

Abstract

Triple negative breast cancer (TNBC) represents a subtype of breast cancer that does not express the three major prognostic receptors of human epidermal growth factor receptor 2 (HER2), progesterone (PR), and estrogen (ER). This limits treatment options and results in a high rate of mortality. We have reported previously on the efficacy of a water-soluble, cationic organometallic compound (Ru-IM) in a TNBC mouse xenograft model with impressive tumor reduction and targeted tumor drug accumulation. Ru-IM inhibits cancer hallmarks such as migration, angiogenesis, and invasion in TNBC cells by a mechanism that generates apoptotic cell death. Ru-IM displays little interaction with DNA and appears to act by a P53-independent pathway. We report here on the mitochondrial alterations caused by Ru-IM treatment and detail the inhibitory properties of Ru-IM in the PI3K/AKT/mTOR pathway in MDA-MB-231 cells. Lastly, we describe the results of an efficacy study of the TNBC xenografted mouse model with Ru-IM and Olaparib monotherapy and combinatory treatments. We find 59% tumor shrinkage with Ru-IM and 65% with the combination. Histopathological analysis confirmed no test-article-related toxicity. Immunohistochemical analysis indicated an inhibition of the angiogenic marker CD31 and increased levels of apoptotic cleaved caspase 3 marker, along with a slight inhibition of p-mTOR. Taken together, the effects of Ru-IM in vitro show similar trends and translation in vivo. Our investigation underscores the therapeutic potential of Ru-IM in addressing the challenges posed by TNBC as evidenced by its robust efficacy in inhibiting key cancer hallmarks, substantial tumor reduction, and minimal systemic toxicity.
Keywords: triple negative breast cancer, in vivo, chemotherapeutics, ruthenium compounds, mechanisms
Introduction
Triple negative breast cancer (TNBC) represents a highly malignant and aggressive subtype of breast cancer with a higher rate of mortality than other subtypes1 affecting disproportionately women of African and Hispanic descent.1 Defined by the lack of estrogen, progesterone, and human epidermal growth factor 2 expression, TNBC accounts for roughly 20% of all breast carcinomas.2 Prognosis following diagnosis is grim, with a median overall survival (OS) of 10.2 months.3 Less than 30% of patients achieve a complete response, with a high recurrence and mortality rate as a result.4 The inability to directly target a hormonal receptor greatly limits the clinical options for the treatment of TNBC.1 Different subtypes of TNBC have also been described, with a classification of six different clusters (basal-like 1, basal-like 2, immunomodulatory, luminal androgen receptor, mesenchymal, and mesenchymal-stem-like) with unique molecular characteristics.5 This leads to a molecular heterogeneity that further complicates clinical options.
Unlike treatment for tumors overexpressing HER2, nonspecific chemotherapy is the standard of care for TNBC as there is only one type of targeted therapy recently approved by the Food and Drug Administration (FDA) in 2020 (antibody-drug conjugate which targets the protein Trop-2).6 As typical hormonal therapies are unavailable, new treatments are in high demand as evidenced by the more than 500 clinical trials completed and ongoing exploring new therapeutic regimens.7 Combinatory therapies are common in these clinical trials, usually with a platinum-based chemotherapeutic (cisplatin, carboplatin, and oxaliplatin) or other conventional agents such as gemcitabine, paclitaxel, and ixabepilone.8 These therapeutics can be combined with others that target specific cellular functions, which can include kinase inhibitors (everolimus), inhibitors of growth factors (erlotibib), antibodies (cetuximab and bevacizumab), cell cycle (abemaciclib and trilaciclib), and inhibitors of histone deacetylase (entinostate and chidamide).4 Targeted therapies directed to cellular pathways have also been explored in clinical trials including Hedgehog (vismodegib), extracellular signal-regulated kinase (ERK) (selumetinib), epidermal growth factor receptor (EGFR) (dasatanib and geftinib), and phosphatidylinositol-3-kinase (PI3K)-protein kinase B (AKT) mammalian target of rapamycin (mTOR) (samotolisib and ipatasertib) pathways.4 While these clinical studies show promise in future TNBC therapies, the exploration of less toxic and cost-efficient chemotherapeutics is relevant.
The potential of new chemotherapeutics for TNBC treatment based on small molecules9 and metal-based compounds10 has been reviewed. The interest in metal-based drugs as prospective TNBC chemotherapeutics comes from successful clinical trial results based on classical platinum adjuvant agents.11 Moreover, patients with BRCA mutated genes and some with specific TNBC subtypes unresponsive to anthracycline-taxane (ACT) seem to benefit from the use of these platinum agents.12 Metallodrugs based on unconventional platinum compounds, and derivatives of metals other than platinum such as gold and ruthenium, have displayed relevant anticancer properties in TNBC cells and mouse models.10,13 Three ruthenium-based compounds have been studied in the clinic for different cancers: two ruthenium(III) compounds NAMI-A (used in phase II trials for the treatment of nonsmall cell lung cancer),14 and BOLD-100 (currently in phase II trials for the treatment of advanced gastrointestinal cancers)15 and a ruthenium(II) derivative TLD-1433 (a photodynamic therapy currently in phase trials for the treatment of nonmuscle invasive bladder cancer).16 Ruthenium(II) compounds specifically display high activity against cisplatin-resistant and metastasized tumors by a mode of action that differs from that of cisplatin (e.g., DNA does not seem the main biological target).13
We have previously reported on a water-soluble, cationic, organometallic Ru(II) compound [(η6-p-cymene)Ru(k-N,O–Ph3P = N–CO–2-N–C5H4)]Cl (Ru-IM, in Figure 1A), that contains an arene, a labile chloro, and a chelating N–O iminophosphorane ligand.17,18Ru-IM displays relevant anticancer activity and a favorable pharmacological profile that makes it highly attractive as a potential TNBC chemotherapeutic.17Ru-IM showed high activity against several cisplatin-resistant cell lines from the 60 cell line National Cancer Institute (NCI) panel, with enhanced cytotoxicity observed against a subsequent selected panel of TNBC cell lines.19 Initial mechanistic studies revealed weak or electrostatic interactions with plasmid and CT DNA and the induction of caspase-dependent apoptosis in leukemia Jurkat cells.17 Further evaluation on TNBC MDA-MB-231 and HCC-1806 cell lines determined an apoptotic mechanism of cell death as well as slight G2/M cell cycle arrest.19 We also demonstrated the impressive antimigratory, anti-invasive, and antiangiogenic properties of Ru-IM in TNBC cell lines.19 Cellular uptake studies showed good overall cellular uptake, with subcellular studies revealing increased mitochondria accumulation (52.7%)20 Preliminary in vivo efficacy studies of Ru-IM in a MDA-MB-231 mouse xenograft model (female NOD.SCID mice) afforded 56% tumor reduction following a 28-day efficacy trial with a dosing schedule of 5 mg/kg every other day.17,18 While tumor growth inhibition in TNBC mouse models has been reported for other metal-based compounds,21−23 tumor shrinkage in these in vivo models is unique to Ru-IM. Further evaluation also showed low systemic toxicity, with preferential accumulation of Ru in tumor tissue.17 Furthermore, a targeted proteomic analysis in TNBC cells treated with Ru-IM involved inhibition of the macrophage colony stimulating factor (M-CSF), while showing that p53 was not affected.19 The change in protein expression levels of M-CSF may imply the potential involvement of the phosphatidylinositol-3-kinase (PI3K)-protein kinase B (Akt) mammalian target of the rapamycin (mTOR) pathway in the mechanistic action of Ru-IM.
Figure 1.

Cell Painting Assay in U-2 OS sarcoma cells demonstrates mitochondrial interaction with Ru-IM. (A) Structure of Ru-IM. (B) Cells were dosed with 1.4 or 2.8 μM Ru-IM and imaged after 24h with multiplexed dyes for nuclei, ER, RNA, mitochondria, and actin. (C) Zoomed representative image of DMSO and 2.8 μM Ru-IM treated cells specific for mitochondria dye.
All the data acquired so far warrant advanced preclinical studies with Ru-IM. Here, we report on experiments to elucidate the role of mitochondrial dysfunction, as well as the potential involvement of the PI3K/AKT/mTOR pathway to shed light on the mode of action of Ru-IM in TNBC cells. Most evaluations were completed in MDA-MB-231 cells as we have previously reported extensively on this cell line. We have evaluated morphological profile changes and changes to mitochondrial function and assessed alterations of protein expression levels of key markers along PI3K/AKT/mTOR in TNBC cancer cells treated with Ru-IM. Moreover, we describe the results of an efficacy study of a TNBC xenografted mouse model with Ru-IM and poly(ADP-ribose) polymerase (PARP) inhibitor Olaparib monotherapy, and in combination treatments including histopathology. Lastly, we have evaluated biomarkers associated with neoplastic growth in tumor tissue of mice treated with Ru-IM, Olaparib, and combined treatments.
Results
Morphological Profile Changes Induced by Ru-IM: Mitochondrial Alteration
As mentioned previously, Ru-IM has a preferential accumulation in mitochondria in MDA-MB-231 cells as demonstrated by ICP-OES and by live-cell imaging of a luminescent BODIPY analog.20 The interaction with Ru-IM and this cell organelle was further investigated to assess whether changes to mitochondrial function were observed with exposure to the ruthenium compound. Mitochondrial dysfunction has been reported for other ruthenium anticancer agents24,25 and may give insight into their underlying mechanism of action. Cell Painting, a high content imaging assay, was used to ascertain the morphological profile of Ru-IM against five multiplexed dyes–actin, RNA, endoplasmic reticulum (ER), mitochondria, and nuclei, as detailed in Figure 1B. This assay can easily profile cell populations in different organelles to determine the detection of cellular phenotypes.26 The human bone osteosarcoma cell line U-2 OS was utilized for testing and is typically used for this assay.26 Eight total doses of Ru-IM were evaluated by a series of dilution factors. At doses of 1.4 and 2.8 μM [whose concentrations are comparable to the IC50 for Ru-IM (3.7 μM)], phenotypic differences from DMSO samples were observed. These dilutions correspond to clear alterations in the mitochondrial channel. Zoomed images of the mitochondrial channel are shown in Figure 1C with DMSO-treated cells seeing standard tubular-shaped mitochondria. In contrast, Ru-IM-treated cells exhibit increased levels of fragmentation, indicative of dysfunctional mitochondria.27 These results reflect interaction with the organelle, confirming preliminary findings in TNBC cell lines.20 Thus, further studies were completed to assess the potential changes to mitochondrial function.
Mitochondrial Dysfunction of ROS Generation and Membrane Potential
Reactive oxygen species (ROS) generation is a measure of the oxidative damage in the cell that involves the production of superoxide, a byproduct usually associated with mitochondrial metabolism and DNA damage.28 Several cellular functions require the production of moderate levels of ROS, including gene expression.28 In cancer cells, the production of ROS is amplified, but it is usually quenched by antioxidant pathways (although a moderate increase in the levels may promote tumor growth and metastatic processes). ROS are, however, known to trigger programmed cell death, and the modulation of ROS can be exploited as a strategy to develop anticancer therapeutics.29
ROS production in MDA-MB-231 cells exerted by Ru-IM was measured using the fluorogenic marker dichlorodihydrofluorescein diacetate (DCF-DA), an oxidant-sensitive dye that converts to fluorescent species dicholorofluorescin (DCF) upon exposure to ROS (Figure 2A).28 Over a 12 h kinetic study in MDA-MB-231 cells with nontreated cells as a negative control and 1 mM hydrogen peroxide (H2O2) as a positive control, Ru-IM showed significant ROS generation upon treatment using the IC50 as the concentration of the complex. This generation of ROS is comparable to that of other ROS-producing ruthenium complexes, such as ruthenium(II) cationic compounds like [Ru-ATZ]+ containing cyclopentadienyl,30 and anionic ruthenium(III) derivative BOLD-100.31 For most anticancer drugs that generate ROS and provoke apoptosis, there is an activation of intrinsic pathways that involves an increase of the mitochondrial permeability, the release of activator factors, and the activation of caspases.29 We have previously demonstrated that Ru-IM exerts canonical or caspase-dependent apoptosis,17,19 which is in accordance with the results described here (accumulation in the mitochondria and generation of ROS). Furthermore, free radical formation can cause damage to the DNA phosphate backbone, providing a potential link between DNA damage and Ru-IM.32 While the use of the DCFDA fluorogenic dye allows insight into ROS species detection within the cell, there is no distinction of the origin of this generation. Hydroxyl radicals, hydrogen peroxide, and superoxide anions all act as types of ROS and are generated in various cellular compartments, which include the mitochondria, plasma membrane, and cytosol.28 To ascertain mitochondrial ROS (mitROS) generation, the antioxidant N-acetyl-l-cysteine (NAC) was employed. The antioxidative properties of NAC come from its generation of hydrogen sulfide (H2S) and subsequent oxidation, which allows for direct scavenging of ROS.33 This process occurs in the mitochondrial membrane, which signifies the potential involvement of this organelle with NAC activity.34 To assess mitROS, 1 mM NAC and Ru-IM at the IC50 concentration were used in a 10 h kinetic study in MDA-MB-231 cells. The results of this study are depicted in Figure 2B. As expected, treatment with NAC on its own (1 mM) resulted in limited ROS generation. The combination of NAC and Ru-IM also resulted in a similar decrease in comparison to that of Ru-IM treatment alone, which demonstrates mitochondrial involvement in ROS generation by this compound. Similar trends have been observed with ruthenium(II) sulfonylethylenediamine and imidazole complexes in the treatment of ovarian35 and lung36 cancer cells, respectively.
Figure 2.

Changes to mitochondrial function in MDA-MB-231 cells observed with exposure to Ru-IM. (A) Ru-IM induces ROS generation as demonstrated by the ROS generation assay in a 12 h kinetic study (IC50 concentration of Ru-IM). Addition of NAC suppresses generation, suggesting the origin of mitochondria. (B) Depolarization of the membrane in MDA-MB-231 cells is observed with treatment of Ru-IM (IC50 concentration) after the 3h incubation period. Data are expressed as the mean ± standard error, ns = not significant, *p < 0.033, **p < 0.002, ***p < 0.001.
The mitochondrial membrane potential (MMP) is an essential component of cellular metabolism. MMP results from the activity of the Krebs cycle, serving as an intermediate form of energy storage in the process of making ATP.33 MMP can also be used to determine mitochondrial viability, as a loss of MMP function can be indicative of apoptosis and decreased cellular function.37 To determine changes in MMP (ΔΨm), the fluorogenic dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidalylcarbocyanine iodide) was utilized. JC-1 aggregates form at high MMP and indicate healthy mitochondrial function. Low MMP forms JC-1 monomers, which are a sign of depolarization.38 By evaluation of the aggregate:monomer ratio, MMP polarization dysfunction may be measured.38 Mitochondrial uncoupling agent carbonyl cyanide m-chlorophenyl hydrazone (CCCP) was used as a negative control, as it is known to depolarize MMP.39 Changes to MMP in MDA-MB-231 cells treated with Ru-IM are depicted in Figure 2B. The JC1-1 aggregate monomer ratio for cells treated with Ru-IM is indicative of ΔΨm depolarization. In comparison to the control, a higher amount of JC-1 monomer indicative of MMP depolarization is found with Ru-IM treatment. This trend has also been reported for other ruthenium compounds active against lung40 and liver41 cancer cell lines.
Potential Involvement of the PI3K/AKT/mTOR Pathway
As previously mentioned, the inhibition of protein expression levels of M-CSF in TNBC cell lines when treated with an IC50 concentration of Ru-IM over 24 h was observed.19 This growth factor is involved in cellular proliferation and survival of monocytes and macrophages.42 M-CSF works upstream of the PI3K/AKT/mTOR pathway, and the Ru-IM mediated inhibition of this cytokine may indicate the involvement of the PI3K/AKT/mTOR pathway as a potential mode of action. This pathway acts as a key regulator of survival during cellular stress and controls many aspects of cellular function, including glucose metabolism, cell cycle arrest, apoptosis, autophagy, DNA repair, and ribosome biogenesis.43 Cancerous cells utilize and overexpress this cellular pathway to continue the limitless cell growth and survival. Roughly 25% of TNBCs have mutations along this pathway. Nine phase II and III clinical trials feature inhibitors along this pathway all in combination with other agents such as capecitabine and paclitaxel (as of January 2024, none of these trials involve metal-based compounds).4
Some of the hallmarks of cancer inhibited by Ru-IM may be explained by its involvement in this pathway. PI3K promotes cellular proliferation, angiogenesis, and migration.43 Similarly, AKT is involved in processes of proliferation and apoptosis.45 Key markers along the pathway may also be involved, such as PTEN (which acts as a negative regulator and tumor suppressor)44 and PDK1 (which regulates growth, invasion, and migration) in PI3K/AKT signaling.43 The aberrant activation of PI3K/AKT has been identified as a key link that modulates multidrug resistance.46 Importantly, the disturbance of mitochondrial function is a common theme throughout this pathway.46−48 AKT has been reported to modulate mitochondria-mediated apoptosis, redox states, dynamic balance, autophagy, and metabolism.47 PI3K regulates mitochondrial homeostasis and has been tied to ROS generation and cell cycle arrest.48 mTOR, in conjunction with the mTORC1 complex, regulates the mitochondrial oxidative metabolism.49 Taken together, the functions of the PI3K/AKT/mTOR pathway can be linked to cancerous growth, and the inhibition of this pathway may explain the mode or modes of action of Ru-IM.
We further evaluated change in protein expression levels of key markers (PI3K, AKT3, PDK1, mTOR, and PTEN) along the PI3K/AKT/mTOR pathway in MDA-MB-231 cells treated with Ru-IM using Western blot (Figure 3). AKT3 represents an isoform of AKT that is thought to play a critical role as a potential target for TNBC.50 Inhibitors for both mTOR and AKT3 have shown success in clinical trials for the treatment of TNBC.43,45,51,52 As the activation of this pathway is dependent upon a phosphorylated signaling cascade, both phosphorylated and nonphosphorylated total protein levels were evaluated. Cell lysates obtained from MDA-MB-231 cells were collected 2 and 24 h after incubation with Ru-IM (IC50 concentration) and compared to the untreated control. Our results indicated inhibition of protein expression levels of p-PI3K, p-AKT3, p-mTOR, and p-PDK1 markers. PTEN levels remained similar to untreated lysates, which was to be expected for this negative regulator. Interestingly, little to no change is observed in total protein for PI3K, AKT3, mTOR, and PDK1. This suggested that the protein expression inhibition observed with Ru-IM treatment occurs only for phosphorylated markers, indicating that pathway activation is necessary to observe changes in overall protein expression levels.
Figure 3.

Inhibition of protein expression levels viewed in phosphorylated markers but not in total protein markers in the MDA-MB-231 cell line with Ru-IM. Changes to protein expression levels in MDA-MB-231 cell line with levels using IC50 concentration of Ru-IM in 2 and 24 h treatment. Data are expressed as the mean ± standard error, ns = not significant, * p < 0.033, ***p < 0.001. (N = 3).
Changes at gene expression levels were also assessed with quantitative polymerase chain reaction (qPCR) using untreated MDA-MB-231 cells compared with cells treated with Ru-IM (IC50 concentration) over 24 h. While a decrease in gene expression was observed in PDK1 and AKT3, expression levels for PI3K, mTOR, and PTEN were elevated (Figure S1). The discrepancy in gene expression level changes can be attributed to alterations in post-transcriptional regulation. As the PI3K/Akt/mTOR signaling cascade influences a multitude of key cellular processes, numerous levels of regulation are necessary to mediate the expression and activity of the genes of this pathway in response to cellular changes.53 Post-transcriptional factors such as alternative splicing,53 microRNA,54 and long noncoding RNA function,55 and epigenetic changes56 can influence mRNA expression levels. Furthermore, it is known that global correlation of transcript and protein is only moderate.57 Although changes to the mRNA expression level vary, the clear decrease of expressional levels of phosphorylated proteins with Ru-IM treatment indicates inhibition of the PI3K/Akt/mTOR pathway as a potential mechanism of action of this ruthenium compound.
In Vivo Efficacy of the TNBC Xenografted Mouse Model with Ru-IM and Olaparib Monotherapy and Combination Treatments
While our group has previously tested Ru-IM compound in vivo,17 current studies further evaluated the mechanism of action of this compound in a TNBC mouse model to establish a correlation with the findings observed in TNBC cells. Immunosuppressed NOD.CB17-Prkdc SCID/J mice were subcutaneously xenografted with MDA-MB-231 cells and allowed to grow until the tumors were palpable. Parameters of the pilot Ru-IM study were reprised, with a 28-day timeline featuring every other day dosing (5 mg/kg).17 However, as chemotherapy in combination with targeted drugs is widely used in the clinic, Olaparib was also included in the study. Olaparib was recently given FDA approval for the treatment of HER2-negative breast cancer for patients with germline BRCA1 or BRCA2 mutations due to the results of phase III clinical study OlympiAD.58 Olaparib acts as a PARP inhibitor, blocking the repair of single-strand DNA breaks.59 This in turn results in synthetic lethality in BRCA-associated cancer cells, which include certain subtypes of TNBC.59 While the OlympiAD trial did not show changes in overall survival for Olaparib (300 mg bid) vs physician’s choice treatment (PCT, consisting of capecitabine, eribulin, or vinorelbine), progression-free survival rates increased from 4.2 months PCT to 7 months with Olaparib.58 Olaparib with PI3Ka inhibitor alpelisib showed synergistic applications with a 6 month progression-free survival (compared to 0.9 months nontreated).58
More recently, as of March 2022, Olaparib has also been approved by the FDA for adjuvant treatment in high risk early breast cancers with the same conditions of HER2- and germline BRCA1/2 mutations, meaning it can be given to patients sooner than before.59 Due to the clinical efficacy of Olaparib, this PARP inhibitor was used as a comparison therapy (and in combination therapy with Ru-IM) for our in vivo efficacy study. The elimination half-life of Olaparib is 11.7 h,60 which is comparable to that of Ru-IM at 12.67 h.17 While the cytotoxic activity of Olaparib is limited in comparison to Ru-IM in MDA-MB-231 cells, an enhanced profile is detected when used in combination therapy (Table S1). The combination of PARP and PI3K/AKT/mTOR inhibitors has been studied in mouse models,61 as well as in a current phase I clinical trial with Olaparib and AKT inhibitor capivasertib in BRCA1/2-deficient and proficient breast tumors.62
As previous testing determined relevant pharmacokinetic parameters17 and maximum tolerated dose for Ru-IM(17) and Olaparib,63 we were able to proceed with an efficacy study accounting for this information. Four groups of five mice were studied and dosed every other day intraperitoneally (i.p.) with vehicle control (saline solution + PEG/DMSO), Olaparib (50 mg/kg), Ru-IM (5 mg/kg), and Ru-IM + Olaparib (5 mg/kg +50 mg/kg) over 28 days. The drug doses and dosing frequency were also chosen based on previous efficacy studies with Ru-IM and Olaparib on MDA-MB-231 xenografted mice models. Mice were closely monitored for behavioral changes (grooming patterns, aggression, etc.) throughout the study. Mice were also weighed regularly throughout the duration of the study to ensure no abnormal changes were observed (Figure S2). Following completion of the efficacy study, mice were sacrificed on day 29 and several biomarkers were evaluated to determine the translation of in vitro findings into an in vivo setting.
Results from the study are summarized in Figure 4. All drug-treated groups showed tumor growth shrinkage, with 41% tumor decrease for Olaparib-treated mice, 59% decrease for Ru-IM, and 65% decrease for the combination-treated mice. In contrast, a 148% tumor growth was observed for mice treated with the vehicle control, which demonstrates untreated growth of TNBC. Similar to our previously reported results, Ru-IM alone demonstrated more than 55% inhibition in tumor growth.17 The tumor shrinkage observed for mice treated with Olaparib is uncommon for MDA-MB-231 xenografted models, but similar results have been recorded previously in the same cell model.64,65 The combination treatment of Olaparib and Ru-IM demonstrated superior (65%) efficacy, suggesting potential synergistic effects in vivo of the use of a PARP inhibitor with Ru-IM.
Figure 4.
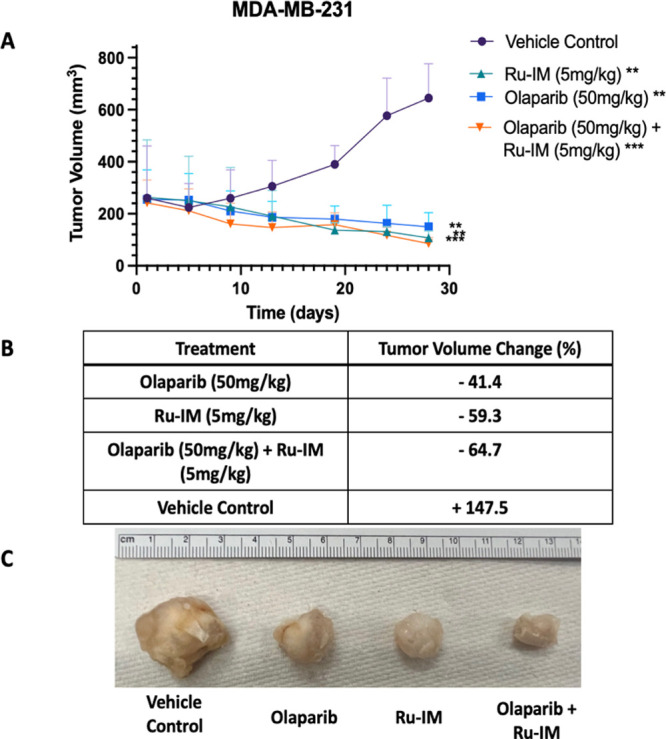
Results of efficacy study testing MDA-MB-231 xenografted mice shows tumor shrinkage associated with Ru-IM therapy. (A) Tumor growth curves for MDA-MB-231 xenografts of 28-day study. Doses were administered every other day. (B) Quantification of percent tumor volume increase or decrease based on treatment type. Data are expressed as the mean ± standard error, **p < 0.002, ***p < 0.001. (C) Ex vivo tumor images at day 28. (N = 5).
Complete Necropsy, Histopathology, and Complete Blood Count Results
To determine the toxicity of Ru-IM to the tested mice, a complete necropsy of each treatment type was completed.17 Forty-eight different sites were evaluated for signs of carcinoma. The results of this study are highlighted in Figure 5 and in Chart S1. Across all treatments, carcinoma was found in the lungs, which is typical for TNBC. Ru-IM monotherapy resulted in signs of carcinoma in 12.5% of sites tested, 20.8% for Olaparib treatment, 31% for the vehicle control, and 10% for the combination of Olaparib and Ru-IM. This shows a decrease in overall cancerous cell invasion and migration for Ru-IM-treated mice, demonstrating a clear translation of the in vitro anticancer properties17,19 of this compound. Very promisingly, the histopathological review shows no evidence of test-article-related toxicity in all submitted tissue samples. This is expected for the Olaparib treatment as previously reported in mouse models59 and given its prevalence in a clinical setting.
Figure 5.

Results of complete necropsy study shows inhibition of cancerous cell migration and invasion. (A) Results of the complete necropsy study evaluating 48 total sites of carcinoma. (B) Hematoxylin and eosin (H&E) staining of key tissues evaluated.
Blood samples were also collected following sacrifice via perfusion. Complete blood count (CBC) panel results are described in Table S3. Blood was collected following a 28 day study via perfusion. Across all treatments, hematocrit values (Hct) that show the composition of red blood cells were considered in the healthy range. Red blood cell count (RBC), hemoglobin (HGB), mean corpuscular volume (MCV), and mean corpuscular hemoglobin (MCH) were well within the reference range for mono- and combination therapies as well as the control group. When delving into values associated with inflammatory markers, which include white blood cell count (WBC#), neutrophil count (NEU#), and lymphocyte count (LYMPH#), a different trend is observed. In this case, mono- and combination treatments continue to be unremarkable, but the vehicle control group shows a marked increase in these markers. This seems to insinuate that an immune response has been observed for the control group but not for treatment groups, suggesting inflammation and metastasis with nontreated mice only.
Immunohistochemical Evaluation of Cell Proliferation, Apoptosis, Angiogenesis, and mTOR/p-mTOR Markers
For further mechanistic analysis of tumor tissue, immunohistochemical (IHC) staining was performed. This immunostaining technique allows for the visualization of antigens in tissue samples.66 An important marker is that of Ki67, a marker of cellular proliferation that is utilized as a prognostic parameter in breast cancer.67 This marker identifies actively dividing cells.67Figure 6A depicts the results of Ki67 immunohistochemical staining in the tumor tissue. As expected, vehicle control tumor tissues were characterized by a high percentage of Ki67+ cells (70.8 ± 8.1.). The percentage was lower in mice treated with Olaparib (51.4 ± 5.3) and even lower in mice treated with Ru-IM alone (28.5 ± 5.7) or with the combination Ru-IM + Olaparib (24.3 ± 2.8) (Figure 6B). This is consistent with the previous report in Olaparib-treated mice with breast68 or ovarian69 cancer. This potentially explains the tumor shrinkage observed with Ru-IM treatment and provides evidence of the inhibition of cancerous growth by this drug.
Figure 6.

Immunohistochemical analysis of cell proliferation, apoptosis, angiogenesis, mTOR, and p-mTOR antigens in tumor tissue of MDA-MB-231 xenografted mice. (A) Immunohistochemical evaluation of cell proliferation marker Ki-67. (B) Quantification of the Ki-67 marker. (C) Immunohistochemical evaluation of cleaved caspase 3 (CC3), CD31, mTOR, and p-mTOR markers. (D) Quantification of CC3, CD31, mTOR, and p-mTOR markers. The signal positive rate was analyzed through the IHC profiler on ImageJ software. Data are expressed as the mean ± standard error ns = not significant; ***p < 0.001.
DAB chromogen was also used to evaluate the markers of cleaved caspase 3 (CC3) (apoptosis), CD31 (angiogenesis), total mTOR, and phosphorylated mTOR (p-mTOR). While we wished to delve into other markers of the PI3K/AKT/mTOR pathway, commercially available antibodies allow for the use of mTOR (both total and phosphorylated) only for the purposes of IHC staining. This chromogen method stains brown for the presence of the tested antigen and remains a purple-blue color for unstained tissue. When observing apoptotic marker CC3 (Figure 6C), tumor tissue showed a signal-positive rate of 60.2% in vehicle control, 78.9% in Olaparib, 86.7% in Ru-IM, and 80.2% in the combinatory Ru-IM + Olaparib-treated tissues (Figure 5D). Ru-IM-treated tissue shows significant activation of this antigen, which fits with what has been observed previously in MDA-MB-231 cells.17,19,68,69 Olaparib and the combinatory group also display significant activation of CC3 (which is to be expected based on previous studies with Olaparib65) but not at such a high level. The evaluation of angiogenic marker CD31 was used to explore antiangiogenic properties of both Olaparib and Ru-IM, in mono- and combination therapies. In this case (Figure 6C), we observe a signal-positive rate of 74.61% in vehicle control, 70.3% in Olaparib, 62.6% in Ru-IM and 49.2% in Ru-IM + Olaparib-treated tissues (Figure 5D). PARP inhibitors have shown some ties to these properties,70 and we have previously described the antiangiogenic role of Ru-IM in human umbilical vein endothelial cells (HUVEC).19 Taken together, we see similar trends of increased apoptosis and inhibition of angiogenesis in both mammalian cell tissue and a xenografted mouse model. This suggests that some of the anticancer properties observed in vitro could be translated in vivo through the evaluation of tumor tissue samples.
As the inhibition of the PI3K/AKT/mTOR pathway has been elucidated as a potential mechanism of action in vitro, we also sought to understand if similar results would be obtained in tumor tissue samples. The evaluation of mTOR in tumor tissues (Figure 6C) led to a signal-positive rate of 64.5% in vehicle control, 65.2% in Olaparib, 62.5% in Ru-IM, and 62.3% in Ru-IM + Olaparib-treated tumor tissue (Figure 6D). The levels of mTOR in treatment groups match that of the endogenous control. When assessing the antigen of p-mTOR, an increase is observed for the vehicle control (71.9%) and a decrease is observed for Ru-IM (51.6%) and Ru-IM + Olaparib (55.63%) treatments. The Olaparib treatment had a similar value to its mTOR counterpart at 64.4%. While the changes between antigen markers compared to the control are significant in the p-mTOR treatment with Ru-IM treatments, the variation between mTOR and p-mTOR expressions does not demonstrate a significant difference. The overall inhibition in expression is 10.9 and 10.7% in Ru-IM and Ru-IM + Olaparib-treated tissues, respectively. Although not a statistically significant differentiation, there is still an inhibition of the active phosphorylated marker. A slight increase in activity is observed for p-mTOR of the vehicle control (7.34%), which is expected given the prevalence of overexpression of this pathway in TNBC.
Conclusions
Metallodrugs have shown preclinical efficacy in TNBC mouse models, but less than a dozen studies have focused on the mechanistic action of these compounds.10,71 In order to translate the potential of these highly efficacious drugs for clinical use, the underlying processes involved in their mode of action must be detailed. The studies described here provide insight into a potential mode of action for Ru-IM in TNBC.
We demonstrate that the previously known accumulation of Ru-IM in mitochondria involves its interaction with this organelle, which, in turn, produces mitochondrial ROS generation and mitochondrial membrane depolarization. It seems clear that mitochondrial dysfunction in TNBC caused by Ru-IM may produce DNA damage and cell death.
A preliminary proteomic study with TNBC cells treated with Ru- IM indicated the inhibition of protein expression levels of M-CSF. M-CSF works upstream of the PI3K/AKT/mTOR pathway, and the Ru-IM-mediated inhibition of this cytokine may indicate the involvement of this pathway. The inhibition of specific cancer hallmarks by Ru-IM such as migration, invasion, angiogenesis, and mitochondrial dysfunction is also indicative of the possible involvement of the PI3K/AKT/mTOR pathway.
Western blots indicated inhibition of protein expression levels of phosphorylated biomarkers along this pathway such as p-PI3K, p-AKT3, p-mTOR, and p-PDK1 markers, while p-PTEN levels remained similar to untreated lysates. Interestingly, little to no change is observed in total protein for PI3K, AKT3, mTOR, and PDK1. This suggests that the protein expression inhibition observed with Ru-IM treatment occurs only for phosphorylated markers, meaning that pathway activation is necessary to observe changes in overall protein expression levels.
Furthermore, an efficacy trial in MDA-MB-231 xenografted mice with Ru-IM, FDA-approved PARP-1 inhibitor Olaparib, and a combination of the two shows impressive tumor reduction (shrinkage) for all groups, especially for those including Ru-IM. The efficacy results for Ru-IM fit very well with those from a preliminary study (2014) indicating high reproducibility. Histopathological analysis demonstrated reduced sites of carcinoma in groups treated with Ru-IM in both mono- and combination therapies and highlighted no systemic toxicity observed in tissue samples. CBC panel results show healthy hematocrit values and signs of inflammation and metastasis only in the nontreated vehicle control group. The inhibition of IHC-stained Ki67+ proliferation, and CD31 angiogenic tumor cells, along with an increase of CC3 apoptotic marker emphasize the translation of in vitro cancer inhibition into an in vivo setting. We also see a translation of inhibition of expression of p-mTOR in tumor tissue, similar to the inhibition observed in Western blots in MDA-MB-231 cells. While this shows promise for the inhibition by Ru-IM of the PI3K/AKT/mTOR pathway, this may just represent a facet of the mechanism of action for the ruthenium drug. Other related pathways and cellular functions affected by Ru-IM such as histone deacetylase or the MAPK pathway may not be discarded at this point.
Taken together, the in vivo efficacy of Ru-IM is quite promising, showing tumor size reduction, no test-article-related toxicity, and no signs of inflammation and metastasis in blood panel results. In addition, we had previously demonstrated preferential accumulation of Ru-IM in the tumor.
Given the morbidity rate of TNBC and its limited treatment options, it is very pertinent to develop more viable and cost-effective options. Ru-IM is extremely potent in TNBC in vitro and, in vivo, has a very attractive pharmacological profile, it is easy and economical to synthesize, and it is water-soluble. Further preclinical efficacy and mechanistic analysis in immunocompetent mice models are warranted for subsequent clinical translation.
Experimental Methods and Materials
Compounds Used
Ru-IM was prepared as described previously.17 Olaparib was purchased from Selleck Chemicals and was used without further purification.
Cell Culture
MDA-MB-231 (HTB-26), a breast adenocarcinoma line derived from metastatic breast mammary gland tissue, was newly obtained for this study from the American Type Culture Collection (ATCC), Manassas, Virginia and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Fisher Scientific, Hampton, NH) with 10% fetal bovine serum (FBS) (Fisher Scientific), 1% minimum essential medium (MEM) nonessential amino acids (NEAA) (Fisher Scientific), and 1% penicillin–streptomycin (PenStrep) (Fisher Scientific). All cells were maintained in a humidified CO2 incubator at 37 °C. PrestoBlue was purchased from Thermo Fisher Scientific (Fair Lawn, NJ).
Reactive Oxygen Species Generation Assay
MDA-MB-231 cells were plated in a 96-well plate with a density of 10,000 cells/well. Following a 24 h seeding time, the cells were washed with Hank’s Balanced Salt Solution (HBSS/Ca/Mg). A DCF-DA solution (100 μM in HBSS/Ca/Mg) was added to the cells, and the cells were incubated at 37 °C at 5% CO2 and 95% air for 30 min. Following incubation, the staining solution was washed off, HBSS was added, and the mixture was further incubated for 30 min. Cells were then treated with negative (no treatment, stained) and positive (1 mM H2O2, known to produce ROS) controls and an IC50 value of Ru-IM (3.7 μM), and kinetic measurements were performed in 1 h increments over 12 h in a Biotek Synergy plate reader at a fluorescence excitation of 485 nm and an emission of 530 nm. All data presented are expressed as the mean ± the standard deviation.
Mitochondrial Membrane Potential Assay
MDA-MB-231 were plated into a 96-well plate with a density of 8000 cells/well. Following a 24 h seeding time, cells were washed twice with PBS. A JC-1 solution (Cayman Chemicals) was then added to the cells, and the cells were incubated for 20 min at 37 °C at 5% CO2 and 95% air. Following incubation, the staining solution was washed off twice with PBS after which compounds at the correct concentrations were added to the wells. This included positive (stained, nontreated) and negative (CCCP-treated) cells. A Biotek Synergy plate reader was used following a 3h incubation at 535 nm for aggregate readings and 595 nm for monomer readings. All data presented are expressed as the mean ± standard deviation.
Cell Painting Assay
To assess morphological profiling of Ru-IM, a cell painting assay was utilized using an osteosarcoma U2-OS cell line. The activity of Ru-IM against the U2-OS cell line was first tested in a 24-h MTT assay (IC50 = 3.7 μM). A dilution series of 8 individual samples (using a 3× dilution) was tested with five cell compartmental dyes consisting of ER, nuclei, mitochondria, RNA, and actin. CellProfiler was used to extract individual morphological features from this data set, with standard data analysis of Python modules (NumPy, Pandas, SciPy, Seaborn) used to aggregate, normalize, and cluster samples into morphological profiles.26,72 This contribution was made through BOLD-100 Therapeutics.
Cell Viability
Cytotoxic profiles (IC50) of all compounds in mono and combination therapies were obtained by measuring the viability of human breast cancer cell MDA-MB-231 with the appropriate culture medium containing the specific compounds (Table S4). Ru-IM was dissolved in H2O, while Olaparib was dissolved in DMSO. Concentrations ranged from 100 to 0.5 μM for all compounds. After 24 h, cell viability was determined by use of the colorimetric cell viability assay PrestoBlue (Thermo Fisher Scientific). IC50 values were fit using GraphPad Prism 7 and the appropriate curve.
Western Blotting
For the assessment of protein expression levels, cells were grown to ∼80% confluency and then dosed with an IC50 concentration of Ru-IM. Lysates were collected following 2 and 24 h incubations and quantified with a Bradford assay. 25 μg of protein was used per sample in sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) at 150 V, followed by a semidry electrotransfer onto a polyvinylidene difluoride (PVDF) membrane for 1 h at 150 mA. Membranes were dried completed and then blocked using Intercept Protein Free Blocking Buffer (LI-COR Biosciences, Lincoln, NE, USA) for 1 h on a shaking platform. Overnight incubation of membranes with appropriate primary antibodies was followed with five membrane washes of TBST before the addition of secondary antibodies. After 1 h incubation, four washes of TBST and one wash of TBS membranes were visualized with 600 and 700 channels on an Odyssey FC Imaging system (LICOR Biosciences).
qPCR Analysis
To determine changes in mRNA expression, cells were grown to ∼80% confluency and then dosed with an IC50 value of Ru-IM. Lysates were collected after 24h, and RNA was extracted from samples using RNeasy Plus Mini Kit with QIAshredder (Qiagen, Hilden, Germany). The integrity of RNA was analyzed by a nanodrop, and it was further purified with sodium acetate when needed. Total RNA was used for cDNA synthesis through SuperScript III First Strand Synthesis System (Thermo Fisher). Real-time-PCR amplification was then carried out with SYBR green on an Applied Biosystems 7500 Real-Time PCR System (ThermoFisher). Reactions were run in technical quadruplicates. Primer sequences are described in Supporting Information. Expression data were normalized to housekeeping gene GAPDH and analyzed using the 2−ΔΔCT method.
Animals and Tumor Models
All animal experiments were performed in accordance with the City University of New York City College and Brooklyn College regulations and by approval of all the responsible authorities (CCNY IACUC Protocol number: 1086, Brooklyn College Protocol Number: 317). Mice were closely monitored for changes in behavior throughout all studies.
In Vivo Efficacy Study of Ru-IM and Olaparib in Mono- and Combination Therapies
NOD.CB17-Prkdc scid/J (nonobese diabetic–severe combined immunodeficiency) female mice aged 8–12 weeks from Jackson Laboratory (Bar Harbor, ME and Sacramento, CA, USA) were used for the xenograft experiments. Mice were injected with 5 × 106 MDA-MB-231 cells per mouse subcutaneously in the right flank suspended 1:1 with Matrigel and phosphate buffer saline (PBS) pH 7.4. Once tumors were palpable, mice were divided into groups (N = 5) based on tumor diameter. The diameter of tumors was measured using an electronic digital caliper, and the tumor volume (TV) was calculated according to the empirical equation TV = (4/3) × π × (L/2) × (L/2) × (D/2), where W is tumor width, L is tumor length, and D is tumor depth. Each group of five MDA-MB-231 transplanted animals received Ru-IM (5 mg per kg per every other day), Olaparib (50 mg per kg per every other day), combination Ru-IM, and Olaparib (5 mg per kg + 50 mg per kg every other day) or vehicle (0.9% NaCl or 10%DMSO/20%PEG/70% saline) intraperitoneally (i.p.). Treatment started when the tumors were palpable (about 6 mm in diameter). Mice were randomized to treatment groups based on their starting tumor burden at 12 weeks of age to ensure equivalent distribution between groups. At the trial end point, the mice were sacrificed, and tumors were measured again after excision and then processed for further analysis.
Histological as well as biochemical evaluations of the blood were conducted. Tumor volumes were graphed for mice compared to vehicle-treated mice based on biweekly external digital caliper measurements. Statistical analysis was determined by one-way ANOVA in GraphPad Prism 7.
Complete Necropsy
Following study completion, mice were sacrificed and two whole bodies per treatment were fixed in formalin with one sagittal cut down the sternum. Mouse samples were given to Memorial Sloan Kettering Cancer Center (MSK-CC) histopathology core for further analysis. Histological analysis was completed with the use of hematoxylin and eosin (H&E) stains looking at 48 different sites of carcinoma. Further H&E imaging was completed at the Center of Discovery and Innovation at CUNY City College.
Immunohistochemistry
Formalin-fixed tissue samples were dehydrated and embedded in paraffin blocks. Five micrometer-sized slices were distributed onto charged microscope slides (Globe Scientific, Mahwah, NJ). Slides were deparaffinized and rehydrated in a series of xylene and ethanol washes. Slides were boiled for 10 min in citrate buffer pH 6.0 for heat-induced antigen retrieval. Slides were then blocked with a 2% milk powder. A 20 min incubation with 10:1 dilution of PGBA buffer (1× phosphate buffer, 1% BSA, 0.002% sodium azide, 0.1% gelatin) and goat serum (Vector laboratories, Newark, CA) was then completed. Cells were incubated at 4C overnight in primary antibodies in PGBA buffer. Slides were washed 3 times in TBS for 5 min/wash and then incubated with secondary antibodies at 1:500 dilution for 2h at RT. Slides were then washed 3 times in TBS for 10 min/wash and then mounted using Prolong gold antifade mounting media with DAPI (ThermoFisher). Imaging was completed using a Zeiss 810 LSM 800. An extra peroxidase step was added to DAB chromogen slides, which were stained using an ImmPACT DAB Substrate Kit (Vector Laboratories). Data represent an average of at least 8 fields per slide.
Antibodies Used
The antibodies used in this study are listed in Table 1.
Table 1. Antibodies Used.
| antibody | company (catalog number) | dilution |
|---|---|---|
| PI3K | Cell Signaling Technology (4249) | 1:1000 |
| p-PI3K | ThermoFisher Scientific (BS-5587R) | 1:1000 |
| PDK1 | Cell Signaling Technology (3062) | 1:1000 |
| p-PDK1 | Cell Signaling Technology (3438) | 1:1000 |
| AKT3 | GeneTex (GTX113312) | 1:1000 |
| p-AKT3 | ThermoFisher Scientific (PA5-12898) | 1:1000 |
| mTOR | Cell Signaling Technology (2983) | 1:1000 |
| p-mTOR | Cell Signaling Technology (2971) | 1:1000 |
| Beta Actin | ThermoFisher Scientific (PA5-16914) | 1:10000 |
| Cleaved Caspase 3 | Cell Signaling Technology (9661) | 1:100 |
| Ki-67 | Abcam (Ab21700) | 1:100 |
| CD31 | R&D Systems (MAB835) | 1:100 |
Acknowledgments
We thank two Brooklyn College group members for synthesizing Ru-IM (Aiman Hafeez and Javier López-Hernández). We gratefully acknowledge Dr. Jorge Morales from CUNY City College of New York (CCNY) for aid with H&E imaging. We thank BOLD-100 Therapeutics for providing the cell painting assay data. We thank Lijie Chen (CCNY) for his invaluable aid in qPCR experiments. The Laboratory of Comparative Pathology from Memorial Sloan Kettering Cancer Center is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.4c00020.
Cell viability assay, CBC panel results, qPCR results, and all sites of histopathological evaluation of in vivo tissue samples (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the National Institute of General Medical Sciences through Grants 2SC1GM127278 (to M.C.) and 1R16GM149396-01 (to M.C. and S.P.), and PSC-CUNY Enhanced Award ENHC-52-14 (to M.C. and K.H.). This work was also supported by NIH/NCI U54CA132378/U54 CA137788 (K.H). N.N. thanks the City University of New York for the Mina Rees Fellowship dissertation grant and the support from the American Cancer Society grant DICRIDG-22-1012253-01. P.C. is supported by NIH/NINDS R35 NS111604 S.P. is supported American Cancer Society (RSG-22-123-01-ET). R.R. thanks the City College of New York for funding from the Graduate Research Training Initiative for Student Enhancement (G-RISE, NIH/NIGMS T32GM136499). We thank the Graduate Research Technology Initiative Fund Round 21 Supplement from CUNY for funds to purchase a plate reader (to M.C.). We are grateful to the Brooklyn College Cancer Center (through the Gray Foundation) for a summer internship (S.K.) as well as the Tow Mentoring Initiative at Brooklyn College (S.K. Fall 2023).
The authors declare the following competing financial interest(s): The authors declare the following financial interests/personal relationships which may be considered as potential competing interests. Maria Contel has patent #US Patent 9555049 issued to Maria Contel, Isabel Marzo, Malgorzata Frik and Benelita T. Elie. Mark Bazett and Brian J Park are employees and/or stock holders of Bold Therapeutics Inc.
Supplementary Material
References
- Bianchini G.; Balko J. M.; Mayer I. A.; Sanders M. E.; Gianni L. Triple-Negative Breast Cancer: Challenges and Opportunities of a Heterogeneous Disease. Nat. Rev. Clin Oncol 2016, 13 (11), 674–690. 10.1038/nrclinonc.2016.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Castro A. C.; Lin N. U.; Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov 2019, 9 (2), 176–198. 10.1158/2159-8290.CD-18-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonotto M.; Gerratana L.; Poletto E.; Driol P.; Giangreco M.; Russo S.; Minisini A. M.; Andreetta C.; Mansutti M.; Pisa F. E.; Fasola G.; Puglisi F. Measures of Outcome in Metastatic Breast Cancer: Insights from a Real-World Scenario. Oncologist 2014, 19 (6), 608–615. 10.1634/theoncologist.2014-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Zhang H.; Merkher Y.; Chen L.; Liu N.; Leonov S.; Chen Y. Recent Advances in Therapeutic Strategies for Triple-Negative Breast Cancer. Journal of Hematology & Oncology 2022, 15 (1), 121. 10.1186/s13045-022-01341-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann B. D.; Bauer J. A.; Chen X.; Sanders M. E.; Chakravarthy A. B.; Shyr Y.; Pietenpol J. A. Identification of Human Triple-Negative Breast Cancer Subtypes and Preclinical Models for Selection of Targeted Therapies. J. Clin Invest 2011, 121 (7), 2750–2767. 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardia A.; Mayer I. A.; Vahdat L. T.; Tolaney S. M.; Isakoff S. J.; Diamond J. R.; O’Shaughnessy J.; Moroose R. L.; Santin A. D.; Abramson V. G.; Shah N. C.; Rugo H. S.; Goldenberg D. M.; Sweidan A. M.; Iannone R.; Washkowitz S.; Sharkey R. M.; Wegener W. A.; Kalinsky K. Sacituzumab Govitecan-Hziy in Refractory Metastatic Triple-Negative Breast Cancer. New England Journal of Medicine 2019, 380 (8), 741–751. 10.1056/NEJMoa1814213. [DOI] [PubMed] [Google Scholar]
- Home | ClinicalTrials.gov. https://clinicaltrials.gov/ (accessed December 1, 2023).
- Gupta G. K.; Collier A. L.; Lee D.; Hoefer R. A.; Zheleva V.; Siewertsz van Reesema L. L.; Tang-Tan A. M.; Guye M. L.; Chang D. Z.; Winston J. S.; Samli B.; Jansen R. J.; Petricoin E. F.; Goetz M. P.; Bear H. D.; Tang A. H. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12 (9), 2392. 10.3390/cancers12092392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam R.; Lam K. W. Recent Progress in Small Molecule Agents for the Targeted Therapy of Triple-Negative Breast Cancer. Eur. J. Med. Chem. 2020, 207, 112812 10.1016/j.ejmech.2020.112812. [DOI] [PubMed] [Google Scholar]
- Nayeem N.; Contel M. Exploring the Potential of Metallodrugs as Chemotherapeutics for Triple Negative Breast Cancer. Chem. - Eur. J. 2021, 27 (35), 8891–8917. 10.1002/chem.202100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S. Cisplatin: The First Metal Based Anticancer Drug. Bioorg Chem. 2019, 88, 102925 10.1016/j.bioorg.2019.102925. [DOI] [PubMed] [Google Scholar]
- Kelland L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7 (8), 573–584. 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- Muhammad N.; Hanif M.; Yang P. Beyond Cisplatin: New Frontiers in Metallodrugs for Hard-to-Treat Triple Negative Breast Cancer. Coord. Chem. Rev. 2024, 499, 215507 10.1016/j.ccr.2023.215507. [DOI] [Google Scholar]
- Leijen S.; Burgers S. A.; Baas P.; Pluim D.; Tibben M.; van Werkhoven E.; Alessio E.; Sava G.; Beijnen J. H.; Schellens J. H. M. Phase I/II Study with Ruthenium Compound NAMI-A and Gemcitabine in Patients with Non-Small Cell Lung Cancer after First Line Therapy. Invest New Drugs 2015, 33 (1), 201–214. 10.1007/s10637-014-0179-1. [DOI] [PubMed] [Google Scholar]
- Bold Therapeutics, Inc. A Phase 1b/2a Dose Escalation Study of BOLD-100 in Combination With FOLFOX Chemotherapy in Patients With Advanced Solid Tumours; Clinical trial registration NCT04421820, 2023; clinicaltrials.gov. https://clinicaltrials.gov/ct2/show/NCT04421820 (accessed April 5, 2023).
- Monro S.; Colón K. L.; Yin H.; Roque J. I.; Konda P.; Gujar S.; Thummel R. P.; Lilge L.; Cameron C. G.; McFarland S. A. Transition Metal Complexes and Photodynamic Therapy from a Tumor-Centered Approach: Challenges, Opportunities, and Highlights from the Development of TLD1433. Chem. Rev. 2019, 119 (2), 797–828. 10.1021/acs.chemrev.8b00211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frik M.; Martínez A.; Elie B. T.; Gonzalo O.; Ramírezde Mingo D.; Sanaú M.; Sánchez-Delgado R.; Sadhukha T.; Prabha S.; Ramos J. W.; Marzo I.; Contel M. In Vitro and in Vivo Evaluation of Water-Soluble Iminophosphorane Ruthenium(II) Compounds. A Potential Chemotherapeutic Agent for Triple Negative Breast Cancer. J. Med. Chem. 2014, 57 (23), 9995–10012. 10.1021/jm5012337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arene ruthenium (II) derivatives containing iminophosphorane ligands and their use in cancer therapy. Google Patents US9555049B2. https://patents.google.com/patent/US9555049B2/en (accessed April 15, 2023).
- Nayeem N.; Yeasmin A.; Cobos S. N.; Younes A.; Hubbard K.; Contel M. Investigation of the Effects and Mechanisms of Anticancer Action of a Ru(II)-Arene Iminophosphorane Compound in Triple Negative Breast Cancer Cells. ChemMedChem. 2021, 16 (21), 3280–3292. 10.1002/cmdc.202100325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miachin K.; Del Solar V.; El Khoury E.; Nayeem N.; Khrystenko A.; Appelt P.; Neary M. C.; Buccella D.; Contel M. Intracellular Localization Studies of Luminescent Analog of an Anticancer Ruthenium Iminophosphorane with High Efficacy in Triple Negative Breast Cancer Mouse Model. Inorg. Chem. 2021, 60 (24), 19152–19164. 10.1021/acs.inorgchem.1c02929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montani M.; Pazmay G. V. B.; Hysi A.; Lupidi G.; Pettinari R.; Gambini V.; Tilio M.; Marchetti F.; Pettinari C.; Ferraro S.; Iezzi M.; Marchini C.; Amici A. The Water Soluble Ruthenium(II) Organometallic Compound [Ru(p-Cymene)(Bis(3,5 Dimethylpyrazol-1-Yl)Methane)Cl]Cl Suppresses Triple Negative Breast Cancer Growth by Inhibiting Tumor Infiltration of Regulatory T Cells. Pharmacol. Res. 2016, 107, 282–290. 10.1016/j.phrs.2016.03.032. [DOI] [PubMed] [Google Scholar]
- Rico S. R. A.; Abbasi A. Z.; Ribeiro G.; Ahmed T.; Wu X. Y.; Silva D. de O. Diruthenium(II,III) Metallodrugs of Ibuprofen and Naproxen Encapsulated in Intravenously Injectable Polymer–Lipid Nanoparticles Exhibit Enhanced Activity against Breast and Prostate Cancer Cells. Nanoscale 2017, 9 (30), 10701–10714. 10.1039/C7NR01582H. [DOI] [PubMed] [Google Scholar]
- Côrte-Real L.; Brás A. R.; Pilon A.; Mendes N.; Ribeiro A. S.; Martins T. D.; Farinha J. P. S.; Oliveira M. C.; Gärtner F.; Garcia M. H.; Preto A.; Valente A. Biotinylated Polymer-Ruthenium Conjugates: In Vitro and In Vivo Studies in a Triple-Negative Breast Cancer Model. Pharmaceutics 2022, 14 (7), 1388. 10.3390/pharmaceutics14071388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.-C.; Lu J.-J.; Jiang N.; Ma X.-R.; Li R.-T.; Ye R.-R. Synthesis, Characterization and Antitumor Mechanism Investigation of Ruthenium(II) Polypyridyl Complexes with Artesunate Moiety. J. Biol. Inorg. Chem. 2021, 26 (8), 909–918. 10.1007/s00775-021-01901-8. [DOI] [PubMed] [Google Scholar]
- Silvestri S.; Cirilli I.; Marcheggiani F.; Dludla P.; Lupidi G.; Pettinari R.; Marchetti F.; Di Nicola C.; Falcioni G.; Marchini C.; Orlando P.; Tiano L.; Amici A. Evaluation of Anticancer Role of a Novel Ruthenium(II)-Based Compound Compared with NAMI-A and Cisplatin in Impairing Mitochondrial Functionality and Promoting Oxidative Stress in Triple Negative Breast Cancer Models. Mitochondrion 2021, 56, 25–34. 10.1016/j.mito.2020.11.004. [DOI] [PubMed] [Google Scholar]
- Bray M.-A.; Singh S.; Han H.; Davis C. T.; Borgeson B.; Hartland C.; Kost-Alimova M.; Gustafsdottir S. M.; Gibson C. C.; Carpenter A. E. Cell Painting, a High-Content Image-Based Assay for Morphological Profiling Using Multiplexed Fluorescent Dyes. Nat. Protoc 2016, 11 (9), 1757–1774. 10.1038/nprot.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.-H.; Oh S.-J.; Yun J.; Shin O. S. Nonstructural Protein NS1 of Influenza Virus Disrupts Mitochondrial Dynamics and Enhances Mitophagy via ULK1 and BNIP3. Viruses 2021, 13 (9), 1845. 10.3390/v13091845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snezhkina A. V.; Kudryavtseva A. V.; Kardymon O. L.; Savvateeva M. V.; Melnikova N. V.; Krasnov G. S.; Dmitriev A. A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxid. Med. Cell. Longevity 2019, 2019, 6175804 10.1155/2019/6175804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy M. P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417 (1), 1–13. 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbaghi G.; Groleau M.-C.; Santos Y. L. de L.; Doucet N.; Déziel E.; Castonguay A. Cationic Ru II Cyclopentadienyl Complexes with Antifungal Activity against Several Candida Species. ChemBioChem. 2020, 21 (21), 3112. 10.1002/cbic.202000254. [DOI] [PubMed] [Google Scholar]
- Bakewell S.; Conde I.; Fallah Y.; McCoy M.; Jin L.; Shajahan-Haq A. N. Inhibition of DNA Repair Pathways and Induction of ROS Are Potential Mechanisms of Action of the Small Molecule Inhibitor BOLD-100 in Breast Cancer. Cancers (Basel) 2020, 12 (9), 2647. 10.3390/cancers12092647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perillo B.; Di Donato M.; Pezone A.; Di Zazzo E.; Giovannelli P.; Galasso G.; Castoria G.; Migliaccio A. ROS in Cancer Therapy: The Bright Side of the Moon. Exp Mol. Med. 2020, 52 (2), 192–203. 10.1038/s12276-020-0384-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keszler A.; Zhang Y.; Hogg N. The Reaction between Nitric Oxide, Glutathione and Oxygen in the Presence and Absence of Protein: How Are S-Nitrosothiols Formed?. Free Radic Biol. Med. 2010, 48 (1), 55–64. 10.1016/j.freeradbiomed.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezeriṇa D.; Takano Y.; Hanaoka K.; Urano Y.; Dick T. P. N-Acetyl Cysteine Functions as a Fast-Acting Antioxidant by Triggering Intracellular H2S and Sulfane Sulfur Production. Cell Chem. Biol. 2018, 25 (4), 447–459.e4. 10.1016/j.chembiol.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F.; Romero-Canelón I.; Habtemariam A.; Song J.-I.; Banerjee S.; Clarkson G. J.; Song L.; Prokes I.; Sadler P. J. Effect of Cysteine Thiols on the Catalytic and Anticancer Activity of Ru(II) Sulfonyl-Ethylenediamine Complexes. Dalton Trans 2022, 51 (11), 4447–4457. 10.1039/D1DT03856G. [DOI] [PubMed] [Google Scholar]
- Chen L.; Li G.; Peng F.; Jie X.; Dongye G.; Cai K.; Feng R.; Li B.; Zeng Q.; Lun K.; Chen J.; Xu B. The Induction of Autophagy against Mitochondria-Mediated Apoptosis in Lung Cancer Cells by a Ruthenium (II) Imidazole Complex. Oncotarget 2016, 7 (49), 80716–80734. 10.18632/oncotarget.13032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorova L. D.; Popkov V. A.; Plotnikov E. Y.; Silachev D. N.; Pevzner I. B.; Jankauskas S. S.; Babenko V. A.; Zorov S. D.; Balakireva A. V.; Juhaszova M.; Sollott S. J.; Zorov D. B. Mitochondrial Membrane Potential. Anal. Biochem. 2018, 552, 50–59. 10.1016/j.ab.2017.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivandzade F.; Bhalerao A.; Cucullo L. Analysis of the Mitochondrial Membrane Potential Using the Cationic JC-1 Dye as a Sensitive Fluorescent Probe. Bio Protoc 2019, 9 (1), e3128 10.21769/BioProtoc.3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koncha R. R.; Ramachandran G.; Sepuri N. B. V.; Ramaiah K. V. A. CCCP-Induced Mitochondrial Dysfunction - Characterization and Analysis of Integrated Stress Response to Cellular Signaling and Homeostasis. FEBS J. 2021, 288 (19), 5737–5754. 10.1111/febs.15868. [DOI] [PubMed] [Google Scholar]
- Tang H.; Guo X.; Yu W.; Gao J.; Zhu X.; Huang Z.; Ou W.; Zhang H.; Chen L.; Chen J. Ruthenium(II) Complexes as Mitochondrial Inhibitors of Topoisomerase Induced A549 Cell Apoptosis. Journal of Inorganic Biochemistry 2023, 246, 112295 10.1016/j.jinorgbio.2023.112295. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wu Q.; Yu G.; Li L.; Zhao X.; Huang X.; Mei W. Polypyridyl Ruthenium(II) Complex-Induced Mitochondrial Membrane Potential Dissipation Activates DNA Damage-Mediated Apoptosis to Inhibit Liver Cancer. Eur. J. Med. Chem. 2019, 164, 282–291. 10.1016/j.ejmech.2018.12.041. [DOI] [PubMed] [Google Scholar]
- Mun S. H.; Park P. S. U.; Park-Min K.-H. The M-CSF Receptor in Osteoclasts and Beyond. Exp Mol. Med. 2020, 52 (8), 1239–1254. 10.1038/s12276-020-0484-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porta C.; Paglino C.; Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014, 4, 64. 10.3389/fonc.2014.00064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massihnia D.; Galvano A.; Fanale D.; Perez A.; Castiglia M.; Incorvaia L.; Listì A.; Rizzo S.; Cicero G.; Bazan V.; Castorina S.; Russo A. Triple Negative Breast Cancer: Shedding Light onto the Role of Pi3k/Akt/Mtor Pathway. Oncotarget 2016, 7 (37), 60712–60722. 10.18632/oncotarget.10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Killock D. AKT Inhibition Improves OS in TNBC. Nat. Rev. Clin Oncol 2020, 17 (3), 135–135. 10.1038/s41571-019-0322-1. [DOI] [PubMed] [Google Scholar]
- Liu R.; Chen Y.; Liu G.; Li C.; Song Y.; Cao Z.; Li W.; Hu J.; Lu C.; Liu Y. PI3K/AKT Pathway as a Key Link Modulates the Multidrug Resistance of Cancers. Cell Death Dis 2020, 11 (9), 1–12. 10.1038/s41419-020-02998-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X.; Shu R.; Yu C.; Fu Z.; Li Z. Mammalian AKT, the Emerging Roles on Mitochondrial Function in Diseases. Aging Dis 2022, 13 (1), 157–174. 10.14336/AD.2021.0729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M.; Wang J.; Wang W.; Liu J.; Wong C.-W. Phosphatidylinositol 3-Kinase Affects Mitochondrial Function in Part through Inducing Peroxisome Proliferator-Activated Receptor γ Coactivator-1β Expression. Br. J. Pharmacol. 2011, 162 (4), 1000–1008. 10.1111/j.1476-5381.2010.01105.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz López K. G.; Toledo Guzmán M. E.; Sánchez E. O.; García Carrancá A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. 10.3389/fonc.2019.01373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin Y. R.; Yoshida T.; Marusyk A.; Beck A. H.; Polyak K.; Toker A. TARGETING AKT3 SIGNALING IN TRIPLE NEGATIVE BREAST CANCER. Cancer Res. 2014, 74 (3), 964–973. 10.1158/0008-5472.CAN-13-2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J.; Wang X.; Proud C. G. mTOR Inhibitors in Cancer Therapy. F1000Res. 2016, 5, F1000. 10.12688/f1000research.9207.1. [DOI] [PMC free article] [PubMed] [Google Scholar]; Faculty Rev-2078.
- Jovanović B.; Mayer I. A.; Mayer E. L.; Abramson V. G.; Bardia A.; Sanders M. E.; Kuba M. G.; Estrada M. V.; Beeler J. S.; Shaver T. M.; Johnson K. C.; Sanchez V.; Rosenbluth J. M.; Dillon P. M.; Forero-Torres A.; Chang J. C.; Meszoely I. M.; Grau A. M.; Lehmann B. D.; Shyr Y.; Sheng Q.; Chen S.-C.; Arteaga C. L.; Pietenpol J. A. A Randomized Phase II Neoadjuvant Study of Cisplatin, Paclitaxel With or Without Everolimus in Patients with Stage II/III Triple-Negative Breast Cancer (TNBC): Responses and Long-Term Outcome Correlated with Increased Frequency of DNA Damage Response Gene Mutations, TNBC Subtype, AR Status, and Ki67. Clin. Cancer Res. 2017, 23 (15), 4035–4045. 10.1158/1078-0432.CCR-16-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewer M.; Findlay S. D.; Postovit L.-M. Post-Transcriptional Regulation in Cancer Progression. J. Cell Commun. Signal 2012, 6 (4), 233–248. 10.1007/s12079-012-0179-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M.; Zhu S.; Xiong S.; Xue X.; Zhou X. MicroRNAs and the PTEN/PI3K/Akt Pathway in Gastric Cancer (Review). Oncol. Rep. 2019, 41 (3), 1439–1454. 10.3892/or.2019.6962. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Zhang Y.; Qin X.; Geng H.; Zuo D.; Zhao Q. PI3K/AKT/mTOR Pathway-Related Long Non-Coding RNAs: Roles and Mechanisms in Hepatocellular Carcinoma. Pharmacol. Res. 2020, 160, 105195 10.1016/j.phrs.2020.105195. [DOI] [PubMed] [Google Scholar]
- Yang Q.; Jiang W.; Hou P. Emerging Role of PI3K/AKT in Tumor-Related Epigenetic Regulation. Seminars in Cancer Biology 2019, 59, 112–124. 10.1016/j.semcancer.2019.04.001. [DOI] [PubMed] [Google Scholar]
- Schwanhäusser B.; Busse D.; Li N.; Dittmar G.; Schuchhardt J.; Wolf J.; Chen W.; Selbach M. Global Quantification of Mammalian Gene Expression Control. Nature 2011, 473 (7347), 337–342. 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Robson M. E.; Tung N.; Conte P.; Im S.-A.; Senkus E.; Xu B.; Masuda N.; Delaloge S.; Li W.; Armstrong A.; Wu W.; Goessl C.; Runswick S.; Domchek S. M. OlympiAD Final Overall Survival and Tolerability Results: Olaparib versus Chemotherapy Treatment of Physician’s Choice in Patients with a Germline BRCA Mutation and HER2-Negative Metastatic Breast Cancer. Ann. Oncol 2019, 30 (4), 558–566. 10.1093/annonc/mdz012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikesdal H. P.; Yndestad S.; Elzawahry A.; Llop-Guevara A.; Gilje B.; Blix E. S.; Espelid H.; Lundgren S.; Geisler J.; Vagstad G.; Venizelos A.; Minsaas L.; Leirvaag B.; Gudlaugsson E. G.; Vintermyr O. K.; Aase H. S.; Aas T.; Balmaña J.; Serra V.; Janssen E. A. M.; Knappskog S.; Lo̷nning P. E. Olaparib Monotherapy as Primary Treatment in Unselected Triple Negative Breast Cancer. Ann. Oncol 2021, 32 (2), 240–249. 10.1016/j.annonc.2020.11.009. [DOI] [PubMed] [Google Scholar]
- Chase D. M.; Patel S.; Shields K. Profile of Olaparib in the Treatment of Advanced Ovarian Cancer. Int. J. Womens Health 2016, 8, 125–129. 10.2147/IJWH.S55906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; He G.; Li H.; Ge Y.; Wang S.; Xu Y.; Zhu Q. Discovery of Novel PARP/PI3K Dual Inhibitors with High Efficiency against BRCA-Proficient Triple Negative Breast Cancer. Eur. J. Med. Chem. 2021, 213, 113054 10.1016/j.ejmech.2020.113054. [DOI] [PubMed] [Google Scholar]
- Schmid P.; Abraham J.; Chan S.; Wheatley D.; Brunt A. M.; Nemsadze G.; Baird R. D.; Park Y. H.; Hall P. S.; Perren T.; Stein R. C.; Mangel L.; Ferrero J.-M.; Phillips M.; Conibear J.; Cortes J.; Foxley A.; de Bruin E. C.; McEwen R.; Stetson D.; Dougherty B.; Sarker S.-J.; Prendergast A.; McLaughlin-Callan M.; Burgess M.; Lawrence C.; Cartwright H.; Mousa K.; Turner N. C. Capivasertib Plus Paclitaxel Versus Placebo Plus Paclitaxel As First-Line Therapy for Metastatic Triple-Negative Breast Cancer: The PAKT Trial. J. Clin Oncol 2020, 38 (5), 423–433. 10.1200/JCO.19.00368. [DOI] [PubMed] [Google Scholar]
- Sun K.; Mikule K.; Wang Z.; Poon G.; Vaidyanathan A.; Smith G.; Zhang Z.-Y.; Hanke J.; Ramaswamy S.; Wang J. A Comparative Pharmacokinetic Study of PARP Inhibitors Demonstrates Favorable Properties for Niraparib Efficacy in Preclinical Tumor Models. Oncotarget 2018, 9 (98), 37080–37096. 10.18632/oncotarget.26354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siraj A. K.; Pratheeshkumar P.; Parvathareddy S. K.; Divya S. P.; Al-Dayel F.; Tulbah A.; Ajarim D.; Al-Kuraya K. S. Overexpression of PARP Is an Independent Prognostic Marker for Poor Survival in Middle Eastern Breast Cancer and Its Inhibition Can Be Enhanced with Embelin Co-Treatment. Oncotarget 2018, 9 (99), 37319–37332. 10.18632/oncotarget.26470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min A.; Im S.-A.; Kim D. K.; Song S.-H.; Kim H.-J.; Lee K.-H.; Kim T.-Y.; Han S.-W.; Oh D.-Y.; Kim T.-Y.; O’Connor M. J.; Bang Y.-J. Histone Deacetylase Inhibitor, Suberoylanilide Hydroxamic Acid (SAHA), Enhances Anti-Tumor Effects of the Poly (ADP-Ribose) Polymerase (PARP) Inhibitor Olaparib in Triple-Negative Breast Cancer Cells. Breast Cancer Research 2015, 17 (1), 33. 10.1186/s13058-015-0534-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magaki S.; Hojat S. A.; Wei B.; So A.; Yong W. H. An Introduction to the Performance of Immunohistochemistry. Methods Mol. Biol. 2019, 1897, 289–298. 10.1007/978-1-4939-8935-5_25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey M. G.; Hynes S. O.; Kerin M. J.; Miller N.; Lowery A. J. Ki-67 as a Prognostic Biomarker in Invasive Breast Cancer. Cancers (Basel) 2021, 13 (17), 4455. 10.3390/cancers13174455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X.; Chen L.; Huang B.; Li X.; Yang L.; Hu X.; Jiang Y.; Shao Z.; Wang Z. Efficacy and Mechanism of the Combination of PARP and CDK4/6 Inhibitors in the Treatment of Triple-Negative Breast Cancer. Journal of Experimental & Clinical Cancer Research 2021, 40 (1), 122. 10.1186/s13046-021-01930-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A. J.; Stubbs M.; Liu P.; Ruggeri B.; Khabele D. The BET Inhibitor INCB054329 Reduces Homologous Recombination Efficiency and Augments PARP Inhibitor Activity in Ovarian Cancer. Gynecol Oncol 2018, 149 (3), 575–584. 10.1016/j.ygyno.2018.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barchiesi G.; Roberto M.; Verrico M.; Vici P.; Tomao S.; Tomao F. Emerging Role of PARP Inhibitors in Metastatic Triple Negative Breast Cancer. Current Scenario and Future Perspectives. Front. Oncol 2021, 11, 769280 10.3389/fonc.2021.769280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Zhao W.; Jin C.; Xiong H. Dual–Target Platinum(IV) Complexes Reverse Cisplatin Resistance in Triple Negative Breast via Inhibiting Poly(ADP–Ribose) Polymerase (PARP–1) and Enhancing DNA Damage. Bioorganic Chemistry 2023, 133, 106354 10.1016/j.bioorg.2023.106354. [DOI] [PubMed] [Google Scholar]
- Cimini B. A.; Chandrasekaran S. N.; Kost-Alimova M.; Miller L.; Goodale A.; Fritchman B.; Byrne P.; Garg S.; Jamali N.; Logan D. J.; Concannon J. B.; Lardeau C.-H.; Mouchet E.; Singh S.; Shafqat Abbasi H.; Aspesi P.; Boyd J. D.; Gilbert T.; Gnutt D.; Hariharan S.; Hernandez D.; Hormel G.; Juhani K.; Melanson M.; Mervin L. H.; Monteverde T.; Pilling J. E.; Skepner A.; Swalley S. E.; Vrcic A.; Weisbart E.; Williams G.; Yu S.; Zapiec B.; Carpenter A. E. Optimizing the Cell Painting Assay for Image-Based Profiling. Nat. Protoc 2023, 18 (7), 1981–2013. 10.1038/s41596-023-00840-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.