

Abstract
Rollout of meningococcal serogroup A conjugate vaccine in Africa started in 2010, aiming to eliminate meningitis outbreaks, in meningitis belt countries. Since then, studies have been conducted, primarily using isolates, to assess the vaccine impact on the distribution of meningococcal strains in the region. Here, we implemented an innovative, culture-free whole-genome sequencing approach on almost 400 clinical specimens collected between 2017 and 2019 from meningococcal meningitis cases in 6 African countries. About 50% of specimens provided high-quality whole-genome sequence data for comprehensive molecular profiling of the meningococcal pathogen. Three major clonal complexes were identified: CC11 associated with serogroup W, CC181 associated with serogroup X, and CC10217 associated with serogroup C, which continues to rise as a predominant clonal complex in the region. Genomic surveillance for meningococcal meningitis can be significantly improved using culture-free methods to increase data representativeness and monitor changes in epidemiological landscape, especially for countries with low culture rate.
Keywords: Neisseria meningitidis, selective whole-genome amplification, culture-free whole-genome sequencing, targeted sequencing, meningococcal meningitis surveillance
Neisseria meningitidis is one of the main pathogens responsible for bacterial meningitis worldwide. The largest recurring meningitis epidemics mainly affect a region in sub-Saharan Africa known as the meningitis belt, which comprises 26 countries, from Senegal in the west to Ethiopia in the east with a total population exceeding 400 million [1]. Factors predisposing the population in this region to bacterial meningitis are not well understood but may include a combination of semiarid ecoclimatic conditions [2] with increased sociocultural activities during the meningitis season [3]. Of the 6 disease-causing meningococcal serogroups identified (A, B, C, W, X, and Y), serogroup A (NmA) had accounted for approximately 90% of cases during epidemics in the African meningitis belt for the last 100 years [4].
The introduction of a meningococcal A conjugate vaccine in 2010 in countries of the meningitis belt drastically reduced NmA cases [5]. However, new epidemic-prone meningococcal strains of other serogroups, such as serogroup C meningococci (NmC), have emerged and have spread across countries within the meningitis belt, causing endemic and epidemic disease in the region [6]. Genomic surveillance, an important component of Defeat Meningitis By 2030 Roadmap [7], is critical to track the emergence and spread of new meningococcal strains and inform disease control interventions. A global meningitis genome partnership was established to improve genomic surveillance coordination and data sharing globally [8].
Historically, sequencing data have been used to trace meningococcal strains through multilocus sequencing typing (MLST) involving 7 housekeeping genes (abcZ, adk, aroE, fumC, gdh, pgm, and pghC) [9], core genome MLST, or single-nucleotide polymorphism (SNP)-based phylogenetic analysis [10, 11]. However, implementation of genomic surveillance is hampered in many countries due to the lack of sequencing capacity and available isolates [10, 12]. Countries that do not have sequencing capacity rely on global or regional reference centers for genomic surveillance. To further improve the representativeness of sequencing data when bacterial isolates are not available, development of culture-independent sequencing methods are needed.
Selective whole-genome amplification (SWGA) is a low-cost method that amplifies and enriches bacterial DNA directly from clinical specimens for successful culture-independent sequencing. SWGA has already been successfully used for the investigation of 2 N. meningitidis outbreaks in Burkina Faso and Togo [13]. Herein, we present an improved and upscaled SWGA method using a new enzyme to enhance genomic surveillance for meningitis in Africa.
METHODS
Specimen and Isolate Collection
A total of 419 cerebrospinal fluid (CSF) specimens from quantitative polymerase chain reaction (qPCR)-confirmed meningitis patients (sodC qPCR [14]) and 109 N. meningitidis isolates un-matched to clinical specimens were included in the study. All were collected through meningitis surveillance in Burkina Faso, Ghana, Chad, Nigeria, Niger, and Togo during 2017–2019 and stored at least 1 year at −80°C after arrival at Centers for Disease Control and Prevention (CDC) before testing. CSF specimens project determination (2017_DBD_Wang_411) states that personal identification information is not shared with CDC.
DNA Extraction From CSFs and Isolates
DNA was extracted from 200 μL of CSF specimens using high-throughput QIAmp 96 DNA Blood kit (Qiagen). For isolates, 10 μL of N. meningitidis stock were streaked on blood agar plates (BD) and grown overnight at 37°C. Bacterial lawn was collected and resuspended in 1 mL of 10 mM Tris buffer (pH 7.5). After centrifugation, the resultant pellet was processed for DNA extraction using the Gentra Puregene Yeast/Bact kit (Qiagen).
Enrichment of N. meningitidis DNA Using Improved Selective Whole-Genome Amplification
SWGA was modified to improve its efficiency, using EquiPhi29 DNA polymerase (ThermoFisher), which provides increased protein thermostability, reaction speed, and product yield over phi29 DNA polymerase used in the previous study [13]. The improved SWGA was performed on total DNA extracted from 399 of 419 specimens. For each specimen, both nondiluted and 10-fold diluted DNA were enriched by SWGA. Specifically, 2.5 μL of extracted DNA was mixed with 2.5 μL of 100 mM KOH solution, vortexed briefly, spun down, and incubated 5 minutes at room temperature. The mixture was neutralized with 5 μL of neutralization solution containing 50 mM of HCl and 100 mM of Tris buffer (pH 7.5). The resulting 10-μL solution was mixed with 4 μL of EquiPhi29 buffer (ThermoFisher), 4 μL of dNTPs mixture (10 mM of each type; Roche), 4 μL of N. meningitidis SWGA heptamers, 2 μL of EquiPhi29 (ThermoFisher), 2 μL of inorganic pyrophosphatase (ThermoFisher), 0.4 μL of Dithiothreitol 0.1 M, and 13.6 μL of double-distilled water. SWGA heptamers consists of the following 25 primers, with asterisks indicating 2 consecutive 3ʹ phosphodiester bonds replaced by phosphorothioate bonds necessary to prevent degradation by EquiPhi29: TTCGG*C*G, CGCCG*A*T, GTCGG*C*G, TCGGC*G*G, AACGG*C*G, CGACG*G*C, CGGCG*A*A, CGGCG*G*T, ACGGC*G*G, GACGG*C*G, CGGCG*C*G, CGTCG*C*C, GCGGC*G*A, CGTGC*C*G, GACGG*C*A, TGCGC*C*G, AACGC*C*G, TGCCG*C*G, TGCGC*G*G, ACGGC*A*T, GCGGC*A*A, GCCGC*C*G, TTGCC*G*A, TGCGG*C*G, TCCGC*C*G. Each primer was dissolved at 1 mM in water, and then all primers were mixed in equimolar proportions. The final reaction mixture was incubated at 46°C for 3 hours. Upon completion, DNA was purified from the reaction mixture using Agencourt AMPure XP beads (Beckman) according to the manufacturer’s instruction, eluted into 50 μL of 10 mM Tris buffer (pH 7.5), quantified by Qubit (Invitrogen), and stored for qPCR, library preparation, and sequencing. The final DNA concentration was in the range 300–1000 ng/ μL.
Enrichment of N. meningitidis DNA was assessed by sodC qPCR [14]. SWGA-enriched DNA samples with cycle threshold (Ct) value less than 16 were selected for whole-genome sequencing (WGS). This cutoff was operatively chosen to ensure the WGS data obtained from a specimen would generate high-quality genome assembly with at least 1400 core genome MLST loci [13] (Supplementary Figure 1). If both nondiluted and 10-fold diluted DNA sample passed this criterion, the DNA sample with the lower Ct was selected for sequencing.
DNA Library Preparation and Sequencing
A total of 214 enriched and 20 unenriched CSF DNA samples, as well as 109 isolate DNA samples were sequenced. One μg of enriched CSF DNA or isolate DNA and 1–100 ng of unenriched CSF DNA were used for library preparation. Each DNA sample was diluted in 55 μL of 10 mM Tris buffer (pH 7.5) and fragmented by Covaris ML230 focused ultrasonicator (Covaris) to obtain an average size of 550 bp. The WGS libraries were prepared by NEBNext DNA Library Prep Kit (New England Biolabs) Ultra I for isolate DNA and Ultra II for enriched or unenriched clinical DNA, using size selection for 400–500 bp fragments and PCR amplification for 6 cycles. Isolate DNA were sequenced with Miseq 500-cycle V2 kits (Illumina) with 32 isolates per run. Seventy-eight enriched and unenriched CSF DNA samples were sequenced with HiSeq 2500 rapid SBS 500-cycle V2 kits (Illumina) and the remaining 156 enriched CSF DNA samples were sequenced on a Novaseq 6000 using SP flow cell and SP SBS cartridge for 500-cycle (Illumina).
Genome Sequence Assembly and Analysis of Genes of Interest
Sequencing reads of isolate DNA specimens were assembled, and the following genes of interest were searched for identification using Bacterial Meningitis Genome Analysis Platform (BMGAP) [15]:
Fourteen molecular typing loci including 7 MLST loci, 4 fine-typing loci (PorA VR1, PorA VR2, FetA, PorB), and 3 vaccine antigen loci (NadA, NhbA, FHbp) as described in PubMLST database [16] (https://pubmlst.org/neisseria).
Essential capsule genes to determine serogroup. A specific serogroup was assigned if all essential capsule genes in the cps locus were present and intact; if 1 or more capsule genes within a given serogroup were fragmented or missing, a serogroup was assigned based on the serogroup’s genetic backbone only [17].
Alleles associated with reduced susceptibility to antibiotics (Supplementary Table 1).
Sequencing data of CSF DNA specimens (enriched and unenriched) were analyzed using an in-house developed computational pipeline as previously described [13] with addition of a search module for alleles associated with reduced susceptibility to antibiotics [18] (Supplementary Table 1).
Unmapped reads are available at SRA BioProject PRJNA776248: the isolates are listed as “Genomic” for their source, and the clinicals are listed as “Metagenomic” for their source. The workflow for bench work and data analysis is shown in Supplementary Figure 2.
Phylogenetic Analysis
The phylogeny trees for the individual clonal complexes (CCs) were produced using full genome assemblies as previously described [6]. Briefly, SNP alignment was created by Snippy version 3.1 (https://github.com/tseemann/snippy) and analyzed by Gubbins [19], which uses RAxML [20] to generate an initial maximum likelihood phylogeny. The efficacy of this method for meningococcal meningitis outbreak investigations was previously shown [11].
Binstrain Analysis
Sequence reads of high-quality assembles (>1400 core genome MLST [cgMLST]), which missed 1 or more of the 7 MLST loci, and those of low-quality ones (<1400) were analyzed using binstrain software version 1.0 [21], which compares the N. meningitidis reads to a SNP database containing a global collection of 222 N. meningitidis genomes, each genome representing a unique sequence type (ST). ST assignment cutoff was determined by the β value of a binomial mixture model used in binstrain analysis. If the β value for a certain ST was greater than 0.7, then the N. meningitidis in the specimen was considered likely to belong to the same ST. The binstrain database was created as described previously for Staphylococcus aureus [22].
RESULTS
Sequence Data Quality
Computational analysis of sequence data from 214 enriched clinical specimens, 20 unenriched clinical specimens, and 109 isolates (see “Methods” section) revealed high-quality de novo assemblies from 84% (180/214), 100% (20/20), and 100% (109/109) of specimens belonging to each of 3 groups described above. High-quality assemblies are those that have more than 1400 intact cgMLST loci, thus with more than 95% of calling accuracy per locus [13]. The other 16% of enriched clinical specimens (34/214) with less than 1400 intact cgMLST loci belong to the low-quality assembly group. The 87% (157/180) of high-quality assembles of enriched clinical specimens and all unenriched specimens and isolates were fully typed based on 7 MLST loci. Additionally, 9% (16/180) of high-quality assembles of enriched clinical specimens, which missed 1 or more of the 7 MLST loci, were successfully fully typed using binstrain analysis. The sequencing reads of low-quality assemblies were typed using binstrain analysis to the CC only. In total, full molecular typing was obtained in 46% (193/419) of all clinical specimens that were sequenced and 100% (109/109) of isolates. Additionally, 10% (41/419) of clinical specimens were typed partially, enabling CC identification. The average size of genome assembled from high-quality enriched specimens (2.11 × 106 ± 90 327) was similar to that of isolates (2.11 × 106 ± 10 894), with average SNP calling accuracy approaching almost 100% for both types of specimens (Table 1).
Table 1.
Comparison of Sequence Data Quality Obtained From Isolates and High-Quality Enriched Specimens
| Genome | cgMLST Count | SNP Calling Accuracy, %a | Contig Count | N50b | Mean Coverage | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CSF (n = 180) | Isolates (n = 109) | CSF (n = 180) | Isolates (n = 109) | CSF (n = 180) | Isolates (n = 109) | CSF (n = 180) | Isolates (n = 109) | CSF (n = 180) | Isolates (n = 109) | CSF (n = 180) | Isolates (n = 109) | |
| Minimum | 1.94 × 106 | 2.09 × 106 | 1405 | 1562 | 95.6 | 98.8 | 63 | 94 | 7781 | 37 341 | 16 | 24 |
| Maximum | 2.55 × 106 | 2.14 × 106 | 1580 | 1581 | 99.2 | 99.% | 718 | 165 | 78 704 | 68 913 | 364 | 127 |
| Mean | 2.11 × 106 | 2.11 × 106 | 1534 | 1569 | 98.2 | 98.9 | 215 | 132 | 35 413 | 52 558 | 222 | 53 |
| ± SD | ± 90 327 | ± 10 894 | ± 44 | ± 3 | ± 0.9 | ± 0.06 | ± 119 | ± 16 | ± 12 759 | ± 6724 | ± 81 | ± 26 |
Abbreviations: cgMLST, core genome multilocus sequencing type; CSF, cerebrospinal fluid; SNP, single-nucleotide polymorphism.
Calling accuracy was assessed using the correlation between accuracy of allele calling for cgMLST loci and the number of loci present [13].
N50 is the shortest contig length that needs to be included for covering 50% of the genome.
Serogroups and Clonal Complexes Among Clinical Specimens and Isolates
Three clonal complexes (CC) of N. meningitidis were identified: CC11, CC10217, and CC181 (Figure 1A). Unique STs were identified within each CC (Figure 1B). The predominant (>90%) STs revealed in the study were ST-11/CC11 detected in all 6 countries, ST-181/CC181 detected in 5 countries and ST-5789/CC181 in Chad, and ST-10217/CC10217 detected in 5 countries and ST-9376/CC10217 in Togo. Meningococcal serogroup was determined in 57% (115/200) of all high-quality assembles of clinical specimens and 100% (109/109) of isolates. An association was observed between serogroups and CCs with CC11 associated with serogroup W, CC181 with serogroup X, and CC10217 with serogroup C (Table 2). CC41/44 was represented by only 1 specimen from Ghana and was associated with serotype B.
Figure 1.
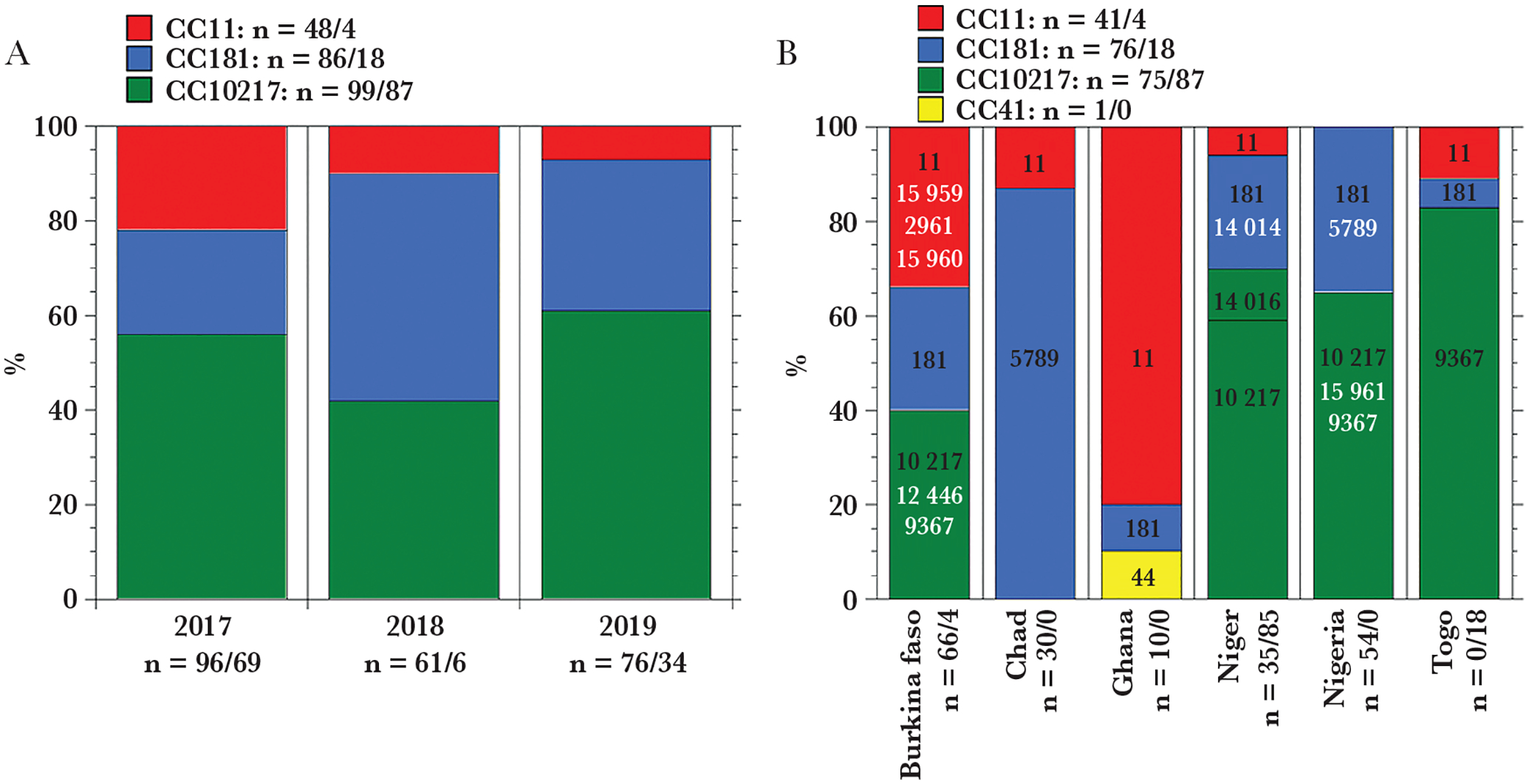
Distribution of meningococcal strains in meningitis belt countries. A, Major clonal complexes found among all sequenced clinical specimens (n = 233) and isolates (n = 109) by year. The additional clinical specimen belongs to CC41 and as a minor representative is not included in the panel. B, All unique sequence types according to pubMLST database [16] (black font, major [> 90%] sequence type; white font, minor [<10%] sequence type) found within each clonal complex in fully typed clinical specimens (n = 193) and isolates (n = 109) by country. The number of specimens found within each clonal, annual, geographic group is designated as n = number of clinical specimens/number of isolates.
Table 2.
Association Between Clonal Complex (CC) and Serogroup Among High-Quality Sequenced Enriched and Unenriched Clinical Specimens (n = 200) and Isolates (n = 109)
| Serogroup | CC | No. of Clinicals, Conclusive/Backbonec (Total) | No. of Isolates, Conclusive/Backbone (Total) |
|---|---|---|---|
| C | CC10217 | 48/15 (63) | 85/2 (87) |
| X | CC181 | 0/46 (46) | 18/0 (18) |
| Wa | CC11 | 0/5 (5) | 4/0 (4) |
| B | CC41/44 | 1/0 (1) | (0) |
| Unclearb | CC10217; CC181; CC11 | (85) | (0) |
The computational pipeline for sequencing data analysis of clinical specimens was not able to distinguish between Y and W serogroups as they differ only by the polymerase gene, csy for NmY and csw for NmW, which share almost 95% sequence similarity. Based on the historical association between clonal complexes C11 and serogroup W, we assign the identified Y backbone for clinical specimens to actual W capsule type.
The number of clinical specimens that did not have enough sequence data for their serogrouping.
Serogroups were designated as conclusive if all essential capsule genes were present and intact; if some of capsule genes within a given serogroup were fragmented or missing the assignment was to the level of serogroup backbone only.
Phylogenetic Analysis of 3 Major Clonal Complexes
CC11
As shown in Figure 1A, CC11 presented in 15% (52/343) of sequenced clinical specimens and isolates. Of these strains, 17% (9/52) were determined to be serogroup W by WGS; the other 83% (all clinical specimens) did not have the serogroup determined due to insufficient sequence data (Table 2). The CC11 phylogeny was built using 48 genomes that had high-quality assemblies in this study (2017–2019) and genomes of 435 isolates collected from meningitis belt countries in 2011–2016, with most (229 of 435) from Burkina Faso. All the CC11 strains from Burkina Faso belonged to at least 6 subclades. At least 4 subclades continued to expand during 2017–2019 from their ancestral strains that emerged in 2011–2012 (Figure 2). Strains from other countries, such as Ghana, Niger, and Chad, formed at least 2 subclades and demonstrated continued expansion in 2017–2019.
Figure 2.
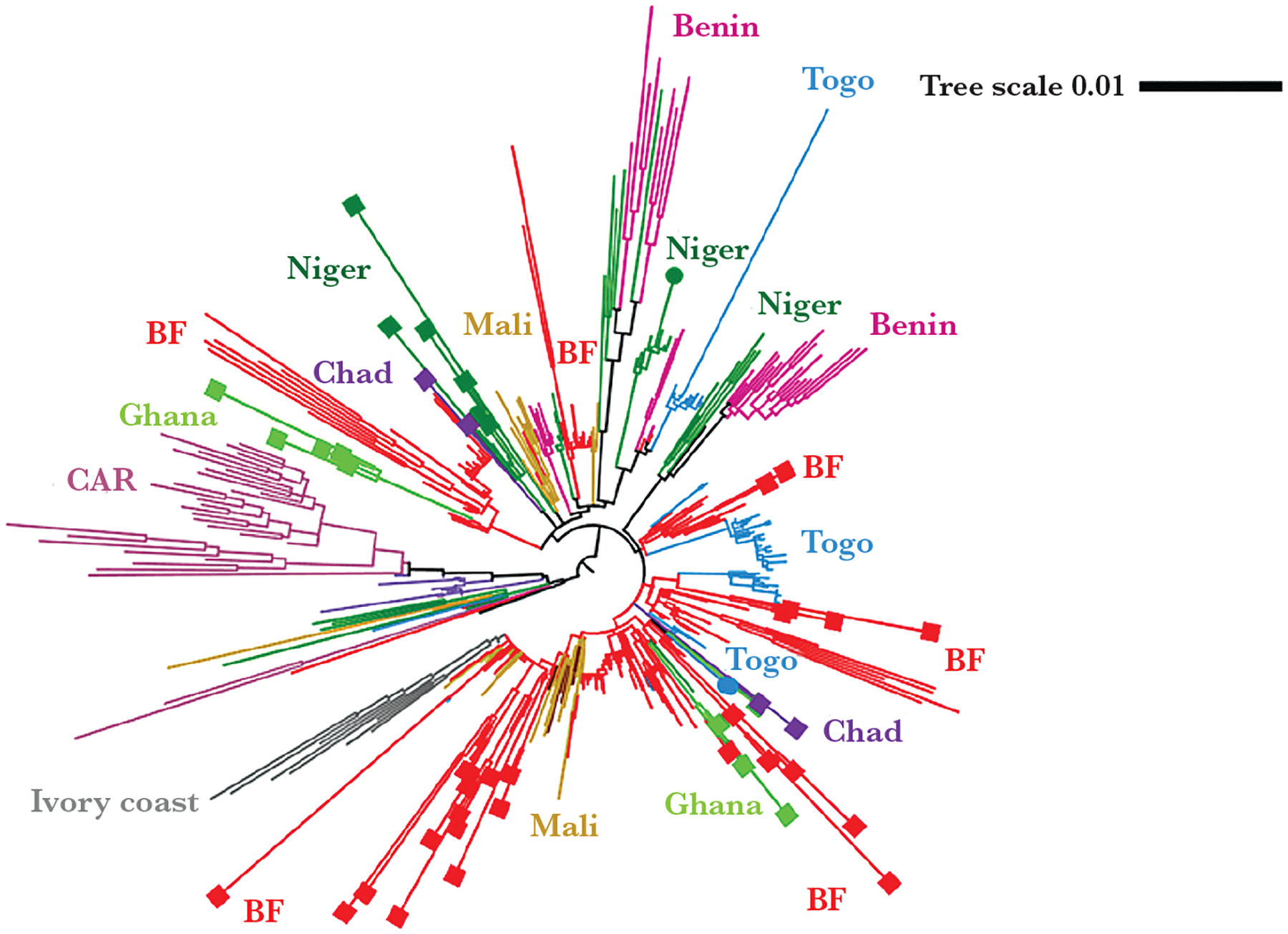
CC11 phylogenetic tree: Major clades/subclades are labeled with the countries and highlighted with corresponding colors. Specimens collected for the current (2017–2019) study are capped with squares for clinicals or circles for isolates. Uncapped specimens are from the previous study (2011–2016). The tree scale is in units of average substitutions per site along that length of the branch. Abbreviations: BF, Burkina Faso; CAR, Central African Republic.
CC181
CC181 represented 30% (104/343) of all sequenced isolates and specimens. Of these, 62% (64/104) were serogroup X by WGS; the other 38% did not have the serogroup determined due to insufficient sequence data (Table 2). The CC181 phylogeny contained 94 high-quality genome assemblies from this study (2017–2019) and 74 African isolates (2011–2016). N. meningitidis strains belong to 2 major subclades: subclade 1, primarily detected in Chad (ST-5789), and subclade 2 (ST-181), primarily detected in West African countries such as Burkina Faso, Niger, and Nigeria (Figure 3). These 2 subclades diverged from ancestral strains that were present in Niger between 1998 and 2005. Subclade 1 has been mostly in Chad through years 2011–2019, while subclade 2 has expanded in Burkina-Faso in 2011–2012, Mali in 2015, and more recently in Niger and Nigeria in 2017–2019. In addition, subclade 2 demonstrated additional expansion specifically in Burkina Faso during 2017–2019, resulting in development of subclade 2.1 (Figure 3).
Figure 3.
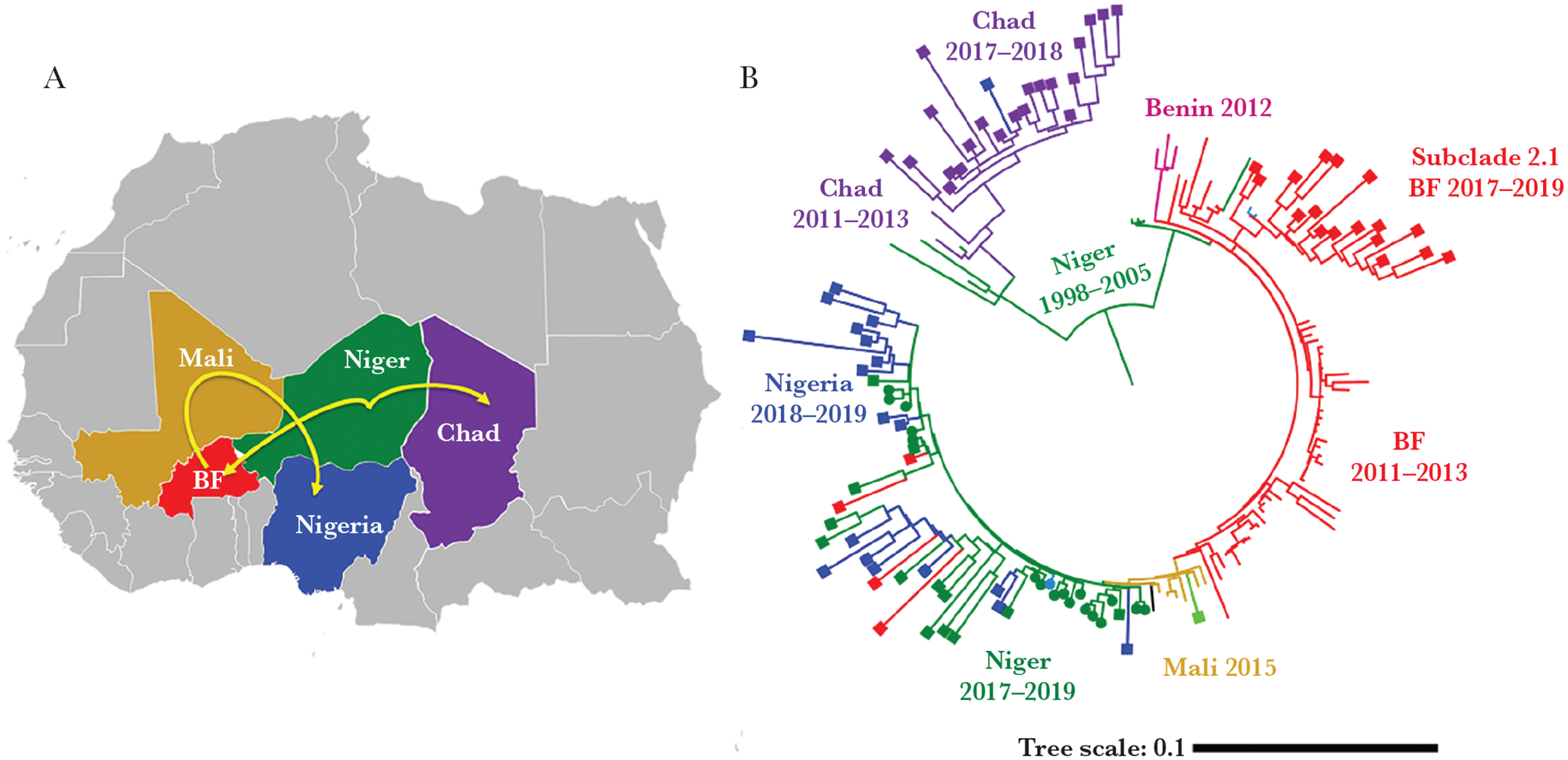
Phylogenesis of CC181 (A) and its plausible paths of expansion in meningitis belt countries (B). Major clades/subclades are labeled with the countries and corresponding years of strains identification. Specimens collected for the current (2017–2019) study are capped with squares for clinicals or circles for isolates. Uncapped specimens are from the previous study (2011–2016). The tree scale is in units of average substitutions per site along that length of the branch. Abbreviation: BF, Burkina Faso.
CC10217
CC10217 represented the most abundant strains in our collection, comprising 55% (186/343) of sequenced isolates and specimens. Of these strains, 80% (150/186) were determined to be serogroup C by WGS with the other 20% not being serogroup determined due to insufficient sequence data (Table 2). The CC10217 phylogeny was constructed using 167 high-quality genome assemblies from this study and 116 African isolates (2011–2016) (Figure 4). Strains from Burkina Faso, Niger, and Nigeria formed more than 1 subclade. Those from Togo and Mali formed 1 subclade for each country.
Figure 4.
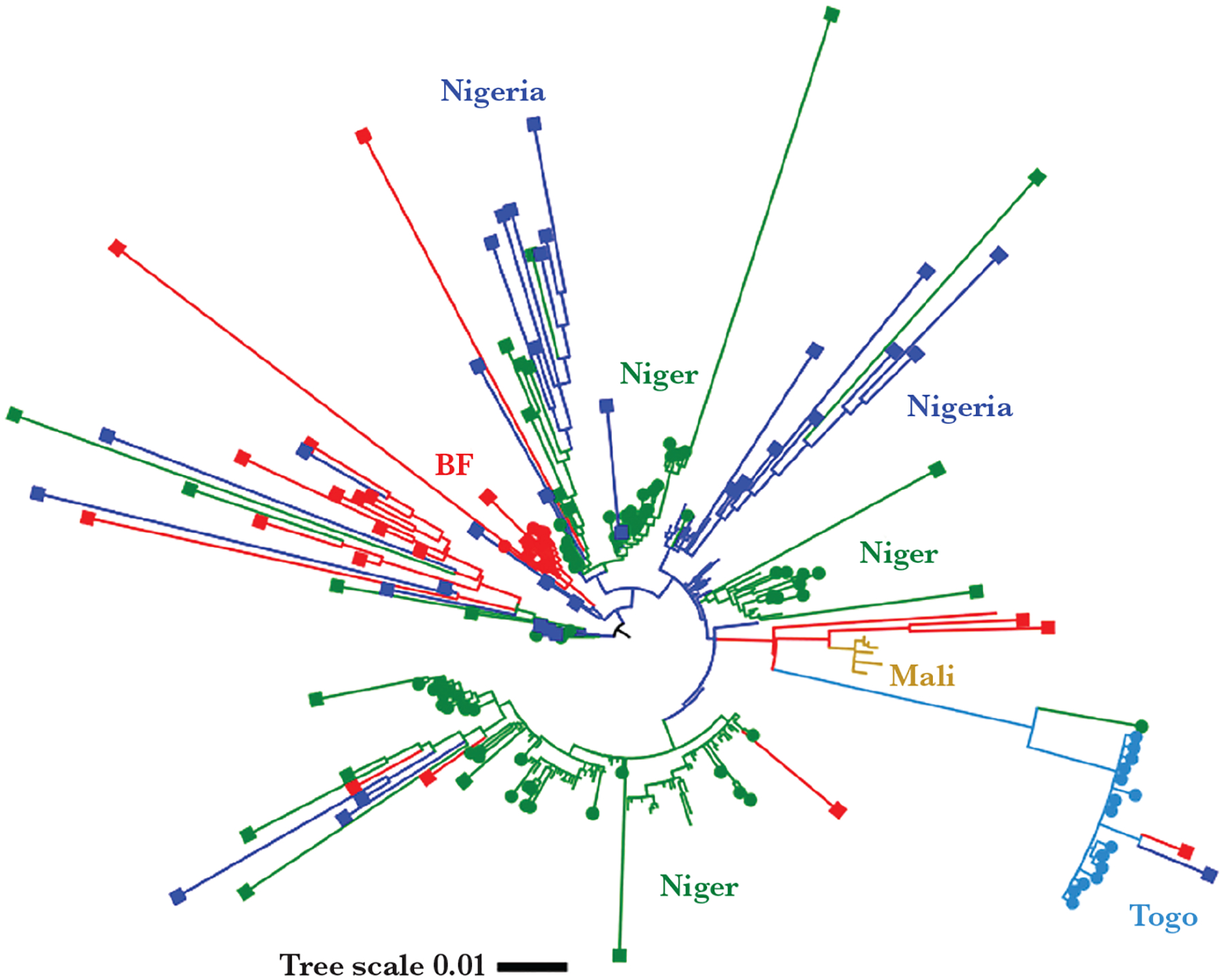
CC10217 phylogenetic tree. Major clades/subclades are labeled with the countries and highlighted with corresponding colors. Specimens collected for the current (2017–2019) study are capped with squares for clinicals or circles for isolates. Uncapped specimens are from the previous study (2011–2016). The tree scale is in units of average substitutions per site along that length of the branch. Abbreviation: BF, Burkina Faso.
Genomic Information Provided by SWGA
The sequences of more than 1400 genes generated using SWGA can be used for comprehensive characterization of meningococci in clinical specimens. To increase typing resolution of N. meningitidis strains, 2 variable regions (VR1 and VR2) of porin A (PorA), porin B (PorB), and an additional VR defined for ferric enterobactin transport (FetA) were analyzed along with conventional MLST loci [23]. Three genes (fHbp, nadA, and nhbA) are of interest because they encode vaccine antigens that are included altogether with PorA antigen in the 2 serogroup B meningococcal vaccines (Bexsero and Trumenba) [24]. The aforementioned loci found among the clinical specimens were compared with the isolates with the same ST; full consistency was found between these 2 specimen types. PorA, FHbp, and NhbA were found in all CCs while NadA was found only in CC11. CC11 also demonstrates a high diversity in PorB loci. Overall, there was an association between CC/ST and vaccine protein and fine-typing profiles (Table 3). Alleles associated with reduced antimicrobial susceptibility of N. meningitidis were identified (Supplementary Table 1) among clinical specimens and isolates. Eight of 180 specimens with high-quality genome assemblies (1 ST11/CC11 from Niger, 4 ST181/CC181 and 3 ST10217/CC10217 from Nigeria) had a gyrA T91I allele, which is associated with reduced susceptibility to ciprofloxacin [25]. No alleles associated with antibiotic resistance were found in the other clinical specimens or isolates.
Table 3.
Fine-Typing and Vaccine Antigen Composition for Each ST in Fully Typed Clinical Specimens (n = 193) and Isolates (n = 109)
| CC (No.a) | ST (No.) | PorA type VR1, VR2 (No.) | PorB type (No.) | FetA (No.) | FHbp (No.) | NadA (No.) | NhbA (No.) |
|---|---|---|---|---|---|---|---|
| CC10217 (73/87) | 10217 (65/69) | 21–15, 16 (63/69) | 3–463 (64/69) | F1–7 (57/69) F1–31 (2) F1–136 (1) | 2.27 (63/69) | Not found (64/69) NadA-2/3.6 (1) | 798 (63/69) |
| 14016 (3/2) 12446 (2) 15961 (1) | 21–15, 16 (6) | 3–463 (6) | F1–7 (6) | 2.27 (6) | Not found (6) | 798 (5) | |
| 9367 (2/16) | 21–15, 16–46 (2/16) | 3–463 (2/16) | F1–7 (2/16) | 3.111 (2/16) | Not found (2/16) | 798 (2/16) | |
| CC11 (44/4) | 11 (44/4) | 5, 2 (44/4) | 2–2 (27/4), 2–39 (5), 2–48 (5), 2–60(3), 2–195 (2), 2–297 (1), 2–337 (1) | F1–1 (44/4) | 1.9 (44/4) | NadA-2/3.6 (44/4) | 96 (41/4) |
| CC181 (75/18) | 181 (53/18) | 5–1, 10–1 (51/18) | 2–231 (49/18) | F1–31 (51/18), F1–153 (1), F1–1 (1) | 1.74 (45/13), 3.499 (1) | Not found (52/18) | 359 (52/18) |
| 5789 (22) | 5–1, 10–1 (21) | 2–205 (20) | F4–23 (21) | 1.391 (21) | Not found (22) | 358 (21) | |
| CC41(1) | 44 (1) | 22, 14–6 (1) | 3–38 (1) | F5–84 (1) | 2.19 (1) | Not found (1) | 43 (1) |
Abbreviation: CC, clonal complex; ST, sequence type.
The number of clinical specimens and isolates found within each group and designated as (No. of clinicals/No. of isolates). The single number in parentheses is designated when only clinical specimens were available for the group. The assignment for the other summable alleles was not possible due to the insufficiency of sequencing data for them.
DISCUSSION
Timely control and prevention of epidemic meningitis in African meningitis belt countries rely on early and rapid detection and characterization of meningitis cases through surveillance programs in the region. Genomic surveillance provides data for in-depth strain characterization using next-generation sequencing; the sequencing data have been used to inform public health interventions, especially during outbreaks. Strain characterization in laboratories typically relies on bacterial culture from the patient specimen. However, obtaining bacterial cultures from meningitis cases is challenging in meningitis belt countries due to the limited laboratory capacity; therefore, the sequencing data from these countries are underrepresented. Therefore, affordable culture-free enrichment approaches are critical for enhancing genome surveillance in the meningitis belt given that most meningococcal cases identified in this region do not have an isolate available. Our work demonstrates that the SWGA-based sequencing procedure significantly increased the number of clinical specimens from meningitis belt countries that could be successfully sequenced for molecular analysis for N. meningitidis. SWGA enrichment increased the total number of sequence reads for meningococcal strains and produced sufficient high-quality molecular data for in-depth strain characterization, including MLST analysis, fine-typing, OMP (outer membrane protein) vaccine antigen typing, and antibiotic-resistance profiling. Moreover, it provides more than 1400 genes for phylogenetic analysis of meningococcal strains. The cost of the EquiPhi29-based sequencing procedure has been further reduced to be comparable with that of isolate sequencing (Supplementary Table 2). Therefore, SWGA can present an effective alternative method for genomic surveillance when bacterial culture rate is low.
In our study, only 20% (109/528) of the samples were isolates, reflecting the low culture rate in the region. Most samples were clinical specimens; only 4% (20/528) of untreated specimens had a high bacterial DNA load (average CtsodC = 16 or approximately 500 000 genomes/μL) allowing for sequencing without enrichment. SWGA enrichment allowed the analysis of specimens with bacterial DNA 1000-fold lower (average CtsodC = 26 or approximately 500 genomes/μL) than those without enrichment, thus increasing the number of clinical specimens that could be sequenced by 10-fold (from 20 to 214) and the total number of samples that could be characterized by 3-fold (109 isolates to 343 isolates/specimens). In total, half of the clinical specimens (200/399) in this study produced high-quality assemblies after SWGA enrichment with the majority of them (97%, 193/200) being fully typed. The other specimens failed at the enrichment step or at generation of high-quality sequencing data for molecular typing due to the limitations imposed by the sensitivity of the procedure requiring at least 500 genomes/μL of its constituent N. meningitidis DNA to guaranty a success rate of 50% for the molecular typing (Supplementary Figure 3).
The SWGA method for enrichment enabled us to study strain distribution in countries of the meningitis belt over time by comparing current strains with previous phylogenetic studies conducted on isolates collected in meningitis belt countries between 2011 and 2016 [6]. During 2011–2016, serogroup W strains belonging to CC11 were predominant in the region (up to 60.6%). During 2017–2019, while the serogroup W CC11 strain has continued to spread across the region, especially in Burkina-Faso, it has decreased to 7% during 2017–2019. In contrast, there was an increase in CC10217 strains that are associated with serogroup C, from 18% of the cases in 2011–2016 to 61% in 2017–2019. CC10217 has been increasingly reported in Niger, Nigeria, and Togo, where CC10217 now comprises 70%–80% of the reported cases. CC10217 has also been associated with outbreaks in Togo and Burkina Faso in 2019 [13], and Benin in 2020 (unpublished data). Multiple STs were identified within this CC (at least 5 STs shown in Figure 1B), indicating its rapid evolution. Serogroup X CC181 represented by 2 STs (ST-5789 and ST-181) was the second most prevalent CC, comprising up to 48% of cases in 2019. Phylogenetic analysis has allowed us to map the spread of CC181 strains across the meningitis belt (Figure 3).
While SWGA-based WGS can generate high-quality data for molecular typing, the serogroup identification rate in these specimens was only 52% (95/180), due to insufficient coverage of the capsule locus by SWGA heptamers and the strict requirement for the integrity of all capsule genes by the computational pipeline [13]. Nevertheless, an association between CCs and serogroups was observed, consistent with the study by Topaz et al [6]. Additionally, we found the MenB vaccine antigen profiling consistent with previous findings [6]. Moreover, we found that some vaccine antigens are associated with certain STs. For example, NhbA peptide 358 was only detected in ST-5789 while NhbA 359 was detected in ST-181.
In conclusion, SWGA-based WGS methods provide information on the genetic relatedness of circulating meningococcal strains as well as for individual genes of interest. Integration of this method into genomic surveillance workflows can improve data quality and representativeness. This is especially important in situations where obtaining bacterial isolates from meningococcal cases is challenging.
Supplementary Material
Acknowledgments.
We thank How Yi Chang, Fang Hu, and Daya Marasini for their assistance with DNA extraction from bacterial isolates and WGS library preparations, and Shalabh Sharma for his assistance with computational analysis of sequence data.
Financial support.
This work was supported by the Centers for Disease Control and Prevention and GAVI, the Vaccine Alliance.
Footnotes
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Supplementary materials consist of data provided by the author that are published to benefit the reader. The posted materials are not copyedited. The contents of all supplementary data are the sole responsibility of the authors. Questions or messages regarding errors should be addressed to the author.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.World Health Organization. Control of epidemic meningitis in countries in the African meningitis belt, 2019. Weekly Epidemiological Record 2020; 95:133–44. [Google Scholar]
- 2.Molesworth AM, Cuevas LE, Connor SJ, Morse AP, Thomson MC. Environmental risk and meningitis epidemics in Africa. Emerg Infect Dis 2003; 9:1287–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Omoleke SA, Alabi O, Shuaib F, et al. Environmental, economic and socio-cultural risk factors of recurrent seasonal epidemics of cerebrospinal meningitis in Kebbi state, north-western Nigeria: a qualitative approach. BMC Public Health 2018; 18:1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greenwood B Manson Lecture. Meningococcal meningitis in Africa. Trans R Soc Trop Med Hyg 1999; 93:341–53. [DOI] [PubMed] [Google Scholar]
- 5.Trotter CL, Lingani C, Fernandez K, et al. Impact of MenAfriVac in nine countries of the African meningitis belt, 2010–15: an analysis of surveillance data. Lancet Infect Dis 2017; 17:867–72. [DOI] [PubMed] [Google Scholar]
- 6.Topaz N, Caugant DA, Taha MK, et al. Phylogenetic relationships and regional spread of meningococcal strains in the meningitis belt, 2011–2016. EBioMedicine 2019; 41:488–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.World Health Organization (WHO). Defeating meningitis by 2030: a global road map. Geneva, Switzerland: WHO, 2020. [Google Scholar]
- 8.Rodgers E, Bentley SD, Borrow R, et al. The global meningitis genome partnership. J Infect 2020; 81:510–20. [DOI] [PubMed] [Google Scholar]
- 9.Maiden MC, Bygraves JA, Feil E, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA 1998; 95:3140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Retchless AC, Fox LM, Maiden MCJ, et al. Toward a global genomic epidemiology of meningococcal disease. J Infect Dis 2019; 220(Suppl 4):S266–73. [DOI] [PubMed] [Google Scholar]
- 11.Whaley MJ, Joseph SJ, Retchless AC, et al. Whole genome sequencing for investigations of meningococcal outbreaks in the United States: a retrospective analysis. Sci Rep 2018; 8:15803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feagins AR, Vuong J, Fernandez K, et al. The strengthening of laboratory systems in the meningitis belt to improve meningitis surveillance, 2008–2018: a partners’ perspective. J Infect Dis 2019; 220(Suppl 4):S175–81. [DOI] [PubMed] [Google Scholar]
- 13.Itsko M, Retchless AC, Joseph SJ, et al. Full molecular typing of Neisseria meningitidis directly from clinical specimens for outbreak investigation. J Clin Microbiol 2020; 58:e01780–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dolan Thomas J, Hatcher CP, Satterfield DA, et al. sodC-based real-time PCR for detection of Neisseria meningitidis. PLoS One 2011; 6:e19361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buono SA, Kelly RJ, Topaz N, et al. Web-based genome analysis of bacterial meningitis pathogens for public health applications using the bacterial meningitis genomic analysis platform (BMGAP). Front Genet 2020; 11:601870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jolley KA, Bray JE, Maiden MCJ. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res 2018; 3:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marjuki H, Topaz N, Rodriguez-Rivera LD, et al. Whole-genome sequencing for characterization of capsule locus and prediction of serogroup of invasive meningococcal isolates. J Clin Microbiol 2019; 57:e01609–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potts CC, Retchless AC, McNamara LA, et al. Acquisition of ciprofloxacin resistance among an expanding clade of beta-lactamase-positive, serogroup Y Neisseria meningitidis in the United States. Clin Infect Dis 2021; 73:1185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croucher NJ, Page AJ, Connor TR, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 2015; 43:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014; 30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joseph SJ, Li B, Ghonasgi T, et al. Direct amplification, sequencing and profiling of Chlamydia trachomatis strains in single and mixed infection clinical samples. PLoS One 2014; 9:e99290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Joseph SJ, Li B, Petit Iii RA, Qin ZS, Darrow L, Read TD. The single-species metagenome: subtyping Staphylococcus aureus core genome sequences from shotgun metagenomic data. PeerJ 2016; 4:e2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jolley KA, Brehony C, Maiden MC. Molecular typing of meningococci: recommendations for target choice and nomenclature. FEMS Microbiol Rev 2007; 31:89–96. [DOI] [PubMed] [Google Scholar]
- 24.Lucidarme J, Comanducci M, Findlow J, et al. Characterization of fHbp, nhba (gna2132), nadA, porA, and sequence type in group B meningococcal case isolates collected in England and Wales during January 2008 and potential coverage of an investigational group B meningococcal vaccine. Clin Vaccine Immunol 2010; 17:919–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong E, Thulin Hedberg S, Abad R, et al. Target gene sequencing to define the susceptibility of Neisseria meningitidis to ciprofloxacin. Antimicrob Agents Chemother 2013; 57:1961–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.