

Aire is an unconventional transcription factor (TF) that enhances the expression of thousands of genes in medullary thymic epithelial cells (mTECs), which promotes clonal deletion or phenotypic diversion of self-reactive T cells1–4. The biological logic of Aire’s target specificity remains largely unknown as, unlike many TFs, it does not bind to a particular DNA-sequence motif. We implemented two orthogonal approaches to investigate Aire’s cis-regulatory mechanisms: construction of a convolutional neural network and leveraging of natural genetic variation via analysis of F1-hybrid mice5. Both approaches nominated Z-DNA and Nfe2•Maf as putative positive influences on Aire’s target choices. Genome-wide mapping studies revealed that Z-DNA-forming and Nfe2l2-binding motifs were positively associated with the inherent ability of a gene’s promoter to generate DNA double-strand breaks (DSBs), and promoters showing strong DSB generation were more likely to enter a poised state with accessible chromatin and already-assembled transcriptional machinery. Consequently, Aire preferentially targeted genes with poised promoters. We propose a model whereby Z-DNA anchors the Aire-mediated transcriptional program by enhancing DSB generation and promoter poising. Beyond resolving a long-standing mechanistic conundrum, these findings suggest previously unexplored routes of manipulating T-cell tolerance.
Deep learning has recently emerged as a powerful tool for uncovering complex genomic-sequence patterns predictive of various functional features in the genome6. Convolutional neural network (CNN), a regularized type of feed-forward network, is one of the most established deep-learning algorithms, having been successfully applied to the analysis of various types of genomic data with impressive results7. The ability of CNN to model complex non-linear dependencies between sequence features of arbitrary lengths makes it an attractive approach for investigating the cis-regulatory elements distinguishing Aire’s target genes.
Another powerful strategy for systematic exploration of cis-regulatory mechanisms is to exploit the natural genetic variation in target-gene cis-regulatory elements in different mouse strains. In particular, the F1-hybrid approach, which eliminates confounding influences of trans factors, has been used successfully in several immunological contexts5,8. This approach seemed ideal for identifying DNA-sequence features underlying Aire’s target specificity by unbiasedly testing the association between genetic variation in each TF-binding motif and allelic imbalances in the chromatin accessibility and expression of Aire-induced genes.
Employing a CNN
To distinguish the extended-promoter sequences of previously collated sets of Aire-induced and expression-matched Aire-neutral genes9, we built and trained a CNN model with dilated convolutional layers and residual skip connections7. This approach followed a relatively new paradigm in the deep-learning field: to pre-train on large-scale datasets to acquire generic knowledge and then transfer the knowledge to specific downstream tasks, termed the fine-tuning process. At that point, inputs to the model were extended-promoter sequences from the C57BL/6J genome, i.e., DNA stretches ±1024 bp from the transcriptional start-sites (TSSs); outputs were predictions as to whether the input sequence was from an Aire-induced or Aire-neutral gene. Our overall, two-stage strategy and diverse quality-control data are depicted in Extended Data Fig. 1 and additional explanations can be found in Supplementary Notes.
To retrieve relevant sequence features learned by the deep CNN, we computed a contribution score to the output prediction (gradient × input) for each input nucleotide using back-propagation. We then extracted the stretch of input sequence with the largest positive contribution scores for each input DNA sequence, thereby highlighting the regions most predictive of Aire-induced genes. These subsequences were scanned for both de novo motifs and known TF-binding motifs. (CA)n repeats (see Methods and Supplementary Notes) and the Nfe2•Maf-binding motif (a specific type of bZIP-family TF-binding motif)10 were enriched in the regions with the largest positive gradients (Fig. 1a; Supplementary Table 1), as exemplified by the contribution-score profiles of four Aire-induced genes (Extended Data Fig. 2a).
Fig. 1 |. The Z-DNA and Nfe2•Maf-binding motifs are salient features of the extended promoters of Aire-induced genes.

a, Motifs enriched in the regions with the largest positive contribution scores. b,c, Heatmaps showing the ISM scores for promoters of two AIGs which contain (CA)n repeats (b) or Nfe2•Maf-binding motifs (c). Each of the three rows shows results for one possible substitution in the order of A->C->G->T from top to bottom. Red indicates decreased predictive probability as AIG and blue indicates the other way after substitution. d, Plots showing the effect of in silico replacement by (CA)n repeats or random sequences of the same length on the predictions of the CNN model. For each ANG, 50 positions spaced evenly across the 1000-bp region upstream of the TSS were individually tested for replacement. The distance between adjacent (CA)8 repeats was 20bp when multiple (CA)8 repeats were used. e, Analogous plots showing the effect of the Nfe2•Maf-binding motif on the predictions. The Nfe2•Maf-binding motif and the Batf3-binding motif belong to the TRE•MARE and CRE•CRE subclass of the bZIP family10, respectively. f, Boxplot comparing the lengths of Z-DNA motifs at the promoters of AIGs (n=1747) versus ANGs (n=1520). g,h, Percentage of AIGs among weakly expressed genes (see Methods) that have Z-DNA motifs of varying lengths (g, the red line), or Z-DNA motifs of varying distances relative to the TSS (h, the red line). The dotted grey lines indicate the percentage of AIGs (n=1563) among all of the weakly expressed genes (n=4537). p-value for panel f was calculated using the Wilcoxon rank sum test (two-tailed), and for panels g and h using the Spearman correlation. AIG: Aire-induced gene; ANG: Aire-neutral gene; TPM: transcripts per million.
In silico saturation mutagenesis (ISM) inspects the influence of every nucleotide in an input sequence on a prediction of interest by analyzing how single-nucleotide substitutions of it impact the prediction6. This approach also identified (CA)n repeats and the Nfe2•Maf-binding motif as features impacting Aire’s target-gene specificity (Fig. 1b,c, Extended Data Fig. 2b). In contrast, simple motif-enrichment analysis could not identify these two DNA features as being relatively enriched in the extended-promoter sequences of Aire-induced compared with Aire-neutral genes (Extended Data Fig. 2c). (CA)n repeats are well-known to be Z-DNA-forming sequences11; in general, alternating purine-pyrimidine tracts are sequences with high potential to form Z-DNA12. So, we also examined whether the Z-DNA scores computed by an independent Z-DNA prediction model, Z-DNABERT13, were sensitive to in silico single-nucleotide mutations in the (CA)n repeats at promoters of Aire-induced genes. This indeed proved to be the case (Extended Data Fig. 2d,e).
To further confirm that Z-DNA-forming and Nfe2•Maf-binding motifs were predictive of Aire-induced loci, we randomly selected hundreds of genes originally classified in our model as Aire-neutral and then, for each gene, replaced part of the input sequence at varying positions with a (CA)n repeat or an Nfe2•Maf-binding motif, which would address whether these motifs were sufficient for the input sequence to be re-classified as being Aire-induced. For (CA)n repeats, various replacement settings were tested, including single, double or triple (CA)8 repeats and a single (CA)16 repeat. The longer and more TSS-proximal (CA)n repeats were more likely to “convert” Aire-neutral genes into Aire-induced genes in silico (Fig. 1d). In comparison, replacement with random sequences of the same length did not significantly influence the model’s predictions. Replacements with Nfe2•Maf-binding motifs resulted in a somewhat smaller impact (Fig. 1e), but this effect was still significant in comparison with replacement by a bZIP-family-binding motif not enriched in the strongly positive gradient regions, i.e., the Batf3-binding motif.
We then identified all of the Z-DNA motifs at promoters of Aire-induced and Aire-neutral genes. The former set had significantly longer Z-DNA motifs than the latter did (Fig. 1f; Supplementary Table 2). To examine which quantitative features of Z-DNA-forming motifs were most predictive of Aire-induced genes, we tested how the length and distance from the TSS of Z-DNA-forming motifs were associated with the likelihood of a weakly expressed gene to become Aire-induced. Specifically, we quantified the percentage of Aire-induced genes among weakly expressed loci that had Z-DNA motifs of varying lengths and of varying distances from TSSs. There was an increasingly higher percentage of Aire-induced genes among the entire pool of weakly expressed loci with longer Z-DNA motifs (Fig. 1g). The relative distances of Z-DNA motifs to the TSSs had a negative impact on Aire’s targeting preferences (Fig. 1h), consistent with results from the in silico sequence-replacement experiments (Fig. 1d).
Leveraging natural genetic variation
As an orthogonal approach to investigating Aire’s cis-regulatory mechanisms, we leveraged natural genetic variation between the B6 and NOD mouse strains to systematically examine the contribution of cis-regulation to Aire-induced gene expression. (NOD rather than the more typical CAST/EiJ (CAST) comparator was chosen because of its propensity to develop autoimmunity.) We generated F1 hybrids by crossing NOD and B6 mice, and performed genome-wide RNA sequencing (RNA-seq) and Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) on mTECs from the two parental strains and their offspring (Extended Data Fig. 3a,b; Supplementary Table 3). We then examined the effects of naturally occurring genetic variants, including single-nucleotide polymorphisms (SNPs) and insertions and deletions (InDels), on the transcription and chromatin accessibility of Aire-induced genes. There are roughly 5.1 million SNPs and InDels in the NOD versus B6 genomes14, allowing us to unbiasedly examine, for each TF-binding motif, which sequence variants were associated with allelic imbalances in chromatin accessibility and expression of nearby Aire-induced genes (Extended Data Fig. 3c). Allelically imbalanced open chromatin regions (OCRs) and gene transcripts were identified using the beta-binomial test. Overall, we identified 3,750 imbalanced OCRs and 1,975 imbalanced gene transcripts (Extended Data Fig. 3d).
There was a substantial positive correlation between the fold-changes of chromatin accessibility of the two parental strains and the allelic imbalances in chromatin accessibility of the F1 hybrids (Extended Data Fig. 3e), suggesting that the differential chromatin accessibilities of mTECs from B6 versus NOD mice were primarily cis-regulated, as has been reported for other cell types5. In addition, 77% of genes expressed differentially between B6 and NOD mTECs were regulated in cis because their expression differences in the two parental strains were recapitulated as allelic imbalances in F1-hybrid mTECs (Extended Data Fig. 3f). More particularly, there was a strong positive correlation between allelic imbalances in chromatin accessibility at OCRs and expression of the nearest Aire-induced gene (Extended Data Fig. 3g), indicating that cis-regulatory variation was a major contributor to the imbalanced expression of Aire-induced genes in F1-hybrid mTECs.
For each motif in the CIS-BP database, we scanned the two alleles of the imbalanced OCRs for matches, and assigned each OCR to the allele with which it had the stronger motif match. To determine whether motif variants of this particular TF were associated with the generation of imbalanced OCRs, we compared the allelic imbalance in the ATAC-seq signals at the OCRs having stronger motif matches with the B6 allele with those having stronger matches with the NOD allele (Extended Data Fig. 3c). This method revealed that genetic variants in binding motifs of the bZIP-family TFs and in (CA)n-containing motifs were significantly associated with allelic imbalances in transcript expression and chromatin accessibility of Aire-induced genes (Fig. 2a). Among the bZIP-family TFs, Nfe2l2’s motif variants had the strongest association with chromatin accessibility, with an average of around 70% of SNP/InDel-overlapping ATAC-seq reads mapping to the allele with the stronger match to the Nfe2l2-binding motif. Notably, Maff, a bZIP-family TF that can form a heterodimer with Nfe2l2 to regulate target-gene expression10, was high amongst those whose binding motifs were associated with imbalanced expression of Aire-induced genes. These two observations suggested that an Nfe2•Maf heterodimer (e.g. Nfe2l2•Maff) might be a positive influencer of Aire-induced gene expression: the OCR allele with a stronger match to the Nfe2•Maf-binding motif was more accessible and the expression of an Aire-induced gene was biased to the allele with a stronger match to the Nfe2•Maf-binding motif (Fig. 2b; Extended Data Fig. 4a,b).
Fig. 2 |. Identification of TF-motif variants associated with allelic imbalances in the chromatin accessibility and expression of Aire-induced genes.

a, TFs associated with allelic imbalance in the gene transcript level and chromatin accessibility of Aire-induced genes. Red dots represent potential positive regulators and blue dots potential negative regulators. TFs with a p-value <0.1 are shown. b, An example of a genetic variant of the Nfe2l2-binding motif associated with imbalanced OCR accessibility and expression of an Aire-induced gene. There is an intact Nfe2l2-binding motif in the OCR of Bpifb1 on the B6 allele, which is disrupted in the NOD allele due to an A to G conversion. c, Motif-enrichment analysis for imbalanced OCRs associated with Aire-induced genes (n=757). d, An example of a genetic variant of a Z-DNA motif associated with allelic imbalance in OCR accessibility and expression of an Aire-induced gene. At the promoter of Marcksl1, the NOD allele contained a longer (CA)n repeat than did the B6 allele due to an insertion. p-values for TFs in panel a were calculated using the Wilcoxon rank sum test (two-tailed). OCR: open chromatin region; TF: transcription factor.
To explore the associations of motifs not included in the CIS-BP database on allelic imbalances, we performed de novo motif analysis to identify sequences enriched in the imbalanced OCRs assigned to Aire-induced genes. Both the Nfe2l2-binding motif and (CA)n repeats were enriched in the imbalanced OCRs of Aire-induced genes (Fig. 2c). As an example for the Z-DNA motif, the NOD allele of the promoter of the Aire-induced gene Marcksl1 had a longer Z-DNA-forming motif, and the expression of Marcksl1 was also biased to the NOD allele in the F1-hybrid mTECs (Fig. 2d; see Extended Data Fig. 4c,d for additional examples). While there were other DNA motifs of potential interest, the convergence on Z-DNA-forming and Nfe2•Maf-binding motifs in the CNN and F1-hybrid data prompted us to focus on these two elements in subsequent experiments.
More Z-DNA, more Aire-induced gene expression
Z-DNA is a noncanonical, left-handed, double-helical form of DNA11. There are tens of thousands of Z-DNA motifs in mammalian genomes13,15, enriched at gene promoters. The lengths of Z-DNA-forming motifs are positively correlated with promoter activities and downstream gene expression, and several studies have demonstrated them to have cis-regulatory activity12,16–20. Z-DNA motifs are hotspots of genetic variation in human populations, and harbor an excess of expression quantitative-trait loci (eQTLs)12,21.
We first verified in vivo Z-DNA formation at gene promoters in the mTECs of Aire-WT mice by performing chromatin immunoprecipitation followed by sequencing (ChIP-seq) using the Z22 antibody22, which recognizes both Z-DNA and Z-RNA (the latter of which was removed during the ChIP-seq procedure) (see Supplementary Table 3 for quality-control data). Genes with higher expression levels generally had stronger Z-DNA signals at their promoters (Fig. 3a), consistent with another study that mapped Z-DNA formation genome-wide19. Notably, for all four gene sets (binned by expression levels), promoters with Z-DNA-forming motifs exhibited stronger Z-DNA signals than did those without such motifs, suggesting that alternating purine/pyrimidine tracts (Z-DNA-forming motifs) indeed more readily formed Z-DNA in vivo. In vivo detection of Z-DNA signals at promoter regions was more dependent on Z-DNA-forming motifs for genes with relatively low expression levels, likely due to the limited availability of energy from negative supercoils induced by active transcription. Given that Aire-induced genes are generally transcribed at low levels in the absence of Aire, Z-DNA-forming motifs in their promoters could have a strong impact on their expression. There was clear enrichment both of Z-DNA in Aire peaks and, vice versa, of Aire in Z-DNA peaks (Fig. 3b), and Aire binding-levels were positively correlated with Z-DNA signals within Z-DNA peaks (Fig. 3c).
Fig. 3 |. Enhancing Z-DNA stability promotes the expression of Aire-induced genes.

a, Profiles of Z-DNA ChIP-seq of mTECs from Aire-WT mice at promoters of genes with different expression levels. TPM < 2: transcripts per million smaller than 2 in Aire-WT mTECs (n=5612); Low, medium, and high: bottom 25% (n=3879), 25%−90% (n=10414), and top 10% (n=1582) of genes with TPM>=2. b, Profiles of Aire ChIP-seq at Z-DNA ChIP-seq peaks (n=6022, top) and vice versa (n=37567, bottom) versus corresponding random GC content-matched and size-matched peaks in mTECs from Aire-WT mice. Enrichment of ChIP-seq signals in both a and b were normalized to corresponding IgG controls using the ngs.plot. c, Boxplot comparing Aire ChIP-seq signals at Z-DNA ChIP-seq peaks that had low, medium or high Z-DNA ChIP-seq signals in mTECs from Aire-WT mice. Low: < 25th percentile (n=1508); Medium: 25th - 75th percentile (n=3008); High: > 75th percentile (n=1506). d, Log2 ratio (M-values) versus log2 average (A-values) plots (MA plots) showing the effect of spermidine treatment on previously assigned Aire-induced and Aire-neutral genes in mTECs from Aire-KO mice. e, Volcano plots of scRNA-seq for Aire-expressing cells from PBS-treated versus spermidine treated mice. AIGs were highlighted in red (top) and ANGs in dark grey (bottom). p-values for panels a-c were calculated using the Wilcoxon rank sum test (two-tailed), and for panels d and e using the Fisher’s exact test (two-tailed). WT: wild-type; KO: knockout. FC: fold change.
To directly test the hypothesis that Z-DNA influenced Aire-induced gene transcription, we enhanced Z-DNA formation in mTECs by intraperitoneal (ip) injection of spermidine, known to stabilize Z-DNA formation in vitro or in vivo20,23,24. Flow-cytometric analyses seven days after administration of spermidine confirmed that Z-DNA formation was indeed increased (Extended Data Fig. 5a). Importantly, this spermidine-injection protocol did not affect the number or composition of the major thymic stromal-cell or thymocyte compartments (Extended Data Fig. 5b–d). RNA-seq analyses showed that injection of spermidine into Aire-KO mice partially rescued the loss of Aire, as indicated by increased expression of a proportion of the previously assigned Aire-induced genes (termed “Aire-inducible” genes in the absence of Aire) among those having relatively higher mean-expression values (Fig. 3d, indicated by arrow, mean expression>16). Specifically, 8.5% of these Aire-inducible genes had fold-changes of >2, while 5.0% had fold-changes of <0.5. In contrast, we did not see such an effect on the activities of expression-matched Aire-neutral genes (Fig. 3d). These findings raised the possibility that enhanced Z-DNA formation enabled the recruitment of factors required for transcriptional induction through some mechanism that bypassed the need for Aire, thereby allowing induction of some Aire-inducible genes in mTECs of Aire-KO mice.
We also injected spermidine into Aire-WT mice. There were 240 more Aire-induced genes (fold-change >2, p-value <0.05) after spermidine injection than after control injection of phosphate-buffered saline (PBS) (Extended Fig. 6a, left). In this context, most of the spermidine-induced genes were expressed at low levels after PBS treatment (Extended Fig. 6a, right), suggesting that Z-DNA formation enabled Aire to upregulate the expression of more genes with low basal levels of transcription.
To better understand the effect of spermidine on Aire-induced gene expression, we performed single-cell RNA-seq (scRNA-seq) on the mTEC compartments of PBS- and spermidine-injected Aire-WT mice (Extended Data Fig. 6b,c; Supplementary Table 4). We focused on the Aire-expressing cell clusters that also showed high expression of major histocompatibility complex (MHC) class II genes (a.k.a. mTEChi) (Extended Data Fig. 6d). Spermidine treatment did not dramatically influence the transcriptome of Aire-expressing cells; however, there was a significant up-regulation of Aire-induced, though not Aire-neutral, genes (Fig. 3e), suggesting a collaboration between Aire and Z-DNA. (In this case, to focus the analysis on Aire-expressing mTECs, Aire-induced genes were defined by comparing the transcriptomes of Aire-GFP+ mTECs from Aire-WT and Aire-KO mice, i.e. the same bulk B6 RNA-seq datasets we used for the F1 analyses).
Z-DNA promotes DSBs at Aire-induced genes
How Z-DNA enhances transcription is not fully clear. Z-DNA structures are inherently fragile and can promote the generation of DSBs, sometimes spreading over a few hundred base-pairs25,26. Though the enzyme responsible for Z-DNA-mediated DSB generation remains uncertain, strong candidates include DNA topoisomerases (TOPs)25. TOP-generated DNA breaks are enriched at cis-regulatory regions, including promoters, enhancers and chromatin loop anchors27. The TOP-reaction cycle sometimes fails to reseal the DNA breaks, leading to their persistence, the recruitment of proteins involved in the DNA-damage response (DDR), and induction of DNA-repair pathways28,29. DNA breaks and DDR proteins regulate multiple aspects of transcription, including promoting initiation. Both DSBs and TOPs can facilitate de novo recruitment of transcriptional machineries, including Pol II and general TFs30–32. Remarkably, the proteins involved in DSB generation and the DDR are both physically and functionally associated with Aire in mTECs9,33,34, prompting the hypothesis that Z-DNA affects Aire’s target choices by promoting DSB generation at the regulatory sequences of Aire-induced genes.
We profiled the DSBs of mTECs from both Aire-WT and Aire-KO mice genome-wide, employing the Breaks Labeling In Situ and Sequencing (BLISS) technique (Supplementary Table 5). In mTECs from Aire-WT mice, BLISS signals were enriched at the Z-DNA peaks delineated by ChIP-seq and, vice versa, Z-DNA signals were enriched at DSB hotspots (Fig. 4a); plus, there was a positive correlation between the Z-DNA and DSB signal strengths (Extended Data Fig. 7a). In the absence of Aire, promoters of Aire-inducible genes contained more Z-DNA-forming motifs had significantly stronger BLISS signals than did those without them (Fig. 4b, left); in contrast, (GA)n repeats, were not positively associated with BLISS signal intensity at the promoters of Aire-inducible genes (Fig. 4b, right). There were stronger BLISS signals at promoters of Aire-inducible genes than those of expression-matched Aire-neutral genes in mTECs from Aire-KO mice (Extended Data Fig. 7b). Thus, Z-DNA was associated with DSB generation at the promoters of Aire-inducible genes independently of Aire.
Fig. 4 |. Z-DNA motifs are positively associated with the generation of DSBs, the strength of which correlates positively with poising of the promoters of Aire-inducible genes.
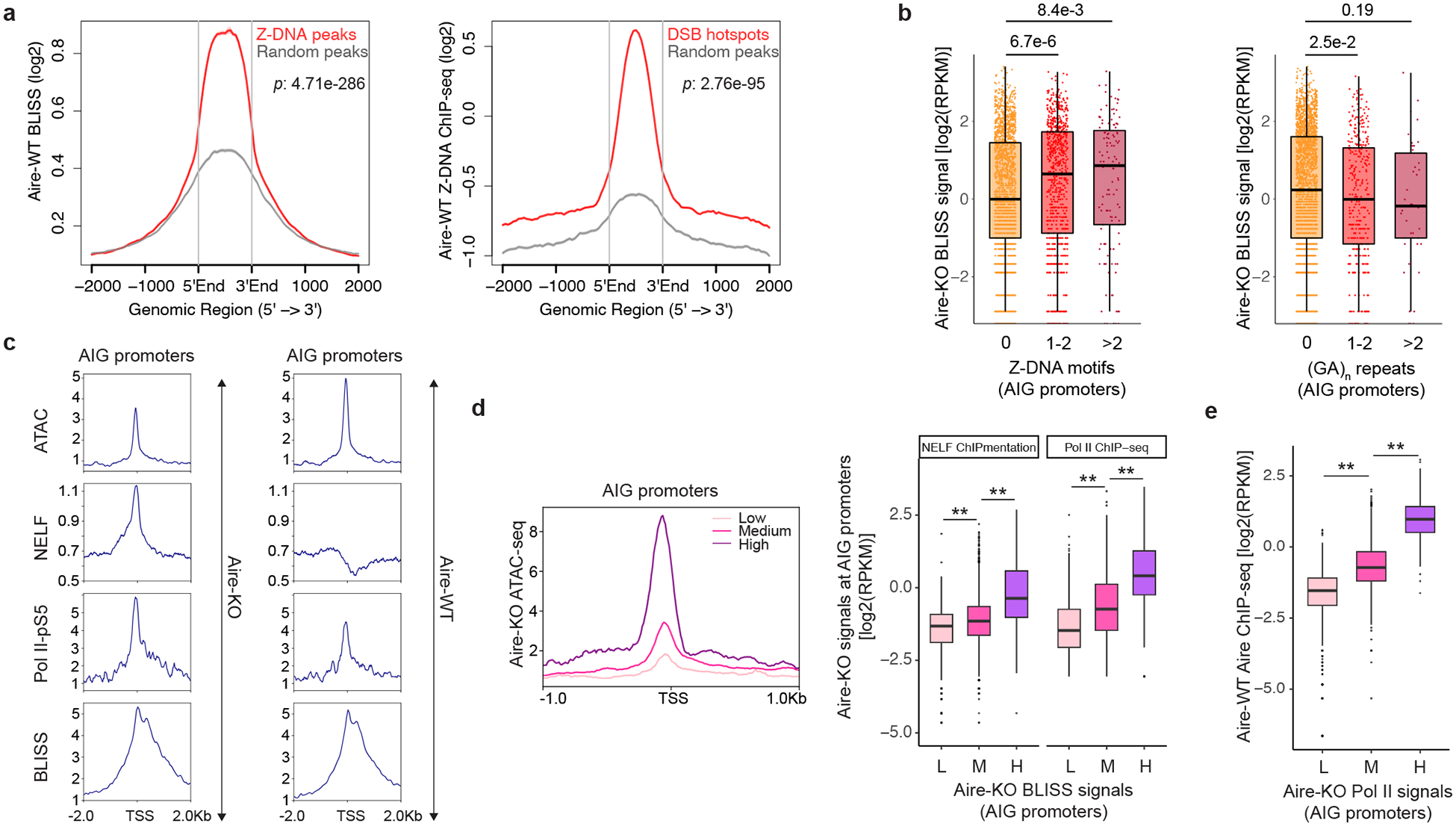
a, Profiles of BLISS at Z-DNA ChIP-seq peaks (n=6022, left) and vice versa (n=21178, right) versus corresponding random GC content-matched and size-matched peaks in mTECs from Aire-WT mice. b, Boxplot comparing Aire-KO BLISS signals between previously assigned AIG promoters with varying numbers of Z-DNA motifs (n=2124, 872 and 131 for the groups ‘0’, ‘1–2’ and ‘>2’ Z-DNA motifs; left) or (GA)n repeats (n=2719, 367 and 41 for the groups ‘0’, ‘1–2’ and ‘>2’ (GA)n repeats; right). c, Profiles of ATAC-seq, NELF ChIPmentation, Pol II-pS5 C&T and BLISS at AIG promoters of mTECs from Aire-KO and WT mice. d, ATAC-seq, ChIP-seq and ChIPmentation data for sets of AIG promoters that had low, medium or high BLISS signals in mTECs from Aire-KO mice. e, Boxplot showing the correlation between Pol II ChIP-seq signals in mTECs from Aire-KO mice and Aire ChIP-seq signals in mTECs from Aire-WT mice at AIG promoters. For panels d and e, L: < 25th percentile; M: 25th - 90th percentile; H: > 90th percentile. p-values for panels a, b, d and e were calculated using the Wilcoxon rank sum test (two-tailed). RPKM: reads per kilobase per million. L: Low, n=747; M: Medium, n=2130; H: High, n=322; *: p-value<1e-10; **: p-value<1e-20. Data for WT and Aire-KO ATAC-seq, WT Aire ChIP-seq came from Ref.9.
To directly test Z-DNA’s function in enhancing DSB generation, we performed BLISS on spermidine-treated Aire-WT mTECs. The DSB hotspots most upregulated by spermidine (fold-change >2, p-value <0.1) were significantly more enriched with Z-DNA-forming motifs than were those unaffected by spermidine (Extended Data Fig. 7c). In contrast, control CTCF-binding motifs exhibited similar enrichment in upregulated and unaffected DSB hotspots (Extended Data Fig. 7c), again arguing that spermidine treatment could enhance Z-DNA formation, which favored DSB generation.
In an independent approach, to unbiasedly investigate what DNA sequence features influenced DSB generation in mTECs, we performed BLISS on F1-hybrid mTECs generated by crossing CAST and B6 mice. Vis-a-vis the B6, the CAST genome has around 20 million genetic variants14. We sought to identify TF-binding motifs whose genetic variants were associated with imbalanced DSB generation on the two alleles of DSB hotspots. CTCF appeared to be a positive regulator of DSB generation as the allele with a stronger match with the CTCF-binding motif had significantly higher DSB signals (Extended Data Fig. 7d). This finding is consistent with studies demonstrating that polymorphisms in CTCF-binding motifs are accompanied by relocation of DSBs at chromatin-loop anchors35, serving to validate our approach. (CA)n repeats were also positive regulators of DSB generation (Extended Data Fig. 7e). The results from this set of studies prompted us to hypothesize that Z-DNA functioned upstream of Aire to regulate the generation of DSBs at promoters of Aire-induced genes.
DSBs and promoter poising prior to Aire action
We profiled chromatin accessibility using ATAC-seq9; DNA DSB generation using BLISS; binding of NELF, an indicator of promoter poising, using ChIPmentation; and binding of Ser5-phosphorylated RNA Pol II (Pol II-pS5), a proxy for Pol II pausing, using Cleavage Under Targets and Tagmentation (CUT&Tag) in mTECs from Aire-KO versus Aire-WT mice. As expected36, promoters of Aire-inducible genes were poised for expression before Aire’s engagement, as indicated by accessible chromatin, NELF binding, and Pol II pausing in its absence (Fig. 4c). The decrease in NELF binding in the presence of Aire was consistent with its ability to release paused Pol II from poised promoters36,37. Promoters of expression-matched Aire-neutral genes were less accessible and showed less Pol II binding than those of Aire-inducible genes in mTECs from Aire-KO mice (Extended Data Fig. 8a). Exemplar profiles are shown for two Aire-inducible genes and one Aire-neutral gene that had low basal expression (Extended Data Fig. 8b).
Previous studies have demonstrated the importance of DSBs and TOPs in assisting the disassembly of nucleosomes and assembly of pre-initiation complexes at gene promoters30–32,38. In agreement, we observed BLISS signals to be strongly correlated with chromatin accessibility, NELF binding and Pol II binding at Aire-inducible-gene promoters in mTECs of Aire-KO mice (Fig. 4d). High BLISS signals in Aire-KO mTECs were also associated with strong recruitment of total Pol II, Aire and MED1 (a key subunit of the Mediator complex required for Pol-II-mediated transcription) in Aire-WT mTECs (Extended Data Fig. 8c), and Aire binding in Aire-WT mTECs was positively correlated with Pol II binding at promoters of Aire-inducible genes in Aire-KO mTECs (Fig. 4e). Thus, prior to the engagement of Aire, strong DSB generation at promoters of Aire-inducible genes is associated with promoter poising and foretells binding of Aire.
Z-DNA-promoted DSBs enhance promoter poising
Topotecan prevents TOP1 from re-ligating the DNA strands it breaks, thereby enhancing the persistence of DNA breaks39. Topotecan treatment of Aire-WT mice significantly increased DSB generation at Aire-binding sites (Fig. 5a) as well as Aire binding at promoters of Aire-induced genes (Fig. 5b). Accordingly, there was a general increase in the expression of Aire-induced, but not Aire-neutral, genes upon topotecan treatment (Fig. 5c). More generally, DSB hotspots upregulated by topotecan contained slightly longer Z-DNA-generating motifs than did those unchanged after topotecan treatment (Fig. 5d).
Fig. 5 |. DSBs facilitate Aire-induced gene expression.

a, BLISS profiles at Aire ChIP-seq peaks (n=37567) +/− topotecan treatment. b, Aire ChIPmentation profiles at AIG promoters (n=3231) +/− topotecan treatment. c, MA plots showing the effect of topotecan on AIGs and ANGs of mTECs from Aire-WT mice. d, Boxplot comparing the lengths of Z-DNA motifs at DSB hotspots unchanged (n=82) or up-regulated (fold-change > 2, p-value < 0.1, n=67) by topotecan in mTECs. e, ATAC-seq profiles at promoters of AIGs (n=3231) in mTECs from Aire-KO mice treated with spermidine versus PBS. p-values in panels a-b and d-e were calculated using the Wilcoxon rank sum test (two-tailed) and in panel c using Fisher’s exact test (one-tailed). DMSO: Dimethyl sulfoxide.
To further substantiate that Z-DNA regulated promoter poising in cis, we profiled chromatin accessibility at the promoters of genes subject to Aire induction in mTECs of Aire-KO mice treated with spermidine. Compared with control injection of PBS, administration of spermidine increased the accessibility of such promoters (Fig. 5e).
Nfe2l2 impacts Aire-induced gene expression
Both the CNN and F1-hybrid approaches pointed to the Nfe2•Maf heterodimer as a second putative regulator of Aire-induced-gene expression. There are three Nfe2-related factors in mice and humans – Nfe2l1, Nfe2l2 and Nfe2l3 – all of which are expressed in mTECs. We focused on Nfe2l2 because floxed-Nfe2l2 mice were available and because Nfe2l2 is known to induce the expression of some of its target genes by stabilizing nearby Z-DNA formation40.
Hence, we examined the correlation between the strength of DSB signals and the presence of Nfe2l2-binding motifs at promoters of Aire-inducible genes in mTECs from Aire-KO mice. DSB hotspots containing more Nfe2l2-binding motifs had significantly stronger DSB signals than did those with fewer such motifs; this was not always true of DSB hotspots with CTCF-binding motifs (Extended Data Fig. 9a). Additionally, the DSB hotspots upregulated in mTECs of spermidine-treated Aire-WT mice (FC > 2, p-value < 0.1) were more enriched with Nfe2l2-binding motifs than were those unaffected by spermidine (Extended Data Fig. 9b).
Z-DNA-forming motifs and Nfe2l2-binding motifs had distinct distribution patterns at DSB hotspots: the former enriched around the centers, the latter concentrated at the boundaries (Extended Data Fig. 9c). This pattern was consistent with previous studies showing that Nfe2l2 can stabilize Z-DNA formation nearby, and that the Z-DNA structure propagates into adjacent segments from the central Z-DNA-forming motifs in a cooperative manner16–18. Nfe2l2 can stabilize Z-DNA formation by recruiting BRG117, a core subunit of the BAF chromatin-remodeling complex. BRG1 stabilizes the energetically unfavorable formation of Z-DNA by using the energy produced by nearby nucleosome ejection16,18. BRG1 was reported to increase the accessibility of cis-regulatory elements of Aire-induced genes, thereby poising these loci for expression41. We re-analyzed published ATAC-seq data for mTECs of mice lacking BRG1 (encoded by Smarca4) specifically in TECs (and skin) vs WT controls41. The OCRs upregulated by BRG1 had longer Z-DNA-forming motifs than did those unaffected by BRG1 expression (Extended Data Fig. 9d; Supplementary Table 6). In addition, de novo motif analysis showed that both (CA)n repeats and Nfe2l2-binding motifs were enriched at OCRs upregulated by BRG1, while the CTCF-binding motif was increased at OCRs unaffected by BRG1 expression (Extended Data Fig. 9e).
We next compared the transcriptomes of mTECs from Foxn1Cre-Nfe2l2flox/flox mice, and Foxn1Cre-Nfe2l2WT/WT littermate controls. We first established that expression of the genes encoding the other two Nfe2-related factors was unaffected in these mice (Extended Data Fig. 9f) and that Nfe2l2-KO mice had thymic stromal-cell compartments of normal number and composition (Extended Data Fig. 9g). RNA-seq analysis revealed that expression of Aire-induced genes with Nfe2l2-binding motifs at their promoters was slightly more depressed in the absence of Nfe2l2 than was expression of Aire-neutral genes (Extended Data Fig. 9h). This mild effect may reflect redundancy between Nfe2l2 and other family members. More of the differentially expressed loci in Nfe2l2-KO mTECs were Aire-induced than Aire-neutral genes (Extended Data Fig. 9i), suggesting that the lack of Nfe2l2 did not generally influence mTEC functions. Notably, the differentially expressed Aire-induced genes were about half upregulated and half downregulated, indicating that Nfe2l2 may not be a general up-regulator of Aire-induced genes, consistent with the limited effect of the Nfe2•Maf motif sequence in the in silico replacement experiment (Fig. 1e). Together, the two findings suggest that other TFs may cooperate with Z-DNA to regulate Aire’s target specificity.
KEGG pathway analysis on the differentially expressed genes identified a diverse set of mainly downregulated gene modules, composed of Aire-induced genes (Extended Data Fig. 10a,b). This finding suggested that Nfe2l2 might at least partially facilitate Aire-induced gene expression in a hierarchical manner whereby Nfe2l2 upregulates the expression of certain TFs that induce expression of groups of functionally related Aire-induced genes. Recently, it was reported that Aire indirectly induces the expression of groups of functionally related genes encoding peripheral-tissue antigens (PTAs) by promoting the generation of thymic mimetic cells, i.e. mTEC subtypes whose transcriptional programs mimic those of particular peripheral cell types42–44. The generation of mimetic mTECs depends on expression of the appropriate lineage-defining TFs, which directly bind to and induce the transcription of sets of PTA genes characteristic of their peripheral counterparts42. Aire did upregulate the expression of many lineage-defining TFs in mTECs (Extended Data Fig. 10c). Importantly, Nfe2l2 deficiency downregulated expression of the signature genes of several mimetic mTEC subtypes, including Tuft 1, Tuft 2 and microfold mimetic mTECs (Extended Data Fig. 10d). The down-regulated KEGG term “taste transduction” (Extended Data Fig. 10b) indeed enriches for genes related to Tuft cells. Moreover, spermidine or topotecan injection led to upregulation of the signature genes of several mimetic mTEC subtypes (Extended Data Fig. 10e,f). The overlapping influence on tuft mTEC signature genes between spermidine treatment, topotecan treatment and loss of Nfe2l2 suggests that Z-DNA, DSBs and Nfe2l2 might cooperate to indirectly regulate the expression of groups of functionally related Aire-induced genes.
Discussion
The logic of Aire’s target specificity has remained a mystery since its discovery as a key regulator of autoimmunity1. Extended Data Fig. 11 illustrates the Z-DNA-anchored model we are proposing and extensive discussion of it can be found in the Supplementary Discussion. Our findings, coupled with published observations, argue that the strength of Aire recruitment depends on the abundance of pre-assembled transcriptional machineries3,9,45. Importantly, Aire-dependent upregulation of a gene’s expression is decoupled from Aire binding to the gene’s promoter – strong Aire binding does not necessarily lead to Aire-dependent upregulation. Since Aire cannot perturb the expression of highly transcribed genes already robustly induced by TFs, the resulting picture is that Aire preferentially upregulates the expression of weakly expressed genes, which has often been incorrectly considered to be equal to Aire preferentially being recruited to weakly expressed or repressed genes. This mindset has greatly impeded identification of the logic underlying Aire’s target specificity.
A growing body of results suggests that Z-DNA, as a form of non-B DNA, is widely involved in transcriptional regulation. Z-DNA-forming motifs are enriched at the promoter regions of a number of genes, influencing their promoter activities and target-gene expression12,18–20,40. It remains to be determined how generally the Aire-focused mechanism we have uncovered operates with other genes and in other cell types.
METHODS
CNN model architecture
Our model was composed of a main body and a task-specific head. The architectures of the main body and pre-train46,47 head were similar to those of the model Basenji27,48, which is one of the state-of-the-art models for learning the cis-regulatory lexicon from mammalian genomes. However, our model’s main body had a shorter input length and a higher nucleotide resolution. At the pre-training stage, we trained a multi-task CNN model on sequences of 2048 bp length tiled across the mouse genome to predict read-coverage profiles across the 2048-bp input sequence for 1643 functional genomic read-outs from the ENCODE49 and FANTOM50 projects. Thus, by modeling activities of genomic regulatory sequences as a function of the underlying DNA sequences, parameters of the main body were optimized during pre-training to encode cis-regulatory DNA-sequence motif information. In detail:
The main body of the model consisted of three parts. The first part used four convolutional blocks to extract relevant DNA sequence motifs. Each convolutional block was comprised of the following layers: 1) Batch normalization, 2) 1D convolution (width 20 in the first block, width 5 in the following blocks), 3) GELU activation51, and 4) Max pool (width 2). The convolutional blocks reduced the dimension from 2048 bp to 128 so that each position vector encoded sequence information of 16 bp. The number of filters in the convolutional layers increased from an initial 384 to 768 in the last block. The second part had five repeated blocks containing dilated convolutional layers with residual skip connections52,53, to spread information and model long-range interactions across the input DNA sequences. The skip connection technique was used to relieve the difficulty in optimization caused by the vanishing gradient problem. Each dilated convolutional block had the following layers: 1) Batch normalization, 2) Dilated 1D convolution (width 3, filter 768), 3) GELU activation, 4) Batch normalization, 5) 1D convolution (width 1, filter 768), 6) GELU activation, 7) Dropout (rate 0.3), and 8) Skip connection (Add with the block input before step 1). The dilation rate was increased by a factor of 2 at each block. The third part of the main body was a 1D convolution (width 1, filter 1536) with dropout (rate 0.05) to further summarize DNA motif patterns around each 16bp region. Thus, the output from the main body was the 128 (length) × 1536 (filters) representation of 16-bp windows across the 2048-bp input DNA sequence.
CNN model pre-train
The model was pre-trained on the same sequencing datasets as Basenji27,48 (https://console.cloud.google.com/storage/browser/basenji_barnyard/data/mouse), and used the same Poisson negative log likelihood loss. Briefly, the 1,643 mouse genome datasets included 228 DNase-seq or ATAC-seq datasets, 308 TF ChIP-seq datasets, 750 histone modification ChIP-seq datasets, and 357 CAGE datasets. We modified the input DNA sequences of Basenji2 by trimming ¼ length on each side of the original input sequence, dividing the kept sequence into 2048-bp subsequences and randomly taking 5 subsequences from the middle half of each sequence. The modified dataset contained 146,475 training, 11,045 validation, 10,085 test sequences for the mm10 mouse genome.
To predict the read coverage profiles for the 1,643 datasets, the pre-train head used a 1D convolution (width 8, stride 8, filter 1643) followed by a Softplus activation function to generate a positive value for every position in the output 16 × 1643 representation of 128-bp windows.
The pre-training model was implemented in TensorFlow (v1.14.0), and was trained on 2 Tesla V100 GPU cards. We used a batch size of 128, and we stopped training when the validation loss had not improved for 15 epochs and then returned to the model that had achieved the smallest validation loss. We used the Adam optimizer with a learning rate 1e-4 and default values for hyperparameters (0.9) and (0.999). We used 5,000 warm-up steps to linearly increase the learning rate from 0 to 1e-4.
CNN model fine-tune
The fine-tuning dataset consisted of 3,231 extended-promoter sequences from Aire-induced genes and 3,121 sequences from expression-matched Aire-neutral genes in the B6 genome. 80% of the dataset was used for training, 15% for validation and 5% for testing. ‘Extended promoters’ were defined as 1024bp upstream of the TSSs to 1024bp downstream of the TSSs. All other ‘promoters’ in the main text were defined as 1000bp upstream of the TSSs to 200bp downstream of the TSSs. We also included two test sets from the NOD genome. One of them had 2,000 sequences that contained SNPs or Indels compared with their B6 counterparts. The other had 261 sequences not annotated in the B6 genome. Both NOD test sets were balanced with half sequences from Aire-induced genes and half from Aire-neutral genes. Each sequence was a DNA stretch +/− 1024 bp around the transcriptional start site. To predict whether the input sequence is from an Aire-induced or Aire-neutral gene, the fine-tune head implemented the following operations to transform the representation output by the main body to the probability of input sequence being from an Aire-induced gene: 1) 1D convolution (width 1, filter 10), 2) Dropout (rate 0.5), 3) GELU activation, 4) Flatten layer, 5) Dense layer (unit 3), 6) GELU activation, 7) Dense layer (unit 1), and 8) Sigmoid activation.
The fine-tuning model was implemented in TensorFlow (v2.3.0) and trained on 1 Tesla V100 GPU cards to minimize the binary cross-entropy loss. In the first stage of fine-tuning, parameters in the main body were frozen when optimizing the parameters in the fine-tune head. We used a batch size of 8 and Adam optimizer with a learning rate 1e-5 and default values for hyperparameters (0.9) and (0.999). The optimal batch size, learning rate and dropout rate were tuned by grid search using validation dataset. We stopped the first-stage training when the validation loss had not improved for 20 epochs and then returned to the model that had achieved the smallest validation loss. Then we unfreezed parameters in the last 1D convolutional layer of the main body and trained for another 10 epochs using a small learning rate 1e-7 to slightly adjust parameters in the main body.
CNN model interpretation
To identify the DNA sequence motifs that contributed most to the prediction accuracy of the model, for each input sample in the dataset, we computed the absolute value of the gradient of the output prediction with regard to each input nucleotide using backpropagation. For one-hot encoded dataset, this is equivalent to the gradient × input score54. Since each position in the representation output by the main body represented a 16-bp window in the input sequence, we aggregated the gradient × input profile for every input sequence by taking the average scores for every 16 bp. Thus, we obtained a vector of 128 elements for every input sequence, containing the contribution scores for the 128 16-bp windows. We then determined the genomic loci that had the two largest positive contribution scores for each input sequence and extracted the DNA sequences centered at these two genomic loci containing ten 16-bp windows. To identify DNA motifs enriched in these large-positive-gradient sequences, we used the XSTREME program (E-value ≤ 0.05, Minimum width 6, Maximum width 20)55 in the MEME suite (v5.4.1)56 to scan these sequences for both de novo motifs and known TF-binding motifs.
Genes originally classified in our model as Aire-neutral genes (n=583) out of 3000 randomly selected genes from the genome were used for the in silico saturation mutagenesis experiment. For ISM, computational mutation was performed for every nucleotide in the 2048-bp input sequence (6,144 substitutions per sequence) to examine how the mutation affected the Aire-induced gene prediction. The effect of the substitution, which was called the ISM score in Fig. 1, was measured as , where is the output of the sigmoid function, the prediction probability, for the unaltered sequence, and for the computationally mutated sequence57. To identify DNA motifs enriched in the regions (50bp in length) with the largest ISM scores, we again used the MEME suite56.
In the sequence replacement experiment, the sequences used were: the Z-DNA motifs - (CA)8 and (CA)16 as well as their reverse complements; a random control sequence of 16bp - CTACCTAACGCCCCTA and its reverse complement; a random control sequence of 32bp - TTGGCCGTTAACGTTTGTCTGCCGGATATTCA and its reverse complement; the Nfe2•Maf-binding motif - ATGACTCAGCA and its reverse complement; the Batf3-binding motif - TGACGTCAC and its reverse complement. When calculating the percentage of genes classified as Aire-induced after replacement with some sequence, we took the union of genes that were re-classified as Aire-induced genes by the neural network due to a replacement by some sequence and by its reverse complement.
In silico mutagenesis using Z-DNABERT
Z-DNABERT computes a Z-DNA score (ranging from 0 to 1) for each nucleotide in the input sequence, reflecting how likely that nucleotide is positioned in a stretch of Z-DNA-forming sequence. (CA)n repeats with high Z-DNA-forming potential (default parameter values: ‘model_confidence_threshold’ set to 0.5; and the ‘minimum_sequence_length’ set to 10bp) were identified for promoters of Aire-induced genes (AIGs) using Z-DNABERT (n=333). Then for every nucleotide in every (CA)n repeat, we performed in silico mutagenesis (ISM) and calculated the ISM score. A positive ISM score indicates that substitution of the original nucleotide leads to a decreased average Z-DNA score across the (CA)n repeat. The input sequence to Z-DNABERT was still the entire AIG promoter sequence, with only one nucleotide in the (CA)n repeat mutated to one of the other three nucleotides each time. This procedure ensured that all of the other local genomic information stayed the same when making predictions.
Example ISM score heatmaps showed that disruptions of (CA)n repeats in most cases led to decreased Z-DNA scores, the one exception being (CA) to (CG) substitutions. This finding was consistent with the fact that Z-DNABERT gave high scores to (CG)n repeats. Distributions of ISM scores for nucleotides at various positions near the boundaries of (CA)n repeats in AIG promoters showed that ISM scores were significantly larger than 0 at all positions examined.
Z-DNA motif identification
Z-DNA motifs were identified as alternating purine-pyrimidine tracts of at least 10 bp. Specifically, G was followed by C or T for at least 10 bp58. For the definition of (CA)n repeats, we used (CA)n repeats to represent (CA/TG)n repeats in the main text since (TG)n on one strand is (CA)n on the other strand. If (TG)n repeats occurred in some examples, we also used ‘(CA)n repeats’ to describe the result. For example, the (CA)n repeat at promoter of Pglyrp4 in Extended Data Fig. 1a was a (TG)n repeat.
Weakly expressed genes
In the analyses shown in Fig. 1g,h, we focused on weakly expressed genes because both previous1,59,60 and the current study found that Aire upregulates the expression primarily of genes that have low basal expression levels prior to its action. We defined ‘weakly expressed genes’ as those with a TPM (transcripts per million) < 0.35 in mTECs from Aire-KO mice (n=4537). We chose the threshold 0.35 because 50% of Aire-induced genes have an Aire-KO TPM < 0.35 (n=1563).
Mice
All mice were housed and bred under specific-pathogen-free conditions at the Harvard Medical School Center for Animal Resources and Comparative Medicine (Institutional Animal Care and Use Committee protocol #IS00001257).
All experimental mice were 4–6-week-old females. The Aire-deficient mice have been described1. Littermates were routinely used for WT and KO comparisons. (C57BL/6J) B6 and (NOD/ShiLtJ) NOD mice with Aire-driven expression of Igrp-GFP (Adig)61 were provided by Dr. Mark Anderson and were appropriately bred to generate Aire-GFP+.Aire+/+ and Aire-GFP+.Aire−/− littermates on the B6, NOD or B6/NOD F1 hybrid backgrounds. CAST/EiJ (CAST) mice were purchased from Jackson Laboratory and bred to B6 mice to generate B6/CAST F1 hybrids. Foxn1cre and Nfe2l2flox/flox mice were also purchased from Jackson Laboratory and were appropriately bred to generate Foxn1Cre-Nfe2l2flox/flox and Foxn1Cre-Nfe2l2+/+ littermates.
For experiments that used spermidine to enhance Z-DNA formation, 4-week-old B6 Aire+/+ and Aire−/− mice were ip-injected with 15mg/kg spermidine (Sigma-Aldrich) in PBS for seven consecutive days. For experiments that used topotecan to stabilize TOP1 binding, 4-week-old B6 Aire+/+ and Aire−/− mice were ip-injected with 0.5mg/kg topotecan (Sigma-Aldrich) dissolved in dimethylsulfoxide (DMSO) for three consecutive days. Mice under different ip injections were randomly allocated into experimental and control groups.
Isolation, sorting and analysis of thymus cells
Thymus tissues from individual female mice were collected in Dulbecco’s Modified Eagle Medium (DMEM; Gibco) supplemented with 2% fetal bovine serum (FBS) and 25mM HEPES (basic medium), and minced with scissors. The fragments were then digested for 20 min with 0.5mg/ml collagenase (Sigma-Aldrich) and 0.2mg/mL DNase (Sigma-Aldrich), then with collagenase/dispase (Roche) and 0.2mg/mL DNase (Sigma-Aldrich) for 15 min. CD45+ cells were depleted by magnetic-activated cell sorting (MACS) with CD45 MicroBeads (Miltenyi). For mice on B6 and B6/CAST background, cells were stained with the following primary antibodies: MHCII-APC (107614, BioLegend), Ly51-PE (108308, BioLegend); CD45-PE/Cy7 (103114, BioLegend). For mice on NOD and B6/NOD background, cells were stained with: MHCII-PE (205308, BioLegend), Ly51-Alexa Fluor 647 (108312, BioLegend); CD45-PE/Cy7 (103114, BioLegend). Cells were stained in the basic medium. 4’,6-diamino-2-phenylindole (DAPI) was added before sorting to exclude dead cells. For F1-hybrid sequencing experiments, DAPI−CD45−Ly51loMHCIIhiGFPhi mTECs from B6 homozygous, NOD homozygous or B6/NOD F1 mice were sorted on a FACSAria sorter (BD). For all other experiments, DAPI−CD45−Ly51loMHCIIhi mTECs were sorted.
For flow cytometric analysis of thymocytes, the following primary antibodies were used: CD4-Brilliant Violet 605 (100548, BioLegend), CD8a-FITC (100706, BioLegend), CD19-APC (115512, BioLegend). For flow cytometric analysis of Z-DNA formation in mTECs, the anti-Z DNA antibody (ab2079, abcam) was used.
Mapping of F1 hybrid sequencing reads
We took an unbiased diploid genome alignment strategy to resolve the origin of each read from an F1-hybrid sequencing experiment5,62,63. Briefly, we obtained the SNP and InDel information for NOD and CAST genome from the mouse-genome project14, and then modified the B6 genome (mm10) to obtain pseudo-NOD and pseudo-CAST genomes. Sequencing reads were aligned in parallel to both the B6 and the pseudo-NOD or pseudo-CAST genomes. Since two alignments for each read were under different coordinate systems, which impeded their direct comparison, we converted the coordinates of alignments in the pseudo-genomes back to the B6 coordinate system. The read was then assigned to the genome where it was uniquely mapped or had a higher mapping score. A read was randomly assigned to the B6 or another genome (NOD or CAST) if it had identical mapping scores for both genomes. The resulting alignment file (BAM) contains genome assignment information for each read, and also has tag indicating how the origin of the read was determined. For genetic-variant-containing reads, they were assigned to a genome in an allele-specific manner. For reads not covering genetic variants, they were randomly assigned to a genome because of the identical mapping scores.
Statistical testing of allelic imbalance
Only reads overlapping any high-confidence SNP or InDel within a gene (RNA-seq), OCR (ATAC-seq) or DSB hotspot (BLISS) were considered when testing for allelic imbalance. We counted the number of variant-overlapping reads from the reference genome (B6), and those from the alternative genome (NOD or CAST), respectively, for every gene, OCR or DSB hotspot, using bcftools64 (v1.9). Allelically imbalanced regions were identified by modeling allele-specific read-count using the beta-binomial distribution62,65. Specifically, we assumed that the probability of having reads being from the reference or alternative genome for region followed the beta-binomial distribution where was the total number of allele-specific reads for region , and indicated the probability of a read being from the reference or alternative genome, and for capturing the overdispersion in read count data. For a perfectly balanced region , equaled (null hypothesis). An allelically imbalanced region was identified if was significantly larger or smaller than . The Benjamini-Hochberg procedure was used to calculate the false discovery rate.
CIS-BP database
The CIS-BP database66 is an online database of TF-binding motifs. It currently hosts data for >700 species, >300 TF families, and a total of >390,000 TFs. The binding-motif data came from >70 sources, including Transfac, JASPAR and HOCOMOCO that are frequently used in TF-binding motif analysis. For Mus Musculus, CIS-BP database contains data for 938 TF-binding motifs.
Motif analysis for allelic imbalance
For imbalanced OCRs and DSB hotspots, the alternative-allele (NOD or CAST) sequences were obtained by modifying the corresponding B6-allele sequences using known SNPs and InDels14. We used FIMO67 from MEME suite (v5.4.1) to scan the peak sequences of both the reference and alternative allele for occurrences of TF-binding motifs (match p-value < 1e-4) in the CIS-BP database66. The coordinates of motifs found in the alternative genome were then converted back to the B6 coordinate system so that the scanning results for two alleles could be merged. Next, we repeated the following analysis for every TF-binding motif: 1) assign each imbalanced peak to the allele where it had a stronger match as indicated by a smaller FIMO match p-value for the particular TF-binding motif; 2) use the non-parametric Wilcoxon rank sum test to examine whether the allelic ratios of ATAC-seq or BLISS reads from peaks having stronger motif matches on the reference allele were significantly different from those of peaks having stronger motif matches on the alternative allele. De novo motif analysis for imbalanced OCRs was conducted using STREME68 (p-value < 0.05 Minimum width 8, Maximum width 20).
RNA-seq library preparation
For each sample, 1000 cells were sorted directly into the lysis buffer [5uL TCL buffer (Qiagen) supplemented with 1% 2-mercaptoethanol (Sigma)]. Libraries were constructed following the Smart-seq2 RNA-seq library preparation protocol, and were then sequenced by the Broad Genomics Platform, following the standard ImmGen ultra-low-input RNA-seq protocol (https://www.immgen.org/).
RNA-seq analysis
For non-F1-hybrid RNA-seq, paired-end RNA-seq reads were aligned to the mm10 genome using STAR69. Samples with fewer than 8,000 genes that have more than ten reads, medium transcript integrity number across all transcripts smaller than 45, or poor intra-replicate correlation were removed from downstream analyses. Pearson correlations between replicates were calculated using TPM (Transcripts Per Million) value output by Kallisto70 (‘quant’, v0.45.1). MA plots were generated using the log2 fold-change and mean of normalized read-count across all samples for each gene calculated by DESeq271 (lowly expressed genes were removed, v1.22.2). Differential expression analyses were performed using DESeq2. For F1-hybrid RNA-seq experiments, quality control was performed using Sickle (default setting, v1.33, https://github.com/najoshi/sickle) to remove low-quality reads. Paired-end RNA-seq reads were aligned to the corresponding reference or alternative genome using Tophat272 (v2.1.1). Only paired-end reads mapped to the single unique genomic location were kept for downstream analysis (SAMtools73, v1.3.1). Allele-specific read-counts were obtained as described above. Gene annotations for the B6 and NOD genome were downloaded from Ensembl release 102 based on the mouse genome assembly GRCm38.
scRNA-seq library preparation
mTEChi and post-Aire mTEClo were sorted from 4–6-week-old female mice based on the gating strategies reported before42. Samples were hashed using the TotalSeq-A anti-mouse (anti-CD45/MHC class I) hashtags (BioLegend). Cells were then submitted to the Broad Institute Genomics Platform for encapsulation and library preparation following 10X Genomics protocols.
scRNA-seq analysis
Sequencing reads were demultiplexed, aligned, and assigned to cells, and transcript-by-cell matrices were generated using Cell Ranger. CITE-seq-count was used to compute the Hash-by-cell matrices. The downstream analyses were largely performed using the Seurat package74 (v 4.3.0.1). Briefly, hash counts were normalized using the centered log-ratio (CLR) transformation, and cells were then assigned hash identities by high expression of a single hash. Cells negative for hashtag signals or with multiple hashes were removed. Next, cells with >10% mitochondrial counts or unique feature counts > 7000 or unique feature count < 200 were removed. Gene expression data were then normalized using the default method “LogNormalize”. The top 2000 variable genes were selected by the “vst” (variance-stabilizing transform) method. After scaling the data, PCA was performed on the top variable genes. The top 50 PCs were retained based on jackstraw and elbow plots for constructing the (shared) nearest-neighbor graph (k.param=20). Cell clustering was first performed using the Louvain algorithm (resolution=1.55). Then, the top 50 PCs were kept for running the UMAP dimensional reduction. Cell clusters expressing canonical T cell, B cell, myeloid, fibroblast, or endothelial markers were removed due to contamination. Cell type annotation was done based on expression of marker genes of “Aire-expressing”, “Transit-amplifying”, “Immature” and “Post-Aire” mTECs.
ATAC-seq library preparation
For each ATAC-seq sample, ~15,000 mTECs from 4–6-week-old female mice were sorted into basic medium (described above) for ATAC-seq library preparation, following the fast-ATAC-seq protocol75,76, which provides high-quality data with low cell input. Briefly, cells were spun down at 4°C and washed with 1mL PBS supplemented with protease inhibitor (Complete EDTA-free protease inhibitor cocktail, Roche). Then 10uL Tn5 transposase mixture was added to resuspend the cell pellet on ice. [10uL Tn5 transposase mixture: 5uL 2x Tagment DNA Buffer, 0.5uL Tagment DNA Enzyme from the Nextera DNA Library Preparation Kit (Illumina), 4.4uL nuclease-free water, 0.2mg/ml digitonin (G9441, Promega)]. Cells were incubated for 30 min at 37°C with agitation. Transposed DNA fragments were purified using the MinElute Reaction Cleanup Kit (QIAGEN). Two rounds of PCR were performed to generate the library as described in the ImmGen ATAC-seq protocol76. After the initial round of PCR with 7 cycles, small DNA fragments were size-selected and purified using the Agencourt AMPure XP beads (Beckman Coulter) followed by a second round of PCR with 10 cycles. Amplified libraries were purified by AMPure XP beads (x1.8 vol.) and sequenced on the NextSeq 500 instrument (Illumina) to generate paired-end reads (41+40bp) at the Harvard Medical School Biopolymers Facility.
ATAC-seq analysis
After removing low-quality reads using Sickle and trimming adapter sequences using cutadapt77 (v1.14), we aligned ATAC-seq reads to the corresponding genome (mm10 reference genome, pseudo-NOD genome or pseudo-CAST genome) by bowtie278 (v2.3.4.3) with the following parameters: -X 1000 --fr --no-mixed --no-discordant. Non-uniquely mapped and mitochondrial DNA reads were removed using a combination of SAMtools functions. PCR duplicates were removed using Picard (‘MarkDuplicates’, v2.8.0, https://broadinstitute.github.io/picard/). OCRs for individual samples were identified using MACS279 (‘callpeak’, v2.1.1.20160309) with the following parameters: --keep-dup all --nomodel --shift −100 --extsize 200 -p 0.05. High-confidence, reproducible OCRs among replicates were then identified using the irreproducible discovery rate (IDR) framework80 (v2.0.2) with a global IDR < 0.05. OCRs overlapping ENCODE blacklisted regions were removed from downstream analyses using BEDTools81 (v2.27.1). To visualize individual ATAC-seq tracks using the Integrative Genomics Viewer (IGV)82, the alignment file (BAM file) was converted to the read-coverage file (BigWig file) using deepTools83 (‘bamCoverage’, v3.0.2). ATAC-seq profiles over various genomic regions (e.g. TSS) were generated using the function ‘plotProfile’ of deepTools or using the ngs.plot84 (v2.47.1). For F1 ATAC-seq, allele-specific read counts for OCRs were obtained as described above.
BLISS library preparation
We followed the suspension-cell BLISS protocol85, which is an adaptation of the original BLISS protocol86 for non-adherent cells. The entire library preparation process took around 4 days. Briefly, on Day 1, for each sample, ~100,000 mTECs from 4–6-week-old female mice were sorted into PBS. Cells were fixed in 4% paraformaldehyde (15710, Electron Microscopy Sciences) for 10 min at room temperature on a roller shaker. Formaldehyde was quenched with glycine. Fixed cells were then washed twice using ice-cold PBS. For permeabilization, fixed cells were subjected to two sequential lysis using lysis buffer 1 (10mM Tris-HCl, 10mM NaCl, 1mM EDTA, 0.2% Triton X-100, pH 8) and lysis buffer 2 (10mM Tris-HCl, 150mM NaCl, 1mM EDTA, 0.3% SDS, pH 8). After permeabilization, DSB ends were blunted using the Quick Blunting Kit (E1201L, NEB) for 1 h at room temperature, which was followed by in situ DSB ligation of BLISS adapters using T4 DNA ligase (M0202M, NEB) for ~16 h (overnight) at 16°C. On Day 2, genomic DNAs were reverse crosslinked and extracted using DNA extraction buffer (10mM Tris-HCl, 100mM NaCl, 50mM EDTA, 1% SDS, 1mg/mL Proteinase K, pH 8) at 55°C for ~18 h (overnight) with shaking. On Day 3, DNA was purified and then sonicated in 1 × TE buffer using the Covaris M220 ultrasonicator with the following setting: peak incident power 50, duty factor 10%, cycles per burst 200, time 70s. After purification by Agencourt AMPure XP beads, DNA was in vitro transcribed using the MEGAscript T7 Transcription Kit (AM1333, ThermoFisher) at 37°C for ~14 h (overnight). On Day 4, genomic DNA was removed using DNase I (RNase-free) (AM2222, ThermoFisher) followed by RNA purification using Agencourt RNAClean XP beads (Beckman Coulter). Libraries were then built via the following steps: Illumina RA3 adapter ligation using T4 RNA Ligase 2, truncated KQ (M0373S, NEB) at 25°C for 2 h; reverse transcription using SuperScript III Reverse Transcriptase (18080044, ThermoFisher) at 50°C for 1 h; library indexing and amplification using NEBNext High-Fidelity 2x PCR Master Mix (M0541S, NEB); DNA purification and size-selection (300–800bp) using Agencourt AMPure XP beads. All primers and adapters were custom synthesized by Integrated DNA Technologies (IDT) based on sequences in TruSeq Small RNA Library Preparation kit (Illumina). BLISS libraries were sequenced on the NextSeq 500 instrument (Illumina) to generate single-end reads (80bp) at the Harvard Medical School Biopolymers Facility.
BLISS analysis
We used Cutadapt (v2.5) to scan BLISS reads for matches of the expected 16bp prefix [8bp unique molecular identifier (UMI) + 8bp sample barcode] in the BLISS adapters, allowing a maximum of one mismatch. Only reads containing the 16bp prefix were kept. Prefixes were then clipped using Cutadapt. Reads were aligned to the corresponding genome (mm10 reference genome or pseudo-CAST genome) using bowtie2 (v2.3.4.3). Only uniquely mapped reads were kept (SAMtools, v1.3.1). PCR duplicates that had the same UMIs (allowing for a maximum of two mismatch) were removed using UMI-tools87 (‘dedup’, v1.0.1). DSBs independently generated at the same genomic location in different cells were kept because they had different UMIs. DSB hotspots, defined as peaks identified by HOMER88 (‘findPeaks’, v4.9) with fold-enrichment > 1.5, p-value < 1e-5, were called for individual samples. DSB hotspots overlapping ENCODE blacklisted regions were removed from downstream analyses using BEDTools (v2.27.1). DSB hotspots in replicates were merged using BEDOPS89 (v2.4.30) functions and only those occurring in both replicates were kept for further analyses. BLISS profiles over various genomic regions were generated as for ATAC-seq. For B6/CAST F1 BLISS, allele-specific read-counts for DSB hotspots were obtained as described above. For differential DSB hotspot analyses, DSB hotspots were called using MACS279 with -p set to 0.1. High-confidence, reproducible peaks among replicates were then identified using the irreproducible discovery rate (IDR) framework80 (v2.0.2) with a global IDR < 0.1. Htseq-count was used to count the number of reads in each DSB hotspot. DESeq2 was then used to perform the differential analyses.
ChIP-seq library preparation
Pol II ChIP-seq libraries were constructed as described previously9, and were sequenced on the HiSeq 2500 instrument (Illumina) to generate single-end reads (50bp). For Z-DNA ChIP-seq, 300k-500k mTECs from 4–6-week-old female mouse were sorted and pooled for each sample. Briefly, cells were fixed with 1% formaldehyde (28906, Thermo Fisher Scientific) for 10 min at room temperature with rotation, followed by quenching using 0.125M glycine for 5 min at room temperature. After washing using ice-cold PBS supplemented with protease inhibitor (Complete EDTA-free protease inhibitor cocktail, Roche), cells were resuspended in lysis buffer (10mM Tris-HCl pH 8.0, 140mM NaCl, 1mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100) supplemented with protease inhibitor on ice for 10 min. Chromatin was then sonicated on the Covaris M220 ultrasonicator with the following settings: peak incident power 50, duty factor 10%, cycles per burst 200, time 8 min. Lysate was centrifuged for 10 min at 14,000g at 4°C. An “input” sample was preserved from the supernatant. The rest of the supernatant was incubated with 2 ug of anti-Z-DNA/Z-RNA90 (Z22) (Ab00783–23.0, Absolute Antibody) or Rabbit IgG (#2729, Cell Signaling) antibodies precoupled to Protein A Dynabeads (Invitrogen) on a rotator overnight at 4°C. On the following day, beads were washed twice sequentially on a pre-cold magnet (Invitrogen) using wash buffer 1 (10mM Tris-HCl pH 8.0, 140mM NaCl, 1mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100), wash buffer 2 (10mM Tris-HCl pH 8.0, 500mM NaCl, 1mM EDTA, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100), wash buffer 3 (10mM Tris-HCl pH 8.0, 250mM LiCl, 1mM EDTA, 0.5% NP-40, 0.5% sodium deoxycholate) and 1 × TE. IPed chromatin material on beads was treated with 1μg DNase-free RNase (Roche) for 30 min at 37°C. Beads were then incubated with 50uL elution buffer (10mM Tris-HCl pH 8.0, 300mM NaCl, 5mM EDTA, 0.4% SDS) containing 2uL Proteinase K (NEB) at 55°C for 1h and 65°C overnight to de-crosslink. On the next day, DNA was purified using MinElute Reaction Cleanup Kit (QIAGEN), and the library was prepared using NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB) following the manufacturer’s protocol. After purification using AMPure XP beads, the libraries were sequenced on the NextSeq 500 instrument (Illumina) to generate paired-end reads (42+42bp).
ChIPmentation library preparation
For ChIPmentation, each library was constructed using ~50,000 mTECs from a 4–6-week-old female mouse were sorted. ChIPmentation libraries were prepared as described elsewhere91 with some adjustments. Briefly, cells were prepared following the same steps as for ChIP-seq library preparation till the wash step except that 1) the antibodies used were anti-Aire (14-5934-82, eBioscience) and anti-NELF (ab170104, abcam); 2) the sonication time was 5min; and 3) the last wash was using 10mM Tris-HCl pH 8.0 instead of 1 × TE. After wash, beads were then resuspended in 25uL tagmentation reaction mix with 1uL Tn5 transposase (Illumina) added, and were incubated at 37°C for 5 min. After removing the tagmentation reaction mix, beads were washed twice with wash buffer 1 and once with 1 × TE. They were then incubated with 50uL elution buffer (10mM Tris-HCl pH 8.0, 300mM NaCl, 5mM EDTA, 0.4% SDS) containing 2uL Proteinase K (NEB) at 55°C for 1h and 65°C for 8h to de-crosslink. DNA was purified using MinElute Reaction Cleanup Kit (QIAGEN). Libraries were indexed and amplified using the NEBNext High-Fidelity 2x PCR Master Mix (M0541S, NEB) and ATAC-seq primers92, followed by purification using AMPure XP beads and sequencing as for ATAC-seq.
ChIPmentation and ChIP-seq analysis
Alignment of ChIPmentation reads was performed as for ATAC-seq reads: trimming low-quality reads, clipping adapters, mapping to the genome, keeping only uniquely mapped reads, and removing PCR duplicates (see above). Read-alignment of ChIP-seq datasets was performed as per ATAC-seq except that the adapter trimming step trimmed different sequences. ChIPmentation and ChIP-seq profiles over various genomic regions were generated also as for ATAC-seq (see above). IgG control for Pol II ChIP-seq of Aire-KO mTECs came from ref.9. ChIPmentation and ChIP-seq signals over promoters, defined as −1000 to 200bp relative to the TSS, of Aire-induced and Aire-neutral genes as in Fig. 4d,e and Extended Data Fig. 8c were calculated using the HOMER (v4.9) script ‘annotatePeaks.pl’. Z-DNA ChIP-seq peaks were identified using MACS279 (‘callpeak’, v2.1.1.20160309) with the following parameters: --keep-dup all -f BAMPE -c ctrl_bam -p 0.1. High-confidence, reproducible peaks among replicates were then identified using the irreproducible discovery rate (IDR) framework80 (v2.0.2) with a global IDR < 0.05. Peaks overlapping ENCODE blacklisted regions were removed from downstream analyses using BEDTools81 (v2.27.1).
CUT&Tag library preparation
Libraries were prepared following the protocol detailed in Ref.93. Briefly, for each sample, 50,000 – 80,000 mTECs from a female mouse were sorted. After washing in wash buffer 1 (20mM HEPES pH 7.5, 150mM NaCl, 0.5mM spermidine, Complete EDTA-free protease inhibitor cocktail), cells were bound to the Concanavalin A beads (Bangs Laboratories), followed by incubation with 1:50 primary antibody [anti-RNA polymerase II phospho S5 (ab5131, abcam)] in wash buffer 1 supplemented with 0.05% digitonin, 2mM EDTA and 0.1% BSA at 4°C overnight. The next day, cells were incubated with 1:100 secondary antibody [guinea pig anti-rabbit IgG (611-201-122, Rockland)] in wash buffer 1 supplemented with 0.05% digitonin at room temperature for 1 h on a rotator. After washing, cells were mixed with 1:200 pA-Tn5 (124601, Addgene; purified in-house) in wash buffer 2 (20mM HEPES pH 7.5, 300mM NaCl, 0.5mM spermidine, Complete EDTA-free protease inhibitor cocktail) plus 0.01% digitonin at room temperature for 1 h on a rotator. Cells were then washed twice, followed by tagmentation in wash buffer 2 plus 0.01% digitonin and 10mM MgCl2 at 37°C for 1 h. DNA was solubilized by adding 16.7mM EDTA, 0.1% SDS and 0.167mg/mL Proteinase K to each sample and incubating at 55°C for 1 h. DNA was phenol-chloroform extracted and amplified using the NEBNext High-Fidelity 2x PCR Master Mix (M0541S, NEB) and ATAC-seq primers92, After purification by AMPure XP beads (x1.3 vol.), libraries were sequenced as for ATAC-seq.
CUT&Tag analysis
Quality control, adapter trimming, read alignment and generation of profile plots were performed as for the ATAC-seq data (see above) except that 1) the bowtie2 parameters used were --local --very-sensitive --no-mixed --no-discordant -X 1000 –fr; and 2) reads were aligned to the E. coli genome in parallel to quantify the number of carry-over reads from pA-Tn5.
Z-DNA and Nfe2l2-binding motif distribution at DSB hotspots
Z-DNA motifs were identified as alternating purine-pyrimidine tracts of at least 10 bp as described above. Nfe2l2 binding motifs in the DSB hotspot were identified using FIMO67 from MEME suite (v5.4.1). The length of each DSB hotspot was bucketized into 100 position bins. For each position bin of all the DSB hotspots, we counted how many Z-DNA and Nfe2l2-binding motifs fell into that bin, and then used the 100 counts to generate the density plots and heatmaps as shown in Extended Data Fig. 9c. Curve fitting was performed using polynomial regression.
BRG1 ATAC-seq analysis
GC-content- and Quantile-normalized ATAC-seq data for mTEClo and mTEChi from BRG1-WT and BRG1-cKO mice were obtained from GEO with the accession code GSE102526. OCRs up-regulated by BRG1 were defined as 1) mTEChi BRG1-WT ATAC-seq / mTEClo BRG1-WT ATAC-seq > 1.5, and 2) mTEChi BRG1-WT ATAC-seq / mTEChi BRG1-cKO ATAC-seq > 1.5. Unchanged OCRs were defined as 1) mTEChi BRG1-WT ATAC-seq / mTEClo BRG1-WT ATAC-seq > 1.5, and 2) 0.9 < mTEChi BRG1-WT ATAC-seq / mTEChi BRG1-cKO ATAC-seq < 1.1.
KEGG enrichment pathway analysis
The KEGG pathway analysis for differentially expressed genes between mTECs from Nfe2l2-KO and control mTECs were performed using the function ‘run_KEGG’ of the R package clusterProfiler. The cutoff for the enriched pathways was 0.05 for the adjusted p-value.
Statistical analysis
De novo motif and known TF-motif-enrichment p-values and E-values were defined and calculated by MEME Suite, p-values for bulk RNA-seq differentially expressed genes were calculated in DESeq2 using the Wald test, adjusted p-values for enriched KEGG pathways were calculated in clusterProfiler using the Benjamini-Hochberg procedure. Boxplots showed the median, the first and third quartiles as boxes. The upper and lower whiskers of boxplots extended from the hinges to ±1.5*IQR (where IQR is the inter-quartile range). Other statistical tests were performed using R as specified in the figure legends.
Extended Data
Extended Data Fig. 1 |. Performance of the pre-trained CNN model.

a, Schematics of the CNN model for pre-training and fine-tuning. The first section of the main body is comprised of convolutional layers to extract relevant DNA sequence motifs. The following section has repeated blocks containing dilated convolutional layers with residual skip connections, to spread information and model long-range interactions in the input DNA sequences. Aire-induced and expression-matched Aire-neutral gene lists have been described3,9. Briefly, Aire-induced genes were defined as Aire+/+/Aire−/− > 2 and Aire-neutral genes were Aire+/+/Aire−/− > 0.9 and < 1.1, based on bulk RNA-seq data from Ref.3. b, Exemplar true versus predicted profiles over a randomly selected sequence from the test set. Profiles for 15 sequencing datasets are shown. c, Boxplot showing the Pearson correlations between the predicted and true sequencing profiles of test set sequences for the sequencing datasets used for the pre-training, including DNase-seq, ATAC-seq and ChIP-seq. d, Model evaluation on four datasets: a test set and validation set from the B6 genome, and two test sets from the NOD genome: (1) containing SNPs/Indels compared with counterparts in the B6 genome, and (2) derived from NOD-specific genes (in order to prevent data leakage during prediction). e, Bar plot comparing the performances of randomly initialized model and pre-trained model on the test set from the B6 genome. SNPs: single-nucleotide polymorphisms; InDels: insertions and deletions; AIG: Aire-induced gene; ANG: Aire-neutral gene.
Extended Data Fig. 2 |. Additional analysis using the fine-tuned CNN model and Z-DNABERT, related to.

Fig. 1. a, Contribution score profiles for AIGs whose largest-positive-gradient regions contained (CA)n repeats (left) or Nfe2•Maf-binding motifs (right). b, Motifs enriched in the regions (50bp in length) with the largest ISM scores. c, Motifs that are relatively more enriched in extended-promoter sequences of Aire-induced genes than Aire-neutral genes (E-value < 0.05). The MEME suite was used to identify the enriched motifs for panel b and c. d, Example ISM score heatmaps for (CA)n repeats in AIG promoters. Each of the three rows shows results for one possible substitution in the order of A->C->G->T from top to bottom. Red (positive ISM score) indicates that substitution of the original nucleotide leads to a decreased average Z-DNA score across the stretch of the (CA)n repeat; Blue (negative ISM score) indicates the other way. e, Boxplot showing the distribution of ISM scores at various positions near the boundaries of (CA)n repeats in AIG promoters. For example, position 2 indicates the second nucleotides from both ends of a (CA)n repeat. p-values for panel e were calculated using the one-sample Wilcoxon Signed Rank Test (one-tailed). AIG: Aire-induced gene.
Extended Data Fig. 3 |. Strain-specific gene expression in mTECs was predominantly driven by cis-regulation.

a,b, Cytofluorometric gating scheme for isolation of mTECs from B6 (a), NOD and F1 (b) mice. c, Rationale of the F1-hybrid analysis. d, Allelically imbalanced gene transcripts and OCRs in B6×NOD F1-hybrid mTECs. Red dots depict significantly imbalanced events with a false discovery rate (FDR) <0.05. e, Correlation between the fold-changes in accessibility for mTEC OCRs in B6 versus NOD mice (x axis) and fold-changes in accessibility for OCRs (n=23256) on the B6 versus NOD allele in mTECs of F1 hybrids (y axis). Red dots depict significantly imbalanced OCRs (n=3750). f, Correlation between the fold-changes in transcript levels for mTECs from B6 versus NOD mTECs (x axis) and transcript fold-changes for the B6 versus NOD allele in mTECs of F1 hybrids (y axis). Only genes significantly differentially expressed in B6 and NOD mTECs are shown (adjusted p-value < 0.05, n=248). g, Correlation between allelic biases in the expression of the nearest AIG (x axis) and in the accessibility of the OCR (y axis). The imbalanced OCR was assigned to an imbalanced AIG (n=248) if 1) the OCR was located within 50,000 bp of the gene’s TSS and 2) the AIG was the OCR’s nearest gene. There were 156 imbalanced OCRs assigned to imbalanced Aire-induced genes. p-values according to panels e and g were from Spearman’s correlation. FC: fold-change. OCR: open chromatin region; TF: transcription factor.
Extended Data Fig. 4 |. Exemplar genetic variants associated with allelic imbalances in chromatin accessibility and gene expression.

a,b, Examples of genetic variants of Nfe2l2-binding motifs associated with imbalanced expression of Aire-induced genes. c,d, Examples of genetic variants of Z-DNA motifs associated with allelic imbalances in the expression of an Aire-induced gene. OCR: open chromatin region; WT: wild-type; Aire-KO: Aire knockout.
Extended Data Fig. 5 |. Effect of spermidine on Z-DNA formation and thymic cell populations.

a, Density plot showing the effect of spermidine vs control PBS injection on Z-DNA intensity in mTECs measured by flow cytometry using an anti-Z-DNA antibody. b, Representative cytofluorimetric plots and quantitative summary for mTECs of WT and Aire-KO mice treated with spermidine or control PBS. c, Cytofluorometric gating scheme for analyses of thymocyte compartments. d, Analogous plots to panel b for thymocyte compartments. Error bars, mean ± s.e.m. from n=3 biological replicates. KO: Aire-KO.
Extended Data Fig. 6 |. Additional analysis of scRNA-seq of PBS-treated versus spermidine-treated mTECs, related to Fig. 3.

a, Log2 ratio (M-values) versus log2 average (A-values) plots showing the effect of spermidine treatment on mTECs from wild-type mice. Red dots depict spermidine-specific Aire-induced genes (FC >2, p-value <0.05). Dark grey dots depict Aire-induced transcripts shared between mice injected with spermidine and PBS. b, Per-replicate UMAPs of scRNA-seq of mTEChi and post-Aire mTEClo for PBS-treated and spermidine-treated Aire-WT mice. Each dot on the UMAPs is a single cell (n=3184). Each number on the UMAPs indicates a cluster identified using Seurat. c, Merged UMAPs of scRNA-seq of mTEChi and post-Aire mTEClo from PBS-treated (n=2 biological replicates) and spermidine-treated (n=2 biological replicates) Aire-WT mice. mTEC subtypes were labeled. d, UMAPs showing the expression of Aire (left) and one MHC Class II gene (right).
Extended Data Fig. 7 |. Additional analysis of BLISS in mTECs, related to Fig. 4.

a, Boxplot comparing BLISS signals at Z-DNA ChIP-seq peaks that had low, medium or high Z-DNA ChIP-seq signals in mTECs from Aire-WT mice. Low: < 25th percentile (n=1508); Medium: 25th - 75th percentile (n=3008); High: > 75th percentile (n=1506). b, Boxplot comparing Aire-KO BLISS signals at promoters of Aire-inducible (n=1563) and expression-matched Aire-neutral genes in Aire-KO mTECs (n=1907). Aire-inducible and Aire-neutral genes were weakly expressed genes (Aire-KO TPM<0.35) whose promoter DSB generation was detected by BLISS in mTECs from Aire-KO mice. c, Boxplot comparing the enrichment of Z-DNA motifs (left) at DSB hotspots upregulated by spermidine (n=97) versus those unaffected by spermidine (n=78). Analogous plot for CTCF-binding motifs is shown on the right. The number of motifs was normalized according to the length of the DSB hotspots. d, Correlation between genetic variation in CTCF-binding motifs and allelic imbalance in DSB generation. Individual lines indicate DSB hotspots with a stronger CTCF-binding motif match on the B6 allele (red, n=60) or on the CAST allele (blue, n=59). e, Analogous plot for (CA)n repeats (n=38 for B6 and n=51 for CAST). p-values for panels a-c were calculated using the Wilcoxon rank sum test (two-tailed), and for panels d and e using the Kolmogorov-Smirnov (KS) test (two-tailed).
Extended Data Fig. 8 |. Promoters of Aire-induced genes were poised for expression prior to the engagement of Aire.

a, Boxplot comparing the ATAC-seq and Pol II ChIP-seq signals at promoters of AIGs (n=1563) versus those at expression-matched ANGs (n=1907) in mTECs from Aire-KO mice. b, Exemplar DNA and chromatin profiles of AIGs poised for expression in mTECs from Aire-KO mice. In comparison, exemplar profiles for an ANG were shown on the right. c, Same as Fig. 4d except WT ATAC-seq and ChIP-seq signals. p-values in panels a and c were calculated using the Wilcoxon rank sum test (two-tailed). C&T: CUT&Tag; L: Low, n=747; M: Medium, n=2130; H: High, n=322; *: p-value<1e-10; **: p-value<1e-20. Data for WT and Aire-KO ATAC-seq, WT Pol II and Aire ChIP-seq came from Ref.9. Data for WT MED1 ChIP-seq came from ref.94.
Extended Data Fig. 9 |. Nfe2l2 may cooperate with Z-DNA to poise Aire-induced genes for expression.

a, Boxplots comparing the Aire-KO BLISS signals at AIG promoter DSB hotspots containing varying numbers of Nfe2l2-binding motifs (left) and CTCF-binding motifs (right). For Nfe2l2-binding motifs: Low: <=1 (n=598); Medium: 2–5 (n=258); High: >5 (n=87). For CTCF-binding motifs: Low: <=1 (n=468); Medium: 2–4 (n=404); High: >4 (n=71). b, Boxplots comparing the enrichment of Nfe2l2-binding motifs at DSB hotspots up-regulated by spermidine (n=159) versus those unaffected by spermidine (n=104). The number of motifs was normalized according to the length of the DSB hotspots. c, Density plots and heatmaps showing distributions of Z-DNA motifs (top) and Nfe2l2-binding motifs (bottom) at DSB hotspots in promoters (n=6884). Grey areas depict 95% confidence intervals. d, Boxplot comparing the lengths of Z-DNA motifs at OCRs unchanged (n=282) or up-regulated (n=356) by BRG1 (See Methods) in mTECs. e, De novo motif analysis for OCRs unchanged versus up-regulated by BRG1. f, MA plot (log2-scale) showing the expressions of Nfe2-related factors in mTECs from Nfe2l2-KO and Ctrl mice. g, Representative cytofluorimetric plots and quantitative summary for mTECs from Nfe2l2-KO and Ctrl mice. Error bars, mean ± s.e.m. from n=3 biological replicates. h, Volcano plots showing the expression of AIGs and ANGs that contain high-confidence Nfe2l2-binding motifs at promoters in mTECs from Nfe2l2-KO and Ctrl mice. i, Differentially expressed AIGs and ANGs (p-value < 0.05) between mTECs from Nfe2l2-KO and Ctrl mice. p-values for panels a-b and d were calculated using the Wilcoxon rank sum test (two-tailed), and for panel h using the Fisher’s exact test (two-tailed). L: Low; M: Medium; H: High; CI: confidence interval. Nfe2l2-KO: Foxn1Cre-Nfe2l2flox/flox; Ctrl: control, Foxn1Cre-Nfe2l2+/+.
Extended Data Fig. 10 |. Manipulation of Z-DNA formation, DSB generation or Nfe2l2 expression affected the expression of signature genes of mTEC mimetic cells.

a, KEGG pathway analysis (adjusted p-value < 0.05) for differentially expressed genes (p-value < 0.05, fold-change > 2, n=745) between mTECs from Nfe2l2-KO and Ctrl mice. b, Network plot showing the significantly enriched downregulated KEGG pathways and the associated genes. c, Expression of lineage-defining TFs42 in WT versus Aire-KO Aire-stage mTECs (log2 scale). d, MA plots (log2 scale) highlighting expression changes of signatures genes of several mimetic mTEC subtypes in mTECs from Nfe2l2-KO versus Ctrl mice. Red dots depict signature genes of the corresponding subtypes. e-f, Analogous plots showing the impact of spermidine and topotecan, respectively. Signature gene lists used were available at https://github.com/dmichelson/mimetic_cells/tree/main/scrnaseq/adult-neonate/mimetic-cell-signatures. p-values for panels d-f were calculated using the Fisher’s exact test (two-tailed).
Extended Data Fig. 11 |. A model of Z-DNA’s influence on Aire target choice.

Independent of Aire, Z-DNA formation is more likely to occur at the promoters of genes having Z-DNA motifs but not under robust TF-mediated transcriptional control. Nfe2l2 and other unknown factors would engage BRG1 or other chromatin remodelers to stabilize the energetically unfavorable Z-DNA formation. Z-DNA would enhance DSB generation at the promoters of genes subject to Aire induction, which would facilitate their poising, thereby promoting the recruitment of and induction of gene expression by Aire.
Supplementary Material
Acknowledgements:
We gratefully thank A. Baysov, J. Lee, I. Magill, and the Broad Genomics Platform for RNA-seq; the HMS Biopolymers Facility for all other sequencings; the HMS Immunology Flow Core; L. Du and the HMS Transgenic Mouse Core; K. Hattori and A. Ortiz-Lopez for experimental assistance; L. Yang and B. Vijaykumar for computational help; C. Laplace for graphics; K. Chowdhary and D. Michelson for discussions; and M. Anderson for providing the NOD mice with Aire-driven expression of Igrp-GFP. We thank Dr Alan Herbert for drawing our attention to the Z22 mAb. This work was supported by NIH grant R01AI088204 (to D.M.). Y. Fang is in part supported by the Harvard Molecules, Cells and Organisms Training Program. K. Bansal is supported by Department of Biotechnology/Wellcome Trust India Alliance Intermediate Fellowship (IA/I/19/1/504276).
Footnotes
Competing interests: The authors declare no competing interests.
Code availability
Code and scripts used in this study can be found at Zenodo (https://doi.org/10.5281/zenodo.10472904).
Data availability
All the sequencing data reported in this article were deposited as a SuperSeries at Gene Expression Omnibus (GEO) under accession GSE224557 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE224557). Specifically, the ATAC-seq, BLISS, ChIP-seq, ChIPmentation, CUT&Tag, bulk RNA-seq and scRNA-seq data are under accessions GSE224551, GSE224552, GSE224553, GSE224554, GSE224555, GSE224556 and GSE253215, respectively. Public datasets used in this article are: ATAC-seq for WT and Aire-KO mTECs from GSE92594; RNA Pol II, Aire and IgG ChIP-seq for WT mTECs from GSE92597; MED1 and IgG ChIP-seq for WT mTECs from GSE180937; ATAC-seq for mTECs from Brg1-WT and Brg1-cKO mice from GSE102526.
REFERENCES
- 1.Anderson MS et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002). [DOI] [PubMed] [Google Scholar]
- 2.Sansom SN et al. Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 24, 1918–1931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meredith M, Zemmour D, Mathis D, & Benoist C Aire controls gene expression in the thymic epithelium with ordered stochasticity. Nat Immunol 16, 942–949 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennecke P et al. Single-cell transcriptome analysis reveals coordinated ectopic gene-expression patterns in medullary thymic epithelial cells. Nat Immunol 16, 933–941 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Veeken J et al. Natural genetic variation reveals key features of epigenetic and transcriptional memory in virus-specific CD8 T cells. Immunity. 50, 1202–1217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Novakovsky G et al. Obtaining genetics insights from deep learning via explainable artificial intelligence. Nat Rev Genet 24, 125–137 (2023). [DOI] [PubMed] [Google Scholar]
- 7.Kelley DR et al. Sequential regulatory activity prediction across chromosomes with convolutional neural networks. Genome Res. 28, 739–750 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Link VM et al. Analysis of genetically diverse macrophages reveals local and domain-wide mechanisms that control transcription factor binding and function. Cell 173, 1796–1809 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bansal K, Yoshida H, Benoist C, & Mathis D The transcriptional regulator Aire binds to and activates super-enhancers. Nat Immunol 18, 263–273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodriguez-Martinez JA et al. Combinatorial bZIP dimers display complex DNA-binding specificity landscapes. Elife. 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rich A, Nordheim A, & Wang AH The chemistry and biology of left-handed Z-DNA. Annu. Rev. Biochem 53, 791–846 (1984). [DOI] [PubMed] [Google Scholar]
- 12.Georgakopoulos-Soares I et al. High-throughput characterization of the role of non-B DNA motifs on promoter function. Cell Genom. 2, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Umerenkov D et al. Z-flipon variants reveal the many roles of Z-DNA and Z-RNA in health and disease. Life Sci Alliance. 6, (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Keane TM et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kouzine F et al. Permanganate/S1 nuclease footprinting reveals non-B DNA structures with regulatory potential across a mammalian genome. Cell Syst. 4, 344–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu R et al. Regulation of CSF1 promoter by the SWI/SNF-like BAF complex. Cell 106, 309–318 (2001). [DOI] [PubMed] [Google Scholar]
- 17.Zhang J et al. BRG1 interacts with Nrf2 to selectively mediate HO-1 induction in response to oxidative stress. Mol. Cell Biol 26, 7942–7952 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu H, Mulholland N, Fu H, & Zhao K Cooperative activity of BRG1 and Z-DNA formation in chromatin remodeling. Mol. Cell Biol 26, 2550–2559 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shin SI et al. Z-DNA-forming sites identified by ChIP-Seq are associated with actively transcribed regions in the human genome. DNA Res. 23, 477–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marshall PR et al. Dynamic regulation of Z-DNA in the mouse prefrontal cortex by the RNA-editing enzyme Adar1 is required for fear extinction. Nat. Neurosci 23, 718–729 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fotsing SF et al. The impact of short tandem repeat variation on gene expression. Nat. Genet 51, 1652–1659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang T et al. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 606, 594–602 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thomas TJ, Gunnia UB, & Thomas T Polyamine-induced B-DNA to Z-DNA conformational transition of a plasmid DNA with (dG-dC)n insert. J Biol Chem. 266, 6137–6141 (1991). [PubMed] [Google Scholar]
- 24.Brooks WH Increased polyamines alter chromatin and stabilize autoantigens in autoimmune diseases. Front Immunol. 4, 91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang G & Vasquez KM Z-DNA, an active element in the genome. Front Biosci. 12, 4424–4438 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Meng Y et al. Z-DNA is remodelled by ZBTB43 in prospermatogonia to safeguard the germline genome and epigenome. Nat Cell Biol 24, 1141–1153 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pommier Y, Sun Y, Huang SN, & Nitiss JL Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol 17, 703–721 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Puc J et al. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell 160, 367–380 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madabhushi R et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pessina F et al. Functional transcription promoters at DNA double-strand breaks mediate RNA-driven phase separation of damage-response factors. Nat Cell Biol 21, 1286–1299 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sperling AS, Jeong KS, Kitada T, & Grunstein M Topoisomerase II binds nucleosome-free DNA and acts redundantly with topoisomerase I to enhance recruitment of RNA Pol II in budding yeast. Proc Natl Acad Sci U S A 108, 12693–12698 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shykind BM et al. Topoisomerase I enhances TFIID-TFIIA complex assembly during activation of transcription. Genes Dev. 11, 397–407 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Abramson J, Giraud M, Benoist C, & Mathis D Aire’s partners in the molecular control of immunological tolerance. Cell 140, 123–135 (2010). [DOI] [PubMed] [Google Scholar]
- 34.Guha M et al. DNA breaks and chromatin structural changes enhance the transcription of autoimmune regulator target genes. J Biol Chem. 292, 6542–6554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canela A et al. Genome Organization Drives Chromosome Fragility. Cell 170, 507–521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giraud M et al. Aire unleashes stalled RNA polymerase to induce ectopic gene expression in thymic epithelial cells. Proc Natl Acad Sci U S A 109, 535–540 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oven I et al. AIRE recruits P-TEFb for transcriptional elongation of target genes in medullary thymic epithelial cells. Mol Cell Biol 27, 8815–8823 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Durand-Dubief M et al. Topoisomerase I regulates open chromatin and controls gene expression in vivo. EMBO J. 29, 2126–2134 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Creemers GJ, Lund B, & Verweij J Topoisomerase I inhibitors: topotecan and irenotecan. Cancer Treat. Rev. 20, 73–96 (1994). [DOI] [PubMed] [Google Scholar]
- 40.Maruyama A, Mimura J, Harada N, & Itoh K Nrf2 activation is associated with Z-DNA formation in the human HO-1 promoter. Nucleic Acids Res. 41, 5223–5234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koh AS et al. Rapid chromatin repression by Aire provides precise control of immune tolerance. Nat Immunol 19, 162–172 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Michelson DA et al. Thymic epithelial cells co-opt lineage-defining transcription factors to eliminate autoreactive T cells. Cell 185, 2542–2558 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Michelson DA & Mathis D Thymic mimetic cells: tolerogenic masqueraders. Trends Immunol 43, 782–791 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Givony T et al. Thymic mimetic cells function beyond self-tolerance. Nature 622, 164–172 (2023). [DOI] [PubMed] [Google Scholar]
- 45.Giraud M et al. An RNAi screen for Aire cofactors reveals a role for Hnrnpl in polymerase release and Aire-activated ectopic transcription. Proc Natl Acad Sci U S A 111, 1491–1496 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kenton JDMWC & Toutanova LK BERT: Pre-training of Deep Bidirectional Transformers for Language Understanding. Proceedings of NAACL-HLT 4171–4186 (2019). [Google Scholar]
- 47.Avsec Z et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat Methods 18, 1196–1203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kelley DR Cross-species regulatory sequence activity prediction. PLoS Comput. Biol 16, e1008050 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moore JE et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Forrest AR et al. A promoter-level mammalian expression atlas. Nature 507, 462–470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hendrycks D & Gimpel K Gaussian Error Linear Units (GELUs). arXiv https://arxiv.org/abs/1606.08415v4 (2020). [Google Scholar]
- 52.Jaganathan K et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 176, 535–548 (2019). [DOI] [PubMed] [Google Scholar]
- 53.He K, Zhang X, Ren S, & Sun J Deep residual learning for image recognition. 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR). 2016 IEEE Conference on Computer Vision and Pattern Recognition, 770–778. 2016. [Google Scholar]
- 54.Simonyan K, Vedaldi A, & Zisserman A Deep inside convolutional networks: visualising image classification models and saliency maps. arXiv https://arxiv.org/abs/1312.6034v2 (2014). [Google Scholar]
- 55.Grant CE & Bailey TL XSTREME: comprehensive motif analysis of biological sequence datasets. bioRxiv https://www.biorxiv.org/content/10.1101/2021.09.02.458722v1 (2021). [Google Scholar]
- 56.Bailey TL, Johnson J, Grant CE, & Noble WS The MEME Suite. Nucleic Acids Research 43, W39–W49 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou J & Troyanskaya OG Predicting effects of noncoding variants with deep learning-based sequence model. Nat Methods 12, 931–934 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cer RZ et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 41, D94–D100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Derbinski J et al. Promiscuous gene expression patterns in single medullary thymic epithelial cells argue for a stochastic mechanism. Proc Natl. Acad Sci U S. A 105, 657–662 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peterson P, Org T, & Rebane A Transcriptional regulation by AIRE: molecular mechanisms of central tolerance. Nat. Rev. Immunol 8, 948–957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gardner JM et al. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 321, 843–847 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Huang S et al. A novel multi-alignment pipeline for high-throughput sequencing data. Database. (Oxford) 2014, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van der Veeken J et al. The transcription factor Foxp3 shapes regulatory T cell identity by tuning the activity of trans-acting intermediaries. Immunity. 53, 971–984 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de Santiago I et al. BaalChIP: Bayesian analysis of allele-specific transcription factor binding in cancer genomes. Genome Biol. 18, 39 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weirauch MT et al. Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Grant CE, Bailey TL, & Noble WS FIMO: scanning for occurrences of a given motif. Bioinformatics. 27, 1017–1018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bailey TL STREME: accurate and versatile sequence motif discovery. Bioinformatics. 37, 2834–2840 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bray NL, Pimentel H, Melsted P, & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 71.Love MI, Huber W, & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim D et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Satija R et al. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 33, 495–502 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Corces MR et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoshida H et al. The cis-Regulatory Atlas of the Mouse Immune System. Cell 176, 897–912 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. journal 10.14806/ej.17.1.200, (2015). [DOI] [Google Scholar]
- 78.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qunhua L, James BB, Haiyan H, & Peter JB Measuring reproducibility of high-throughput experiments. The Annals of Applied Statistics 5, 1752–1779 (2011). [Google Scholar]
- 81.Quinlan AR & Hall IM BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramirez F et al. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shen L, Shao N, Liu X, & Nestler E ngs.plot: Quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC. Genomics 15, 284 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gothe HJ et al. Spatial Chromosome Folding and Active Transcription Drive DNA Fragility and Formation of Oncogenic MLL Translocations. Mol. Cell 75, 267–283 (2019). [DOI] [PubMed] [Google Scholar]
- 86.Yan WX et al. BLISS is a versatile and quantitative method for genome-wide profiling of DNA double-strand breaks. Nat. Commun 8, 15058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Smith T, Heger A, & Sudbery I UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heinz S et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Neph S et al. BEDOPS: high-performance genomic feature operations. Bioinformatics. 28, 1919–1920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Möller A et al. Monoclonal antibodies recognize different parts of Z-DNA. J Biol Chem. 257, 12081–12085 (1982). [PubMed] [Google Scholar]
- 91.Schmidl C, Rendeiro AF, Sheffield NC, & Bock C ChIPmentation: fast, robust, low-input ChIP-seq for histones and transcription factors. Nat. Methods 12, 963–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Buenrostro JD et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bansal K et al. Aire regulates chromatin looping by evicting CTCF from domain boundaries and favoring accumulation of cohesin on superenhancers. Proc Natl Acad Sci U S A 118, e2110991118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the sequencing data reported in this article were deposited as a SuperSeries at Gene Expression Omnibus (GEO) under accession GSE224557 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE224557). Specifically, the ATAC-seq, BLISS, ChIP-seq, ChIPmentation, CUT&Tag, bulk RNA-seq and scRNA-seq data are under accessions GSE224551, GSE224552, GSE224553, GSE224554, GSE224555, GSE224556 and GSE253215, respectively. Public datasets used in this article are: ATAC-seq for WT and Aire-KO mTECs from GSE92594; RNA Pol II, Aire and IgG ChIP-seq for WT mTECs from GSE92597; MED1 and IgG ChIP-seq for WT mTECs from GSE180937; ATAC-seq for mTECs from Brg1-WT and Brg1-cKO mice from GSE102526.