

Abstract
The CD3/T cell receptor (TCR) complex is responsible for antigen-specific pathogen recognition by T cells, and initiates the signaling cascade necessary for activation of effector functions. CD3 agonistic antibodies are commonly used to expand T lymphocytes in a wide range of clinical applications, including in adoptive T cell therapy for cancer patients. A major drawback of expanding T cell populations ex vivo using CD3 agonistic antibodies is that they expand and activate T cells independent of their TCR antigen specificity. Therapeutic agents that facilitate expansion of T cells in an antigen-specific manner and reduce their threshold of T cell activation are therefore of great interest for adoptive T cell therapy protocols. To identify CD3-specific T cell agonists, several RNA aptamers were selected against CD3 using Systematic Evolution of Ligands by EXponential enrichment combined with high-throughput sequencing. The extent and specificity of aptamer binding to target CD3 were assessed through surface plasma resonance, P32 double-filter assays, and flow cytometry. Aptamer-mediated modulation of the threshold of T cell activation was observed in vitro and in preclinical transgenic TCR mouse models. The aptamers improved efficacy and persistence of adoptive T cell therapy by low-affinity TCR-reactive T lymphocytes in melanoma-bearing mice. Thus, CD3-specific aptamers can be applied as therapeutic agents which facilitate the expansion of tumor-reactive T lymphocytes while conserving their tumor specificity. Furthermore, selected CD3 aptamers also exhibit cross-reactivity to human CD3, expanding their potential for clinical translation and application in the future.
Keywords: MT: Oligonucleotides: Therapies and Applications, CD3, aptamer, cancer immunotherapy, adoptive T cell therapy
Graphical abstract
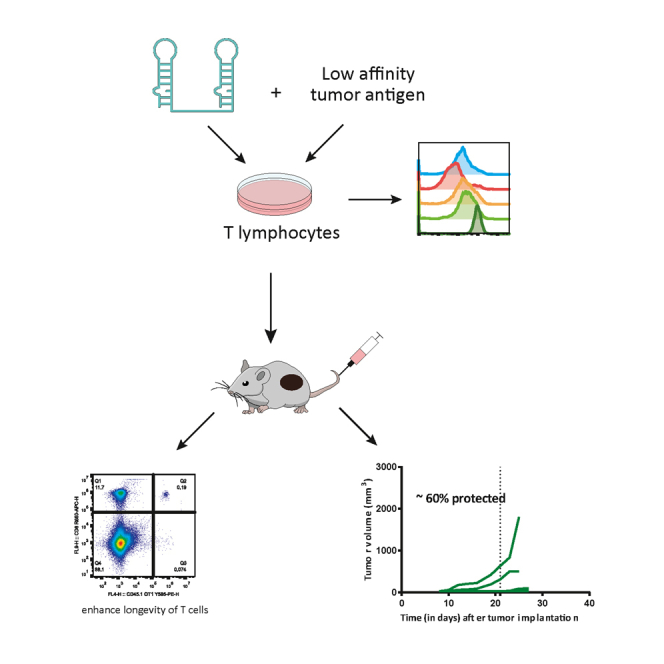
Menon and colleagues described an approach to expand T cells in an antigen-dependent manner for adoptive T cell therapy in cancer, targeting CD3δε with a multivalent aptamer. The infused T cells expanded in the presence of the CD3δε aptamer show enhanced longevity and effector functions, leading to reduced tumor growth.
Introduction
Adoptive T cell therapy is a cancer immunotherapy approach that involves ex vivo enrichment and reinfusion of genetically modified T cells (such as chimeric antigen receptor [CAR]-T cells or engineered T cell receptor [TCR]-T cells) and tumor-reactive tumor-infiltrating lymphocytes (TILs). Devising effective CAR-T cell therapy modalities is a very active area of research with a considerable clinical impact, in particular in the treatment of various hematological neoplasia.1 However, CAR-T cell-based approaches have not yet led to similarly successful clinical outcomes in the treatment of solid tumors due to a variety of factors including lack of tumor-specific targets as well as the immunosuppressive complexity of the tumor microenvironment.2 Furthermore, CAR-T cell therapy is technically cumbersome, requiring the isolation, genetic modification, and ex vivo expansion of each patient’s endogenous T cells before their reinfusion into the patient. Thus, modifying T cells to express artificial receptors that engage better with tumor targets involves processes that are laborious, time-consuming, and associated with high costs.
Other adoptive T cell therapies, such as those involving TILs, rely on boosting the pre-existing tumor-reactive TCR repertoire via tumor-extracted antigen preparations and/or recombinant interleukin-2 (rIL-2), providing a broader range of reactivity toward tumor-specific antigens, potentially improving fitness of tumor-specific T cells to enhance their survival and infiltration into the tumor milieu.3 Challenges associated with the use of TILs concern their often highly limited number upon isolation due to their scarcity at the tumor site, the difficulty of their isolation, as well as tedious ex vivo expansion protocols required to obtain adequate numbers of tumor-reactive lymphocytes needed for reinfusion. The ex vivo expansion of tumor-specific TILs is a major bottleneck in this T cell therapy because in most cases, tumor-antigen TCR recognition might be of low affinity, and the identity of their cognate antigens is generally unknown. As a consequence, the current method used to expand TILs involves polyclonal activation using agonistic CD3 antibodies. Alternatively, in case sufficient tumor tissue available, tumor lysate is used for T cell priming. Although these methods may successfully achieve the expansion of tumor-reactive T lymphocytes, underrepresented T cells harboring weak affinity TCRs may be lost or underrepresented without antigen-specific stimulation. The presence of certain markers in T lymphocytes can be of help to identify some of the potential reactive lymphocytes4 as well as from HLA unbiased genetic neoantigen screening.5 Regardless, antigen specificities of TILs are unknown in most cases and additional strategies may be applied for augmenting T cell priming, even when some specificities are known.
Developing small, artificial ligands to modulate and mold CD3/TCR conformations to improve antigen may turn out highly beneficial in efforts to improve the expansion of TILs in the presence of suboptimal amount tumor antigens.6 CD3 is a multimeric complex formed by four different chains (ε, δ, γ, ζ) that are non-covalently assembled with the TCR receptor on the cell surface of the T cell. Most of the agonistic antibodies for CD3 are directed against CD3ε. Herein we describe a group of CD3/TCR-specific oligonucleotide aptamers that were selected against recombinant tethered CD3δε dimers that functionally enhanced the antigen-dependent activation of low-affinity tumor-reactive T lymphocytes.
CD3 aptamers may present several advantages in their ability to expand tumor-reactive lymphocytes compared with other small molecules such as T cell-reactive single-chain variable fragments (scFVs) or nanobodies. The selection of aptamers is not influenced by the antigenicity or epitope immunodominance of the target protein. This characteristic likely expands the potential recognition sites of aptamers for any given protein. Once optimized for affinity, aptamers can be chemically synthesized at good manufacturing practices (GMP) grade for large-scale production with minimal cost, little batch-to-batch variability, and lyophilized for long-term storage and easy distribution compared with antibodies or recombinant proteins that require proper conformation for activity, are more susceptible to denaturation, and cumbersome to produce. Aptamers display lower half-lives than proteins in serum, with a maximum of 24–48 h, and thus degrade after the process of T cell expansion. If desired, aptamers can be neutralized with the use of a universal antidote in case of detected side effects after infusion. Last but not least, aptamers are weakly immunogenic, reducing the chances of eliciting a neutralizing anti-drug antibody response, which is likely to happen after several reinfusions of recombinant therapeutic proteins. These characteristics make aptamers customizable, versatile, and translatable for in vitro as well as in vivo therapies.7
Results
CD3 aptamer selection
To identify aptamers agonistic to the murine CD3 complex, we performed Systematic Evolution of Ligands by EXponential enrichment (SELEX) against a recombinant mouse protein consisting of the extracellular domains of the CD3 delta and epsilon subunits dimerized with an Fc tag (rmCD3δε-Fc). The human immunoglobulin (Ig)G1 recombinant protein containing an Fc tag was used for counterselection. Both proteins were mounted on Protein A-Sepharose beads and used as the scaffolding medium on which SELEX was performed.
Aptamer selection was carried out with an RNA library modified with 2′ Fluoropyrimidines that affords them resistance to RNAses and increases their half-life in vivo. The RNA library was incubated with hIgG1- Fc beads and RNA species that did not bind to the counterselection beads were collected. After the counterselection, the negatively selected RNA library was incubated with the target rmCD3δε-Fc protein-coated beads, and the RNA species bound to the beads were isolated (Figure 1A). Further restriction in each SELEX round was introduced by successively decreasing the concentration of aptamer library with the target protein (Figure 1B).
Figure 1.

Selection of aptamer by HT-SELEX
(A) SELEX procedure scheme. (B) Six rounds of SELEX were performed. During each round of SELEX, the concentration of the aptamer library was decreased by a factor of 3, by increasing the volume of the binding step. (C) Several candidate aptamers were enriched after selection. The variable regions of the 11 most enriched aptamers are depicted above. Shared motifs in the aptamer sequences are highlighted. (D) Motifs shared among candidate aptamers were enriched in the Round 6 SELEX library. (E) Phylogenetic tree depicting the distribution of candidate CD3 aptamers. Graphical representation of candidate aptamers according to degree of similarities and differences in their nucleotide sequences. Similar species are clustered in families represented by each arm of the tree.
After six rounds of selection, the affinity of library to the rmCD3δε-Fc was in low μmolar range (Figure S1), the library was reaching a considerable enrichment as measured by Sanger sequencing (Figure S2). Thus, the enriched DNA library from R5 and R6 was sequenced using Ion Torrent high-throughput sequencing (Figure S3). Clustal Omega and FASTAptamer were used to rank and cluster the most enriched hits and candidate aptamers were resolved. Aptamers 1, 2, 3, 4, 5, 6, 7, 9, 11, 12, and 40 were chosen for preliminary characterization (Figure 1C).
Candidate aptamers were analyzed for their nucleotide composition, and several motifs, or recurring, shared nucleotide sequences were identified. Their distribution in the candidate aptamers (Figure 1D) as well as in the SELEX library as a whole was analyzed (Figure 1E).
Candidate aptamers were highly diverse—different motifs characterized different aptamers. Two to four potential iterations of motifs were shared among the aptamers (Figure 1C). Aptamers 2, 3, and 11 contained three shared motifs each, the highest accumulated number of motifs among all the candidate aptamers (Figure 1C). CGATTGATTC, GGCCTT, and GTCCCCTCGT were the top three motifs enriched in the Round 6 SELEX library (Figure 1D). CGATTGATTC and GTCCCCTCGT stood out because they were each 10 base pairs (bp) in length (i.e., approximately one-fourth of the total aptamer length). Aptamers 1 and 2 shared both these motifs, so that approximately half of their nucleotide bases were identical (Figures 1C and S4). The preferential representation of these motifs in the aptamers as well as the entire SELEX library suggested that these might have been conserved species that played an essential role in binding to the target CD3δε protein.
Motifs CCTACC and AACTC were the most abundantly shared motifs among the candidate aptamers (Figure 1D). Aptamers 3, 7, 9, and 11 were particularly similar, sharing two motifs each (Figure 1C). Aptamers 2, 3, and 11 contained three shared motifs each, the highest accumulated number of motifs found in all the candidate aptamers (Figure 1C). Aptamers 1 and 5 had only one of the shared motifs each (Figure 1C).
Aptamers were graphically represented according to their degree of similarity and differences from each other in the entire SELEX library by grouping according to their motifs (Figure 1E). Candidate aptamers 1, 2, 3, 5, and 12 were located in non-overlapping phylogenetic groups, indicating the diversity in the candidate aptamer pool (Figure 1E).
Binding of CD3 aptamers to target receptor
Aptamers 1, 2, 3, 4, 5, 6, 7, 9, 11, and 12 were screened via various methods for specific binding to the recombinant murine CD3-Fc protein. The nitrocellulose filter binding assay, also known as the P32 double-filter assay, was used as the first screen to assess the various aptamers for their ability to bind CD3 target protein and weed out aptamers that bound to the counterselection protein human IgG1 (hIgG1) (Figure 2A). Radiolabeled aptamers 1, 3, 5, and 12 were found to bind the target protein specifically as judged by their robust binding to the target protein and lack of binding to the control hIgG1 protein used for counterselection. These aptamers were chosen for further characterization. Aptamers 7, 9, and 11 showed concomitant binding to hIgG1, making them non-exclusive binders of rmCD3δε (Figure 2A) and were hence excluded from further analysis. We chose to continue the characterization with aptamers 1, 3, 5, and 12 as they seem to be quite different among them when their conserved motifs are compared, and they were easier to produce at higher yields.
Figure 2.

CD3 aptamer binding characterization in vitro
(A) Binding of P32 radiolabeled CD3 aptamers to rmCD3 target protein and hIgG1 control protein in a nitrocellulose binding assay. CD3 candidate aptamers were radioactively labeled, and co-incubated with serially diluted CD3 control protein or hIgG1 target protein. Aptamers 1, 3, 5, and 12 were determined to be specific binders to the target protein rmCD3. (B) Aptamers 1, 3, 5, and 12 bind to rmCD3 target protein as determined by surface plasmon resonance. (C) Binding of fluorescently labeled CD3 aptamers to recombinant murine-CD3δε target protein-coated beads. (D) Binding of fluorescently labeled CD3 aptamers to recombinant human-CD3δε target protein-coated beads and not to human IgG1 nor to murine-CD3γε target protein-coated beads.
We next quantified target-specific binding of aptamers 1, 3, 5, and 12 via surface plasmon resonance (Figure 2B). Biotinylated monomeric aptamer at 1-μM concentration was immobilized onto the wells of a streptavidin-coated sensor chip. We then added the analyte, i.e., the recombinant rmCD3δε-Fc protein in solution at different concentrations to determine the equilibrium constant of their binding to the immobilized aptamers. Fc-tagged control protein was also included to determine the level of nonspecific binding.
We calculated the KD, or binding affinity, of each aptamer and observed that aptamer 5 was the strongest binder (37.9 nM), followed by aptamer 1 (91.3 nM), aptamer 12 (206 nM), and aptamer 3 (430 nM) (Figure 2B). The differences in KD seem to be contributed by the kd, or rate of dissociation, of each of the aptamers. Aptamer 1 appeared to induce the highest resonance units produced upon interaction with the target protein, suggesting its superior proclivity to associate with the target protein.
We further confirmed binding of the aptamers at the protein level via flow cytometry assays using Tosylactivated Dynabeads coated with CD3δε-Fc thorough covalent bound with accessible Lys of the recombinant protein (Figure 2C). To fluorescently label the aptamers, we added a 23 bp long nucleotide extension to our monomeric aptamers which was complementary to an oligonucleotide probe containing the Cy5 fluorophore. After probe-hybridization we purified labeled species to perform flow cytometry. A randomized sequence of same length and constant flanking sequence was also fluorescently tagged and used as a negative control aptamer for all measurements. Furthermore, the Cy5-oligonucleotide probe alone was included in the assay to assess the degree of nonspecific binding between fluorophore and bead. As shown in Figure 2C, aptamers 1, 3, 5, and 12 bound to rmCD3δε-coated and rhCD3δε-coated beads, but not to beads functionalized with control protein IgG1 or rmCD3γε-coated beads (Figure 2D).
To determine the binding of the aptamer to the CD3 native complex expressed on the surface of T cells, we performed flow cytometry assays with the murine EL-4 T cell line, applying Cy5-labeled monomeric forms of the CD3 aptamers (Figure S5). As monomers, none of the CD3 aptamers supported specific binding to EL-4 cells when compared with the control aptamer. We reasoned that CD3δε as part of a complete and properly assembled TCR/CD3 complex on the cell surface adopts a conformation that deviates from that featured by the recombinant soluble CD3δε dimer, which would explain a lower aptamer-affinity, or the density of the CD3 complex in the cell was lower than in rmCD3δε-coated beads. It has been extensively reported that aptamers selected against a recombinant soluble protein can display lower affinity when tested against the native protein expressed on the cell membrane.8 To increase probe avidity we tetramerized our aptamers by hybridizing the extended monomeric aptamers with the complementary probe modified at its 3′ end with a biotin group instead of the Cy5 probe, followed by incubation with phycoerythrin (PE)-conjugated streptavidin (Figure S6). With these reagents we were able to show specific binding to the EL-4 cells (Figure S5). The same strategy of biotin- and tetravalent streptavidin-mediated pMHC-tetramerization is routinely applied to track T cells in an epitope-specific manner, as TCR-peptide-major histocompatibility complex (pMHC) affinities are typically insufficient to support stable TCR-pMHC binding.9 Binding was also observed with the use of the human TCR/CD3+ Jurkat T cell line (Figure S7).
We also stained primary splenocytes freshly isolated from lymph nodes of C57/BL6 mice with tetramerized CD3 aptamers (Figure S8). All the CD3 aptamers show binding to CD8 T cells above the background observed with the tetramerized control aptamer. CD19+ B cells in the same cell preparation showed some binding of CD3-selected tetramerized aptamers, though lower than to T cells (Figure S8). In order to better clarify this result, we used an additional assay to confirm CD3 binding specificity.
Through CRISPR-Cas9-mediated targeting of the CD3δ, CD3ε, and Cd3γ genes followed by fluorescence-activated cell sorting and expansion of TCR-negative cells, we were able to generate EL-4 CD3δKO, EL-4 CD3εKO, and EL-4 CD3γKO cell lines. We performed flow cytometry assays to compare the patterns of CD3 aptamer binding observed with wild-type (WT) EL-4 cells and the CD3KO EL-4 cells (Figure 3A).
Figure 3.

CD3 aptamer binding to CD3-expressing cells
(A) Binding of CD3 aptamers to wild-type EL-4 cells and different CD3 chain KO cells. (B) Quantitative representation of the specificity of binding of CD3 aptamers to CD3+ WT EL-4 cells as compared with CD3δKO, EL-4 CD3εKO, and EL-4 CD3γKO cell lines. We calculated the percentage of reduction in stain index of each of the aptamers in each of the CD3KO cell lines, as compared with their stain index in CD3+ WT cells. All stain indexes were normalized to the signal of the control aptamer. A reduction in stain index in the CD3KO cell line compared with the WT cell line is indicative of specific binding (mean ± SEM). n = 2–3. Two-way ANOVA was performed as compared with the basal stain index. ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05. Anti-CD3 antibodies 17A2 and 2C11 were used as positive control.
We concurrently stained WT and CD3KO EL-4 cells with a sub-saturating dose of Streptavidin-tetramerized biotinylated CD3 aptamers. When overlaying the staining of WT EL-4 cells and CD3KO EL-4 cells, we noticed a reduction in staining with aptamer 12 in CD3εKO and CD3γKO cells. Aptamers 1, 3, and 5 showed varying levels of specificity to the three CD3 subunits, but to a lower extent than aptamer 12 (Figure 3B).
Staining of the knockout (KO) cell lines with the tetramerized aptamers gave rise to various degrees of reduction in binding when compared with staining of T cells. This may indicate that the aptamers recognized the CD3 complex at the cell surface with some nonspecific background related to weak binding to other cell surface components.
CD3 aptamers elicit T cell activation and reinvigorate weak TCR-pMHC engagers
We next studied the functional potential of the CD3 aptamer to activate T cells in vitro. We first measured changes in intracellular second messenger calcium, one of the first downstream signaling events ensuing engagement of the TCR/CD3 complex by pMHC or stimulatory antibodies. For this we carried out live-cell ratiometric calcium imaging experiments to monitor activation levels of T cells upon engagement with CD3 agonistic aptamers. More specifically, we decorated glass-supported planar lipid bilayers (SLBs)—synthetic plasma membranes—with monomeric CD3 aptamers titrated at different densities and monitored the extent of calcium flux induced in murine 5c.c7 TCR-transgenic T cells. The use of fluid-phase SLBs resulted in laterally mobile SLB-anchored proteins, which served to stimulate T cells and the distribution of which could be followed over time.
We sought to construct two types of SLBs, one with which we could study the aptamers’ endogenous activation capacity (Figure 4A, aptamers and pMHC with B7-1 and ICAM-1), and a second in which we could study how the aptamers influence signaling in the presence of a suboptimal T cell stimulus in the form of the cognate ligand of the 5c.c7 TCR-transgenic T cells, i.e., the moth cytochrome c peptide embedded within the murine MHC class II protein IEk (IEk/MCC, Figure 4B, aptamers without pMHC, but with B7 and ICAM). In addition, SLBs contained ICAM-1 for adhesion and B7-1 for co-stimulation.
Figure 4.

CD3 aptamers boost and induce T cell activation
(A) Two types of CD3 aptamer-containing SLBs were created; one containing biotinylated CD3 aptamers (attached via ABS635P-labeled monovalent streptavidin), pMHC, B7-1, and ICAM-1 (A) to measure the capacity of CD3 aptamers to induce calcium flux in the presence of suboptimal pMHC T cell stimulus, and one without pMHC (B) to study the aptamers’ endogenous activation capacity. (C) Dose-response curves for T cell activation of CD3 aptamers ± suboptimal pMHC stimulus. Apt Ctrl, Apt 1, and Apt 12 were decorated at various ligand densities on SLBs with or without suboptimal pMHC. The percentage of activated cells was determined for each condition and normalized to the positive control (100% T cell activation). SLBs ranked from lowest to highest activation thresholds: Apt 12 + pMHC < Apt 1 + pMHC < Apt 12 < Apt 1 < Apt Ctrl + pMHC < Apt Ctrl. Dose-response curves were analyzed as described in Hellmeier et al.10 to extract activation thresholds (D and E). For all calcium flux experiments, 13 ligand densities were assayed. n = 13 SLBs. A two-sided Bootstrap ratio test hoc was performed, the extracted values are extracted from a fit, and error bars represent the 95% CI. ∗∗∗p value < 0.001.
We recorded the changes in intracellular calcium in 5c.c7 TCR-transgenic T cells in the SLBs decorated with various surface densities of CD3 aptamers with pMHC suboptimal stimulus (Figure 4C). The ligand densities at half-maximum response (activation threshold) were determined as described in Hellmeier et al..10
Indeed, CD3 aptamers 1 and 12, when tethered on an SLB, lowered the activation threshold both in absence and presence of a suboptimal pMHC stimulus (Figures 4C–4E), as compared with a control aptamer based on a scrambled sequence generated from aptamer 1—of same length and composition (Figures 4C–4E). This suggests a specific T cell activating potential mediated by the CD3 aptamers.
To test the valency of CD3 aptamers needed to induce T cell proliferation, we created various constructs based on CD3 aptamer 1—a monomer, a dimer, and various oligomers (Figures 5A and 5B). The CD3 dimer consisted of a 155-bp-long aptamer transcribed in vitro as a single transcript bringing together two CD3 monomers. Two monomers were arranged side by side, adjacent to each other, separated by a 3-bp long nucleotide linker in the construct (Figure S9).
Figure 5.

Multimeric CD3 aptamers enhance proliferation of suboptimally activated T cells
(A) Design of monomers, dimers, and multimers of the CD3 aptamers. (B) Monomers, dimers, and multimers of the CD3 aptamer 1 were co-incubated with naive CD8+ T cells with or without suboptimal anti-CD3 antibody stimulus in CFSE dilution-based proliferation assay by flow cytometry. (C) Representative images of CFSE dilution observed in CD8 T cells after 72 h, and (D) percentage of proliferation (mean ± SEM; triplicates). Ordinary one-way ANOVA with a Bonferroni post hoc test was performed. The data are representative of three independent experiments. (E) Dimers of CD3 Apt 1 and 12 significantly enhance ICOS and CD69 expression in CD8 T cells in vitro. Naive CD8 T cells were suboptimally activated with an anti-CD3 antibody and co-incubated with CD3 aptamers. CD69 and ICOS expression after 72 h of stimulation is shown. n = 3 from the same experiment, although independent experiments were repeated with similar results more than two times. Data are represented as mean ± SEM. Ordinary one-way ANOVA with a Bonferroni post hoc test was performed. (F) Quantification of CD69 expression (mean ± SEM; triplicates). (G) Quantification of ICOS expression (mean ± SEM; triplicates). Ordinary one-way ANOVA with a Bonferroni post hoc test was performed. Data shown are representative of three independent experiments.
To arrive at higher-order multimers of the CD3 aptamers, a single-stranded DNA (ssDNA) scaffold was used. CD3 aptamer monomers and dimers were transcribed with a 23-bp extension complementary to the scaffold, which consisted of five concatenated repetitions of 23-bp-long sequences, each one complementary to the extensions added to modify the CD3 aptamers. In this fashion, we were able to oligomerize five CD3 monomers or dimers onto the same nucleic acid support via hybridization. Hence, in addition to monomeric and dimeric CD3 aptamers, we also constructed multimeric CD3 oligomers by scaffolding monomeric CD3 aptamers. Moreover we produced multimers of the CD3 dimers as shown in Figure 5A.
Next, we assessed the capacity of CD3 aptamers to induce T cell proliferation in presence or absence of suboptimal amounts of a stimulatory anti-CD3 antibody. The affinity of the dimeric aptamer was determined by OCTET and was 25.2 nM for Apt1 dimer and 218 nM for Apt12 dimer (Figure S10). The results of the carboxyfluorescein succinimidyl ester (CFSE)-based proliferation assays clearly indicated that monomeric aptamers were not able to induce proliferation of primary T cells in vitro, regardless of whether CD3 antibody had been present or absent (Figure 5B). Remarkably, after aptamer dimerization or oligomerization via ssDNA-mediated scaffolding, T cell proliferation occurred in both an aptamer and CD3 antibody-dependent fashion. The mere presence of multimeric aptamers or CD3-antibody alone failed to induce T cell proliferation, suggesting that the multiple valency of the CD3 aptamer was critical for function, albeit in the presence of anti-CD3 antibodies.
Given the potency of dimeric aptamers as well as their ease of production, we focused on the dimeric versions of CD3 aptamers 1 and 12 for further studies. We also generated a scrambled aptamer Ctrl dimer to use as control. As shown in Figures 5C and 5D, dimeric CD3 aptamers 1 and 12 enhanced CD3-mediated proliferation of polyclonal T cells in vitro, when compared with the control aptamer. After a 72-h stimulation, they also promoted expression of CD69, an early activation marker, and of ICOS, a late-stage activation marker of T cells (Figures 5E–5G). In a similar and physiologically relevant manner, CD3 aptamers were also able to enhance the proliferative ability of transgenic T cells suboptimally stimulated by their cognate antigenic stimulus. OT-1 TCR-transgenic T cells were suboptimally stimulated with the high-affinity SIINFEKL peptide (∼0.6 nM) and low-affinity SIIGFEKL peptides (∼56 nM).11 SIINFEKL and SIIGFEKL both activated OT-1 TCR-transgenic T cells, but their half-maximal proliferation induction capacity was approximately three orders of magnitude apart. In the presence of aptamers 1 and 12, OT-1 T cells showed enhanced proliferation as determined by CFSE dilution (Figure 6A). The capacity of aptamer 1 to further support the activation of OT-1 TCR-transgenic T cells via the low-affinity peptide SIIGFEKL was further confirmed by 3H-thymidine incorporation assays (Figure 6B) and interferon (IFN)-γ production (Figure 6C).
Figure 6.

Dimeric CD3 aptamers enhance proliferation of suboptimally activated CD8+ OT-1 T cells
(A) Naive CD8 + OT-1 T cells were co-incubated with varying concentrations of SIINFEKL (high-affinity OT-1 peptide) and SIIGFEKL (low-affinity OT-1 peptide) and constant dose of CD3 aptamers. Rate of proliferation after 72 h was assayed via CFSE dilution. Proliferative effect of CD3 aptamers was only observed when there was suboptimal proliferation induced by the agonistic peptides. CD3 aptamers 1 and 12 enhanced the proliferative index of OT-1 T cells stimulated with both peptides, but with different patterns. Representative images of this assay are shown above. Independent experiments were repeated with similar results n > 3 times. (B) Naive OT-1 splenocytes were suboptimally activated with SIINFEKL (high-affinity OT-1 peptide) and SIIGFEKL (low-affinity OT-1 peptide), and CD3 aptamers and thymidine 3H incorporation was determined by scintillation. Data are represented as mean of n = 4 technical replicates of one experiment (mean ± SEM). One-way ANOVA followed by a Bonferroni post hoc test was performed. “SIIGFEKL + Apt Ctrl” vs. “SIIGFEKL + Apt 1” p value = 0.0002. “SIIGFEKL” vs. “SIIGFEKL + Apt 1” p value <0.0001. (C) Naive OT-1 splenocytes were suboptimally activated as in (B), and supernatant was collected to measured IFN-γ secretion by ELISA. Data are represented as mean of n = 4 technical replicates of one experiment (mean ± SEM). One-way ANOVA followed by a Bonferroni post hoc test was performed.
We also measured the effect of CD3 aptamers on the proliferation of TCR-transgenic PMEL cells, T cells that react with EGSRNQDWL with an intermediate affinity (∼4.5 nM)11 to endogenous murine gp100 (mgp100) tumor peptides presented on murine melanoma cells. We suboptimally activated PMEL splenocytes with their endogenous murine gp100 peptide, with or without co-incubation with CD3 agonistic aptamers. Again, the presence of both aptamers 1 and 12 dimers enhanced proliferation of PMEL T cells compared with the aptamer control, as was indicated by increased 3H-thymidine incorporation in these groups (Figure S11).
Results suggest that CD3 aptamers enhance proliferation and activation of both low-affinity and high-affinity TCRs. With these two CD3 aptamers we might be able to target a large repertoire of T cells with a wide range of affinities for various tumor antigens. The versatility of the CD3 aptamers to function in these TCR-pMHC affinity ranges indicates that they could be potent tools to enhance already existing T cell responses or reinvigorate T cells by providing them with a CD3-based stimulus.
CD3 aptamers enhance in vivo antitumor activity in adoptive cell transfer therapy models
In vitro experiments led us to hypothesize that CD3 aptamers might be able to lower the antigen threshold for stimulating tumor-reactive T cells. We next sought to study the consequences of CD3 aptamer treatment on the in vivo antitumor response in murine tumor models. To address whether CD3 aptamers could be used to expand T cell populations for enhancing the adoptive transfer of T cells for cancer immunotherapy, we stimulated and expanded T cells ex vivo with low- or high-affinity TCR ligands and tested their protective capacity against a high-affinity ligand-expressing tumor. To this end we suboptimally activated OT-1 T cells in the presence of the low-affinity SIIGFEKL peptide (10 nM) in the presence of CD3 aptamer control or CD3 aptamer 1 (Figure 7A). In a parallel experiment, OT-1 T cells were stimulated with 1 pM of the high-affinity SIINFEKL peptide in the presence of CD3 aptamer control or CD3 aptamer 12 (Figure 7A). We also activated OT-1 T cells with an optimal concentration of affinity SIINFEKL peptide to produce highly activated effector OT-1 T cells that were used as a positive control of the study.
Figure 7.

CD3 aptamers enhance antitumor activity of adoptive cell transfer therapies in vivo
(A) Scheme of experiment. OT-1 T cells were activated suboptimally with SIINFEKL and aptamer 12, or SIIGFEKL and aptamer 1, respectively. After in vitro expansion, these tumor-reactive T cells (0.5 million cells/mouse for SIINFEKL groups; 1 million cells/mouse for SIIGFEKL groups) were adoptively transferred into mice bearing B16OVA tumors, and tumor growth was measured. (B) Individual tumor growth curves of each experimental group. On day 21 after tumor implantation, the proportion of tumor-bearing mice was calculated. Aptamer 1 enhanced the efficacy of SIIGFEKL-activated “low-affinity” OT-1 T cells in vivo. Aptamer 12 enhanced the antitumor efficacy of SIINFEKL-activated “high-affinity” OT-1 T cells in vivo. n = 4–6 mice per group. (C) Adoptively transferred T cells pre-treated with CD3 aptamer 1 exhibit enhanced in vivo longevity at day 21 in tumor inoculated mice as in (B). Representative flow cytometry data showing enhanced accumulation of OT-1 T cells in mice that received adoptive transfer of SIIGFEKL-activated OT-1 cells pre-treated with or without CD3 aptamers. (D) Quantification of OT-1 T cell accumulation in the blood of mice, upon transfer after each of the in vitro treatments (mean ± SEM). ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05.
As a cancer immunotherapy treatment, activated OT-1 T cells were adoptively transferred into mice bearing B16OVA melanoma tumors, and tumor growth was monitored (Figure 7B). Twenty-one days after tumor implantation, and 18 days following adoptive T cell transfer, most mice treated with OT-1 T cells activated with 10 nM of SIINFEKL (positive control), as expected, were protected, with five of six mice remaining tumor free (Figure 7B). Mice that received OT-I T cells expanded with the low-affinity peptide SIIGFEKL at 10 nM either alone or with the control aptamer showed no delay in tumor growth, with only one of six mice protected. Strikingly, the inclusion of aptamer 1 dimers in the activation cocktail with SIIGFEKL led to a higher protection rate with four of six mice being protected (Figure 7B). In the same manner, OT-1 T cells suboptimally activated with 1 pM of SIINFEKL in the presence of aptamer 12 dimer showed enhanced antitumor response in vivo with four of six mice remaining tumor protected (Figure 7B).
In a separate experiment, we also observed that co-treatment with aptamer 1 during the activation step before adoptive transfer of T cells pre-conditioned them for enhanced longevity and survival in vivo, with persistence in blood up to 3 weeks after adoptive transfer and approximately 2 weeks after tumor implantation (Figures 7C and 7D).
We hence conclude that murine CD3 aptamers can be used as therapeutic agents to deliver agonistic signals to murine T cells. In vitro, they surely enhance the activation and proliferation of murine antigen-specific T cells, giving rise to enhanced expansion, longevity, and in vivo antitumor response.
Discussion
Anti-CD3 agonistic antibodies are important tools used for the expansion of polyclonal T cells in vitro. Herein we identify a panel of aptamers that recognize CD3 complex and enhance T cell responses toward suboptimal levels of anti-CD3 antibodies or pMHC. Thus, the aptamers described allow the expansion of clonal reactive T cells driven by specific pMHC ligands. Importantly, in vitro treatment with CD3 aptamers provides a more robust antitumor response and T cell persistence in preclinical models of adoptive T cell antitumor therapy.
The CD3 complex of the TCR is an excellent target for adjuvanting T cell activation because of its inherent nature as the exclusive signaling machinery of the TCR complex. Compared with targeting the antigen-specific alpha-beta TCR chains to engage and activate the T cell, the CD3 complex provides an intervention site that is modular, fine tunable and an invariant target shared by all T lymphocytes.12
The CD3 protein complex consists of homologous CD3δε and CD3γε subunits that could both serve as potential targets for extracellular binding and agonism of CD3. We chose to identify aptamers to especially target the extracellular CD3δε subunit because of several additional advantages it may provide for T cell immunotherapy. First, the CD3δε consists of the delta and epsilon chains, both of which play more important roles in T cell development and function when compared with the CD3 gamma chain. Human immunodeficiencies involving the deletion of the CD3 epsilon or CD3 delta have more severe SCID symptoms than those compared with gamma chain deficiencies, suggesting a hierarchical d > g relationship within CD3 subunits.13,14,15,16 Second, a unique and necessary functional role of the CD3δε subunit is further supported by studies that demonstrate extracellular regions of the CD3δε are necessary for ERK/MAPK signaling17 as well as zeta-chain phosphorylation requisite for T cell activation.18 Introduction of a “tail-less” CD3δε subunit into CD3delta−/− mice with compromised zeta phosphorylation and MAPK signaling restored normal T cell function and maturation. This specifically implicates the role of a cryptic extracellular domain on the CD3δε necessary for its agonistic activity, and may serve as a target for aptamer docking as well.17,19 Last, during intracellular TCR/CD3 assembly, after the alpha-beta TCR chains, CD3δε seems to be the second subunit that incorporates into the growing TCR complex. The alpha-beta chains pair with the CD3δε subunit before the gamma-epsilon and zeta-zeta dimers, perhaps hinting at a hierarchical CD3δε > CD3γε relationship in TCR assembly and function.20
The capacity to expand autologous T cells in vitro for reinfusion spearheaded the way for the development of adoptive T cell therapy in cancer patients with TILs. However, successful expansion of TILs is not always guaranteed, and largely depends on initial numbers of isolated TILs, their overall fitness, and TCR specificity. Of note, tumors are often enriched in bystander T lymphocytes that are not tumor reactive.21 Thus, a pan-TCR T cell mitogenic signal such as the one provided by CD3 agonistic antibody induces proliferation of all T cells regardless of tumor specificity. Since tumor-resident bystander T cells often display a less exhausted phenotype,21 their presence may well bias anti-CD3-driven T cell expansions toward tumor non-reactivity, which would pose a serious concern in T cell-based adoptive therapies.
The advances in cancer genomics and proteomics allow for the identification of intrinsic tumor-specific neoantigens that can be used to expand autologous reactive TILs for each patient. In this context, it will be highly desirable to selectively enhance the proliferation of antigen-specific T cells with the use of a CD3 engager such as the aptamer dimers presented in our study.
CD3 aptamers may serve as a therapeutic agent to expand antigen-specific T cell populations as they enhance the proliferation and activation of tumor-reactive T cells in vitro in the context of appropriate tumor-specific antigenic stimulus. Further investigations will help to evaluate how these aptamers fare in expanding different TIL populations, and whether the expansion timeline may be shortened for faster development of TILs. Moreover, CD3 aptamers can be prepared in vitro by bulk chemical synthesis in a highly feasible and economical fashion compared with the preparation of cell-derived products such as antibodies.
Herein, we isolated anti-CD3 aptamers able to bind to the CD3 complex using HT-SELEX. The binding of CD3 on T cells was reduced when different chains of the CD3 complex (δ, ε, and γ) were knocked out using CRISPR-Cas9 (Figure 3). Intriguingly, loss of binding was not completely abrogated in the different CD3 KO cell lines, as it would have been expected; the CD3 complex needs to be properly folded with all the chain and the TCR to be expressed on the cell membrane.22 In fact, the antibodies showed a complete loss of CD3 binding with either (δ, ε, or γ) of the CD3 KO cell lines. This difference with the aptamers might be explained (1) if there is some cross-reactivity with other receptors with shared binding motifs (aptatopes); (2) by the expression of residual misfolded CD3 complex on cell membrane that can still be recognized by the aptamers but not by the antibodies; and (3) if the aptamers are small and might be internalized to some extent by free uptake reaching the misfolded CD3 complex in the ER of the T cells. Further experimentation will be required to address these possibilities, nonetheless the fact that CD3 aptamers lead to T cell activation when combined with a TCR-pMHC stimulus or by themselves, when tethered on an SLB, makes them amenable to use for adoptive T cell expansion in vitro (Figure 4). As a matter of fact, by expanding suboptimally activated T cells in the presence of CD3 aptamers 1 and 12, we observed enhanced proliferation and activation of T cell populations, as determined by the increased expression of CD69 and ICOS as well as proliferative index of these T cells (Figure 5). Tumor-reactive T cells that may be described to simulate “low-affinity” (OT1-SIIGFEKL) as well as “high-affinity” (OT-1-SIINFEKL) T cells, were both expanded in vitro, showing the versatility of the CD3 aptamers at targeting T cells of a diverse repertoire and affinities (Figure 6). CD3 aptamers do not seem to compromise the quality of the expanded T cells. In fact, they enhance their state of activation as well as in vivo longevity after adoptive transfer. CD3 aptamer 1 pre-treated adoptively transferred T cells could be detected in the blood stream of mice even after 3 weeks of adoptive transfer. More importantly, CD3 aptamer expanded T cells showed enhanced antitumor capacity in vivo. Tumor-bearing mice that received CD3 aptamer pre-treated OT-1 cells showed a reduction and delay in tumor growth compared with the control groups (Figure 7). Aptamer 1 enhanced the antitumor activity of low-affinity SIIGFEKL-activated OT-1 cells, and aptamer 12 enhanced the antitumor capacity of high-affinity SIINFEKL-activated OT-1 cells. Thus, using either of the aptamers by itself, or a combination of them, we can customize the targeting of T cell clones with known specificity and TCR affinities to expand them favorably and efficiently, augmenting their antitumor in vivo capacity.
The ability of the CD3 aptamer to exert its functional effects in a TCR-specific manner in stimulus-experienced cells makes it particularly suitable to reinvigorate antigen-experienced T cells in the tumor microenvironment while mitigating the risk of expanding bystanders or naive non-tumor-reactive T cells.
Previous studies have used LIGS-SELEX to identify human anti-CD3 DNA aptamers that bind to the same epitope of OKT3 and UCHT1 antibodies.23 These CD3 DNA aptamers when multimerized can also trigger T cell activation, which so far has not been evaluated for adoptive cell therapy T cell expansion.24 Our group has more experience with RNA aptamer selection and that is one the reasons we perform 2’F-RNA CD3 aptamer SELEX. We also believe that RNA structure shows even more diverse structure than DNA, which might be reflected in a larger chemical space increasing the chances of selecting aptamers that can exert a higher range of interactions to a chosen target. In nature, functional RNA-structured elements dominate over DNA, for instance ribosome, tRNAs, lncRNA, riboswitches, ribozymes, etc.25 One could argue that this is because DNA in cells has evolved to store the genetic information required to be as dsDNA, and ssDNA in nature is mostly avoided. However, there are other factors that indicate a higher structure complexity of RNA: (1) The presence of 2′OH in the ribose of RNA gives more hindrance to the molecule and pushes the ribose ring toward a C3-endo pucker26 with the possibility of forming other non-canonical interactions that increase the structure diversity.27 (2) Uracil base binds mainly to adenine as thymidine does, but uracil is more permissive interacting with other bases in certain circumstances by wobble base pairing giving to the RNA a higher range of possible structures.28 We have detected CD3 aptamer binding to human CD3+ Jurkat cells, indicating certain human-mouse cross-reactivity of our aptamers. The specificity of this binding was determined by observed non-binding of CD3 aptamers to CD3 null Jurkat CD3ε KO cells. The suggested binding to human T cells makes the CD3 aptamers a potential tool to translate into the clinic and improves the scope and impact of this study. Further characterization of functionality is needed before we can evaluate its potential as a human therapeutic that could also elicit T cell activation in humans.
These results suggest that the anti-CD3 aptamers cooperate with the TCR signal to enhance the T cell stimulus either directly by targeting a CD3 aptatope on cells, or indirectly via allosteric regulation. The CD3 antibodies perhaps bind to the CD3 protein complex on cells, causing a conformational change exposing epitopes that allows for increased biding of aptamer onto the cells. One possibility is that CD3 antibodies may induce clustering of the TCR/CD3 complexes, allowing for more avid interactions of the CD3 and aptamers that increase the binding signal. The CD3 antibodies may fix the T cell in a pre-activation state that makes binding with aptamers more amenable. Interestingly, the CD3 aptamers needed to be multimerized or immobilized in order to trigger the activation signal. This phenomenon has been reported with other agonistic aptamers as well,29 suggesting that the signal is mediated through receptor clustering. A simple dimerization of the CD3 aptamer appears to be enough to exert agonistic activity. However, further optimization of the aptamer constructs, including the linker size, aptamer orientation, and valency, is likely to improve agonistic capacity.
Materials and methods
SELEX
To select CD3-specific RNA aptamers, six rounds of high-throughput SELEX were performed against the recombinant mouse CD3 delta-epsilon Fc-tagged protein (Mouse CD3 delta/CD3 epsilon Heterodimer protein cat. CT025-M2303H SB Sino Biological). Every round of SELEX was performed using a pool of randomized, diverse 2′Fluoropyrimidine modified RNA aptamers: the 72-bp-long RNA library. This RNA library was generated via in vitro transcription of a 98-bp-long DNA library.
For the first round of SELEX, the CD3 DNA template oligo was PCR amplified with the forward primer CD3Sel5 as well as the reverse primer CD3Sel3 to create the DNA library. CD3δε Sel5 (Forward Primer) GGGGAATTCTAATACGACTCACTATAGGGAGAGAAGGATAGGG, CD3δε Sel3 (Reverse Primer) GGGAAAGGAGGTATAAGGAA, DNA Library Template (98 bp) GGGAAAGGAGGTATAAGGAA -35N-GGGATAGGAAGAGAGGGATATCACTCAGCATAATCTTAAGGGG. This 98-bp-long DNA library consisted of a 35-nucleotide (35N)-long variable region flanked by two constant regions used as guides for PCR amplification. The DNA library was transcribed in vitro to produce 2′-Fluoropyrimidine modified RNA library used for the first round of SELEX. This RNA library was 72 bp long, and determined the length and composition of the RNA aptamers being selected. RNA libraries for each successive round of SELEX were generated from the pool of aptamers enriched in the preceding round of SELEX. Half of the aptamers rescued from each round of SELEX were reverse transcribed into DNA libraries to be used for all rounds of SELEX succeeding the first round.
For every round of SELEX, the DNA Taq Polymerase kit (Invitrogen, Ref# 10342053) was used following the manufacturer’s instructions. PCR amplification was optimized and limited to 22 cycles in each round to avoid amplification bias while generating the libraries. 2-Fluoropyrimidine modified RNA libraries used for each round of SELEX were generated by in vitro transcription of the template DNA library using the DuraScribe T7 RNA Polymerase in vitro transcription kit (Lucigen, Ref# DS010925) according to the manufacturer’s instructions. After in vitro transcription, RNA species were purified via PAGE purification (non-denaturing conditions) and eluted in RNA-ase free water (Sigma). RNA libraries thus generated were used for binding screens against the recombinant mouse CD3 delta-epsilon protein in each round of SELEX.
The recombinant mouse CD3 delta-epsilon Fc-tagged protein (Sino Biologicals, Ref# CT025M2303H50) was immobilized onto Protein G-coated Sepharose beads (GE Healthcare Bio-science) and served as the scaffold where selection of the aptamers could take place. SELEX was carried out in physiological binding buffer—150 mM NaCl, 2 mM CaCl2, 20 mM HEPES, and 0.01% BSA—at pH 7.4 at 37°C for 30 min for each round. For every round of SELEX, 1 nmol of RNA library was used and restriction was increased in each round by increasing binding volume and while simultaneously reducing the aptamer library concentration by a factor of 3.
Before each round of SELEX, the aptamer library was counterselected against the Fc tag using human IgG1 (containing the Fc tag) protein (Sigma) coated beads, producing an RNA library potentially devoid of aptamers binding to the control FC tag. The RNA library was incubated for 30 min at 37°C in binding buffer with the hIgG1-Fc control beads, and the supernatant was collected by removing the beads from the library by pelleting. The counterselected RNA library was incubated with the target rmCD3δε beads for 30 min in binding buffer, and the positively selected bead-bound aptamer fraction was isolated by pelleting the beads via centrifugation. The beads were washed three times in Wash Buffer: 150 mM NaCl, 2 mM CaCl2, 20 mM HEPES (pH = 7.4) to remove unbound species. To favor the selection of aptamers with higher binding stability we include in the SELEX procedure three washes of 5 min each of the bound aptamer fraction. CD3δε Bead-bound aptamer species were extracted via phenol:chloroform:isoamyl alcohol (25:24:1) fractionation, followed by chloroform purification. This fraction was then precipitated using linear acrylamide and sodium acetate, and then recovered RNA was retrotranscribed, PCR amplified, and in vitro transcribed to produce the library for the successive rounds of SELEX.
After every round of SELEX, we enriched the RNA libraries with the most affine binders to the target rmCD3δε protein, while removing species that bound non-specifically to the counterselected hIgG1FC protein. Further restriction in each SELEX round was introduced by successively increasing the volume of incubation with the target rmCD3δε protein. The extracted RNA was then reverse transcribed and PCR amplified to generate the DNA library needed to generate the RNA library for the next round of SELEX.
Aptamer species used for the study were generated in the following manner. All primers as well as oligonucleotides used for the study are listed Table S1. To generate CD3δε monomeric aptamers—“CD3δε Apt”—DNA template was PCR amplified using CD3δε Sel3 and CD3δε Sel 5 primers.
P32-labeled monomeric aptamers were also generated in a similar manner, but by lacing half of the ATP needed in the reaction with αP32-ATP (PerkinElmer, Ref# BLU003H250UC).
For aptamer functionalization by hybridization, CD3δε aptamers were extended with 23 nucleotides on its 3ʹ end to create “CD3δε Apt-Extn” aptamers. They were generated by amplifying each aptamer template with the CD3δε Sel 5 forward primer and the CD3δε Sel 3 Extn reverse primer.
To generate biotinylated and Cy5 tagged CD3δε monomeric aptamers, “CD3δε Apt-Extn” were hybridized with a complementary chemically synthesized (Sigma) oligonucleotide with a Biotin or a Cy5 tagged on its 5′ end. These probes were hybridized to the complementary RNA aptamer at a 1:1.25 ratio. The nucleotide mixture was heated for 5 min at 85°C, and cooled gradually till 4°C was reached, reducing the temperature 2°C each time in 30-s increments.
PE-labeled tetrameric aptamers were generated by incubating monomeric biotinylated aptamer with 0.1 μL of Streptavidin-PE per 10 pmol (1:4) of aptamer right before the flow cytometry experiments.
CD3δε dimer DNA template was produced from a 206-bp DNA template cloned into the ampicillin-resistant pUC57 plasmid via Gene synthesis by Gen Script (Cloning site: EcoRV/EcoRV; Cloning direction: Opposite direction to promoter; Promoter: LacZ). The cassette cloned into PUC57 encoded DNA templates for two separate aptamers that could be generated from the same DNA template—“CD3δε dimer” and “CD3δε dimer with Extn” a modified version of the dimer that includes a 23-nucleotide extension on the 3′ end of the aptamer that allows for its hybridization with fluorescent or biotinylated tags. To produce “CD3δε dimers,” plasmid DNA was with Not I enzyme (New England Biosciences) to produce a linear DNA template recognizable by the T7 RNA polymerase (Table S2). Transcription products were purified by chloroform purification, and then combined with 1/10 volume sodium acetate and 2.5 volumes of 100% RNAase free ethanol (Sigma). Reaction mixtures were incubated overnight at −80°C. The next day, they were thawed for 30 min at −20°C, and later pelleted at 4°C, 14,000 RPM, to sediment transcribed RNA. Pelleted RNA was rinsed twice in ice-cold 70% ethanol and left to dry until the pellets were clear. They were resuspended in solvent of choice, and quantified.
The binding of the CD3 aptamers to the recombinant protein was initially determined using double-filter nitrocellulose binding assay.
Surface plasma resonance
Aptamer binding affinity to CD3 protein was determined by surface plasmon resonance using the ProteOn XPR36. Biotinylated aptamers were immobilized onto independent channels of streptavidin-coated sensor chips (Bio-Rad). CD3-Fc or control Fc protein was injected in running buffer consisting of PBS and 0.005% (v/v) Tween 20, pH 7.4, at a flow rate of 30 μL/min.
OCTET
Octet N1 System and Octet SAX2 streptavidin biosensors (ref. 18–5136) were purchased by Sartorius. All the interaction analyses were conducted at room temperature (25°C) in PBS +/+ buffer. Biosensors were hydrated in PBS +/+ for 10 min immediately before use. Biotinylated aptamers were first attached to the sensor. In the optimization, loading test, 500 nM showed to be the best aptamer loading concentration. Afterward, association and dissociation steps were run at different mouse CD3 delta-epsilon protein concentrations. Before testing, protein was incubated for 15 min with yeast tRNA to control nonspecific binding. The condition of the assays was the following: 30 s initial base line, 250 s of loading of the aptamer to the sensor, 100 s of baseline, 250 s of association, and 300 s of dissociation.
Flow cytometry
For all aptamer binding flow cytometry studies, the control was a non-binding randomized sequence or scrambled aptamer of the same length and format of the aptamers being tested. They were produced, modified, and/or labeled in the same manner as the CD3 aptamers being tested in the assays.
For the binding assays using protein-coated Dynabeads, 150,000 Dynabeads were incubated with 15 pmol of Cy5-tagged monomeric CD3 aptamers in BBF 1x for 30 min at 37°C. Beads were washed twice in BBF 1x and the proportion of aptamer-bound beads were quantified via cytometry.
For testing aptamer staining to EL-4 cells and Jurkat cells, 200,000 cells were incubated with 30 pmol of Cy5 or PE-labeled aptamers in 100 μL of PBS +/+ for 20 min at 37°C and 5% CO2. Cells were washed twice with PBS +/+ and passed through the cytometer without fixation.
Primary lymphocytes were isolated from lymph nodes of naive C57BL/6 mice; 500,000 cells were incubated with 75 pmol of Cy5 or PE-labeled aptamers in 100 μL of PBS +/+ for 20 min at 37°C, 5% CO2. Cells were washed twice with PBS +/+ and samples were acquired in the cytometer without fixation.
EL-4 WT, EL-4 CD3δKO, EL-4 CD3εKO, and EL-4 CD3γKO cells were characterized for their level of CD3 expression using anti-mouse CD3 antibody clones 2C11 and 17A2 at a dilution of 1:100. Cells were stained in PBS-EDTA-BSA buffer (PBS −/−, 10% BSA, 5 mM EDTA) and incubated for 20 min at 37°C before being washed and fixed in 2% PFA before acquisition.
To calculate the percentage change in reduction of stain index of each aptamer in each of the CD3KO cell lines, we calculated the normalized signal for each sample by subtracting the signal of the control aptamer from the test aptamers. Then we calculated the stain index of each aptamer as a percentage of increased or decreased normalized signal with the following formula:
Specificity binding assays using EL-4 CD3δKO, EL-4 CD3εKO, EL-4 CD3γKO, and Jurkat CD3εKO cells were performed using sub-saturating doses of aptamer. A total of 200,000 cells were incubated with 10 pmol of aptamer in a final volume of 50 μL PBS +/+. Cells were incubated for 20 min at 37°C, 5% CO2 before being washed twice with PBS +/+ and acquired in the cytometer without fixation.
Generation of EL-4 CD3δKO, EL-4 CD3εKO, and EL-4 CD3γKO cell lines
To test the extent of specific binding illustrated by our aptamers, we sought to knock out CD3 expression in EL-4 cells using CRISPR-Cas9278-based non-viral genetic engineering. We transfected EL-4 cells with the PX458 plasmid279 (Addgene, Ref # 48138) that allows for concomitant delivery of both the single guide RNA (sgRNA) and Cas9 nuclease into the cells. The PX458 plasmid encodes a cassette for both the in situ transcription of the sgRNA using the T7 polymerase and the translation and production of the exonuclease Cas9. Once both components of the CRISPR toolbox are in the cell, the desired genome editing events take place, if conditions are optimal. The sgRNA sequences cloned in PX458 were as follows: CD3ε CACCGAGGGCACGTCAACTCTACAC, CD3δ CACCGCATGCATCTAGATGGAACGG, CD3γ CACCGAACAATGCCAAAGACCCTCG.
Preparation, decoration, and characterization of SLBs
Supported lipid bilayers (SLBs) were prepared for calcium flux experiments as described in Hellmeier et al.10 In brief, small unilamellar POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine) vesicles containing 2% Ni-DOGS NTA (1,2-dioleoyl-sn-glycero-3-[N(5-amino-1- carboxypentyl) iminodiaceticacid] succinyl [nickel salt]), for the attachment of His-tagged proteins were incubated on plasma cleaned, hydrophilic glass coverslips, followed by extensive rinsing with PBS.
Proteins used for SLB decoration were AF555-labled 2xHis6-tagged pMHC (IEk-MCC) at <0.3 molecules per μm2 (for suboptimal stimulation), and His10-tagged B7-1 (at 100 molecules per μm2) and His10-tagged ICAM-1 (at 100 molecules per μm2) (both from Sino Biological).
CD3 aptamers for SLB decoration were prepared via in vitro transcription and biotinylated by hybridizing with a biotin probe. Biotinylated CD3 aptamers were incubated with a His-tagged monomeric streptavidin site specifically labeled with ABS635P (Abberior Star Dyes)30 at a 1:3 M ratio. Thus, each aptamer was attached to one molecule of monovalent streptavidin, one molecule of fluorescent dye, and could diffuse on the SLB as a discrete, monomeric element.
For SLB decoration, all proteins were mixed in PBS and incubated on the SLB for 60 min. Positive control SLBs contained pMHC at 150 molecules per μm2 and ICAM-1 and B7-1 at 100 molecules per μm2, negative controls contained ICAM-1 and B7-1 only.
SLBs were decorated with the CD3 aptamers from approximately 0.05 to 1,000 molecules per μm2. Each bilayer prepared was characterized to quantify ligand density by relating the bulk membrane brightness to the brightness of a single fluorophore, and to confirm homogeneous spread of the lipid bilayer containing freely diffusing SLB-attached molecules.
Calcium flux assays
The levels of intracellular calcium were determined as described in Hellmeier et al.10 After in vitro expansion with agonistic peptide, 106 5c.c7 T cells were incubated in T cell media supplemented with 5 μg mL−1 Fura-2 AM (11524766, ThermoFisher Scientific) for 20 min at 24°C. Excessive Fura-2 AM was removed by washing 3x with HBSS +2% FBS. T cells were diluted with HBSS +2% FBS to get a final concentration of 5∗103 cells μL−1; 105 cells were transferred to the Lab-Tek chamber and image acquisition was started immediately after T cells landed on the functionalized SLBs. Fura-2 AM was excited using a monochromatic light source (Polychrome V, TILL Photonics), coupled to a Zeiss Axiovert 200M equipped with a 10× objective (Olympus), 1.0× tube lens, and an Andor iXon Ultra EMCCD camera. A long-pass filter (T400lp, Chroma) and an emission filter were used (510/80ET, Chroma). Imaging was performed with excitation at 340 nm and 380 nm, to capture the signal of Fura-2 AM in the absence and presence of calcium, respectively. Experiments were carried out at 24°C.
We utilized ImageJ software to generate ratio and sum images of 340 nm/380 nm. Cellular positions and tracks were recorded and utilized for intensity extraction based on the ratio image. The intensity traces were normalized to the initial value at time point zero. These traces were then classified as either “activating” or “non-activating” based on an arbitrary activation threshold ratio of 0.4. The selection of this activation threshold was determined by comparing individual traces of a positive control (ICAM-1 100 μm−2, B7-1 100 μm−2, His-pMHC 150 μm−2) and a negative control (ICAM-1 100 μm−2, B7-1 100 μm−2).
To generate dose-response curves, we conducted multiple calcium measurements, typically exceeding 100 cells in a defined region of interest, with each measurement performed at a specific ligand density. The percentage of activated cells was evaluated for each measurement and normalized to the positive control.
Proliferation assays
T cells were isolated from the spleens of naive C57BL6 WT mice or OT-1/CD45.1 mice, using the MIltenyi negative selection isolation kit (Mouse CD8+ Isolation Kit, Ref # 130-104-075); OT-1 and PMEL splenocytes were isolated after red blood cell lysis. Cells were suspended in PBS at 2,000,000 cells/mL and stained in 5 μM CFSE (Sigma, Ref# 21888) in PBS, at room temperature for 5 min. Then the cells were washed 2X in a PBS-BSA-EDTA buffer, counted, and used for proliferation assays at 50,000 to 200,000 cells/well; 100 pmol of CD3 aptamers or control aptamer (in PBS −/−) was added to each well. Cells were seeded in wells coated overnight with activated in 96-well CD3-coated plates (0.5–1 μg/mL purified anti-CD antibody clone 2C11, BD Biosciences) or with appropriate dose of SIIGFEKL, SIINFEKL, or mgp100 (GenCust, custom ordered). Cells were allowed to proliferate for 72 h, and CFSE dilution was measured via flow cytometry.
For H3 thymidine proliferation assays, the same proliferation protocol was followed but instead unstained cells were used. After 72 h, 0.5 μCi of H3 thymidine was added to each well, and the extent of proportion of thymidine-incorporated cells was assayed 24 h later using a scintillation counter.
Mice
WT C57/BL6 and Balb/C mice were purchased from Charles River and OT-1 and PMEL transgenic mice were bred in house. All in vivo studies were performed with 6- to 8-week-old mice according to institutional ethical board regulations at the animal facility of the Center for Applied Medical Research, Pamplona, Spain. 5c.c7 mice used for calcium flux experiments were housed and sourced from the Technical University of Vienna and Medical University of Vienna as a collaboration project with Drs. Gerhard Schutz, Eva Sevcsik, Joschka Hellmeier, and Johannes Huppa.
Adoptive transfer studies
To assess the effect of CD3 aptamers on enhancing antitumor efficacy of adoptively transferred OT-1 T cells in vivo, naive C57/BL6 mice were implanted subcutaneously with 150,000 B16/OVA cells on day 0. Concurrently, splenocytes were harvested from naive OT-1/CD45.1+ mice, purified, and resuspended in cell culture medium at 5 million cells/mL. Cells were supplemented with 10−8 M of SIIGFEKL or 10−12 M SIINFEKL for suboptimally activating the OT-1 splenocytes. Cells were co-incubated with CD3 dimer aptamers Ctrl, 1, or 12 at 600 pmol/mL or 5000,000 cells, and left to proliferate for 72 h. On day 1, the cultures were supplemented with cell culture medium equivalent to double the initial culture volume. On day 2, the cultures were supplemented with cell culture medium equivalent to the starting culture volume. A group of OT-1 cells were activated with 10−8 M SIINFEKL alone to produce highly activated cells serving as positive controls for the experiment in the same manner. On day 3, cells were harvested, counted, and viability was assessed using Trypan Blue exclusion. A total of 1,000,000 10−8 M SIIGFEKL ± CD3 aptamer activated cells were injected intravenously into B16OVA-bearing mice. A total of 500,000 10−12 M SIINFEKL ± CD3 aptamer activated cells were injected intravenously into B16OVA-bearing mice. A total of 1,000,000 10−8M SIINFEKL-activated cells were injected intravenously into B16OVA-bearing mice. Tumor growth was monitored.
To assess the effect of CD3 aptamers on enhancing longevity and survival of adoptively transferred OT-1 cells in vivo, splenocytes were harvested from naive OT-1/CD45.1 mice and purified. SIIGFEKL ± CD3 aptamer cells were prepared in the same manner as described above. OT-1 cells activated with a high dose of SIINFEKL alone were also prepared as positive controls. After successful proliferation, 1,000,000 activated OT-1 T cells were adoptively transferred into naive, C57/BL6 mice on day 0. One week later, mice were implanted subcutaneously with 150,000 B16OVA cells to provide an in vivo antigenic boost. On day 16 after adoptive transfer, mice were bled, and peripheral blood mononuclear cells were purified. The proportion of CD45.1+CD8+ cells was quantified via flow cytometry. On day 23 after adoptive transfer, mice were euthanized, and CD45.1+CD8+ populations in the spleen were quantified.
T cell activation peptides
SIINFEKL, SIIGFEKL, SPSYVYHQF (HA-1), and EGSRNQDWL (murine gp100) peptides used for T cell activation assays were purchased from GeneCust (France) with more than 95% purity.
Statistical analysis
One-way ANOVA with a Bonferroni post hoc test with a 95% confidence interval was performed to gauge statistical significance in in vitro assays. For the SLB a two-sided Bootstrap ratio test hoc was performed. For the tumor studies, mean tumor size and SEM were calculated for each time point, and two-way ANOVA with a Bonferroni post hoc test was performed. Data are presented as mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Data and code availability
All of the data and material that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
Instituto Salud Carlos III financed with Feder Funds PI20-01132 (“A way to make Europe”) for F.P. This project has received funding from the European Union Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Action grant agreement no. 721358 and under the H2020-FETOPEN “DESTINATION” grant agreement no. 899833.
Author contributions
Conceptualization, F.P. and A.P.M.; Resources, F.P.; Writing—Original Draft, A.P.M., F.P.; Writing—Review and Editing, F.P., A.P.M., D.M.-C., A.Z., H.V., B.M., M.B., J.H., E.V., F.N., J.B.H., and H.M.v.S.; Visualization, F.P. and D.M.-C.; Experimentation: Aptamer selection, A.P.M, H.V., M.B.; Aptamer in vitro characterization: A.P.M., B.M., A.Z., F.N., J.H., T.P., M.H., T.W.; Material and guide experimentation in Glass-supported planar lipid bilayers: E.V., G.J.S., and J.B.H.; Funding Acquisition, F.P. and H.M.v.S.; Supervision, F.P. All authors have read and agreed to the published version of the manuscript.
Declaration of interests
F.P. is associate editor in this journal.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2024.102198.
Supplemental information
References
- 1.Cappell K.M., Kochenderfer J.N. Long-term outcomes following CAR T cell therapy: what we know so far. Nat. Rev. Clin. Oncol. 2023;20:359–371. doi: 10.1038/s41571-023-00754-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu Q., Li J., Zheng H., Yang S., Hua Y., Huang N., Kleeff J., Liao Q., Wu W. Adoptive cellular immunotherapy for solid neoplasms beyond CAR-T. Mol. Cancer. 2023;22:28. doi: 10.1186/s12943-023-01735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brummel K., Eerkens A.L., de Bruyn M., Nijman H.W. Tumour-infiltrating lymphocytes: from prognosis to treatment selection. Br. J. Cancer. 2023;128:451–458. doi: 10.1038/s41416-022-02119-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chatani P.D., Lowery F.J., Parikh N.B., Hitscherich K.J., Yossef R., Hill V., Gartner J.J., Paria B., Florentin M., Ray S., et al. Cell surface marker-based capture of neoantigen-reactive CD8 + T-cell receptors from metastatic tumor digests. J. Immunother. Cancer. 2023;11 doi: 10.1136/jitc-2022-006264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cattaneo C.M., Battaglia T., Urbanus J., Moravec Z., Voogd R., de Groot R., Hartemink K.J., Haanen J.B.A.G., Voest E.E., Schumacher T.N., Scheper W. Identification of patient-specific CD4+ and CD8+ T cell neoantigens through HLA-unbiased genetic screens. Nat. Biotechnol. 2023;41:783–787. doi: 10.1038/s41587-022-01547-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menon A.P., Moreno B., Meraviglia-Crivelli D., Nonatelli F., Villanueva H., Barainka M., Zheleva A., van Santen H.M., Pastor F. Modulating T Cell Responses by Targeting CD3. Cancers. 2023;15:1189. doi: 10.3390/cancers15041189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shigdar S., Schrand B., Giangrande P.H., de Franciscis V. Aptamers: Cutting edge of cancer therapies. Mol. Ther. 2021;29:2396–2411. doi: 10.1016/j.ymthe.2021.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sola M., Menon A.P., Moreno B., Meraviglia-Crivelli D., Soldevilla M.M., Cartón-García F., Pastor F. Aptamers Against Live Targets: Is In Vivo SELEX Finally Coming to the Edge? Mol. Ther. Nucleic Acids. 2020;21:192–204. doi: 10.1016/j.omtn.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rius C., Attaf M., Tungatt K., Bianchi V., Legut M., Bovay A., Donia M., Thor Straten P., Peakman M., Svane I.M., et al. Peptide-MHC Class I Tetramers Can Fail To Detect Relevant Functional T Cell Clonotypes and Underestimate Antigen-Reactive T Cell Populations. J. Immunol. 2018;200:2263–2279. doi: 10.4049/jimmunol.1700242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellmeier J., Platzer R., Eklund A.S., Schlichthaerle T., Karner A., Motsch V., Schneider M.C., Kurz E., Bamieh V., Brameshuber M., et al. DNA origami demonstrate the unique stimulatory power of single pMHCs as T cell antigens. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2016857118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Immune Epitope Database and Analysis Resource. 2003. https://www.iedb.org/home_v3.php
- 12.Mariuzza R.A., Agnihotri P., Orban J. The structural basis of T-cell receptor (TCR) activation: An enduring enigma. J. Biol. Chem. 2020;295:914–925. doi: 10.1074/jbc.REV119.009411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer A., de Saint Basile G., Le Deist F. CD3 deficiencies. Curr. Opin. Allergy Clin. Immunol. 2005;5:491–495. doi: 10.1097/01.all.0000191886.12645.79. [DOI] [PubMed] [Google Scholar]
- 14.Dave V.P. Role of CD3ε-mediated signaling in T-cell development and function. Crit. Rev. Immunol. 2011;31:73–84. doi: 10.1615/critrevimmunol.v31.i1.70. [DOI] [PubMed] [Google Scholar]
- 15.Recio M.J., Moreno-Pelayo M.A., Kiliç S.S., Guardo A.C., Sanal O., Allende L.M., Pérez-Flores V., Mencía A., Modamio-Høybjør S., Seoane E., Regueiro J.R. Differential biological role of CD3 chains revealed by human immunodeficiencies. J. Immunol. 2007;178:2556–2564. doi: 10.4049/jimmunol.178.4.2556. [DOI] [PubMed] [Google Scholar]
- 16.de Saint Basile G., Geissmann F., Flori E., Uring-Lambert B., Soudais C., Cavazzana-Calvo M., Durandy A., Jabado N., Fischer A., Le Deist F. Severe combined immunodeficiency caused by deficiency in either the delta or the epsilon subunit of CD3. J. Clin. Invest. 2004;114:1512–1517. doi: 10.1172/JCI22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Delgado P., Fernández E., Dave V., Kappes D., Alarcón B. CD3delta couples T-cell receptor signalling to ERK activation and thymocyte positive selection. Nature. 2000;406:426–430. doi: 10.1038/35019102. [DOI] [PubMed] [Google Scholar]
- 18.Weissman A.M., Frank S.J., Orloff D.G., Merćep M., Ashwell J.D., Klausner R.D. Role of the zeta chain in the expression of the T cell antigen receptor: genetic reconstitution studies. EMBO J. 1989;8:3651–3656. doi: 10.1002/j.1460-2075.1989.tb08539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Werlen G., Hausmann B., Palmer E. A motif in the alphabeta T-cell receptor controls positive selection by modulating ERK activity. Nature. 2000;406:422–426. doi: 10.1038/35019094. [DOI] [PubMed] [Google Scholar]
- 20.Call M.E., Pyrdol J., Wiedmann M., Wucherpfennig K.W. The organizing principle in the formation of the T cell receptor-CD3 complex. Cell. 2002;111:967–979. doi: 10.1016/s0092-8674(02)01194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.-L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H., et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 22.Hall C., Berkhout B., Alarcon B., Sancho J., Wileman T., Terhorst C. Requirements for cell surface expression of the human TCR/CD3 complex in non-T cells. Int. Immunol. 1991;3:359–368. doi: 10.1093/intimm/3.4.359. [DOI] [PubMed] [Google Scholar]
- 23.Zumrut H.E., Batool S., Argyropoulos K.V., Williams N., Azad R., Mallikaratchy P.R. Integrating Ligand-Receptor Interactions and In Vitro Evolution for Streamlined Discovery of Artificial Nucleic Acid Ligands. Mol. Ther. Nucleic Acids. 2019;17:150–163. doi: 10.1016/j.omtn.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freage L., Jamal D., Williams N.B., Mallikaratchy P.R. A Homodimeric Aptamer Variant Generated from Ligand-Guided Selection Activates the T Cell Receptor Cluster of Differentiation 3 Complex. Mol. Ther. Nucleic Acids. 2020;22:167–178. doi: 10.1016/j.omtn.2020.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spitale R.C., Incarnato D. Probing the dynamic RNA structurome and its functions. Nat. Rev. Genet. 2023;24:178–196. doi: 10.1038/s41576-022-00546-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harp J.M., Lybrand T.P., Pallan P.S., Coates L., Sullivan B., Egli M. Cryo neutron crystallography demonstrates influence of RNA 2′-OH orientation on conformation, sugar pucker and water structure. Nucleic Acids Res. 2022;50:7721–7738. doi: 10.1093/nar/gkac577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denning E.J., MacKerell A.D. Intrinsic Contribution of the 2′-Hydroxyl to RNA Conformational Heterogeneity. J. Am. Chem. Soc. 2012;134:2800–2806. doi: 10.1021/ja211328g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varani G., McClain W.H. The G·U wobble base pair. EMBO Rep. 2000;1:18–23. doi: 10.1093/embo-reports/kvd001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McNamara J.O., Kolonias D., Pastor F., Mittler R.S., Chen L., Giangrande P.H., Sullenger B., Gilboa E. Multivalent 4-1BB binding aptamers costimulate CD8+ T cells and inhibit tumor growth in mice. J. Clin. Invest. 2008;118:376–386. doi: 10.1172/JCI33365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Platzer R., Rossboth B.K., Schneider M.C., Sevcsik E., Baumgart F., Stockinger H., Schütz G.J., Huppa J.B., Brameshuber M. Unscrambling fluorophore blinking for comprehensive cluster detection via photoactivated localization microscopy. Nat. Commun. 2020;11:4993. doi: 10.1038/s41467-020-18726-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All of the data and material that support the findings of this study are available from the corresponding author upon reasonable request.