

Abstract

To study the relationship between polymorphism and catalytic activities of lanthanide coordination polymers in the cycloaddition reactions of CO2 with epoxides, the monoclinic and triclinic polymorphs of [LnIII(NH3–Glu)(ox)]·2H2O, where LnIII = LaIII (I), PrIII (II), NdIII (III), SmIII(IV), EuIII (V), GdIII (VI), TbIII (VII), and DyIII (VIII), NH3–Glu– = NH3+ containing glutamate, and ox2– = oxalate, were synthesized and characterized. Factors determining polymorphic preference, the discrepancy between the two polymorphic framework structures, potential acidic and basic sites, thermal and chemical stabilities, active surface areas, void volumes, CO2 sorption/desorption isotherms, and temperature-programmed desorption of NH3 and CO2 are comparatively presented. Based on the cycloaddition of CO2 with epichlorohydrin in the presence of tetrabutylammonium bromide under solvent-free conditions and ambient pressure, catalytic activities of the two polymorphs were evaluated, and the relationship between polymorphism and catalytic performances has been established. Better performances of the monoclinic catalysts have been revealed and rationalized. In addition, the scope of monosubstituted epoxides was experimented and the outstanding performance of the monoclinic catalyst in the cycloaddition reaction of CO2 with allyl glycidyl ether under ambient pressure has been disclosed.
Short abstract
Monoclinic and triclinic polymorphs of lanthanide–glutamate–oxalate coordination polymers were synthesized and characterized. Single-crystal structures were included. Factors determining polymorphic preference are discussed. Based on the CO2 cycloaddition reactions with a scope of monosubstituted epoxides, a relationship between polymorphism and catalytic performance has been established.
1. Introduction
To mitigate the rising concentration of atmospheric CO2 and its adverse effects, CO2 capture and utilization has emerged as one of the most sustainable outlooks.1 The use of CO2 as a C1 feedstock to produce commodity chemicals is particularly appealing, as they comply with the 100% atom economy.2,3 Among various possibilities, the production of cyclic carbonates from cycloaddition reactions of CO2 with epoxides has been extensively explored. Because CO2 is thermodynamically stable and kinetically inert, majority of the research work is on the development of high-performance catalysts for use at or near ambient temperature and pressure.4−7 The recently emerging trend is the research on bifunctional catalysts.8 In terms of practicality, thermal and chemical stability as well as an ability to be recycled are also demanded.9−12
Coordination polymers (CPs), which are also known as metal–organic frameworks (MOFs), have been well acknowledged for their catalytic potential.13−15 Through appropriate choices of inorganic and organic building units, their structures and properties can be tailor-made at the molecular level. CPs of the transition metals and lanthanides (LnIII-CPs) have been extensively explored, the latter of which tend to provide better performance at ambient pressure.16−19 This is partially due to the hard acidic nature of the vacant coordination sites of LnIII, which can be generated simply through thermal treatment or the so-called activation step.20 At slightly elevated temperatures, the small solvent ligands can be removed to provide the vacant coordination sites. These sites can interact with the epoxide O atom to promote the subsequent nucleophilic attack and ring opening.21 As the nucleophile is essential, the presence of nucleophile sources, e.g., tetrabutylammonium bromide (TBABr), tetrapropylammonium bromide (TPABr), and tetramethylammonium bromide (TMABr), in the catalytic reaction is unavoidable.19 Thus far, reports on the use of LnIII-CPs without prior activation are noticeably limited and the reported performances are moderate (Table S1).22−26 Ligand substitution is reportedly responsible for the exemption of the prior activation.22−26 It was only recently that the noteworthy performance manifested through the turnover number (TON) and turnover frequency (TOF) of 7142 and 1786 h–1, respectively, could be achieved from [EuIII2(Habtc)2(DMSO)4]·DMSO·H2O (H4abtc = 3,3′,5,5′-azobenzenetetracarboxylic acid, DMSO = dimethyl sulfoxide).18 Regarding the organic building units, they also play important role in promoting the catalytic efficiency of CPs. With respect to LnIII-CPs, the majority of the organic building units are polycarboxylates, e.g., 2-sulfoterephthalic acid, 2-aminoterephthalic acid, and 3,3′,5,5′-azobenzenetetracarboxylic acid.8,27,28 The introduction of acidic and basic sites, e.g., –SO3H, –NH2, –N=N–, and –COOH,8,18 can be achieved through both appropriate choice of molecular backbones and postmodification.
The catalytic performance of CPs depends not only on the choices of structural building units but also the arrangement of these units in the framework structures.19,29−31 Contrary to the transition metal-based CPs, the arrangement of these units in LnIII-CPs can vary significantly depending on subtle changes in the synthesis conditions.19,29−31 Polymorphism or framework isomerism, which is the ability of a compound to display different framework structures,32 is therefore frequently observed in LnIII-CPs, although their polymorphic phase transition is often undetected.32−35 Examples of polymorphism in LnIII-CPs include the 2D and 3D polymorphs of [Ln(Tz*)3] (Ln = Gd–Lu and Tz*– = 1,2,3-triazolate),36 the low- and high-temperature polymorphs of [Eu(BDC)(NO3)(DMF)2]n (BDC2– = benzenedicarboxylate, DMF = dimethylformamide),32 and the α-, β-, and γ-phases of [Ln2(hfipbb)3] (Ln = Sc, Y, La–Yb and hfipbb2– = hexafluoroisopropylidenebisbenzoate).35 In view of crystal engineering, polymorphism allows the investigation of the structure–property relationship, which is crucial to the rational design of crystal structures. Literature reporting the relationship between polymorphism and catalytic activities is nonetheless rare. One of the few examples is the different catalytic activities of two polymorphic hcp-UiO-66 and fcc-UiO-66.37 The better activity of hcp-UiO-66 than fcc-UiO-66 in alcoholysis of styrene oxide was reported to originate from a dissimilar framework assembly. The other example is the higher electrocatalytic activities of the two-dimensional Co-MOF (CTGU-5) than its polymorphic three-dimensional CTGU-6.38 The difference was rationalized by the more open framework structure of CTGU-5 and the presence of coordinating water molecules. Noticeably, there is no previous report on the relationship between polymorphism and catalytic activities of LnIII-CPs, particularly in cycloaddition reactions of CO2.
Herein, the monoclinic (m) and triclinic (t) polymorphs of [LnIII(NH3–Glu)(ox)]·2H2O (LnIII = LaIII (I), PrIII (II), NdIII (III), SmIII(IV), EuIII (V), GdIII (VI), TbIII (VII), and DyIII (VIII), NH3–Glu– = NH3+ containing glutamate, ox2– = oxalate) were synthesized and characterized. Glutamate was chosen due to the presence of the –NH2 group, which should be beneficial to the catalysis, whereas oxalate, which is a small dicarboxylate with a tendency to adopt a chelating mode of coordination, was selected to promote framework stability. Their single-crystal structures were elucidated from which the distinct structural differences have been revealed. Thermal and chemical robustness, Brunauer–Emmett–Teller (BET) active surface areas, framework void volumes, CO2 sorption/desorption isotherms, and temperature-programmed desorption of NH3 and CO2 were studied. Their catalytic activities in the cycloaddition reactions of CO2 and a scope of monosubstituted epoxides under solvent-free conditions and atmospheric CO2 pressure were then explored. Founded on experimental evidence, the relationship between polymorphism and catalytic performance has been established. In addition, a substantial catalytic performance in the reaction with allyl glycidyl ether (AGE) was demonstrated.
2. Experimental Section
Details of materials, characterization methods, experimental setting, and single-crystal structure determination39−44 are provided as the Supporting Information. Crystallographic data and refinement details are summarized in Table S2.
2.1. Synthesis of I–VIII
The corresponding LnIII(NO3)3·6H2O (0.100 mmol) was dissolved in 3.00 mL of deionized water to give solution A. Separately, monosodium glutamate (NaHGlu, 0.100 mmol, 16.9 mg) and di-ammonium oxalate [(NH4)2ox, 0.100 mmol, 14.2 mg] were dissolved in 7.00 mL of deionized water to give solution B. Solutions A and B were then mixed in a 10 mL glass vial, closed carefully, and kept in a hot-air oven at a specified temperature and time as summarized in Table 1.
Table 1. Summary of Synthesis Conditions and Corresponding Products with Their Percentage Yields (% Yield) and CHN Analysis Results.
| synthesis
conditions |
elemental analysis |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 60 °C, 4 days |
80 °C, 4 days |
80 °C, 1 days |
C (% wt) |
H (% wt) |
N (% wt) |
|||||||
| LnIII source (weight used in synthesis) | product | % yield | product | % yield | product | % yield | Calcda | Expb | Calcda | Expb | Calcda | Expb |
| LaIII(NO3)3·6H2O (43.3 mg) | It | 76 | LaIII-oxc | LaIII-oxc | 20.6 | 20.7 | 2.70 | 2.90 | 3.43 | 3.43 | ||
| PrIII(NO3)3·6H2O (43.5 mg) | IIt | 79 | IIt | 77 | IIt | 77 | 20.5 | 20.8 | 2.68 | 2.86 | 3.41 | 3.46 |
| NdIII(NO3)3·6H2O (43.8 mg) | IIIt + IIIm | IIIt | 76 | IIIt | 76 | 20.3 | 20.8 | 2.66 | 2.87 | 3.39 | 3.45 | |
| SmIII(NO3)3·6H2O (44.4 mg) | IVm | 78 | IVt | 75 | IVt | 78 | 20.0 | 21.1 | 2.62 | 3.21 | 3.34 | 3.77 |
| EuIII(NO3)3·6H2O (44.6 mg) | Vm | 76 | Vt + EuIII-oxc | Vt | 78 | 19.9 | 19.8 | 2.61 | 3.11 | 3.32 | 3.37 | |
| GdIII(NO3)3·6H2O (45.1 mg) | VIm | 78 | GdIII-oxc | VIt | 76 | 19.7 | 19.7 | 2.56 | 3.06 | 3.28 | 3.52 | |
| TbIII(NO3)3·6H2O (45.3 mg) | VIIm | 79 | TbIII-oxc | VIIt | 77 | 19.6 | 19.4 | 2.57 | 2.94 | 3.27 | 3.44 | |
| DyIII(NO3)3·6H2O (45.7 mg) | VIIIm | 79 | DyIII-oxc | VIIIt + DyIII-oxc | 19.4 | 17.9 | 2.55 | 2.63 | 3.24 | 3.07 | ||
Two crystallizing water molecules per formula, i.e., [LnIII(NH3–Glu)(ox)]·2H2O, which is the most consistent with the CHN analysis results, were assumed in the calculations.
The experiments were carried out using pure samples.
Based on PXRD experiments, they were isostructure to [NdIII2(ox)3(H2O)2]·H2O.45
2.2. Catalytic Experiments
The crystals were ground manually using a mortar and pestle and used as is without particle size control. Typically, the tested catalyst was weighed into a 10 mL two-neck round-bottom flask into which epichlorohydrin (ECH, 20 mmol) and TBABr (0.25 mmol) were subsequently added. The flask was then sealed and equipped with a CO2 balloon. The catalytic reaction was carried out under atmospheric CO2 pressure and solvent-free conditions at 80 °C for 3.5–4 h with continuous stirring and regular filling of CO2. To stop the reaction, the flask was cooled down to room temperature using an ice bath, and the remaining CO2 was slowly released. Progress of the reactions was followed by using 1H NMR spectroscopy. The triplicate results were produced for every experiment unless specified otherwise. Yields of the reactions were determined using mesitylene as an internal standard. In addition to ECH, 1,2-epoxyoctane (1,2-EO), glycidol (Gly), butyl glycidyl ether (BGE), benzyl glycidyl ether (BnGE), 1,2-epoxy-3-phenoxypropane (EPP), styrene oxide (SO), and AGE were examined.
Recyclability and robustness of the catalyst were additionally studied using Vm and ECH as the modeled catalyst and epoxide. To regenerate the catalyst, it was recovered from the reaction by filtration followed by washing with CHCl3 several times, drying in air, and regrinding. Several batches were carried out for each cycle, particularly the earlier ones to ensure that the recovered catalysts from each cycle were sufficient for further experiments until the 10th cycle. Stability of the samples was assured using powder X-ray diffraction (PXRD) experiments.
3. Results and Discussion
3.1. Synthesis and Single-Crystal Structures of I–VIII
According to the PXRD experiments (Figure 1) and single-crystal X-ray diffraction data (Table S2), the triclinic (t) and monoclinic (m) polymorphs of I–VIII could be synthesized as pure phases, although their purity depended on the synthesis temperature and time. At 60 °C and 4 days, the monoclinic phases were mostly yielded except for LaIII (I), PrIII (II), and NdIII (III), which provided the triclinic phrases. Increasing the temperature from 60 to 80 °C at the same synthesis time of 4 days resulted in the formation of the triclinic phases for almost all LnIII except II (PrIII), III (NdIII), and IV (SmIII), which provided the oxalate frameworks isostructural to [NdIII2(ox)3(H2O)2]·H2O.45 Shortening of the synthesis time from 4 days to 1 day at 80 °C led to the formation of the triclinic phases for also almost every LnIII except LaII (I). According to these results, the monoclinic frameworks can be considered as the low-temperature phases while the triclinic and the oxalate frameworks are the high-temperature counterparts. Notably, the crystals of monoclinic samples were substantially larger than those of the triclinic samples (Figure S1). In addition, the synergy between the synthesis temperature and time with lanthanide contraction can be assumed. While the light LnIII, i.e., LaIII (I), PrIII (II), and NdIII (III), preferred the triclinic structures, the heavier LnIII, i.e., SmIII(IV), EuIII (V), GdIII (VI), TbIII (VII), and DyIII (VIII), showed a tendency to provide both the low- and high-temperature phases.
Figure 1.

PXRD patterns of the as-synthesized I–VIII yielded from the reactions conducted at (a) 60 °C for 4 days, (b) 80 °C for 4 days, and (c) 80 °C for 1 day, compared with the simulated patterns of It, Vm, and [NdIII2(ox)3(H2O)2]·H2O45
3.1.1. Description of Triclinic Structures
The single-crystal structure is described based only on the data of It because the crystals of IIt–VIIt were inappropriate for full data collection. Their structures were alternatively concluded from a preliminary check of the unit cell parameters and PXRD experiments.
The asymmetric unit of It comprises one crystallographically unique LaIII, two halves of independent ox2–, and a molecule of glutamate with its amine group protonated (NH3–Glu–) (Figure 2). The protonation of the –NH2 group has been expected because of the acidic synthesis condition. The presence of the Bronsted –NH3 group should be beneficial to the catalysis. The N atom of NH3–Glu– is disordered over two crystallographic sites with approximately equal site occupancy. The carbon skeleton of NH3–Glu– is arranged with approximately identical ∠C1–C2–C3–C4 (168°) and ∠C2–C3–C4–C5 (169°) torsion angles and the C1···C5 distance of ca. 4.9 Å (Figure S2). Viewed along the molecular skeleton, the two terminal carboxyl groups almost coincide with each other, reflected through the small ∠O1–C1–C5–O4 (9.0°) and ∠O2–C1–C5–O3 (4.8°) torsion angles. All atoms of ox2– are notably aligned in the molecular plane.
Figure 2.

Views of (a) an extended asymmetric unit, the (b) local coordination environment of the edge-shared TPRS-{LaIIIO8}2 dimer and coordination modes of the organic linkers, the (c) three-dimensional framework of It, and the (d) corresponding net topology (hydrogen atoms are omitted for clarity. Symmetry operations: (i) −x, 1 – y, 1 – z; (ii) −x, −y, 1 – z; (iii) −x, 1 – y, −z; (iv) −1 + x, y, z).
The LaIII is 9-fold coordinated to two ox2– and four NH3–Glu–. The O atoms define a distorted tricapped trigonal prismatic coordination geometry and therefore the TPRS-{LaIIIO9} coordination entity (Figure 2). The two ox2– are positioned next to each other, making a dihedral angle of ca. 81°. Through the –O– and –OCO– bridges of four NH3–Glu–, every two TPRS-{LaIIIO9} coordination entities are fastened into an edge-shared TPRS-{LaIIIO8}2 dimer. Each of the dimeric units is further linked to the other six equivalents via ox2– (μ2:η1η1η1η1) and NH3–Glu– (μ4:η2η1η1η1). The linkage results in a 3D framework of [LnIII(NH3–Glu)(ox)]. As anticipated, ox– stabilizes the framework by using its commonly adopted chelating coordination mode. The framework contains ca. 17% void calculated using PLATON.46 This corresponds to the 1D channel with an opening of 2.6 × 3.9 Å2. Within the channel, two crystallizing water molecules (O1w and O2w) are located. These water molecules show site disordering attributable to their involvement in the hydrogen bonding interactions with –NH3+ of NH3–Glu– and the carboxyl O atoms of both NH3–Glu– and ox2– (Table S3).
3.1.2. Description of Monoclinic Structures
The single-crystal data of IVm–VIIIm were elucidated (Table S2), revealing their isostructures and similar asymmetric structures to It. Their asymmetric structures comprise one unique LnIII, two halves of independent ox2–, and a molecule of NH3–Glu– (Figure 3). The structural building units are also identical to those of It, which are the edge-shared TPRS-{LnIIIO8}2 dimer and the organic linkers of the same coordination modes. In addition, the derived 3D frameworks are closely similar to It from a quick glance.
Figure 3.

Views of an (a) extended asymmetric unit, the (b) local coordination environment of the edge-shared TPRS-{EuIIIO8}2 dimer and coordination modes of the organic linkers, (c) three-dimensional framework of Vm, and the (d) corresponding net topology (hydrogen atoms are omitted for clarity). Symmetry operations: (i) 1 – x, 2 – y, 1 – z; (ii) 1 – x, y, 0.5 – z; (iii) 1.5 – x, 1.5 – y, 1 – z; (iv) x, 1 – y, −0.5 + z; (v) 1.5 – x, 0.5 + y, 1.5 – z).
Discrepancies between the two polymorphic framework structures are, however, distinct and principally originate from the organic linkers. First, the terminal carboxyl groups of NH3–Glu– in Vm (as a representative of IVm–VIIIm) do not coincide with each other (Figure S2). The ∠O1–C1–C5–O4 (39.4°) and ∠O2–C1–C5–O3 (31.7°) torsion angles are large. This is contrary to It case although the arrangement of their molecular skeleton is similar, i.e., ∠C1–C2–C3–C4 = 180°, ∠C2–C3–C4–C5 = 176°, and the C1···C5 distance = ca. 5.0 Å in Vm. Second, the carboxyl O6 atom of one ox2– is not coplanar with the other atoms but slightly projecting out of the molecular plane (Figure S2). Third, the dihedral angle between two ox2– of Vm is ca. 75° (Figure 3a), which is slightly smaller than that of It (Figure 2a). Fourth and probably the most important is the spatial arrangement of four NH3–Glu– linkers about the TPRS-{LnIIIO8}2 dimer. Each dimer of It is linked to six equivalents (Figure 2b) whereas that of Vm (Figure 3b) is connected to eight. This difference is reflected explicitly in the simplified framework topologies. Using the TPRS-{LnIIIO8}2 dimer as a node, the framework structure of It can be simplified to a 6-connected pcu or sqc1 net with a 412.63 point symbol (Figure 2d) while that of Vm to an 8-connected sqc117 net with a 36.412.58.62 point symbol (Figure 3d). It is therefore conclusive that the structural discrepancy between the two polymorphic frameworks is genuine and driven by dissimilarity in the spatial conformation of the organic linkers and their linking patterns.
Calculated using PLATON,46 the framework void of Vm is ca. 19%. The 1D channel with an aperture of 4.2 × 5.9 Å2 is accountable for this void. Noticeably, these values are slightly larger than those of It. The crystallizing water molecules residing inside the channel are extremely disordered and could not be precisely located from the single-crystal data. This is also contrary to the It case. The treatments of the single-crystal data using SQUEEZE routine47 were necessary, resulting in the estimation of 2.2 to 6.6 water molecules per formula (Table S2). These numbers were, however, larger than the numbers derived from the elemental analyses (ca. 2 water molecules). The thermogravimetric/differential thermal analysis (TG/DTA) experiments of It and Vm (as the representatives of the two polymorphs) were therefore conducted (Figures S3 and S4) from which the assumption of 2 water molecules per formula was concluded. Identical chemical formulas of both the triclinic and monoclinic structures, i.e., [LnIII(NH3–Glu)(ox)]·2H2O, were accordingly decided. The Fourier transform infrared (FT-IR) spectra displayed the characteristic vibrational features of the expected functional groups (Figure S5 and Table S4).
3.2. Thermal Robustness of It and Vm
Based on the TG/DTA experiments, similar weight losses were observed for both It and Vm, which were used as the representatives of the two polymorphs (Figures S3, S4). The patterns showed two major weight losses alike. The first weight loss occurred over the 150–220 °C range, signifying the removal of the crystallizing water molecules, followed by the second substantial loss at ca. 350 °C. Disregarding similarity in the weight loss patterns, the PXRD experiments suggested otherwise (Figure S6). The framework structure of It remained intact even after heating at 350 °C whereas that of Vm started to transform to other phases after heating at 220 °C. Better thermal robustness of the triclinic framework, whose structural building units are organized more orderly, can therefore be assumed. At 80 °C, which would be the temperature used in the catalysis experiments, the stability of the two polymorphs was nonetheless certain because the framework structure of Vm remained stable up to 150 °C.
3.3. Chemical Robustness of Vt and Vm
Since chemical robustness is an important property of catalysts, the stabilities of Vt (EuIII) and Vm (EuIII), which would be employed in the catalysis experiments to avoid any influence from different LnIII, in water and common organic solvents were studied. The experimented organic solvents were toluene, tetrahydrofuran (THF), hexane, methyl alcohol (MeOH), ethyl alcohol (EtOH), ethyl acetate (EtOAc), acetonitrile, chloroform (CHCl3), dichloromethane (CH2Cl2), benzene, and acetone. After soaking the ground crystals of Vt (EuIII) and Vm (EuIII) in the solvents for 1 day at room temperature, their PXRD patterns were collected and showed the retaining of all the diffraction peaks (Figure S7). The robustness of both Vt and Vm toward these common solvents at room temperature is therefore conclusive.
3.4. Sorption/Desorption Isotherms of N2 and CO2 of Vt and Vm
The N2 sorption/desorption isotherms of Vt and Vm are similar (Figure S8), exhibiting the characteristic profiles of microporous frameworks. However, the BET surface area and void volume of Vm (5.037 m2·g–1 and 1.157 cm3·g–1) were approximately 3 times larger than those of Vt (1.566 m2·g–1 and 0.3599 cm3·g–1) based on the measurement at 77 K. These results are seemingly contradicting with the slight difference in the calculated voids of the two polymorphs. This contradiction, on the other hand, implies the better access of N2 into the framework of Vm than Vt. This implication is in good accordance with the larger channel aperture of Vm (4.2 × 5.9 Å2) relative to that of It (2.6 × 3.9 Å2). With respect to the kinetic diameter of N2 (3.64 Å),48 N2 should not be able to enter the It framework.
Intriguingly, the CO2 uptake capacities of Vm (1.5163 mL·g–1 at 195 K and 1.4419 mL·g–1 at 298 K) were also approximately 3 times larger than those of Vt (0.39601 mL·g–1 at 195 K and 0.33431 mL·g–1 at 298 K) (Figure S9). Considering the kinetic diameter of CO2 (3.30 Å),48 these substantial differences clearly demonstrate the molecular sieve property of the frameworks. Furthermore, both polymorphs showed the retention of CO2 upon the desorption (Figure S9), suggesting the strong interaction between CO2 and the frameworks. The interaction may occur at both the acidic –NH3+ group and the lone-pair electron-containing O atoms of the carboxyl groups. If the desorption isotherms at 195 and 298 K are compared, then the dissimilarity of the two polymorphs is magnified. While the retention of CO2 at 298 K of Vt was significantly less than that of 195 K, the retention of CO2 of Vm at both temperatures was approximately equal. This suggests a stronger interaction of CO2 with Vm than Vt. In line with these results, a better catalytic performance should be expected from Vm.
3.5. Framework Acidity and Basicity
The acidity and basicity of Vt and Vm were additionally evaluated using the temperature-programmed desorption of NH3 (NH3-TPD) and CO2 (CO2-TPD) (Figure S10). The NH3-TPD profile of Vt showed a vivid desorption peak at a lower temperature (ca. 147 °C) than that of Vm, which showed only a small shoulder at ca. 194 °C. A tendency to desorb at temperatures higher than 200 °C can however be assumed. Complying with these results, stronger interaction with NH3 and therefore stronger acidity should be expected from Vm. This is considered to be beneficial to its catalytic function.
The CO2-TPD experiments did not provide any significant information as the desorption of CO2 commenced at ca. 140 °C, which is close to the limitation of the employed instrument (200 °C). The desorption of CO2 from Vt was nonetheless more significant than Vm up to 200 °C. Projecting to temperatures higher than 200 °C, the release of CO2 from both Vt and Vm can be hypothesized. This implies strong interaction between CO2 and the frameworks especially in the Vm case, which are in good accordance with the CO2 sorption/desorption isotherms.
3.6. Catalytic Activities of Vt and Vm
Catalytic activities of Vt and Vm were evaluated based on the CO2 cycloaddition with ECH in the presence of TBABr (Table 2). The catalysts were used as powders and without prior activation unless stated otherwise. The amount used in each batch of the experiments was notably diminutive, i.e., 0.025 mol % (ca. 2.1 mg) with respect to the ECH substrate, and 1/50 of TBABr (1.25 mol % or 80.5 mg). As references, the catalytic activities of TBABr (without the catalysts, entry 1), the catalysts (without TBABr, entries 2 and 3) and Eu(NO3)·6H2O (entry 4) were also determined. Based on the integrated areas of 1H NMR spectra, the performance of the catalysts was evaluated via the percentage conversion of ECH to chloromethyl ethylene carbonate (%conversion) and selectivity (%selectivity). The byproduct from the reactions was 1-bromo-3-chloro-2-propanol. Exemplified 1H NMR spectra are provided as the Supporting Information (Figures S11–S27). Percentage yields of the products (%yield) were also determined by using mesitylene as an internal standard. The TON and TOF values were then calculated based on either % conversion and % selectivity or % yield.
| 1 |
| 2 |
| 3 |
Table 2. Catalytic Performance of Non-activated Catalysts (unless Specified Otherwise) in Catalyzing CO2 Cycloaddition with ECH at 80 °C for 3.5 h under Ambient CO2 Pressure and Solvent-less Conditions.

| entry | catalyst | % conversiona | % yield | % selectivitya | TONb | TOF (h–1)b |
|---|---|---|---|---|---|---|
| 1 | None (use only TBABr) | 53(±1) | 96 | |||
| 2 | Vm (without TBABr) | trace | ||||
| 3 | Vt (without TBABr) | trace | ||||
| 4 | Eu(NO3)3·6H2O | 79 | 74 | 96 | 2960 | 846 |
| 5 | IVm | 80 | 77 | 97 | 3080 | 880 |
| 6 | Vm | 84 | 84 | 95 | 3360 | 960 |
| 7 | Vmc | 81 | 81 | 95 | 3240 | 926 |
| 8 | Vm crystal | 79 | 77 | 95 | 3080 | 880 |
| 9 | VIm | 81 | 79 | 97 | 3160 | 903 |
| 10 | VIIm | 81 | 81 | 97 | 3240 | 926 |
| 11 | VIIIm | 82 | 82 | 94 | 3280 | 937 |
| 12 | It | 76 | 74 | 95 | 2960 | 846 |
| 13 | IIt | 76 | 74 | 95 | 2960 | 846 |
| 14 | IIIt | 75 | 73 | 96 | 2920 | 834 |
| 15 | IVt | 77 | 74 | 95 | 2960 | 846 |
| 16 | Vt | 77 | 74 | 94 | 2960 | 846 |
| 17 | Vtc | 77 | 73 | 95 | 2920 | 834 |
| 18 | VIt | 79 | 77 | 95 | 3080 | 880 |
| 19 | VIIt | 78 | 77 | 95 | 3080 | 880 |
Averaged from triplicate results with ±1 to ±3 standard deviation (based on 1H NMR results).
Calculated
from % yield: 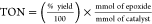
With prior activation (soaking in MeOH overnight and then heating at 80 °C for 30 min under N2).
At 80 °C and 3.5 h, IVm–VIIIm (entry 5, 6, 9–11) provided comparable activities, i.e., 80–84% conversion with 94–97% selectivity. The %yields of 77–84 were achieved, and the corresponding TON and TOF were 3080–3360 and 880–960 h–1, respectively. Similarly, It–VIIt (entry 12–16, 18, 19) showed indistinguishable results, i.e., 75–79% conversion, 94–96% selectivity, and 73–77% yields, which correspond to the TON and TOF of 2920–3080 and 834–880 h–1, respectively. The absence of lanthanide contraction can thus be presumed. Intriguingly, these values are in the same range as the values obtained from Eu(NO3)2·6H2O. The salt was, however, completely soluble under the experimented condition and therefore acted as a homogeneous catalyst. As the existing species of Eu(NO3)2·6H2O in an aqueous medium is [EuIII(H2O)n]3+, the catalysis may involve directly with EuIII through the substitution of the hydrating water molecules with the epoxide,18,22−26 which is contrary to the LnIII-CPs reported herein.
Since all of the catalysts were experimented with without prior activation and there is no small ligand coordinating with LnIII, the catalysis should proceed without any direct involvement with LnIII. This assumption is consistent with the fact that closely similar results were observed from the same polymorphic catalysts. Additionally, the attempt to activate the catalyst before use (entries 7 and 17) did not lead to any significant differentiation. Accordingly, the important functional groups in the catalysis should be the Bronsted acidic –NH3+ group of NH3–Glu– and the lone-pair electron-containing O atoms of the organic linkers (Figures 4 and S28). The epoxide O atom may interact with the Bronsted acidic –NH3+ group of NH3–Glu–, inducing the attack of the nucleophilic Br– provided from TBABr. The CO2 bearing both negative (on O atoms) and positive (on C atom) partial charges can interact with both the nearby carboxylate O atom and the Bronsted acidic –NH3+. The proximity of the activated intermediate and CO2 can assumingly promote the subsequent CO2 addition and ring closing.
Figure 4.

Locations of potential acidic (blue) and basic (red) sites in the frameworks of (a) Vt and (b) Vm and their neighboring environment.
Because the active surface area can be influential to catalytic activities, the catalytic activity of Vm crystals (entry 8) was assessed. The crystals of Vm were selected owing to their large sizes. Compared with the activity of the ground crystals (entry 6), it is apparent that the conversion yielded from the crystals was slightly inferior. The effect of surface area has therefore manifested. Contrarily, this effect was suggested to be insignificant from the small standard deviations of the averaged conversions and selectivity in the cases of the ground crystals although their sizes were not carefully controlled (Figure S1).
If the two polymorphs are compared (Figure 5), the monoclinic polymorphs evidently offer slightly better activities. The performance of Vm (entry 6, 84% yield) was, for instance, better than that of Vt (entry 16, 74% yield). This can be accounted for by the larger surface area and void volume as well as the stronger acidity and basicity of Vm. Since these differences are brought about through structural dissimilarity between the two polymorphs, the relationship between polymorphism and catalytic activity has been demonstrated.
Figure 5.

Comparison of (a) percentage yields and (b) TOF of two polymorphic catalysts based on the CO2 cycloaddition reactions with ECH at 80 °C for 3.5 h under ambient CO2 pressure.
3.7. Catalytic Performance of Vm
Because Vm showed the best performance in catalyzing the reaction with ECH, further experiments were performed using this catalyst. The prolongation of the reaction from 3.5 to 4 h at 80 °C (Table 3) led to the improved conversion and TON value although the TOF value (863 h–1) was slightly reduced as a penalty of the longer reaction time. Compared with the other LnIII-CP-based catalysts (Table S1), the performances of Vm were nonetheless remarkable.
Table 3. Catalytic Performance of Non-activated Vm in the CO2 Cycloaddition with Various Epoxides at 80 °C for 4 h under Ambient CO2 Pressure and Solvent-less Conditions.


Could not be calculated due to the unidentified byproduct.
Average values from triplicate results.
Calculated from the average % conversion
and % selectivity: 
Vm could also be easily recovered and reused. Over 10 successive cycles of catalysis and regeneration, the conversions and selectivity provided by Vm were approximately unaltered, varying between 83–85 and 94–96%, respectively (Figure S29a). Its robustness was demonstrated via the preservation of the framework structure and crystallinity according to the PXRD experiments (Figure S29b).
In addition to ECH, Vm was additionally attempted with the other monosubstituted epoxides (Table 3 and Figures S30–S36). While poor to moderate results were observed from the other epoxides, 67% conversion with an absolute selectivity could be yielded from AGE. The corresponding TON and TOF values of 2680 and 670 h–1, respectively, were inspiring. The superior catalytic efficiency in the reaction of AGE to those of SO, EPP, and BnGE may be attributed to steric hindrance imparted from the bulky substituted chains of SO, EPP, and BnGE. Compared with 1,2-EO and BGE possessing similar structures, the presence of electron-rich moieties on the substituted chain of AGE may be responsible for its superiority in a similar manner to the presence of halide in ECH.49,50
The attempt to elevate the reaction temperature from 80 to 90 °C led to a leap of the conversion to 84% with the improved TON and TOF values to 3360 and 840 h–1, respectively. With reference to mesitylene, an excellent yield of 83% was also affirmed. Compared with the other LnIII-CPs operated under similar conditions (Table S5), the performance of Vm was assuredly remarkable and superior to the other previously reported LnIII-CPs. It was literally comparable to those of the heterometallic 3d-4f CPs, e.g., [Dy2Zn4(μ3-OH)2L4(AcO)2(NO3)2(DMF)2]·2(CH3OH) (TOF = 783 h–1),51 Tb2Zn2(μ3-OH)2L4(NO3)4 (TOF = 700 h–1), and Tb2Zn4(μ3-OH)2L4(OAc)6(NO3)2 (TOF = 668 h–1).52 This is a rare example of the efficient catalysis of the CO2 cycloaddition with AGE, which does not involve the vacant coordination sites of the metal but the other acidic and basic sites on the organic linkers, i.e., the Bronsted–NH3+ group of NH3–Glu– and the basic O atoms of the carboxyl groups in this study.
4. Conclusions
The low-temperature monoclinic (m) and high-temperature triclinic (t) polymorphs of I–VIII were synthesized and characterized. The synergistic influences of the lanthanide contraction and synthesis parameters on the formation of these polymorphic frameworks were described. The thermodynamically stable oxalate frameworks were identified, in addition to the two polymorphs. Based on the single-crystal data, similarities and dissimilarities of the two polymorphic framework structures were presented. These dissimilarities were apparently manifested in diverse thermal stability but not chemical robustness. With a larger aperture into the framework void, the monoclinic polymorph (represented by Vm) allowed better accessibility to both N2 and CO2 and therefore larger BET surface area and effective void volume than the triclinic polymorph (represented by Vt). Stronger interactions with NH3 and CO2 implying stronger acidity and basicity could also be concluded for the monoclinic polymorph. The as-described structural features and properties of the monoclinic polymorphs evidently led to superiority in catalytic performance to the triclinic polymorphs. The Bronsted acidic–NH3+ and Lewis basic O atoms of the organic linkers are responsible for effective catalysis. This assumption is supported by the independence of the catalytic activities on the lanthanide contraction and the prior activation. Based on the experiments using Vm, the catalyst could be reused for at least 10 cycles without deterioration in its crystallinity and performance. In addition to ECH, Vm also exhibited inspiring catalytic activity in the reaction of AGE.
Acknowledgments
This research was supported by Chiang Mai University and the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (grant number B40G660031).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.4c00095.
Details on materials and methods, crystallographic data, and other results including additional crystallographic presentations, FT-IR spectra and band assignments, 1H NMR spectra, TD/DTA results, hydrogen bonding interactions, sorption/desorption isotherms of N2 and CO2, NH3-TPD, CO2-TPD, PXRD spectra, and additional catalysis data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Fu L.; Ren Z.; Si W.; Ma Q.; Huang W.; Liao K.; Huang Z.; Wang Y.; Li J.; Xu P. Research progress on CO2 capture and utilization technology. J. CO2 Util. 2022, 66, 102260. 10.1016/j.jcou.2022.102260. [DOI] [Google Scholar]
- Gulzar A.; Gulzar A.; Ansari M. B.; He F.; Gai S.; Yang P. Carbon dioxide utilization: A paradigm shift with CO2 economy. Chem. Eng. J. Adv. 2020, 3, 100013. 10.1016/j.ceja.2020.100013. [DOI] [Google Scholar]
- Rehman A.; Saleem F.; Javed F.; Ikhlaq A.; Ahmad S. W.; Harvey A. Recent advances in the synthesis of cyclic carbonates via CO2 cycloaddition to epoxides. J. Environ. Chem. Eng. 2021, 9, 105113. 10.1016/j.jece.2021.105113. [DOI] [Google Scholar]
- Pander M.; Janeta M.; Bury W. Quest for an efficient 2-in-1 MOF-based catalytic system for cycloaddition of CO2 to epoxides under mild conditions. ACS Appl. Mater. Interfaces 2021, 13 (7), 8344–8352. 10.1021/acsami.0c20437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L.; Chen X. L.; Liu L.; Cai M.; Yan R. K.; Cui H. L.; Yang H.; Wang J. J. Catalytic performance of MOFs containing trinuclear lanthanides clusters in the cycloaddition reaction of CO2 and epoxide. J. CO2 Util. 2022, 65, 102235. 10.1016/j.jcou.2022.102235. [DOI] [Google Scholar]
- Das R.; Manna S. S.; Pathak B.; Nagaraja C. M. Strategic design of Mg-centered porphyrin metal-organic framework for efficient visible light promoted fixation of CO2 under ambient conditions: combined experimental and theoretical investigation. ACS Appl. Mater. Interfaces 2022, 14, 33285–33296. 10.1021/acsami.2c07969. [DOI] [PubMed] [Google Scholar]
- Das R.; Ezhil T.; Palakkal A. S.; Muthukumar D.; Pillai R. S.; Nagaraja C. M. Efficient chemical fixation of CO2 from direct air under environment friendly co-catalyst and solvent-free ambient conditions. J. Mater. Chem. A 2021, 9, 23127–23139. 10.1039/D1TA05138E. [DOI] [Google Scholar]
- Thi Nguyen Q.; Na J.; Lee Y. R.; Baek K. Y. Boosting catalytic performance for CO2 cycloaddition under mild condition via amine grafting on MIL-101-SO3H catalyst. J. Environ. Chem. Eng. 2024, 12, 111852. 10.1016/j.jece.2023.111852. [DOI] [Google Scholar]
- Li C.; Lv H.; Yang K.; Zhang X. Robust fluorine-functionalized {Ln5}-organic frameworks for excellent catalytic performance on cycloaddition of CO2 with epoxides and Knoevenagel condensation. ACS Appl. Mater. Interfaces 2023, 15 (29), 35052–35061. 10.1021/acsami.3c06804. [DOI] [PubMed] [Google Scholar]
- He H.; Zhu Q. Q.; Zhao J.; Sun H.; Chen J.; Li C.; Du M. Rational construction of an exceptionally stable MOF catalyst with metal-adeninate vertices toward CO2 cycloaddition under mild and cocatalyst-free conditions. Chem.—Eur. J. 2019, 25, 11474–11480. 10.1002/chem.201901471. [DOI] [PubMed] [Google Scholar]
- Cokoja M.; Wilhelm M. E.; Anthofer M. H.; Herrmann W. A.; Kühn F. E. Synthesis of cyclic carbonates from epoxides and carbon dioxide by using organocatalysts. ChemSusChem 2015, 8, 2436–2454. 10.1002/cssc.201500161. [DOI] [PubMed] [Google Scholar]
- Sun J.; Cheng W.; Fan W.; Wang Y.; Meng Z.; Zhang S. Reusable and efficient polymer-supported task-specific ionic liquid catalyst for cycloaddition of epoxide with CO2. Catal. Today 2009, 148, 361–367. 10.1016/j.cattod.2009.07.070. [DOI] [Google Scholar]
- Pal T. K.; De D.; Bharadwaj P. K. Metal-organic frameworks for the chemical fixation of CO2 into cyclic carbonates. Coord. Chem. Rev. 2020, 408, 213173. 10.1016/j.ccr.2019.213173. [DOI] [Google Scholar]
- Bavykina A.; Kolobov N.; Khan I. S.; Bau J. A.; Ramirez A.; Gascon J. Metal-organic frameworks in heterogeneous catalysis: Recent progress, new trends, and future perspectives. Chem. Rev. 2020, 120, 8468–8535. 10.1021/acs.chemrev.9b00685. [DOI] [PubMed] [Google Scholar]
- Guillerm V.; Weseliński Ł. J.; Belmabkhout Y.; Cairns A. J.; D’Elia V.; Wojtas L.; Adil K.; Eddaoudi M. Discovery and introduction of a (3,18)-connected net as an ideal blueprint for the design of metal-organic frameworks. Nat. Chem. 2014, 6, 673–680. 10.1038/nchem.1982. [DOI] [PubMed] [Google Scholar]
- Ugale B.; Dhankhar S. S.; Nagaraja C. M. Exceptionally stable and 20-connected lanthanide metal-organic frameworks for selective CO2 capture and conversion at atmospheric pressure. Cryst. Growth Des. 2018, 18, 2432–2440. 10.1021/acs.cgd.8b00065. [DOI] [Google Scholar]
- Le D. H.; Loughan R. P.; Gładysiak A.; Rampal N.; Brooks I. A.; Park A. H. A.; Fairen-Jimenez D.; Stylianou K. C. Lanthanide metal-organic frameworks for the fixation of CO2 under aqueous-rich and mixed-gas conditions. J. Mater. Chem. A 2022, 10, 1442–1450. 10.1039/D1TA09463G. [DOI] [Google Scholar]
- Sinchow M.; Konno T.; Rujiwatra A. Reversible structural transformation and catalytic potential of lanthanide-azobenzenetetracarboxylates. Inorg. Chem. 2022, 61, 10383–10392. 10.1021/acs.inorgchem.2c00963. [DOI] [PubMed] [Google Scholar]
- Sinchow M.; Semakul N.; Konno T.; Rujiwatra A. Lanthanide coordination polymers through design for exceptional catalytic performances in CO2 cycloaddition reactions. ACS Sustainable Chem. Eng. 2021, 9, 8581–8591. 10.1021/acssuschemeng.1c01955. [DOI] [Google Scholar]
- Hu Y.; Abazari R.; Sanati S.; Nadafan M.; Carpenter-Warren C. L.; Slawin A. M. Z.; Zhou Y.; Kirillov A. M. A dual-purpose Ce(III)-organic framework with amine groups and open metal sites: Third-order nonlinear optical activity and catalytic CO2 fixation. ACS Appl. Mater. Interfaces 2023, 15, 37300–37311. 10.1021/acsami.3c04506. [DOI] [PubMed] [Google Scholar]
- Qiao N.; Xin X. Y.; Guan X. F.; Zhang C. X.; Wang W. M. Self-assembly bifunctional tetranuclear Ln2Ni2 clusters: Magnetic behaviors and highly efficient conversion of CO2 under mild conditions. Inorg. Chem. 2022, 61, 15098–15107. 10.1021/acs.inorgchem.2c02180. [DOI] [PubMed] [Google Scholar]
- Dong J.; Cui P.; Shi P. F.; Cheng P.; Zhao B. Ultrastrong alkali-resisting lanthanide-zeolites assembled by [Ln60] nanocages. J. Am. Chem. Soc. 2015, 137, 15988–15991. 10.1021/jacs.5b10000. [DOI] [PubMed] [Google Scholar]
- Dong J.; Xu H.; Hou S. L.; Wu Z. L.; Zhao B. Metal-organic frameworks with Tb4 clusters as nodes: luminescent detection of chromium (VI) and chemical fixation of CO2. Inorg. Chem. 2017, 56, 6244–6250. 10.1021/acs.inorgchem.7b00323. [DOI] [PubMed] [Google Scholar]
- Xu C.; Liu Y.; Wang L.; Ma J.; Yang L.; Pan F.; Kirillov A.; Liu W. New lanthanide(III) coordination polymers: Synthesis, structural features, and catalytic activity in CO2 fixation. Dalton Trans. 2017, 46, 16426–16431. 10.1039/C7DT03574H. [DOI] [PubMed] [Google Scholar]
- Song T. Q.; Dong J.; Yang A. F.; Che X. J.; Gao H. L.; Cui J. Z.; Zhao B. Wheel-like Ln18 cluster organic frameworks for magnetic refrigeration and conversion of CO2. Inorg. Chem. 2018, 57, 3144–3150. 10.1021/acs.inorgchem.7b03142. [DOI] [PubMed] [Google Scholar]
- Si X.; Pan X.; Xue J.; Yao Q.; Huang X.; Duan W.; Qiu Y.; Su J.; Cao M.; Li J. Robust acid-base Ln-MOFs: Searching for efficient catalysts in cycloaddition of CO2 with epoxides and cascade deacetalization-knoevenagel reactions. RSC Adv. 2022, 12, 33501–33509. 10.1039/D2RA06545B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janicki R.; Mondry A.; Starynowicz P. Carboxylates of rare earth elements. Coord. Chem. Rev. 2017, 340, 98–133. 10.1016/j.ccr.2016.12.001. [DOI] [Google Scholar]
- You L. X.; Ren B. Y.; He Y. K.; Wang S. J.; Sun Y. G.; Dragutan V.; Xiong G.; Ding F. Structural features of lanthanide coordination polymers with catalytic properties. J. Mol. Struct. 2024, 1304, 137687. 10.1016/j.molstruc.2024.137687. [DOI] [Google Scholar]
- Sinchow M.; Chuasaard T.; Yotnoi B.; Rujiwatra A. Diversity in framework architecture of lanthanide-2,5-pyridinedicarboxylate-sulfate coordination polymers. J. Solid State Chem. 2019, 278, 120902. 10.1016/j.jssc.2019.120902. [DOI] [Google Scholar]
- Fan W.; Cheng Y.; Feng M.; Liu P.; Wang L.; Liu Y.; Cao Q. E.; Zheng L. Y. Lanthanide metal-organic framework isomers with novel water-boosting lanthanide luminescence behaviors. ACS Appl. Mater. Interfaces 2023, 15, 41977–41991. 10.1021/acsami.3c10272. [DOI] [PubMed] [Google Scholar]
- Hanna S. L.; Debela T. T.; Mroz A. M.; Syed Z. H.; Kirlikovali K. O.; Hendon C. H.; Farha O. K. Identification of a metastable uranium metal-organic framework isomer through non-equilibrium synthesis. Chem. Sci. 2022, 13, 13032–13039. 10.1039/D2SC04783G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulakh D.; Varghese J. R.; Wriedt M. The importance of polymorphism in metal-organic framework studies. Inorg. Chem. 2015, 54 (17), 8679–8684. 10.1021/acs.inorgchem.5b01311. [DOI] [PubMed] [Google Scholar]
- Monni N.; Baldoví J. J.; García-López V.; Oggianu M.; Cadoni E.; Quochi F.; Clemente-León M.; Mercuri M. L.; Coronado E. Reversible tuning of luminescence and magnetism in a structurally flexible erbium-anilato MOF. Chem. Sci. 2022, 13, 7419–7428. 10.1039/D2SC00769J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z.; Zhao F.; Fan L.; Zhao W.; Chen B.; Chen X.; Zhou S. F.; Xiao J.; Zhan G. Improved hydrolytic robustness and catalytic performance of flexible lanthanide-based metal-organic frameworks: A matter of coordination environments. Mater. Des. 2020, 194, 108881. 10.1016/j.matdes.2020.108881. [DOI] [Google Scholar]
- Monge A.; Gándara F.; Gutiérrez-Puebla E.; Snejko N. Lanthanide, Y and Sc MOFs: Where amazing crystal structures meet outstanding material properties. CrystEngComm 2011, 13, 5031–5044. 10.1039/c0ce00891e. [DOI] [Google Scholar]
- Rybak J. C.; Tegel M.; Johrendt D.; Muller-Buschbaum K. Polymorphism of isotypic series of homoleptic 1,2,3-triazolate MOFs [Ln(Tz*)3] containing the heavy lanthanides Gd-Lu: From open to dense frameworks of varying dimensionality. Z. Kristallogr. 2010, 225, 187–194. 10.1524/zkri.2010.1242. [DOI] [Google Scholar]
- Guo J.; Xue X.; Li F.; Zhao M.; Xing Y.; Song Y.; Long C.; Zhao T.; Liu Y.; Tang Z. Modulation of the assembly fashion among metal-organic frameworks for enantioretentive epoxide activation. Nanoscale Horiz. 2024, 9, 118–122. 10.1039/D3NH00419H. [DOI] [PubMed] [Google Scholar]
- Wu Y. P.; Zhou W.; Zhao J.; Dong W. W.; Lan Y. Q.; Li D. S.; Sun C.; Bu X. Surfactant-assisted phase-selective synthesis of new cobalt MOFs and their efficient electrocatalytic hydrogen evolution reaction. Angew. Chem., Int. Ed. 2017, 56 (42), 13001–13005. 10.1002/anie.201707238. [DOI] [PubMed] [Google Scholar]
- Rikagu Oxford diffraction . CrysAlisPRO Software System (Version 1.171.39.46); Rigaku cooperation: Oxford, U.K., 2018, p 26. [Google Scholar]
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. J. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Spek A. L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/S0021889802022112. [DOI] [Google Scholar]
- Blatov V. A.; Shevchenko A. P.; Proserpio D. M. Applied topological analysis of crystal structures with the program package ToposPro. Cryst. Growth Des. 2014, 14, 3576–3586. 10.1021/cg500498k. [DOI] [Google Scholar]
- Huskić I.; Arhangelskis M.; Friščić T. Solvent-free ageing reactions of rare earth element oxides: From geomimetic synthesis of new metal-organic materials towards a simple, environmentally friendly separation of scandium. Green Chem. 2020, 22, 4364–4375. 10.1039/D0GC00454E. [DOI] [Google Scholar]
- Spek A. L. Structure validation in chemical crystallography. Acta Crystallogr. 2009, 65, 148–155. 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. 2015, 71, 9–18. 10.1107/S2053229614024929. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Xie Y.; Cui H.; Alothman Z. A.; Alduhaish O.; Lin R. B.; Chen B. An ultramicroporous metal-organic framework with dual functionalities for high sieving separation of CO2 from CH4 and N2. Chem. Eng. J. 2022, 446, 137101. 10.1016/j.cej.2022.137101. [DOI] [Google Scholar]
- Monteiro W. F.; Vieira M. O.; Laschuk E. F.; Livotto P. R.; Einloft S. M. O.; de Souza M. O.; Ligabue R. A. Experimental-theoretical study of the epoxide structures effect on the CO2 conversion to cyclic carbonates catalyzed by hybrid titanate nanostructures. J. CO2 Util. 2020, 37, 20–28. 10.1016/j.jcou.2019.11.024. [DOI] [Google Scholar]
- Chuasaard T.; Sinchow M.; Chiangraeng N.; Nimmanpipug P.; Rujiwatra A. Enhanced catalysis of CO2 cycloaddition at ambient pressure through rational design of interpenetrating ZnII/LnIII heterometallic coordination polymers. J. CO2 Util. 2024, 80, 102686. 10.1016/j.jcou.2024.102686. [DOI] [Google Scholar]
- Gao G.; Wang L.; Zhang R.; Xu C.; Yang H.; Liu W. Hexanuclear 3d-4f complexes as efficient catalysts for converting CO2 into cyclic carbonates. Dalton Trans. 2019, 48, 3941–3945. 10.1039/C8DT05048A. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Wang L.; Xu C.; Yang H.; Chen W.; Gao G.; Liu W. Anion-induced 3d-4f luminescent coordination clusters: Structural characteristics and chemical fixation of CO2 under mild conditions. Dalton Trans. 2018, 47, 7159–7165. 10.1039/C8DT01292J. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.