

SUMMARY
During infections, host cells are exposed to pathogen-associated molecular patterns (PAMPs) and virulence factors that stimulate multiple signaling pathways that interact additively, synergistically, or antagonistically. The net effect of such higher-order interactions is a vital determinant of the outcome of host-pathogen interactions. Here, we demonstrate one such complex interplay between bacterial exotoxin- and PAMP-induced innate immune pathways. We show that two caspases activated during enterohemorrhagic Escherichia coli (EHEC) infection by lipopolysaccharide (LPS) and Shiga toxin (Stx) interact in a functionally antagonistic manner; cytosolic LPS-activated caspase-11 cleaves full-length gasdermin D (GSDMD), generating an active pore-forming N-terminal fragment (NT-GSDMD); subsequently, caspase-3 activated by EHEC Stx cleaves the caspase-11-generated NT-GSDMD to render it nonfunctional, thereby inhibiting pyroptosis and interleukin-1β maturation. Bacteria typically subvert inflammasomes by targeting upstream components such as NLR sensors or full-length GSDMD but not active NT-GSDMD. Thus, our findings uncover a distinct immune evasion strategy where a bacterial toxin disables active NT-GSDMD by co-opting caspase-3.
In brief
Wright et al. report a bacterial immune subversion strategy in which Shiga toxin (Stx), an exotoxin produced by enterohemorrhagic Escherichia coli (EHEC), disables active NT-GSDMD by co-opting caspase-3 and limits noncanonical inflammasome responses.
Graphical Abstract

INTRODUCTION
Secreted exotoxins are some of the most well-characterized virulence factors of pathogenic bacteria.1 Bacterial exotoxins cause immense damage to host cells through multiple mechanisms. During an infection, in addition to exotoxin exposure, host cells encounter an array of pathogen components that activate innate immune surveillance pathways. While a toxin itself may be sufficient to inflict damage on the host, the interplay between toxin-mediated responses and innate immune responses is also consequential. However, many of these complex interactions are poorly characterized.
Shiga toxin (Stx), a well-characterized exotoxin produced by the food-borne pathogen enterohemorrhagic Escherichia coli (EHEC) causes hemorrhagic colitis and potentially fatal hemolytic uremic syndrome (HUS) in humans.2–4 HUS, characterized by hemolysis, thrombocytopenia, and renal failure, develops primarily in pediatric and geriatric patients and is the leading infectious cause of renal failure in children worldwide.4 While EHEC remains in the intestinal lumen or attached to the intestinal epithelium, Stx released at the mucosal surface crosses the epithelial barrier, reaches the systemic circulation, and mediates disease.2,5,6 Stx is encoded by a lysogenic bacteriophage, and antibiotics induce phage and Stx production, precluding the use of antibiotics as a treatment.4,7 Thus, there is an acute need for new treatments for EHEC disease. However, a poor understanding of disease-relevant EHEC-host interactions remains a barrier to developing new treatments.
During EHEC infection, similar to Stx, EHEC lipopolysaccharide (LPS) becomes systemic and interacts with a wide range of cells.2,5,8 The noncanonical inflammasome is a critical innate immune component that detects intracellular LPS via inflammatory caspases (caspase-11 in mice and caspase-4 in humans).9–13 Active caspase-11/−4 directly cleaves its substrate gasdermin D (GSDMD) into N-terminal (NT) and C-terminal (CT) fragments.14,15 Owing to its pore-forming property, NT-GSDMD mediates pyroptotic cell death and also triggers NLRP3 inflammasome-mediated caspase-1 activation and interleukin (IL)-1β secretion.14,16,17 We have previously shown that during EHEC infection, caspase-11 senses LPS that enters the host cell cytosol via outer membrane vesicles, leading to noncanonical inflammasome activation, GSDMD cleavage, cell death, and IL-1 cytokine responses.18 Importantly, our recent study revealed that Stx suppresses GSDMD-mediated noncanonical inflammasome responses to EHEC.19 While this finding assigned a previously undescribed function to Stx, the molecular mechanism by which Stx inhibits GSDMD was not clear. This study reveals that Stx activates caspase-3 and active caspase-3 directly targets and inactivates active NT-GSDMD generated by caspase-11, impairing noncanonical inflammasome responses. This finding unravels a unique innate immune evasion strategy where a bacterial toxin co-opts a predominantly apoptotic caspase to restrain inflammasome responses, expanding our understanding of integrative response of host cells to coincident signals from bacterial pathogens.
RESULTS AND DISCUSSION
Stx induces further processing of the active NT domain of GSDMD
Stx-mediated noncanonical inflammasome suppression occurs at the level of GSDMD cleavage.19 Stx treatment prior to LPS transfection or EHEC infection results in a marked reduction in the level of the pore-forming NT domain of GSDMD (NT-GSDMD) in bone-marrow-derived macrophages (BMDMs) (Figures 1A and 1B).19 Similarly, infection with an E. coli BL21 strain that expresses Stx (BL21/pStx) in an isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible manner also results in a decrease in GSDMD cleavage compared to infection with an empty-vector-containing strain (BL21/pEmpty) or H2O-treated BL21/pStx (Figure 1C). Intriguingly, we observed that this decrease in NT-GSDMD is accompanied by the appearance of a new ~23 kDa GSDMD fragment (p23 GSDMD) in the lysates of cells exposed to Stx during LPS transfection or infection with EHEC or E. coli BL21 (Figures 1A–1C). While infection with Stx-deficient EHEC (ΔStx) did not result in the appearance of p23 GSDMD, complementing ΔStx-infected cells with purified Stx generated p23 GSDMD, indicating that Stx is essential for the generation of p23 GSDMD during EHEC infection (Figure 1B). Furthermore, the appearance of p23 GSDMD was dependent on the enzymatic activity of Stx, as treatment with the catalytically inactive Stx toxoid did not result in the generation of this p23 fragment (Figure 1A).
Figure 1. Stx induces further processing of caspase-11-generated NT-GSDMD.
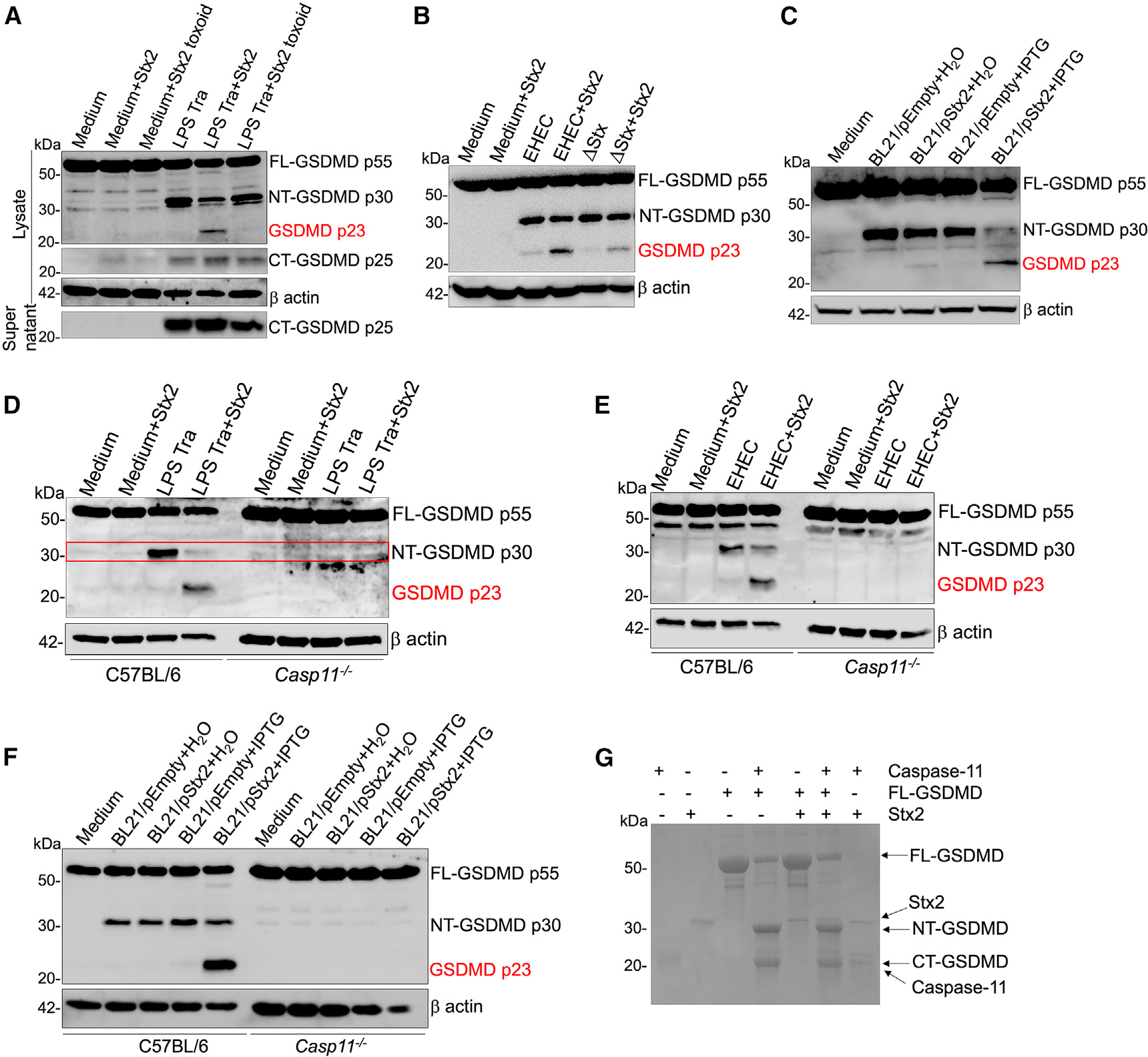
(A–F) GSDMD cleavage in the lysates of Pam3CSK4-primed C57BL/6 or Casp11−/− BMDMs treated with 4 μg/mL purified Stx2 or enzymatically inactive Stx2 (toxoid) at the time of transfection (Tra) with LPS (1 μg/106 cells) complexed with lipofectamine 2000 for 16 h (A and D) or infection with EHEC (B and E) or H2O- or IPTG-treated BL21/pEmpty or BL21/pStx2 (C and F) for 16 h. Note the appearance of a p23 fragment of GSDMD in indicated conditions.
(G) Coomassie-blue-stained SDS-PAGE gel of recombinant GSDMD subjected to cleavage by recombinant active caspase-11 (p20/p10) in the presence or absence of Stx2. Data are representative of 3 experiments.
Notably, the p23 GSDMD fragment was detected specifically by an antibody that detects only full-length (FL)- and NT-GSDMD, but not CT-GSDMD, suggesting that it belongs to the NT region of GSDMD. Consistently, an antibody specific for CT-GSDMD did not detect any p23-like additional fragments in Stx-treated cells (Figure 1A). Further, Stx treatment prior to LPS transfection specifically reduced the levels of NT-GSDMD but not CT-GSDMD (Figure 1A). The p23 fragment could thus be derived from either the NT region of uncleaved FL-GSDMD or the cleaved NT-GSDMD itself. Notably, the p23 fragment did not appear in cells exposed to Stx alone but rather appeared only when cells were exposed to Stx in the presence of noncanonical inflammasome activators such as EHEC, E. coli BL21, or LPS transfection (Figures 1A–1C). This indicated that the mere presence of FL-GSDMD was not sufficient for the appearance of p23 GSDMD, but the generation of NT-GSDMD was a prerequisite. Consistent with this idea, in Casp11−/− cells where FL-GSDMD is present but NT-GSDMD is not generated upon treatment with noncanonical inflammasome activators, Stx did not induce the appearance of p23 GSDMD (Figures 1D–1F), affirming that caspase-11-generated NT-GSDMD is the precursor for p23 GSDMD. Together, these observations indicate that upon exposure to Stx, NT-GSDMD is further processed to a nonfunctional p23 GSDMD, which could underlie the reduction in NT-GSDMD levels. Next, we conducted a cell-free in vitro GSDMD cleavage assay to test if Stx directly cleaves NT-GSDMD to generate p23 GSDMD. While the incubation of FL-GSDMD with active caspase-11 generated NT-GSDMD, the addition of Stx did not reduce NT-GSDMD levels or generate p23 GSDMD (Figure 1G). This result suggested the involvement of an intermediate factor, likely a protease activated by Stx, in the generation of p23 GSDMD from NT-GSDMD.
Stx-induced processing of NT-GSDMD occurs coincidentally with caspase-3 activation
Stx-induced responses and lethality are highly cell-type specific. In sensitive cells such as HeLa cells, Stx is known to induce responses such as endoplasmic reticulum stress and mitochondrial cytochrome c release leading to caspase-8- and caspase-9-mediated activation of the cysteine protease caspase-3.20–22 Interestingly, we and others have shown that innate immune cells such as primary human and mouse macrophages and monocytes are not susceptible to Stx-mediated lethality.8,19,23,24 Therefore, it is not clear whether Stx activates caspase-3 in primary macrophages. Notably, we observed that treatment with purified Stx2 (Figures 2A and S1A), infection with IPTG-treated BL21/pStx (Figure 2B), or infection with EHEC (Figures 2C and S1A) resulted in caspase-3 cleavage in BMDMs. Initiator caspases, caspase-8 and caspase-9, were also cleaved by Stx and EHEC infection (Figure S1A), and pretreatment with caspase-8 or −9 inhibitors reduced Stx- and EHEC-induced caspase-3 cleavage, indicating that caspase-3 activation by Stx in BMDMs is mediated by caspase-8 and −9 (Figures S1B and S1C). Interestingly, Stx-induced caspase-3 activation did not appear to result in apoptosis in BMDMs as assessed by Apotracker Green25 staining (Figures S2A and S2B).
Figure 2. Caspase-3 activation corresponds to the appearance of p23 GSDMD.
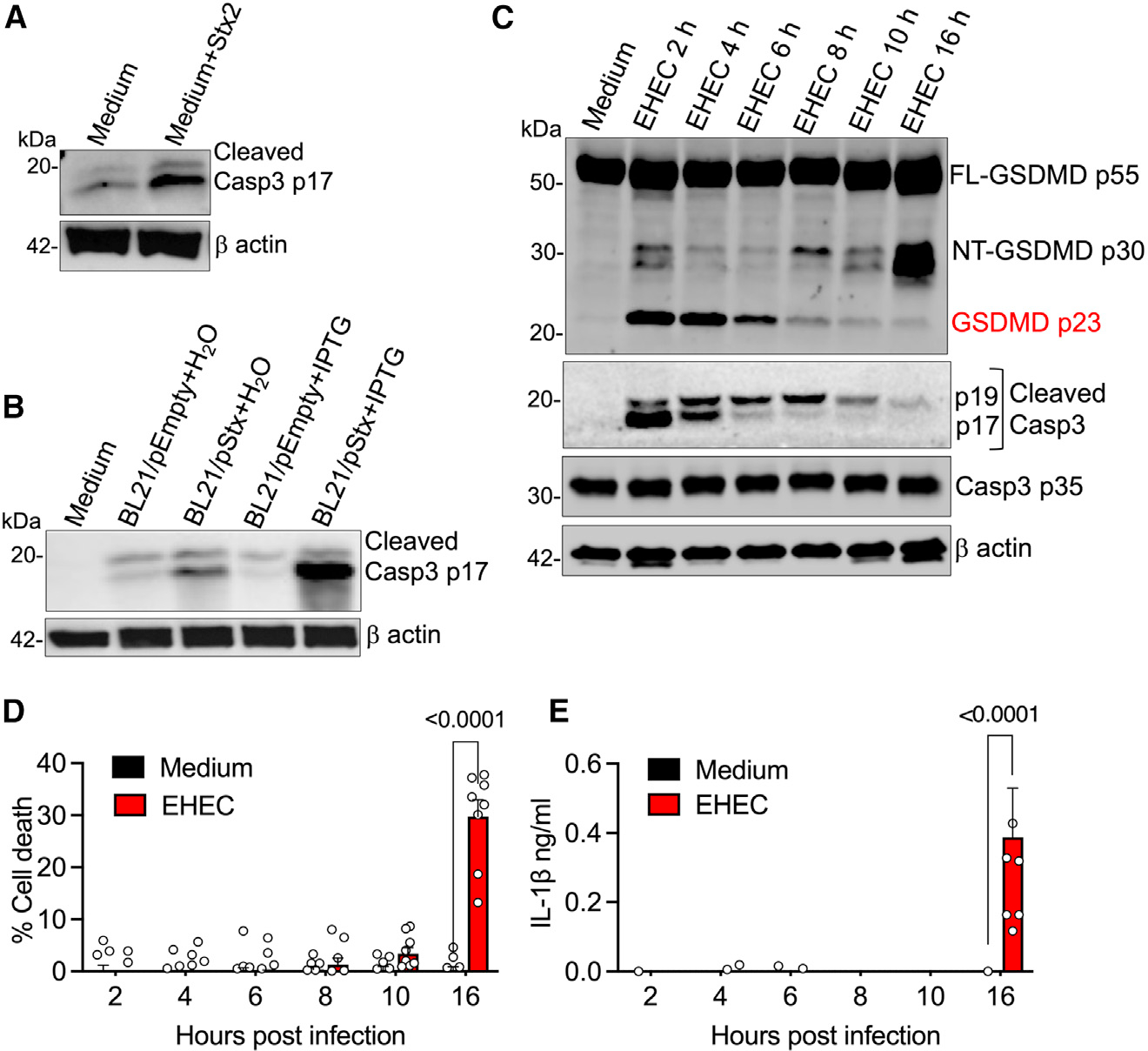
(A–C) Caspase-3 (A–C) and GSDMD (C) cleavage in the lysates of BMDMs treated with Stx2 (4 μg/mL) or infected with H2O- or IPTG-treated BL21/pEmpty or BL21/pStx2 for 6 h or infected with EHEC for the indicated time.
(D and E) Pyroptosis and IL-1β secretion at the indicated times post-infection with EHEC as assessed by lactate dehydrogenase (LDH) assay and ELISA, respectively, in the supernatants of BMDMs. Mean ± SEM of combined data from 3 biological replicates, p < 0.05 is considered significant; two-way ANOVA with Tukey’s post-test.
(A–C) Data are representative of 3 experiments. See also Figure S1 and Figure S2.
Subsequently, to examine whether EHEC-induced caspase-3 activation corresponds to the appearance of p23 GSDMD, we infected BMDMs with EHEC and monitored the kinetics of caspase-3 activation and GSDMD cleavage. Remarkably, we observed a strong activation of caspase-3 early in EHEC infection with a concomitant appearance of the p23 GSDMD fragment (Figure 2C). Intriguingly, the levels of both active caspase-3 and p23 GSDMD diminished correlatively at later stages of EHEC infection (Figure 2C), likely due to degradation of Stx in the cells. This unsustained caspase-3 activation may account for the lack of apoptosis in EHEC-infected BMDMs. More importantly, this decrease in caspase-3 and p23 GSDMD levels corresponded with an increase in the functional NT-GSDMD p30 fragment (Figure 2C) and subsequent cell death and IL-1β secretion (Figures 2D and 2E). Taking these results together, it appears that although NT-GSDMD is generated early in EHEC infection, it is processed rapidly to the nonfunctional p23 GSDMD likely by caspase-3, thereby suppressing cell death and IL-1 responses. In contrast, as the infection proceeds, active caspase-3 level declines and, correspondingly, NT-GSDMD then remains intact and functional, leading to effective downstream responses such as cell death and IL-1β release.
Stx-activated caspase-3 mediates inhibition of noncanonical inflammasome responses
The above observations give rise to a compelling hypothesis that during EHEC infection, Stx-activated caspase-3 cleaves and inactivates NT-GSDMD generated by caspase-11, leading to the suppression of inflammasome responses. We tested this using caspase-3-deficient BMDMs. Briefly, BMDMs from wildtype (WT) or Casp3−/− mice were infected with EHEC in the presence or absence of Stx2 or E. coli BL21 strains or were treated with Stx2 prior to LPS transfection, and inflammasome responses were assessed 16 h later. As expected, infection with EHEC and E. coli BL21/pStx2+IPTG and Stx treatment prior to LPS transfection led to the generation of the p23 GSDMD fragment in WT cells (Figures 3A–3C). Stx addition resulted in a marked decrease in NT-GSDMD, cell death, and IL-1β secretion in WT cells (Figures 3A–3D). Remarkably, this Stx-mediated decrease in the levels of NT-GSDMD, cell death, and IL-1β was rescued in Casp3−/− BMDMs (Figures 3A–3D). More importantly, the p23 GSDMD fragment did not appear in the lysates of EHEC- or BL21/pStx+IPTG-infected or Stx2+LPS-transfected Casp3−/− BMDMs in contrast to WT BMDMs (Figures 3A–3C). The noncanonical inflammasome-suppressive ability is shared by multiple Stx variants.19 The above-described studies were conducted with Stx2, a more potent variant of Stx. To test the role of caspase-3 in inflammasome suppression by other variants of Stx, we pretreated WT or Casp3−/− BMDMs with Stx1 or Stx2d prior to LPS transfection and assessed GSDMD cleavage. Like Stx2, Stx1 and Stx2d induced activation of caspase-3, reduction in NT-GSDMD levels, and generation of p23 GSDMD in WT cells transfected with LPS (Figures 3E and 3F). In contrast, no decrease in NT-GSDMD or the appearance of p23 GSDMD was observed in Casp3−/− cells, demonstrating that caspase-3 is essential for GSDMD inhibition by multiple Stx variants. Together, these results support our hypothesis and identify caspase-3 as the primary mediator of Stx-induced generation of the p23 GSDMD fragment and suppression of NT-GSDMD-driven noncanonical inflammasome responses during EHEC infection.
Figure 3. Caspase-3 is essential for Stx-mediated inflammasome suppression.
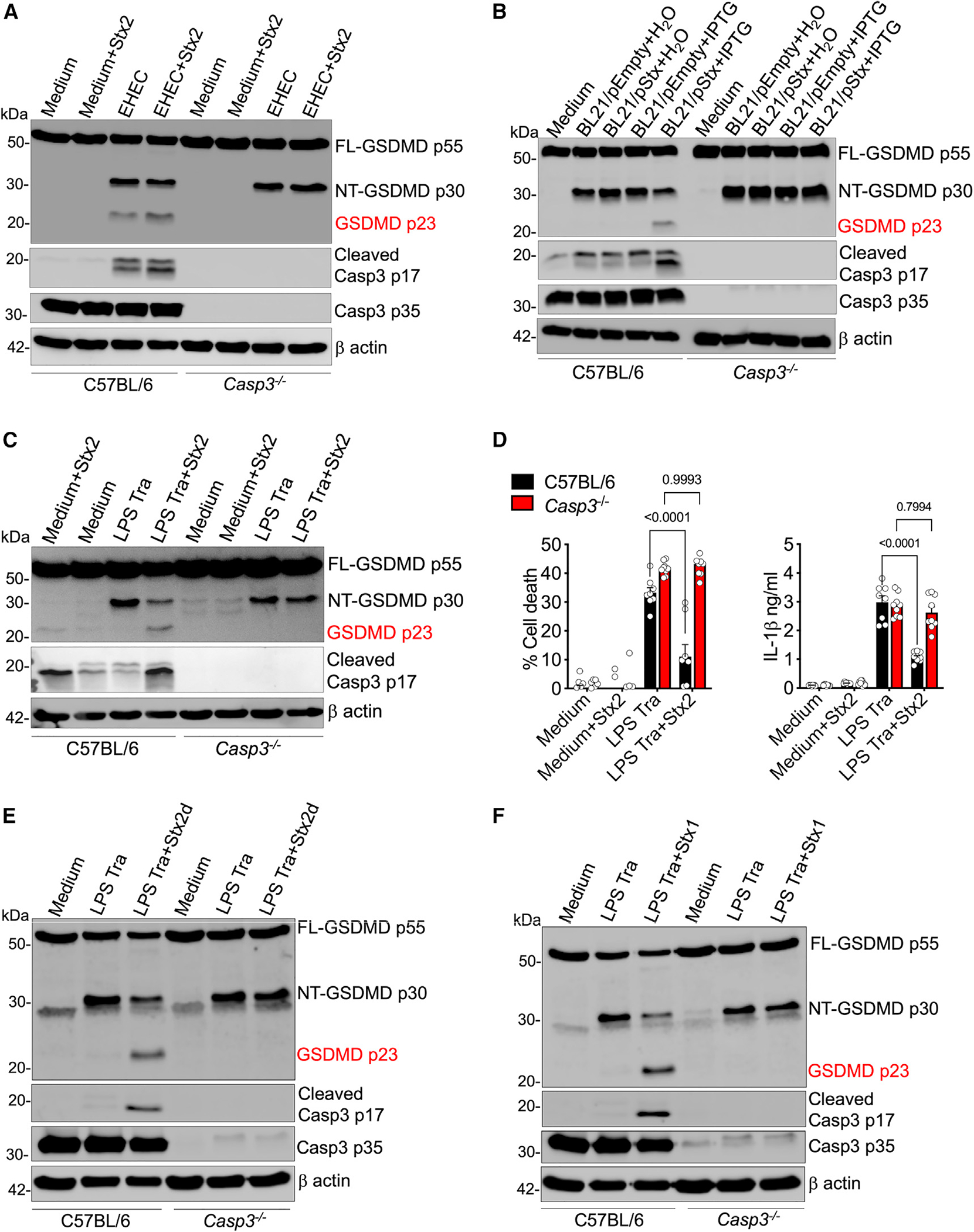
(A–C, E, and F) GSDMD and caspase-3 cleavage in the lysates of Pam3CSK4-primed C57BL/6 or Casp3−/− BMDMs treated with Stx2 (4 μg/mL) at the time of EHEC infection (A) or infected with H2O- or IPTG-treated BL21/pEmpty or BL21/pStx2 (B) or treated with 4 μg/mL of Stx2, Stx2d, or Stx1 at the time of LPS Tra (C, E, and F) for 16 h.
(D) Pyroptosis and IL-1β secretion at 16 h as assessed by LDH assay and ELISA, respectively, in the supernatants of BMDMs treated as in (C). Mean ± SEM of combined data from 3 biological replicates, p < 0.05 is considered significant; two-way ANOVA with Tukey’s post-test.
(A–C, E, and F) Data are representative of 3 experiments.
Caspase-3 cleaves and inactivates caspase-11-generated NT-GSDMD
Recent studies have shown that caspase-3 is capable of cleaving FL-GSDMD at a site (D88 in mouse and D87 in human) distinct from that of the caspase-1/−11/−4 cleavage site (D275), resulting in a prominent p43 GSDMD fragment (Figure 4A).26,27 In our studies, Stx treatment alone did not induce the generation of a p43 fragment, and the p23 fragment only appeared when cells were exposed to Stx in the presence of a noncanonical inflammasome activator (Figures 1 and 3). Nonetheless, based on the caspase-3 cleavage site, D88, located within the NT region of the GSDMD domain as identified previously, the direct cleavage of NT-GSDMD by caspase-3 at D88 is predicted to generate a ~23 kDa fragment (NT-GSDMD 89–275 amino acids) (Figure 4A), which is the exact size of the fragment observed in cells treated with Stx and caspase-11 activators (LPS transfection or E. coli). Consequently, we tested if caspase-3 directly cleaves NT-GSDMD. Purified NT-GSDMD is highly unstable, making it unsuitable for cell-free cleavage assays. Therefore, we employed an inducible NT-GSDMD system28; here, Gsdmd−/− immortalized BMDMs (iBMDMs) express BFP-tagged NT-GSDMD or FL-GSDMD in a doxycycline (Dox)-inducible manner (hereafter referred to as iBMDM-NT-GSDMD-BFP and iBMDM-FL-GSDMD-BFP, respectively). Dox stimulation (0.5 μg/mL for 4 h) resulted in robust expression of FL-GSDMD (Figure 4C) or NT-GSDMD (Figure 4D). FL-GSDMD was detected as a ~81 kDa fragment (55 kDa FL-GSDMD +26 kDa BFP tag) (Figures 4B and 4C) and NT-GSDMD as a 56 kDa fragment (30 kDa NT-GSDMD +26 kDa BFP tag) (Figures 4B and 4D). As expected, the inducible expression of NT-GSDMD, but not FL-GSDMD, resulted in cell death (Figure 4E).
Figure 4. Caspase-3 cleaves NT-GSDMD at D88.
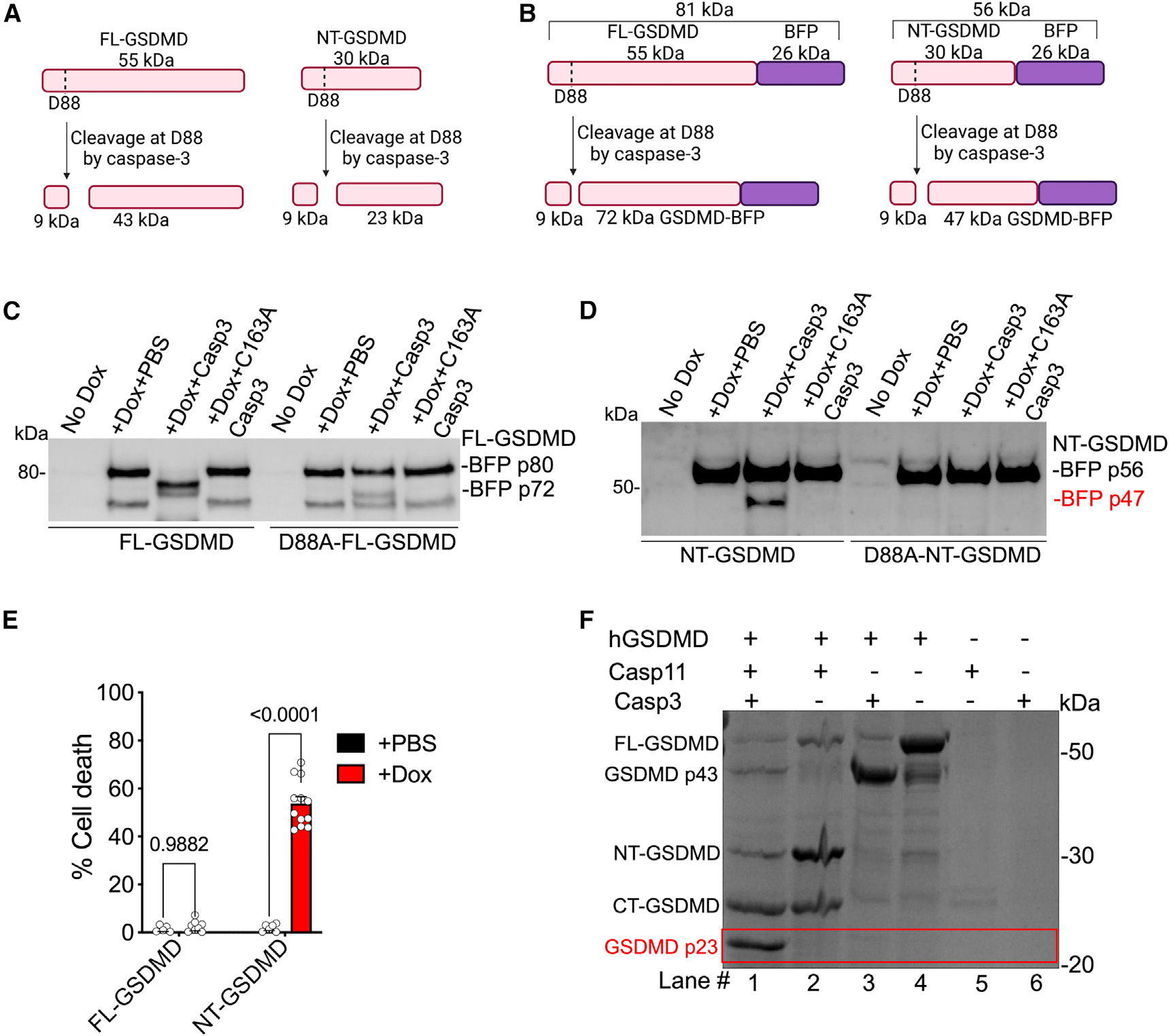
(A and B) Caspase-3 cleavage site (D88) and predicted cleaved fragments for FL-GSDMD or NT-GSDMD (A) and dox-inducible BFP-tagged FL-GSDMD or BFP-tagged NT-GSDMD (B) (created with BioRender.com).
(C and D) Lysates of Dox-treated Gsdmd−/− iBMDMs expressing FL-GSDMD-BFP or NT-GSDMD-BFP were incubated with 3 units of recombinant active or inactive (C163A) caspase-3 at 37C for 3 h and the cleavage of BFP-tagged FL-GSDMD (C) or NT-GSDMD (D) was assessed. Note in (D) the appearance of a p47 band corresponding to caspase-3-cleaved NT-GSDMD-BFP.
(E) Cell death in Gsdmd−/− iBMDMs expressing NT-GSDMD-BFP or FL-GSDMD-BFP treated with 0.5 μg/mL Dox for 4 h. Mean ± SEM of combined data from 3 biological replicates, p < 0.05 is considered significant; two-way ANOVA with Tukey’s post-test.
(F) Coomassie-blue-stained SDS-PAGE gel of recombinant human GSDMD subjected to cleavage by recombinant active caspase-3 and/or active caspase-11 as indicated by “+.”
(C, D, and F) Data are representative of 3 experiments.
To test if caspase-3 can cleave NT-GSDMD in a semi-cell-free system, lysates from iBMDM-NT-GSDMD-BFP or iBMDM-FL-GSDMD-BFP and their corresponding D88A mutant versions—Dox-induced to express the corresponding GSDMD domains—were incubated with recombinant active caspase-3 for 3 h, and GSDMD-BFP cleavage was assessed. The cleavage of 56 kDa NT-GSDMD (with a CT BFP tag) by caspase-3 at D88 will generate a BFP-tagged 47 kDa fragment and an untagged 9 kDa fragment (Figure 4B). In contrast, caspase-3 cleavage of 81 kDa FL-GSDMD will generate 72 and 9 kDa fragments (Figure 4B). In line with previous studies,26,27 caspase-3 cleaved FL-GSDMD, but not FL-GSDMD with a D88A mutation, generating a 72 kDa fragment (Figure 4C). Remarkably, consistent with our prediction, we detected a 47 kDa GSDMD-BFP fragment in the NT-GSDMD lysates treated with active caspase-3 but not the catalytically inactive C163A caspase-3 (Figure 4D). Furthermore, caspase-3 did not cleave the NT-GSDMD with a D88A mutation. These data demonstrate that caspase-3 cleaves NT-GSDMD at D88.
Finally, to determine if caspase-3 directly cleaves NT-GSDMD generated by caspase-11, we used a cell-free GSDMD cleavage assay. Purified human FL-GSDMD or FL-GSDMD plus active caspase-11 (to generate NT-GSDMD) was incubated with active caspase-3, and the cleavage pattern of GSDMD was assessed on an SDS-PAGE gel. As expected, incubation of FL-GSDMD with caspase-11 generated NT-GSDMD and CT-GSDMD (Figure 4F, lane #2). Strikingly, the addition of active caspase-3 to this reaction with FL-GSDMD and caspase-11 led to a marked reduction in NT-GSDMD and the corresponding appearance of p23 GSDMD (Figure 4F, lane #1). Remarkably, in the absence of caspase-11-generated NT-GSDMD, the p23 band was not generated by caspase-3 (Figure 4F, lane #3), demonstrating that caspase-3 is indeed directly cleaving NT-GSDMD generated by caspase-11.
Notably, while caspase-3 cleaves FL-GSDMD in a cell-free system (Figures 4C and 4F, lane #3),26,27 we observed only the cleavage of NT-GSDMD, but not FL-GSDMD, by Stx-activated caspase-3 during noncanonical inflammasome activation in cells (no p43 in Figures 1A–1C and 3A–3C). Furthermore, in our GSDMD cleavage assay, we observed a dramatic decrease in the caspase-3-FL-GSDMD interaction product, p43 GSDMD, when caspase-11 was added to the GSDMD+caspase-3 reaction (Figure 4F, lane #3 vs. #1) with the accompanying appearance of NT- and CT-GSDMD. These data suggest that when both active caspase-11 and active caspase-3 are present, perhaps owing to the stronger affinity of caspase-11 for FL-GSDMD,29 FL-GSDMD is preferably cleaved by caspase-11, limiting its availability for caspase-3. It is also possible that CT-GSDMD blocks caspase-3’s access to the NT domain in FL-GSDMD, as recent structural studies have revealed that CT-GSDMD keeps FL-GSDMD in an autoinhibitory conformation by folding back onto NT-GSDMD.17,30 However, once NTGSDMD is generated by caspase-11, caspase-3 binds and cleaves NT-GSDMD, generating the inactive p23 NT-GSDMD.
This study uncovers an essential role for Stx-activated caspase-3 in direct processing and inactivation of NT-GSDMD generated upon caspase-11 sensing of cytosolic LPS leading to the suppression of inflammasome responses. Inhibition of these noncanonical inflammasome responses will likely aid in EHEC colonization of the host and pathogenesis, and therefore, caspase-3 deficiency presumably enhances resistance to EHEC infection. Bacterial inflammasome inhibitors typically target the inflammasome sensors or the FL-GSDMD.31–36 In contrast, Stx-induced caspase-3 targets active NT-GSDMD generated by caspase-11. To the best of our knowledge, such proteolysis of cleaved NT-GSDMD by bacterial effectors has not been described previously. Thus, the unique bacterial mechanism targeting active GSDMD uncovered here expands our understanding of microbial strategies to subvert innate immune defense. This study’s findings provide significant insights into how the interplay between coincidental signaling networks activated by EHEC via Stx and LPS shape host defense responses at the cellular level. Our finding also expands our fundamental understanding of cellular checkpoints that control active NT-GSDMD in general, with implications beyond microbial infections.
Limitations of the study
While this study has demonstrated that Stx-activated caspase-3 cleaves and inactivates NT-GSDMD, the following limitations should be noted. Our study primarily used murine cells, and we have not assessed the role of caspase-3 in Stx-mediated noncanonical inflammasome suppression in human cells. Similarly, whether Stx-activated caspase-3 plays a role in limiting noncanonical inflammasome responses in vivo during EHEC disease is also not known. Furthermore, since caspase-8 and caspase-9 inhibitors are known to cross-react with caspase-3, further studies are required to genetically validate the role of caspase-8 and −9 in Stx-induced caspase-3 activation.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sivapriya Kailasan Vanaja (kailasanvanaja@uchc.edu).
Materials availability
Plasmids and cells lines generated in this study are available from the lead contact upon request.
Data and code availability
All data reported in the paper are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Mice
C57BL/6 mice from NCI Charles River, Casp11−/− mice (kind gift of Vishva Dixit and Kate Fitzgerald), and Casp3−/− mice (Stock number 006233; The Jackson Laboratories) were bred and maintained in specific pathogen–free conditions at the UConn Health animal facility. Casp3−/− mice were generated by crossing heterozygous male and female mice. Deletion of Casp11 and Casp3 was confirmed by PCR analysis of tail genomic DNA.37,38 Both male and female mice were used for this study. Animal protocols were carried out in accordance with the guidelines set forth by the Institutional Animal Care and Use Committee at UConn Health.
Cell culture
Bone-marrow derived macrophages (BMDMs) from C57BL/6, Casp11−/−, and Casp3−/− mice were generated by culturing bone marrow cells from mice in DMEM with 10% FCS and 20% MCF-containing L929 supernatant at 37°C. The cell culture medium was replaced with fresh medium on days 3 and 6. Cells were used for experiments on days 8–11.18,19,39,40 Gsdmd−/− iBMDMs expressing BFP-tagged versions of GSDMD were cultured in complete DMEM supplemented with 10% FBS and penicillin-streptomycin.
METHOD DETAILS
Bacterial strains and growth conditions
Bacterial strains used in this study include EHEC strain EDL933, Stx-deficient EHEC (ΔStx), E. coli BL21(DE3)/pLysS carrying an empty pET21a vector (E. coli BL21/pEmpty), and E. coli BL21(DE3)/pLysS carrying pET21a-Stx2a (E. coli BL21/pStx).19 All bacterial strains were grown in Luria Bertani (LB) broth at 37°C overnight unless otherwise mentioned. For macrophage infections, overnight grown E. coli cultures were reinoculated at 1/20 dilution into fresh LB and grown until OD600 is ~1.5. To induce Stx expression, overnight grown E. coli BL21/pStx2 or E. coli BL21/pEmpty was reinoculated into fresh LB media containing ampicillin (for pET21a) and chloramphenicol (for pLysS). After 2 h of growth, the cultures were treated with 0.5 mM IPTG or water for 5 h and used for infecting macrophages at a multiplicity of infection (MOI) of 50.
Cell culture stimulations
For bacterial infections, BMDMs primed with Pam3CSK4 (tlrI-pms; Invivogen) for 3 h or left unprimed were infected with MOI = 50 of EHEC or E. coli BL21 strains. After 1 h of infection, media was replaced with 100 μg/mL gentamicin-containing media. Cell supernatants and lysates were collected at 16 h of infection and inflammasome responses including GSDMD cleavage, cell death, and IL-1β secretion were assessed. For LPS transfection, Pam3CSK4-primed BMDMs were transfected with 1 μg/106 cells of ultrapure LPS (L3024; Sigma-Aldrich) using Lipofectamine 2000 (1668019; ThermoFisher Scientific) and supernatants and lysates were collected after 16 h of treatment. In experiments with Stx treatment, BMDMs were treated with 4 μg/mL of purified Stx2a (Stx2), catalytically inactive Stx2a (Stx2 toxoid), Stx2d, or Stx1 (Tufts Phoenix Laboratories) at the time of infection with EHEC or LPS transfection.
ELISA and cell death assay
IL-1β levels were assessed by Ready-SET-Go! enzyme-linked immunosorbent assay (ELISA) kit (88–7013A-88; ThermoFisher Scientific) according to the manufacturer’s instructions. Cell death was assessed by measuring LDH levels in the supernatant with the LDH cytotoxicity kit (MK401; Takara) according to the manufacturer’s instructions.
Immunoblotting and antibodies
Cell lysates were prepared with RIPA lysis buffer containing protease inhibitor cocktail (1861279; ThermoFisher Scientific) and immunoblotting was performed with the following antibodies: gasdermin D (ab209845; Abcam), C-terminal gasdermin D (ab228824; Abcam), cleaved caspase-3 (9664; Cell Signaling Technologies), caspase-3 (9662; Cell Signaling Technologies), caspase-8 (4790; Cell Signaling Technologies), caspase-9 (9508; Cell Signaling Technologies), BFP (MA5–15257; Invitrogen), TetR (631131; Takara Bio), and β-actin (3700S; Cell Signaling Technologies). Corresponding HRP-conjugated secondary antibodies (Jackson ImmunoResearch) were diluted in PBS-Tween containing 5% w/v nonfat milk. Immunoblot images were captured and analyzed with Azure 800.
Apoptosis assay
WT BMDMs were infected with EHEC (MOI = 50) or treated with 4 μg/mL of Stx2 or 1 μM of doxorubicin (15007; Cayman Chemicals) for 5 h. Live cells were stained with ApoTracker Green25 (800 nM; 427402; BioLegend) in colorless DMEM (21063029; ThermoFisher Scientific) for 30 min in the dark before imaging with a ZOE Fluorescent Cell Imager (Bio-Rad).
Initiator caspase inhibition
WT BMDMs were treated with 20 μM specific inhibitors of caspase-8, Z-IETD-FMK (inh-ietd; InvivoGen), or caspase-9, Z-LEHD-FMK (FMK008; R&D Systems), or DMSO 30 min before EHEC infection (MOI = 50) or Stx2 treatment (4 μg/mL). Cells were lysed in RIPA lysis buffer containing protease inhibitor after 5 h. Caspase-3 cleavage was assessed by Western blotting.
Generation of immortalized BMDMs expressing doxycycline-inducible GSDMD proteins
Doxycycline-inducible cell lines were generated through retroviral transduction as previously described.28 Briefly, HEK 293T cells were transfected with the Tet3G trans-activator plasmid or pRetroX-GSDMD-BFP plasmids (FL or NT)28 along with VSV-glycoprotein and gag-pol using the Lipofectamine 2000 reagent. Virus-containing supernatants were collected 3 and 4 days post transfection, passed through a 0.22 μm filter, and pelleted at 20,000g for 2 h. Viral pellets were resuspended in DMEM (10% Tet-free FBS [631106; Takara Bio], 1% pen-strep) supplemented with 2 μg/mL polybrene and then added to Gsdmd−/− immortalized BMDMs (iBMDMs) before centrifuging at 2000g for 45 min at 37°C. Selection antibiotics were added to cells (40 μg/mL geneticin [ant-gn5; Invivogen] for Tet3G or 0.4 μg/mL puromycin [ant-pr-1; Invivogen] for pRetroX) one day post transduction and cell death was monitored for 5 days. Transduction efficiency was established by adding 0.5 μg/mL doxycycline (D3072; Sigma-Aldrich) for 4 h and monitoring cell death by LDH assay as well as GSDMD-BFP and Tet3G expression by immunoblotting. Transduced cells were maintained in Tet-free FBS-containing media with geneticin and puromycin.
For D88A cleavage-resistant GSDMD mutants, site directed mutagenesis (SDM) was performed on the GSDMD-BFP-pRetroX plasmids using the Q5 high fidelity DNA polymerase (M0491; New England Biolabs) followed by Dpn1 digestion (R0176; New England Biolabs). Plasmids were transformed into Stellar E. coli (636763; Takara Bio) and grown on ampicillin-containing LB agar. Colonies were selected, grown overnight in ampicillin-containing LB broth, and plasmids were purified using the Miniprep kit (27104; Qiagen) according to the manufacturer’s instructions. Purified plasmids were sequenced to confirm the D88A mutation. D88A-GSDMD-BFP plasmids were transduced into Tet3G-expressing Gsdmd−/−iBMDMs as above, and transduction was confirmed by LDH assay and immunoblotting.
Purification of caspase-3
Active caspase-3 was purified as previously described.41 Briefly, pET22b-6xHis-tagged WT and C163A caspase-3, generated by SDM as described above, were transformed into E. coli BL21 and grown overnight in ampicillin-containing LB broth. The following day, primary cultures were inoculated into fresh ampicillin-containing LB broth and grown at 37°C until OD600 0.8. To induce protein expression, 0.5 mM IPTG was added to cultures, which were shaken for 16 h at 20°C. Following IPTG induction, bacteria were pelleted then lysed in lysis buffer (25 mM Tris-HCl pH 8.0, 150 mM NaCl, 25 mM imidazole, 5 mM 2-mercaptoethanol) with sonication, and centrifuged to remove insoluble fractions. Bacterial supernatants containing the recombinant proteins were purified using Ni-NTA agarose beads (30210; Qiagen) according to manufacturer’s instructions. Protein expression was confirmed by Western blotting.
Assessing cleavage of FL-GSDMD-BFP and NT-GSDMD-BFP by caspase-3
Immortalized BMDMs expressing WT and D88A mutants of both FL- and NT-GSDMD-BFP were treated with PBS or doxycycline (0.5 μg/mL) for 4 h at 37°C. Media was removed and cells were rinsed with PBS before the addition of NP40 lysis buffer (50 mM HEPES, 150 mM NaCl, 1% NP40) without protease inhibitor. Following 30 min of gentle shaking on ice, lysates were collected and centrifuged at 7000g for 10 min at 4°C and the supernatant was used for subsequent cleavage reactions. GSDMD lysates were mixed with 2x caspase reaction buffer (100 mM HEPES, 2 mM EDTA, 200 mM NaCl, 100 mM DTT, 20% glycerol) and 2.5 μM purified WT or C163A caspase-3 for 4 h at 37°C. Reactions were stopped by adding SDS loading buffer followed by boiling at 95°C for 5 min. Cleavage of GSDMD was assessed via SDS-PAGE immunoblotting.
In vitro gasdermin D cleavage assays
To examine the cleavage of GSDMD, about 7.5 μM purified human GSDMD was incubated with 2.5 μM purified caspase-11 (p22/p10 form) at 37°C for 30 min in buffer A [20 mM tris-HCl (pH 8.0) and 150 mM NaCl] with or without 2 or 4 μg of purified Stx2. The cleavage was stopped by adding SDS loading buffer to the reaction mixture, followed by boiling at 95°C for 5 min. The samples were analyzed by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie Brilliant Blue staining.
For assessing cleavage of GSDMD by caspase-11 in the presence of caspase-3, 10 μM purified GSDMD was incubated with 0.5 μM purified caspase-11 (p22/p10 form) and/or 0.5 μM purified caspase-3 (p12/p17 form) at 16°C for 4 h. The cleavage was stopped by adding SDS loading buffer to the reaction mixture, followed by boiling at 95°C for 5 min. The samples were analyzed by SDS–SDS-PAGE and Coomassie Brilliant Blue staining.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed for statistical significance by two-way analysis of variance (ANOVA) followed by Tukey’s post-test with GraphPad Prism 10.1.1 software. p values of less than 0.05 were considered significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-GSDMD (Clone EPR 19828) | Abcam | Cat# ab209845; RRID:AB_2783550 |
| Rabbit polyclonal anti-GSDMD c terminal | Abcam | Cat# ab228824 |
| Rabbit polyclonal anti-Caspase-3 | Cell Signaling Technology | Cat# 9662; RRID:AB_331439 |
| Rabbit monoclonal anti-Caspase-8 (Clone D35G2) | Cell Signaling Technology | Cat#4790; RRID:AB_10545768 |
| Mouse monoclonal anti-Caspase-9 (Clone C9) | Cell Signaling Technology | Cat# 9508; RRID:AB_2068620 |
| Mouse monoclonal anti-RFP (Clone RF5R) | ThermoFisher Scientific | Cat# MA5–15257; RRID:AB_10999796 |
| Mouse monoclonal anti-TetR (Clone 9G9) | Takara Bio | Cat# 631132 |
| Mouse monoclonal anti-β actin (Clone 8H10D10) | Cell Signaling Technology | Cat# 3700; RRID:AB_2242334 |
| HRP-conjugated Anti-mouse | Jackson ImmunoResearch | Cat# 115035166; RRID:AB_2338511 |
| HRP-conjugated Anti-rabbit | Jackson ImmunoResearch | Cat# 711035152; RRID:AB_10015282 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Escherichia coli O157:H7 EDL933 (EHEC) | Vanaja Lab | N/A |
| ΔStx E. coli O157:H7 EDL933 | Vanaja Lab | N/A |
| E. coli BL21 | Vanaja Lab | N/A |
| E. coli BL21(DE3)/pLysS/pET21 | Vanaja Lab | N/A |
| E. coli BL21(DE3)/pLysS/pET21-Stx2 | Vanaja Lab | N/A |
| Stellar competent E. coli | Takara Bio | Cat# 636763 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Shiga toxin 2 (Stx2) | Tufts Phoenix Laboratory | N/A |
| Shiga toxin 2d (Stx2d) | Tufts Phoenix Laboratory | N/A |
| Shiga toxin 1 (Stx1) | Tufts Phoenix Laboratory | N/A |
| Stx2 toxoid | Tufts Phoenix Laboratory | N/A |
| LPS E. coli O111:B4 | Sigma-Aldrich | Cat# L3024–25MG |
| Pam3CSK4 | Invivogen | Cat# tlrI-pms |
| Lipofectamine 2000 reagent | ThermoFisher Scientific | Cat# 1668019 |
| Halt™ Protease Inhibitor Cocktail (100X) | ThermoFisher Scientific | Cat# 1861279 |
| IPTG solution | ThermoFisher Scientific | Cat# R1171 |
| Dimethyl sulfoxide (DMSO) | Sigma-Aldrich | Cat# D2650 |
| Caspase-8 Inhibitor Z-IETD-FMK | Invivogen | Cat# inh-ietd |
| Caspase-9 Inhibitor Z-LEHD-FMK | R&D Systems | Cat# FMK008 |
| Apotracker™ Green | BioLegend | Cat# 427402 |
| NuPAGE LDS sample buffer (4X) | ThermoFisher Scientific | Cat# NP0007 |
| Trans-Blot Turbo Transfer System | Bio-Rad | Cat# 1704271 |
| Clarity ECL HRP Substrate | Bio-Rad | Cat# 170–5060S |
| ACK Lysis buffer | ThermoFisher Scientific | Cat# A1049201 |
| Ni-NTA Agarose | Qiagen | Cat# 30210 |
| Purified active caspase-3 | Vanaja Lab | N/A |
| Purified C163A caspase-3 | Vanaja Lab | N/A |
| Purified active caspase-11 | Ruan Lab, UConn Health | N/A |
| Purified mouse GSDMD | Ruan Lab, UConn Health | N/A |
| Purified human GSDMD | Ruan Lab, UConn Health | N/A |
| Tet System Approved FBS | Takara Bio | Cat# 631106 |
| G418 (Geneticin) | Invivogen | Cat# ant-gn-5 |
| Puromycin | Invivogen | Cat# ant-pr-1 |
| Q5® High-Fidelity DNA Polymerase | New England Biolabs | Cat# M0491 |
| DpnI | New England Biolabs | Cat# R0176 |
| Doxycycline Hydrochloride, Ready Made Solution | Sigma-Aldrich | Cat# D3072 |
| QIAprep Spin Miniprep Kit | Qiagen | Cat# 27104 |
| DMEM | ThermoFisher Scientific | Cat# 11995073 |
| Colorless DMEM | ThermoFisher Scientific | Cat# 21063029 |
| Doxorubicin (hydrochloride) | Cayman Chemicals | Cat# 15007 |
|
| ||
| Critical commercial assays | ||
|
| ||
| LDH cytotoxicity detection kit | Takara Bio | Cat# MK401 |
| Mouse IL-1β ELISA kit | ThermoFisher Scientific | Cat# 88–7013A-88 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| HEK 293T | Rathinam Lab, UConn Health | N/A |
| Gsdmd−/− iBMDMs | Generated in this study | N/A |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:000664 |
| Mouse: Casp11−/− | Kayagaki et al.37 and Genentech | N/A |
| Mouse: Casp3−/− | The Jackson Laboratory | RRID:IMSR_JAX:006233 |
|
| ||
| Oligonucleotides | ||
| Casp11−/− genotyping Primer 1: CCCTGGAAAAATCGATGACT | Integrated DNA Technologies and Kayagaki et al.37 | Mouse listed above |
|
Casp11−/− genotyping Primer 2: TGAAATGCATGTACTGAGAGCAAGG |
Integrated DNA Technologies and Kayagaki et al.37 | Mouse listed above |
| Casp11−/− genotyping Primer 3: CAATTGACTTGGGGATTCTGG | Integrated DNA Technologies and Kayagaki et al.37 | Mouse listed above |
|
Casp3−/− genotyping Primer 1: GGGAAACCAACAGTAGTCAGTCCT |
Integrated DNA Technologies and Wang et al.38 | Mouse listed above |
|
Casp3−/− genotyping Primer 2: GCGAGTGAGAATGTGCATAAATTC |
Integrated DNA Technologies and Wang et al.38 | Mouse listed above |
|
Casp3−/− genotyping Primer 3: TGCTAAAGCGCATGCTCCAGACTG |
Integrated DNA Technologies and Wang et al.38 | Mouse listed above |
|
| ||
| Recombinant DNA | ||
|
| ||
| pRETROX Tre3G/GSDMD-FL-tagBFP | Evavold et al.28 | N/A |
| pRETROX Tre3G/GSDMD-NT-tagBFP | Evavold et al.28 | N/A |
| pRETROX Tre3G/D88A-FL-GSDMD-tagBFP | Generated in this study | N/A |
| pRETROX Tre3G/D88A-NT-GSDMD-tagBFP | Generated in this study | N/A |
| pET22b-6xHis-tagged caspase-3 | Generated in this study | N/A |
| pET22b-6xHis-tagged C163A caspase-3 | Generated in this study | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 10.1.1 | N/A | |
Highlights.
Shiga toxin (Stx) activates caspase-3 in macrophages
Caspase-3 inactivates the functional N-terminal GSDMD generated by caspase-11
Caspase-3 inactivation of NT-GSDMD curtails noncanonical inflammasome responses
Interplay between caspase-3 and caspase-11 governs the fate of infected cells
ACKNOWLEDGMENTS
We thank Drs. Jonathan Kagan and Charles Evavold for Tet3G transactivator plasmid and pRetroX-GSDMD-BFP plasmids and Drs. Vishwa Dixit and Kate Fitzgerald for Casp11−/− mice. This work was supported by the National Institutes of Health (R01AI132850 and R56 AI132850 to S.K.V., R01AI119015 to V.A.R., and R01AI158435 to J.R.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.114004.
REFERENCES
- 1.Schmitt CK, Meysick KC, and O’Brien AD (1999). Bacterial toxins: friends or foes? Emerg. Infect. Dis. 5, 224–234. 10.3201/eid0502.990206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Proulx F, Seidman EG, and Karpman D (2001). Pathogenesis of Shiga Toxin-Associated Hemolytic Uremic Syndrome. Pediatr. Res. 50, 163–171. 10.1203/00006450-200108000-00002. [DOI] [PubMed] [Google Scholar]
- 3.Bryan A, Youngster I, and McAdam AJ (2015). Shiga Toxin Producing Escherichia coli. Clin. Lab. Med. 35, 247–272. 10.1016/j.cll.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Tarr PI, Gordon CA, and Chandler WL (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086. 10.1016/s0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 5.Schüller S (2011). Shiga toxin interaction with human intestinal epithelium. Toxins 3, 626–639. 10.3390/toxins3060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorpe CM (2004). Shiga Toxin—Producing Escherichia coli Infection. Clin. Infect. Dis. 38, 1298–1303. 10.1086/383473. [DOI] [PubMed] [Google Scholar]
- 7.Williams DM, Sreedhar SS, Mickell JJ, and Chan JCM (2002). Acute Kidney Failure. Arch. Pediatr. Adolesc. Med. 156, 893–900. 10.1001/archpedi.156.9.893. [DOI] [PubMed] [Google Scholar]
- 8.Lee M-S, Cherla RP, Jenson MH, Leyva-Illades D, Martinez-Moczygemba M, and Tesh VL (2011). Shiga toxins induce autophagy leading to differential signalling pathways in toxin-sensitive and toxin-resistant human cells. Cell Microbiol. 13, 1479–1496. 10.1111/j.1462-5822.2011.01634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, and Shao F (2014). Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192. 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 10.Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, Miyake K, Zhang J, Lee WP, Muszynski A, et al. (2013). Noncanonical Inflammasome Activation by Intracellular LPS Independent of TLR4. Science 341, 1246–1249. 10.1126/science.1240248. [DOI] [PubMed] [Google Scholar]
- 11.Aachoui Y, Leaf IA, Hagar JA, Fontana MF, Campos CG, Zak DE, Tan MH, Cotter PA, Vance RE, Aderem A, and Miao EA (2013). Caspase-11 Protects Against Bacteria That Escape the Vacuole. Science 339, 975–978. 10.1126/science.1230751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Casson CN, Yu J, Reyes VM, Taschuk FO, Yadav A, Copenhaver AM, Nguyen HT, Collman RG, and Shin S (2015). Human caspase-4 mediates noncanonical inflammasome activation against gram-negative bacterial pathogens. Proc. Natl. Acad. Sci. USA 112, 6688–6693. 10.1073/pnas.1421699112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casson CN, Copenhaver AM, Zwack EE, Nguyen HT, Strowig T, Javdan B, Bradley WP, Fung TC, Flavell RA, Brodsky IE, and Shin S (2013). Caspase-11 activation in response to bacterial secretion systems that access the host cytosol. PLoS Pathog. 9, e1003400. 10.1371/journal.ppat.1003400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 15.Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671. 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, and Lieberman J (2016). Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535, 153–158. 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang D-C, and Shao F (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116. 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 18.Vanaja SK, Russo AJ, Behl B, Banerjee I, Yankova M, Deshmukh SD, and Rathinam VAK (2016). Bacterial Outer Membrane Vesicles Mediate Cytosolic Localization of LPS and Caspase-11 Activation. Cell 165, 1106–1119. 10.1016/j.cell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Havira MS, Ta A, Kumari P, Wang C, Russo AJ, Ruan J, Rathinam VA, and Vanaja SK (2020). Shiga toxin suppresses noncanonical inflammasome responses to cytosolic LPS. Sci. Immunol. 5, eabc0217. 10.1126/sciimmunol.abc0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tesh VL (2010). Induction of apoptosis by Shiga toxins. Future Microbiol. 5, 431–453. 10.2217/fmb.10.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujii J, Matsui T, Heatherly DP, Schlegel KH, Lobo PI, Yutsudo T, Ciraolo GM, Morris RE, and Obrig T (2003). Rapid Apoptosis Induced by Shiga Toxin in HeLa Cells. Infect. Immun. 71, 2724–2735. 10.1128/iai.71.5.2724-2735.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johansson KE, Ståhl AL, Arvidsson I, Loos S, Tontanahal A, Rebetz J, Chromek M, Kristoffersson A-C, Johannes L, and Karpman D (2019). Shiga toxin signals via ATP and its effect is blocked by purinergic receptor antagonism. Sci. Rep. 9, 14362. 10.1038/s41598-019-50692-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Setten P, Monnens L, Verstraten R, van den Heuvel L, and van Hinsbergh V (1996). Effects of verocytotoxin-1 on nonadherent human monocytes: binding characteristics, protein synthesis, and induction of cytokine release. Blood 88, 174–183. 10.1182/blood.v88.1.174.174. [DOI] [PubMed] [Google Scholar]
- 24.Paton JC, and Paton AW (2006). Shiga toxin ‘goes retro’ in human primary kidney cells. Kidney Int. 70, 2049–2051. 10.1038/sj.ki.5001954. [DOI] [PubMed] [Google Scholar]
- 25.Barth ND, Subiros-Funosas R, Mendive-Tapia L, Duffin R, Shields MA, Cartwright JA, Henriques ST, Sot J, Goñi FM, Lavilla R, et al. (2020). A fluorogenic cyclic peptide for imaging and quantification of drug-induced apoptosis. Nat. Commun. 11, 4027. 10.1038/s41467-020-17772-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Taabazuing CY, Okondo MC, and Bachovchin DA (2017). Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem. Biol. 24, 507–514.e4. 10.1016/j.chembiol.2017.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, et al. (2018). Pathogen blockade of TAK1 triggers caspase-8–dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evavold CL, Hafner-Bratkovič I, Devant P, D’Andrea JM, Ngwa EM, Borsic E, Doench JG, LaFleur MW, Sharpe AH, Thiagarajah JR, and Kagan JC (2021). Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 184, 4495–4511.e19. 10.1016/j.cell.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang J, Liu Z, Wang C, Yang R, Rathkey JK, Pinkard OW, Shi W, Chen Y, Dubyak GR, Abbott DW, and Xiao TS (2018). Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc. Natl. Acad. Sci. USA 115, 6792–6797. 10.1073/pnas.1800562115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ruan J, Xia S, Liu X, Lieberman J, and Wu H (2018). Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557, 62–67. 10.1038/s41586-018-0058-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ulland TK, Ferguson PJ, and Sutterwala FS (2015). Evasion of inflammasome activation by microbial pathogens. J. Clin. Invest. 125, 469–477. 10.1172/jci75254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ta A, and Vanaja SK (2021). Inflammasome activation and evasion by bacterial pathogens. Curr. Opin. Immunol. 68, 125–133. 10.1016/j.coi.2020.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luchetti G, Roncaioli JL, Chavez RA, Schubert AF, Kofoed EM, Reja R, Cheung TK, Liang Y, Webster JD, Lehoux I, et al. (2021). Shigella ubiquitin ligase IpaH7.8 targets gasdermin D for degradation to prevent pyroptosis and enable infection. Cell Host Microbe 29, 1521–1530.e10. 10.1016/j.chom.2021.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li Z, Liu W, Fu J, Cheng S, Xu Y, Wang Z, Liu X, Shi X, Liu Y, Qi X, et al. (2021). Shigella evades pyroptosis by arginine ADP-riboxanation of caspase-11. Nature 599, 290–295. 10.1038/s41586-021-04020-1. [DOI] [PubMed] [Google Scholar]
- 35.Pallett MA, Crepin VF, Serafini N, Habibzay M, Kotik O, Sanchez-Garrido J, Di Santo JP, Shenoy AR, Berger CN, and Frankel G (2017). Bacterial virulence factor inhibits caspase-4/11 activation in intestinal epithelial cells. Mucosal Immunol. 10, 602–612. 10.1038/mi.2016.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi T, Ogawa M, Sanada T, Mimuro H, Kim M, Ashida H, Akakura R, Yoshida M, Kawalec M, Reichhart J-M, et al. (2013). The Shigella OspC3 effector inhibits caspase-4, antagonizes inflammatory cell death, and promotes epithelial infection. Cell Host Microbe 13, 570–583. 10.1016/j.chom.2013.04.012. [DOI] [PubMed] [Google Scholar]
- 37.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. (2011). Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121. 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y-J, Liu M-G, Wang J-H, Cao W, Wu C, Wang Z-Y, Liu L, Yang F, Feng Z-H, Sun L, et al. (2020). Restoration of Cingulate Long-Term Depression by Enhancing Non-apoptotic Caspase 3 Alleviates Peripheral Pain Hypersensitivity. Cell Rep. 33, 108369. 10.1016/j.celrep.2020.108369. [DOI] [PubMed] [Google Scholar]
- 39.Ta A, Ricci-Azevedo R, Vasudevan SO, Wright SS, Kumari P, Havira MS, Surendran Nair M, Rathinam VA, and Vanaja SK (2023). A bacterial autotransporter impairs innate immune responses by targeting the transcription factor TFE3. Nat. Commun. 14, 2035. 10.1038/s41467-023-37812-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Russo AJ, Vasudevan SO, Méndez-Huergo SP, Kumari P, Menoret A, Duduskar S, Wang C, Pérez Sáez JM, Fettis MM, Li C, et al. (2021). Intracellular immune sensing promotes inflammation via gasdermin D–driven release of a lectin alarmin. Nat. Immunol. 22, 154–165. 10.1038/s41590-020-00844-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C, Shivcharan S, Tian T, Wright S, Ma D, Chang J, Li K, Song K, Xu C, Rathinam VA, and Ruan J (2023). Structural basis for GSDMB pore formation and its targeting by IpaH7.8. Nature 616, 590–597. 10.1038/s41586-023-05832-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in the paper are available from the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.