

Abstract
Objective
Most families with heritable neuromuscular disorders do not receive a molecular diagnosis. Here we evaluate diagnostic utility of exome, genome, RNA sequencing, and protein studies and provide evidence‐based recommendations for their integration into practice.
Methods
In total, 247 families with suspected monogenic neuromuscular disorders who remained without a genetic diagnosis after standard diagnostic investigations underwent research‐led massively parallel sequencing: neuromuscular disorder gene panel, exome, genome, and/or RNA sequencing to identify causal variants. Protein and RNA studies were also deployed when required.
Results
Integration of exome sequencing and auxiliary genome, RNA and/or protein studies identified causal or likely causal variants in 62% (152 out of 247) of families. Exome sequencing alone informed 55% (83 out of 152) of diagnoses, with remaining diagnoses (45%; 69 out of 152) requiring genome sequencing, RNA and/or protein studies to identify variants and/or support pathogenicity. Arrestingly, novel disease genes accounted for <4% (6 out of 152) of diagnoses while 36.2% of solved families (55 out of 152) harbored at least one splice‐altering or structural variant in a known neuromuscular disorder gene. We posit that contemporary neuromuscular disorder gene‐panel sequencing could likely provide 66% (100 out of 152) of our diagnoses today.
Interpretation
Our results emphasize thorough clinical phenotyping to enable deep scrutiny of all rare genetic variation in phenotypically consistent genes. Post‐exome auxiliary investigations extended our diagnostic yield by 81% overall (34–62%). We present a diagnostic algorithm that details deployment of genomic and auxiliary investigations to obtain these diagnoses today most effectively. We hope this provides a practical guide for clinicians as they gain greater access to clinical genome and transcriptome sequencing.
Introduction
Neuromuscular disorders (NMDs) are a heterogenous group of muscle and nerve pathologies with primarily genetic etiologies. 1 Individuals and families without a genetic diagnosis for a suspected heritable disorder lack certainty surrounding disease prognosis and risk of recurrence, 2 and are precluded from accessing time‐sensitive family planning, clinical trial enrolment, and targeted therapies. 3
Prior to 2012, the diagnostic workflow for NMD diagnostics was driven by clinical phenotype, muscle histopathology, immunostaining, 4 and in some instances, western blot. 5 These findings were correlated with phenotypically concordant NMD gene(s) which were then sequenced to identify and segregate variants in an affected individual and their family. 6 The advent of massively parallel sequencing (MPS) has led to an explosion in genes associated with monogenic conditions, 7 with over 640 genes now associated with NMDs. 8 Given the high degree of phenotypic overlap for many neuromuscular presentations, MPS has become a first tier diagnostic investigation. Over the past decade, implementation of MPS targeted‐capture NMD gene panels, 9 , 10 or exome sequencing for NMD diagnosis, 11 , 12 , 13 , 14 has reduced turn‐around times and costs 15 while increasing diagnostic rates from ~24% by traditional methods 6 , 16 to 30–60%. 9 , 10 , 11 , 12 , 13 , 14
Research application of genome sequencing 17 , 18 and RNA sequencing (RNA‐Seq) 19 , 20 , 21 for individuals with likely monogenic disease undiagnosed by exome sequencing approaches, commonly identifies pre‐mRNA splice‐altering variants, structural variants, and/or variants in novel or newly discovered disease genes as the elusive basis for disease. 19 , 22 , 23 Consequently, genome sequencing and RNA studies are beginning to be implemented in the clinic for exome‐negative monogenic conditions. 24 , 25 , 26 However, genome and/or RNA testing is an additional expense that places further strain on limited clinical budgets for genomic testing, despite accelerating demand. There remains uncertainty regarding in which order to deploy genome or RNA sequencing or both, and whether for a trio or singleton, for optimal diagnostic and cost benefits.
Over the past 10 years, we have iteratively assessed the diagnostic utility of exome, genome, and transcriptome technologies in an ethnically diverse cohort of 247 families with suspected monogenic NMD, for whom contemporary genetic testing of the time failed to yield a molecular diagnosis. Herein we retrospectively review the diagnostic odyssey for our 152 out of 247 families diagnosed by research‐led genomic testing (62%), identifying exactly which MPS platform and auxiliary investigation(s) provided evidence critical to confer each molecular diagnosis. While our path to diagnosis was regularly convoluted, adopting a contemporary lens of what is possible in present‐day genomics, we present our expert team's recommended MPS diagnostic algorithm for NMD diagnosis.
Methods
Ascertainment criteria
In total, 247 families with genetically undiagnosed NMDs were referred by neurologists and clinical geneticists to the Kids Neuroscience Centre Biospecimen Bank between 2012 and 2021. Referrals were primarily from Australia and New Zealand and broadly representative of the diverse ethnic demographics of these countries. Ascertainment was based on a strong clinical suspicion of a genetic neuromuscular disorder with one or more clinical investigations consistent with primary involvement of nerves and/or muscle: serum creatine kinase and other biochemical investigations, muscle biopsy, electromyography, nerve conduction studies, and/or muscle magnetic resonance imaging, as previously described. 11 , 13 Triage criteria for research‐led exome and genome sequencing required; (1) negative prescreening for known/suspected genetic causes including at least one of: candidate gene sequencing (including myotonic dystrophy screening), multiplex ligation‐dependent probe amplification, microarray, MPS gene panels, immunohistochemistry (dystrophin, sarcogylcans), and exome sequencing (from 2018 only). 11 , 13 No participant had genome or RNA‐Seq prior to this study. (2) For autosomal dominant pedigrees, the Broad Institute required DNA to be available from the family trio, or from one parent and two or more affected children, or large pedigrees where linkage analysis restricted variant searches to 1% of the genome. This study was approved by the Children's Hospital at Westmead Human Research Ethics Committee (10/CHW/45 or 2019/ETH11736) and all participants, and/or their legal guardians, provided informed, written consent.
Exome and genome sequencing
Exomic sequencing was performed for all 247 families: 216 by the Broad Institute of MIT and Harvard Genomics Platform, USA (117 singletons, 78 trios, and 21 duos/quads). 11 , 13 Sixty‐four out of 216 of these cases also received targeted‐capture MPS NMD gene panel (NMD panel v1‐5) at PathWest Laboratory, Nedlands, Australia. 9 Thirty‐one cases were referred with candidate variant(s) of uncertain significance identified by clinical exome or NMD gene panel. Genome sequencing was performed for 47 families (27 singletons, 19 trios, and 1 duo) through Illumina HiSeq X Ten v2 PCR‐free short‐read (150 nt) genome sequencing (from 2016) to a mean target coverage of >30x through the Broad Institute, USA. 27 A small subset of patients underwent external clinical screening for repeat expansions in DMPK/CNBP and/or microarray in parallel with our sequencing studies.
RNA and protein studies
Poly‐A pulldown RNA‐Seq (75 nt or 100 nt paired reads) was conducted for 47 out of 247 cases using RNA extracted from patient skeletal muscle as described previously. 19 , 28 Muscle RNA isolation and rRNA‐depleted RNA‐Seq (150 nt paired reads) was performed for 5 out of 247 cases as described. 28 cDNA synthesis and RT‐PCR are described previously. 25 , 29 Immunohistochemistry (IHC) and/or western blotting (WB) methodologies are described in previous work. 11 , 23 , 29
Variant curation and interpretation
Exome and genome sequencing data were filtered using Seqr. 30 Variants with an allele frequency of ≥1% (in ExAC 31 or gnomAD 32 ), deemed too common to be likely causal variants for rare disorders, were filtered out. Staged variant curation: (1) Clinician‐defined phenotypically concordant gene list; (2) All known NMD genes 33 ; (3) Disease genes in Online Mendelian Inheritance in Man 34 (OMIM); (4) Non‐OMIM genes. NMD and OMIM gene lists were updated annually to account for newly discovered disease genes. When trio sequencing data or family history were available, variants were filtered by inheritance pattern(s) consistent with the pedigree.
Variant prioritization strategy
(1) Gene‐phenotype concordance via multidisciplinary expert clinical review; (2) Previous reports as pathogenic/likely pathogenic in the literature or in variant databases (ClinVar 35 or Leiden Open Variation Database 36 ); (3) Occurring in trans with a previously reported pathogenic/likely pathogenic variant; (4) Predicted by in silico tools to be deleterious or splice‐altering (PolyPhen2 37 ; NNsplice 38 ; in silico tools available in seqr 30 ; Alamut Visual Biosoftware® from 2017; SpliceAI 39 from 2019).
Variant confirmation
Sanger sequencing was performed (in‐house or in a diagnostic laboratory) to confirm and segregate variants in some families, prior to availability of accredited MPS pipelines and practice standards that did not require Sanger segregation of identified variants in clinical testing.
Variant classification
From 2012 to 2016, likely causality of identified variants was reported in “Research Reports” to the referring clinician, based upon our expert opinion and/or published findings. From ~2016, we sought formal classification of variants from accredited diagnostic scientists and genetic pathologists according to the American College of Medical Genetics and Genomics (ACMG) guidelines. 33 Identification of a novel disease‐gene association, 40 , 41 , 42 , 43 , 44 , 45 or a novel gene‐phenotype expansion 46 , 47 , 48 , 49 , 50 , 51 , 52 was defined by peer‐reviewed publication.
Bioinformatic detection of pathogenic repeat expansions
Whole genome and exome sequenced samples were analyzed for known pathogenic repeat expansions of short tandem repeats with the bioinformatic tools ExpansionHunter and exSTRa, using default parameters. Repeat expansion lengths estimated by ExpansionHunter to be larger than a locus‐specific threshold were prioritized as candidate pathogenic variants. The database of known pathogenic repeat expansion loci can be found at https://github.com/bahlolab/exSTRa/blob/abdf75d89f095f7b7f8573d22206edd735d8914b/inst/extdata/repeat_expansion_disorders_hg38.txt.
SpliceAI delta‐score threshold evaluation
Proband genome sequencing data were annotated with precomputed SpliceAI 39 scores (window length ± 50 nt) through Seqr 30 or an in‐house pipeline and rare, segregating variants returned. Rare variants were defined as having allele frequency ≤1% and number of alternate allele homozygotes <5 as listed in gnomAD. 32 Segregating variants were those consistent with the expected inheritance and disease presentation in the pedigree. Receiver‐operator characteristic/precision‐recall curves were created with the pROC and PRROC R packages.
Splicing outlier analysis from RNA‐Seq with FRASER
FRASER (v1.0.2) 53 was deployed to identify splicing outliers in three probands with skeletal muscle RNA‐Seq and genome sequencing data. Fifty control skeletal muscle RNA‐Seq samples were sourced from the Genotype‐Tissue Expression (GTEx) project. 54 FRASER was run for each proband, comparing to the 50 GTEx controls, with default configuration. Analysis of FRASER data was conducted using the FRASER Bioconductor R package. High confidence splice‐junction outliers were defined by an adjusted p‐value <0.05 and an effect‐size/delta‐psi ≥0.3 as used in the FRASER manuscript.
Rare segregating variants from trio genome sequencing data were incorporated to filter high confidence splice‐junctions identified by FRASER. Variants were defined as proximal to a splicing outlier if they occurred within 250 nt either side of the donor or acceptor splice site of an outlier junction. Rare variants were defined as having allele frequency <1% and number of alternate allele homozygotes <5 as listed in gnomAD. 32 Segregating variants were those consistent with the expected inheritance and disease presentation in the pedigree.
Results
A cohort of 247 families with NMD
From 2012 to 2021, we ascertained a cohort of 291 individuals affected with NMD from 247 families, for whom available genetic testing had not yielded a molecular diagnosis. The cohort comprises 32·4% (80 out of 247) congenital‐onset (<2 years), 30·8% (76 out of 247) pediatric‐onset (2–12 years), and 36·8% (91 out of 247) adolescent (13–17 years) or adult‐onset (≥18 years) NMD (Figure 1Ai). Most families had a single affected individual (92·7%; 229 out of 247), 6·5% (16 out of 247) had two, one family had three, and another family four affected individuals. Singletons (sequencing data only available for proband) comprise 58·3% (144 out of 247) of the cohort and 6·1% (15 out of 247) of families had consanguineous pedigrees.
Figure 1.
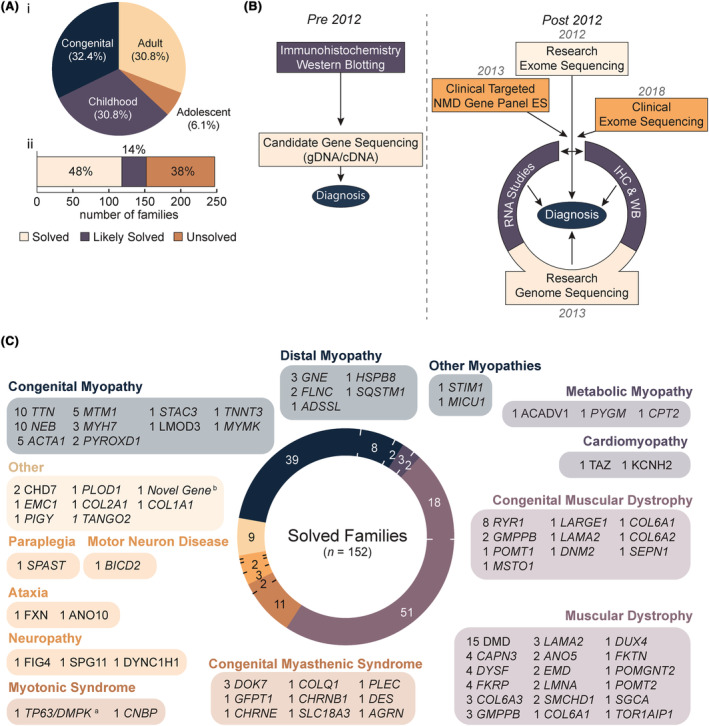
Overview of research‐led diagnostic NMD cohort. (A) Age of onset of the 247 probands in our neuromuscular disease (NMD) cohort (i), with two‐thirds presenting with congenital (<2 years; n = 80) or pediatric‐onset (2–12 years; n = 76) disorders. A majority of the cohort is considered either solved (48%) or likely solved (14%) (ii). (B) Schematic of NMD diagnostic pipeline employed by our laboratory prior to, and after, 2012 when massively parallel sequencing was adopted as a first‐line investigation. (C) Research‐led massively parallel sequencing revealed a diverse set of genetic etiologies within NMD sub‐groups. The number of families in our cohort linked to each causal gene is listed. Genes are divided into disease groups according to observed phenotype and classification in the gene table of neuromuscular disorders. 8 aNote, it is the collective opinion of our multidisciplinary team that digenic TP63 and DMPK variants are the likely causal basis for CMD in the proband. The proband is now deceased and there are no additional family members to gather further evidence supporting a digenic disorder. We therefore consider the family “likely solved” and are not pursuing further investigations. bA high confidence candidate novel disease gene in an advanced stage of functional validation. Three other genes that form part of the same complex are existing OMIM genes with a related phenotype. ES, exome sequencing; IHC, immunohistochemistry; WB, western blot.
We deployed research‐led genome sequencing, RNA studies (RNA‐Seq and/or RT‐PCR) or protein studies (IHC and/or western blot) on a case‐by‐case basis (Fig. 1B) subject to (a) biospecimen availability; (b) one or more VUS's identified in a phenotypically concordant gene; (c) the predicted functional impact of candidate variant(s) on pre‐RNA splicing and/or upon the levels or size of the encoded protein; (d) no candidate variants identified but a clinical diagnosis (or high clinical suspicion) of a specific condition with one or only a few associated disease genes (e.g., dystrophinopathy and nemaline myopathy). To date, we have identified the causal (solved and clinically diagnosed) or likely causal variant(s) (research reported as likely solved, explained below) for 61·5% (152 out of 247) of families (Figure 1Aii, Table S1 for variant information). Variants in 73 genes were identified (Fig. 1C), of which the top 6 most frequent, DMD, TTN, NEB, RYR1, ACTA1, and MTM1, account for 32% of diagnoses. A further 32% of diagnoses were in genes observed only once in the cohort.
Variants we categorize as ‘likely causal’ are those presumed by our expert, multidisciplinary center as the likely basis for the affected individual's condition and are unable to (yet) be afforded a clinical diagnosis via ACMG criteria due to one case involving a novel disease gene candidate under pursuit; one with a presumed novel digenic disorder; fifteen without variant segregation (family member DNA not available); two deemed by expert NMD clinicians as likely to have two, separate, genetic disorders; eleven with missense variants (or in‐frame deletions) which remain variants of uncertain significance (VUS) in clinically consistent genes; and two cases with “high confidence” candidate splice‐altering variants in a phenotypically concordant gene though biospecimens are unavailable to provide functional evidence of pathogenicity.
Novel genes and novel phenotypes comprise <9% of diagnoses
We identified causal variants in six novel disease genes that account for 3·9% (6 out of 152) of diagnoses (Fig. 2). 40 , 41 , 42 , 43 , 44 , 45 On average, it took 2–5 years (range 11–52 months) from identification‐to‐publication of a novel gene‐disease association, requiring; international collaboration to identify similarly‐affected individuals with segregating variants in the same gene, RNA and/or protein investigations using clinical specimens, and cell and/or animal disease models. The complexity of research validation of a novel gene‐disease association is highlighted by PYROXD1, a gene previously undescribed in the literature. Confirmation of biallelic PYROXD1 variants as novel basis for congenital myopathy in five unrelated affected families required three cell and animal models of disease and 52 months of multidisciplinary research. 45 Seven novel phenotypes (4·6%; 7 out of 152) for known human disease genes which expand the published clinical spectrum were also identified in the cohort and are outlined in Figure 2. 46 , 47 , 48 , 49 , 50 , 51 , 52
Figure 2.
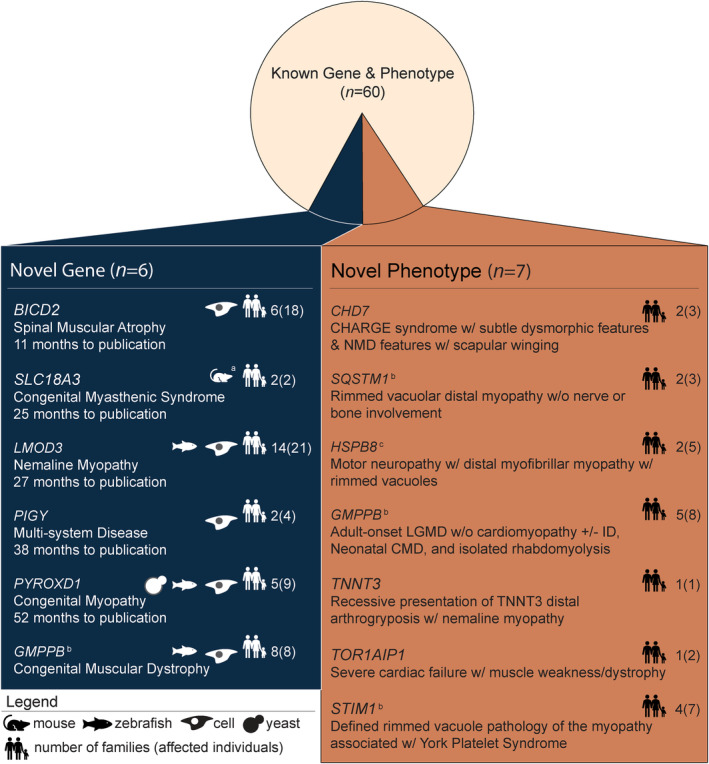
Novel disease genes and phenotypes identified in the NMD cohort. Six novel genes were identified from the cohort: BICD2, 42 SLC18A3, 43 LMOD3, 44 PIGY, 41 PYROXD1, 45 and GMPPB. 40 a SLC18A3 was published citing two previously reported mouse models. 55 , 56 The average time to publication for novel genes was 2·5 years. Seven novel phenotypes in known neuromuscular disorder genes were reported from our cohort: CHD7, 51 SQSTM1, 46 HSPB8, 48 STIM1, 50 GMPPB, 47 TNNT3, 52 and TOR1AIP1. 49 Families from our cohort with novel disease genes and phenotypes were reported either by our group, by other groupsb (timeline not available), or in collaborationc. CMD, congenital muscular dystrophy; GDD, global developmental delay; ID, intellectual disability; LGMD, limb girdle muscular dystrophy; NM, nemaline myopathy; NMD, neuromuscular disease.
Diagnoses involving splice‐altering variants (36% diagnoses) were nine times more common than novel disease genes (4% diagnoses)
Splice‐altering and structural variants in known NMD genes constitute 28·4% of causal variants (63 out of 222; Figure 3Ai) underpinning diagnoses for 36·2% (55 out of 152) of solved families, due to the prevalence of biallelic recessive NMDs (61·2%; 93 out of 152; Figure 3Aii). To account for ascertainment bias since 2018 due to our expanding expertise with splicing variants, scrutiny of our pre‐2018 NMD cohort shows splicing variants underpin 30·1% (40 out of 133) of diagnoses. Further, 2 out of 6 (33%) families with diagnoses involving novel disease genes had one or more causal splice‐altering and/or structural variant(s).
Figure 3.
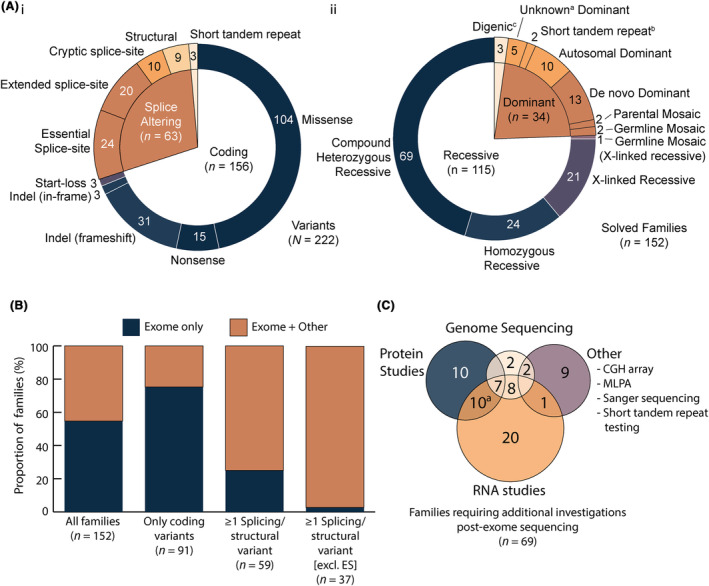
Variants identified in the cohort by variant type, inheritance, and the investigations required to confirm them as causal. (A) The nature of different classes of pathogenic variants identified in our NMD cohort (i) and the pattern of inheritance (ii). Heterozygous variants for which segregation or family history was not available are labeled as unknown dominanta. bTrinucleotide repeat disorder testing was conducted in a diagnostic genetic laboratory. cNote the digenic inheritance count includes the unconfirmed TP63/DMPK family addressed in Figure 1. (B) Proportion of families diagnosed by exome sequencing alone or exome sequencing and additional investigations (+ Other) stratified by the variant types identified. (C) Venn diagram of additional investigations required for diagnosis post‐exome. aIncludes one case which underwent RNA studies, protein studies, and MLPA. AD, autosomal dominant; CGH, comparative genomic hybridization; ES, essential splice‐site; MLPA, multiplex ligation‐dependent probe amplification.
96% of identified splice‐altering variants were detected above SpliceAI delta score 0.1
Given the high proportion of splicing variants in our cohort, we evaluated the optimal SpliceAI delta score for use as a screening tool. Analysis of SpliceAI scores for all rare segregating variants in 606 known NMD genes for 50 cases shows a delta score ≥0·1 captures 96% causal splice‐altering variants (Fig. 4B–D, Case 35 the exception [see Table S1]) while yielding a manageable average number of 4·8 noncausal variants above this threshold per case, for further prioritization. We therefore recommend a SpliceAI 43 delta score threshold of ≥0·1 as a pragmatic, high sensitivity cutoff, to assist curation of splice‐altering variants (Fig. 4C,D).
Figure 4.
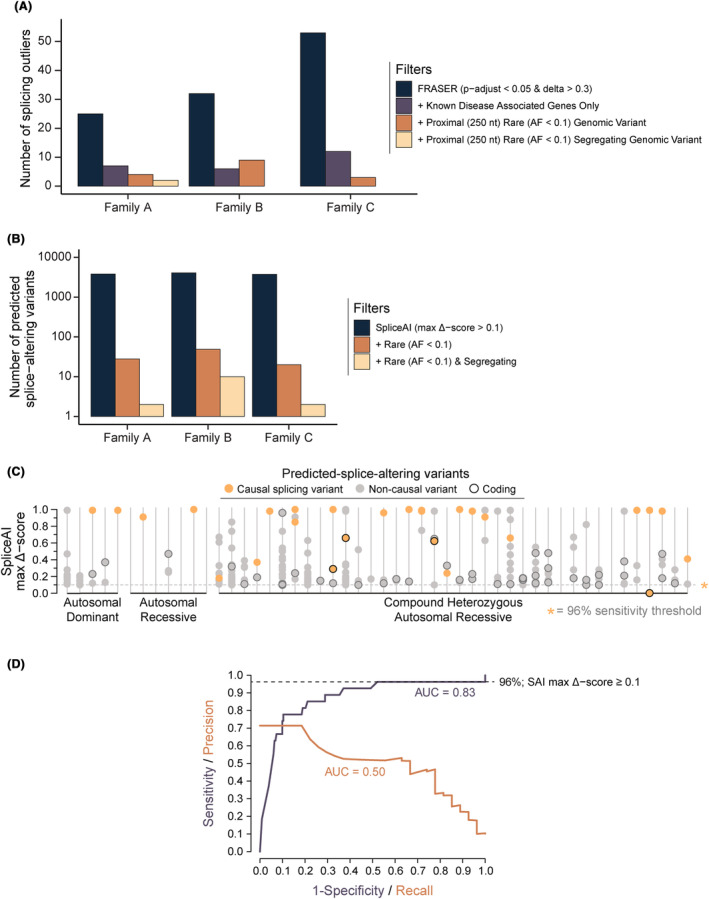
Trio genome sequencing data filters out highly scored but noncausal findings from in silico splicing analyses. (A) Deployment of FRASER to identify splicing outliers in 3 families with RNA‐Seq data available. Analyzing statistically significant splicing outliers identified by FRASER using the recommended thresholds (range 25–55 outliers per sample/case; adjusted p‐value <0.05 & effect‐size/delta‐psi ≥0.3) alongside genomic sequencing data was essential to reduce the large number of noncausal findings. In practice, by confirming the presence of a rare (allele frequency <0.1 and number of alternate allele homozygotes <5), segregating variant identified by genome sequencing proximal (±250 nt) to a splicing outlier all but two splicing outliers across the three families were excluded, substantially reducing the time required to manually validate candidate outliers (in Integrative Genomics Viewer 30 ). (B) Number of variants identified in genome sequencing data across all genes using SpliceAI delta score of ≥0.1 as threshold for a prediction of splice‐altering outcomes in the same three families as in S1a. Inclusion of trio segregation data reduced noncausal predicted‐splice‐altering variants by greater than 10‐fold. (C) Segregating rare variants from exome or genome data for 50 solved families with SpliceAI maximum delta scores ≥0.1. On average, families had 4.8 rare (allele frequency <0.1 & number of alternate allele homozygotes <5), noncausal predicted‐splice‐altering rare segregating variants in known NMD genes (n = 606). Searches were conducted in each family using the segregation of the known causal variant. For compound heterozygous recessive noncausal predicted‐splice‐altering variants, confirmation of a second rare variant in trans was not sought. Therefore, 4.8 noncausal predicted‐splice‐altering segregating variants per family is likely an overestimation. (D) Receiver‐operator characteristic curve showing a SpliceAI maximum delta score threshold of ≥0.1 results in a sensitivity of 96% for the detection of splice‐altering causal variants in the 50 solved families in S1c. Poor performance across the precision‐recall curve is indicative of the high number of noncausal segregating variants predicted to be splice‐altering by SpliceAI.
Post‐exomic auxiliary studies required for diagnosis in 45% of solved families
Exomic sequencing (targeted NMD panel or exome) diagnosed 54·6% (83 out of 152) of solved families with causal coding or essential splice‐site variants. The remaining 45·4% (69 out of 152) diagnoses required RNA or protein studies and/or genome sequencing (Fig. 3B,C; Table 1). For the 59·8% (91 out of 152) of solved families with coding variants only, 23 out of 91 required at least one auxiliary investigation beyond exomic sequencing; most commonly RNA studies to test splice‐altering impact. For the 38·8% (59 out of 152) of families with at least one splicing or structural variant in a known NMD gene, 44 required additional investigations while 15 carried essential splice site variants and were classified as pathogenic without further functional evidence (based on pathogenic very strong criteria 52 ). Among 37 (likely) causal extended splice site variants, only one was reported without RNA studies (muscle biopsy unavailable) and it remains a VUS.
Table 1.
Diagnostic outcomes from additional studies post‐exomic sequencing.
| Post‐exome investigation | Outcome from additional studies | No. of families | % of diagnosed families | % required for diagnosis |
|---|---|---|---|---|
| Protein studies | Identified reduced levels, activity or altered size of protein | 27 |
18·4% (n = 28/152) |
96% (n = 27/28) |
| No meaningful contribution to date | 1 | |||
| RNA studies | Demonstrated aberrant splicing from variant/s | 34 |
34·9% (n = 53/152) |
86% (n = 46/53) |
| Excluded aberrant splicing from coding variant/s | 6 | |||
| Confirmed structural variant at transcript level | 4 | |||
| No meaningful contribution to date | 9 | |||
| Genome sequencing | Identified causal deep intronic a variant/s | 5 |
17·1% (n = 26/152) |
73% (n = 19/26) |
| Identified causal extended splice site variant/s | 3 | |||
| Identified structural variant | 6 | |||
| Variant/s was present in exome but missed during analysis | 3 | |||
| Variant/s identified due to better coverage than exome | 2 | |||
| Variant/s confirmed as mosaic by genome | 1 | |||
| Variant/s in novel gene not reported until after genome | 1 | |||
| Used to exclude other candidate genes and/or variant/s | 5 | |||
| Other | Sanger sequencing identified variant/s missed by exome | 1 |
8·6% (n = 13/152) |
100% (n = 13/13) |
| Sanger sequencing demonstrated variant/s was mosaic | 1 | |||
| Structural variant identified by MLPA (3) or CGH array (3) | 6 | |||
| DMPK trinucleotide repeat disorder identified b | 1 | |||
| FSHD testing positive | 3 | |||
| Repeat expansion in FXN intron 1 | 1 |
Post‐exome investigations and outcomes for variant interpretation. The second to last column denotes the percentage of all diagnosed families subject to each investigation. The final column depicts the proportion of investigations that informed or assisted diagnosis, based on our expert opinion. Note that families undiagnosed after exome underwent on average 1.5 additional investigations, thus, families have been counted multiple times in this table.
CGH, comparative genomic hybridization; FSHD, facioscapulohumeral muscular dystrophy; MLPA, multiplex ligation‐dependent probe amplification.
Deep intronic variant ≥50 nt from intron‐exon junction.
Via bioinformatic analysis of genome/exome sequencing.
Contemporary exomic sequencing would diagnose ~65% of solved families
Over the last decade, new genes associated with NMD presentations have been dynamically incorporated into NMD gene panel lists. 8 The current version of the Australian targeted‐capture NMD panel (PathWest, Table S2) or exome sequencing, capturing 50 nt of the flanking intron, would detect (but not definitively classify) the causal variant(s) in 87.5% (133 out of 152) of families, and diagnose 65.8% (100 out of 152) of families; due collectively to iterative expansion of NMD genes in the panel, improved coverage of coding and near‐coding intronic sequences (~50 nt flanking each exon), improved copy number variant calling and improved bioinformatic mapping methods to identify variants within TTN and NEB triplicated repeat regions. 9 Variants in only 19 out of 152 solved families would remain undetected; 8 due to the genes not yet being covered by the panel and 11 due to a deep intronic variant >50 nt into the intron (5 families; intronic depth range 151–30954 nt), a complex structural variant (5), or a short tandem repeat expansion (1).
Discussion
This study retrospectively evaluates diagnostic outcomes of different MPS modalities deployed over 2012–2021 for 247 NMD families who remained genetically undiagnosed by contemporary genomic diagnostics of the time. Exome, genome, transcriptome, and protein studies collectively diagnosed 61·5% (152 out of 247) of referred families. Targeted NMD panel or exome sequencing (capturing 50 intronic nucleotides at the exon‐intron junction) deployed today would detect ~87% of causal variants, including 75% of identified splicing and structural variants. Auxiliary studies were required for 45·4% (69 out of 152) diagnoses, primarily to resolve the functional impact of splicing or structural variants and/or to demonstrate reduced levels and/or altered size of the encoded protein (Fig. 3C). For a small subset of cases, auxiliary studies were performed to exclude other possible candidate variants, and/or to support likely pathogenicity of novel disease genes. Variants disrupting pre‐mRNA splicing account for more than one‐third of diagnoses and exceed novel disease gene discovery by 9:1.
Phenotype‐driven deeper scrutiny of known associated genes was a cornerstone behind many diagnoses. For example, for 14 cases with exomic detection of a single variant in a phenotypically concordant, recessive NMD gene (“single hit”)—the second pathogenic variant was identified by microarray (in an external diagnostic laboratory) (2 out of 14) or genome sequencing and/or RNA studies (12 out of 14). Further, for 7 exome‐negative cases involving 11 affected males with suspected dystrophinopathy (persistently elevated creatine kinase, myalgia, with or without weakness), we identified structural and splice‐altering variants in DMD via genome (6 out of 7) and RNA‐Seq (1 out of 7). 23
Building on evidence‐based outcomes from our large NMD cohort, Figure 5 provides our expert team's best recommendation on how and when to deploy genome sequencing, RNA, and protein studies in different contexts. We recommend starting either with a targeted NMD gene panel or exome sequencing, depending on regional availability and pretesting clinical hypotheses, in accordance with the different strengths of each technique. A targeted NMD panel provides increased coverage for improved copy number variation and mosaic variant detection, due to the approximately sixfold reduction in the number of genes. 9 Alternatively, exome sequencing enables stepwise interrogation of known NMD genes, followed by other Mendelian genes which is expressly useful when the phenotype involves global developmental features in addition to neuromuscular involvement.
Figure 5.
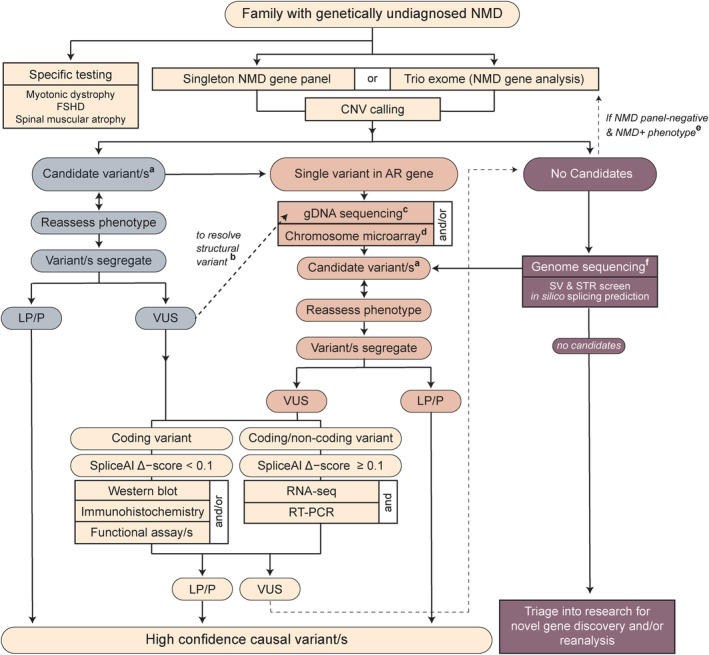
Proposed diagnostic algorithm for neuromuscular disorders. Our recommendation for genomic testing in neuromuscular disorders (NMD) begins with a high coverage, targeted‐capture NMD panel or exome sequencing interrogated for a regularly updated list of NMD disease genes. Depending on whether candidate variants are found in a phenotypically concordant genea, further resolution of VUSb or finding a second, likely‐splice altering or structural variant through gDNA sequencingc or microarrayd, may be necessary. When no candidate variants are detected from the initial screen, trio exome sequencing is only recommended when the presentation suggests other multi‐system abnormalities, with the potential for a causal gene not covered on an NMD panele. Instead, we recommend genome sequencing; singleton for X‐linked or consanguineous pedigrees, and trio genome for suspected autosomal recessive or de novo dominant pedigreesf. From either the panel, exome, or genome branches of the algorithm, variants are divided into suspected splice‐altering (SpliceAI delta score ≥0.1) or not (≤0.1) and functional studies conducted as detailed. We acknowledge that immunostaining, western blot, or functional assays to resolve pathogenicity of VUS are services not available to most tertiary centers. However, RNA testing is likely to become increasingly available over the next 5 years. Our previous work outlines clinically endorsed strategies for RNA testing. 25 Due to the complexity of RNA and known caveats with alignment of short sequencing reads, we recommend confirming all reportable findings detected by RNA‐Seq by RT‐PCR. 25 This algorithm provides a comprehensive interrogation of massively parallel sequencing data for coding, splicing, and structural variants in NMD. AR, autosomal recessive; CNV, copy number variant; FSHD, facioscapulohumeral muscular dystrophy; LP/P, likely pathogenic/pathogenic; NMD, neuromuscular disorder; RT‐PCR, reverse transcription PCR; STR, short tandem repeat; SV, structural variant; VUS, variant of uncertain significance.
We acknowledge that many clinical departments do not have access to laboratory support for immunostaining, western blot, or functional assays to resolve pathogenicity of VUS described within our Figure 5 algorithm. However, RNA diagnostics is emerging onto the diagnostic horizon and is likely to become increasingly available. RNA studies as a single adjunct investigation gives the greatest incremental increase in diagnostic yield from 34% to 42%: our maximal current diagnostic yield of 62% required a combination of additional genomic sequencing, RNA and/or protein‐based investigations.
As a molecular diagnosis rests on a segregating DNA variant, one requires both gDNA and RNA sequencing information to identify, and confirm the pathogenicity of, splice‐altering variants. The tricky part is deciding which test to conduct first. Singleton genome sequencing is often informative for consanguineous or X‐linked recessive pedigrees. However, singleton genome is rarely informative for non‐consanguineous recessive pedigrees, which are most likely to be compound heterozygous (74% of autosomal recessive families in our cohort; 69 out of 93; Figure 3Ai), or de novo dominant pedigrees, due to great challenges in identifying de novo variants and in filtering intronic variants. In our laboratory, the specificity of the phenotype for “single hit” or “exome negative” cases (see Fig. 5) guides our testing strategy. For cases involving a phenotype highly specific for a genetic condition associated with only one or a few genes, especially when another diagnostic test is strongly indicative of that genetic disorder, we recommend singleton genome sequencing with targeted curation of the gene(s) associated with this condition (significantly lower cost than whole genome curation), combined with, or followed by, RNA testing of an appropriate biospecimen (as determined via guidelines in 25 ) and segregation studies. Even if a variant is not identified by genome sequencing, we recommend conducting RNA testing to exclude any abnormalities in the strongly associated candidate genes(s), due to known shortcomings in short read sequencing alignment, particularly for structural variants, as we discuss further below. For less specific phenotypes with high genetic heterogeneity, we recommend whole genome sequencing with curation of all NMD genes as the better first step.
Our experience utilizing FRASER (Find Rare Splicing Events in RNA‐Seq; v1.0.2) 53 to identify splicing outliers from muscle RNA‐Seq was technically and resource intensive and likely beyond the capacity of most clinical diagnostic laboratories. For example, running FRASER for 26 samples took 4 days super‐computing time and ~ 8 h per case of subsequent processing on a standard computer prior to analysis of the reported data. The scale of the reported output is typically too large to interpret without first filtering for splicing outliers proximal to (± 250 nt) a segregating, rare variant identified by genome sequencing (see three example families in Fig. 4A). Though variant calling from RNA‐Seq is reported by others, 57 we have had limited success with this approach, consider it too unreliable for variant calling in a diagnostic context and recommend genomic sequencing. Our opinion is that agnostic interrogation of transcriptomic RNA‐Seq data is currently most effective when it is possible to filter for splicing outliers that lie proximal to a rare variant (identified first via genome sequencing).
Due to the high rate of novel disease gene discovery, we concordantly advocate diagnostic benefits of annual or bi‐annual re‐analysis of available sequencing data for undiagnosed probands using updated gene lists. 18 For cases sequenced on early exome platforms, or platforms with low coverage of some NMD genes, or pipelines that did not capture (or assess) 50 intronic nucleotides, exomic re‐sequencing is a worthwhile consideration. Novel disease discovery is being enhanced substantially by aggregation of genotype/phenotype data from affected families through international collaboration, such as the Matchmaker Exchange, 58 and with the increase in MPS and data sharing, the ability to identify and characterize novel disease genes is likely to improve. 7 , 17
There are important caveats in short‐read sequencing that must be acknowledged. Repetitive regions of the genome, including those involved in repeat expansion disorders, remain elusive and are common causes of NMDs that require considered testing beyond the algorithm in Figure 5. So too, short‐read sequencing cannot always identify, align reads to, or resolve the specific nature of, structural variants. 59 Stranded sequencing protocols present a substantial advantage over non‐stranded protocols for observing abnormalities in read orientation within a strongly associated disease gene. Most of our structural variants were identified only via manual, targeted scrutiny of a suspected causal gene, with soft‐clipped reads in the genomic and/or transcriptomic data sometimes the only hallmark. Resolving the structural variant was often a complex, manual process, moving iteratively back and forth between RT/PCR and short‐read gDNA and RNA sequencing data. We believe that long‐read sequencing is likely to play an important role in future diagnostic algorithms for neuromuscular disorders, as has been demonstrated by resolving structural variants 60 , 61 and short tandem repeats. 62 Orthogonal structural variant calling approaches including optical genome mapping may also be considered for future adoption. 63
Splice‐altering and structural variants may be particularly prevalent in NMDs due to the long length and high number of exons present in common NMD associated genes such as DMD, TTN, NEB, RYR1, and myosins. Two previously published cohorts of exome‐negative NMD cases subject to RNA‐Seq similarly report ~30–35% causal splice‐altering or structural variants. 19 , 21 Our collective findings suggest it is highly likely that DNA variants that alter pre‐mRNA splicing are a common basis for disease among our remaining 95 out of 247 exome‐negative families, including in novel disease genes we are yet to identify, and therefore our current research is focused upon genomic sequencing and/or RNA studies for 19 out of 95 families prioritized based on a severe presentation in childhood and at least one specimen available for RNA studies. The considerable number of variants altering pre‐mRNA splicing in our cohort is particularly relevant given the growing application of precision splice‐switching oligonucleotide therapeutics. We also recognize the possibility that some undiagnosed families may have neuromuscular conditions with a nongenetic or polygenic etiology.
Our study demonstrates that the incorporation of additional genome, transcript, and protein investigations after exomic sequencing can substantially augment diagnostic yield in NMDs from 40% to >60%.
Massively parallel sequencing in NMDs has accelerated discovery of novel NMD genes, improved detection of pathogenic structural variants, and transformed our ability to detect pathogenic splice‐altering variants. With splice‐altering and/or structural variants so prevalent among our diagnosed families, our overarching message is a simple one: “go back to the gene(s) where you think the problem is and have a really good look.” Good clinical phenotyping followed by scrutiny of all coding and non‐coding rare genetic variation in phenotypically consistent genes will be key to enhancing diagnostic yield in NMD. We hope that our evidence‐based diagnostic algorithm, devised from lessons learned and diagnostic outcomes from a decade of MPS testing in NMDs, may provide a practical guide for NMD clinicians as they prospectively negotiate emerging access to genome and transcriptome sequencing, to maximize diagnostic yield in NMDs to 60% or more.
Author Contributions
R.G. Marchant Concept and design of the study, acquisition, and analysis of data, and drafting the manuscript/figures for intellectual content. S.J. Bryen Concept and design of the study, acquisition and analysis of data, and drafting the manuscript/figures for intellectual content. M. Bahlo Acquisition and analysis of data A. Cairns Acquisition and analysis of data. K.R. Chao Acquisition and analysis of data. A. Corbett Acquisition and analysis of data. M.R. Davis Acquisition and analysis of data V.S. Ganesh Acquisition and analysis of data. Funded study. R. Ghaoui Acquisition and analysis of data. K.J. Jones Acquisition and analysis of data. A.J. Kornberg Acquisition and analysis of data. M. Lek Acquisition and analysis of data. Funded study. C. Liang Acquisition and analysis of data. D.G. MacArthur Concept and design of the study. Funded study. E.C. Oates Acquisition and analysis of data. A. O'Donnell‐Luria Acquisition and analysis of data. Funded study. G.L. O'Grady Acquisition and analysis of data. I.A. Osei‐Owusu Acquisition and analysis of data. H. Rafehi Acquisition and analysis of data. S.W. Reddel Acquisition and analysis of data. R.H. Roxburgh Acquisition and analysis of data. M.M. Ryan Acquisition and analysis of data. S.A. Sandaradura Acquisition and analysis of data. L.W. Scott Acquisition and analysis of data. E. Valkanas Acquisition and analysis of data. B. Weisburd Acquisition and analysis of data. H. Young Acquisition and analysis of data. F.J. Evesson Concept and design of the study, acquisition and analysis of data, and drafting the manuscript/figures for intellectual content. L.B. Waddell Concept and design of the study, acquisition and analysis of data, and drafting the manuscript/figures for intellectual content. S.T. Cooper Concept and design of the study. Funded study. Acquisition and analysis of data and drafting the manuscript/figures for intellectual content.
Conflict of Interest
S.T. Cooper is director of Frontier Genomics Pty Ltd (Australia). S.T. Cooper receives no remuneration (salary or consultancy fees) for this role. Frontier Genomics Pty Ltd has no current financial interests that will benefit from publication of this data. S.T. Cooper is a named inventor on intellectual property owned jointly by the University of Sydney and Sydney Children's Hospitals Network. This IP relates to splicing variant detection and interpretation and is licensed by Frontier Genomics Pty Ltd. S.T. Cooper is named inventor on Australian Patent No. 2019379868 and Australian Provisional Patent No. 2019900836. The remaining co‐authors declare no competing interests.
Supporting information
Table S1.
Acknowledgments
In memorial of A/Professor Nigel Clarke. We thank the families involved in this study for their invaluable contributions to this research. We also thank the clinicians and health care workers involved in their assessment and management. Special thanks to long‐term comrades Nigel Laing, Gina Ravenscroft, Robert Bryson‐Richardson as part of our Australasian Neuromuscular Research Network, and to Beryl Cummings, Taru Tukiainen, Alysia Lovgren, Katie Larkin, Jaime Chang, and other members from the Broad Institute of MIT and Harvard Center for Mendelian Genomics (CMG). We are extremely grateful to the Broad Center for Mendelian Genomics team for a wonderful collaboration that has enabled privileged early access of our undiagnosed, Australasian NMD cohort to the latest advances in parallel sequencing and analytical technologies. S.T. Cooper had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. S.T. Cooper received support from National Health and Medical Research Council (NHMRC) of Australia Fellowships APP1048816 and APP1136197, NHMRC Project Grant APP1080587 and NHMRC Ideas Grants APP1106084, APP2002640. S.J. Bryen received a Muscular Dystrophy Association of New South Wales Sue Connor postgraduate training scholarship. E.C. Oates received a NHMRC ECR Fellowship APP1090428. G.L. O'Grady was supported by an NHMRC postgraduate scholarship APP1056285. R. Ghaoui was supported by NHMRC grant APP1074954 and Muscular Dystrophy New South Wales. M. Bahlo was supported by an NHMRC Leadership 1 grant APP1195236. H. Rafehi was supported by an NHMRC Emerging Leadership 1 grant APP1194364. V.S. Ganesh was supported by NIH/NHGRI T32HG010464. Sequencing and analysis provided by the Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) was funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1HG008900 and in part by National Human Genome Research Institute grant R01HG009141 to D. MacArthur and A. O'Donnell‐Luria. Open access publishing facilitated by The University of Sydney, as part of the Wiley ‐ The University of Sydney agreement via the Council of Australian University Librarians.
Funding Statement
This work was funded by National Human Genome Research Institute grants R01HG009141 and T32HG010464; National Health and Medical Research Council grants APP1048816, APP1056285, APP1074954, APP1080587, APP1090428, APP1106084, APP1136197, APP1194364, APP1195236, and APP2002640; Muscular Dystrophy NSW; National Eye Institute ; National Heart, Lung and Blood Institute grant UM1HG008900.
Data Availability Statement
Data compiled and analyzed in this study are available upon request to the corresponding author or through dbGaP phs001272 under the Broad Institute Center for Mendelian Genomics projects “AnVIL_CMG_Broad_Muscle_INMR‐Cooper_WES,” “AnVIL_CMG_Broad_Muscle_KNC_WES,” and “AnVIL_CMG_Broad_Muscle_KNC_WGS” for 220 families (63 undiagnosed). Genomic data for 27 families (7 undiagnosed) with clinical exome or genome sequencing cannot be shared.
References
- 1. Laing NG. Genetics of neuromuscular disorders. Crit Rev Clin Lab Sci. 2012;49(2):33‐48. doi: 10.3109/10408363.2012.658906 [DOI] [PubMed] [Google Scholar]
- 2. Fraiman YS, Wojcik MH. The influence of social determinants of health on the genetic diagnostic odyssey: who remains undiagnosed, why, and to what effect? Pediatr Res. 2021;89(2):295‐300. doi: 10.1038/s41390-020-01151-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Beecroft SJ, Lamont PJ, Edwards S, et al. The impact of next‐generation sequencing on the diagnosis, treatment, and prevention of hereditary neuromuscular disorders. Mol Diagn Ther. 2020;24(6):641‐652. doi: 10.1007/s40291-020-00495-2 [DOI] [PubMed] [Google Scholar]
- 4. North KN, Wang CH, Clarke N, et al. Approach to the diagnosis of congenital myopathies. Neuromuscul Disord. 2014;24(2):97‐116. doi: 10.1016/j.nmd.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Anderson LVB, Davison K. Multiplex Western blotting system for the analysis of muscular dystrophy proteins. Am J Pathol. 1999;154(4):1017‐1022. doi: 10.1016/S0002-9440(10)65354-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peat RA, Smith JM, Compton AG, et al. Diagnosis and etiology of congenital muscular dystrophy. Neurology. 2008;71(5):312‐321. doi: 10.1212/01.wnl.0000284605.27654.5a [DOI] [PubMed] [Google Scholar]
- 7. Bamshad MJ, Nickerson DA, Chong JX. Mendelian gene discovery: fast and furious with No end in sight. Am J Hum Genet. 2019;105(3):448‐455. doi: 10.1016/j.ajhg.2019.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cohen E, Bonne G, Rivier F, Hamroun D. The 2022 version of the gene table of neuromuscular disorders (nuclear genome). Neuromuscul Disord. 2021;31(12):1313‐1357. doi: 10.1016/j.nmd.2021.11.004 [DOI] [PubMed] [Google Scholar]
- 9. Beecroft SJ, Yau KS, Allcock RJN, et al. Targeted gene panel use in 2249 neuromuscular patients: the Australasian referral center experience. Ann Clin Transl Neurol. 2020;7(3):353‐362. doi: 10.1002/acn3.51002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large lgmd cohort of 4656 patients. Ann Clin Transl Neurol. 2018;5(12):1574‐1587. doi: 10.1002/acn3.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ghaoui R, Cooper ST, Lek M, et al. Use of whole‐exome sequencing for diagnosis of limb‐girdle muscular dystrophy: outcomes and lessons learned. JAMA Neurol. 2015;72(12):1424. doi: 10.1001/jamaneurol.2015.2274 [DOI] [PubMed] [Google Scholar]
- 12. Herman I, Lopez MA, Marafi D, et al. Clinical exome sequencing in the diagnosis of pediatric neuromuscular disease. Muscle Nerve. 2021;63(3):304‐310. doi: 10.1002/mus.27112 [DOI] [PubMed] [Google Scholar]
- 13. O'Grady GL, Lek M, Lamande SR, et al. Diagnosis and etiology of congenital muscular dystrophy: we are halfway there: diagnosis of CMD. Ann Neurol. 2016;80(1):101‐111. doi: 10.1002/ana.24687 [DOI] [PubMed] [Google Scholar]
- 14. Reddy HM, Cho KA, Lek M, et al. The sensitivity of exome sequencing in identifying pathogenic mutations for LGMD in the United States. J Hum Genet. 2017;62(2):243‐252. doi: 10.1038/jhg.2016.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schofield D, Alam K, Douglas L, et al. Cost‐effectiveness of massively parallel sequencing for diagnosis of paediatric muscle diseases. NPJ Genom Med. 2017;2(1):4. doi: 10.1038/s41525-017-0006-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Clement EM, Feng L, Mein R, et al. Relative frequency of congenital muscular dystrophy subtypes: analysis of the UK diagnostic service 2001–2008. Neuromuscul Disord. 2012;22(6):522‐527. doi: 10.1016/j.nmd.2012.01.010 [DOI] [PubMed] [Google Scholar]
- 17. Cohen ASA, Farrow EG, Abdelmoity AT, et al. Genomic answers for children: dynamic analyses of >1000 pediatric rare disease genomes. Genet Med. 2022;24(6):1336‐1348. doi: 10.1016/j.gim.2022.02.007 [DOI] [PubMed] [Google Scholar]
- 18. Elliott AM, Adam S, du Souich C, et al. Genome‐wide sequencing and the clinical diagnosis of genetic disease: the CAUSES study. Hum Genet Genomics Adv. 2022;3(3):100108. doi: 10.1016/j.xhgg.2022.100108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9(386):eaal5209. doi: 10.1126/scitranslmed.aal5209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Frésard L, Smail C, Ferraro NM, et al. Identification of rare‐disease genes using blood transcriptome sequencing and large control cohorts. Nat Med. 2019;25(6):911‐919. doi: 10.1038/s41591-019-0457-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gonorazky HD, Naumenko S, Ramani AK, et al. Expanding the boundaries of RNA sequencing as a diagnostic tool for rare Mendelian disease. Am J Hum Genet. 2019;104(3):466‐483. doi: 10.1016/j.ajhg.2019.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Douglas AGL, Baralle D. Translating RNA splicing analysis into diagnosis and therapy. OBM Genet. 2021;5(1):125. doi: 10.21926/obm.genet.2101125 [DOI] [Google Scholar]
- 23. Waddell LB, Bryen SJ, Cummings BB, et al. WGS and RNA studies diagnose noncoding DMD variants in males with high Creatine kinase. Neurol Genet. 2021;7(1):e554. doi: 10.1212/NXG.0000000000000554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stark Z, Schofield D, Martyn M, et al. Does genomic sequencing early in the diagnostic trajectory make a difference? A follow‐up study of clinical outcomes and cost‐effectiveness. Genet Med. 2019;21(1):173‐180. doi: 10.1038/s41436-018-0006-8 [DOI] [PubMed] [Google Scholar]
- 25. Bournazos AM, Riley LG, Bommireddipalli S, et al. Standardized practices for RNA diagnostics using clinically accessible specimens reclassifies 75% of putative splicing variants. Genet Med. 2022;24(1):130‐145. doi: 10.1016/j.gim.2021.09.001 [DOI] [PubMed] [Google Scholar]
- 26. Farnaes L, Hildreth A, Sweeney NM, et al. Rapid whole‐genome sequencing decreases infant morbidity and cost of hospitalization. NPJ Genom Med. 2018;3(1):10. doi: 10.1038/s41525-018-0049-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gofin Y, Wang T, Gillentine MA, et al. Delineation of a novel neurodevelopmental syndrome associated with PAX5 haploinsufficiency. Hum Mutat. 2022;43(4):461‐470. doi: 10.1002/humu.24332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bryen SJ, Ewans LJ, Pinner J, et al. Recurrent TTN metatranscript‐only c.39974–11T>G splice variant associated with autosomal recessive arthrogryposis multiplex congenita and myopathy. Hum Mutat. 2020;41(2):403‐411. doi: 10.1002/humu.23938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bryen SJ, Joshi H, Evesson FJ, et al. Pathogenic abnormal splicing due to Intronic deletions that induce biophysical space constraint for spliceosome assembly. Am J Hum Genet. 2019;105(3):573‐587. doi: 10.1016/j.ajhg.2019.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pais LS, Snow H, Weisburd B, et al. seqr : a web‐based analysis and collaboration tool for rare disease genomics. Hum Mutat. 2022;43:698‐707. doi: 10.1002/humu.24366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Exome Aggregation Consortium , Lek M, Karczewski KJ, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285‐291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434‐443. doi: 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Amberger JS, Bocchini CA, Scott AF, Hamosh A. OMIM.Org: leveraging knowledge across phenotype–gene relationships. Nucleic Acids Res. 2019;47(D1):D1038‐D1043. doi: 10.1093/nar/gky1151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018;46(D1):D1062‐D1067. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fokkema IFAC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32(5):557‐563. doi: 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- 37. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248‐249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comput Biol. 1997;4(3):311‐323. doi: 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- 39. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell. 2019;176(3):535‐548.e24. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 40. Carss KJ, Stevens E, Foley AR, et al. Mutations in GDP‐mannose Pyrophosphorylase B cause congenital and limb‐girdle muscular dystrophies associated with Hypoglycosylation of α‐Dystroglycan. Am J Hum Genet. 2013;93(1):29‐41. doi: 10.1016/j.ajhg.2013.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ilkovski B, Pagnamenta AT, O'Grady GL, et al. Mutations in PIGY : expanding the phenotype of inherited glycosylphosphatidylinositol deficiencies. Hum Mol Genet. 2015;24(21):6146‐6159. doi: 10.1093/hmg/ddv331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oates EC, Rossor AM, Hafezparast M, et al. Mutations in BICD2 cause dominant congenital spinal muscular atrophy and hereditary spastic paraplegia. Am J Hum Genet. 2013;92(6):965‐973. doi: 10.1016/j.ajhg.2013.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. O'Grady GL, Verschuuren C, Yuen M, et al. Variants in SLC18A3, vesicular acetylcholine transporter, cause congenital myasthenic syndrome. Neurology. 2016;87(14):1442‐1448. doi: 10.1212/WNL.0000000000003179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yuen M, Sandaradura SA, Dowling JJ, et al. Leiomodin‐3 dysfunction results in thin filament disorganization and nemaline myopathy. J Clin Invest. 2014;124(11):4693‐4708. doi: 10.1172/JCI75199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. O'Grady GL, Best HA, Sztal TE, et al. Variants in the oxidoreductase PYROXD1 cause early‐onset myopathy with internalized nuclei and Myofibrillar disorganization. Am J Hum Genet. 2016;99(5):1086‐1105. doi: 10.1016/j.ajhg.2016.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bucelli RC, Arhzaouy K, Pestronk A, et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology. 2015;85(8):665‐674. doi: 10.1212/WNL.0000000000001864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cabrera‐Serrano M, Ghaoui R, Ravenscroft G, et al. Expanding the phenotype of GMPPB mutations. Brain. 2015;138(4):836‐844. doi: 10.1093/brain/awv013 [DOI] [PubMed] [Google Scholar]
- 48. Ghaoui R, Palmio J, Brewer J, et al. Mutations in HSPB8 causing a new phenotype of distal myopathy and motor neuropathy. Neurology. 2016;86(4):391‐398. doi: 10.1212/WNL.0000000000002324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghaoui R, Benavides T, Lek M, et al. TOR1AIP1 as a cause of cardiac failure and recessive limb‐girdle muscular dystrophy. Neuromuscul Disord. 2016;26(8):500‐503. doi: 10.1016/j.nmd.2016.05.013 [DOI] [PubMed] [Google Scholar]
- 50. Markello T, Chen D, Kwan JY, et al. York platelet syndrome is a CRAC channelopathy due to gain‐of‐function mutations in STIM1. Mol Genet Metab. 2015;114(3):474‐482. doi: 10.1016/j.ymgme.2014.12.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. O'Grady GL, Ma A, Sival D, et al. Prominent scapulae mimicking an inherited myopathy expands the phenotype of CHD7‐related disease. Eur J Hum Genet. 2016;24(8):1216‐1219. doi: 10.1038/ejhg.2015.276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sandaradura SA, Bournazos A, Mallawaarachchi A, et al. Nemaline myopathy and distal arthrogryposis associated with an autosomal recessive TNNT3 splice variant. Hum Mutat. 2018;39(3):383‐388. doi: 10.1002/humu.23385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mertes C, Scheller IF, Yépez VA, et al. Detection of aberrant splicing events in RNA‐seq data using FRASER. Nat Commun. 2021;12(1):529. doi: 10.1038/s41467-020-20573-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lonsdale J, Thomas J, Salvatore M, et al. The genotype‐tissue expression (GTEx) project. Nat Genet. 2013;45(6):580‐585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lara A, Damasceno DD, Pires R, et al. Dysautonomia due to reduced cholinergic neurotransmission causes cardiac remodeling and heart failure. Mol Cell Biol. 2010;30(7):1746‐1756. doi: 10.1128/MCB.00996-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Prado VF, Martins‐Silva C, de Castro BM, et al. Mice deficient for the vesicular acetylcholine transporter are myasthenic and have deficits in object and social recognition. Neuron. 2006;51(5):601‐612. doi: 10.1016/j.neuron.2006.08.005 [DOI] [PubMed] [Google Scholar]
- 57. Yépez VA, Gusic M, Kopajtich R, et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022;14(1):38. doi: 10.1186/s13073-022-01019-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Philippakis AA, Azzariti DR, Beltran S, et al. The matchmaker exchange: a platform for rare disease gene discovery. Hum Mutat. 2015;36(10):915‐921. doi: 10.1002/humu.22858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Colin E, Duffourd Y, Chevarin M, et al. Stepwise use of genomics and transcriptomics technologies increases diagnostic yield in Mendelian disorders. Front Cell Dev Biol. 2023;11:1021920. doi: 10.3389/fcell.2023.1021920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miller DE, Sulovari A, Wang T, et al. Targeted long‐read sequencing identifies missing disease‐causing variation. Am J Hum Genet. 2021;108(8):1436‐1449. doi: 10.1016/j.ajhg.2021.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bruels CC, Littel HR, Daugherty AL, et al. Diagnostic capabilities of nanopore long‐read sequencing in muscular dystrophy. Ann Clin Transl Neurol. 2022;9(8):1302‐1309. doi: 10.1002/acn3.51612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mitsuhashi S, Nakagawa S, Sasaki‐Honda M, Sakurai H, Frith MC, Mitsuhashi H. Nanopore direct RNA sequencing detects DUX4‐activated repeats and isoforms in human muscle cells. Hum Mol Genet. 2021;30(7):552‐563. doi: 10.1093/hmg/ddab063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Mantere T, Neveling K, Pebrel‐Richard C, et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am J Hum Genet. 2021;108(8):1409‐1422. doi: 10.1016/j.ajhg.2021.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.
Data Availability Statement
Data compiled and analyzed in this study are available upon request to the corresponding author or through dbGaP phs001272 under the Broad Institute Center for Mendelian Genomics projects “AnVIL_CMG_Broad_Muscle_INMR‐Cooper_WES,” “AnVIL_CMG_Broad_Muscle_KNC_WES,” and “AnVIL_CMG_Broad_Muscle_KNC_WGS” for 220 families (63 undiagnosed). Genomic data for 27 families (7 undiagnosed) with clinical exome or genome sequencing cannot be shared.