

Targeting NPEPPS, which induces cisplatin resistance by controlling intracellular drug concentrations, is a potential strategy to improve patient responses to platinum-based therapies and lower treatment-associated toxicities.
Abstract
There is an unmet need to improve the efficacy of platinum-based cancer chemotherapy, which is used in primary and metastatic settings in many cancer types. In bladder cancer, platinum-based chemotherapy leads to better outcomes in a subset of patients when used in the neoadjuvant setting or in combination with immunotherapy for advanced disease. Despite such promising results, extending the benefits of platinum drugs to a greater number of patients is highly desirable. Using the multiomic assessment of cisplatin-responsive and -resistant human bladder cancer cell lines and whole-genome CRISPR screens, we identified puromycin-sensitive aminopeptidase (NPEPPS) as a driver of cisplatin resistance. NPEPPS depletion sensitized resistant bladder cancer cells to cisplatin in vitro and in vivo. Conversely, overexpression of NPEPPS in sensitive cells increased cisplatin resistance. NPEPPS affected treatment response by regulating intracellular cisplatin concentrations. Patient-derived organoids (PDO) generated from bladder cancer samples before and after cisplatin-based treatment, and from patients who did not receive cisplatin, were evaluated for sensitivity to cisplatin, which was concordant with clinical response. In the PDOs, depletion or pharmacologic inhibition of NPEPPS increased cisplatin sensitivity, while NPEPPS overexpression conferred resistance. Our data present NPEPPS as a druggable driver of cisplatin resistance by regulating intracellular cisplatin concentrations.
Significance:
Targeting NPEPPS, which induces cisplatin resistance by controlling intracellular drug concentrations, is a potential strategy to improve patient responses to platinum-based therapies and lower treatment-associated toxicities.
Introduction
Platinum-based chemotherapy has been successful in testicular, ovarian, bladder, head and neck, and lung cancers and remains a standard of care for many patients despite the advent of immunotherapy (1–3). However, dose-dependent side effects and chemoresistance have reduced the suitability and effectiveness of these drugs. The discovery of more effective agents and the development of strategies that improve the efficacy of platinum-based regimens are approaches that can address these issues. Here, we describe results from the latter approach, using bladder cancer as a clinically relevant, and translationally tractable tumor model.
Bladder cancer accounts for over 573,000,000 new diagnoses and 212,000 deaths worldwide annually (4, 5). Cisplatin-based combination chemotherapy remains the first-line, standard of care for metastatic bladder cancer, providing a 5% to 10% cure rate. However, up to 30% of patients are ineligible for cisplatin-based treatment (6) and are commonly offered immune-checkpoint therapies (ICT; ref. 7). Of the patients treated with ICT, the response rate is about 30% (8). Recent results from the CheckMate 901 trial showed a nearly 3-fold increase in the duration of complete response when nivolumab was given in combination with cisplatin-based chemotherapy in the metastatic setting (9). Cisplatin-based combination chemotherapy is also standard of care in the neoadjuvant (NAC) setting for the management of localized muscle-invasive bladder cancer (MIBC; refs. 10, 11). However, NAC adoption has been slow due to cisplatin toxicity and patient eligibility, along with the relatively small survival benefit of 5% to 15% over immediate cystectomy (12). Importantly, in both the metastatic and NAC settings, patient selection and therapeutic efficacy remain critical unresolved challenges (13). These results support the relevance and promise of platinum-based therapies and highlight the potential benefit of extending these benefits to a greater number of patients.
Large-scale efforts have performed whole-genome loss-of-function screening across hundreds of cancer cell lines using CRISPR- and shRNA-based libraries to define pan-cancer and context-specific genetic dependencies (14, 15). Although these efforts defined genetic dependencies in cancer, a limitation is that cells were grown under basal growth conditions in the absence of any treatment. Those studies were also performed in cells that had not acquired resistance to any treatments. To better understand the functional drivers of therapeutic resistance in the context of platinum drug treatment, here we performed CRISPR screens in the presence and absence of the therapy of interest (16–18), and in cells that have acquired resistance to the treatment itself (Fig. 1A). Complementing these results with multiomic profiling of treatment-responsive and -resistant cells allowed us to prioritize gene candidates for the best potential future clinical utility. Using these data, which are made available as a public resource (https://bioinformatics.cuanschutz.edu/BLCA_GC_Omics/), we identified puromycin-sensitive aminopeptidase, NPEPPS, as a novel driver of cisplatin resistance and validated this finding in vitro and in vivo. We then show that NPEPPS has its effect by directly regulating the intracellular concentrations of cisplatin. To determine the translational relevance of this discovery, tumor-derived organoids generated from patients before and after cisplatin-based chemotherapy were used to validate and strengthen our results from bladder cancer cells and xenografts. In cell lines, xenografts, and organoids, we show that pharmacologic inhibition of NPEPPS by tosedostat, a clinically used small molecule that inhibits NPEPPS, phenocopies genetic depletion of NPEPPS. Taken together, our data support NPEPPS as a therapeutic target and provide a compelling rationale for combining NPEPPS inhibition with cisplatin to improve bladder cancer patient outcomes.
Figure 1.

Project overview and synthetic lethal screen results. A, Human bladder cancer cell lines were made resistant to cisplatin, gemcitabine, or gemcitabine plus cisplatin through dose escalation. All cell lines were profiled using -omic technologies. The gemcitabine plus cisplatin-resistant cells were subjected to a pooled CRISPR screen to identify synthetic lethal gene-to-drug relationships. B, Aggregate gene set enrichment results for the synthetic lethal screen ranked by log2-fold change across all cell lines reveal DNA damage response and repair pathways. Each tick mark represents a gene in the associated pathway. The bars at the right are normalized enrichment scores (NES), with the FDR-corrected P values reported in the bars. C, The intersection across the CRISPR screen results identified 46 common synthetic lethal genes; all counts and gene annotations are reported in Supplementary Fig. S2. D, The percentage change in the aggregate of the sgRNAs targeting the 46 commonly synthetic lethal genes are reported across PBS or gemcitabine plus cisplatin treatment arms of the CRISPR screen. Cell lines are coded with the same colors throughout all figures.
Materials and Methods
Cell culture
All human bladder cancer cell lines as reported in the Key Resource Table were obtained from the Resistant Cancer Cell Line (RCCL) collection and were grown in Iscove's Modified Dulbecco's Medium (IMDM) with 10% fetal bovine serum (FBS). Cells were passaged every two to three days. Resistance to gemcitabine and cisplatin was confirmed at the reported resistance doses from the RCCL (Supplementary Table S1; Supplementary Fig. S1). Lentivirus production utilized 293FT cells (Thermo Fisher), which were maintained in DMEM (high glucose) supplemented with 0.1 mmol/L nonessential amino acids (NEAA), 6 mmol/L L-glutamine, 1 mmol/L sodium pyruvate, and 500 μg/mL geneticin (G418) with 10% FBS added. Cells were routinely monitored for Mycoplasma and confirmed negative multiple times during this study using MycoAlert (Lonza). All cells were grown at 37°C with 5% CO2 in a humidified incubator.
All molecular characterization efforts (RNA sequencing, whole-exome sequencing, and mass spectrometric proteomics) were performed on cells from independent passages and in drug-free, complete media to identify stable molecular changes rather than treatment-induced transient responses. Cells were routinely passaged through drug-containing media at the resistant doses (Supplementary Table S1) to confirm resistance was maintained and early passage cells were utilized whenever possible.
RNA sequencing
Sample preparation
All cell lines were grown for several passages in the absence of antibiotics, gemcitabine, or cisplatin. Cell pellets were snap frozen from subconfluent dishes from 3 separate passages (replicates) for each of the 20 cell lines sequenced (5 cell lines, each with 4 derivatives: parental, Gem-resistant, Cis-resistant, GemCis-resistant). RNA was extracted using the RNAeasy Plus Kit (Qiagen). Cells were lysed and passed through the QIAShredder column (Qiagen) according to the manufacturer's protocol. gDNA elimination columns (Qiagen) were used to remove any residual gDNA from the purified RNA. RNA integrity was assessed on the High Sensitivity ScreenTape Assay on the TapeStation 2200 (Agilent) and only samples with a RIN score of 8 or higher were used for sequencing. RNA library preparation was performed using the Universal Plus mRNA–seq +UDI kit (Nugen) according to the manufacturer's specifications. Each library was sequenced to a minimum of 40 million clusters or 80 million 150 bp paired-end reads on a NovaSeq 6000 instrument (Illumina) at the University of Colorado Cancer Center Genomics Shared Resource.
Data processing
Illumina adapters and the first 12 base pairs of each read were trimmed using BBDuk and reads <50 bp post trimming were discarded. Reads were aligned and quantified using the STAR aligner against the Ensembl human transcriptome (GRCh38.p12 genome, release 96). Ensembl genes were mapped to HGNC gene symbols using HGNC and Ensembl BioMart. Gene counts were generated using the sum of counts for transcripts of the same gene. Lowly expressed genes were removed if the mean raw count <1 or mean CPM (counts per million) <1 for the entire data set. Reads were normalized to CPM using the edgeR R package. Differential expression was calculated using the voom function in the limma R package. In addition to two-group comparisons, single drug comparisons for all cell lines were generated with cell line as a covariate (Supplementary Table S2).
Alignment and transcript quantification
STAR –runThreadN 12 –runMode genomeGenerate –sjdbGTFfile Homo_sapiens.GRCh38.96.gtf –genomeFastaFiles Homo_sapiens.GRCh38.dna_sm.primary_assembly.fa
STAR –readFilesIn Read1.fastq.gz Read2.fastq.gz –readFilesCommand zcat –runThreadN 6 –alignEndsProtrude 13 ConcordantPair –outFilterScoreMinOverLread 0.66 –outFilterMatchNminOverLread 0.66 –outSAMtype BAM SortedByCoordinate –quantMode GeneCounts
Pathway analysis
Gene set enrichment analysis (GSEA) was performed using the full list of genes ranked by fold change for the indicated comparison and the fgsea R package (19) using gene sets from the Molecular Signatures Database (v7.0; ref. 20). General plots were generated with the ggplot2 and ggpubr R packages (21). Heat maps were generated with the ComplexHeatmap R package following z-score transformation (22).
Proteomics
Sample preparation
All cell lines were grown for several passages in the absence of antibiotics, gemcitabine, or cisplatin, then seeded at 100,000 to 200,000 cells per well, and grown for 48 hours in IMDM + 10% FBS. Approximately 48 hours after seeding cells, the supernatant was aspirated and cells were washed 3 times with cold phosphate-buffered saline (PBS). Cells were lysed in 100 μL of 8 mol/L urea, 50 mmol/L Tris-HCl, pH 8.0. Lysates were transferred to prechilled 1.5 mL microcentrifuge tubes and centrifuged at 15,000 RCF for 10 minutes to pellet. The supernatant was then transferred to a clean, prechilled tube and frozen. Lysate replicates were collected in triplicate from different passages. Cell pellets were lysed in 8 mol/L urea supplemented with 0.1% Rapigest MS compatible detergent. DNA was sheared using probe sonication, and protein concentration was estimated by BCA (Pierce, Thermo Scientific). A total of 30 μg protein per sample was aliquoted, and samples were diluted to <2 mol/L urea concentration using 200 mmol/L ammonium bicarbonate while also undergoing reduction with DTT (10 mmol/L) and then alkylation with IAA (100 mmol/L). The pH of diluted protein lysates was verified as between 7 and 8, and samples were digested with sequencing grade trypsin/Lys-C enzyme (Promega) in the presence of 10% acetonitrile for 16 hours at 37°C. Samples were acidified adding formic acid to 1%, and speed vac dehydration was used to evaporate acetonitrile. Peptides were desalted on C18 tips (Nest group) and dried to completion. Prior to MS, peptides were resuspended in 0.1% formic acid solution at 0.5 μg/μL concentration with 1:40 synthetic iRT reference peptides (Biognosys).
Data acquisition
Peptides were analyzed by liquid chromatography coupled with mass spectrometry in data-independent acquisition (DIA) mode essentially as described previously (23). Briefly, 4 μL of digested samples was injected directly unto a 200-cm micropillar array column (uPAC, Pharmafluidics) and separated over 120 minutes reversed phase gradient at 1,200 nL/minute and 60°C. The gradient of aqueous 0.1% formic acid (A) and 0.1% formic acid in acetonitrile (B) was implemented as follows: 2% B from 0 to 5 minutes, ramp to 4% B at 5.2 minutes, linear ramp to 28% B at 95 minutes, and ramp to 46% B at 120 minutes. After each analytical run, the column was flushed at 1,200 nL/minute and 60°C by injection of 50% methanol at 95% B for 25 minutes followed by a 10-minute ramp down to 2% B and a 5-minute equilibration to 2% B. The eluting peptides were electro-sprayed through a 30-mm bore stainless steel emitter (EvoSep) and analyzed on an Orbitrap Lumos using DIA spanning the 400 to 1,000 m/z range. Each DIA scan isolated a 4 m/z window with no overlap between windows, accumulated the ion current for a maximum of 54 seconds to a maximum AGC of 5E5, activated the selected ions by HCD set at 30% normalized collision energy, and analyzed the fragments in the 200 to 2,000 m/z range using 30,000 resolution (m/z = 200). After analysis of the full m/z range (150 DIA scans), a precursor scan was acquired over the 400 to 1,000 m/z range at 60,000 resolution.
Peptide library generation
To construct a comprehensive peptide ion library for the analysis of human bladder cancer, we combined several data sets, both internally generated and external publicly available data resources were utilized. First, we utilized a previously published (24) human bladder tumor proteomics experiment by downloading raw files from the online data repository (ProteomeXchange, PXD010260) and searching them through our internal pipeline for data-dependent acquisition MS analysis (25) against the UniProt human reviewed canonical sequence database, downloaded July 2019, using internal peptides to perform retention time alignment (26). To this library, we appended a sample-specific library generated from DIA-Umpire extraction of pseudospectra from one full set of replicates from the experimental bladder tumor cell lines. A final, combined consensus spectrast library containing all peptide identifications made between the internal and external data set was compiled and decoy sequences were appended.
Data analysis
Peptide identification was performed as previously described in (25). Briefly, we extracted chromatograms and assigned peak groups using openSWATH against the custom bladder cancer peptide assay library described above. False discovery rate for peptide identification was assigned using PyProphet and the TRIC algorithm was used to perform feature-alignment across multiple runs of different samples to maximize data completeness and reduce peak identification errors. Target peptides with a false discovery rate (FDR) of identification <1% in at least one data set file, and up to 5% across all data set files were included in the final results. We used SWATH2stats to convert our data into the correct format for use with downstream software MSstats. Each data file was intensity normalized by dividing the raw fragment intensities by the total MS2 signal. MSstats was used to convert fragment-level data into protein-level intensity estimates via the “quantData” function, utilizing default parameters with the exception of data normalization, which was set to “FALSE.” For plotting purposes, protein intensities were VSN normalized, log-transformed, and replicate batch effects were removed using the removeBatchEffect function in the limma R package. The limma package was also used to calculate differential protein expression. Multiple hypothesis correction was performed using the Benjamini–Hochberg method.
Whole-exome sequencing
Sample preparation
All cell lines were grown for several passages in the absence of antibiotics, gemcitabine, or cisplatin. Cell pellets were snap frozen from subconfluent dishes for each of the 20 cell lines sequenced (5 cell lines, each with 4 derivatives: parental, Gem-resistant, Cis-resistant, GemCis-resistant). gDNA isolation was performed using the Puregene cell and tissue kit (Qiagen) with the addition of RNase A Solution (Qiagen) according to the manufacturer's instructions. gDNA was quantified using a Qubit 4.0 and then sheared using a Covaris S220 Sonicator to 200 bp. Libraries were constructed using the Sure Select All Exon v6 library kit (Agilent) following the XT library preparation workflow. Completed libraries were run on the 4200 Tape Station (Agilent) using D1000 screen tape. Libraries were quantitated using the Qubit, diluted to 4 nmol/L prior to verification of cluster efficiency using qPCR, and then sequenced on the NovaSeq 6000 instrument (Illumina; 150 bp, paired-end) at the University of Colorado Cancer Center Genomics Shared Resource. The mean insert size across all cell lines was 177.8 bp and the mean coverage was 193.7× with > 96.8% at >30×. Individual call line quality control metrics are reported in Supplementary Table S3.
Data processing
The analysis pipeline was developed using Nextflow. For the raw fastq files, Fastqc was used to assess overall quality. For computational efficiency, raw sequence reads were partitioned using BBMap (partition.sh) into 40 partitions. They then were aligned to the GRCh38 reference genome (including decoy sequences from the GATK resource bundle) using the BWA-MEM short read aligner and merged back into single BAM files using Samtools. The resulting BAM files were deduplicated using Samblaster, and sorted using Samtools. These duplicate-marked bams were further passed through the GATK Base Quality Score Recalibration to detect systematic errors made by the sequencing machine when it estimates the accuracy of base calls. The dbSNP (version 146), the 1000 Genome Project Phase 1, and the Mills and 1000G gold-standard sets were used as databases of known polymorphic sites to exclude regions around known polymorphisms from analysis. After alignment, Samtools, Qualimap, and Picard tools were run to acquire various metrics to ensure there were no major anomalies in the aligned data.
Alignment
bwa mem -K 100000000 -R “read_group” -t 64 -M ref_fasta read_1 read_2
Marking duplicates
samtools sort -n -O SAM sample_bam | samblaster -M –ignoreUnmated
Base quality score recalibration
gatk BaseRecalibrator -I sample_bam -O sample.recal.table -R ref_fasta –known-sites known_sites
Whole-exome sequencing variant calling
We used Mutect2 from the GATK toolkit for SNVs and short indels. Mutect2 is designed to call somatic variants and makes no assumptions about the ploidy of samples. It was run in tumor-only mode to maximize the sensitivity albeit at the risk of high false positives. We used tumor-only mode to call variants for each cell line separately. Mutect2 workflow is a two-step process. In the first step, it operates in high sensitivity mode to generate intermediate callsets that are further subjected to filtering to generate the final variant calls. Annotation of variants was performed using Annovar with the following databases: refGene, cytoBand, exac03, avsnp150, clinvar_20190305, gnomad211_exome, dbnsfp35c, cosmic90. Intergenic variants were removed along with variants that were identified at greater than 0.001% of the population according to ExAC or gnomAD, or had a depth <20.
Mutect2 raw callset
gatk Mutect2 -R ref_fasta -I bam_tumor -tumor Id_tumor –germline-resource germline_resource -O raw_vcf
Mutect2 filtering:
gatk FilterMutectCalls -V raw_vcf –stats raw_vcf_stats -R ref_fasta -O filtered_mutect2_vcf
Copy-number calling using GATK
Base quality score recalibrated bams were used as the input. The covered regions for the exome kit were converted into bins (defining the resolution of the analysis) for coverage collection. Read counts, which form the basis of copy-number variant detection, were collected for each bin. The read counts then go through denoising, modeling segments, and calling the final copy ratios.
Preprocess intervals
gatk PreprocessIntervals –intervals intervals_bed_file –padding 0 –bin-length 0 -R ref_fasta –interval-merging-rule OVERLAPPING_ONLY -O preprocessed_intervals_list
Collect read counts
gatk CollectReadCounts -I sample_bam -L preprocessed_intervals} –interval-merging-rule OVERLAPPING_ONLY -O sample.counts.hdf5
Denoise read counts
gatk DenoiseReadCounts -I sample.counts.hdf5 –standardized-copy-ratios sample_std_copy_ratio –denoised-copy-ratios sample_denoised_copy_ratio
Model segments
gatk ModelSegments –denoised-copy-ratios denoised_copy_ratio –output-prefix id_sample -O output_dir
Call copy ratio segments
gatk CallCopyRatioSegments -I sample.modelled_segments -O sampled.called.segments
Cell line authentication using whole-exome sequencing
Variant calls from the Mutect2 pipeline were filtered for each cell line to identify high-confidence variants according to the filtering criteria above. These high-confidence variants were then compared with the variants reported for all cell lines in the DepMap (https://depmap.org/portal/) for the Cancer Cell Line Encyclopedia (CCLE_mutations_hg38.csv, sample_info.csv) and COSMIC (CosmicCLP_MutantExport.tsv) as measured by the Jaccard index, the intersection of variants divided by the union of variants. Cells listed in CCLE or COSMIC were rank-ordered for each bladder cancer cell line in this study according to the Jaccard index. Results are reported in Supplementary Table S4.
Cell-line drug treatments
Gemcitabine (Sigma) and cisplatin (Sigma) stocks were resuspended in 0.9% PBS solution. All stock solutions were stored protected from light and kept frozen until use. For cell culture dose response, cells were seeded in 96-well tissue culture plates with 500 to 2,000 cells per well depending on the growth rate and duration of the experiment. Cells were seeded and allowed to attach overnight, followed by replacing the media with fresh, prewarmed media just prior to treatment. Drug dilutions were performed serially and using complete media (IMDM + 10% FBS) and the associated drug treatments. Growth inhibition was measured using confluence estimates over time on the IncuCyte ZOOM (Essen Bioscience) over varying amounts of time depending on each experiment. Dose-response curves were generated with Prism v9.3.1 using a variable slope, four-parameter nonlinear regression model. Comparison between treatment groups was done between IC50 values using the sum-of-squares F test. Details for timing and replicates for each experiment are included in their respective figure legends.
Antibodies and Western blotting
Whole-cell lysates were prepared from cultured cells using RIPA lysis and extraction buffer (Thermo Scientific). Lysates from xenograft tissues were prepared using tissue protein extraction reagent (T-PER) and glass tissue homogenizer. All lysates were prepared on ice and with the addition of Halt protease and phosphatase inhibitor cocktail and EDTA (Thermo Fisher). Protein concentration of lysates was quantified with BCA protein assay (Pierce, Thermo Fisher). All lysates were prepared with 4× LI-COR Loading buffer with 50 mmol/L DTT added and boiled for 10 minutes prior to gel loading. All Western blots were run using PROTEAN TGX precast 4% to 15% or 4% to 20% gradient gels (Bio-Rad) and transferred to either 0.2 μm or 0.44 μm nitrocellulose membranes. Transfer was done for 1.5 to 2 hours in cold TrisGlycine buffer (Bio-Rad) with 20% methanol prior to blocking for 1 hour at room temperature in 5% BSA in TBS-T. Primary antibodies were diluted and incubated overnight at 4°C on a rocker. Membranes were washed 3 or 4 times in fresh TBST prior to a 1-hour room temperature incubation in an appropriate secondary antibody. Membranes were washed 3 to 4 times in TBST, developed with enhanced SuperSignal West Pico Plus or SuperSignal West Fempto (Thermo Fisher), and imaged using Li-Cor Odyssey Fc instrument. Densitometry was performed using LI-COR Image Studio software. Statistical comparisons using densitometry measurements were done using a one-way ANOVA with Tukey post hoc to control for the experiment-wise error rate.
Primary antibodies include GAPDH (Cell Signaling Technology, 5174S; 1:1,000 dilution), H2AX (Thermo Fisher, MA1-2022; 1:1,000 dilution), and NPEPPS (Thermo Fisher, PA5-83788; 1:1,000 dilution). Secondary antibodies include Goat anti-Rabbit IgG, Whole Molecule Polyclonal Secondary Antibody, HRP (FisherScientific, ICN55689, 1:20,000 dilution), and Anti-Mouse IgG (whole molecule)–Peroxidase antibody produced in rabbit (Millipore Sigma, A9044-2ML; 1:20,000 dilution).
shRNA-mediated knockdown experiments
Lentiviral production and transduction were carried out by the University of Colorado Cancer Center Functional Genomics Shared Resources. Plasmids from The RNAi Consortium (TRC) collection (TRC construct numbers TRCN0000073838, TRCN0000073839, and TRCN0000073840) were used for targeting NPEPPS were selected based on predicted knockdown efficiency; nontargeting controls used were SHC002 (shCtrl1) and SHC016 (shCtrl2). 2 μg of target shRNA construct and 2 μg of 3:1 ratio of psPAX2 (Addgene) and pMD2.G (Addgene) were transfected into HEK293FT cells using 2 μg of Polyethylenimine (Polysciences). Lentiviral particle containing media were filtered using a 0.45-μm cellulose acetate syringe filter and used for transduction. Puromycin selection was performed at doses used for CRISPR library screening or in some cases, cells were reselected with higher doses of puromycin (10 μg/mL), to ensure the complete elimination of nontransduced cells. Selected cells were frozen at early passage, and early passage cells were used for all experiments.
Intracellular cisplatin measurements using CyTOF
Cell lines were cultured for several passages in IMDM + 10% FBS. Prior to the experiment, cells were cultured in IMDM10 to be 50% to 80% confluence overnight and then treated the next day with 10 μmol/L cisplatin or PBS and then dissociated after 4 hours of treatment. For dissociation, cells were washed twice with room temperature PBS and then incubated with PBS + 0.05% Trypsin-EDTA for 10 to 15 minutes. Cells were neutralized with IMDM10 and then fully dissociated into single-cell suspension by gentle pipetting. After dissociation, cells were counted by Trypan blue staining and then placed in separate tubes at 3 × 105 cells. Individual samples were then fixed, permeabilized, and labeled using unique barcodes using the Cell-ID 20-plex Pd Barcoding kit (Fluidigm) according to the manufacturer's protocol. Barcoded samples were pooled across cell line conditions and cisplatin concentration, incubated with Cell-ID Intercalator-Ir, mixed with equilibration beads, and acquired on a Helios mass cytometer (Fluidigm). Post-acquisition data were normalized to equilibration beads and debarcoded, using the bead-normalization and single-cell-debarcoder packages from the Nolan Laboratory GitHub page (https://github.com/nolanlab). Relative cisplatin intensity (defined by 195Platinum isotopic mass intensity) was analyzed among nucleated 191Iridium+ 193Iridium+ events defined by Boolean gating within FlowJo 10.7.1.
Whole-genome CRISPR screening
Plasmid library expansion and quality control
Whole-genome CRISPR screening was performed using the Human CRISPR Knockout Pooled Library (Brunello)–1 vector system (Addgene and a gift from John Doench to the Functional Genomics Facility at the University of Colorado Anschutz Medical Campus). Two distinct plasmid expansions were performed. The library distribution was assessed using next-generation sequencing to determine the impact on the overall library was modest following reexpansion. Library width was calculated as previously described (27, 28) by dividing the 10th percentile of the library distribution by the 90th percentile using the log2 average expression of all sgRNAs in the library and found to be 6.7 and 7.13 for batches 1 and 2, respectively. All quality control metrics for each sample are reported in Supplementary Table S5. Different screening parameters were used based on the cell line screened; these are summarized in Supplementary Table S6.
Lentivirus production and titration
For the two plasmid batches, two distinct protocols for lentivirus production were utilized. The first batch was generated by using polyethylenimine, linear (PEI; Polysciences) and was used for the T24-GemCis and TCCSUP-GemCis screens. The second used lipofectamine 3000 and was applied for the 253J-GemCis, KU1919-GemCis, and 5637-GemCis screens. For the first batch, 293FT cells were seeded at a density of 36,800 cells/cm2 into a 4-layer CELLdisc (Greiner) using DMEM + 10% FBS along with antibiotic and antimycotic solution. Transfection mix consisting 47.6 μg pMD2G (Addgene), 95.2 μg of psPAX2 (Addgene), and 190.5 μg of Brunello whole-genome knockout library (Addgene) was mixed with 448 μL PEI (1 mg/mL) and 3 mL Opti-MEM, vortexed for 30 seconds and allowed to incubate at room temperature for 20 minutes. Fresh media containing transfection mix were added to the CELLdisc using up to 270 mL of media. The next day media were changed to 280 mL fresh media followed by a 48-hour incubation. After this 48-hour incubation, the viral supernatant was harvested and filtered through a cellulose acetate filter system (Thermo Scientific) and frozen at −80°C.
The first method had low functional virus titer, so we implemented a different virus production method for subsequent screens. In the second batch of virus production, we utilized lipofectamine 3000 instead of PEI, eliminated the use of multilayer flasks, and centrifuged to remove debris as opposed to filtering. Briefly, 293FT cells were plated in T225 flasks to be 80% confluent after 24 hours. Two hours before transfection, media were changed, and 40 mL of fresh media was used per T225 flask. The lipofectamine 3000 protocol was followed according to the manufacturer's instructions and scaled based on the volume of the virus being prepared. For each T225 flask, 2 mLOptiMEM was mixed with 40 μg of Brunello whole-genome library plasmid, 30 μg of psPAX2 and 20 μg of pMD2.G, and 180 μL of P3000. This mix was added to a tube containing 2 mL Opti-MEM and 128 μL Lipofectamine 3000, which was scaled according to the number of T225 flasks being prepared. Transfection mix was mixed thoroughly by pipetting up and down slowly, and allowed to incubate at room temperature for 15 minutes. Transfection mix was then added dropwise to the plates of 293FT cells with gentle swirling and incubated overnight (∼16 hours). The following morning, the media were changed and 60 mL of fresh media were added to each T225 flask. This was allowed to incubate overnight and replaced the following morning. This first lentiviral supernatant was stored at 4°C to be pooled with a subsequent 48-hour collection. Upon collection, viral supernatants had 1 mol/L HEPES added at 1%. Following the second virus collection, supernatants were pooled and centrifuged at 1250 rpm for 5 minutes to pellet debris. Lentivirus was stored in polypropylene tubes as polystyrene is known to bind lentivirus, and all tubes were flash-frozen in liquid nitrogen and stored at −80°C. Despite the changes to the lentiviral production protocols, functional lentiviral titers were not improved using these changes to the methodology, but feel it is worth noting these changes in protocol to account for any possible variability associated with this change.
Lentivirus was titered functionally based on protocols adapted from the Broad Institute's Genetic Perturbation Platform's public web portal (https://portals.broadinstitute.org/gpp/public/).
Screening parameter optimization
All screening parameters for each cell line including cell polybrene and puromycin sensitivity, screening coverage, technical and biological replicates performed, and gemcitabine and cisplatin treatment concentrations are reported in Supplementary Table S6.
DNA isolation
Cell pellets of 2e7 were snap-frozen in liquid nitrogen in 1.5-mL tubes and stored at −80°C before extraction. When possible, at least 8e7 cells were used for 4 separate genomic DNA isolations, which were pooled to account for any variation in pellet size. DNA isolation was performed using the Puregene cell and tissue kit (Qiagen) with the addition of RNase A Solution (Qiagen) according to the manufacturer's instructions. DNA concentration was measured in quadruplicate using either a nanodrop spectrophotometer (Thermo), Qubit dsDNA assay (Life Technologies), and the average DNA content per cell was determined.
Library preparation
The minimum number of cell equivalents of gDNA to maintain equal coverage was used for library preparation. In all screens, the minimum coverage based on cell number was multiplied by the average gDNA content per cell for each cell line to determine the minimum number for 10 μg PCR reactions needed to maintain coverage. A minimum coverage of 500-fold per sgRNA in the library was targeted for each independent sample or replicate, but this was increased in some cases where screening was carried out with greater depth (see Supplementary Table S6 for coverage and replicate information).
Library preparation was performed using primers sequences designed by the Broad Institute's Genetic Perturbation Platform (https://portals.broadinstitute.org/gpp/public/) and utilized a pool of eight P5 primers to introduce a stagger in reads associated with each library and sample-specific P7 primer that contained a unique sample index sequence for each time point, replicate, or treatment condition to be sequenced in the same pool (Supplementary Table S7). All library preparation primers were resuspended at 100 μmol/L.
Each library preparation PCR reaction contained the following components: 1 μL Herculase II Fusion Enzyme (Agilent), 2.5 μL Deoxynucleotide (dNTP) Solution Mix (New England Biolabs), 0.5 μL P5 primer pool, 0.5 μL P7 index primer, 20 μL 5× Reaction Buffer (Agilent), 10 μg of gDNA and nuclease-free water to bring the total reaction volume to 100 μL. Samples underwent 23 cycles of thermal cycling followed by a quality assessment by electrophoresis on 2% agarose gel to ensure consistent library amplification across multiple wells and samples for each plate.
Each unique library had 10 μL pooled from all PCR reactions performed on that unique sample and mixed thoroughly. Fifty to 100 μL of the pooled library preparation reactions was used to perform magnetic bead-based purification and elimination of any residual free primer using a 0.8× ratio SPRIselect beads (Beckman Coulter) according to the manufacturer's instructions. Libraries were then assessed for appropriate amplicon size and complete elimination of free primer peaks using the High Sensitivity ScreenTape Assay on the Tape Station2200 (Agilent) and quantified using the qPCR-based quantification in order to ensure only NGS-compatible amplicon was quantified using the Library Quant ROX Low Kit (Kapa Biosystems) on a QuantStudio 6 Realtime PCR System (Thermo Fisher). Following qPCR quantification, all libraries were normalized to a standard concentration (typically 20–40 nmol/L) depending on the lowest concentration library to be pooled, and then requantified by qPCR to ensure all samples were within ∼10% to 20% of the pool mean target concentration. After confirming accurate library quantification and normalization, samples were pooled at an equimolar ratio and submitted for sequencing. Libraries were sequenced on the NovaSeq 6000 instrument (Illumina; 150 bp, paired-end) at the University of Colorado Cancer Center Genomics Shared Resource.
CRISPR screening bioinformatic pipeline and analysis
sgRNA counts were extracted directly from R1 raw sequence reads using a custom perl script that uses regular expression string matching to exactly match the sgRNA sequence flanked by 10 bases of vector sequence. The vector sequence was allowed to have one error before and after the sgRNA sequence. sgRNAs were tabulated for each sample based on the sgRNA sequence (Supplementary Table S8). The sgRNA IDs of the Brunello library were updated to current HGNC gene names using the Total Approved Symbols download from HGNC, accessed on September 1, 2020 (https://www.genenames.org/download/statistics-and-files/). Transcript IDs were matched when possible, and when matches were not found, past symbols and aliases were updated to current names. Finally, 5 sgRNAs with missing updated gene names were manually curated using literature searches. Library distribution was calculated using the caRpools R package (29). The DESeq2 R package (30) was used to calculate the differential abundance of genes (Supplementary Table S9). Gene counts were generated using the sum of counts for sgRNAs of the same gene. Synthetic lethality compared GemCis day 19 and GemCis day 25 versus PBS day 19 and PBS day 25 with the day as a covariate. In the comparison integrating all cell lines, the cell line was additionally modeled as a covariate. Gene essentiality was calculated by comparing PBS day 25 to PBS day 0 and in the integrated all cell lines comparison; the cell line was modeled as a covariate. Common synthetic lethal genes were defined as being statistically significantly differentially lost (FDR < 0.05 and Log2 FC < 0) in each of the 5 cell lines (Supplementary Table S10). GSEA was performed using the fgsea R package run with 10,000 permutations (19) with the KEGG and Reactome gene sets from MSigDB (20). Heat maps were generated with the ComplexHeatmap R package following z-score transformation (22). Other plots were generated using the ggplot2 R package.
Xenograft experiment
Six-week-old, female NU/J mice (Jackson Labs) were allowed to acclimate for at least one week prior to initiating any experiments. Mice had free access to food and water in pathogen-free housing and cared for in accordance NIH guidelines, and all experiments were performed under protocols approved by the University of Colorado Denver Institutional Animal Care and Use Committee.
For KU1919-GemCis xenografts, cells that had been stably transduced with nontargeting control (shCtrl1, SHC002) and NPEPPS (shN39, TRCN0000073839) shRNA constructs. Mice were divided into groups of 22 and 23 for the nontargeting control and NPEPPS shRNA constructs, respectively. Mice were injected with 4e6 cells in phenol red- and serum-free RPMI mixed with equal volume Matrigel Matrix (Corning) to total 100 μL volume. Tumors were allowed to engraft for 9 days following injection and mice were randomized based on tumor volume within each shRNA condition into groups of 11 or 12 to be treated with a combination of gemcitabine plus cisplatin or DPBS. Treatment was initiated 13 days after inoculation with dosing adjusted based on individual mouse weight.
Cisplatin (Sigma) and gemcitabine hydrochloride (BOC Sciences) were both resuspended in 0.9% PBS and stored protected from light at −80°C as individual aliquots. Before treatment, fresh aliquots of gemcitabine and cisplatin were thawed and diluted to their final concentration with 1× DPBS (Gibco). Mice were treated three times weekly on a Monday, Wednesday, and Friday schedule for four weeks total. All mice in the gemcitabine plus cisplatin-treated groups were given 50 mg/kg gemcitabine and 2 mg/kg cisplatin that were mixed and administered as a single intraperitoneal injection, whereas control mice were administered an equivalent volume of DPBS.
Mouse health was monitored daily, and all tumor volume measurements and weights were measured 3× weekly schedule. Tumor volume was calculated using the formula (L × W2)/2, for which L is the length of the long axis and W is the width of the axis perpendicular to the long axis measurement. All measurements were performed using digital calipers. Animals were humanely euthanized with CO2 followed by cervical dislocation when tumors reached a predetermined endpoint of 2 cm3 or when weight loss exceeded 15% body weight. Mice that were removed from the study due to weight loss were censored in the survival analyses.
Linear mixed-effects model of tumor growth
Linear mixed-effects models were used to model longitudinal observations of xenograft tumor growth volumes normalized by their corresponding baseline volume. Mixed-effects models from the R package lme4 and Satterthwaite's approximation for degrees of freedom for the fixed effects from lmerTest were used for model fitting and inspection in the R statistical software (4.0.3). Volume changes compared with the baseline were log2-transformed. The final model was structured as:
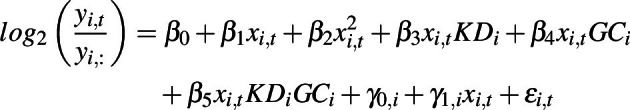 |
where β is the fixed effects capturing population-level trends, γ is the normally distributed random effects capturing individual-level variation, ε is the i.i.d. normally distributed residual term, i is the unique individual identifier, t notes the time points, xi,t ∈ {2, 4, 5, 7, 9, 11, 14, 16, 18, 21, 23, 25, 28} depicted days since initiating interventions, yi,: is tumor volume at baseline prior to treatments upon randomization, and yi,t were the observed tumor volumes over the treatment period measured in mm3. The model was fit using restricted maximum likelihood and built iteratively until the underlying model assumptions and model convergence criteria were met. To this end, a quadratic growth term (β2) was added on top of the linear growth term (β1) and intercept (β0), allowing slightly nonlinear relative growth patterns to be captured by the otherwise linear model. Binary indicators KDi ∈ {0, 1} and GCi ∈ {0, 1} were used to model knockdown of NPEPPS, GemCis treatment, or the combination. The corresponding model terms were captured in β3, β4 and β5, respectively. Finally, the model allows for individual-specific random effects for intercept (γ0,i) and linear growth slope (γ1,i). Shapiro–Wilk test was used to examine the underlying normality assumption for γ0,i and γ1,i with P = 0.1373 and P = 0.8901, respectively, indicating that these random effects followed underlying assumptions of normality. After inspection of the residual plots, this final model was deemed suitable for population-level statistical inference via the fixed effects. These population-level estimates are as follows: β0 (intercept) = 0.05054, SE = 0.08422, t = 0.600, P = 0.55091; β1 (linear slope) = 0.1236, SE = 0.01493, t = 8.276, P = 8.9e−12; β2 (quadratic slope) = 0.00308, SE = 0.0002242, t = 13.740, P < 2e−16; β3 (knockdown) = −0.0605, SE = 0.01821, t = −3.322, P = 0.00178; β4 (GC) = −0.1063, SE = 0.01821, t = −5.837, P = 5.5e−7; β5 (knockdown + GC) = −0.1233, SE = 0.01791, t = −6.884, P = 1.5e−8.
Patient tumor-derived organoids
Culture of the patient tumor-derived organoids
Human bladder tissue was obtained from the Erasmus MC Bladder Cancer Center, Rotterdam, the Netherlands, and the Amphia Ziekenhuis, Breda, the Netherlands. Bladder tumor-derived organoids from biopsies obtained through TURBT or cystectomy were isolated and cultured using methods developed by Mullenders and colleagues (31) with the addition of conditioned media for recombinant Respondin (2.4% v/v) and WNT3A (2.5% v/v). Briefly, bladder tissues were washed with advanced DMEM/F12 (Gibco) supplemented with 10 mmol/L HEPES (Gibco), 1% GlutaMax (Gibco), and 100 μg/mL primocin (InvivoGen), henceforth Ad+++. Tissue was minced and incubated at 37°C with the digestion solution (collagenase 2.5 mg/mL in EBSS), and isolated cells were passed through 70 μmol/L strainer (Falcon), washed with Ad+++, and seeded in 50 μL drops of BME (R&D Systems) containing 10,000 to 15,000 cells in 24-well suspension plates (Greiner). Bladder tumor organoids were cultured in a culture medium containing Ad+++ supplemented with 1 × B-27 (Gibco), 1.25 mmol/L N-acetylcysteine (Sigma), 10 mmol/L nicotinamide, 20 μmol/L TGFβ receptor inhibitor A83-01, 100 ng/mL recombinant human FGF10 (PeproTech), 25 ng/mL recombinant human FGF7 (PeproTech), 12.5 ng/mL recombinant human FGF2 (PeproTech), 10 μmol/L Y27632 Rho Kinase (ROCK) Inhibitor (Sigma) and conditioned media for recombinant Respondin (2.5% v/v) and Wnt3A (2.5% v/v). The medium was changed every three days. Organoids were passaged at a 1:3 to 1:6 ratio every 7 days using cell dissociation solution nonenzymatic (Sigma) and plated in fresh BME matrix droplets.
Lentiviral transduction of bladder cancer patient-derived organoids
Lentiviral vectors containing the desired shRNA sequences were amplified from bacterial glycerol stocks obtained in-house from the Erasmus Center for Biomics and part of the MISSION shRNA library. The same shRNA constructs used for bladder cancer cell lines, and described above, were used for patient-derived organoids (PDO). For overexpression, pLenti-C-Myc-DDK-P2A-Puro Lentiviral Gene-Expression Vectors were acquired from Origene (NPEPPS RC209037L3, empty vector PS100092). For lentiviral particle generation, in total, 5e6 HEK293T cells were plated in a 10-cm dish and transfected with 12.5 μg of plasmids mix. In total, 4.5 μg of pCMVΔR8.9 (envelope), 2 μg of pCMV-VSV-G (packaging), and 6 μg of shRNA vector were mixed in 500 μL serum-free Opti-MEM and combined with 500 μL Opti-MEM containing 125 μL of 10 mmol/L polyethyleneimine (PEI, Sigma). The resulting 1 mL mixture was added to HEK293T cells after 15 minutes of incubation at room temperature. The transfection medium was removed after 12 hours and replaced with a fresh RPMI medium. Virus-containing medium was harvested and replaced with fresh medium at 48 and 72 hours after transfection. After each harvest, the collected medium was filtered through a cellulose acetate membrane (0.45 μm pore), and viral particles were concentrated by ultracentrifugation at 20,000 rounds per minute for 1 hour at 4°C with pellets resuspended and pooled in a final volume of 2 mL adv. DMEM for each condition. Medium containing viral particles was used either directly or stored at −80°C. For lentiviral transduction of bladder cancer organoid cells, organoids were harvested and dissociated into single cells using a cell dissociation solution. For each condition (shRNA control/shRNA NPEPPS/NPEPPS-OE/empty vector), 1 × 106 cells were collected and gently resuspended in 1 mL of concentrated lentivirus. Cell–virus mixture was then divided over two wells of a prewarmed 24-well plate and subsequently sealed with parafilm. Plates were spinoculated at 600 × g at 25°C for 1 hour. Parafilm was removed immediately after spinoculation, and plates were incubated for 6 to 8 hours at 37˚C 5% CO2. Transduced cells were washed 1 time with 10 mL advanced DMEM spinning down at 200 × g for 10 minutes and seeded in domes at a density of 150 cells per μL BME. Once matured, organoids were treated with 2 μg/mL puromycin for 72 hours. Puromycin-selected organoids were then passaged and given 6 days of recovery time prior to experimental procedures.
Ex vivo drug testing
Organoids were collected 4 to 7 days after passaging and dissociated to single cells using a cell dissociation solution, assisted by mechanical dissociation. All assays were performed in 96-well suspension plates (Greiner bio-one; 655 185), seeding 10,000 cells per well in 100 μL bladder organoid medium containing 15% BME. Drugs were added when mature organoids formed after two to three days, and drug treatment was performed in triplicate. For ex vivo cisplatin response: cisplatin (Sigma; PHR1624; reconstituted in NaCl) was added in six doses in the range of 0.1 to 40 μmol/L. Caspase-3 and -7 activity was measured following three days of treatment (Caspase-Glo #G8093, Promega), whereas cell viability was obtained after six days of treatment (CellTiter-Glo 3D #G9681). Plates were read on a SpectraMax I3 plate reader. Viability data were normalized using organoid wells treated with vehicle control (1.2% PBS; Medchemexpress; HY-14807; reconstituted in DMSO). Organoids were harvested after six days of treatment and dissociated into single cells using a cell dissociation solution, assisted by mechanical dissociation. Three quarters of the cells were used to obtain cell viability as previously described (treatment), whereas one quarter of the cells were reseeded in 200 μL BOM + 15% BME. Following reseeding, organoids were allowed to regrow for another six days, after which, cell viability (reseeding) was again obtained. Cell viability was normalized using organoids treated with vehicle control (0.02% DMSO, 1.2% PBS).
SNaPshot mutation analysis
Tumor and matched organoid DNA was isolated using the QIAmp DNA Mini-Kit (Qiagen) according to the manufacturer's protocol. The presence of hotspot mutations in the TERT promoter sequence (chr5:1,295,228C>T, chr5:1,295,248G>A, and chr5:1,295,250C>T [GRCh37/hg19]), FGFR3 (R248Q/E, S249C, G372C, Y375C, A393E, K652E/M), and PIK3CA (E542K, E545G/K, and H1047R) was assessed on tumor and organoid DNA by SNaPshot mutation analysis with the same methods as previously described (32–34).
Organoid phenotyping and tumor histology
Tissue processing and hematoxylin–eosin (H&E) staining were performed using standard procedures. For H&E staining of organoids, wells of BME-embedded organoids were fixated with 4% formalin (Sigma) and 0.15% glutaraldehyde (produced in-house) at room temperature for 2 hours. Fixated BME and organoids were washed with PBS and engulfed in 2.5% Low-Melting Agarose (Sigma) before paraffin embedding. H&E staining was performed on 4 μm paraffin sections of both tumor and organoid tissue. Stained whole-slides, as well as prior 3D organoid cultures, were imaged by bright-field microscopy (Olympus IX70).
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (35) partner repository with the data set identifier PXD024742. The whole-exome sequencing data have been deposited in the BioProject database with project identifier PRJNA714778. The RNA-seq data have been deposited in the GEO database with data set identifier (GSE171537). The CRISPR screen sequencing data have been deposited in the GEO database with data set identifier (GSE179799). The copy-number data have been deposited in the ArrayExpress database with identifier (E-MTAB-10353). The RNA-seq, proteomics, CRISPR screen, and genetic alteration data are made available through a custom build R Shiny app at: https://bioinformatics.cuanschutz.edu/BLCA_GC_Omics/. All other raw data generated in this study are available upon request from the corresponding author.
Results
We acquired the five human bladder cancer cell lines, KU1919, 5637, T24, TCCSUP, and 253J, from the RCCL collection (36, 37). These lines were all derived from tumors of patients with MIBC and were selected to cover a range of demographic, clinicopathologic, and genomic features in putative bladder cancer drivers as reported in The Cancer Genome Atlas (38) and variants reported in ClinVar (Table 1; Supplementary Tables S11 and S12; ref. 39). For each, we obtained the parental lines (-Par) and derivatives made resistant through dose escalation to cisplatin (-Cis), gemcitabine (-Gem), and the combination of gemcitabine plus cisplatin (-GemCis; Fig. 1A; Supplementary Table S1). We confirmed resistance to the associated drugs for all resistant derivatives in comparison with the parental lines and found them to be consistent with those reported by the RCCL (Supplementary Fig. S1; refs. 36, 37).
Table 1.
Clinicopathologic characteristics and genetic drivers for five muscle-invasive bladder cancer cell lines.

Genome-wide CRISPR screens identify 46 common synthetic lethal genes
To study the connection between drug resistance and gene expression, we performed whole-genome loss-of-function screens in each of the five GemCis-resistant cell line derivatives. After transduction of the Brunello CRISPR-Cas9 knockout library (40), we passaged the cells for 10 days to clear essential genes, then split them into PBS or gemcitabine plus cisplatin treatment groups (Fig. 1A). Each screen was performed at a drug concentration that allowed the GemCis-resistant cells to grow unrestricted, but significantly inhibited the growth of the parental lines (Supplementary Table S1). Screening parameters for each cell line are reported in Supplementary Table S6. We counted sgRNAs 9 and 15 days after the start of treatment (19 and 25 days after transduction). As expected, similar experimental conditions clustered together when correlations were measured across treatment conditions and cell lines (Supplementary Fig. S2).
We defined genes as “synthetic lethal” with gemcitabine plus cisplatin treatment as those for which the combined cognate sgRNA counts were significantly lower (moderated t test, FDR < 0.05) in the gemcitabine plus cisplatin-treated arm compared with the PBS arm when including both days 19 and 25 in the statistical model (Supplementary Table S9). We identified 235 synthetic lethal genes that were statistically significant in KU1919-GemCis, 888 for T24-GemCis, 2099 for TCCSUP-GemCis, 2369 for 253J-GemCis, and 511 for 5637-GemCis. Next, we performed GSEA (41) on the full ranked list of genes according to their synthetic lethality. For this analysis, we created one ranked gene list by including each of the five cell types in the statistical model directly. As expected, and as a validation of the screen itself, we found that the top-ranked pathways were dominated by processes such as DNA repair, Fanconi anemia, nucleotide excision repair, double-stranded break repair, base-excision repair, and DNA damage bypass mechanisms (Fig. 1B; Supplementary Table S13). These results are consistent with the known roles of DNA damage detection and repair in cisplatin resistance (42, 43). In addition, the overall ranking of genes from our CRISPR screen showed enrichment for manually curated genes associated with platinum resistance in cancer (Supplementary Table S9; ref. 44).
From these results, we identified 46 genes that were commonly synthetic lethal across all five cell lines (Fig. 1C; Supplementary Fig. S3A). Consistent with the overall findings (Fig. 1B), 41 of the 46 common synthetic lethal genes fell into one or more putative DNA damage response and repair pathways, including homologous recombination, double-stranded break repair, nuclear excision repair, and Fanconi anemia (Supplementary Fig. S3B; Supplementary Table S14). These genes showed a range of growth patterns over the full length of the CRISPR screen. As illustrated in Fig. 1D, some genes showed patterns of increased cell growth in PBS treatment, then reduced growth in gemcitabine plus cisplatin treatment. Other genes had very little impact on cell growth in PBS treatment, but then reduced growth when treated with gemcitabine plus cisplatin. Finally, some genes reduced cell growth in PBS treatment and further reduced growth with gemcitabine plus cisplatin treatment. Overall, these results provide a robust list of known and novel genes involved in chemotherapy resistance that can potentially be targeted therapeutically to improve treatment response.
Complementary multiomic profiling prioritizes NPEPPS as the top driver of treatment resistance
Pretreatment multiomic profiling has been shown to reveal the biological impact of synthetic lethal hits (45). Accordingly, we performed RNA sequencing and mass spectrometry–based proteomic profiling on cell lysates of all cell lines grown in drug-free media (Fig. 1A). We leveraged transcriptome and proteome profiles from parental to matched drug-resistant derivative lines (-Gem, -Cis, and -GemCis) to prioritize the 46 common synthetic lethal genes that drive resistance in the context of gemcitabine plus cisplatin treatment resistance.
Taking this approach, the transcriptomics data revealed 1,557 significantly upregulated genes across the Gem-resistant lines, 1,897 in the Cis-resistant lines, and 1,530 in the GemCis-resistant lines (moderated t test, FDR < 0.05; Supplementary Table S2). The proteomics data revealed 9 significantly upregulated proteins across the Gem-resistant cell lines, 1 in the Cis-resistant cell lines, and 10 in the GemCis-resistant cell lines (moderated t test, FDR < 0.25; Supplementary Table S15). Given the lower number of significant proteins and the relevance of transcript expression in predicting genetic dependency (45), we first investigated the overlap between the CRISPR screen results and the transcriptomes from each of the resistant cell line derivatives compared with the parental cells. Few genes were significantly and consistently upregulated across the resistant derivatives in the list of 46 commonly synthetic lethal genes (Fig. 2A), but the most significantly and consistently upregulated genes were involved in DNA damage response and repair mechanisms, including ERCC6, XPA, REV1, POLH, ERRC8, PRIMPOL, NBN, and members of the Fanconi anemia pathway. We identified puromycin-sensitive aminopeptidase, NPEPPS, to be the most consistently upregulated gene across the resistant derivatives at the RNA level (Fig. 2A and B). Similarly, we found NPEPPS to be consistently and significantly upregulated at the protein level (Fig. 2C). NPEPPS was also a top synthetic lethal hit (Fig. 2D; Supplementary Table S9). Consistent with the proteomics results, immunoblotting for NPEPPS revealed that it was upregulated in the Cis-resistant and GemCis-resistant lines, with the Gem-resistant lines showing variable upregulation (Fig. 2E).
Figure 2.

NPEPPS is identified as a commonly upregulated and synthetic lethal hit. A, Differential gene expression of 43 common synthetic lethal genes as measured by RNA-seq across all cell lines (43 of 46 genes mapped between RNA-seq and the CRISPR screen), comparing the treatment-resistant derivative (Gem-, Cis-, GemCis-resistant) with the associated parental cell line. Asterisks indicate a statistically significant result (moderated t test; *, FDR < 0.05). The bar plot on top is the aggregate count of significant results across all 15 comparisons. Genes are ranked by the count of statistically significant upregulated hits. B–D, RNA-seq (moderated t test compared with parentals; *, FDR < 0.05; B), mass spectrometry proteomics (moderated t test compared with parentals, *, FDR < 0.25; C), and CRISPR screen results for NPEPPS (mean ± SD; moderated t test; *, FDR < 0.05; D). E, Representative immunoblots and densitometry quantification for independent triplicates (mean ± SEM) for NPEPPS in all cell lines. Comparisons using a one-way ANOVA were made to the parental cell lines (*, FDR < 0.05).
NPEPPS drives cisplatin resistance in vitro and in vivo
To test our prioritization that NPEPPS regulates sensitivity to gemcitabine plus cisplatin treatment in GemCis-resistant bladder cancer cells, and to parse its role in both cisplatin and gemcitabine resistance, we generated stable NPEPPS shRNA knockdowns in the KU1919-GemCis and T24-GemCis cell lines. We found that NPEPPS knockdown preferentially increased cisplatin, but not gemcitabine sensitivity (Fig. 3A and B). Knockdown of NPEPPS delayed the growth of cells but did not have major effects on cell growth rates (Supplementary Fig. S4A). siRNA targeting of NPEPPS in the KU1919-GemCis cell line and shRNA and/or siRNA in T24-GemCis and 253J-GemCis cells confirmed our results that NPEPPS loss preferentially sensitizes cells to cisplatin (Supplementary Fig. S4B and S4C). Additionally, we used a gRNA from the CRISPR screen library to show that knockout of NPEPPS and the associated dose-response matches our findings from shRNA and siRNA-mediated depletion of NPEPPS (Supplementary Fig. S4D). Conversely, overexpression of NPEPPS in KU1919 and T24 parental lines increased resistance to cisplatin, but not gemcitabine (Fig. 3C and D). These results support NPEPPS as a regulator of sensitivity to gemcitabine plus cisplatin through its effect on regulating the cellular response to cisplatin.
Figure 3.

Genetic inhibition of NPEPPS resensitizes GemCis-resistant cells in vitro and in vivo. A and B, KU1919-GemCis (full immunoblot reported in Supplementary Fig. S4B) or T24-GemCis cells with knockdown of NPEPPS treated with increasing doses of cisplatin or gemcitabine. A total of three technical replicates per dose (mean ± SEM). C and D, KU1919 or T24 parental cells with overexpression of NPEPPS treated with increasing doses of cisplatin or gemcitabine. A total of three technical replicates per dose (mean ± SEM). Independent experiments are reported in Supplementary Fig. S4. P values comparing IC50 values using the sum-of-squares F test. E and F, Intracellular cisplatin levels in KU1919 and T24 cells were measured after 4 hours of 10 μmol/L cisplatin treatment using CyTOF, with the number of live cells analyzed as indicated. Group comparisons were made for triplicate experiments by normalizing intracellular cisplatin levels to the GemCis-resistant or parental cells and compared using a one-way ANOVA (*, FDR < 0.05; **, FDR < 0.01; ***, FDR < 0.001; ns, nonsignificant). G, Tumor volume (mean ± SEM) of KU1919-GemCis xenografts measured over time and across four treatment groups considering nontargeting shRNA controls (shCtrl1), shRNA targeting NPEPPS (shN39), PBS vehicle control (PBS), or gemcitabine plus cisplatin treatment (GemCis). H, Survival analysis of xenograft models with a defined endpoint of a tumor volume > 2 cm3. The log-rank test was applied to test significance.
To evaluate NPEPPS’s impact on intracellular cisplatin concentrations, we directly measured intracellular cisplatin using the metal ion detection capabilities of cytometry by time-of-flight, CyTOF (46). We measured intracellular cisplatin after 4 hours of treatment at 10 μmol/L in the same KU1919 and T24 cells evaluated for dose response. Using KU1919 as the illustrative example, KU1919-GemCis cells (median Pt 195 = 102) showed lower cisplatin concentration compared with KU1919 parental cells (median Pt 195 = 565). Control knockdown had little effect (median Pt 195 = 121), but NPEPPS knockdown shifted the intracellular levels of cisplatin to the parent lines (median Pt 195 = 375), suggesting that NPEPPS depletion increased intracellular cisplatin (Fig. 3E; Supplementary Fig. S5A). These findings were replicated in the T24 cell lines (Fig. 3E; Supplementary Fig. S5B). In the overexpression setting (Supplementary Fig. S6A), and using KU1919 as the example, KU1919-GemCis cells (median Pt 195 = 763) again showed lower cisplatin concentration compared with KU1919 parental cells (median Pt 195 = 1,706) with the overexpression control cells (median Pt 195 = 1,738) showing little difference, but the NPEPPS-overexpressing cells (median Pt 195 = 1,203) showing a decrease in intracellular cisplatin (Fig. 3F; Supplementary Fig. S6B). We found similar results with T24 cells (Fig. 3F; Supplementary Fig. S6C). These data are consistent with the dose-response results reported in Fig. 3A–D.
To test if NPEPPS depletion sensitizes tumor cells to cisplatin-based chemotherapy in vivo, we established subcutaneous xenografts using the KU1919-GemCis cells with either NPEPPS shRNA knockdown or nontargeting shRNA control. When tumors reached roughly 200 mm3, mice were randomized into four groups: shCtrl1 with PBS (n = 11), shCtrl1 with gemcitabine plus cisplatin (n = 11), shN39 with PBS (n = 11), and shN39 with gemcitabine plus cisplatin (n = 12). Treatment was delivered through intraperitoneal injection, with PBS or gemcitabine plus cisplatin administered three times weekly for four weeks. Tumor volumes were monitored until they reached the predetermined endpoint of 2 cm3. NPEPPS knockdown alone and gemcitabine plus cisplatin treatment alone had a significant impact on tumor growth compared with vehicle-treated, shRNA controls. The combination of NPEPPS knockdown and gemcitabine plus cisplatin treatment led to a stronger and more significant impact on tumor growth, including two mice that had stable and small tumors; these mice were sacrificed after 55 days but showed no adverse events (Fig. 3G). We further analyzed tumor growth using linear mixed-effects models aimed at capturing trends in tumor volume change in relation to pretreatment baseline tumor volume across the four groups (Supplementary Fig. S7A and S7B). According to this model, tumor growth inhibition by NPEPPS knockdown (P = 0.00178), GemCis treatment (P = 5.49e−7), or the combination of NPEPPS knockdown and gemcitabine plus cisplatin treatment (P = 1.47e−8) were all consistent effects over the treatment period (Fig. 3G and H). We validated NPEPPS knockdown in the pre-xenograft inoculate cells and after tumors were removed from mice upon reaching the 2 cm3 endpoint (Supplementary Fig. S7C). Survival analysis using tumor volume as the endpoint showed that mice treated with gemcitabine plus cisplatin had a 14-day survival advantage. Similarly, the knockdown of NPEPPS resulted in a 14-day survival advantage. Mice treated with gemcitabine plus cisplatin and with NPEPPS knockdown tumors had a 25-day survival advantage, a statistically significant improvement (log-rank test, P < 0.0001; Fig. 3H; Supplementary Fig. S7D).
NPEPPS regulates cisplatin response in MIBC PDOs
We next evaluated the role of NPEPPS in regulating cisplatin response in ex vivo expanded 3D primary cultures of MIBC tissue as PDOs. PDOs are commonly used as an intermediate step to the clinical investigation of novel therapeutics as they more closely recapitulate the human tumor as supported by a high degree of clinical reliability and predictability of drug response in validation trials (47–50). We generated PDOs from MIBC patients undergoing transurethral resection of a bladder tumor (T, n = 5) or radical cystectomy (C, n = 2; Fig. 4A; Supplementary Table S16). Five PDOs were generated prior to cisplatin-based chemotherapy (preChemo), one PDO was generated after the patient received cisplatin-based chemotherapy (postChemo), and one PDO was generated from a patient who did not receive cisplatin-based chemotherapy (noChemo).
Figure 4.

NPEPPS depletion sensitizes ex vivo models of bladder cancer to cisplatin. A, Clinical course of MIBC for patients including the point at which the patient tumor-derived organoid lines were generated (black arrow). Notation is the patient number followed by the tumor source, either TURBT, transurethral resection of bladder tumor (T), or radical cystectomy (C). B, Representative brightfield images of organoids together with H&E staining of patient tumor-PDO pairs illustrating patient-specific growth patterns. Most PDOs exhibited round and dense structures, as represented by 1C-postChemo (Chemo, cisplatin-based chemotherapy regimen), although there was notable variation in growth and morphology. Scale bar, 100 μm. C,Ex vivo response of all seven PDOs treated with increasing concentrations of cisplatin. Cell viability, as a percentage of untreated control, was measured using CellTiter-Glo. The fitted dose-response curves represent viability corresponding to three biological replicate experiments and data are represented as mean ± SEM. D, Experimental workflow for lentiviral, shRNA-mediated NPEPPS depletion in PDOs and representative images after puromycin selection. E,NPEPPS expression was evaluated by RT-PCR in shNPEPPS and shCtrl PDO lines normalized to cyclophilin. Error bars, mean ± SD. F, Representative brightfield images of the control and NPEPPS-depleted 1C-postChemo PDO treated with the indicated cisplatin concentrations. Scale bar, 400 μm. G, IC50 values estimated from dose curves for cell viability measured through CellTiter-Glo (biological triplicates; mean ± SEM). H, Relative caspase-3 and -7 activity in cisplatin-treated shCtrl and shNPEPPS PDOs. Caspase activity was measured by Caspase-Glo and normalized to untreated PDOs. (biological triplicates; mean ± SEM). I, J, and K, Replicate experimental conditions as in F, G, and H, but with the 2T-preChemo PDO. L, Intracellular cisplatin levels were measured after 24 hours of 5 μmol/L cisplatin treatment using CyTOF, with the number of live cells analyzed as indicated. *, P < 0.05; **, P < 0.01; ns, nonsignificant.
The tumor origin of the PDOs was confirmed by the detection of matching bladder cancer–specific mutations in tumor–organoid pairs (Fig. 4A). As expected with this patient-derived platform, PDOs displayed distinct and tumor-specific phenotypes with notable variations in cell density and roundness (Fig. 4B). NPEPPS was robustly expressed in all organoids (Supplementary Fig. S8). We performed dose response with cisplatin to determine ex vivo responses. PDOs displayed a range of responses to cisplatin, with a median cisplatin IC50 value of 7.1 μmol/L, a minimum of 2.2 μmol/L, and a maximum of 37.4 μmol/L (Fig. 4C).
To investigate the effect of NPEPPS depletion on cisplatin resistance, we selected three PDO lines, two pretreatment and one posttreatment, with a range of cisplatin responses (1C-postChemo, 2T-preChemo, and 5T-preChemo), and performed shRNA-mediated knockdown of NPEPPS (Fig. 4D and E; Supplementary Fig. S9A) followed by their treatment with cisplatin for six days. NPEPPS depletion lowered the cisplatin IC50 values by 50% or more in all of the tested PDOs, though not statistically significant. Furthermore, we observed significantly increased caspase-3 and -7 activity following three days of cisplatin treatment in shRNA-mediated NPEPPS depletion compared with control PDOs, suggesting that NPEPPS depletion increases cisplatin-mediated apoptosis (Fig. 4F–K; Supplementary Fig. S9A–S9D). Finally, we found that intracellular cisplatin levels in the shRNA control organoids are increased with the knockdown of NPEPPS (Fig. 4L; Supplementary Fig. S9E). Collectively, these results demonstrate that genetic inhibition of NPEPPS sensitizes PDOs to cisplatin, which is consistent with our findings in the bladder cancer cell lines.
Next, we tested whether NPEPPS independently increases cisplatin resistance. We selected treatment-naïve (7C-noChemo), pre- (4T-preChemo), and posttreatment (1C-postChemo) PDO lines that are sensitive to cisplatin and exogenously expressed NPEPPS via lentiviral transduction (Fig. 5A). NPEPPS overexpression was confirmed by RT-qPCR, showing a 10-fold overexpression for 7C-noChemo, 3-fold overexpression for 1C-postChemo (Fig. 5B), and a 25-fold overexpression for 4T-preChemo (Supplementary Fig. S9F). Cisplatin dose response was measured and we observed a decrease in cisplatin-mediated apoptotic blebbing and loss of structure in NPEPPS-overexpressing PDOs compared with empty vector control PDOs. Furthermore, from the estimated IC50 values, we observed increased resistance to cisplatin associated with increased NPEPPS expression. Moreover, less apoptosis was observed in NPEPPS-overexpressed PDOs treated with 15 μmol/L and 25 μmol/L cisplatin, as measured by caspase-3 and -7 (Fig. 5C–H; Supplementary Fig. S9G–S9I). Again, we found that NPEPPS controlled intracellular cisplatin concentrations, with the NPEPPS-overexpressing cells showing decreased levels of cisplatin compared with the empty vector control cells (Fig. 5I; Supplementary Fig. S9J). Taken together, these results suggest that NPEPPS regulates cisplatin response in patient tumor-derived PDOs, highlighting NPEPPS as an attractive therapeutic target across cisplatin-based chemotherapy settings.
Figure 5.

Ex vivo NPEPPS overexpression enhances cisplatin resistance. A, Schematic representation of the experimental procedure for NPEPPS overexpression in PDOs and representative images after puromycin selection. Scale bar, 400 μm. B,NPEPPS expression was evaluated by RT-PCR in empty vector and NPEPPS overexpression PDO lines normalized to cyclophilin. Error bars, mean ± SD. C, Representative brightfield images of empty vector control and NPEPPS-overexpressing 7C-noChemo PDO treated with the indicated cisplatin concentrations. Scale bar, 400 μm. D, IC50 values estimated from dose curves for cell viability measured through CellTiter-Glo (biological triplicates; mean ± SEM). E, Relative caspase-3 and -7 activity in cisplatin-treated empty vector control and NPEPPS-overexpressing PDOs. Caspase activity was measured by Caspase-Glo and normalized to untreated PDOs (biological triplicates; mean ± SEM). F,G, and H, Replicate experimental conditions as in C, D, and E, but with the 1C-postChemo PDO. I, Intracellular cisplatin levels were measured after 24 hours of 5 μmol/L cisplatin treatment using CyTOF, with the number of live cells analyzed as indicated. *, P < 0.05; **, P < 0.01; ns, nonsignificant.
Pharmacologic inhibition of NPEPPS improves cisplatin response in MIBC organoids
To assess the pharmacologic efficacy of targeting NPEPPS, we tested cisplatin treatment combined with the NPEPPS inhibitor tosedostat (51). We focused on the three preChemo, TURBT-derived PDOs with a range of clinical responses to test the efficacy of cisplatin-based chemotherapy in combination with tosedostat (Fig. 6A). From our initial evaluation of cisplatin sensitivity (Fig. 4C), the most sensitive PDO in this group is 6T-preChemo (IC50 = 7.1 μmol/L), followed by 2T-preChemo (IC50 = 10.5 μmol/L), and the most resistant is 3T-preChemo (IC50 = 37.4 μmol/L).
Figure 6.

NPEPPS inhibitor tosedostat overcomes cisplatin resistance ex vivo. A, Clinical response to cisplatin-based chemotherapy in three MIBC patients. Pathologic preoperative chemotherapy response is annotated according to pathologic stage following radical cystectomy or metastatic biopsy. Response is illustrated by pre- (top) and posttreatment (bottom) computerized tomography scans. Red arrows, bladder wall thickening and subsequence response. B, Experimental design for treating PDOs with cisplatin and tosedostat alone or in combination. PDOs were withdrawn from treatment, dissociated into single cells, and reseeded after 6 days. Cell viability was measured using CellTiter-Glo at days 6 and 12. C,Ex vivo response of PDOs treated with the indicated concentrations of cisplatin with or without the addition of tosedostat. After 6 days, viability was measured by CellTiter-Glo (biological triplicates; mean ± SEM). D, Cisplatin response in reseeded organoids treated at the indicated concentrations of drug, with viability measured by CellTiter-Glo (biological triplicates; mean ± SEM). E, CyTOF results for KU1919-Parental or -GemCis cells treated with 1 μmol/L tosedostat or DMSO for 72 hours, followed by 10 μmol/L cisplatin for 4 hours. Median values of three replicates were normalized to the vehicle control treatment. One-way ANOVA; *, P < 0.05; **, P < 0.005; ns, nonsignificant.
Given that these PDOs are representative molecular (Fig. 4A), morphologic (Fig. 4B), and pharmacologic (Fig. 4C) models of patient tumors, we treated the three PDOs with cisplatin, tosedostat, or the combination of cisplatin plus tosedostat (Fig. 6B) based on previously identified plasma concentrations (51–56). Responses to 2 and 5 μmol/L cisplatin were consistent with our previous results (Fig. 4C), with 6T-preChemo being the most sensitive to cisplatin (Fig. 6C; Supplementary Fig. S10A). Tosedostat alone did not result in significant changes in viability for the 6T-preChemo and 2T-preChemo PDOs, but resulted in a significant reduction in cell viability for 3T-preChemo, potentially highlighting a role for tosedostat as a monotherapy in some contexts (Fig. 6C; Supplementary Fig. S10A). Significant reductions in cell viability were observed for all PDOs when cisplatin and tosedostat were combined after 6 days of treatment (Fig. 6C; Supplementary Fig. S10A). Importantly, cisplatin response of 6T-preChemo was not compromised but was further enhanced with the addition of tosedostat.
Next, we assessed the ability of drug-treated PDOs to reestablish, which is analogous to a clonogenic assay, and tested for cellular potential to outgrow following treatment (Fig. 6B and D). After reseeding PDOs treated with 5 μmol/L cisplatin, 6T-preChemo showed an absence of organoid formation or outgrowth, whereas robust regrowth was observed for PDOs derived from tumors with residual or progressive disease (2T-preChemo and 3-preChemo; Fig. 6D; Supplementary Fig. S10B). Cisplatin plus tosedostat combination treatment eliminated PDO regrowth for all lines (Fig. 6D). We additionally tested if tosedostat affected intracellular cisplatin concentrations similar to NPEPPS depletion. We found that priming bladder cancer cells with 1 μmol/L tosedostat for 72 hours followed by 10 μmol/L of cisplatin for 4 hours phenocopied NPEPPS depletion. Compared with the vehicle-treated control for tosedostat, intracellular cisplatin in the KU1919-Parental cells was increased nearly 1.9-fold on average with intracellular cisplatin in the KU1919-GemCis cells increased even further to nearly 2.5-fold increase on average (Fig. 6E; Supplementary Fig. S11). Taken together, these findings provide strong evidence that tosedostat enhances cisplatin activity in clinically relevant and translatable experimental models of human bladder cancer, most likely by regulating intracellular cisplatin concentrations.
Discussion
NPEPPS has been suggested to play a role in a range of cellular processes including promoting autophagy, regulating cell-cycle progression, and antigen processing (57–60). The majority of what is known about NPEPPS has been from studies in the brain, where it targets the degradation of polyglutamine sequences and misfolded protein aggregates associated with a number of neurodegenerative diseases, including Alzheimer's disease, Huntington's disease, and Parkinson's disease (58, 61–64). As reported in gnomAD, NPEPPS is a highly conserved gene and is constrained based on several metrics of intolerance to genetic variation in the population (65). NPEPPS is also ubiquitously expressed across human tissues (66). However, despite these features, genetic modification in mice is tolerable, though mice are slower growing, more sickly, and sterile (60, 67), and as we have shown from our CRISPR screen results and follow-up experiments, knockout is not essential for growth in bladder cancer cells (Fig. 2D; Supplementary Fig. S4).
Our multiomic data sets all pointed to NPEPPS as a mechanism of platinum drug resistance and we believe this is due to the unique design of the CRISPR screen, which was performed in treatment-resistant cells, and synthetic lethality was measured in the presence of the treatment for which the cells are resistant. A total of 41 of the 46 screen hits were in DNA damage response and repair genes (Fig. 1; Supplementary Fig. S3). These results show that the screen design can robustly identify general mechanisms of cellular response to platinum drugs. In addition, the experimental context is also critical in identifying genes that are uniquely relevant to treatment-resistant cells. We expect that if we had performed the screen in parental lines, NPEPPS would have been missed. Although there is further work required to validate the clinical translation of our findings, the screen results can be mined for additional candidate genes or even to investigate drug combinations.
This work is not without its limitations. NPEPPS could have effects on treatment response outside of platinum drugs. For example, NPEPPS is upregulated in the Gem-resistant cell lines (Fig. 2), and whereas we show that genetic NPEPPS loss is specific to cisplatin response (Fig. 3; Supplementary Fig. S4), NPEPPS upregulation could be part of broader cellular stress responses. Further studies will be needed to test other NPEPPS-mediated mechanisms of stress response. We note that all of our data support a cell-autonomous effect of NPEPPS. As we indicated above, NPEPPS has been linked to mechanisms of immune response and noncell autonomous effects of NPEPPS were not tested here. Finally, we show that NPEPPS changes intracellular platinum drug concentrations. The mechanism by which NPEPPS regulates these intracellular levels requires future studies.
Despite these limitations, all of our data suggest that NPEPPS is a viable therapeutic target. Broadly, aminopeptidases have been therapeutically targeted as potential cancer treatments (68). More specifically, NPEPPS is a zinc-containing M1 aminopeptidase. Tosedostat was developed as a target of M1 aminopeptidases and the intracellular metabolized product CHR-79888 is the most potent inhibitor of NPEPPS reported (51, 69). There have been a total of 11 clinical trials with tosedostat as reported in clinicaltrials.gov (54, 55, 69–71). The focus of its application has been on leukemias and myelomas, with several applications in solid tumors. The few clinical trials completed have reported tosedostat as being well tolerated by patients but with modest effect as a single-agent cancer treatment. A few examples of tosedostat in combination with cytarabine, azacitidine, capecitabine, or paclitaxel have been tried, but there are no reports of tosedostat being tested in combination with platinum-based chemotherapy.
An exciting potential application of NPEPPS inhibition is to provide alternative treatment options for bladder cancer patients. Our in vivo experiments were done with gemcitabine plus cisplatin to replicate the regimen that patients receive. Because we show that NPEPPS is primarily acting on cisplatin, a translational application would be adding tosedostat to the gemcitabine plus cisplatin regimen with a dose reduction in cisplatin. This combination could potentially reduce the toxic side effects of cisplatin while still improving overall efficacy. Additionally, many patients are ineligible for cisplatin-based chemotherapies, leaving them with less effective options, such as carboplatin. Combining tosedostat with carboplatin could provide a more effective and less toxic drug combination option for cisplatin-ineligible patients. Future work would require validation that NPEPPS regulates carboplatin response similar to cisplatin. A further area of novel development would be the impact of NPEPPS inhibition on ICT with its known effect on MHC class I antigen presentation on dendritic cells (60). ERAP1 and ERAP2, other M1 aminopeptidases in the same family as NPEPPS, have been linked to boosting T-cell and NK cell-mediated immune response in cancer (72); however the impact of NPEPPS on antigen presentation in tumor cells is yet to be investigated. Interestingly, low ERAP2 was associated with improved response to anti–PD-L1 in luminal bladder cancer (73). The impact of NPEPPS inhibition on immunotherapies or in combination with platinum drugs will be the subject of future studies.
In conclusion, our finding that NPEPPS regulates cisplatin-based chemoresistance is both novel and actionable. PDOs have shown predictive value in supporting personalized medicine in several tumor types (47–50, 74, 75). Our data with PDOs provide a strong preclinical rationale for pursuing the clinical application of tosedostat in combination with cisplatin-based chemotherapies (2).
Supplementary Material
Supplementary Figures and Supplementary Table legends.
Supplementary Tables 1-7
Supplementary Table 8
Supplementary Tables 9-16
Acknowledgments
We would like to thank Megan Tu, Colin Sempeck, Ana Chauca-Diaz, Jason Duex, and Charles Owens for their help throughout this project. We would also like to thank Dania Manalo-Mae and the Cedars-Sinai Proteomics and Metabolomics Core Facility for the technical handling of the proteomic experiments. This work was generously supported by the Anschutz Foundation to J.C. Costello, CA268055 to D. Theodorescu and J.C. Costello, FICAN Cancer Researcher by the Finnish Cancer Institute to T.D. Laajala, Erasmus MC mRACE grant 111296 to T.C. Zuiverloon, Erasmus MC fellowship project 107088 to T.C. Zuiverloon, and training grants GM007635 supported R.T. Jones, A.e.g., L.E. Feldman, and C. Deiter, and GM008497 supported R.T. Jones. This work utilized the Functional Genomics Facility, Biostatistics and Bioinformatics Shared Resource, Genomics Shared Resource, and Flow Cytometry Shared Resource supported by CA046934.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Disclosures
R.T. Jones reports a patent for 18/237,705 pending. A. Goodspeed reports a patent for 18/237,705 pending. T. Mahmoudi reports grants from HUB Organoids BV outside the submitted work. T.C. Zuiverloon reports a patent for 18/237,705 pending. D. Theodorescu reports a patent for 18/237,705 pending. J.C. Costello reports nonfinancial support from PrecisionProfile and OncoRx Insights outside the submitted work; in addition, J.C. Costello has a patent for 18/237,705 pending. No disclosures were reported by the other authors.
Authors' Contributions
R.T. Jones: Conceptualization, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft. M. Scholtes: Data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. A. Goodspeed: Conceptualization, data curation, software, formal analysis, investigation, visualization, writing–original draft, writing–review and editing. M. Akbarzadeh: Formal analysis, validation, investigation. S. Mohapatra: Formal analysis, validation, investigation. L.E. Feldman: Data curation, formal analysis, validation, investigation. H. Vekony: Formal analysis, validation, investigation, methodology. A. Jean: Formal analysis, validation, investigation. C.B. Tilton: Formal analysis, validation, investigation. M.V. Orman: Formal analysis, validation, investigation. S. Romal: Formal analysis, validation. C. Deiter: Data curation, validation, investigation. T.W. Kan: Formal analysis, validation. N. Xander: Data curation, formal analysis, validation, investigation, visualization. S.P. Araki: Formal analysis, validation, investigation. M. Joshi: Resources, investigation, methodology. M. Javaid: Data curation, software, formal analysis. E.T. Clambey: Formal analysis, visualization, methodology. R. Layer: Software, formal analysis, supervision, investigation. T.D. Laajala: Data curation, software, formal analysis, visualization, methodology. S.J. Parker: Data curation, formal analysis, supervision, methodology. T. Mahmoudi: Conceptualization, resources, formal analysis, supervision, investigation, methodology, writing–review and editing. T.C.M. Zuiverloon: Conceptualization, resources, data curation, funding acquisition, validation, investigation, methodology, writing–original draft, writing–review and editing. D. Theodorescu: Conceptualization, resources, supervision, funding acquisition, investigation, methodology, writing–original draft, writing–review and editing. J.C. Costello: Conceptualization, resources, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, writing–review and editing.
References
- 1. Dilruba S, Kalayda GV. Platinum-based drugs: past, present and future. Cancer Chemother Pharmacol 2016;77:1103–24. [DOI] [PubMed] [Google Scholar]
- 2. Rottenberg S, Disler C, Perego P. The rediscovery of platinum-based cancer therapy. Nat Rev Cancer 2021;21:37–50. [DOI] [PubMed] [Google Scholar]
- 3. Tran L, Xiao J-F, Agarwal N, Duex JE, Theodorescu D. Advances in bladder cancer biology and therapy. Nat Rev Cancer 2021;21:104–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 5. Dyrskjøt L, Hansel DE, Efstathiou JA, Knowles MA, Galsky MD, Teoh J, et al. Bladder cancer. Nat Rev Dis Primers 2023;9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galsky MD, Pal SK, Lin S-W, Ogale S, Zivkovic M, Simpson J, et al. Real-world effectiveness of chemotherapy in elderly patients with metastatic bladder cancer in the United States. Bladder Cancer 2018;4:227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Galsky MD, Arija JÁA, Bamias A, Davis ID, De Santis M, Kikuchi E, et al. Atezolizumab with or without chemotherapy in metastatic urothelial cancer (IMvigor130): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2020;395:1547–57. [DOI] [PubMed] [Google Scholar]
- 8. Lopez-Beltran A, Cimadamore A, Blanca A, Massari F, Vau N, Scarpelli M, et al. Immune checkpoint inhibitors for the treatment of bladder cancer. Cancers 2021;13:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Heijden MS, Sonpavde G, Powles T, Necchi A, Burotto M, Schenker M, et al. Nivolumab plus gemcitabine-cisplatin in advanced urothelial carcinoma. N Engl J Med 2023;389:1778–89. [DOI] [PubMed] [Google Scholar]
- 10. Grossman HB, Natale RB, Tangen CM, Speights VO, Vogelzang NJ, Trump DL, et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med 2003;349:859–66. [DOI] [PubMed] [Google Scholar]
- 11. Vale CL. Neoadjuvant chemotherapy in invasive bladder cancer: update of a systematic review and meta-analysis of individual patient data: advanced bladder cancer (ABC) meta-analysis collaboration. Eur Urol 2005;48:202–6. [DOI] [PubMed] [Google Scholar]
- 12. Witjes JA, Bruins HM, Cathomas R, Compérat EM, Cowan NC, Gakis G, et al. European association of urology guidelines on muscle-invasive and metastatic bladder cancer: summary of the 2020 guidelines. Eur Urol 2020;79:82–104. [DOI] [PubMed] [Google Scholar]
- 13. Patel VG, Oh WK, Galsky MD. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J Clin 2020;70:404–23. [DOI] [PubMed] [Google Scholar]
- 14. Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data 2014;1:140035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, et al. Defining a cancer dependency map. Cell 2017;170:564–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Goodspeed A, Jean A, Costello JC. A whole-genome CRISPR screen identifies a role of MSH2 in cisplatin-mediated cell death in muscle-invasive bladder cancer. Eur Urol 2019;75:242–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huang A, Garraway LA, Ashworth A, Weber B. Synthetic lethality as an engine for cancer drug target discovery. Nat Rev Drug Discovery 2020;19:23–38. [DOI] [PubMed] [Google Scholar]
- 18. Olivieri M, Cho T, Álvarez-Quilón A, Li K, Schellenberg MJ, Zimmermann M, et al. A genetic map of the response to DNA damage in human cells. Cell 2020;182:481–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sergushichev AA. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. Biorxiv 2016;060012. [Google Scholar]
- 20. Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, Mesirov JP. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011;27:1739–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wickham H. Ggplot2: elegant graphics for data analysis. New York: Springer; 2009. [Google Scholar]
- 22. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32:2847–9. [DOI] [PubMed] [Google Scholar]
- 23. Robinson AE, Binek A, Venkatraman V, Searle BC, Holewinski RJ, Rosenberger G, et al. Lysine and arginine protein post-translational modifications by enhanced DIA libraries: quantification in murine liver disease. J Proteome Res 2020;19:4163–78. [DOI] [PubMed] [Google Scholar]
- 24. Berle M, Ghila L, Vethe H, Chaudhry A, Garberg H, Beisland C, et al. Novel protein signatures suggest progression to muscular invasiveness in bladder cancer. PLoS One 2018;13:e0206475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Parker SJ, Venkatraman V, Van Eyk JE. Effect of peptide assay library size and composition in targeted data-independent acquisition-MS analyses. Proteomics 2016;16:2221–37. [DOI] [PubMed] [Google Scholar]
- 26. Parker SJ, Rost H, Rosenberger G, Collins BC, Malmström L, Amodei D, et al. Identification of a set of conserved eukaryotic internal retention time standards for data-independent acquisition mass spectrometry. Mol Cell Proteomics 2015;14:2800–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imkeller K, Ambrosi G, Boutros M, Huber W. gscreend: modelling asymmetric count ratios in CRISPR screens to decrease experiment size and improve phenotype detection. Genome Biol 2020;21:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, et al. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 2017;12:828–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Winter J, Breinig M, Heigwer F, Brügemann D, Leible S, Pelz O, et al. caRpools: an R package for exploratory data analysis and documentation of pooled CRISPR/Cas9 screens. Bioinformatics 2016;32:632–4. [DOI] [PubMed] [Google Scholar]
- 30. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mullenders J, de Jongh E, Brousali A, Roosen M, Blom JPA, Begthel H, et al. Mouse and human urothelial cancer organoids: a tool for bladder cancer research. Proc Natl Acad Sci USA 2019;116:4567–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Allory Y, Beukers W, Sagrera A, Flández M, Marqués M, Márquez M, et al. Telomerase reverse transcriptase promoter mutations in bladder cancer: high frequency across stages, detection in urine, and lack of association with outcome. Eur Urol 2014;65:360–6. [DOI] [PubMed] [Google Scholar]
- 33. Hurst CD, Zuiverloon TCM, Hafner C, Zwarthoff EC, Knowles MA. A SNaPshot assay for the rapid and simple detection of four common hotspot codon mutations in the PIK3CA gene. BMC Res Notes 2009;2:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Junker K, van Oers JMM, Zwarthoff EC, Kania I, Schubert J, Hartmann A, et al. Fibroblast growth factor receptor 3 mutations in bladder tumors correlate with low frequency of chromosome alterations. Neoplasia 2008;10:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perez-Riverol Y, Csordas A, Bai J, Bernal-Llinares M, Hewapathirana S, Kundu DJ, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res 2019;47:D442–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vallo S, Michaelis M, Rothweiler F, Bartsch G, Gust KM, Limbart DM, et al. Drug-resistant urothelial cancer cell lines display diverse sensitivity profiles to potential second-line therapeutics. Transl Oncol 2015;8:210–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vallo S, Köpp R, Michaelis M, Rothweiler F, Bartsch G, Brandt MP, et al. Resistance to nanoparticle albumin-bound paclitaxel is mediated by ABCB1 in urothelial cancer cells. Oncol Lett 2017;13:4085–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017;171:540–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46:D1062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Korotkevich G, Sukhov V, Sergushichev A. Fast gene set enrichment analysis. Biorxiv 2019;060012. [Google Scholar]
- 42. Drayton RM, Catto JWF. Molecular mechanisms of cisplatin resistance in bladder cancer. Expert Rev Anticancer Ther 2012;12:271–81. [DOI] [PubMed] [Google Scholar]
- 43. Galluzzi L, Senovilla L, Vitale I, Michels J, Martins I, Kepp O, et al. Molecular mechanisms of cisplatin resistance. Oncogene 2012;31:1869–83. [DOI] [PubMed] [Google Scholar]
- 44. Huang D, Savage SR, Calinawan AP, Lin C, Zhang B, Wang P, et al. A highly annotated database of genes associated with platinum resistance in cancer. Oncogene 2021;40:6395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dempster JM, Krill-Burger J, Warren A, McFarland JM, Golub TR, Tsherniak A. Gene expression has more power for predicting in vitro cancer cell vulnerabilities than genomics. Biorxiv 2020;2020.02.21.959627. [Google Scholar]
- 46. Chang Q, Ornatsky OI, Koch CJ, Chaudary N, Marie-Egyptienne DT, Hill RP, et al. Single-cell measurement of the uptake, intratumoral distribution and cell cycle effects of cisplatin using mass cytometry. Int J Cancer 2015;136:1202–9. [DOI] [PubMed] [Google Scholar]
- 47. Tiriac H, Belleau P, Engle DD, Plenker D, Deschênes A, Somerville TDD, et al. Organoid profiling identifies common responders to chemotherapy in pancreatic cancer. Cancer Discov 2018;8:1112–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ooft SN, Weeber F, Dijkstra KK, McLean CM, Kaing S, van Werkhoven E, et al. Patient-derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med 2019;11:eaay2574. [DOI] [PubMed] [Google Scholar]
- 49. Yao Y, Xu X, Yang L, Zhu J, Wan J, Shen L, et al. Patient-derived organoids predict chemoradiation responses of locally advanced rectal cancer. Cell Stem Cell 2020;26:17–26. [DOI] [PubMed] [Google Scholar]
- 50. de Witte CJ, Espejo Valle-Inclan J, Hami N, Lõhmussaar K, Kopper O, Vreuls CPH, et al. Patient-derived ovarian cancer organoids mimic clinical response and exhibit heterogeneous inter- and intrapatient drug responses. Cell Rep 2020;31:107762. [DOI] [PubMed] [Google Scholar]
- 51. Reid AHM, Protheroe A, Attard G, Hayward N, Vidal L, Spicer J, et al. A first-in-man phase I and pharmacokinetic study on CHR-2797 (tosedostat), an inhibitor of M1 aminopeptidases, in patients with advanced solid tumors. Clin Cancer Res 2009;15:4978–85. [DOI] [PubMed] [Google Scholar]
- 52. Chargi N, Molenaar-Kuijsten L, Huiskamp LFJ, Devriese LA, de Bree R, Huitema ADR. The association of cisplatin pharmacokinetics and skeletal muscle mass in patients with head and neck cancer: the prospective PLATISMA study. Eur J Cancer 2022;160:92–9. [DOI] [PubMed] [Google Scholar]
- 53. Urien S, Lokiec F. Population pharmacokinetics of total and unbound plasma cisplatin in adult patients. Br J Clin Pharmacol 2004;57:756–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cortes J, Feldman E, Yee K, Rizzieri D, Advani AS, Charman A, et al. Two dosing regimens of tosedostat in elderly patients with relapsed or refractory acute myeloid leukaemia (OPAL): a randomised open-label phase 2 study. Lancet Oncol 2013;14:354–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Löwenberg B, Morgan G, Ossenkoppele GJ, Burnett AK, Zachée P, Dührsen U, et al. Phase I/II clinical study of tosedostat, an inhibitor of aminopeptidases, in patients with acute myeloid leukemia and myelodysplasia. J Clin Oncol 2010;28:4333–8. [DOI] [PubMed] [Google Scholar]
- 56. Hanada K, Nishijima K, Ogata H, Atagi S, Kawahara M. Population pharmacokinetic analysis of cisplatin and its metabolites in cancer patients: possible misinterpretation of covariates for pharmacokinetic parameters calculated from the concentrations of unchanged cisplatin, ultrafiltered platinum and total platinum. Jpn J Clin Oncol 2001;31:179–84. [DOI] [PubMed] [Google Scholar]
- 57. Constam DB, Tobler AR, Rensing-Ehl A, Kemler I, Hersh LB, Fontana A, et al. Puromycin-sensitive aminopeptidase. Sequence analysis, expression, and functional characterization. J Biol Chem 1995;270:26931–9. [DOI] [PubMed] [Google Scholar]
- 58. Menzies FM, Hourez R, Imarisio S, Raspe M, Sadiq O, Chandraratna D, et al. Puromycin-sensitive aminopeptidase protects against aggregation-prone proteins via autophagy. Hum Mol Genet 2010;19:4573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saric T, Graef CI, Goldberg AL. Pathway for degradation of peptides generated by proteasomes: a key role for thimet oligopeptidase and other metallopeptidases. J Biol Chem 2004;279:46723–32. [DOI] [PubMed] [Google Scholar]
- 60. Towne CF, York IA, Neijssen J, Karow ML, Murphy AJ, Valenzuela DM, et al. Puromycin-sensitive aminopeptidase limits MHC class I presentation in dendritic cells but does not affect CD8 T cell responses during viral infections. J Immunol 2008;180:1704–12. [DOI] [PubMed] [Google Scholar]
- 61. Karsten SL, Sang T-K, Gehman LT, Chatterjee S, Liu J, Lawless GM, et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 2006;51:549–60. [DOI] [PubMed] [Google Scholar]
- 62. Kudo LC, Parfenova L, Ren G, Vi N, Hui M, Ma Z, et al. Puromycin-sensitive aminopeptidase (PSA/NPEPPS) impedes development of neuropathology in hPSA/TAU(P301L) double-transgenic mice. Hum Mol Genet 2011;20:1820–33. [DOI] [PubMed] [Google Scholar]
- 63. Schönlein C, Löffler J, Huber G. Purification and characterization of a novel metalloprotease from human brain with the ability to cleave substrates derived from the N-terminus of beta-amyloid protein. Biochem Biophys Res Commun 1994;201:45–53. [DOI] [PubMed] [Google Scholar]
- 64. Yanagi K, Tanaka T, Kato K, Sadik G, Morihara T, Kudo T, et al. Involvement of puromycin-sensitive aminopeptidase in proteolysis of tau protein in cultured cells, and attenuated proteolysis of frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) mutant tau. Psychogeriatrics 2009;9:157–66. [DOI] [PubMed] [Google Scholar]
- 65. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science 2017;357:eaan2507. [DOI] [PubMed] [Google Scholar]
- 67. Osada T, Ikegami S, Takiguchi-Hayashi K, Yamazaki Y, Katoh-Fukui Y, Higashinakagawa T, et al. Increased anxiety and impaired pain response in puromycin-sensitive aminopeptidase gene-deficient mice obtained by a mouse gene-trap method. J Neurosci 1999;19:6068–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hitzerd SM, Verbrugge SE, Ossenkoppele G, Jansen G, Peters GJ. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids 2014;46:793–808. [DOI] [PubMed] [Google Scholar]
- 69. Krige D, Needham LA, Bawden LJ, Flores N, Farmer H, Miles LEC, et al. CHR-2797: an antiproliferative aminopeptidase inhibitor that leads to amino acid deprivation in human leukemic cells. Cancer Res 2008;68:6669–79. [DOI] [PubMed] [Google Scholar]
- 70. van Herpen CM, Eskens FA, de Jonge M, Desar I, Hooftman L, Bone EA, et al. A phase Ib dose-escalation study to evaluate safety and tolerability of the addition of the aminopeptidase inhibitor tosedostat (CHR-2797) to paclitaxel in patients with advanced solid tumours. Br J Cancer 2010;103:1362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mawad R, Becker PS, Hendrie P, Scott B, Wood BL, Dean C, et al. Phase II study of tosedostat with cytarabine or decitabine in newly diagnosed older patients with acute myeloid leukaemia or high-risk MDS. Br J Haematol 2016;172:238–45. [DOI] [PubMed] [Google Scholar]
- 72. Compagnone M, Cifaldi L, Fruci D. Regulation of ERAP1 and ERAP2 genes and their disfunction in human cancer. Hum Immunol 2019;80:318–24. [DOI] [PubMed] [Google Scholar]
- 73. Lim YW, Chen-Harris H, Mayba O, Lianoglou S, Wuster A, Bhangale T, et al. Germline genetic polymorphisms influence tumor gene expression and immune cell infiltration. Proc Natl Acad Sci USA 2018;115:E11701–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sachs N, de Ligt J, Kopper O, Gogola E, Bounova G, Weeber F, et al. A living biobank of breast cancer organoids captures disease heterogeneity. Cell 2018;172:373–86. [DOI] [PubMed] [Google Scholar]
- 75. Lee SH, Hu W, Matulay JT, Silva MV, Owczarek TB, Kim K, et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell 2018;173:515–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Supplementary Table legends.
Supplementary Tables 1-7
Supplementary Table 8
Supplementary Tables 9-16
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (35) partner repository with the data set identifier PXD024742. The whole-exome sequencing data have been deposited in the BioProject database with project identifier PRJNA714778. The RNA-seq data have been deposited in the GEO database with data set identifier (GSE171537). The CRISPR screen sequencing data have been deposited in the GEO database with data set identifier (GSE179799). The copy-number data have been deposited in the ArrayExpress database with identifier (E-MTAB-10353). The RNA-seq, proteomics, CRISPR screen, and genetic alteration data are made available through a custom build R Shiny app at: https://bioinformatics.cuanschutz.edu/BLCA_GC_Omics/. All other raw data generated in this study are available upon request from the corresponding author.