

Abstract
The majority of low‐grade isocitrate dehydrogenase‐mutant (IDHmt) gliomas undergo malignant progression (MP), but their underlying mechanism remains unclear. IDHmt gliomas exhibit global DNA methylation, and our previous report suggested that MP could be partly attributed to passive demethylation caused by accelerated cell cycles. However, during MP, there is also active demethylation mediated by ten‐eleven translocation, such as DNA hydroxymethylation. Hydroxymethylation is reported to potentially contribute to gene expression regulation, but its role in MP remains under investigation. Therefore, we conducted a comprehensive analysis of hydroxymethylation during MP of IDHmt astrocytoma. Five primary/malignantly progressed IDHmt astrocytoma pairs were analyzed with oxidative bisulfite and the Infinium EPIC methylation array, detecting 5‐hydroxymethyl cytosine at over 850,000 locations for region‐specific hydroxymethylation assessment. Notably, we observed significant sharing of hydroxymethylated genomic regions during MP across the samples. Hydroxymethylated CpGs were enriched in open sea and intergenic regions (p < 0.001), and genes undergoing hydroxymethylation were significantly associated with cancer‐related signaling pathways. RNA sequencing data integration identified 91 genes with significant positive/negative hydroxymethylation‐expression correlations. Functional analysis suggested that positively correlated genes are involved in cell‐cycle promotion, while negatively correlated ones are associated with antineoplastic functions. Analyses of The Cancer Genome Atlas clinical data on glioma were in line with these findings. Motif‐enrichment analysis suggested the potential involvement of the transcription factor KLF4 in hydroxymethylation‐based gene regulation. Our findings shed light on the significance of region‐specific DNA hydroxymethylation in glioma MP and suggest its potential role in cancer‐related gene expression and IDHmt glioma malignancy.
Keywords: 5‐hydroxymethyl cytosine (5hmC), glioma, hydroxymethylation, isocitrate dehydrogenase (IDH), malignant progression
The current study observed region‐specific DNA hydroxymethylation along the malignant progression (MP) of isocitrate dehydrogenase (IDH)‐mutant gliomas, which significantly correlated with the expression of cancer‐related genes. The mechanism linking hydroxymethylation to gene expression remains to be elucidated, yet the potential involvement of the transcription factor KLF4 was indicated. Our findings shed light on the significance of region‐specific DNA hydroxymethylation in glioma MP and suggest its potential role in cancer‐related gene expression and IDH‐mutant glioma malignancy.
Abbreviations
- 2HG
2‐hydroxyglutaric acid
- 5caC
5‐carboxylcytosine
- 5hmC
5‐hydroxymethylcytosine
- 5mC
5‐methylcytosine
- CoHMCs
common hydroxymethylated CpGs
- FPKM
fragments per kilobase million
- GSEA
Gene Set Enrichment Analysis
- HGGs
high‐grade gliomas
- IDH
isocitrate dehydrogenase
- IDHmt
IDH‐mutant
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- LGGs
low‐grade gliomas
- MP
malignant progression
- OxBS
oxidative bisulfite
- RNAseq
RNA sequence
- TCGA
The Cancer Genome Atlas
- TET
ten‐eleven translocation
- TSS
transcription start site
- WHO
World Health Organization
1. INTRODUCTION
Gliomas are classified by the WHO as Grades 1–4, with varying prognoses depending on the grade. During the natural course or treatment of LGGs, MP causes the tumor to become more aggressive, leading to its transformation into high‐grade tumors. This transformation leads to a poor patient prognosis, and is commonly observed in IDHmt diffuse gliomas, such as astrocytoma and oligodendroglioma. 1 Although CDKN2A/B homozygous deletion is strongly correlated with the malignancy of IDHmt astrocytoma and is a diagnostic criterion in the WHO2021 classification, the mechanism of MP is still not fully understood. 2 , 3
Wild‐type IDH produces α‐ketoglutaric acid, whereas mutant IDH in glioma cells produces 2HG. 2HG inhibits TET, an enzyme essential for TET‐mediated DNA demethylation, which converts 5mC to 5hmC through oxidation (Figure S1). 4 , 5 , 6 The conversion from 5mC to 5hmC, termed hydroxymethylation, generates 5hmC, which serves as a pivotal intermediate in DNA demethylation. Recently, 5hmC has been identified as a critical epigenetic mark in various cell functions. 6 , 7 , 8 Further oxidation generates 5‐formylcytosine and 5caC, leading to demethylation. 9 , 10 Inhibition of this process by 2HG causes tumor DNA hypermethylation, increasing 5mC and reducing 5hmC. IDHmt gliomas exhibit a widespread abnormal CpG hypermethylation, known as the “glioma with CpG island methylator phenotype.” 11 , 12
During MP, hypermethylated DNA undergoes selective demethylation in specific regions, affecting the expression of genes involved in cell growth. 13 Although some studies suggest that demethylation is a possible cause of MP, 14 , 15 , 16 our previous study suggested that the demethylation was mostly the consequence of MP, because it was mainly observed in late‐replicating regions and was thought to be caused by passive demethylation due to a delay in methylation maintenance during accelerated cell division. 13 Indeed, DNA demethylation associated with cell division in late‐replicating regions has also been reported in other cancer types. 16 Conversely, TET‐mediated active demethylation, such as hydroxymethylation, has been reported in certain genomic regions among high‐grade IDHmt gliomas and has potentially been suggested to influence gene expression. 17 These backgrounds led us to hypothesize that hydroxymethylation in specific genomic regions could trigger MP.
In the present study, we aimed to investigate the role of DNA hydroxymethylation in glioma MP by quantitatively analyzing genome‐wide 5hmC levels in primary and malignantly progressed IDHmt astrocytomas.
2. MATERIALS AND METHODS
2.1. Patient and ethical guideline
We collected frozen tumor samples from glioma patients who had undergone tumorectomy at the University of Tokyo Hospital between 2001 and 2010. In this study, based on WHO2021 classification, 18 we considered MP/transformation as the transition from IDHmt astrocytoma (WHO Grade 2) to IDHmt astrocytoma (WHO Grade 3 or 4). We selected five patients (10 samples) who met the criteria of initial Grade 2 diagnosis and recurrence as Grade 3 or 4 astrocytoma (indicating MP), IDHmt astrocytic glioma, and sufficient DNA (≥2 μg) for OxBS. Patient breakdown including molecular characteristics is shown in Table 1. IDH1/2 mutation presence was confirmed by Sanger sequencing using the primers listed in Table S1. The copy number information was obtained by analyzing the methylation array data using the tool run by the German Cancer Research Center (https://www.molecularneuropathology.org/mnp/).
TABLE 1.
Included patients.
| Patient ID | Sample | Age at onset | Sex | Pathological diagnosis (WHO 2016) | Pathological diagnosis (WHO 2021) | Post operative treatment on initial onset | IDH1/2_status | 1p/19q_codel | CDKN2A/B | CDK4 | Tumor purity |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HM1 | HM1_LGG | 34 | M | DA | Astrocytoma, IDH‐mutant, CNS WHO G2 | 60Gy_Radiation | IDH1_R132H | None | Intact | Intact | 0.66 |
| HM1_HGG | 43 | M | sGBM | Astrocytoma, IDH‐mutant, CNS WHO G4 | Homozygous deletion | Amplified | 0.76 | ||||
| HM2 | HM2_LGG | 39 | M | DA | Astrocytoma, IDH‐mutant, CNS WHO G2 | None | IDH1_R132H | None | Intact | Intact | 0.76 |
| HM2_HGG | 40 | M | sGBM | Astrocytoma, IDH‐mutant, CNS WHO G4 | Intact | Amplified | 0.68 | ||||
| HM3 | HM3_LGG | 36 | F | DA | Astrocytoma, IDH‐mutant, CNS WHO G2 | None | IDH1_R132H | None | Intact | Intact | 0.84 |
| HM3_HGG | 38 | F | AA | Astrocytoma, IDH‐mutant, CNS WHO G3 | Heterozygous deletion | Intact | 0.88 | ||||
| HM4 | HM4_LGG | 56 | M | DA | Astrocytoma, IDH‐mutant, CNS WHO G2 | None | IDH1_R132H | None | Intact | Intact | 0.80 |
| HM4_HGG | 57 | M | AA | Astrocytoma, IDH‐mutant, CNS WHO G4 | Homozygous deletion | Intact | 0.81 | ||||
| HM5 | HM5_LGG | 30 | M | DA | Astrocytoma, IDH‐mutant, CNS WHO G2 | None | IDH1_R132H | None | Intact | Intact | 0.82 |
| HM5_HGG | 37 | M | AA | Astrocytoma, IDH‐mutant, CNS WHO G4 | Homozygous deletion | Intact | >0.9 |
Note: LGG: Before malignant transformation, HGG: After malignant transformation.
Abbreviations: 1p/19q_codel, chromosomal 1p/19q co‐deletion; AA, anaplastic astrocytoma; DA, diffuse astrocytoma; HGG, high‐grade glioma; IDH, isocitrate dehydrogenase; LGG, low‐grade glioma; sGBM, secondary glioblastoma; WHO, World Health Organization.
2.2. Extraction of DNA/RNA and OxBS process
DNA and RNA were extracted from frozen tumor samples using the QIAamp DNA Mini Kit (Qiagen, Germany) and RNeasy Mini kit (Qiagen, Germany), respectively. RNA quality with RNA integrity number values of >7 were selected for next‐generation sequencing. For the OxBS process, the TrueMethyl Array Kit (Cambridge Epigenetix, UK) was used.
2.3. Methylation array, RNA sequence, and tumor purity
Methylation arrays were performed on 250 ng of OxBS‐processed and BS‐processed DNAs using the Infinium Methylation EPIC BeadChip (Illumina, San Diego, USA).
For the RNAseq, we prepared the library using the TrueSeq Stranded mRNA LT Sample Prep Kit (Illumina, San Diego, USA) and sequenced it using a HiSeq 2000 instrument (Illumina, San Diego, USA) as 100‐bp paired‐end reads. Using the Burrows–Wheeler Aligner, resulting FASTQ sequences were aligned to both GRCh37/hg19 and ucsc_gene hg19 reference. 19 The alignment results were then integrated, selecting the hits with higher mapping scores for each mapping (genome, transcript). Nonmapped reads underwent local realignment using the Smith‐Waterman algorithm, resulting in the Binary Alignment/Map file for each sample. The gene expression levels were quantified in FPKM using Cufflinks. 20 The FPKM data were used for the gene expression analysis throughout the study. The tumor purity was detected using integrated genotyper software (karkinos, http://github.com/genome‐rcast/karkinos) from the sequenced data.
2.4. Data process of methylation array
The β‐value of each CpG probe was calculated using the software GenomeStudio (Illumina, San Diego, USA). 21 Among the 865,918 probe data obtained, the rs/ch probes, and those near single nucleotide polymorphisms were removed. Probes with missing β‐values were also excluded, resulting in a total of 776,021 probes for analysis. Probes belonging to TSS200, TSS1500, and 5′UTR regions were treated as promoter probes based on the Illumina manifest. 21 The BS/OxBS method calculated two β‐values per probe, which were as follows: value A (total methylation; 5mC + 5hmC) from the BS‐processed DNA and value B (5mC) from the OxBS‐processed DNA. Value C (5hmC) was calculated as C = A − B. For probes where both A and B had the minimum value, C tended to be negative due to an error, and C = 0.0000001 was substituted for C < 0. These methods were used in several previous reports. 17 , 22 , 23
Our method, hm‐ratio, is a numerical value of 0 to 1 calculated from the β‐value, and is calculated as follows: hm‐ratio = C/A. However, when A is a minimum value, the hm‐ratio value is mathematically unreliable. This issue was resolved by excluding probes with an A value of ≤−1.5 SD (standard deviation) from the analysis across all samples; this was done after calculating the mean of A values of all probes in each sample and computing the SD using the Excel STDEV.P formula. Δhm‐ratio was calculated by subtracting the hm‐ratio before and after MP for each probe in each patient. Probes with a Δhm‐ratio >0 indicated increased hm‐ratio, suggesting the presence of hydroxymethylation. The EPIC probe, targeting 850,000 CpGs, included intergenic regions. The “closest” function of bedtools v2.29.0 24 was used to annotate probes in the intergenic regions with the closest gene.
2.5. ChIP data of H3K27ac
In the present study, we utilized H3K27ac modification as a reference for genetic regulatory regions. We analyzed external H3K27ac ChIP data, MGH15m, which is an IDHmt glioma (astrocytoma Grade 3) reported by the Massachusetts General Hospital (GSM1824808). 25
2.6. siRNA and real‐time PCR
U87 cells were seeded in six‐well culture dishes with DMEM (#11594486, Gibco, Waltham, USA) supplemented with 10% fetal bovine serum and incubated at 37°C in a 5% CO2 atmosphere. Upon reaching 60% confluency, they were transfected with KLF4 siRNA (siKLF4_A: #s17793, siKLF4_B: #s17795, Ambion, Waltham, USA) using lipofectamine 2000 (Thermo Fisher, Waltham, USA) as per the manufacturer's instructions. After 48 h, total RNA was isolated using TRIzol (Invitrogen, Carlsbad, USA). Then, RNA was reverse‐transcribed using Superscript III (Invitrogen) following the manufacturer's protocol, and the obtained cDNAs were used for PCR. Real‐time PCR was performed with the SYBR Green kit to analyze the KLF4, SOX5, PLEKHA5, FRS2, and ACTB mRNA levels. The relative expression levels compared with that of control ACTB were calculated using CFX Maestro (Bio‐Rad Laboratories, Hercules, USA). The real‐time PCR primers are listed in Table S1.
2.7. Statistical analysis
R‐3.5.0 26 and JMP Pro 13.2.0 (SAS Institute, Cary, USA) were used for various statistical analyses. In all tests, p < 0.05 was considered statistically significant, unless otherwise stated. The q‐value is the Benjamini–Hochberg adjusted p‐value.
3. RESULTS
3.1. Analysis using hm‐ratio identified widespread hydroxymethylation during the MP of astrocytoma
In the 5hmC analysis using the methylation array, the relationship between demethylation and 5hmC was conventionally assessed based on the absolute β‐value changes of 5hmC. 17 , 22 , 23 However, in the present study, we utilized the change of the hm‐ratio (5hmC/(5mC + 5hmC)) to improve the accuracy of detecting hydroxymethylated regions. These ratio‐based methods have been reported recently and allow the detection of 5hmC produced from 5mC during the process of TET‐mediated demethylation, even when the absolute amounts of 5mC and 5hmC decrease during MP. 7 , 8 Compared with conventional methods, the hm‐ratio approach enables more precise identification of hydroxymethylated regions. Figure S2A shows a scatter plot of probes with increased hm‐ratio during MP in one patient (HM1), whereas Figure S2B displays the absolute β‐value of 5hmC for the same probes. Probes enclosed in the red triangle in Figure S2B indicate decreased absolute β‐values of 5hmC during MP. These probes cannot be identified as hydroxymethylated regions without the hm‐ratio method. For this patient, 8.9% of the probes with an increased hm‐ratio overlapped with those with a decreased 5hmC value (red dots in Figure S2C). When the other four patients were included, 0.9%–8.9% of the hm‐ratio gaining probes (average 4.1%) overlapped with the probes with decreased 5hmC values (Table S2). These data demonstrated the advantage of using the hm‐ratio in this study.
3.2. Location of hydroxymethylated regions during MP is shared among patients
Our study samples are summarized in Table 1. We quantified genome‐wide 5mC and 5hmC in primary and malignantly progressed IDHmt astrocytomas using the OxBS method, followed by the methylation array. We calculated the Δhm‐ratio for each probe by comparing the hm‐ratio before and after MP in each patient, with a positive Δhm‐ratio indicating hydroxymethylation during MP. We identified the top 50% probes with a positive Δhm‐ratio from each patient (Top50%Δhm‐ratio_probes) and extracted the probes shared by all five patients for further analysis (Figure S3).
These shared probes amounted to 3120, which was significantly higher than the expected coincidence (831 probes) (p < 2.2e‐16, binomial test) (Figures 1A, S3). We named these probes CoHMCs. Heat maps showing 5mC, 5hmC, and hm‐ratio before and after MP in the CoHMC probes are displayed in Figure 1B–D. The distribution of β‐values of 5mC and 5hmC, and hm‐ratio of all analyzed probes in primary (LGGs) and malignantly progressed (HGGs) samples are also shown in Figure S4A–C. These 3120 probes are mapped to genes, with some genes containing multiple CoHMCs, corresponding to 2647 genes.
FIGURE 1.
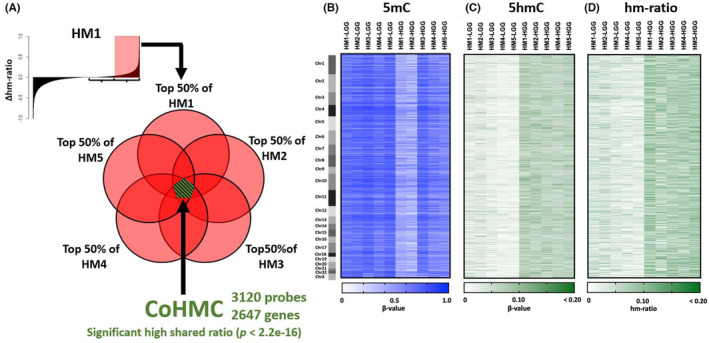
Changes in methylation levels before and after malignant progression (MP) in each patient. (A) Schematic illustration of common hydroxymethylated CpGs (CoHMC). A histogram of the Δhm‐ratio of patient HM1 is shown as an example. All probes were sorted by the Δhm‐ratio to create the histogram. (B–D) Heat maps of 3120 CoHMC probes with the β‐value of 5‐methylcytosine (5mC), 5‐hydroxymethylcytosine (5hmC), and hm‐ratio. Patient ID: HM1‐5. Low‐grade glioma (LGG) and high‐grade gliomas (HGG) represent primary and post‐MP samples.
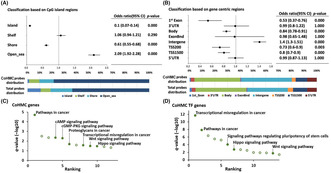
Comparing the genome‐wide distribution of 3120 CoHMC probes to that of all probes analyzed in this study showed CoHMC enrichment in open sea regions and depletion in islands based on CpG island classification (Island, Shelf, Shore, Open sea; Figure 2A). Regarding gene‐centric regions (1st Exon, 3′UTR, Body, ExonBnd, Intergene, TSS200, TSS1500, 5′UTR), CoHMC was enriched in the intergenic regions (Figure 2B).
FIGURE 2.
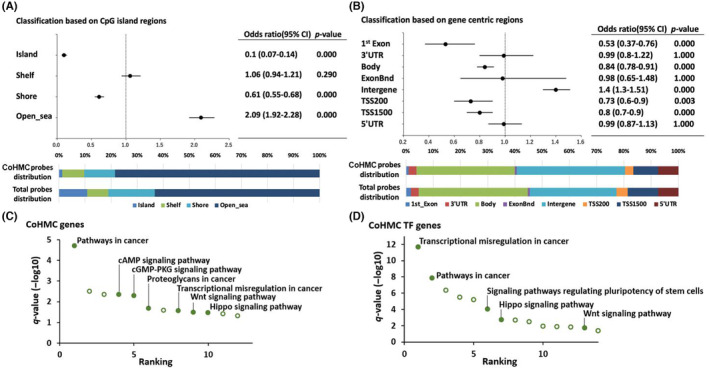
Locations and genes undergoing hydroxymethylation during malignant progression. (A, B) Distribution enrichment analysis of common hydroxymethylated CpGs (CoHMC) 3120 probes with CpG island region and gene‐centric region systems (Fisher's exact test). Total probe distribution was used as reference distribution data. (C, D) Kyoto Encyclopedia of Genes and Genomes pathway analysis of CoHMC‐related all genes and transcription factor‐coding genes. Cancer‐related pathways are represented by filled dots and annotated.
3.3. Hydroxymethylation is frequent in cancer‐related genes
The KEGG pathway analysis of the 2647 CoHMC‐containing genes revealed significant enrichment in “Pathways in cancer” (q = 1.96e‐05). Additionally, the “cyclic AMP pathway” (q = 0.004) and “Hippo signaling pathway” (q = 0.03) showed enrichment (Figure 2C). Both the “cyclic AMP pathway” and the “Hippo signaling pathway” are involved in cell cycle and proliferation. Among the 2647 genes, 140 genes encoded transcription factors. 27 Among these genes, the pathways related to cancer, such as “Transcriptional misregulation in cancer” (q = 2.06e‐12), “Pathways in cancer” (q = 1.50e‐8), and “Signaling pathways regulating pluripotency of stem cells” (q = 9.56e‐5), were significantly enriched (Figure 2D). The details are presented in Table S3. These findings demonstrated that hydroxymethylation is frequent in cancer‐related genes during MP. We also tested the same approach as used in Figure 1A, utilizing the absolute β‐value changes of 5hmC instead of the hm‐ratio. We detected probes demonstrating an increase in 5hmC levels during the MP ((5hmC of HGG) − (5hmC of LGG) >0) in each sample. Subsequently, we identified the top 50% of these probes within each sample and extracted 1233 probes shared across all samples. The 1233 probes belonged to 1153 genes, and when subjected to KEGG pathway analysis, these 1153 genes did not exhibit any significant pathway enrichment. These results suggested the validity of analyzing the hm‐ratio‐altered regions in this study.
3.4. Genes involved in the cell cycle are upregulated during MP
For gene expression analysis, RNAseq was performed on the primary and malignantly progressed glioma specimens from all five pairs. A previous study has indicated changes in the expression levels of cell‐cycle‐related genes during MP. 14 We conducted GSEA 28 of oncogenic signatures using all gene expression data before and after MP in our samples. Consequently, the top three gene sets showing significant enrichment were all related to the cell‐cycle process, such as RB_DN.V1_UP, EIF4E_UP, and MYC_UP.V1_UP (Figure 3A,B). 29 , 30 , 31 Those results suggested that our samples experienced cell‐cycle acceleration during MP.
FIGURE 3.
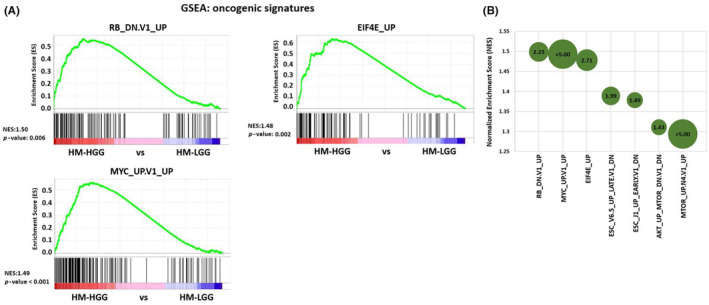
Gene Set Enrichment Analysis (GSEA) of high‐grade gliomas (HGG) compared with low‐grade glioma (LGG). (A) GSEA analysis of oncogenic signatures in the current cases. HGG (after malignant progression [MP]) compared with LGG (before MP) suggests upregulation of oncogenic signature gene sets involved in cell‐cycle processes (e.g., RB_DN.V1_UP, EIF4E_UP, MYC_UP.V1_UP) during MP. (B) Balloon chart displaying GSEA enrichment score and the significance of HGG compared with LGG. Balloon size represents −log10(p‐value), with the value written inside each balloon. Gene sets are plotted in order of normalized enrichment score.
3.5. Positive and negative correlations between hydroxymethylation and gene expression
Among the 2647 CoHMC‐containing genes, 322 genes (corresponding to 380 CoHMC probes) showed significant gene expression changes during MP (Student's t‐test, p < 0.05). We examined the correlation between the hm‐ratio change of each probe and the FPKM change of the mapped genes. Pearson correlation coefficient r was calculated for all probes. As a representative probe for example, the cg12484738 probe (mapped to gene JMJD1C) exhibited a strong negative correlation (r = −0.886) between gene expression and hydroxymethylation at this CpG (Figure 4A). In addition, the adjacent probes showed similar Δhm‐ratio values (Figure S5).
FIGURE 4.
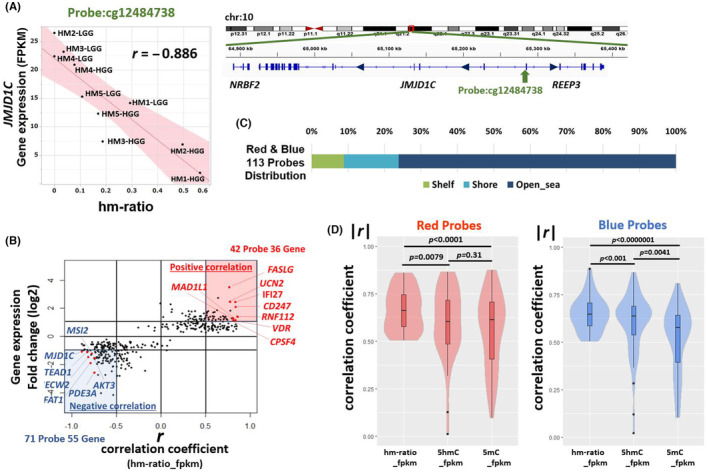
Correlation between hydroxymethylation and gene expression. (A) Correlation between hm‐ratio and gene expression of JMJD1C. The hm‐ratio value corresponds to cg12484738, an common hydroxymethylated CpGs (CoHMC) probe mapped onto JMJD1C. Ten samples (high‐grade gliomas [HGG] and low‐grade glioma (LGG) of HM1‐5 patients) are included in the correlation plot. (B) Scatter plot of 380 CoHMC probes with a significant expression change during malignant progression. Red area: correlation coefficient r ≥ 0.5 and at least a two‐fold increase in expression. Blue area: r ≤ −0.5 and at least a 0.5‐fold decrease. Genes with a strong correlation are labeled. Vertical axis: log2 fold change (fragments per kilobase million [FPKM] mean of HGG/FPKM mean of LGG). (C) Distribution in the classification of CpG island regions of red/blue 113 probes. (D) Violin plots of the absolute values of the correlation coefficient (|r|) between hm‐ratio and FPKM, 5‐hydroxymethylcytosine (5hmC) β‐value and FPKM, and 5‐methylcytosine (5mC) β‐value and FPKM for each probe in the red and blue areas (red and blue probes). p‐values were calculated by paired t‐test. We adopted absolute values to simply compare the strength of correlation between gene expression change and each parameter.
From these 380 probes, we focused on the probes that showed a significant correlation between hm‐ration change and gene expression change (probes in red and blue areas) (Figure 4B). The red area in Figure 4B represents probes with a correlation coefficient r of ≥0.5 and at least a two‐fold increase in expression (mean FPKM) during MP. The blue area represents probes with an r of ≤−0.5 and at least a 0.5‐fold decrease in expression during MP. The red area included 42 probes and 36 genes, whereas the blue area included 71 probes and 55 genes. These 113 red and blue probes are listed in Table S4. These probes can be interpreted as the ones that particularly show a strong correlation between hm‐ratio and gene expression in CoHMC. Distributional analysis revealed that 76.1% of these probes were located in open sea regions (Figure 4C, Table S4), and none were found in islands. The data in Figure 2A, along with this information, suggest that changes in hm‐ratio predominantly occur in the open sea and the hydroxymethylation levels in open sea regions are likely to influence gene expression. We analyzed the correlation between the hm‐ratio and FPKM as in Figure 4A, and similarly, between the 5hmC‐β‐value or 5mC‐β‐value and FPKM for each probe in the red and blue areas. While correlations were found between the β‐values of 5hmC or 5mC and gene expression level (FPKM), the hm‐ratio exhibited the highest association with FPKM when comparing absolute values of the correlation coefficient (Figure 4D). Based on the expected gene upregulation due to demethylation, we anticipated that the expression of these genes would be upregulated. However, interestingly, genes in the blue area were downregulated despite hydroxymethylation, deviating from the general gene expression pattern associated with demethylation.
3.6. Hydroxymethylation in the specific gene region is associated with gene downregulation
In the present study, contrary to conventional expectations, specific gene expression levels were downregulated following hydroxymethylation, which is a form of demethylation. Previous studies have also reported similar findings, but the mechanism remains unclear. 17 Correlations between 5hmC β‐value, 5mC β‐value, and FPKM were analyzed for 113 probes in the red and blue areas of Figure 4B (Figure S6A). In the red area, 95% of probes showed a negative correlation between gene expression and 5mC, but a positive correlation between gene expression and 5hmC. In contrast, in the blue area, 94% of probes showed a positive correlation between gene expression and 5mC and a negative correlation between gene expression and 5hmC. This result defies the conventional explanation of gene expression regulation through promoter methylation, suggesting potential downregulation of gene expression due to CpG demethylation. Then, we examined the gene regions where these blue probes were enriched. A previous report showed that gene expression is downregulated when the gene‐body region is demethylated. 32 Probes in the blue area tended to be enriched in the open sea region (p = 0.039), gene‐body (p = 0.435), and intergenic region (p = 0.548; Figure S6B). Therefore, hydroxymethylation in these regions is associated with the downregulated expression of genes in the blue area.
3.7. CoHMCs in gene regulatory regions were enriched with KLF4 binding motif
Next, we analyzed the probes that are mapped to the promoter region (TSS200, TSS1500, 5′UTR) and H3K27ac peak region of MGH15m (a tumor sample of IDHmt astrocytoma Grade 3)‐ChIP sequencing data among the probes in the red and blue areas of Figure 4B. In other words, we extracted the probes mapped to the gene regulatory regions. In total, 49 probes were mapped to the H3K27ac‐enriched regions. Motif analysis using HOMER v4.10 33 revealed a significant enrichment of the KLF4 transcription factor binding motif in these 49 regions (% of Targets = 15.91%, % of background = 0.08%, p = 1e‐14, match score = 0.61) (Figure S6C). As KLF4 has a unique feature of binding to sites with 5mC and being released with 5hmC, the downregulation of its target genes in gene regulatory regions following hydroxymethylation may be explained by this feature. Table S5 displays genes with a negative correlation to hydroxymethylation and KLF4 binding motif near the demethylated probe.
To examine the methylation changes in gene regions with the KLF4 binding motif during MP, we extracted the probes with the KLF4 core binding sequence (5′‐GGGCGKG‐3′) on the subject cytosine of the EPIC probes analyzed. As a result, 4226 probes were extracted and were compared with the β‐value of 5mC and 5hmC. Interestingly, probes with the KLF4 core sequence exhibited higher 5hmC levels in HGG compared with that in LGG. Conversely, randomly chosen 4226 probes did not show a significant difference (Figure S6D). Considering the characteristics of KLF4 mentioned earlier, our data suggest that KLF4 may be released from its binding site during MP. To validate this, we knocked down KLF4 using siRNA in the U87 cell line and assessed the expression changes of nine genes with negative correlation to hydroxymethylation and the KLF4 binding motif near the demethylated probe (Table S5). The results indicated that the expression of certain genes was downregulated, suggesting that KLF4 regulates their expression (Figure S6E). As we considered that the KLF4 binding sites in IDHmt high‐grade glioma cell lines are already in a predominantly 5hmC state, which is unsuitable for KLF4 binding, we utilized U87 (IDHwt, high‐grade glioma cell lines) for this experiment.
3.8. Expression change of hydroxymethylation‐related genes can contribute to tumor malignancy
We further investigated the impact of expression changes in red/blue genes on tumor malignancy. Pathway analysis revealed that red genes (36 genes) were enriched in the “Positive regulation of cell‐cycle process” pathway (q = 0.0384), whereas blue genes (55 genes) showed enrichments in the “cGMP‐PKG signaling pathway” (q = 0.0025), “Proteoglycans in cancer” (q = 0.0025), and “cAMP signaling pathway” (q = 0.0025) (Figure 5A). The enrichment of the “Positive regulation of cell‐cycle process” pathway in red genes suggests that hydroxymethylation and upregulation of these genes may accelerate the cell cycle. In this study, we observed the expression changes in cell‐cycle‐related genes in glioma cells during MP (Figure 3A,B). For the enriched pathways in blue genes, the cGMP‐PKG and cAMP signaling pathways have antineoplastic effects in various cancer types, whereas proteoglycans in cancer contribute to the stabilization of cancer cells and inhibition of metastasis and invasion. 34 , 35 , 36 The downregulation of these pathways due to hydroxymethylation of blue genes may substantially increase tumor malignancy.
FIGURE 5.
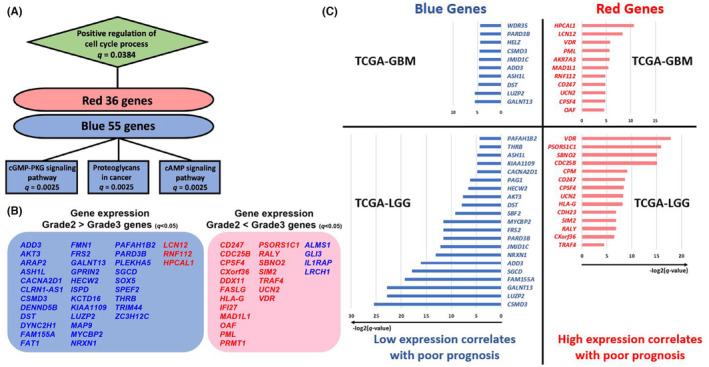
Multifaceted evaluation of genes highly correlated with hydroxymethylation (red/blue genes) (A) Enriched gene pathways in red and blue area genes. Squares represent the Kyoto Encyclopedia of Genes and Genomes pathways, and diamonds represent the GO‐Biological Process. (B) Illustration of red/blue gene expression in The Cancer Genome Atlas (TCGA)‐low‐grade gliomas (LGGs) Grade 2/3 gliomas. Red genes in red text, blue genes in blue. Left blue area shows genes highly expressed in Grade 2 glioma, right red area for Grade 3. Significance determined by t‐test, q < 0.05. Genes with no significant expression difference between Grades 2 and 3 were not listed. (C) Red/blue genes in TCGA‐LGG/GBM with prognosis‐correlated expression changes. Left: blue genes, decreased expression correlates with poor prognosis. Right: red genes, increased expression correlates with poor prognosis. Horizontal axis shows significance (−log2(q‐value), Log rank test).
3.9. Correlation between changes in expression of CoHMC‐containing genes and prognosis
We utilized publicly available clinical data to evaluate whether red/blue genes change their expression with the change in grade, and how they correlate with prognosis. 37 , 38 We analyzed TCGA‐LGG clinical data (n = 538; IDH1/2 wt/mut = 132/406), which includes Grade 2 and 3 gliomas, to assess expression changes of red/blue area genes between tumor grades. We found that 52.8% (19/36) of red genes and 60.0% (33/55) of blue genes showed significant upregulation and downregulation (t‐test, q < 0.05), respectively, with grade progression (Figure 5B, Table S6). These results were generally in line with our expectations, with a minority of genes exhibiting expression changes opposite to what was anticipated. Genes not shown in Figure 5B had no significant expression change with grade progression.
In addition, we assessed whether the expression of red/blue genes correlated with glioma prognosis using TCGA‐GBM (n = 153; IDH1/2 wt/mut = 144/9) and TCGA‐LGG data. In red genes, 11 in TCGA‐GBM and 14 in TCGA‐LGG showed a significant association (Log rank test, q < 0.05) between high expression and poor prognosis. Similarly, among blue genes, 10 in TCGA‐GBM and 21 in TCGA‐LGG demonstrated a significant association (Log rank test, q < 0.05) between low expression and poor prognosis (Figure 5C). The findings derived from the analysis of publicly available clinical data were consistent with our results and interpretations concerning the red/blue genes.
4. DISCUSSION
In this study, we used the OxBS method combined with a methylation array to analyze hydroxymethylation and utilized hm‐ratio for data analysis. We identified biologically significant hydroxymethylated regions during the MP stage of IDHmt astrocytomas. Previous studies on gliomas have provided limited insights into the 5hmC status or hydroxymethylation, and no report has specifically examined the 5hmC changes during MP in gliomas. 17 , 22 , 23 , 39 , 40 By comparing the primary and malignantly progressed‐clinical glioma samples obtained from five patients by the hm‐ratio analysis, we found commonly hydroxymethylated regions with the accumulation of 5hmC across samples. This suggests that hydroxymethylation preferentially occurs in these loci during MP, which is likely driven by specific biological factors. Glowacka et al. analyzed 5hmC in 21 IDHmt or IDHwt HGG samples and reported marked enrichments of 5hmC in the open sea region, particularly in pathogenic glioma genes, and the level of 5hmC possibly regulates gene expression. 17 We obtained similar results, observing an increased hm‐ratio in the open sea region during MP (Figure 2A), accompanied by gene expression changes. Pathway analysis revealed the enrichment of multiple cancer‐related pathways in hydroxymethylated genes during MP (Figure 2C,D).
Genes with upregulated (red genes) or downregulated (blue genes) expression correlated with hydroxymethylation. Pathway analysis indicated that red genes were associated with increased tumor malignancy, whereas blue genes had potential antineoplastic effects. Further exploration using the TCGA database revealed that many of these red and blue genes were associated with the clinical prognosis of HGG and LGG. These findings indicate a potential contribution of hydroxymethylation to the MP of glioma.
Enrichment of the KLF4 binding motif was observed in hydroxymethylated gene regulatory regions (Figure S6C). KLF4, a zinc‐finger transcription factor, plays crucial roles in cell proliferation, differentiation, and embryonic development. It acts as a tumor suppressor in various cancer types, except breast cancer. 41 , 42 , 43 KLF4 exhibits strong binding to 5mC and weak binding to 5hmC and 5caC, enabling it to function as a pioneer factor that accesses heterochromatin regions and recruits TET for hydroxymethylation. 44 , 45 , 46 KLF4's binding strength correlates with cellular differentiation status, being stronger in neuro progenitor cells and weaker in embryonic stem cells, implying its role in gene expression regulation during cell differentiation. 45 , 46 , 47 , 48 , 49 , 50 This unique nature of KLF4 sheds light on the downregulation of hydroxymethylated genes in our study, as the conversion of 5mC to 5hmC during MP may lead to KLF4's release.
Słabuszewska‐Jóźwiak et al. analyzed epigenetic alterations of the placenta, revealing the role of DNA hydroxymethylation in controlling the expression of specific genes. 8 They conducted a genome‐wide analysis of changes in the proportion of 5hmC within methylated CpGs. Mellén et al. analyzed the gene expression regulatory mechanisms in postmitotic neurons, revealing that DNA hydroxymethylation can be equated to “functional” demethylation, showcasing its pivotal role in governing gene expression. 7 Within their study, they utilized a 5hmC/5mC ratio for analysis, evaluating the binding specificity of distinctive proteins interacting with 5mC. These precedent studies demonstrate the potential significance of 5hmC as a crucial epigenetic mark in gene expression regulation, while also indicating that our utilization of the hm‐ratio in hydroxymethylation analysis can be considered a non‐radical approach.
The present study has several limitations. The small sample size is one study limitation. Due to the DNA requirements of the OxBS method, obtaining a large number of primary and malignantly progressed IDHmt glioma pairs with sufficient DNA was challenging. Additionally, using a methylation array instead of whole‐genome methylation sequencing may limit the analysis of methylation status. Higher resolution observations could have provided more detailed insights into the relationship between 5hmC status and gene expression. In this study, the hydroxymethylation‐induced gene expression changes are mainly derived from statistical and observational findings. Future biological intervention experiments will likely enhance the reliability of these results. Despite these limitations, we believe that this study contributes to understanding the role of hydroxymethylation in the MP process of glioma, which lacks detailed mechanistic understanding.
In conclusion, we observed region‐specific hydroxymethylation during MP in glioma. Our analysis revealed the involvement of cancer‐related genes in these regions, and the expression of specific genes was altered by hydroxymethylation, potentially influenced by specific transcription factor binding, such as KLF4. This phenomenon may contribute to the MP of the glioma and represent a potential therapeutic target for the IDHmt glioma. Further research, including in vitro and vivo studies, is awaited.
AUTHOR CONTRIBUTIONS
Taijun Hana: Conceptualization; data curation; formal analysis; investigation; methodology; visualization; writing – original draft. Akitake Mukasa: Conceptualization; funding acquisition; investigation; methodology; project administration; supervision; writing – review and editing. Masashi Nomura: Conceptualization; methodology; supervision. Genta Nagae: Conceptualization; methodology; supervision; writing – review and editing. Shogo Yamamoto: Data curation. Kenji Tatsuno: Data curation. Hiroki Ueda: Data curation. Shiro Fukuda: Data curation. Takayoshi Umeda: Data curation. Shota Tanaka: Data curation; supervision. Takahide Nejo: Data curation. Yosuke Kitagawa: Data curation. Erika Yamazawa: Data curation. Satoshi Takahashi: Data curation. Tsukasa Koike: Data curation. Yoshihiro Kushihara: Data curation. Hirokazu Takami: Supervision. Shunsaku Takayanagi: Supervision. Hiroyuki Aburatani: Conceptualization; methodology; project administration; supervision; writing – review and editing. Nobuhito Saito: Project administration.
FUNDING INFORMATION
This work was funded by the Japan Society for the Promotion of Science KAKENHI Grant Number 17H04300 (AM) and 20H03792 (AM).
CONFLICT OF INTEREST STATEMENT
Akitake Mukasa and Hiroyuki Aburatani are Associate Editors of Cancer Science. Aside from that, the authors have no conflicts of interest to declare regarding this project.
ETHICS STATEMENT
Approval of the research protocol by an Institutional Reviewer Board: Human sample handling and analysis in this study followed the ethical guidelines of The University of Tokyo Hospital and the Declaration of Helsinki (1964). The study was conducted in accordance with approved project G10028 titled “Gene analysis of brain tumors and elucidation of its clinicopathological significance” by the Ethics Committee of the University of Tokyo.
Informed Consent: Written informed consent was obtained from all eligible patients.
Registry and the Registration Number of the study/trial: Project G10028.
Animal Studies: N/A.
Supporting information
Figure S1:
Figure S2:
Figure S3:
Figure S4:
Figure S5:
Figure S6:
Table S1:
Table S2:
Table S3:
Table S4:
Table S5:
Table S6:
ACKNOWLEDGMENTS
We sincerely thank Reiko Matsuura, Mayu Omata, and Hiroko Meguro for their invaluable support in the experimental procedures.
Hana T, Mukasa A, Nomura M, et al. Region‐specific DNA hydroxymethylation along the malignant progression of IDH‐mutant gliomas. Cancer Sci. 2024;115:1706‐1717. doi: 10.1111/cas.16127
Contributor Information
Akitake Mukasa, Email: mukasa-nsu@umin.ac.jp.
Hiroyuki Aburatani, Email: haburata-tky@umin.ac.jp.
REFERENCES
- 1. Kleinman GM, Schoene WC, Walshe TM 3rd, Richardson EP Jr. Malignant transformation in benign cerebellar astrocytoma. Case report. J Neurosurg. 1978;49:111‐118. [DOI] [PubMed] [Google Scholar]
- 2. Brat DJ, Aldape K, Colman H, et al. cIMPACT‐NOW update 5: recommended grading criteria and terminologies for IDH‐mutant astrocytomas. Acta Neuropathol. 2020;139:603‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Appay R, Dehais C, Maurage CA, et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH‐mutant gliomas. Neuro Oncol. 2019;21:1519‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Takai H, Masuda K, Sato T, et al. 5‐Hydroxymethylcytosine plays a critical role in glioblastomagenesis by recruiting the CHTOP‐methylosome complex. Cell Rep. 2014;9:48‐60. [DOI] [PubMed] [Google Scholar]
- 5. Chen Z, Shi X, Guo L, Li Y, Luo M, He J. Decreased 5‐hydroxymethylcytosine levels correlate with cancer progression and poor survival: a systematic review and meta‐analysis. Oncotarget. 2017;8:1944‐1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Madzo J, Liu H, Rodriguez A, et al. Hydroxymethylation at gene regulatory regions directs stem/early progenitor cell commitment during erythropoiesis. Cell Rep. 2014;6:231‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mellén M, Ayata P, Heintz N. 5‐hydroxymethylcytosine accumulation in postmitotic neurons results in functional demethylation of expressed genes. Proc Natl Acad Sci USA. 2017;114:E7812‐e7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Słabuszewska‐Jóźwiak A, Malinowska M, Kloska A, et al. Global changes of 5‐mC/5h‐mC ratio and methylation of adiponectin and leptin gene in placenta depending on mode of delivery. Int J Mol Sci. 2021;22:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Branco MR, Ficz G, Reik W. Uncovering the role of 5‐hydroxymethylcytosine in the epigenome. Nat Rev Genet. 2011;13:7‐13. [DOI] [PubMed] [Google Scholar]
- 10. Booth MJ, Branco MR, Ficz G, et al. Quantitative sequencing of 5‐methylcytosine and 5‐hydroxymethylcytosine at single‐base resolution. Science (New York, NY). 2012;336:934‐937. [DOI] [PubMed] [Google Scholar]
- 11. Malta TM, de Souza CF, Sabedot TS, et al. Glioma CpG Island methylator phenotype (G‐CIMP): biological and clinical implications. Neuro Oncol. 2018;20:608‐620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Turcan S, Rohle D, Goenka A, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nomura M, Saito K, Aihara K, et al. DNA demethylation is associated with malignant progression of lower‐grade gliomas. Sci Rep. 2019;9:1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bai H, Harmanci AS, Erson‐Omay EZ, et al. Integrated genomic characterization of IDH1‐mutant glioma malignant progression. Nat Genet. 2016;48:59‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Souza CF, Sabedot TS, Malta TM, et al. A distinct DNA methylation shift in a subset of glioma CpG Island Methylator phenotypes during tumor recurrence. Cell Rep. 2018;23:637‐651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou W, Dinh HQ, Ramjan Z, et al. DNA methylation loss in late‐replicating domains is linked to mitotic cell division. Nat Genet. 2018;50:591‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Glowacka WK, Jain H, Okura M, et al. 5‐Hydroxymethylcytosine preferentially targets genes upregulated in isocitrate dehydrogenase 1 mutant high‐grade glioma. Acta Neuropathol. 2018;135:617‐634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro‐Oncology. 2021;23:1231‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Durbin R. Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics (Oxford, England). 2009;25:1754‐1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dai H, Han G, Yan Y, et al. Transcript assembly and quantification by RNA‐Seq reveals differentially expressed genes between soft‐endocarp and hard‐endocarp hawthorns. PLoS One. 2013;8:e72910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bibikova M, Barnes B, Tsan C, et al. High density DNA methylation array with single CpG site resolution. Genomics. 2011;98:288‐295. [DOI] [PubMed] [Google Scholar]
- 22. Fernandez AF, Bayon GF, Sierra MI, et al. Loss of 5hmC identifies a new type of aberrant DNA hypermethylation in glioma. Hum Mol Genet. 2018;27:3046‐3059. [DOI] [PubMed] [Google Scholar]
- 23. Johnson KC, Houseman EA, King JE, von Herrmann KM, Fadul CE, Christensen BC. 5‐Hydroxymethylcytosine localizes to enhancer elements and is associated with survival in glioblastoma patients. Nat Commun. 2016;7:13177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics (Oxford, England). 2010;26:841‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Flavahan WA, Drier Y, Liau BB, et al. Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature. 2016;529:110‐114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Team RC . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing 2016.
- 27. Chawla K, Tripathi S, Thommesen L, Laegreid A, Kuiper M. TFcheckpoint: a curated compendium of specific DNA‐binding RNA polymerase II transcription factors. Bioinformatics (Oxford, England). 2013;29:2519‐2520. [DOI] [PubMed] [Google Scholar]
- 28. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545‐15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bild AH, Yao G, Chang JT, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353‐357. [DOI] [PubMed] [Google Scholar]
- 30. Larsson O, Li S, Issaenko OA, et al. Eukaryotic translation initiation factor 4E induced progression of primary human mammary epithelial cells along the cancer pathway is associated with targeted translational deregulation of oncogenic drivers and inhibitors. Cancer Res. 2007;67:6814‐6824. [DOI] [PubMed] [Google Scholar]
- 31. Lara MF, García‐Escudero R, Ruiz S, et al. Gene profiling approaches help to define the specific functions of retinoblastoma family in epidermis. Mol Carcinog. 2008;47:209‐221. [DOI] [PubMed] [Google Scholar]
- 32. Ball MP, Li JB, Gao Y, et al. Targeted and genome‐scale strategies reveal gene‐body methylation signatures in human cells. Nat Biotechnol. 2009;27:361‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heinz S, Benner C, Spann N, et al. Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tang C, Liu D, Fan Y, et al. Visualization and bibliometric analysis of cAMP signaling system research trends and hotspots in cancer. J Cancer. 2021;12:358‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tuttle TR, Mierzwa ML, Wells SI, Fox SR, Ben‐Jonathan N. The cyclic GMP/protein kinase G pathway as a therapeutic target in head and neck squamous cell carcinoma. Cancer Lett. 2016;370:279‐285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ahrens TD, Bang‐Christensen SR, Jørgensen AM, et al. The role of proteoglycans in cancer metastasis and circulating tumor cell analysis. Front Cell Dev Biol. 2020;8:749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Goldman MJ, Craft B, Hastie M, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675‐678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Uhlen M, Zhang C, Lee S, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 39. Zhang F, Liu Y, Zhang Z, et al. 5‐hydroxymethylcytosine loss is associated with poor prognosis for patients with WHO grade II diffuse astrocytomas. Sci Rep. 2016;6:20882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Azizgolshani N, Petersen CL, Chen Y, et al. DNA 5‐hydroxymethylcytosine in pediatric central nervous system tumors may impact tumor classification and is a positive prognostic marker. Clin Epigenetics. 2021;13:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context‐dependent oncogene. Nat Cell Biol. 2005;7:1074‐1082. [DOI] [PubMed] [Google Scholar]
- 42. McCarthy N. Look both ways. Nat Rev Cancer. 2005;5:917. [Google Scholar]
- 43. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663‐676. [DOI] [PubMed] [Google Scholar]
- 44. Liu Y, Olanrewaju YO, Zheng Y, et al. Structural basis for Klf4 recognition of methylated DNA. Nucleic Acids Res. 2014;42:4859‐4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Spruijt CG, Gnerlich F, Smits AH, et al. Dynamic readers for 5‐(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146‐1159. [DOI] [PubMed] [Google Scholar]
- 46. Sardina JL, Collombet S, Tian TV, et al. Transcription factors drive Tet2‐mediated enhancer demethylation to reprogram cell fate. Cell Stem Cell. 2018;23:727‐741. [DOI] [PubMed] [Google Scholar]
- 47. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet. 2016;17:551‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wan J, Su Y, Song Q, et al. Methylated cis‐regulatory elements mediate KLF4‐dependent gene transactivation and cell migration. elife. 2017;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Aksoy I, Giudice V, Delahaye E, et al. Klf4 and Klf5 differentially inhibit mesoderm and endoderm differentiation in embryonic stem cells. Nat Commun. 2014;5:3719. [DOI] [PubMed] [Google Scholar]
- 50. Oyinlade O, Wei S, Kammers K, et al. Analysis of KLF4 regulated genes in cancer cells reveals a role of DNA methylation in promoter‐ enhancer interactions. Epigenetics. 2018;13:751‐768. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1:
Figure S2:
Figure S3:
Figure S4:
Figure S5:
Figure S6:
Table S1:
Table S2:
Table S3:
Table S4:
Table S5:
Table S6: