, or DUPA-nigericin-NC. Renilla values were acquired using the Renilla Glo Luciferase kit (Promega) per the manufacturer’s instructions by using a GloMax plate reader (Promega). Cells transfected with 6 nM (or otherwise indicated in the respective figure legends) miR-34a mimic were used as a positive control.
Cell proliferation assay
A sulforhodamine B (SRB, Sigma) assay was used to measure cell proliferation as previously reported.43 In brief, LNCAP cells were seeded onto a poly-D-lysine-coated 96-well plate followed by transfection with 10 nM miR-34a, let-7a, let-7b, or NC (pre-miR NC #2). At the indicated time points, cells were fixed using 10% tricholoroacetic acid in complete media. Cells were then stained with 0.4% (wt/vol) SRB in 1% acetic acid for 1 h at 37°C followed by washing unbound dye by five washes with 1% acetic acid. We used 10 mM unbuffered Tris base to extract protein-bound dye, and absorbance at 510 nm was measured using a GloMax Multi+ spectrophotometer (Promega). Absorbance values are a proxy for cell mass.
Biodistribution and in vivo efficacy studies
All experimental protocols were approved byt eh Purdue Animal Care adn Use Committtee and were in compliance with the National Instutes of Health (NIH) guidelines for animal use. To determine the biodistribution of DUPA-conjugates, LNCaP cells (5 × 106) were implanted in the flank of 6-week-old NRG mice (purchased from the Purdue animal facility) using Matrigel (Corning) at a 1:1 dilution. DUPA-NIR dye was injected systemically into each mouse followed by imaging using the Spectral Ami instrument. After the last time point, mice were euthanized, and the organs were imaged ex vivo. DUPA-NIR fluorescence intensity in the tumors and other organs was quantified using Aura analysis software. To determine the efficacy of DUPA-miR-34a, DUPA-nigericin-miR-34a DUPA-FM-miR-34a, and DUPA-nigericin-FM-miR-34a, NRG mice were implanted with LNCaP cells (5 × 106) in the right flank using Matrigel (Corning) at a 1:1 dilution. After the tumors reach approximately 150 mm3, mice were randomized and treated with 4 nmol of each conjugate or corresponding NCs. The tumor volume of each mouse was measured using a vernier caliper every 3 days and was calculated using the following formula: Tumor volume: (Length × Width2)/2. The body weight of each mouse was measured throughout the study.
Statistical analysis
Statistical analyses were performed using the Prism statistical package (GraphPad Software, version 10). The two-tailed Student’s t test was used to determine the statistical difference between two groups. One-way or two-way ANOVA was used to compare the differences between multiple groups and multiple comparisons were corrected using Dunnett’s post hoc test or Tucky’s post hoc test. Data are presented as means ± SD or means ± SEM as specified in the figure legends. Statistically significant p values are as indicated in the corresponding figure legends.
Data and code availability
The minimum datasets necessary to interpret, verify, and extend the research in the article will be made available from the corresponding author, A.L.K., upon request.
Acknowledgments
This study was funded in part by the National Institutes of Health (R01CA205420 and R01CA226259) to A.L.K., an LCRP Idea Development Award from the Department of Defense (W81XWH-22-1-1085) to A.L.K., an Early Investigator Research Award from the Department of Defense (W81XWH-21-1-0181) to I.S.S., and a grant from the NIH to the Purdue Institute for Cancer Research (P30CA023168). A.M.A. was supported by an Andrew’s Fellowship from Purdue University and a SIRG Graduate Research Assistantship from the Purdue Institute for Cancer Research. E.A.O. was the recipient of a Fulbright Award from Ecuador. K.E.O. was supported by a T32GM125620 from the National Institutes of Health. Graphical abstract was generated using BioRender.com.
Author contributions
A.L.K. conceived the project; E.A.O., A.M.A., I.S.S., and A.L.K. designed the study; A.M.A., I.S.S., S.G.I., and K.E.O. performed the cell-based and in vivo studies; E.A.O. and K.S. conducted some of the cell-based experiments; A.P.S. conducted the histological evaluation; A.M.A., I.S.S., S.G.I., K.S., K.E.O., P.S.L., and A.L.K. analyzed the data; K.S. and P.S.L. were involved in DUPA ligand synthesis, and MALDI and LC-MS verification; A.M.A., I.S.S., and A.L.K. wrote the manuscript; A.M.A., I.S.S., S.G.I., and A.L.K. edited the manuscript.
Declaration of interests
A.L.K. and P.S.L. are inventors on U.S. patent US20190298843A1 submitted by Purdue University that covers methods and uses of ligand-targeted delivery of miRNAs. A.L.K., A.M.A., and I.S.S. are inventors on U.S. provisional patent 63/454,177 submitted by Purdue University that covers methods and uses of modified miR-34a. A.L.K. is a co-founder of LigamiR Therapeutics, Incorporated, which develops ligand-conjugated microRNAs for therapeutic purpose.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102193.
Contributor Information
Ikjot S. Sohal, Email: isohal@purdue.edu.
Andrea L. Kasinski, Email: akasinski@purdue.edu.
Supplemental information
References
- 1.Orellana E.A., Kasinski A.L. MicroRNAs in Cancer: A Historical Perspective on the Path from Discovery to Therapy. Cancers. 2015;7:1388–1405. doi: 10.3390/cancers7030842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Myoung S., Kasinski A.L. RSC Drug Discovery Series. 2019. Strategies for Safe and Targeted Delivery of MicroRNA Therapeutics; pp. 386–415. [DOI] [Google Scholar]
- 3.Lin S., Gregory R.I. MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer. 2015;15:321–333. doi: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Brien J., Hayder H., Zayed Y., Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. 2018;9:402. doi: 10.3389/fendo.2018.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kasinski A.L., Slack F.J. MicroRNAs en route to the clinic: progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer. 2011;11:849–864. doi: 10.1038/nrc3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Johnson C.D., Esquela-Kerscher A., Stefani G., Byrom M., Kelnar K., Ovcharenko D., Wilson M., Wang X., Shelton J., Shingara J., et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007;67:7713–7722. doi: 10.1158/0008-5472.CAN-07-1083. [DOI] [PubMed] [Google Scholar]
- 7.Liu C., Kelnar K., Liu B., Chen X., Calhoun-Davis T., Li H., Patrawala L., Yan H., Jeter C., Honorio S., et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011;17:211–215. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li W.J., Wang Y., Liu R., Kasinski A.L., Shen H., Slack F.J., Tang D.G. MicroRNA-34a: Potent Tumor Suppressor, Cancer Stem Cell Inhibitor, and Potential Anticancer Therapeutic. Front. Cell Dev. Biol. 2021;9:640587. doi: 10.3389/fcell.2021.640587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Misso G., Di Martino M.T., De Rosa G., Farooqi A.A., Lombardi A., Campani V., Zarone M.R., Gullà A., Tagliaferri P., Tassone P., Caraglia M. Mir-34: A New Weapon Against Cancer? Mol. Ther. Nucleic Acids. 2014;3:e194. doi: 10.1038/mtna.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slabáková E., Culig Z., Remšík J., Souček K. Alternative mechanisms of miR-34a regulation in cancer. Cell Death Dis. 2017;8:e3100. doi: 10.1038/cddis.2017.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orellana E.A., Tenneti S., Rangasamy L., Lyle L.T., Low P.S., Kasinski A.L. FolamiRs: Ligand-targeted, vehicle-free delivery of microRNAs for the treatment of cancer. Sci. Transl. Med. 2017;9:eaam9327. doi: 10.1126/scitranslmed.aam9327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scaranti M., Cojocaru E., Banerjee S., Banerji U. Exploiting the folate receptor α in oncology. Nat. Rev. Clin. Oncol. 2020;17:349–359. doi: 10.1038/s41571-020-0339-5. [DOI] [PubMed] [Google Scholar]
- 13.Orellana E.A., Abdelaal A.M., Rangasamy L., Tenneti S., Myoung S., Low P.S., Kasinski A.L. Enhancing MicroRNA Activity through Increased Endosomal Release Mediated by Nigericin. Mol. Ther. Nucleic Acids. 2019;16:505–518. doi: 10.1016/j.omtn.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abdelaal A.M., Kasinski A.L. Ligand-mediated delivery of RNAi-based therapeutics for the treatment of oncological diseases. NAR Cancer. 2021;3:zcab030. doi: 10.1093/narcan/zcab030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denmeade S.R., Isaacs J.T. Development of prostate cancer treatment: The good news. Prostate. 2004;58:211–224. doi: 10.1002/pros.10360. [DOI] [PubMed] [Google Scholar]
- 16.Katzenwadel A., Wolf P. Androgen deprivation of prostate cancer: Leading to a therapeutic dead end. Cancer Lett. 2015;367:12–17. doi: 10.1016/j.canlet.2015.06.021. [DOI] [PubMed] [Google Scholar]
- 17.Östling P., Leivonen S.K., Aakula A., Kohonen P., Mäkelä R., Hagman Z., Edsjö A., Kangaspeska S., Edgren H., Nicorici D., et al. Systematic analysis of microRNAs targeting the androgen receptor in prostate cancer cells. Cancer Res. 2011;71:1956–1967. doi: 10.1158/0008-5472.CAN-10-2421. [DOI] [PubMed] [Google Scholar]
- 18.Li Q., Liu B., Chao H.P., Ji Y., Lu Y., Mehmood R., Jeter C., Chen T., Moore J.R., Li W., et al. LRIG1 is a pleiotropic androgen receptor-regulated feedback tumor suppressor in prostate cancer. Nat. Commun. 2019;10:5494. doi: 10.1038/s41467-019-13532-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamura S., Saini S., Majid S., Hirata H., Ueno K., Deng G., Dahiya R. MicroRNA-34a Modulates c-Myc Transcriptional Complexes to Suppress Malignancy in Human Prostate Cancer Cells. PLoS One. 2012;7:e29722. doi: 10.1371/journal.pone.0029722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W.J., Tang D.G., Liu X., Dougherty E.M. MicroRNA-34a, Prostate Cancer Stem Cells, and Therapeutic Development. Cancers. 2022;14:4538. doi: 10.3390/cancers14184538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mills I.G. Maintaining and reprogramming genomic androgen receptor activity in prostate cancer. Nat. Rev. Cancer. 2014;14:187–198. doi: 10.1038/nrc3678. [DOI] [PubMed] [Google Scholar]
- 22.Zhang H., Liu X., Wu F., Qin F., Feng P., Xu T., Li X., Yang L. A novel prostate-specific membrane-antigen (PSMA) targeted micelle-encapsulating Wogonin inhibits prostate cancer cell proliferation via inducing intrinsic apoptotic pathway. Int. J. Mol. Sci. 2016;17:676. doi: 10.3390/ijms17050676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sengupta S., Asha Krishnan M., Chattopadhyay S., Chelvam V. Comparison of prostate-specific membrane antigen ligands in clinical translation research for diagnosis of prostate cancer. Cancer Rep. 2019;2:e1169. doi: 10.1002/cnr2.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H., Rajasekaran A.K., Moy P., Xia Y., Kim S., Navarro V., Rahmati R., Bander N.H. Constitutive and antibody-induced internalization of prostate-specific membrane antigen. Cancer Res. 1998;58:4055–4060. [PubMed] [Google Scholar]
- 25.Boinapally S., Ahn H.H., Cheng B., Brummet M., Nam H., Gabrielson K.L., Banerjee S.R., Minn I., Pomper M.G. A prostate-specific membrane antigen (PSMA)-targeted prodrug with a favorable in vivo toxicity profile. Sci. Rep. 2021;11:7114. doi: 10.1038/s41598-021-86551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorff T.B., Fanti S., Farolfi A., Reiter R.E., Sadun T.Y., Sartor O. The Evolving Role of Prostate-specific Membrane Antigen–Based Diagnostics and Therapeutics in Prostate Cancer. Am. Soc. of Clin. Oncol. Educ. Book. 2019;39:321–330. doi: 10.1200/edbk_239187. [DOI] [PubMed] [Google Scholar]
- 27.Giraudet A.-L., Kryza D., Hofman M., Moreau A., Fizazi K., Flechon A., Hicks R.J., Tran B. PSMA targeting in metastatic castration-resistant prostate cancer: where are we and where are we going? Ther. Adv. Med. Oncol. 2021;13 doi: 10.1177/17588359211053898. 17588359211053898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flores O., Santra S., Kaittanis C., Bassiouni R., Khaled A.S., Khaled A.R., Grimm J., Perez J.M. PSMA-targeted theranostic nanocarrier for prostate cancer. Theranostics. 2017;7:2477–2494. doi: 10.7150/thno.18879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li W.J., Wang Y., Liu X., Wu S., Wang M., Turowski S.G., Spernyak J.A., Tracz A., Abdelaal A.M., Sudarshan K., et al. Developing Folate-Conjugated miR-34a Therapeutic for Prostate Cancer: Challenges and Promises. Int. J. Mol. Sci. 2024;25:2123. doi: 10.3390/ijms25042123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li W.J., Wang Y., Liu X., Wu S., Wang M., Turowski S.G., Spernyak J.A., Tracz A., Abdelaal A.M., Sudarshan K., et al. Developing folate-conjugated miR-34a therapeutic for prostate cancer treatment: Challenges and promises. bioRxiv. 2023;2023 doi: 10.1101/2023.11.25.568612. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dong Q., Meng P., Wang T., Qin W., Qin W., Wang F., Yuan J., Chen Z., Yang A., Wang H. MicroRNA let-7a inhibits proliferation of human prostate cancer cells In Vitro and In Vivo by targeting E2F2 and CCND2. PLoS One. 2010;5:e10147. doi: 10.1371/journal.pone.0010147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wagner S., Ngezahayo A., Murua Escobar H., Nolte I. Role of miRNA Let-7 and Its Major Targets. BioMed Res. Int. 2014;2014:376326. doi: 10.1155/2014/376326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nadiminty N., Tummala R., Lou W., Zhu Y., Shi X.B., Zou J.X., Chen H., Zhang J., Chen X., Luo J., et al. MicroRNA let-7c is downregulated in prostate cancer and suppresses prostate cancer growth. PLoS One. 2012;7:e32832. doi: 10.1371/journal.pone.0032832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nadiminty N., Tummala R., Lou W., Zhu Y., Zhang J., Chen X., eVere White R.W., Kung H.J., Evans C.P., Gao A.C. MicroRNA let-7c suppresses androgen receptor expression and activity via regulation of myc expression in prostate cancer cells. J. Biol. Chem. 2012;287:1527–1537. doi: 10.1074/jbc.M111.278705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kularatne S.A., Zhou Z., Yang J., Post C.B., Low P.S. Design, synthesis, and preclinical evaluation of prostate-specific membrane antigen targeted 99mTc-radioimaging agents. Mol. Pharm. 2009;6:790–800. doi: 10.1021/mp9000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Venkatesh C., Shen J., Putt K.S., Low P.S. Evaluation of the reducing potential of PSMA-containing endosomes by FRET imaging. Cancer Drug Resist. 2021;4:223–232. doi: 10.20517/cdr.2020.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kularatne S.A., Wang K., Santhapuram H.-K.R., Low P.S. Prostate-Specific Membrane Antigen Targeted Imaging and Therapy of Prostate Cancer Using a PSMA Inhibitor as a Homing Ligand. Mol. Pharm. 2009;6:780–789. doi: 10.1021/mp900069d. [DOI] [PubMed] [Google Scholar]
- 38.Rangasamy L., Chelvam V., Kanduluru A.K., Srinivasarao M., Bandara N.A., You F., Orellana E.A., Kasinski A.L., Low P.S. New Mechanism for Release of Endosomal Contents: Osmotic Lysis via Nigericin-Mediated K +/H + Exchange. Bioconjug. Chem. 2018;29:1047–1059. doi: 10.1021/acs.bioconjchem.7b00714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Abdelaal A.M., Sohal I.S., Iyer S., Sudarshan K., Kothandaraman H., Lanman N.A., Low P.S., Kasinski A.L. A first-in-class fully modified version of miR-34a with outstanding stability, activity, and anti-tumor efficacy. Oncogene. 2023;42:2985–2999. doi: 10.1038/s41388-023-02801-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nana-Sinkam S.P., Croce C.M. Clinical Applications for microRNAs in Cancer. Clin. Pharmacol. Ther. 2013;93:98–104. doi: 10.1038/clpt.2012.192. [DOI] [PubMed] [Google Scholar]
- 41.Leopold P.L. Lysosomes: Biology, Diseases, and Therapeutics. 2016. Endosomal Escape Pathways for Delivery of Biologics; pp. 383–407. [DOI] [Google Scholar]
- 42.Lönn P., Kacsinta A.D., Cui X.S., Hamil A.S., Kaulich M., Gogoi K., Dowdy S.F. Enhancing Endosomal Escape for Intracellular Delivery of Macromolecular Biologic Therapeutics. Sci. Rep. 2016;6:32301. doi: 10.1038/srep32301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orellana E.A., Kasinski A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio. Protoc. 2016;6:e1984. doi: 10.21769/bioprotoc.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The minimum datasets necessary to interpret, verify, and extend the research in the article will be made available from the corresponding author, A.L.K., upon request.