

Abstract
Membrane proteins play critical physiological roles as receptors, channels, pumps, and transporters. Despite their importance, however, low expression levels often hamper the experimental characterization of membrane proteins. We present an automated and web‐accessible design algorithm called mPROSS (https://mPROSS.weizmann.ac.il), which uses phylogenetic analysis and an atomistic potential, including an empirical lipophilicity scale, to improve native‐state energy. As a stringent test, we apply mPROSS to the Kv1.2–Kv2.1 paddle chimera voltage‐gated potassium channel. Four designs, encoding 9–26 mutations relative to the parental channel, were functional and maintained potassium‐selective permeation and voltage dependence in Xenopus oocytes with up to 14‐fold increase in whole‐cell current densities. Additionally, single‐channel recordings reveal no significant change in the channel‐opening probability nor in unitary conductance, indicating that functional expression levels increase without impacting the activity profile of individual channels. Our results suggest that the expression levels of other dynamic channels and receptors may be enhanced through one‐shot design calculations.
Keywords: membrane protein design, membrane protein expression, PROSS, voltage‐gated potassium channels
1. INTRODUCTION
Membrane proteins (MPs) are gatekeepers to the cell with critical roles in signal transduction and metabolism. Despite their importance, MPs often exhibit marginal stability and low functional expression levels (Marinko et al., 2019). The functional expression of MPs in the eukaryotic plasma membrane depends on their stability (Schlebach et al., 2015) as well as on a multilayered trafficking and quality‐control system embedded in the endoplasmic reticulum and the Golgi apparatus (Marinko et al., 2019). MPs may be mislocalized or targeted for degradation at any stage of biogenesis due to signals that are only partly understood (Marinko et al., 2019).
Methods for improving MP functional expression have had a transformative impact on research but required intense experimental effort. For instance, breakthroughs in determining the structure and function of voltage‐gated potassium (Kv) channels and G‐protein coupled receptors demanded iterative mutagenesis and experimental screening to increase heterologous expression levels (Long et al., 2007b; Magnani et al., 2016; Rosenbaum et al., 2007; Serrano‐Vega et al., 2008; Zhou et al., 2001). Other approaches for improving MP expressibility included deep mutational scanning and genetic recombination of natural homologs (Bedbrook et al., 2019; Scott et al., 2013; Shibata et al., 2013), but these approaches demand medium to high‐throughput screening which is impractical for most MPs (Magnani et al., 2016). Computational design methods have also been developed, but their reliability has so far been limited, demanding multiple iterations of design and experiment (Barth & Senes, 2016; Bedbrook et al., 2017; Bhattacharya et al., 2014; Dodevski & Plückthun, 2011; Popov et al., 2018).
Here, we ask whether the heterologous expression levels of a MP can be improved through one‐shot design calculations that improve native‐state energy. Recently, our lab developed the PROSS algorithm for increasing the stability and heterologous expression levels of water‐soluble proteins (Goldenzweig et al., 2016; Weinstein, Goldenzweig, et al., 2020). PROSS combines phylogenetic analysis and Rosetta atomistic calculations to design dozens of mutations that stabilize the native state. In a benchmark, PROSS improved thermal stability or expressibility in most proteins and required testing fewer than five constructs per target (Peleg et al., 2021). In some cases, PROSS improved thermal stability by ≥20°C and boosted heterologous expression levels by several orders of magnitude without compromising activity (Goldenzweig et al., 2016). PROSS has been applied successfully to optimize therapeutic enzymes (Hettiaratchi et al., 2020), vaccine immunogens (Campeotto et al., 2017; Malladi et al., 2020), soluble receptors (Kriegel et al., 2021), and recently, dramatically increased the expression levels of previously uncharacterized enzymes starting from AI‐based model structures (Barber‐Zucker et al., 2022). At its core, however, PROSS uses a water‐soluble energy function that is inappropriate for MP design. To develop a membrane version of PROSS (mPROSS), we changed the energy function to one that accounts for the membrane environment (Weinstein et al., 2019) by using the dsTβL empirical scale of amino acid insertion into the plasma membrane (Elazar, Weinstein, Biran, et al., 2016; Elazar, Weinstein, Prilusky, et al., 2016). Furthermore, the new energy function addresses both water‐soluble and membrane domains to account for the large extra‐membrane domains often observed in MPs.
As a stringent test, we apply mPROSS to a model Kv channel, the paddle chimera of Kv1.2–Kv2.1 (Long et al., 2007b). We chose this synthetic construct as it is one of the only Kv channels characterized by crystallographic analysis at high resolution (Long et al., 2007b). Furthermore, the paddle chimera exhibits 20‐fold lower K+ current densities than the wild‐type Kv1.2, suggesting that its functional expression levels may be impaired (Karbat et al., 2019). Kv channels are highly selective, mostly homotetrameric channels found throughout the animal kingdom (Moran et al., 2015). They have important roles in the physiology of many organs and in various autoimmune and cardiac muscular and neurological pathologies (Chandy & Norton, 2017; Pardo & Stühmer, 2014). Moreover, Kv channels are highly dynamic, undergoing large conformational rearrangements that control channel opening in response to changes in the electric potential across the plasma membrane. Many natural and synthetic toxins target ion channels, and the paddle chimera is an important model for investigating these interactions (Finol‐Urdaneta et al., 2020). Furthermore, Kv channels can be investigated at the single‐molecule level, providing detailed information on functionality that complements bulk experiments. We tested four mPROSS designs encoding 9–26 mutations relative to the parental channel and found that they exhibited higher expressibility in Xenopus oocytes and increased conductance while preserving voltage sensitivity and channel‐opening probability and conductance. The results demonstrate that automated design calculations can be applied even to a large, oligomeric, dynamic, and structurally complex MP to improve its heterologous functional expression levels without impacting its functional properties. Moreover, the results suggest that the premisses that underlie successful stability design in soluble proteins (Goldenzweig & Fleishman, 2018) may extend to MPs. mPROSS can be applied, in principle, to any MP through a web server that is freely available to academic users (https://mPROSS.weizmann.ac.il).
2. RESULTS
2.1. Improving native‐state energy in membrane proteins
mPROSS combines phylogenetic analysis with Rosetta atomistic calculations to design variants that exhibit improved native‐state energy. Starting from the amino acid sequence of the target MP, we search the non‐redundant (nr) sequence database (Sayers et al., 2022) for homologs and at each position, eliminate mutations that are rarely observed in the natural diversity. The protein is then embedded in a virtual membrane (Alford et al., 2015; Barth et al., 2007) using the Orientations of Proteins in Membranes (OPM) algorithm (Lomize et al., 2012) and relaxed in Rosetta. Next, we use Rosetta atomistic design calculations to eliminate destabilizing single‐point mutations (Table S1). Last, we use Rosetta combinatorial sequence design to compute several low‐energy multipoint mutants that may incorporate dozens of mutations relative to the parental protein (Figure 1a). In all atomistic design calculations, regions that are critical for activity are immutable (Table S3). Furthermore, we disallow mutations at positions that exhibit Gly, Pro, or positions immediately preceding Pro, because they may have roles in determining MP structure and dynamics (Yohannan et al., 2004).
FIGURE 1.
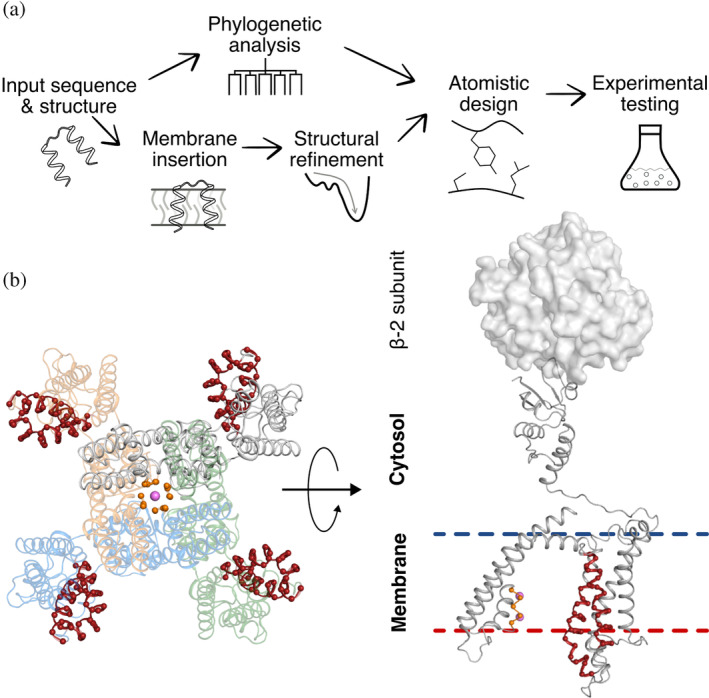
Steps in applying mPROSS to the Kv channel. (a) Scheme of the mPROSS algorithm. mPROSS starts with a phylogenetic analysis of homologs of the target protein and inserts the protein into a virtual membrane. The structure is minimized and subjected to combinatorial design calculations, generating designs with different mutational loads for experimental analysis. (b) Overview of the paddle chimera structure, including the β‐2 soluble subunits responsible for attenuating channel dynamics (PDB entry: 2R9R). The selectivity filter is indicated in orange, and the voltage sensor is in red. The left‐hand side shows the tetrameric arrangement of the channel looking from the extracellular domain toward the fourfold symmetry axis of the channel. Pink spheres indicate K+ ions. The right‐hand side shows a side view of one protomer, and a single β‐2 subunit is shown as a molecular surface.
To account for the physical heterogeneity of the membrane environment, mPROSS uses the ref2015_memb energy function in all atomistic calculations (Weinstein et al., 2019). In the water‐soluble regions, ref2015_memb is identical to the standard ref2015 energy function (Park et al., 2016; Weinstein et al., 2019), whereas within the membrane, it recapitulates the experimentally determined MP insertion scale dsTβL (Elazar, Weinstein, Prilusky, et al., 2016; Weinstein et al., 2019). The dsTβL amino acid insertion scale exhibits membrane‐depth‐dependent lipophilicity. For instance, the hydrophobic amino acids Leu, Ile, and Phe are highly favored near the membrane midplane. In contrast, polar amino acids, such as Asn and Asp, are disfavored in the membrane, as are the helix‐distorting amino acids, Gly and Pro. The basic amino acids Arg and Lys exhibit asymmetric potentials, favoring their localization to the membrane inner leaflet, following the “positive‐inside” rule (von Heijne, 1989). We recently demonstrated that ref2015_memb accurately predicts structures of homomeric single‐pass MPs ab initio (Weinstein et al., 2019) and is effective in de novo design of receptor‐like transmembrane domains of defined oligomeric state and geometry (Elazar et al., 2022). Here, we extend it to design low‐energy variants of large and complex MPs.
As a benchmark, we applied mPROSS to 20 nonredundant (<80% sequence identity (Duran & Meiler, 2018)) multi‐spanning membrane proteins (Figure 2, Table S2). These proteins range in size from 103 to 492 amino acids and span multiple functional classes, including channels, transporters, enzymes, and receptors.
FIGURE 2.
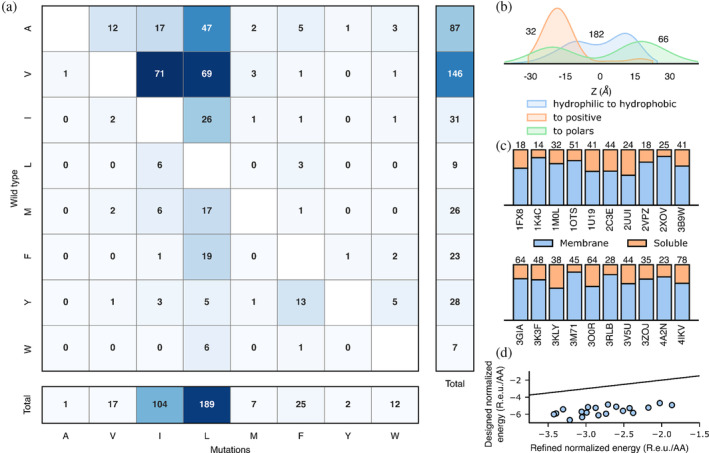
mPROSS introduces mutations that increase lipophilicity and the “positive‐inside” rule. For each of the 20 proteins in the benchmark, we analyzed a single mPROSS design that incorporates approximately 15% mutations, the maximal fraction of mutations we recommend for testing. (a) The number of hydrophobic‐to‐hydrophobic mutations within the membrane domain. (b) Normalized distributions of mutations across the membrane. The horizontal axis represents the distance to the membrane midplane, with negative and positive values representing the intra‐ and extracellular domains, respectively. Numbers denote the number of mutations in each distribution. (c) Fraction of mutations in the membrane or soluble domain in each protein. The number of mutations is noted above the bars, and Protein Data Bank (PDB) entries are noted below the bars. (d) Energies of refined and designed proteins. Line marks x = y. R.e.u./AA denotes the total Rosetta energy divided by protein length.
Near the membrane midplane, mPROSS introduces predominantly hydrophobic‐to‐hydrophobic and hydrophilic‐to‐hydrophobic mutations, and mutations to aromatic identities (51%, 30%, and 9%, respectively, Figure 2a). Moreover, most mutations increase side chain lipophilicity, mainly by introducing Leu, the most lipophilic amino acid (Elazar, Weinstein, Prilusky, et al., 2016). In contrast, mutations in the soluble domain are of a mixed character, as expected (Figure 2b,c, Figure S1). Furthermore, only a small fraction of mutations in the membrane are to polar residues (7%), mostly at positions buried within the protein where they would be shielded from lipid and predominantly to the mildly polar Thr. Strikingly, mutations to the positive Arg and Lys identities mostly occur in the inner‐membrane leaflet or intracellular domain (90%), following the “positive‐inside” rule (Figure 2b) (Gavel et al., 1991; von Heijne, 1986, 1989). The overall effect of introducing dozens of mPROSS mutations is a significant improvement in the Rosetta energy relative to the parental protein (Figure 2d). We conclude that mutations introduced by mPROSS may improve membrane localization and enforce its native topology.
2.2. Paddle chimera designs exhibit high conductance levels
To avoid mutations that impact channel function, we disabled design in the voltage‐sensor domain (Appendices S3 and S4, Figure 1b), the K+‐selectivity filter, and the inner vestibule. Additionally, we restricted positions in the tetrameric interfaces to maintain the oligomeric structure (Table S3). mPROSS automatically generates 18 designs, of which we chose four that exhibit a large number of mutations (designs 12, 16, 17, and 18; these were renumbered for clarity in the remainder of the manuscript to D1–D4, respectively). Following visual inspection, we removed up to 10 mutations from each design (Tables S3 and S4). The four designs introduced 9–26 mutations (5%–13% of mutable positions; Figure 3 and Appendix S2) and were subjected to experimental analysis. We note that as in other applications of PROSS (Goldenzweig et al., 2016), the Rosetta atomistic energies correlate with the number of designed mutations (Table S4) but may not directly correlate with stability or expression levels. Because Rosetta energies do not accurately predict expressibility, we typically test experimentally 3–5 design, as we did here.
FIGURE 3.
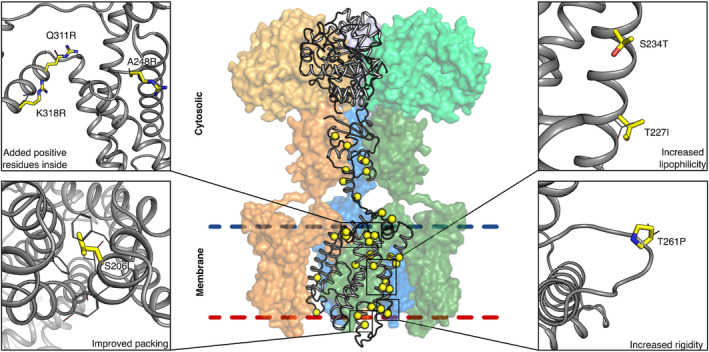
Designed mutations improve protein lipophilicity and native‐state molecular interactions. mPROSS designs both the membrane and cytosolic domains. Mutations increase the lipophilicity of membrane‐exposed positions, add positively charged residues in the inner leaflet (following the “positive‐inside” rule (von Heijne, 1989)), rigidify loops, and improve interhelix packing. The protein mainchain is shown in gray, and mutations are shown in thick yellow sticks or spheres. Only one protomer is shown in cartoon representation, and the other three are represented as colored molecular surfaces. The β‐2 subunits are shown in lighter colors (top).
To determine the yield of functional channels, we recorded channel activity and characterized voltage dependence of the parental channel and four designs in Xenopus laevis oocytes using a single voltage step from a holding potential of −80 to −20 mV for 120 ms. Remarkably, all designs exhibited higher current amplitudes than the parental channel (Figure 4a), and designs D1 and D2 exhibited significantly elevated amplitudes (5‐ and 14‐fold relative to the paddle chimera, respectively). Because designs D3 and D4 conducted currents similar to D2 (Table S6), we focused our subsequent analysis on D1 and D2.
FIGURE 4.
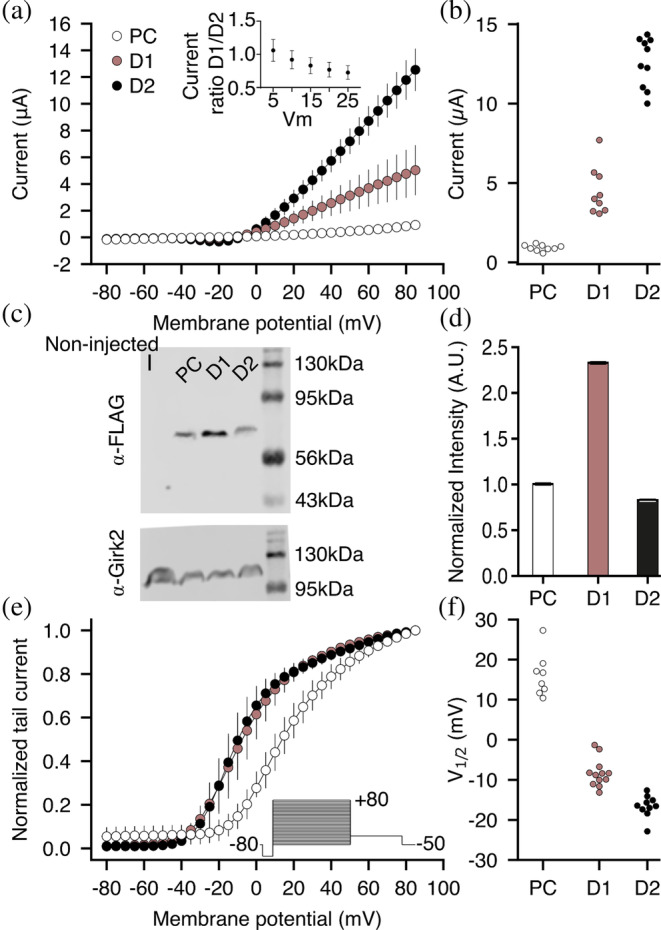
Current–voltage relationship of the Kv1.2–Kv2.1 paddle chimera and its designs. (a) Currents of the paddle chimera (PC) and designs are plotted against varying test voltages. Each curve represents recordings from n ≥ 8 oocytes. Error bars represent standard deviations. (Inset) Ratios of currents observed for D1 and D2 at voltages in the range 5–25 mV. (b) Average currents (paddle chimera: 0.91 ± 0.19 μA; D1: 4.65 ± 1.52 μA; D2: 12.50 ± 1.63 μA). (c) Western blot analysis of total membrane expression. The blots were labeled with anti‐FLAG (top) or anti‐GIRK2 (as control; bottom) antibodies. (d) Normalized blot intensities. The experiment was performed in triplicate. Error bars represent the standard deviation. (e) Normalized tail currents were recorded at −50 mV as a function of the test pulse voltage (see inset). Measured tail current amplitudes were fitted with a single‐component Boltzmann equation (solid lines), from which V 1/2 of activation was inferred. (f) Inferred V 1/2 values (paddle chimera: 16.10 ± 5.39 mV; D1: −8.33 ± 3.49 mV; and D2: −16.61 ± 2.75 mV).
We sought to understand the large difference in current amplitudes between D1 and D2 using mutational analysis. The two designs differ by six mutations, precluding a complete analysis of all combinations of mutations and prompting us to nominate mutations based on structural analysis. We noticed that the mutation Thr271Arg is present in both designs and faces the membrane inner leaflet (all position numbers according to Long et al. (2007b) unless indicated otherwise). We hypothesized that the designed Arg could interact with the phospholipid head groups to stabilize the open state. To test this hypothesis, we reverted the mutation Thr271Arg on the D2 background. The revertant exhibited voltage sensitivity similar to D2, however, refuting a substantial role for this mutation in stabilizing the open state (Figure S2 and Table S5). We then examined several other mutations that differ between D1 and D2 that we deemed likely to affect conductance. On the D2 background, we individually reverted Phe261Leu and Val280Leu (within the voltage‐sensing domain), Met340Leu (on the S4–S5 linker), and Tyr151Phe, Arg133Lys and Glu75Asp (within the cytosolic tetramerization domain). Remarkably, none of the revertants recapitulated D1 currents and some had currents higher than D2. These single substitution analyses indicate that the increased current in D2 likely results from the interaction of multiple designed mutations. They also highlight a strength of the mPROSS strategy of designing multipoint mutations by demonstrating that, individually, mutations may exhibit only a limited impact on expressibility (Goldenzweig & Fleishman, 2018).
The increase in current levels we observed for all designs can stem from higher levels of functional channels at the plasma membrane, elevated single‐channel conductance, or higher opening probability. To decide among these options, we estimated the total protein translation levels from immunoblots of oocyte total membrane fractions finding that D2 expressed at similar levels to the parental channel, whereas D1 expressed close to twofold higher levels (Figure 4c,d). This observation does not reflect the differences seen in current levels (Figure 4a). Because the total membrane fraction includes proteins located at various intracellular membranes of the biogenesis pathway (e.g., ER and Golgi) and currents are measured only from functional channels at the plasma membrane, we can safely suggest that the increase in current levels is not due to higher protein translation levels. However, the ratios of current levels (D1/D2) at potentials where the opening probability is lower than the maximum (2–25 mV) are not significantly different (Figure 4a, inset), suggesting that the twofold difference in current densities between D1 and D2 may be due to a change in the number of functional channels at the plasma membrane. We also tested whether the increased current amplitudes arise from a hyperpolarizing shift in the voltage dependence of the channels. We recorded currents elicited by voltage steps from –80 mV to +80 mV in 90 mM KCl‐containing solution and determined the relative conductance from the resulting tail currents at –50 mV. D1 and D2 exhibited only a slight shift in voltage dependence relative to the parental protein (Figure 4e,f). Nevertheless, the current amplitudes of the designed channels were higher than those of the paddle chimera across the entire voltage range, including at positive potentials where the opening probability is maximal. These data suggest that the shift in voltage dependence is not the main cause for the apparent increase in current amplitudes observed for the designs.
Finally, we tested whether the increased currents were due to a change in the unitary conductance or the channel opening probability. We recorded single‐channel currents under the cell‐attached configuration of the patch‐clamp technique in oocytes. Single‐channel conductance for the parental channel and D2 were similar (10.4 ± 0.4 and 9.7 ± 2.3 pS, respectively; Figure 5). Furthermore, no change was observed in the opening probability between these channels, with Po of wild type and D2 of 0.009 ± 0.004 and 0.011 ± 0.005 at 0 mV, respectively. Taken together, these results rule out the possibility that the designed mutations increase potassium fluxes or stabilize the channel open state and suggest, again, that the increase in ion conductance is due to a higher abundance of functional channels in the plasma membrane.
FIGURE 5.
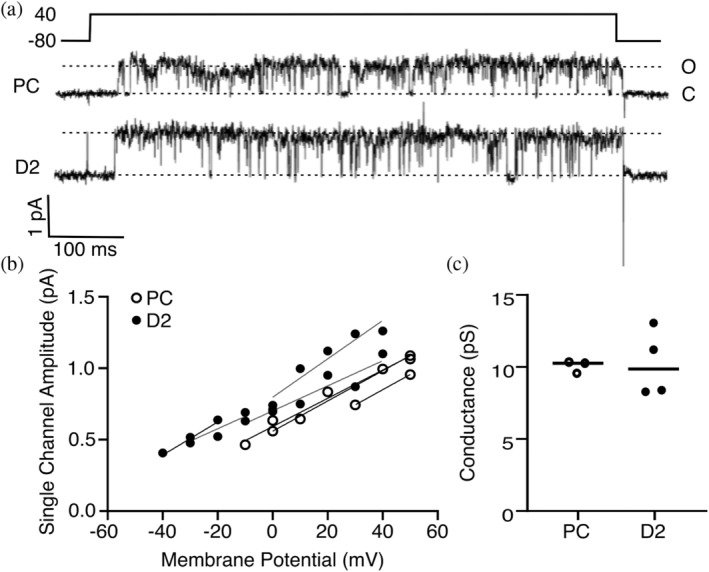
Design D2 behaves similarly to the parental channel at the single‐channel level. (a) Representative current traces were elicited at +40 mV from a holding potential of −80 mV under cell‐attached configuration in Xenopus oocytes for the parental channel chimera and D2. The upward deflection indicates the open state. (b) Individual single‐channel amplitudes at different potentials from all recorded patches. (c) Channel conductance derived from the current to voltage slopes shown in (b).
3. DISCUSSION
Eukaryotic MP biogenesis is a complex process that involves multiple quality‐control steps (Marinko et al., 2019). Despite this complexity, our results provide a case study that validates a one‐shot computational design approach that introduces dozens of mutations selected only according to native‐state energy and phylogenetic propensity. Some designs exhibit substantially higher functional expression levels than the parental protein without exhibiting observable activity differences at the single‐protein level. Increased functional expression may be due to a higher level of plasma membrane localization or to a greater fraction of natively folded and active channels by stabilizing interactions with membrane lipids or within the designs. It is also possible that the designs alter the protein folding pathway, limiting off‐pathway misfolding and slowing the kinetics of unfolding (Goldenzweig & Fleishman, 2018). Further experimental analysis will be required to decide between these possibilities. In a companion paper, we demonstrate that mPROSS can also increase the stability and expression levels while retaining almost wild‐type activity levels in a human membrane enzyme based on its AlphaFold2 model structure (Zelnik et al., 2023). In that case, two homologous human ceramide synthases were subjected to modeling and design, and for one of these, a large increase in expression was observed while a few designs exhibited some decrease in activity at the highest mutation levels. These results show that despite the uncertainties in using structure models of dynamic MPs as targets for atomistic design calculations, mPROSS may in some cases extend to proteins that have not been successfully subjected to experimental structure determination. We also note the low effort required to experimentally screen a few mPROSS designs compared to previous successful engineering efforts that have required testing dozens if not hundreds of mutants (Dodevski & Plückthun, 2011; Magnani et al., 2016; Rosenbaum et al., 2007; Sarkar et al., 2008; Serrano‐Vega et al., 2008). The low expressibility of membrane proteins is a critical limitation of studies in cell signaling, metabolism, and drug discovery, and mPROSS may have a critical role in accelerating research in these areas. Taken together with the broad success of the PROSS algorithm in designing stable and expressible soluble proteins (Campeotto et al., 2017; Goldenzweig et al., 2016; Hettiaratchi et al., 2020; Peleg et al., 2021; Weinstein, Khersonsky, & Fleishman, 2020), we conclude that protein expression may depend, to a large extent, on native‐state energy, whether in soluble or membrane‐embedded proteins.
4. METHODS
4.1. mPROSS algorithm
mPROSS is identical to PROSS2, except where explicitly mentioned. The mPROSS web server contains an FAQ and a tutorial that explain how to use the method.
4.2. Phylogenetic analysis
The phylogenetic analysis is identical to PROSS2 (Weinstein, Goldenzweig, et al., 2020). Briefly, sequences are collected using BLASTP (Altschul et al., 1990; Mahram & Herbordt, 2010) against the non‐redundant database. Only hits with sequence identity >35% and coverage >70% are kept. Sequences are then aligned using MUSCLE (Edgar, 2004) and clustered using CD‐HIT (Li & Godzik, 2006). The MSA is then segmented by secondary structure elements. The sub‐MSA for each sequence is pruned of sequences with different gaps than the query sequence. A sub‐PSSM (Position Specific Scoring Matrix) is then created for each segment, and all resulting sub‐PSSMs are concatenated to create a PSSM for the complete protein sequence (Altschul et al., 2009). Any mutation with PSSM score <0 or absent from the MSA is eliminated from consideration. Additionally, the PSSM score is used to bias the energy function to favor mutations that are more common in the natural diversity.
4.3. Constraints
To avoid introducing deleterious mutations, certain positions are restricted from design. The first and last three positions of each chain, including next to missing density gaps, are automatically restricted, and all non‐canonical residues are automatically restricted. If the user specifies interacting proteins or small molecule or ions ligands, all positions within 8 or 5 Å are restricted, respectively. The user can also specify additional positions to restrict from design.
4.4. Rosetta calculations
All RosettaScripts and flags are in Appendix S1.
Protein structures are inserted into a virtual membrane (Alford et al., 2015) using the OPM algorithm (Lomize et al., 2012). Structures are then refined using the Rosetta full‐atom membrane protein energy function, ref2015_memb, and a “softened” version of ref2015_memb, where van der Waals repulsion is weakened (Weinstein et al., 2019) to enable crossing energy barriers. Five refined structures and the best scoring one are used in all subsequent structural calculations.
A virtual mutational scan, where each non‐restricted position is mutated to each possible amino acid, is conducted using the Rosetta FilterScan mover. Amino acids absent from the MSA in each position are avoided in the mutational scan. The virtual mutation scan applies a set of thresholds and weights (Table S1), generating a set of mutations that pass the ∆∆G threshold. Each set is considered a sequence space for combinatorial design.
The amino acid sequence of the refined structure is then optimized within each sequence space. Due to the high convergence we observe in PROSS design calculations (Goldenzweig et al., 2016), we only carry out two independent trajectories and use the best‐scoring model for each sequence space.
5. EXPERIMENTAL TESTING
5.1. Gene cloning
Gene fragments encoding the paddle chimera and designed channels, codon‐optimized for expression in Xenopus, were ordered from Twist Biosciences. A backbone vector containing the 3′ and 5′ segments of the Kv1.2 gene (including the UTR regions) in pUC57‐Kan was ordered from Genscript. The final constructs were assembled using golden‐gate cloning (Pryor et al., 2020), taking advantage of BsaI sites engineered into the gene fragments and the backbone vector. The final constructs encode for the full‐length paddle chimera with a 5′ 10‐histidine tag and a thrombin cleavage site, cloned into the SacII/EcoRI sites of pUC57‐Kan.
5.2. Two‐electrode voltage clamp
cRNAs encoding for the Kv1.2–Kv2.1 paddle chimera and the various designs were transcribed using T7 mMESSAGE mMACHINE Transcription Kit (Ambion) and stored as stock solutions at −80°C. Xenopus laevis female frogs surgery and oocyte isolation and injection were according to a published protocol (Karbat et al., 2019). Potassium currents from the injected Xenopus oocytes were measured using a two‐electrode voltage clamp technique with a Gene Clamp 500 amplifier (Molecular Devices). Some measurements that required high external K+ were conducted in a 90 K solution containing 90 mM KCl, 2 mM MgCl2, and 10 mM HEPES at pH 7.4. Data were sampled at 10 kHz and filtered at 5 kHz using a Digidata 1550A device controlled by pCLAMP 10.5 (Molecular Devices). Capacitance transients and leak currents were removed by subtracting a scaled control trace utilizing a P/4 protocol.
5.3. Western blot
Injected oocytes were checked for expression using the two‐electrode voltage‐clamp technique (above). Oocytes expressing paddle chimera or designed channels were collected and lyzed using Tris buffer (pH 8) containing protease inhibitors (APExBIO, catalog No. K1007). The lyzed oocytes were manually homogenized and centrifuged at 1500g for 15 min. The supernatant was collected and centrifuged again at 80,000g for 1 h to isolate the membrane fraction. The pellet was resuspended in lysis buffer, and protein concentration was determined using the Bradford assay. A total of 20 μg protein was separated by 12% SDS–PAGE and then transferred onto nitrocellulose paper. Membranes were incubated with mouse monoclonal anti‐flag antibody (abcam, catalog no. ab125243) for 16 h. Post‐incubation, membranes were washed with Dulbecco's phosphate‐buffered saline (Merck) with an HRP‐conjugated anti‐mouse secondary antibody at 1:1000 (Jackson ImmunoResearch Laboratories). The blot was developed using Odyssey® XF Imaging System (LI‐COR Biosciences). For the controls, anti‐GIRK2 (Alomone lab, catalog no. APC‐006) and HRP conjugated anti‐rabbit secondary antibodies were added to label intrinsic membrane proteins expressed in Xenopus oocytes. Controls confirm that the same amount of protein was loaded in each well.
5.4. Single‐channel recordings
Single‐channel currents from devitalized injected oocytes were measured by the cell‐attached patch‐clamp technique using an Axopatch 200B amplifier (Molecular Devices) (Hamill et al., 1981). The bath solution was 90 K (see above), and the pipette solution contained ND96 supplemented with 50 𝜇M GdCl3. Current traces were elicited by step depolarization pulses for the various voltages (−30 to +40) from a holding potential of −80 mV for 600 ms (Clampex 10.4, Molecular Devices), and were low‐pass filtered at 1 kHz, digitized at 10 kHz (Digidata 1550A, Molecular Devices). Leak and transient currents were subtracted using current traces with no openings. Po was calculated as percent time spent in the open state at 100 steps to 0 mV. Patches that contained more than one channel were corrected accordingly. Data analysis was performed using Clampfit 11.2 (Molecular Devices) and Prism 9.4 (GraphPad).
5.5. Animal handling
All procedures employed on experimental animals, including their transportation, routine care, and use in experiments, were conducted in accordance with the Israel animal welfare law and guidelines, NIH guidelines, the Animal Welfare Act, the ethical standards and guidelines of FP7, H2020 with the EU directive 86/609/EEC, as well as the revised directive 2010/63/EU on the protection of animals used for scientific purposes. The procedures were approved by the Weizmann Institute IACUC.
AUTHOR CONTRIBUTIONS
Jonathan Jacob Weinstein: Conceptualization; investigation; writing – original draft; methodology; visualization; writing – review and editing; software; formal analysis. Chandamita Saikia: Investigation; validation; visualization; writing – review and editing; formal analysis. Izhar Karbat: Investigation; writing – original draft; methodology; validation; visualization; writing – review and editing; formal analysis. Adi Goldenzweig: Investigation. Eitan Reuveny: Conceptualization; investigation; funding acquisition; writing – original draft; methodology; validation; visualization; writing – review and editing; formal analysis; project administration; supervision; resources. Sarel Jacob Fleishman: Conceptualization; investigation; funding acquisition; writing – original draft; methodology; validation; visualization; writing – review and editing; software; formal analysis; project administration; supervision; resources.
Supporting information
APPENDIX S1. Supporting information.
ACKNOWLEDGMENTS
We thank Nir Fluman for critical discussions. Research in the Fleishman lab was supported by the Israel Science Foundation (1844), the European Research Council through a Consolidator Award (815379), the Dr. Barry Sherman Institute for Medicinal Chemistry, and a donation in memory of Sam Switzer. Research in the Reuveny lab was supported by the Israeli Science Foundation (349/22), the Wilner Family Fund, and the Gladys Monroy and Larry Marks Center for Brain Disorders. E. R. is the incumbent of the Charles H. Hollenberg Professorial Chair.
Weinstein JJ, Saikia C, Karbat I, Goldenzweig A, Reuveny E, Fleishman SJ. One‐shot design elevates functional expression levels of a voltage‐gated potassium channel. Protein Science. 2024;33(6):e4995. 10.1002/pro.4995
Jonathan Jacob Weinstein, Chandamita Saikia, and Izhar Karbat contributed equally to this work.
Review Editor: Aitziber L. Cortajarena
Contributor Information
Eitan Reuveny, Email: e.reuveny@weizmann.ac.il.
Sarel Jacob Fleishman, Email: sarel@weizmann.ac.il.
REFERENCES
- Alford RF, Leman JK, Weitzner BD, Duran AM, Tilley DC, Elazar A, et al. An integrated framework advancing membrane protein modeling and design. PLoS Comput Biol. 2015;11(9):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W. Basic local alignment search tool. J Mol. 1990;215(3):403–410. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Michael Gertz E, Agarwala R, Schäffer AA, Yi‐Kuo Y. PSI‐BLAST pseudocounts and the minimum description length principle. Nucleic Acids Res. 2009;37(3):815–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber‐Zucker S, Mindel V, Garcia‐Ruiz E, Weinstein JJ, Alcalde M, Fleishman SJ. Stable and functionally diverse versatile peroxidases designed directly from sequences. J Am Chem Soc. 2022;144(8):3564–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P, Schonbrun J, Baker D. Toward high‐resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 2007;104(40):15682–15687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth P, Senes A. Toward high‐resolution computational design of the structure and function of helical membrane proteins. Nat Struct Mol Biol. 2016;23(6):475–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedbrook CN, Yang KK, Elliott Robinson J, Mackey ED, Gradinaru V, Arnold FH. Machine learning‐guided Channelrhodopsin engineering enables minimally invasive optogenetics. Nat Methods. 2019;16(11):1176–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedbrook CN, Yang KK, Rice AJ, Gradinaru V, Arnold FH. Machine learning to design integral membrane Channelrhodopsins for efficient eukaryotic expression and plasma membrane localization. PLoS Comput Biol. 2017;13(10):e1005786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Lee S, Grisshammer R, Tate CG, Vaidehi N. Rapid computational prediction of thermostabilizing mutations for G protein‐coupled receptors. J Chem Theory Comput. 2014;10(11):5149–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeotto I, Goldenzweig A, Davey J, Barfod L, Marshall JM, Silk SE, et al. One‐step design of a stable variant of the malaria invasion protein RH5 for use as a vaccine immunogen. Proc Natl Acad Sci U S A. 2017;114(5):998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandy KG, Norton RS. Peptide blockers of Kv1.3 channels in T cells as therapeutics for autoimmune disease. Curr Opin Chem Biol. 2017;38(June):97–107. [DOI] [PubMed] [Google Scholar]
- Dodevski I, Plückthun A. Evolution of three human GPCRs for higher expression and stability. J Mol Biol. 2011;408(4):599–615. [DOI] [PubMed] [Google Scholar]
- Duran AM, Meiler J. Computational design of membrane proteins using Rosetta membrane. Protein Sci. 2018;27(1):341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar A, Chandler NJ, Davey AS, Weinstein JY, Nguyen JV, Trenker R, et al. De novo‐designed transmembrane domains tune engineered receptor functions. eLife. 2022;11:e75660. 10.7554/eLife.75660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar A, Weinstein J, Biran I, Fridman Y, Bibi E, Fleishman SJ. Mutational scanning reveals the determinants of protein insertion and association energetics in the plasma membrane. eLife. 2016;5:e12125. 10.7554/eLife.12125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elazar A, Weinstein J, Prilusky J, Fleishman SJ. The interplay between hydrophobicity and the positive‐inside rule in determining membrane‐protein topology. Proc Natl Acad Sci. 2016;113:10340–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finol‐Urdaneta RK, Belovanovic A, Micic‐Vicovac M, Kinsella GK, McArthur JR, Al‐Sabi A. Marine toxins targeting Kv1 channels: pharmacological tools and therapeutic scaffolds. Mar Drugs. 2020;18(3):173. 10.3390/md18030173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavel Y, Steppuhn J, Herrmann R, von Heijne G. The ‘positive‐inside rule’ applies to thylakoid membrane proteins. FEBS Lett. 1991;282(1):41–46. [DOI] [PubMed] [Google Scholar]
- Goldenzweig A, Fleishman S. Principles of protein stability and their application in computational design. Annu Rev Biochem. 2018;87:105–129. 10.1146/annurev-biochem-062917-012102 [DOI] [PubMed] [Google Scholar]
- Goldenzweig A, Goldsmith M, Hill SE, Gertman O, Laurino P, Ashani Y, et al. Automated structure‐ and sequence‐based design of proteins for high bacterial expression and stability. Mol Cell. 2016;63(2):337–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch‐clamp techniques for high‐resolution current recording from cells and cell‐free membrane patches. Pflugers Archiv: Eur J Physiol. 1981;391(2):85–100. [DOI] [PubMed] [Google Scholar]
- Hettiaratchi MH, O'Meara MJ, O'Meara TR, Pickering AJ, Letko‐Khait N, Shoichet MS. Reengineering biocatalysts: computational redesign of chondroitinase ABC improves efficacy and stability. Sci Adv. 2020;6(34):eabc6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbat I, Altman‐Gueta H, Fine S, Szanto T, Hamer‐Rogotner S, Dym O, et al. Pore‐modulating toxins exploit inherent slow inactivation to block K+ channels. Proc Natl Acad Sci U S A. 2019;116(37):18700–18709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegel M, Wiederanders HJ, Alkhashrom S, Eichler J, Muller YA. A PROSS‐designed extensively mutated estrogen receptor α variant displays enhanced thermal stability while retaining native allosteric regulation and structure. Sci Rep. 2021;11(1):10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd‐hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. [DOI] [PubMed] [Google Scholar]
- Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long SB, Tao X, Campbell EB, MacKinnon R. Atomic structure of a voltage‐dependent K+ channel in a lipid membrane‐like environment. Nature. 2007b;450(7168):376–382. [DOI] [PubMed] [Google Scholar]
- Magnani F, Serrano‐Vega MJ, Shibata Y, Abdul‐Hussein S, Lebon G, Miller‐Gallacher J, et al. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat Protoc. 2016;11(8):1554–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahram A, Herbordt MC. Fast and accurate NCBI BLASTP: acceleration with multiphase FPGA‐based prefiltering. Proceedings of the 24th ACM international conference on supercomputing, ICS ‘10. New York, NY, USA: Association for Computing Machinery; 2010. p. 73–82. [Google Scholar]
- Malladi SK, Schreiber D, Pramanick I, Sridevi MA, Goldenzweig A, Dutta S, et al. One‐step sequence and structure‐guided optimization of HIV‐1 envelope gp140. Curr Res Struct Biol. 2020;2:45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinko JT, Huang H, Penn WD, Capra JA, Schlebach JP, Sanders CR. Folding and misfolding of human membrane proteins in health and disease: from single molecules to cellular proteostasis. Chem Rev. 2019;119(9):5537–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran Y, Barzilai MG, Liebeskind BJ, Zakon HH. Evolution of voltage‐gated ion channels at the emergence of metazoa. J Exp Biol. 2015;218(Pt 4):515–525. [DOI] [PubMed] [Google Scholar]
- Pardo LA, Stühmer W. The roles of K(+) channels in cancer. Nat Rev Cancer. 2014;14(1):39–48. [DOI] [PubMed] [Google Scholar]
- Park H, Bradley P, Jr PG, Liu Y, Mulligan VK, Kim DE, et al. Simultaneous optimization of biomolecular energy functions on features from small molecules and macromolecules. J Chem Theory Comput. 2016;12(12):6201–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg Y, Vincentelli R, Collins BM, Chen K‐E, Livingstone EK, Weeratunga S, et al. Community‐wide experimental evaluation of the PROSS stability‐design method. J Mol Biol. 2021;433(13):166964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov P, Peng Y, Shen L, Stevens RC, Cherezov V, Liu Z‐J, et al. Computational design of thermostabilizing point mutations for G protein‐coupled receptors. Elife. 2018;7:e34729. 10.7554/elife.34729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor JM, Potapov V, Kucera RB, Bilotti K, Cantor EJ, Lohman GJS. Enabling one‐pot golden gate assemblies of unprecedented complexity using data‐optimized assembly design. PLoS One. 2020;15(9):e0238592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SGF, Thian FS, Kobilka TS, et al. GPCR engineering yields high‐resolution structural insights into beta2‐adrenergic receptor function. Science. 2007;318(5854):1266–1273. [DOI] [PubMed] [Google Scholar]
- Sarkar CA, Dodevski I, Kenig M, Dudli S, Mohr A, Hermans E, et al. Directed evolution of a G protein‐coupled receptor for expression, stability, and binding selectivity. Proc Natl Acad Sci U S A. 2008;105(39):14808–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayers EW, Bolton EE, Rodney Brister J, Canese K, Chan J, Comeau DC, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022;50(D1):D20–D26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlebach JP, Narayan M, Alford C, Mittendorf KF, Carter BD, Li J, et al. Conformational stability and pathogenic misfolding of the integral membrane protein PMP22. J Am Chem Soc. 2015;137(27):8758–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DJ, Kummer L, Tremmel D, Plu A. Stabilizing membrane proteins through protein engineering. Curr Opin Chem Biol. 2013;17(3):427–435. [DOI] [PubMed] [Google Scholar]
- Serrano‐Vega MJ, Magnani F, Shibata Y, Tate CG. Conformational thermostabilization of the β1‐adrenergic receptor in a detergent‐resistant form. Proc Natl Acad Sci U S A. 2008;105(3):877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Gvozdenovic‐Jeremic J, Love J, Kloss B, White JF, Grisshammer R, et al. Optimising the combination of thermostabilising mutations in the neurotensin receptor for structure determination. BBA‐Biomembranes. 2013;1828:1293–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. The distribution of positively charged residues in bacterial inner membrane proteins correlates with the trans‐membrane topology. EMBO J. 1986;5(11):3021–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Control of topology and mode of assembly of a polytopic membrane protein by positively charged residues. Nature. 1989;341(6241):456–458. [DOI] [PubMed] [Google Scholar]
- Weinstein JY, Elazar A, Fleishman SJ. A lipophilicity‐based energy function for membrane‐protein modelling and design. PLoS Comput Biol. 2019;15(8):e1007318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JJ, Goldenzweig A, Hoch S‐Y, Fleishman SJ. PROSS 2: a new server for the design of stable and highly expressed protein variants. Bioinformatics. 2020;37(1):123–125. 10.1093/bioinformatics/btaa1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein J, Khersonsky O, Fleishman SJ. Practically useful protein‐design methods combining phylogenetic and atomistic calculations. Curr Opin Struct Biol. 2020;63:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yohannan S, Faham S, Yang D, Whitelegge JP, Bowie JU. The evolution of transmembrane helix kinks and the structural diversity of G protein‐coupled receptors. Proc Natl Acad Sci U S A. 2004;101(4):959–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelnik ID, Mestre B, Weinstein JJ, Dingjan T, Izrailov S, Ben‐Dor S, et al. Computational design and molecular dynamics simulations suggest the mode of substrate binding in ceramide synthases. Nat Commun. 2023;14(1):2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Morais‐Cabral JH, Kaufman A, MacKinnon R. Chemistry of Ion Coordination and Hydration Revealed by a K+ Channel‐Fab Complex at 2.0 A Resolution. Nature. 2001;414(6859):43–48. 10.2210/pdb1k4c/pdb [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1. Supporting information.