

Abstract
RAF kinases are key components of the RAS‐MAPK signaling pathway, which drives cell growth and is frequently overactivated in cancer. Upstream signaling activates the small GTPase RAS, which recruits RAF to the cell membrane, driving a transition of the latter from an auto‐inhibited monomeric conformation to an active dimer. Despite recent progress, mechanistic details underlying RAF activation remain unclear, particularly the role of RAS and the membrane in mediating this conformational rearrangement of RAF together with 14‐3‐3 to permit RAF kinase domain dimerization. Here, we reconstituted an active complex of dimeric BRAF, a 14‐3‐3 dimer and two KRAS4B on a nanodisc bilayer and verified that its assembly is GTP‐dependent. Biolayer interferometry (BLI) was used to compare the binding affinities of monomeric versus dimeric full‐length BRAF:14‐3‐3 complexes for KRAS4B‐conjugated nanodiscs (RAS‐ND) and to investigate the effects of membrane lipid composition and spatial density of KRAS4B on binding. 1,2‐Dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS) and higher KRAS4B density enhanced the interaction of BRAF:14‐3‐3 with RAS‐ND to different degrees depending on BRAF oligomeric state. We utilized our reconstituted system to dissect the effects of KRAS4B and the membrane on the kinase activity of monomeric and dimeric BRAF:14‐3‐3 complexes, finding that KRAS4B or nanodiscs alone were insufficient to stimulate activity, whereas RAS‐ND increased activity of both states of BRAF. The reconstituted assembly of full‐length BRAF with 14‐3‐3 and KRAS on a cell‐free, defined lipid bilayer offers a more holistic biophysical perspective to probe regulation of this multimeric signaling complex at the membrane surface.
Keywords: biolayer interferometry, in vitro reconstitution, KRAS, MAPK signaling, mechanisms of regulation, nanodiscs, RAF kinase
1. INTRODUCTION
RAF kinase activation is a complex regulatory step within the fundamental RAS‐RAF‐MEK‐ERK (RAS‐MAPK) pathway that controls growth, differentiation, division, and proliferation. Five RAF family members are present in vertebrates: ARAF, BRAF and CRAF (RAF1) in addition to the pseudokinases KSR1/2. RAF kinases share three conserved regions: CR1 comprising the RAS‐binding domain (RBD) and cysteine‐rich domain (CRD) involved in membrane interaction; CR2, a Ser/Thr‐rich region comprising a 14‐3‐3 binding phosphosite and CR3, consisting of the C‐terminal kinase domain (Lavoie & Therrien, 2015). CR2 lies within a more divergent stretch of ~170 residues linking CR1 and CR3, which is predicted to be intrinsically disordered. BRAF also possesses a short N‐terminal helical BRAF‐specific region (BRS) (Lavoie et al., 2018).
The RAS‐MAPK signaling pathway is aberrantly activated in many cancers due to direct mutation of its components including receptor tyrosine kinases (e.g., EGFR), RAS GTPases (especially KRAS), or RAF kinases (predominantly BRAF). BRAF(V600E) is the most prevalent oncogenic RAF mutation (particularly in melanoma) and has been a major focus of drug targeting efforts. The V600E mutation mimics phosphorylation of residues T599 and S602 within the activation segment of the BRAF kinase domain by creating a salt bridge between E600 and K507. This stabilizes the activation segment in an extended conformation which favors kinase domain lobe closure, resulting in constitutive activation of BRAF. (Lavoie & Therrien, 2015). First‐generation V600E‐specific ATP‐competitive inhibitors such as vemurafenib were revealed to induce resistance in patients by stabilizing a closed‐lobe conformation in the BRAF(V600E) kinase domain, which promotes heterodimerization with and resultant ‘paradoxical activation’ of CRAF, thereby restoring MAPK signaling (Lavoie et al., 2013). Although various strategies have been developed to circumvent this phenomenon (Durrant & Morrison, 2018), newer inhibitor types are still vulnerable to acquired resistance mechanisms such as feedback reactivation of other MAPK signaling components. Besides highlighting the need for novel approaches to target mutant RAF as well as wild‐type RAF hyperactivated by upstream signaling perturbations, this underlines the importance of better understanding the mechanistic details of RAF activation.
Biochemical and structural studies over the past three decades have revealed many important aspects of RAF kinase regulation, but key questions remain regarding the process by which RAS binding activates RAF. In its quiescent state, RAF exists as an inactive monomer in the cytosol, stabilized in an auto‐inhibited conformation in which the kinase domain dimerization interface (DIF) and the membrane‐binding surface of the CRD are buried in its core, while the RBD is inferred to be somewhat exposed; a 14‐3‐3 dimer further stabilizes this conformation via phosphorylation‐dependent binding to an inhibitory site within CR2 (pS365 in BRAF; pS259 in CRAF) and a constitutively phosphorylated site within the distal C‐terminus (pS729 in BRAF; pS621 in CRAF) (Martinez Fiesco et al., 2022; Park et al., 2019, 2023). Upon growth factor stimulation, upstream RTK signaling recruits the exchange factor SOS to the cell membrane where it mediates the GTP‐loading of prenylated RAS, inducing conformational changes in RAS switch regions that allow them to bind the RBD, recruiting RAF to the membrane. Via processes that are not fully understood, this triggers the relief of auto‐inhibition and subsequent dimerization of RAF kinase domains, resulting in full activation of RAF, thus enabling it to phosphorylate MEK1/2 (Lavoie & Therrien, 2015). CryoEM structures of autoinhibited and active BRAF (Kondo et al., 2019; Martinez Fiesco et al., 2022; Park et al., 2019, 2023) suggest that specific conformational changes must collectively occur to drive the transition between these two states: 14‐3‐3 must be displaced from pS365 and prevented from rebinding by dephosphorylation of the latter, and the CRD must be extracted to allow interaction with the membrane and expose the kinase domain DIF. RAF dimerization requires spatial proximity of exposed kinase domains and rearrangement of 14‐3‐3 to bridge two pS729 sites across the two protomers. It remains uncertain how exactly these events take place in a coordinated manner.
One central unresolved aspect in this picture is how exactly the recruitment of RAF to the membrane by RAS leads to RAF activation. RAS:RBD‐CRD co‐crystal structures have been solved (Cookis & Mattos, 2021; Tran et al., 2021), and the interaction of RAS and the RBD‐CRD with the membrane have been modeled based on NMR restraints (Fang et al., 2020) and computational approaches (Li et al., 2018; Nguyen et al., 2022; Travers et al., 2018). Due to the difficulty of studying membrane‐anchored RAS, there have been no crystallographic or cryoEM studies of RAF in conjunction with RAS on a membrane. However, in recent years our group and others have demonstrated the use of nanodisc bilayers to examine KRAS behavior on membranes, successfully obtaining NMR structures of nanodisc‐tethered KRAS bound to RAF RBD and RBD‐CRD domains (Fang et al., 2020; Mazhab‐Jafari et al., 2015). Nevertheless, to date the interaction between full‐length BRAF and membrane‐anchored RAS has remained virtually unexamined, precluding observation of key intra‐ and inter‐molecular regulatory interactions.
In this study, we reconstituted the signaling complex comprising purified recombinant BRAF bound to 14‐3‐3 and KRAS4B‐tethered nanodiscs (RAS‐ND). Size‐exclusion chromatography demonstrated that the assembly of this complex is GTP‐dependent. Biolayer interferometry (BLI) measurements revealed that the affinity of 14‐3‐3‐bound BRAF complexes for RAS‐ND is dependent on BRAF oligomeric state, membrane lipid composition and density of RAS on the nanodisc. BRAF activity was noticeably enhanced in the presence of RAS‐ND compared to RAS or nanodisc alone. These studies provide valuable insights into how interactions with RAS and the membrane promote RAF activation.
2. RESULTS
2.1. Purification of dimeric and monomeric BRAF:14‐3‐3 species from baculovirus‐infected Sf9 insect cells
To facilitate studies of the interactions between BRAF, RAS, and the membrane, we first sought to generate full‐length BRAF:14‐3‐3 complexes. A cDNA encoding full‐length wild‐type human BRAF (WT‐BRAF) with an N‐terminal maltose‐binding protein (MBP) tag (Figure 1a) was cloned into the baculovirus expression vector pDest381 and transformed into the Escherichia coli strain DE32 to generate recombinant bacmid (Mehalko & Esposito, 2016), from which baculovirus was generated in Spodoptera frugiperda (Sf9) insect cells. Recombinant MBP‐BRAF was expressed in Sf9 cells via baculovirus‐mediated transfection and the lysate was incubated with amylose resin. After washing, elution with maltose yielded a ~130 kDa band on SDS‐PAGE (Figure 1b), which was confirmed by tandem mass spectrometry (MS/MS) to be the MBP‐BRAF fusion protein. Two distinct lower molecular weight bands (~30 kDa) were observed to co‐elute (Figure 1b) and were identified by MS/MS as the two endogenous S. frugiperda 14‐3‐3 isoforms (ε and ζ, see Appendix S1).
FIGURE 1.

Recombinant expression and purification of BRAF. (a) Schematic of BRAF domain architecture and expression construct consisting of full‐length human BRAF with a TEV protease‐cleavable N‐terminal maltose‐binding protein (MBP) tag and a C‐terminal 3xFLAG tag. BRS, BRAF specific region; CRD, cysteine‐rich domain; CR2, conserved region 2; RBD, RAS‐binding domain. Phosphorylation sites bound by 14‐3‐3 (Ser365 and Ser729) are indicated. (b) SDS‐PAGE analysis of fractions from amylose resin affinity purification of MBP‐tagged BRAF (131 kDa) from baculovirus‐infected Sf9 cells. The 30 and 35 kDa bands were identified by MS/MS as endogenous Spodoptera frugiperda 14‐3‐3 ε and ζ isoforms. Example shown is from purification of BRAF(S365A). E1–E5, fractions eluted with 10 mM maltose; FT, flowthrough after binding amylose resin; P, pellet; SN, supernatant. (c) Size exclusion chromatograms of MBP‐BRAF (WT, purple; S365A, green) in complex with 14‐3‐3 on AdvanceBio SEC 130Å, 4.6 × 300 mm, 2.7 μm, LC column. BRAF(WT) eluted as two peaks in positions consistent with the size of the active dimeric complex (2 MBP‐BRAF: 2 14‐3‐3, ~327 kDa) and the auto‐inhibited monomer (1 MBP‐BRAF: 2 14‐3‐3, ~196 kDa) based on calibration of the column with protein standards. BRAF(S365A) eluted as a single peak consistent with the active dimeric complex.
Phosphoproteomic analysis of MBP‐BRAF revealed that of the two 14‐3‐3 binding sites, S729 was highly phosphorylated (25 out of 27 counts of peptides containing S729 were phosphorylated at this site) consistent with the known role of pS729 in stabilizing both auto‐inhibited and active BRAF (Park et al., 2019). Phosphorylation of the auto‐inhibitory site S365 was more heterogeneous (8 of 14 peptide counts contained pS365), suggesting submaximal RAS‐MAPK pathway activation under the given Sf9 growth conditions. Varying degrees of phosphorylation were also observed for several other documented regulatory sites including the inhibitory‐feedback ERK phosphorylation sites S151 (43 of 67 counts were phosphorylated), T401 (3 of 13), pS750 (1 of 8), and S753 (3 of 46) (Ritt et al., 2009) and the activating PAK/CK2 phosphorylation site S446 (Ritt et al., 2007) (10 of 19), while only unphosphorylated peptides were detected for the activating MLK3 phosphorylation sites in the kinase domain, T599 and S602 (7 and 3 counts, respectively), consistent with observations reported elsewhere (Lavoie & Therrien, 2015).
Size exclusion chromatography (SEC) of the amylose resin eluate produced two overlapping peaks (Figure 1c), both containing MBP‐BRAF and 14‐3‐3 (Figure S1). Based on the chromatograms of protein standards, the elution volumes correspond to the molecular masses of dimeric and monomeric MBP‐BRAF in complex with 14‐3‐3 (Figure 1c). The ratio of 14‐3‐3 to BRAF was lower in the first versus the second peak (Figure S1), correlating with the stoichiometry of the BRAF dimer (2 BRAF:2 14‐3‐3) versus monomer (1 BRAF:2 14‐3‐3). Given that S365 phosphorylation is understood to stabilize the auto‐inhibited monomer, this mixture of oligomeric states is consistent with the heterogeneous phosphorylation levels of S365 in this sample.
Mutation of the S365 auto‐inhibitory phosphorylation site to alanine (S365A‐BRAF) resulted in a single earlier‐eluting peak on SEC corresponding to the position of the dimeric wild‐type MBP‐BRAF:14‐3‐3 complex (Figures 1c and S1). Given the variability of S365 phosphorylation in wildtype BRAF, we chose to use the S365A‐BRAF mutant to assemble an active BRAF complex bound to membrane‐tethered KRAS4B.
2.2. Assembly of a nanodisc‐bound BRAF:14‐3‐3:KRAS4B complex
To assemble KRAS4B–BRAF complexes on a membrane with highly controllable variables, we elected to use nanodiscs: lipid bilayers of defined diameter encapsulated and solubilized by membrane scaffold protein (MSP). Nanodiscs allow precise specification of lipid composition, and the average number of KRAS per nanodisc. KRAS4B was conjugated to a lipid with a maleimide‐conjugated phosphoethanolamine head group (PE‐MCC) in pre‐assembled nanodiscs (Figure 2a) as described previously (Mazhab‐Jafari et al., 2015). Briefly, KRAS4B 1–185 (containing a C118S mutation to eliminate surface‐exposed cysteines such that only the farnesylation site Cys185 reacts with maleimide) was purified and loaded with GMPPNP and nanodiscs were assembled using MSP1E3D1 and a 4:1 ratio of DOPC to 1,2‐dioleoyl‐sn‐glycero‐3‐phospho‐L‐serine (DOPS) (to mimic the inner plasma membrane leaflet) with an experimentally optimized fraction of PE‐MCC (Figure 2a, Step 1). KRAS4B was then covalently linked to PE‐MCC via the thiol of Cys185 (Figure 2a, Step 2) (Gureasko et al., 2008).
FIGURE 2.

Assembly of BRAF:14‐3‐3:KRAS complex on a nanodisc. (a) Nanodisc assembly schematic: Empty ~12 nm nanodiscs were assembled from the membrane scaffold protein MSP1E3D1 and a mixture of phospholipids (1:4 ratio of DOPS:DOPC plus an experimentally optimized amount of PE‐MCC). GMPPNP‐loaded KRAS4B was tethered to nanodiscs via a thiol‐maleimide reaction linking Cys185 to PE‐MCC to attach an average of 2 RAS per nanodisc face. KRAS‐conjugated nanodiscs (RAS‐ND) were incubated with freshly purified BRAF(S365A):14‐3‐3 complexes in a 1:1 [RAS]:[BRAF] ratio, then passed through a SEC column to isolate BRAF:14‐3‐3:RAS‐ND. Note that the BRAF construct used here retains the MBP tag. (b) SEC of BRAF:14‐3‐3:RAS‐ND. GMPPNP‐loaded RAS‐ND incubated with BRAF(S365A):14‐3‐3 (black line) form a complex that co‐elutes earlier than either species alone (BRAF:14‐3‐3—blue, RAS‐ND—red) on a Superdex 200 10/300 GL column. SDS‐PAGE of SEC fractions shows the distribution of bands corresponding to MBP‐BRAF, RAS, MSP and the two endogenous Sf9 14‐3‐3 isoforms, indicating that the ~9 mL peak contains a BRAF:14‐3‐3:RAS‐ND complex. Unbound RAS‐ND (~12.5 mL) was present in slight excess in this example. (c) SEC of BRAF:14‐3‐3 mixed with GDP‐ versus GMPPNP‐loaded RAS‐ND. Co‐elution of RAS‐ND with BRAF:14‐3‐3 requires RAS to be in the activated state (black line). Chromatogram with GDP‐loaded RAS represents a superposition of the peaks corresponding to BRAF:14‐3‐3 and RAS‐ND eluting separately (purple line).
We aimed to assemble nanodiscs with one activated KRAS4B:BRAF complex (i.e., two KRAS4B bound to a BRAF dimer) on each face (Figure 2a). By varying PE‐MCC percentages and KRAS4B:MSP conjugation ratios to control the stoichiometry of RAS on nanodiscs (Figure S3), we populated nanodiscs with an average of two KRAS4B on each membrane leaflet (i.e., [RAS]/[MSP] = 2). MSP1E3D1 produces nanodiscs with an approximate diameter of 12 nm if no transmembrane proteins are incorporated (Denisov et al., 2007), which should be sufficient to accommodate two KRAS4B:RBD‐CRD assemblies on each side of the nanodisc with spatial allowance for potential conformational rearrangements on the membrane surface.
To form a 2:2 BRAF:KRAS4B complex, we incubated MBP‐BRAF(S365A):14‐3‐3 with nanodisc‐tethered, GMPPNP‐loaded KRAS4B (RAS‐ND) in a BRAF:KRAS4B ratio of 1:1 (Figure 2a, Step 3). SEC of this mixture produced a single peak that eluted just after (9 mL) but distinct from the void volume (8 mL), and earlier than either of its two constituent subcomplexes (MBP‐BRAF:14‐3‐3, 10.5 mL; RAS‐ND, 12 mL) (Figure 2b). SDS‐PAGE analysis of these fractions confirmed that the 9‐mL peak contained MBP‐BRAF, 14‐3‐3, KRAS4B, and MSP1E3D1 (Figure 2b), indicating the formation of a complex comprising all three species on the nanodisc. To confirm whether this assembly involved a nucleotide‐dependent BRAF:KRAS4B interaction, we repeated the incubation and SEC using GDP‐loaded KRAS4B. The chromatogram revealed a broad peak (10.5–12 mL) (Figure 2c) consistent with the superposition of the peaks produced when BRAF:14‐3‐3 and RAS‐ND were run separately, demonstrating that formation of the reconstituted complex requires GTP, and supporting the biological relevance of this assembly.
We then investigated whether the monomeric and dimeric forms of BRAF(WT):14‐3‐3 form complexes with KRAS4B‐GMPPNP tethered to nanodiscs. We incubated BRAF(WT) eluate from amylose resin with Ras‐ND; the SEC chromatogram of this mixture showed that the peak corresponding to dimeric MBP‐BRAF:14‐3‐3 shifted, co‐eluting earlier with KRAS4B‐GMPPNP and MSP in a manner similar to BRAF(S365A), while the position of the monomeric species remained unchanged (Figure S4). This suggests that auto‐inhibited monomeric BRAF either does not interact with RAS‐ND or does not interact with sufficient affinity to survive SEC.
2.3. Biophysical studies of factors affecting binding affinity of full‐length BRAF for membrane‐anchored KRAS4B
Binding constants in the nanomolar range have been previously recorded for the interaction between RAS isoforms and RAF RBD and RBD‐CRD domains in solution (Fischer et al., 2007; Herrmann et al., 1995; Smith & Ikura, 2014; Tran et al., 2021). However, these values are likely to be altered when full‐length, 14‐3‐3‐bound BRAF interacts with KRAS4B on a bilayer due to the entropic and enthalpic effects of conformational changes and additional interaction interfaces being formed and disrupted, as well as steric constraint induced by anchoring RAS on the membrane. In the cell, this interaction may also potentially be influenced by RAS dimerization/nanoclustering and local variations in membrane lipid composition. However, no quantitative binding data have yet been reported for full‐length BRAF:14‐3‐3 interacting with membrane‐anchored KRAS.
Prompted by the differences in behavior of monomeric and dimeric BRAF:14‐3‐3 in forming complexes with RAS‐ND, we set out to quantify their respective binding affinities for membrane‐anchored KRAS4B via BLI assays. KRAS4B(GMPPNP)‐conjugated nanodiscs were immobilized on Ni‐NTA sensors via the His tag on MSP, thereby attaching the nanodisc in an orientation that would potentially leave both faces of the nanodiscs accessible to bind BRAF (Figure 3a). The monomeric fraction of BRAF(WT) and the dimeric fraction of BRAF(S365A) were purified by SEC as MBP‐BRAF:14‐3‐3 complexes (henceforth referred to as “M‐BRAF” and “D‐BRAF” respectively for simplicity). Sensors coated with RAS‐ND were dipped into wells containing increasing concentrations of BRAF and binding affinities were quantified via kinetic fitting of association/dissociation curves (Figure 3b). Nonspecific binding of BRAF to empty sensors or to sensors coated with GDP‐loaded RAS‐nanodiscs was negligible (Figure S5a).
FIGURE 3.

Biophysical characterization of the interaction between full‐length BRAF:14‐3‐3 complexes and membrane‐tethered KRAS4B. (a) Experimental set‐up for biolayer interferometry (BLI) assays: RAS‐ND were immobilized onto Ni‐NTA sensors via the 6xHis tag on MSP1E3D1, exposing both faces of the nanodisc for binding. Sensors were dipped into analyte wells containing MBP‐tagged monomeric (M−) or dimeric (D−) BRAF in complex with 14‐3‐3. (b) Representative BLI sensorgrams obtained from GMPPNP‐loaded RAS‐ND binding to two‐fold serial dilutions of BRAF from 12.5 to 400 nM (processed aligned data, blue; kinetic fitting, red). Example shown is for D‐BRAF binding to RAS‐ND with 20% DOPS and a [RAS]/[MSP] ratio of 2. (c) Presence of multiple RAS per nanodisc surface enhances binding of D‐BRAF but not M‐BRAF. Association constants (K A) for D‐BRAF and M‐BRAF binding to RAS‐ND (20% DOPS) at [RAS]/[MSP] ratios of 0.5, 1 or 2 (equivalent to the average number of RAS per nanodisc face). (d) DOPS enhances affinity of BRAF for RAS‐ND. Association constants (K A) for D‐BRAF and M‐BRAF binding to RAS‐ND (2 RAS per face) containing 20% or 0% DOPS. (Values: mean ± 1S.D., n = 4. *p < 0.05, **p < 0.01, ***p < 0.001; statistical analysis by two‐way ANOVA followed by Tukey's post test (c) or Fisher's LSD (d).)
D‐BRAF exhibited a stronger binding affinity than M‐BRAF for Ras‐ND by ~2–4‐fold in all conditions tested (Figure 3c,d, Tables S1 and S2). This finding is consistent with recent cryoEM structures of BRAF, which illustrate that RBD samples both exposed and occluded conformations in the monomer whereas the lack of resolution observed for N‐terminal regions in the dimer suggests that the RBD is substantially more exposed (Martinez Fiesco et al., 2022; Park et al., 2019, 2023). The weaker binding of M‐BRAF suggests that the RBD may be less readily available to RAS‐ND in the auto‐inhibited state, while the binding of D‐BRAF to RAS‐ND may be further enhanced here due to avidity arising from the bivalent interaction between the BRAF dimer and two RAS on the nanodisc.
To account for the possibility that the N‐terminal MBP tag may perturb BRAF:RAS‐ND binding, we prepared BRAF:14‐3‐3 complexes in which MBP was removed by TEV protease (Figure S2) and conducted control BLI assays measuring their binding to RAS‐ND (20% DOPS, 2 RAS per face; Figure S5b). K D values obtained for tagless BRAF (mean ± SD for dimer: 25.5 ± 4.1 nM, n = 4; monomer: 35.5 ± 0.4 nM, n = 2) were similar to those for MBP‐BRAF (Table 1), with negligible or marginally significant differences (dimer: p = 0.164, monomer: p = 0.0464). Tagless BRAF exhibited higher nonspecific binding to empty sensors compared to MBP‐BRAF (Figure S5b), and was less stable, thus background‐subtracted binding data was less reproducible. Considering that steric hindrance of RBD‐CRD by the MBP tag is unlikely due to the predicted flexibility of BRAF residues 1–40 preceeding the BRS, and of linker residues added between MBP and BRAF, we opted to use MBP‐BRAF for all further assays unless otherwise specified.
TABLE 1.
Summary of binding (dissociation constants, K D) and kinetic parameters (association rates, k ON and dissociation rates, k OFF) derived from BLI data for MBP‐BRAF dimer (D‐BRAF) versus monomer (M‐BRAF) binding to nanodiscs with variable DOPS concentrations and number of attached KRAS molecules (mean values ± 1SD, n = 4).
| DOPS% | [RAS]/[MSP] | ⟨K D⟩ (nM) | ⟨k ON⟩ (M−1 s−1 × 10−4) | ⟨kOFF⟩ (s−1 × 103) | |||
|---|---|---|---|---|---|---|---|
| M‐BRAF | D‐BRAF | M‐BRAF | D‐BRAF | M‐BRAF | D‐BRAF | ||
| 0 | 2 | 177.0 ± 8.7 | 57.4 ± 4.2 | 2.11 ± 0.12 | 6.90 ± 0.33 | 3.72 ± 0.26 | 3.95 ± 0.16 |
| 20 | 2 | 68.2 ± 20.0 | 18.5 ± 7.4 | 2.40 ± 0.50 | 10.49 ± 2.04 | 1.60 ± 0.43 | 1.85 ± 0.43 |
| 1 | 93.3 ± 9.0 | 28.6 ± 2.7 | 2.54 ± 0.35 | 9.41 ± 0.18 | 2.35 ± 0.12 | 2.69 ± 0.22 | |
| 0.5 | 65.5 ± 27.8 | 29.1 ± 2.2 | 4.42 ± 1.58 | 10.65 ± 1.02 | 2.66 ± 0.51 | 3.40 ± 0.40 | |
RAS dimerization and nanoclustering has been shown to be important for downstream MAPK signaling (Plowman et al., 2005; Spencer‐Smith et al., 2017), although the exact functional role(s) of this phenomenon remain unclear. To examine whether the density of KRAS4B molecules on the membrane impacts binding affinity for BRAF, we varied the amount of KRAS4B bound to the nanodisc by manipulating the fraction of PE‐MCC in the nanodisc lipid mixture and the RAS:MSP stoichiometric ratio during conjugation (Figure S3). BLI measurements showed that RAS density affected BRAF binding in a manner dependent on BRAF oligomeric state. The presence of multiple RAS on each nanodisc face (i.e., [RAS]/[MSP] = 2) enhanced the binding affinity of D‐BRAF by ~1.6‐fold compared to lower conjugation ratios that favor a single RAS per leaflet ([RAS]/[MSP] ≤ 1), with K D values decreasing from 29.1 to 18.5 nM as the RAS:MSP ratio increased from 0.5 to 2 (Tables 1, S1; Figure 3c). In contrast, RAS density had no obvious effect on the binding affinity of M‐BRAF.
For further insight, we analyzed the kinetics of these interactions (Tables 1 and S1) and found that increased RAS density negligibly affected on‐rates, but resulted in a decrease in off‐rates, which is consistent with enhancement of binding due to avidity associated with a BRAF dimer bound to two RAS. BRAF oligomeric state significantly affected only on‐rates, which were 2–5 fold faster for D‐BRAF relative to M‐BRAF, consistent with exposure of the CRD and improved accessibility of the RBD.
BRAF‐membrane interactions are predominantly driven by the CRD binding to negatively charged membrane lipids, especially phosphatidylserine (Fang et al., 2020; Ghosh et al., 1996). To investigate the role of lipid composition on the assembly of RAS‐BRAF complexes on a bilayer, we compared binding affinities of D‐ and M‐BRAF for RAS‐NDs prepared with or without 20% DOPS incorporated into the nanodisc bilayer. We found that DOPS influenced binding to a degree that varied by BRAF oligomeric state (Tables 1 and S2; Figure 3d). Binding of dimeric BRAF increased ~3‐fold upon inclusion of DOPS, with a reduction in K D from 57.4 to 18.5 nM, which was expected as structural evidence implies that the CRD is fully solvent‐exposed in this conformation (Kondo et al., 2019) and thus available to mediate lipid‐dependent membrane binding. DOPS also appeared to enhance binding of monomeric BRAF, with a K D for DOPC‐only RAS‐NDs of 177.0 nM compared to 68.2 nM with 20% DOPS. Although the difference in binding (K A = 1/K D) remained below the threshold for significance (Table S2), such a trend would potentially suggest that the recruitment of auto‐inhibited BRAF to membrane‐bound KRAS4B is sufficient to induce exposure of the CRD. Kinetic constants derived from the BLI data (Tables 1 and S2) show that DOPS noticeably increased the on‐rate for D‐BRAF but not M‐BRAF, consistent with an initial electrostatic interaction with lipids that is mediated by the CRD only when it is already in an exposed state. DOPS further enhanced the overall affinity of BRAF for KRAS‐ND by decreasing the off‐rates for both M‐ and D‐BRAF (~2‐fold for each state). These results support a scenario in which RAS binding to the RBD initiates CRD exposure to further stabilize BRAF on the membrane.
2.4. Effect of nanodisc‐tethered KRAS4B on the kinase activity of BRAF in vitro
To compare the kinase activities of our monomeric and dimeric BRAF:14‐3‐3 complexes and investigate whether these activity levels were affected in the presence of RAS and/or the membrane, we performed in vitro MEK phosphorylation assays with M‐ and D‐BRAF, alone or pre‐incubated with KRAS4B, RAS‐ND or empty nanodiscs (Figure 4). Both M‐ and D‐BRAF samples possessed kinase activity, as phosphorylated Ser217/221 of MEK was detected in Western blots but absent in negative control reactions (Figures 4b and S6a). Basal kinase activity of M‐BRAF alone was significantly lower compared to D‐BRAF, which in turn was less active than V600E‐BRAF (Figure 4b, Table S3). After incubation with RAS‐ND (RAS:MSP = 2, 20% DOPS), M‐BRAF exhibited moderate enhancement of kinase activity (~3‐fold), while the high activity of V600E‐BRAF was not further enhanced by incubation with RAS‐ND. Incubation with RAS only or empty nanodiscs alone did not significantly affect kinase activity of M‐ or D‐BRAF (Table S3). Control assays using M‐ and D‐BRAF without the MBP tag (Figure S6b) showed similar activity levels to MBP‐tagged BRAF under equivalent conditions.
FIGURE 4.
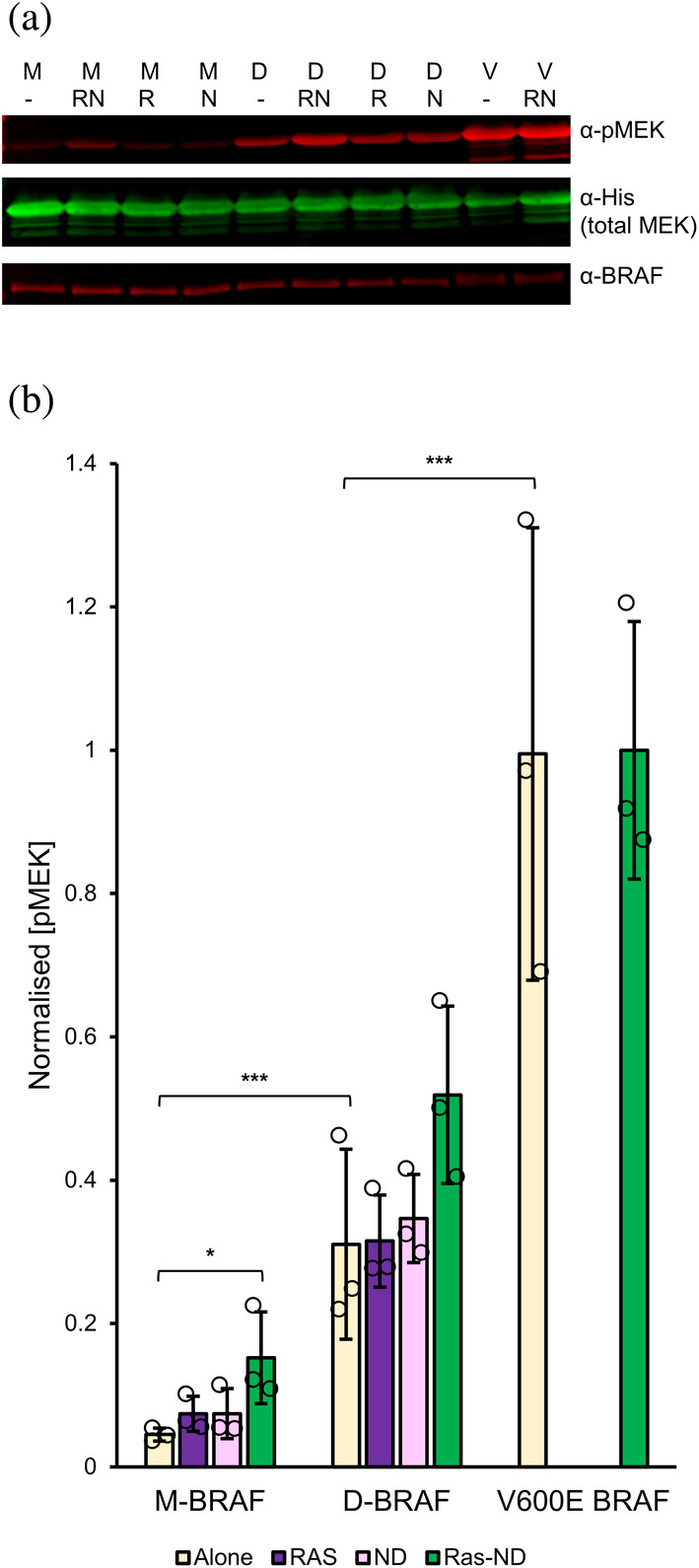
Functional characterization of the effect of membrane‐tethered RAS on in vitro kinase activity of BRAF:14‐3‐3 complexes. (a) Representative Western blot probing phosphorylated (anti‐pSer217/221) and total (anti‐His) recombinant His‐tagged MEK levels following incubation with monomeric (M−), dimeric (D−) or V600E (V−) BRAF, alone (−) or in the presence of RAS (R), empty nanodiscs (N), or RAS‐conjugated nanodiscs (RN). Nanodiscs contained 20% DOPS and 2 RAS per face. (b) RAS‐nanodiscs enhance the kinase activity of both M‐ and D‐BRAF compared to RAS or empty nanodisc alone. Quantification of BRAF activity based on MEK phosphorylation as in panel A. Normalized pMEK refers to the pMEK/total MEK band intensity ratios, normalized against the maximum values in the sample set (V600E + RAS‐ND). (Values: mean ± 1S.D., n = 3. *p < 0.05, **p < 0.01, ***p < 0.001; statistical analysis by unpaired t‐tests comparing treated groups against BRAF alone for each oligomeric state and between M/D/V alone.)
These assay results are consistent with previous evidence showing that the role of RAS in mediating membrane localization of RAF is crucial for activation (Leevers et al., 1994). Our finding that M‐BRAF activity is slightly enhanced in the presence of RAS‐nanodisc, but remains lower than D‐BRAF alone, confirms the requirement for additional cellular factors to fully reconstitute BRAF activation in vitro. Our reconstituted system did not incorporate the SHOC2:MRAS:PP1C (SMP) complex, which is understood to enhance the transition of RAF to the activated dimeric state by mediating dephosphorylation of the CR2 14‐3‐3 binding site (S365 in BRAF) (Liau et al., 2022; Rodriguez‐Viciana et al., 2006). However, the enhancement of M‐BRAF kinase activity by RAS‐ND indicates that this transition complex is partially activated in the absence of SMP‐mediated dephosphorylation, perhaps due to transient relaxation of the autoinhibited conformation.
RAS‐ND did not significantly enhance the activity of D‐BRAF in vitro (Table S3), which is compatible with current knowledge of BRAF activation predicting that dimerized BRAF kinase domains should be fully active already. The ~2‐fold difference in activity between D‐BRAF and V600E‐BRAF (whose kinase domain is constitutively active regardless of oligomerization status) might suggest that the former represents maximum possible activation of dimerization‐dependent BRAF, considering evidence for asymmetrical activation within the RAF kinase dimer whereby the distal C‐terminus of one protomer inhibits the other (Kondo et al., 2019).
3. DISCUSSION
Intensive research efforts over the past decades have yielded by general consensus an outline of the basic mechanism of RAS‐dependent activation of BRAF (Lavoie & Therrien, 2015; Simanshu & Morrison, 2022). However, it remains unclear how the sequence of events initiated by recruitment of RAF to the membrane by RAS drive the conformational changes in the former, resulting in relief of auto‐inhibition and subsequent dimerization. This stems partly from a lack of biophysical/biochemical information regarding the interaction between full‐length RAF and RAS in the presence of biological membranes. Structural and biophysical studies of RAF kinases have mostly omitted RAS and/or used truncations containing isolated RAF domains. Recent cryo‐EM structures of BRAF have revealed key features regarding the overall architecture of the full‐length molecule, confirming the role of 14‐3‐3 in stabilizing its active and auto‐inhibited states through distinct interactions. Nevertheless, the N‐terminal RBD and CRD domains remain unresolved in structures of dimeric BRAF. Some ambiguity also persists as to whether engagement by RAS of the RBD in auto‐inhibited BRAF is sterically incompatible with 14‐3‐3:pS365 binding, potentially destabilizing the latter (Martinez Fiesco et al., 2022; Park et al., 2023). Additionally, despite the importance of membrane localization for RAS function, studies involving recombinantly purified RAS have frequently been limited to using truncated GTPase domain that is not farnesylated, neglecting the presence of the membrane. However, in recent years nanodisc technology has been successfully applied as an experimental approach to obtain insights into RAS behavior on membranes, including by our group (Fang et al., 2020; Lee et al., 2020; Mazhab‐Jafari et al., 2015). We therefore extended this approach to study full‐length, 14‐3‐3‐bound BRAF, making use of the highly tunable nature of RAS‐conjugated nanodiscs to probe in more detail the roles of RAS and the membrane in facilitating the conformational changes in BRAF that bring about its activation.
We reconstituted a stable complex in vitro consisting of a 14‐3‐3‐bound BRAF dimer bound to two KRAS4B‐GMPPNP on a lipid nanodisc. The complex recapitulates the biological characteristics of the RAF:RAS signaling complex in its ability to phosphorylate MEK and its dependence on GTP‐loaded KRAS4B for assembly. We observed that only BRAF isolated as a dimer from Sf9, as opposed to monomeric BRAF, was able to form a SEC‐stable reconstituted complex with RAS‐ND. Prompted by this finding, we used a novel BLI set‐up incorporating all components of the reconstituted complex to quantify experimentally for the first time binding affinities between membrane‐anchored KRAS4B and full‐length monomeric and dimeric BRAF:14‐3‐3 complexes. Consistent with SEC, this data revealed a weaker interaction for monomeric BRAF compared to dimeric BRAF, presumably due to the occluded CRD and partly inaccessible RBD in auto‐inhibited BRAF. Interestingly, a recent study of full‐length BRAF:14‐3‐3 binding to KRAS in solution by microscale thermophoresis measured lower affinity K D values (>100 nM) which differed little between monomeric, dimeric, and BRS‐RBD‐CRD‐only BRAF (Park et al., 2023). The higher affinity K D values we obtained for D‐BRAF binding RAS‐ND are likely due to the energetic contribution of interactions between the nanodisc lipids and the CRD, which is more exposed in the BRAF dimer compared to the monomer.
Manipulating the amount of RAS bound to the nanodisc face allowed us to simulate RAS dimerization/nanoclustering on the membrane. It is unclear exactly how RAS dimers/nanoclusters contribute to downstream signaling. One obvious mechanism would be via increasing proximity of open RAS‐bound BRAF monomers to promote dimerization of their kinase domains. Evidence also suggests that dimerization may stabilize a RAS orientation on the membrane that favors effector binding (Lee et al., 2020). Our BLI data showed that increased RAS density on nanodiscs enhanced affinity only for dimeric BRAF, but not for monomeric BRAF. This effect was driven primarily by slower dissociation from RAS‐ND and amplified by an overall higher initial binding rate of the dimer compared to monomeric BRAF, as would be expected from a bivalent molecule bridging two adjacent RAS, with both RBD and CRD already fully exposed for binding. The apparent independence of the on‐rate with respect to RAS density suggests that the presence of multiple RAS on the membrane acts to enhance RAF binding primarily through avidity, rather than by allosterically increasing the availability/affinity of the RAS effector binding site through RAS dimerization. Although monomeric BRAF also exhibited a slightly reduced off‐rate with increasing RAS density, this did not noticeably affect its binding affinity due to its overall lower on‐rate, which is likely due to steric hindrance of RBD‐CRD in the auto‐inhibited conformation and thus relatively insensitive to alterations on the RAS‐membrane side. Although in a biological signaling context BRAF would typically be recruited by RAS to the membrane as a monomer, the avidity that comes into effect after dimerization of RAS‐bound RAF could assist in prolonging the lifetime of the assembled dimeric RAF–RAS complex on the membrane.
Phosphatidylserine is known to co‐localize preferentially with KRAS nanoclusters (Zhou et al., 2017). As a negatively charged lipid, it has also been shown to enhance CRD binding to the membrane (Fang et al., 2020). Our BLI results bore out this finding, showing that the incorporation of DOPS in the RAS‐nanodisc enhanced BRAF binding. Given that known lipid interactions of BRAF are governed primarily by the CRD, these results may have implications for BRAF conformation within its oligomeric states. CryoEM structures suggest that the CRD is well‐exposed in dimeric BRAF, but buried and inaccessible in the auto‐inhibited monomer. This is consistent with our findings that DOPS enhanced binding of dimeric BRAF, although the decreased off‐rate of monomeric BRAF in the presence of DOPS suggests that the CRD may be at least transiently exposed in the latter. While the bivalent interaction of the 14‐3‐3 dimer with RAF is likely strong due to avidity, temporary monovalent association of 14‐3‐3 is possible given the weaker affinity of 14‐3‐3 for individual pSer sites on RAF, particularly the lower‐affinity CR2 site (Hekman et al., 2004) (Figure 5, Step 1). It is possible that membrane localization of BRAF upon RAS:RBD engagement may facilitate capture of the CRD by the membrane during transient dissociation and rebinding of 14‐3‐3 to either pS365 or pS729, trapping BRAF on the RAS‐nanodisc in a semi‐open monomeric intermediate state where 14‐3‐3 remains in equilibrium binding to both pSer sites, transiently occluding the kinase domain dimerization interface (Figure 5, Step 2).
FIGURE 5.

Model for RAF activation by RAS at the membrane. (1) RAS engagement of the RBD recruits RAF to the cell membrane. Bivalency of dimeric 14‐3‐3 interaction with RAF maintains the 14‐3‐3:RAF complex due to avidity, but the weaker affinity of 14‐3‐3 for individual pSer sites on RAF allows 14‐3‐3 to transiently detach from each site. (2) During RAS binding, transient partial dissociation of 14‐3‐3 releases CRD from within auto‐inhibited BRAF to be captured by the adjacent membrane. (3) A dynamic binding equilibrium between 14‐3‐3 and CR2 pSer may hinder kinase domain dimerization of adjacent RAS‐bound RAF protomers, maintaining RAF in a semi‐open monomeric state. (4) Exposure of the kinase domain dimerization interface via detachment of 14‐3‐3 from CR2 pSer of adjacent RAS/membrane‐bound RAF protomers allows RAF dimerization and activation. After CR2 pSer becomes exposed, PP1C‐mediated dephosphorylation enhances activation by irreversibly exposing the dimerization interface.
One key question we sought to examine was whether the presence of membrane‐anchored RAS constitutes a minimal requirement for BRAF activation in vitro. Our kinase assay results suggest that while RAS and the membrane together have a stronger effect on BRAF activity than each component alone, full activation of M‐BRAF requires additional biochemical factors. Elimination of 14‐3‐3 binding to pS365 is clearly integral to BRAF dimerization and subsequent activation, as evidenced by our observation that the S365A mutation is sufficient to drive BRAF dimerization and enhance its activity in solution. This is further supported by recent structural and functional studies highlighting the importance of the SHOC2:MRAS:PP1C complex (which specifically dephosphorylates BRAF pS365) in MAPK signaling (Liau et al., 2022). The 14‐3‐3:pS365 binding equilibrium likely disfavors exposure of the BRAF dimerization interface, preventing most RAS‐bound BRAF from progressing past the semi‐open monomeric stage in the absence of pS365 dephosphorylation by PP1C (Figure 5, Step 3). However, given that RAS‐ND enhanced M‐BRAF activity but not to the extent of matching D‐BRAF, and considering our BLI results, it is possible that membrane‐tethered RAS may facilitate transient PP1C‐independent BRAF dimerization via CRD‐membrane interactions stabilizing neighboring BRAF protomers in a semi‐open monomeric intermediate state, leading to modest enhancement of total BRAF activity levels. PP1C would be expected to push this equilibrium irreversibly towards dimerization by abolishing the rebinding of 14‐3‐3 to pS365 and is likely required to permit more substantial levels of downstream signaling (Figure 5, Step 4). Nonetheless, the mild activating effect observed with RAS‐ND suggests that targeting the SMP complex therapeutically may not completely inhibit RAF activation.
In summary, we assembled for the first time an in vitro reconstitution of the membrane‐bound active dimeric BRAF:14‐3‐3:KRAS signaling complex using purified full‐length proteins on a lipid nanodisc and examined factors influencing the formation of this complex through biophysical and biochemical means. While our data broadly supports the model for BRAF activation that has emerged through consensus from recent studies (Simanshu & Morrison, 2022), we underscore that, rather than being a linear series of steps, the structural interactions leading up to RAF activation are probably in dynamic equilibrium, making for multiple possible intermediates prior to the formation of a fully active signaling complex of RAS and RAF.
We also demonstrated the use of RAS‐conjugated nanodiscs as a tool for investigating quasi‐native RAS:RAF complexes. Compared to other synthetic membrane systems, our approach has certain advantages for simulating the microenvironment of the RAS nanocluster: greater stability of the covalent RAS‐membrane linkage compared to farnesylation, along with the spatial constraint of the nanodisc, which allows more precise control of RAS distribution. This could make dimerization of RAS‐bound RAF more likely compared to larger membrane surfaces more sparsely populated by RAS. Conversely, nanodiscs cannot reproduce other cellular aspects such as membrane curvature, nor the ability of RAS to diffuse to form larger nanoclusters. Besides the lack of pS365 dephosphorylation, it is possible that these limitations may have prevented us from reproducing full activation of BRAF in vitro. Nevertheless, we see potential for this system to be leveraged further in future studies, for instance functional assays to distinguish the effects of RAS dimerization versus simple spatial proximity, as a platform for cryoEM (though inherent conformational heterogeneity poses significant challenges in structural resolution) or adapted to study interactions on a membrane between other small GTPases and their full‐length RAS effectors.
4. MATERIALS AND METHODS
4.1. BRAF expression and purification
4.1.1. BRAF cloning
A cDNA encoding human BRAF isoform 1 (NCBI Reference Sequence NP_004324.2) with a C‐terminal 3xFLAG tag was obtained as a gift from M.‐S. Tsao (Princess Margaret Cancer Centre, Toronto, Canada) in a pLenti CMV Puro DEST vector. This cDNA was amplified by PCR using primers incorporating flanking 5′ SalI and 3′ NotI restriction sites and a stop codon. The PCR product and pENTR1a gateway vector were digested with SalI and NotI and ligated. Separately, a sequence encoding maltose‐binding protein (MBP) was amplified from pMAL‐c5X via PCR with primers that added an N‐terminal translation initiation sequence (L21) to enhance protein expression in insect cells (Sano et al., 2002), a C‐terminal TEV protease cleavage site and flanking 5′ HindIII +3′ SalI restriction sites. The PCR product was digested and ligated upstream of BRAF into pENTR1a, which had been modified using Q5 mutagenesis to introduce a 5′ HindIII site. S365A and V600E single point mutants of MBP‐BRAF were generated via Quikchange mutagenesis (Agilent). The entire protein‐coding sequence was verified by Sanger sequencing after each cloning step. A Gateway LR reaction (Reece‐Hoyes & Walhout, 2018) was then performed to transfer MBP‐BRAF from pENTR1A to the pDest381 vector for baculovirus expression.
4.1.2. BRAF expression
We used a baculovirus‐based system provided by Esposito et al. (NCI RAS Initiative, Frederick, MD) (Mehalko & Esposito, 2016), which was modified to express MBP‐BRAF in Spodoptera frugiperda (Sf9) insect cells. Briefly, the E. coli strain DE32 was transformed with pDest381 containing the MBP‐BRAF insert to generate bacmids, which were isolated by DNA precipitation and transfected into Sf9 cells with Cellfectin II (Invitrogen) to produce baculovirus. The resulting P1 virus was amplified through two more rounds of infection (P2, P3) to optimize viral titer. For large‐scale expression, 1–2 L of Sf9 was cultured to 1.0 million cells/mL in I‐MAX medium (Wisent Inc.) with 1% penicillin–streptomycin solution and 2.5% fetal bovine serum (Gibco), then infected with 4% P3 virus, and incubated for 72 h at 27°C with gentle shaking at 90 rpm. Cells were harvested by centrifugation at 3000g for 5 min, washed with PBS, flash‐frozen with liquid N2 and stored at −80°C.
4.1.3. Purification of BRAF:14‐3‐3 complexes
Pellets of transfected Sf9 cells were resuspended in 50 mL BRAF buffer (50 mM Tris pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM MgCl2, 10 μM ZnCl2, 0.5 mM DTT) supplemented with cOmplete protease inhibitor cocktail (Roche) and DNAseI. Cells were lysed by sonication and the lysate was clarified by centrifugation at 27,000g for 40 min. The supernatant was incubated with amylose resin (1 mL resin per 50 mL lysate, pre‐equilibrated with BRAF buffer) at 4°C with gentle rocking for 1.5 h. Bound resin was then transferred to a gravity column and washed with 10 mL BRAF buffer. MBP‐BRAF was eluted from the resin in 250 μL fractions using BRAF buffer supplemented with 10 mM maltose.
4.1.4. SEC of BRAF:14‐3‐3 alone
MBP‐BRAF(WT):14‐3‐3 or MBP‐BRAF(S365A):14‐3‐3 fractions eluted from amylose resin were each either concentrated to 500 μL and run on a Superdex 200 10/300 (GE Healthcare) SEC column, or concentrated to 100–200 μL and run on an AdvanceBio SEC 130 Å, 4.6 × 300 mm, 2.7 μm, LC column with a 50 mm guard column (Agilent Technologies), both in BRAF buffer. Molecular weights of peaks were estimated based on calibration with protein standards. The MBP‐BRAF(WT) peak eluting at ~2.75 mL and the MBP‐BRAF(S365A) peak eluting at ~2.45 mL from the AdvanceBio column (designated M‐ and D‐BRAF respectively) were used for subsequent BLI and kinase assays due to better separation of the two peaks on this column, while data from the Superdex column was used to analyze SEC co‐elution experiments with Ras‐ND.
To produce BRAF:14‐3‐3 complexes lacking the MBP tag for use as controls, MBP‐BRAF:14‐3‐3 eluted from amylose resin was mixed with TEV protease in a 1:20 ratio of TEV to total protein by mass, incubated at 4°C for 16 h, then concentrated to 500 μL and run on an AdvanceBio SEC 300Å, 7.8 × 300 mm, 2.7 μM, LC column with a 50 mm guard column (Agilent Technologies) in BRAF buffer. The BRAF(WT) peak eluting at ~8 mL and the BRAF(S365A) peak eluting at ~7.2 mL were collected as the cleaved fractions of M‐ and D‐BRAF, respectively.
4.1.5. Tandem mass spectrometry (MS/MS) analysis of BRAF and Sf9 14‐3‐3 isoforms
Tryptic digest and subsequent MS/MS of SDS–PAGE gel bands were performed to identify BRAF and Sf9 isoforms and to analyze phosphorylation sites in BRAF. Refer to Appendix S1.
4.2. Assembly and SEC analysis of BRAF:14‐3‐3:RAS‐nanodisc complexes
4.2.1. KRAS and MSP1E3D1 expression and purification
We followed previously described protocols for expression and purification of KRAS4B (residues 1–185, C118S) (Mazhab‐Jafari et al., 2015) and His‐tagged MSP1E3D1 (Denisov et al., 2007). Refer to Appendix S1.
4.2.2. Preparation of RAS‐conjugated nanodiscs
Lipid stocks in aqueous buffer were prepared and nanodiscs were assembled based on previously established methods (Gureasko et al., 2008; Mazhab‐Jafari et al., 2015), which were modified to vary the nanodisc lipid composition and RAS conjugation ratio. Refer to Appendix S1.
4.2.3. SEC analyses of BRAF:14‐3‐3 with KRAS4B‐nanodisc
Fractions eluted from amylose resin containing BRAF(S365A):14‐3‐3 (typically ~1 mg/mL in 0.75–1 mL) were incubated with RAS‐nanodiscs containing either GMPPNP‐loaded or GDP‐loaded RAS (where [RAS]/[MSP] = 2 and DOPC:DOPS = 4:1) at a 1:1 RAS:BRAF ratio for 1 h at 4˚C. Following this, each mixture was separated on a Superdex 200 10/300 column in BRAF buffer. SEC analysis of BRAF(WT):14‐3‐3 with RAS(GMPPNP)‐ND was also carried out following the same protocol.
4.3. Biolayer interferometry assays
4.3.1. Assay set‐up and data acquisition
RAS‐nanodiscs (comprising [RAS]/[MSP] = 2, 1 or 0.5 with DOPS, and [RAS]/[MSP] = 2 without DOPS) were immobilized on Ni‐NTA biosensors via the His tag on MSP, and their interactions with M‐BRAF and D‐BRAF were measured via BLI using an Octet RED‐384 (ForteBio/Sartorius) instrument and analyzed using ForteBio software. Refer to Appendix S1.
4.4. In vitro BRAF kinase activity assays
4.4.1. Expression and purification of MEK substrate
We obtained a plasmid containing a previously described His‐tagged MEK1 (residues 63–393) construct (Lavoie et al., 2018) as a gift from F. Sicheri (Lunenfeld‐Tanenbaum Research Institute, Toronto, Canada). We introduced the inactivating mutation K97R to prevent MEK autophosphorylation and followed previously established protocols for its expression and purification. Refer to Appendix S1.
4.4.2. Kinase assay reactions
Reactions were carried out in kinase assay buffer (50 mM Tris pH 7.5, 150 mM NaCl, 10 mM MgCl2, 0.5 mM TCEP, 1 mM sodium vanadate, 10 mM NaF). Freshly purified BRAF samples (monomeric BRAF(WT) and dimeric BRAF(S365A) fractions isolated by SEC on the AdvanceBio column, or BRAF‐V600E without SEC) were incubated alone or with RAS, empty nanodisc, or RAS‐nanodisc for 1 h at 4°C, with BRAF in an equimolar ratio with RAS where applicable. Samples were then diluted to 0.1 μM BRAF. For each reaction, 2 μL of 0.1 μM BRAF was pre‐mixed with 5 μL of 4 μM ATP. Reactions were initiated through addition of 13 μL of 5 μM MEK, incubated for 30 min at 25°C, then quenched by adding 4× SDS‐PAGE sample loading buffer and heating for 10 min at 60°C. Additional control reactions were performed with V600E alone in the absence of ATP or BRAF, and with monomeric/dimeric BRAF lacking the MBP tag, alone and with RAS‐ND.
4.4.3. Western blotting and quantification
pMEK, total MEK, and BRAF levels in reaction samples were detected by Western blotting. Normalized pMEK levels were obtained via densitometric analysis using ImageJ (Schneider et al., 2012). Refer to Appendix S1.
AUTHOR CONTRIBUTIONS
Ningdi F. Liu: Writing – original draft; conceptualization; investigation; methodology; formal analysis; visualization; data curation; validation; writing – review and editing. Masahiro Enomoto: Conceptualization; methodology; writing – review and editing; formal analysis; validation. Christopher B. Marshall: Conceptualization; writing – review and editing; project administration; funding acquisition; methodology; formal analysis; validation; writing – original draft. Mitsuhiko Ikura: Conceptualization; funding acquisition; writing – review and editing; project administration; supervision; methodology; validation; writing – original draft.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
Appendix S1. Supporting information.
ACKNOWLEDGMENTS
We thank D. Esposito (NCI RAS Initiative, Frederick, MD, USA) for the baculovirus expression system; M.‐S. Tsao (Princess Margaret Cancer Centre, Toronto, Canada) for the plasmid encoding BRAF cDNA; F. Sicheri (Lunenfeld‐Tanenbaum Research Institute, Toronto, Canada) for the plasmid encoding MEK1 kinase domain; and L. Wybenga Groot of the SPARC Biocentre (Sick Kids Hospital, Toronto, Canada) for mass spectrometric data collection. This work was supported by the Canadian Cancer Society Research Institute (Grant 703209 to M.I.), Princess Margaret Foundation (M.I.), Canadian Institutes of Health Research (FDN‐1542284 to M.I.). M.I. holds a Canada Research Chair in cancer structural biology.
Liu NF, Enomoto M, Marshall CB, Ikura M. Reconstitution and characterization of BRAF in complex with 14‐3‐3 and KRAS4B on nanodiscs. Protein Science. 2024;33(6):e5016. 10.1002/pro.5016
Review Editor: Hideo Akutsu
Contributor Information
Christopher B. Marshall, Email: cmarshal@uhn.ca.
Mitsuhiko Ikura, Email: mitsu.ikura@uhn.ca.
REFERENCES
- Cookis T, Mattos C. Crystal structure reveals the full Ras–Raf interface and advances mechanistic understanding of Raf activation. Biomolecules. 2021;11(7):996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov IG, Baas BJ, Grinkova YV, Sligar SG. Cooperativity in cytochrome P450 3A4: linkages in substrate binding, spin state, uncoupling, and product formation. J Biol Chem. 2007;282(10):7066–7076. [DOI] [PubMed] [Google Scholar]
- Durrant DE, Morrison DK. Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br J Cancer. 2018;118(1):3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Z, Lee K‐Y, Huo K‐G, Gasmi‐Seabrook G, Zheng L, Moghal N, et al. Multivalent assembly of KRAS with the RAS‐binding and cysteine‐rich domains of CRAF on the membrane. Proc Natl Acad Sci. 2020;117(22):12101–12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Hekman M, Kuhlmann J, Rubio I, Wiese S, Rapp UR. B‐ and C‐RAF display essential differences in their binding to RAS: the isotype‐specific N terminus of B‐RAF facilitates RAS binding. J Biol Chem. 2007;282(36):26503–26516. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM. Raf‐1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf‐1 in 12‐O‐tetradecanoylphorbol‐13‐acetate‐stimulated Madin‐Darby canine kidney cells. J Biol Chem. 1996;271(14):8472–8480. [DOI] [PubMed] [Google Scholar]
- Gureasko J, Galush WJ, Boykevisch S, Sondermann H, Bar‐Sagi D, Groves JT, et al. Membrane‐dependent signal integration by the Ras activator son of sevenless. Nat Struct Mol Biol. 2008;15(5):452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman M, Wiese S, Metz R, Albert S, Troppmair J, Nickel J, et al. Dynamic changes in C‐Raf phosphorylation and 14‐3‐3 protein binding in response to growth factor stimulation: differential roles of 14‐3‐3 protein binding sites. J Biol Chem. 2004;279(14):14074–14086. [DOI] [PubMed] [Google Scholar]
- Herrmann C, Martin GA, Wittinghofer A. Quantitative analysis of the complex between p21ras and the Ras‐binding domain of the human Raf‐1 protein kinase. J Biol Chem. 1995;270(7):2901–2905. [DOI] [PubMed] [Google Scholar]
- Kondo Y, Ognjenović J, Banerjee S, Karandur D, Merk A, Kulhanek K, et al. Cryo‐EM structure of a dimeric B‐Raf:14‐3‐3 complex reveals asymmetry in the active sites of B‐Raf kinases. Science. 2019;366(6461):109–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Sahmi M, Maisonneuve P, Marullo SA, Thevakumaran N, Jin T, et al. MEK drives BRAF activation through allosteric control of KSR proteins. Nature. 2018;554(7693):549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol. 2015;16(5):281–298. [DOI] [PubMed] [Google Scholar]
- Lavoie H, Thevakumaran N, Gavory G, Li JJ, Padeganeh A, Guiral S, et al. Inhibitors that stabilize a closed RAF kinase domain conformation induce dimerization. Nat Chem Biol. 2013;9(7):428–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KY, Fang Z, Enomoto M, Gasmi‐Seabrook G, Zheng L, Koide S, et al. Two distinct structures of membrane‐associated homodimers of GTP‐ and GDP‐bound KRAS4B revealed by paramagnetic relaxation enhancement. Angew Chem Int Ed. 2020;59(27):11037–11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leevers SJ, Paterson HF, Marshall CJ. Requirement for Ras in Raf activation is overcome by targeting Raf to the plasma membrane. Nature. 1994;369(6479):411–414. [DOI] [PubMed] [Google Scholar]
- Li Z‐L, Prakash P, Buck M. A “tug of war” maintains a dynamic protein‐membrane complex: molecular dynamics simulations of C‐Raf RBD‐CRD bound to K‐Ras4B at an anionic membrane. ACS Cent Sci. 2018;4(2):298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau NPD, Johnson MC, Izadi S, Gerosa L, Hammel M, Bruning JM, et al. Structural basis for SHOC2 modulation of RAS signalling. Nature. 2022;609(7926):400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Fiesco JA, Durrant DE, Morrison DK, Zhang P. Structural insights into the BRAF monomer‐to‐dimer transition mediated by RAS binding. Nat Commun. 2022;13(1):486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazhab‐Jafari MT, Marshall CB, Smith MJ, Gasmi‐Seabrook GMC, Stathopulos PB, Inagaki F, et al. Oncogenic and RASopathy‐associated K‐RAS mutations relieve membrane‐dependent occlusion of the effector‐binding site. Proc Natl Acad Sci. 2015;112(21):6625–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehalko JL, Esposito D. Engineering the transposition‐based baculovirus expression vector system for higher efficiency protein production from insect cells. J Biotechnol. 2016;238:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen K, López CA, Neale C, Van QN, Carpenter TS, Di Natale F, et al. Exploring CRD mobility during RAS/RAF engagement at the membrane. Biophys J. 2022;121(19):3630–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rawson S, Li K, Kim BW, Ficarro SB, Del PGG, et al. Architecture of autoinhibited and active BRAF–MEK1–14‐3‐3 complexes. Nature. 2019;575(7783):545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Rawson S, Schmoker A, Kim B‐W, Oh S, Song K, et al. Cryo‐EM structure of a RAS/RAF recruitment complex. Nat Commun. 2023;14(1):4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plowman SJ, Muncke C, Parton RG, Hancock JF. H‐ras, K‐ras, and inner plasma membrane raft proteins operate in nanoclusters with differential dependence on the actin cytoskeleton. Proc Natl Acad Sci U S A. 2005;102(43):15500–15505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reece‐Hoyes JS, Walhout AJM. Gateway recombinational cloning. Cold Spring Harb Protoc. 2018;2018(1):pdb.top094912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B‐Raf signaling. Mol Cell Biol. 2009;30(3):806–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritt DA, Zhou M, Conrads TP, Veenstra TD, Copeland TD, Morrison DK. CK2 is a component of the KSR1 scaffold complex that contributes to Raf kinase activation. Curr Biol. 2007;17(2):179–184. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Viciana P, Oses‐Prieto J, Burlingame A, Fried M, McCormick F. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M‐Ras effector to modulate Raf activity. Mol Cell. 2006;22(2):217–230. [DOI] [PubMed] [Google Scholar]
- Sano K‐I, Maeda K, Oki M, Maéda Y. Enhancement of protein expression in insect cells by a lobster tropomyosin cDNA leader sequence. FEBS Lett. 2002;532(1–2):143–146. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu DK, Morrison DK. A structure is worth a thousand words: new insights for RAS and RAF regulation. Cancer Discov. 2022;12(4):899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Ikura M. Integrated RAS signaling defined by parallel NMR detection of effectors and regulators. Nat Chem Biol. 2014;10(3):223–230. [DOI] [PubMed] [Google Scholar]
- Spencer‐Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, et al. Inhibition of RAS function through targeting an allosteric regulatory site. Nat Chem Biol. 2017;13(1):62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TH, Chan AH, Young LC, Bindu L, Neale C, Messing S, et al. KRAS interaction with RAF1 RAS‐binding domain and cysteine‐rich domain provides insights into RAS‐mediated RAF activation. Nat Commun. 2021;12(1):1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers T, López CA, Van QN, Neale C, Tonelli M, Stephen AG, et al. Molecular recognition of RAS/RAF complex at the membrane: role of RAF cysteine‐rich domain. Sci Rep. 2018;8:8461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Prakash P, Liang H, Cho K‐J, Gorfe AA, Hancock JF. Lipid‐sorting specificity encoded in K‐Ras membrane anchor regulates signal output. Cell. 2017;168(1–2):239–251.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information.