

Abstract
INTRODUCTION
Lewy body disease (LBD) is a common primary or co‐pathology in neurodegenerative syndromes. An alpha‐synuclein seed amplification assay (αSyn‐SAA) is clinically available, but clinical performance, especially lower sensitivity in amygdala‐predominant cases, is not well understood.
METHODS
Antemortem CSF from neuropathology‐confirmed LBD cases was tested with αSyn‐SAA (N = 56). Diagnostic performance and clinicopathological correlations were examined.
RESULTS
Similar to prior reports, sensitivity was 100% for diffuse and transitional LBD (9/9), and overall specificity was 96.3% (26/27). Sensitivity was lower in amygdala‐predominant (6/14, 42.8%) and brainstem‐predominant LBD (1/6, 16.7%), but early spread outside these regions (without meeting criteria for higher stage) was more common in αSyn‐SAA‐positive cases (6/7, 85.7%) than negative (2/13, 15.4%).
DISCUSSION
In this behavioral neurology cohort, αSyn‐SAA had excellent diagnostic performance for cortical LBD. In amygdala‐ and brainstem‐predominant cases, sensitivity was lower, but positivity was associated with anatomical spread, suggesting αSyn‐SAA detects early LBD progression in these cohorts.
Highlights
A cerebrospinal fluid alpha‐synuclein assay detects cortical LBD with high sensitivity/specificity.
Positivity in prodromal stages of LBD was associated with early cortical spread.
The assay provides precision diagnosis of LBD that could support clinical trials.
The assay can also identify LBD co‐pathology, which may impact treatment responses.
Keywords: alpha synuclein, cerebrospinal fluid, dementia with Lewy bodies, Lewy body disease, seed amplification assay
1. BACKGROUND
Lewy body disease (LBD) is the underlying or contributing neuropathology in many neurodegenerative presentations seen in the behavioral neurology clinic, especially syndromes strongly associated with primary α‐synucleinopathies, for example, dementia with Lewy bodies (DLB) and Parkinson's disease dementia (PDD). LBD is also common as a co‐pathology in syndromes where the primary pathology is Alzheimer's disease (AD), with some degree of comorbid LBD reported in upwards of 50% of AD cases. 1 Unfortunately, the sensitivity of a clinical diagnosis is relatively low for LBD pathology, 2 , 3 suggesting a high proportion of cognitively impaired patients with clinically relevant LBD pathology goes unrecognized. 4 Inadequate clinical detection is especially prevalent when LBD is present as co‐pathology in a patient with high levels of AD neuropathology, and this has been associated with decreased prevalence of core LBD clinical features like hallucinations and fluctuations. 5 , 6 Therefore, increased detection is critical both to improve precision diagnosis for primary LBD, which would facilitate clinical trials in syndromes with underlying α‐synucleinopathies, and to improve the detection of LBD co‐pathology in AD‐related syndromes, where it may impact outcomes of disease‐modifying therapies. 7
Recent developments of cerebrospinal fluid (CSF) α‐synuclein (αSyn) seed amplification assays (SAAs) have proven to be highly sensitive and specific to the presence of seed‐competent αSyn in Parkinson's disease (PD), 8 , 9 and autopsy validation studies have shown excellent diagnostic performance for αSyn‐SAA detection of cortical LBD, but lower sensitivity in amygdala‐ and brainstem‐predominant LBD, 10 , 11 which may represent precursor stages of LBD cortical spread. Interestingly, in patients with isolated rapid eye movement sleep behavior disorder (iRBD), thought to be a prodromal symptom in αSyn‐related clinical syndromes, the sensitivity of αSyn‐SAA is 90% or higher, 12 , 13 further increasing the need to better understand lower sensitivity in amygdala‐ and brainstem‐predominant LBD.
Therefore, we investigated the diagnostic performance and clinicopathological correlations of a clinically available αSyn‐SAA (SYNTap Biomarker Test) in CSF from autopsied patients with ante mortem clinical evaluation through a single center's observational studies, with a focus on comparison between clinical and neuropathological characteristics in detected and undetected cases, especially in cases with amygdala‐ and brainstem‐predominant LBD.
2. METHODS
2.1. Participant characteristics
This retrospective autopsy study included participants of observational studies seen between 2009 and 2017 at the University of California, San Francisco (UCSF) Memory and Aging Center (MAC). Autopsies were performed by the Neurodegenerative Disease Brain Bank (NDBB) at the UCSF Alzheimer Disease Research Center between 2011 and 2021. All participants provided written informed consent at time of recruitment and underwent a comprehensive clinical research evaluation, including cognitive testing and neurological examination. 14 Studies were approved by the Institutional Review Board at UCSF. A total of N = 879 available autopsies were queried, and 28.2% had LBD neuropathology. Of these participants with LBD, N = 29 had available ante mortem CSF. To compare the prevalence of clinical features, cases without LBD with ante mortem CSF were chosen to match clinical syndromes (N = 27; Table 1 and Table S1). Clinical syndrome was diagnosed based on available data at the time of clinical evaluation by an experienced behavioral neurologist or formal consensus panel following established criteria, updated when necessary to follow contemporary nomenclature (LV). 6 , 15 , 16 , 17 , 18 , 19 All available clinical data were reviewed to determine ante mortem symptoms at the closest time point to CSF collection (NS, SL, DLF).
TABLE 1.
Clinical characteristics. Statistical comparisons were performed with linear or logistic regression as appropriate.
| Total | Non‐LBD | Diffuse / Transitional | Amygdala‐predominant | Brainstem‐ predominant | αSyn‐SAA negative | αSyn‐SAA positive | |
|---|---|---|---|---|---|---|---|
| N = 56 | N = 27 | N = 9 | N = 14 | N = 6 | N = 39 | N = 17 | |
| Demographics | |||||||
| Age at onset, y | 60 ± 9 | 59 ± 9 | 64 ± 10 | 60 ± 12 | 59 ± 5 | 59 ± 9 | 64 ± 11 |
| Age at death, y | 70 ± 9 | 69 ± 8 | 75 ± 9 | 69 ± 12 | 68 ± 6 | 68 ± 9 | 73 ± 9 |
| CSF‐autopsy interval, y | 3.8 ± 2.1 | 3.5 ± 1.4 | 3.2 ± 1.8 | 4.9 ± 2.9 † | 3.7 ± 2.9 | 3.9 ± 2.1 | 3.5 ± 2.2 |
| Female, No., % | 26, 46% | 17, 63% | 3, 33% | 5, 36% | 1, 17% | 21, 54% | 5, 29% |
| Education, y | 17 ± 3 | 17 ± 2 | 18 ± 2 | 17 ± 3 | 16 ± 3 | 17 ± 2 | 18 ± 3 |
| APOE 𝜀4 carrier, No., % | 25, 47% | 11, 42% | 2, 22% | 10, 83% † | 2, 33% | 18, 50% | 7, 41% |
| Clinical severity | |||||||
| MMSE | 21.9 ± 6.0 | 22.5 ± 6.1 | 21.8 ± 6.7 | 22.1 ± 5.1 | 19.4 ± 7.0 | 22.3 ± 5.8 | 21.2 ± 6.2 |
| CDR, global | 1.0 ± 0.6 | 0.9 ± 0.6 | 1.3 ± 0.8 | 0.9 ± 0.4 | 1.2 ± 0.7 | 1.0 ± 0.6 | 1.1 ± 0.7 |
| CDR, box score | 5.6 ± 3.5 | 5.2 ± 3.3 | 6.9 ± 4.5 | 5.1 ± 2.6 | 6.6 ± 4.8 | 5.7 ± 3.4 | 5.5 ± 3.9 |
| CDR+NACC FTLD, global | 1.3 ± 0.7 | 1.2 ± 0.7 | 1.6 ± 0.7 | 1.2 ± 0.5 | 1.4 ± 0.7 | 1.3 ± 0.7 | 1.3 ± 0.7 |
| CDR+NACC FTLD, box score | 7.5 ± 4.2 | 6.8 ± 3.7 | 9.1 ± 5.4 | 6.8 ± 3.4 | 9.3 ± 5.5 | 7.5 ± 4.0 | 7.6 ± 4.6 |
| Clinical symptoms | |||||||
| Parkinsonism, No., % | 36, 64% | 19, 70% | 7, 78% | 7, 50% | 3, 50% | 25, 64% | 11, 65% |
| RBD, No., % | 9, 16% | 5, 19% | 2, 22% | 1, 7% | 1, 17% | 5, 13% | 4, 24% |
| Dysautonomia, No., % | 5, 10% | 4, 17% | 1, 11% | 0, 0% | 0, 0% | 4, 11% | 1, 6% |
| Fluctuations, No., % | 9, 17% | 3, 13% | 3, 33% | 3, 21% | 0, 0% | 6, 17% | 3, 18% |
| Anxiety, No., % | 20, 36% | 8, 30% | 5, 55% | 6, 43% | 1,17% | 13, 33% | 7, 41% |
| Depression, No., % | 23, 41% | 11, 41% | 4, 44% | 6, 43% | 2, 33% | 17, 44% | 6, 35% |
| Hallucinations, No., % | 6, 11% | 0, 0% | 4, 44% | 1, 7% | 1, 17% | 1, 3% * | 5, 29% * |
| Neuropathology | |||||||
| Brain weight, g | 1132 ± 143 | 1111 ± 135 | 1229 ± 145 † | 1114 ± 143 | 1121 ± 143 | 1105 ± 142 * | 1192 ± 129 * |
| ADNC | 1.8 ± 1.2 | 1.5 ± 1.2 | 2.2 ± 1.2 | 2.7 ± 0.8 † | 0.8 ± 0.4 | 1.7 ± 1.2 | 2.2 ± 1.1 |
| AD pathology, No., % ** | 27, 48% | 9, 33% | 6, 66% | 12, 85% † | 0, 0% | 16, 41% | 11, 65% |
| FTLD‐tau, No., % | 20, 36% | 12, 44% | 3, 33% | 1, 7% † | 4, 67% | 16, 41% | 4, 24% |
| FTLD‐TDP, No., % | 13, 23% | 7, 26% | 0, 0% | 4, 29% | 2, 33% | 11, 28% | 2, 12% |
Abbreviations: LBD, Lewy body disease; CSF, cerebrospinal fluid; MMSE, Mini Mental State Examination; CDR, Clinical Dementia Rating Scale; NACC, National Alzheimer's Coordinating Center; FTLD, frontotemporal lobar degeneration; RBD, rapid eye movement sleep behavior disorder; ADNC, Alzheimer's disease neuropathological change (0 = none, 1 = low, 2 = intermediate, 3 = high); TDP, TAR DNA‐binding protein.
Significantly different from non‐LBD (p < 0.05).
Significantly different from each other (p < 0.05).
Presence of AD neuropathology defined as ADNC of intermediate or high
2.2. Autopsy assessment
Postmortem brain tissue was processed and analyzed in the NDBB according to previously described standard protocols, including stage of AD neuropathological change (ADNC) and presence of co‐pathology. 20 , 21 , 22 , 23 , 24 Of note, multiple neuropathological staging systems are available for LBD; here, McKeith et al. criteria were used with the incorporation of “amygdala‐predominant” as described in Leverenz et al. 20 , 21 In brief, four categories are reported based on the distribution of αSyn immunohistochemical staining: brainstem‐predominant (roughly equivalent to Braak stages 1 to 3), transitional‐limbic (limbic, roughly equivalent to Braak stage 4), diffuse neocortical (roughly equivalent to Braak stages 5 to 6). 25 We also had the category amygdala‐predominant, which does not follow the traditional Braak staging system but is a relatively common finding in early‐onset AD. 26 Following prior convention, cases with cortical LBD in the transitional and diffuse stages were combined for analyses. 10 , 11
2.3. CSF analysis
After lumbar puncture, CSF was collected by gravity in polypropylene tubes, aliquoted (0.5 mL), and stored at −80°C until use, according to standard protocol. CSF was sent to Amprion's Clinical Laboratory Improvement Amendment/College of American Pathologists (CLIA/CAP) certified laboratory for qualitative αSyn‐SAA analysis (SYNTap Biomarker Test), as previously described. 3 , 10 Assays were performed by operators blinded to sample identity, and results were returned prior to unblinding.
2.4. Statistical analysis
Overall diagnostic performance for αSyn‐SAA to detect pathological LBD was calculated, including sensitivity and specificity for subgroups of LBD pathology. Statistical comparisons were made both among neuropathological cohorts and between αSyn‐SAA status on various demographic features, clinical symptoms, measures of clinical severity, and neuropathologic characteristics using linear or logistic regression as appropriate. Statistical analyses were performed using Stata version 17.0 (StataCorp).
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional sources, for example, PubMed. Accurate detection of Lewy body disease (LBD) neuropathology is urgently needed, and a clinically available alpha‐synuclein seed amplification assay (αSyn‐SAA) has been validated in recent publications. However, numerous clinicopathological questions remain, especially low sensitivity in amygdala‐ and brainstem‐predominant cases.
Interpretation: We found excellent diagnostic performance to detect cortical LBD in this autopsy‐confirmed behavioral neurology cohort. We also describe an association between positivity in amygdala‐ and brainstem‐predominance stages and early spread into the cortex, suggesting the αSyn‐SAA assay accurately detects LBD, even when co‐pathology.
Future directions: The diagnostic performance needs to be confirmed in larger and more diverse cohorts, preferably in a real‐world clinical setting. Additional contexts of use should be explored, especially prognostic and theragnostic applications, such as the impact of LBD on disease‐modifying therapies in AD.
3. RESULTS
3.1. Diagnostic performance of αSyn‐SAA to detect LBD
In the entire cohort (N = 56), αSyn‐SAA was 55.2% sensitive (95% confidence interval [CI] 35.7% to 73.6%) and 96.3% specific (95% CI 81.0% to 99.9%). The total prevalence of LBD pathology in the UCSF NDBB was 28.2%, resulting in an estimated positive predictive value (PPV) of 85.4% (95% CI 45.4% to 97.6%) and an estimated negative predictive value (NPV) of 84.5% (95% CI 78.4% to 89.2%) for autopsy‐confirmed LBD pathology of any type (Figure 1). In subgroup analyses, αSyn‐SAA had perfect sensitivity to detect diffuse (4/4) and transitional (5/5) LBD and detected αSyn in 3/3 cases where LBD was considered the primary neuropathology. Consistent with prior reports, 10 , 11 sensitivity was lower in amygdala‐predominant LBD co‐pathology (6/14, 42.8%), and sensitivity was lower for brainstem‐predominant LBD co‐pathology (1/6, 16.7%). One false positive result was present in 27 non‐LBD cases (3.7%), in which the primary pathology was AD (the participant also lacked clinical features of DLB). In four participants with a clinical diagnosis of DLB, αSyn‐SAA was positive in three cases due to diffuse or transitional LBD. In the αSyn‐SAA negative DLB case, the participant had brainstem‐predominant LBD, but the primary pathology was TAR DNA‐binding protein 43 (TDP‐43), type B due to a pathogenic variant of C9ORF72.
FIGURE 1.
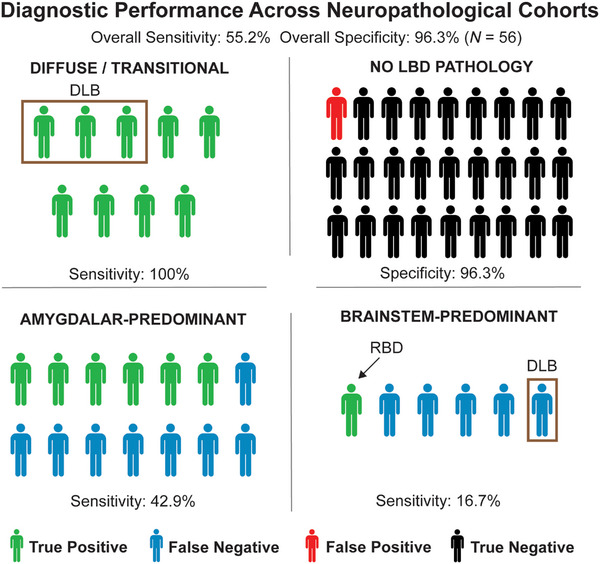
Diagnostic performance of CSF αSyn‐SAA by LBD Neuropathological Stage. Color code by diagnostic status at figure bottom. Patients inside brown box were diagnosed with DLB while alive (sensitivity 13.8%, specificity 80%). Arrow highlights patient with brainstem‐predominant LBD who also had RBD. See Table S1 for full clinicopathological details.
Parkinsonism (frequently atypical) was a clinical feature in 36 participants, including six participants with progressive supranuclear palsy Richardson's syndrome (PSP‐RS) and 11 participants with corticobasal syndrome (CBS). In this mixed/atypical parkinsonism cohort, αSyn‐SAA had a 68.8% sensitivity (95% CI 41.3% to 89.0%) and 100% specificity (95% CI 83.2% to 100.0%) to detect underlying LBD. In the cohort of patients with a clinical diagnosis of PSP‐RS, αSyn‐SAA positively detected LBD in one case caused by diffuse LBD and AD co‐pathology (a rare PSP‐RS mimic), whereas 5/5 cases were true negatives. Within patients with a clinical diagnosis of CBS, αSyn‐SAA was positive in 1/1 case with diffuse LBD co‐pathology, 0/1 case with amygdala‐predominant LBD, and 1/2 cases with brainstem‐predominant LBD, whereas 7/7 cases were true negatives.
3.2. Comparison of antemortem clinical features
Compared to non‐LBD, amygdala‐predominant cases were more likely to carry an APOE ε4 allele (83% vs 42%, p < 0.05), and they also had a higher average stage of AD neuropathological change (ADNC 2.7 ± 0.8 vs 1.5 ± 1.2, p < 0.05), with an overall higher prevalence of AD co‐pathology (defined as ADNC intermediate or high; 85% vs 33%, p < 0.05). Notably, in amygdala‐predominant LBD, the interval between CSF collection and autopsy was over a year longer than all other groups (eg, 4.9 ± 2.9 years vs 3.5 ± 1.4 years for non‐LBD, p < 0.05), which may decrease sensitivity as LBD pathology can develop in the interim. Diffuse/transitional cases showed lower brain weights when compared to non‐LBD cases (1229 ± 145 g vs 1111 ± 135 g). In patients with a positive αSyn‐SAA compared to negative tests, hallucinations were more common (29% vs 3%, p < 0.05) and brain weights were higher (1192 ± 129 g vs 1105 ± 142 g, p < 0.05). Otherwise, no difference was seen between LBD subgroups on clinical severity or a range of clinical symptoms (Table 1).
In the amygdala‐predominant cohort, αSyn‐SAA positivity was associated with an older age at onset (63 ± 14 years vs 58 ± 11 years in the negative αSyn‐SAA group, p < 0.05) and pathologic spread beyond the amygdala (83.3% vs 25.0%, p < 0.05; Figure 2, Table S2), where the burden of LBD was concentrated in the amygdala but present to a trace degree in other structures (but not enough to change the LBD stage and still not conforming to Braak PD staging). 25 Syndromic features of DLB thought to be highly specific for LBD, including visual hallucinations and RBD, were only seen in participants with a positive test but were rare overall (N = 1 for each). In the brainstem‐predominant cohort, only one case in six was αSyn‐SAA positive, but intriguingly this participant reported RBD and had LBD pathology spread to the anterior cingulate cortex, differing from the other five cases that did not show LBD spread beyond the brainstem.
FIGURE 2.
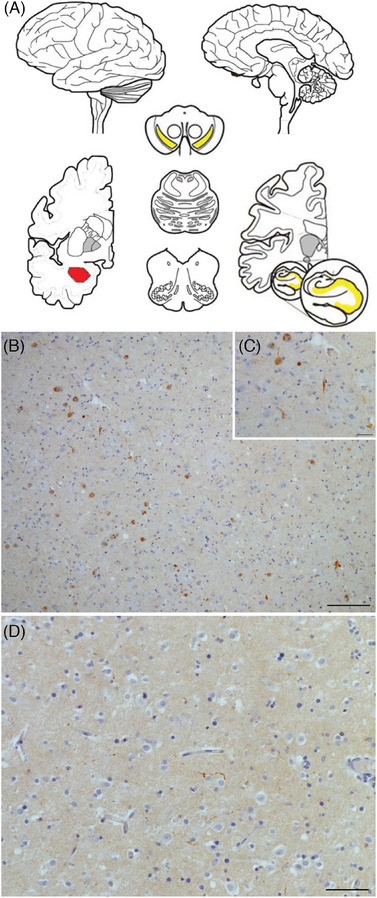
CSF αSyn‐SAA positivity is associated with spread in amygdala‐predominant LBD. (A) Schematic showing amygdala‐predominant LBD concentrated in amygdala (red); it may be confined there, or it may be found to a lesser degree in mesial temporal lobe structures (hippocampus or entorhinal cortex) or substantia nigra (yellow), adapted from Attems et al. (2021). 31 Immunohistochemical staining for all αSyn species in a representative case shows abundant Lewy bodies and Lewy neurites in the amygdala at low (10×, B) and high (40×, C) magnification with little LBD in the entorhinal cortex (20×, D). Scale bars: 100 μm (B), 25 μm (C), and 50 μm (D).
4. DISCUSSION
In this retrospective autopsy validation study of clinical syndromes and neuropathological entities commonly encountered in the field of behavioral neurology, we find that a clinically available CSF αSyn‐SAA (SYNTap Biomarker Test) had excellent diagnostic performance to detect cortical LBD, including diffuse and transitional stages, with a high degree of specificity in the cohort overall. The sensitivity of αSyn‐SAA in subjects with cortical pathology is similar to prior studies, with this assay showing excellent diagnostic performance for the detection of LBD, even in prodromal stages of αSyn with isolated RBD. 9 , 10 Comparison across assays should be considered cautiously given differences in diagnostic performance, but, reassuringly, other αSyn‐SAAs have similar profiles. 11 , 12 One point to note is that CSF collection occurred earlier in the disease course of amygdala‐predominant patients (i.e., longer CSF‐autopsy interval), which may negatively impact diagnostic performance in this subgroup.
In amygdala‐ and brainstem‐predominant cases, we found comparatively low sensitivity, in line with prior reports, 10 , 11 but we found that detection in these groups was associated with early αSyn pathology spread beyond the initial site of involvement. This finding supports the hypothesis from prior work using this and a related assay that αSyn‐SAA may have a threshold of detection dependent on the number of pathogenic seeds in the CSF, which reflects the extent of pathological deposition. 10 , 27 Alternatively, as this assay primarily detects pathogenic “seed‐competent” αSyn fragments, amygdala‐predominant LBD may comprise a heterogeneous profile of αSyn species, with one subgroup (detected by αSyn‐SAA) reflecting an early LBD stage transitioning to more diffuse spread.
In this cohort, we found that the DLB clinical syndrome and even highly specific core clinical characteristics (eg, RBD and visual hallucinations) were poorly predictive of underlying LBD, primarily due to low prevalence, whereas clinical symptoms like parkinsonism, dysautonomia, fluctuations, and depression/anxiety were more common overall but non‐specific for underlying LBD neuropathology, also consistent with prior reports. 2 The low sensitivity of clinical criteria is a particular problem in the face of evidence, suggesting that up to 50% of patients with AD may have comorbid LBD and 23% of all patients with cognitive symptoms may have clinically relevant LBD, 1 , 4 which may impact the outcome of treatment with newly approved anti‐amyloid antibodies. 7
In the modern era, the field of behavioral neurology is tasked with predicting underlying neuropathology based on patients presenting clinical symptoms, a complex endeavor given the high prevalence of co‐pathologies in neurocognitive disorders of age, 26 , 28 made somewhat easier by the recent implementation of several tests in the clinic that reliably identify AD neuropathology. However, a reliable, predictive marker for clinically relevant αSyn is of particular importance as LBD is the second most common neurodegenerative pathology to cause dementia. 29 The advent of new clinical diagnostic tools for LBD is well timed in an era when precision diagnosis supported by biomarker confirmation is increasingly preferred, 30 but appropriate clinical use of a LBD‐specific biomarker has yet to be defined for patients with cognitive, behavioral, and/or motor symptoms that may be related to LBD.
Future analyses of the use of αSyn‐SAA in clinical contexts likely would benefit from a prospective design. Here, a retrospective analysis limited the degree to which clinical symptoms could be specifically queried, which can impact assessments of prevalence. Our assessment of RBD, for example, relied on participant report, rather than on polysomnography or structured questionnaire, and it is possible that careful ante mortem phenotyping of sleep and autonomic symptoms would yield different results, as RBD was previously reported to strongly correlate with αSyn‐SAA positivity. 9 Prospective design with incorporation of modern structured collection tools could yield different results. Additionally, given the years‐long gap between CSF collection and autopsy, sensitivity may be underestimated. The advent of PET biomarkers for αSyn would greatly (and reciprocally) facilitate future validation efforts.
Our study has additional limitations, including small sample size, though it is larger than many autopsy studies and comparable to prior validation efforts, and our cohort benefits from extensive ante mortem phenotyping not typically available in autopsy studies. For all studies validating an in vivo marker to autopsy, any variability in biomarker‐to‐autopsy interval will impact assessment of diagnostic performance, and here that interval was longer for amygdala‐predominant LBD. As this is a research cohort drawn from observational studies, the findings may not generalize to that of a routine clinic population, where co‐pathologies may be more common. Additionally, referral bias may also impact prevalence calculations, which should be interpreted cautiously. Findings on diagnostic performance also need replication in an ethnically and racially diverse cohort given the lack of diversity in this dataset (Table S1). Another consideration is that newer LBD diagnostic criteria now include an olfactory‐only stage, 31 which for practical reasons was not applied retroactively in this study. Future work with αSyn‐SAA may utilize a quantitative assay to reflect subtler prodromal neurochemical abnormalities.
In conclusion, we report excellent diagnostic performance for a clinically available αSyn‐SAA to detect cortical LBD pathology, validated against autopsy, in a behavioral neurology cohort not enriched for DLB diagnoses. Differing diagnostic performance in amygdala‐ and brainstem‐predominant cohorts may be affected by the degree of spread from the initial site of involvement, with αSyn‐SAA more likely to detect cases with early cortical spread.
CONFLICT OF INTEREST STATEMENT
L.V. is a site principal investigator (PI) for Biogen‐sponsored clinical trials in AD. P.A.L. reported grants from the Alzheimer's Association Part the Cloud program; serving as PI for trials sponsored by Woolsey Pharmaceuticals, Transposon, Alector, and AbbVie. J.C.R. is a site PI for clinical trials sponsored by Eli Lilly and Eisai. A.M.S. has provided consultation to Alector, Lilly/Prevail, Passage Bio, and Takeda. J.L. and L.C.M. are employees of Amprion, Inc. A.L.B. reported grants from Rainwater Charitable Foundation during the conduct of the study and stocks/options from Alector, Arvinas, Arkuda, Truebinding, and AZTherapies; personal fees from the AIDS Clinical Trials Group, Boehringer Ingelheim, GSK, Denali Therapeutics, Oligomerix, Merck, Roche, Transposon, Wave, Oscotec, and Alzprotect; and grants from Biogen, Eisai, Regeneron, Bluefield Project, the Alzheimer's Association, and CurePSP. Other authors have no relevant disclosures. Author disclosures are available in the supporting information
CONSENT STATEMENT
All participants provided written informed consent at time of recruitment.
Supporting information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
The authors thank the patients and their families who participated in this research, as well as staff and investigators at the University of California, San Francisco (UCSF) Memory and Aging Center (MAC). This publication was made possible by grant number P01AG019724 and P30AG062422 from the National Institute on Aging. Funding was provided by National Institutes for Health: P30AG062422, P01AG019724, U01AG057195, K23AG073514 (LV), K23AG059888 (JCR), K24AG053435 (LTG), T32AG023481 (DLF, NS, SL), as well as the Rainwater Charity Foundation, the Alzheimer's Association (L.V.), AlzOut (J.C.R.), Shenandoah Fund (L.V., J.C.R.) , and the John Douglas French Alzheimer's Foundation (J.C.R.). The funders had no role in study design or analysis.
Samudra N, Fischer DL, Lenio S, et al. Clinicopathological correlation of cerebrospinal fluid alpha‐synuclein seed amplification assay in a behavioral neurology autopsy cohort. Alzheimer's Dement. 2024;20:3334–3341. 10.1002/alz.13799
REFERENCES
- 1. Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha‐synuclein immunohistochemistry. Brain Pathol. 2000;10(3):378‐384. doi: 10.1111/j.1750-3639.2000.tb00269.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nelson PT, Jicha GA, Kryscio RJ, et al. Low sensitivity in clinical diagnoses of dementia with Lewy bodies. J Neurol. 2010;257(3):359‐366. doi: 10.1007/s00415-009-5324-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Middleton JS, Hovren HL, Kha N, et al. Seed amplification assay results illustrate discrepancy in Parkinson's disease clinical diagnostic accuracy and error rates. J Neurol. Published online August 17, 2023. doi: 10.1007/s00415-023-11810-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Quadalti C, Palmqvist S, Hall S, et al. Clinical effects of Lewy body pathology in cognitively impaired individuals. Nat Med. 2023;29(8):1964‐1970. doi: 10.1038/s41591-023-02449-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863‐1872. doi: 10.1212/01.wnl.0000187889.17253.b1 [DOI] [PubMed] [Google Scholar]
- 6. McKeith IG, Boeve BF, Dickson DW, et al. Diagnosis and management of dementia with Lewy bodies: fourth consensus report of the DLB Consortium. Neurology. 2017;89(1):88‐100. doi: 10.1212/WNL.0000000000004058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. VandeVrede L, La Joie R, Horiki S, et al. Co‐pathology may impact outcomes of amyloid‐targeting treatments: clinicopathological results from two patients treated with aducanumab. Acta Neuropathol. 2023;146(5):777‐781. doi: 10.1007/s00401-023-02631-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Concha‐Marambio L, Pritzkow S, Shahnawaz M, Farris CM, Soto C. Seed amplification assay for the detection of pathologic alpha‐synuclein aggregates in cerebrospinal fluid. Nat Protoc. 2023;18(4):1179‐1196. doi: 10.1038/s41596-022-00787-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Siderowf A, Concha‐Marambio L, Lafontant DE, et al. Assessment of heterogeneity among participants in the Parkinson's Progression Markers Initiative cohort using α‐synuclein seed amplification: a cross‐sectional study. Lancet Neurol. 2023;22(5):407‐417. doi: 10.1016/S1474-4422(23)00109-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arnold MR, Coughlin DG, Brumbach BH, et al. α‐Synuclein Seed Amplification in CSF and brain from patients with different brain distributions of pathological α‐Synuclein in the context of Co‐Pathology and Non‐LBD diagnoses. Ann Neurol. 2022;92(4):650‐662. doi: 10.1002/ana.26453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall S, Orrù CD, Serrano GE, et al. Performance of αSynuclein RT‐QuIC in relation to neuropathological staging of Lewy body disease. Acta Neuropathol Commun. 2022;10(1):90. doi: 10.1186/s40478-022-01388-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iranzo A, Fairfoul G, Ayudhaya ACN, et al. Detection of α‐synuclein in CSF by RT‐QuIC in patients with isolated rapid‐eye‐movement sleep behaviour disorder: a longitudinal observational study. Lancet Neurol. 2021;20(3):203‐212. doi: 10.1016/S1474-4422(20)30449-X [DOI] [PubMed] [Google Scholar]
- 13. Concha‐Marambio L, Weber S, Farris CM, et al. Accurate detection of α‐Synuclein seeds in Cerebrospinal Fluid from isolated rapid eye movement sleep behavior disorder and patients with Parkinson's disease in the DeNovo Parkinson (DeNoPa) Cohort. Mov Disord. 2023;38(4):567‐578. doi: 10.1002/mds.29329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Perry DC, Brown JA, Possin KL, et al. Clinicopathological correlations in behavioural variant frontotemporal dementia. Brain. 2017;140(12):3329‐3345. doi: 10.1093/brain/awx254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology. 1984;34(7):939‐944. doi: 10.1212/wnl.34.7.939 [DOI] [PubMed] [Google Scholar]
- 16. Gorno‐Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76(11):1006‐1014. doi: 10.1212/WNL.0b013e31821103e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134(9):2456‐2477. doi: 10.1093/brain/awr179. Pt. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology. 2013;80(5):496‐503. doi: 10.1212/WNL.0b013e31827f0fd1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Höglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the movement disorder society criteria. Mov Disord. 2017;32(6):853‐864. doi: 10.1002/mds.26987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. McKeith IG. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimers Dis. 2006;9(3):417‐423. doi: 10.3233/jad-2006-9s347. Suppl. [DOI] [PubMed] [Google Scholar]
- 21. Leverenz JB, Hamilton R, Tsuang DW, et al. Empiric refinement of the pathologic assessment of Lewy‐related pathology in the dementia patient. Brain Pathol. 2008;18(2):220‐224. doi: 10.1111/j.1750-3639.2007.00117.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1‐11. doi: 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mackenzie IRA, Neumann M, Bigio EH, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117(1):15‐18. doi: 10.1007/s00401-008-0460-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic‐predominant age‐related TDP‐43 encephalopathy (LATE): consensus working group report. Brain. 2019;142(6):1503‐1527. doi: 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Braak H, Del Tredici K, Rüb U, de Vos RAI, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24(2):197‐211. doi: 10.1016/s0197-4580(02)00065-9 [DOI] [PubMed] [Google Scholar]
- 26. Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late‐onset Alzheimer's disease. Brain. 2021;144(7):2186‐2198. doi: 10.1093/brain/awab099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bentivenga GM, Mammana A, Baiardi S, et al. Performance of a seed amplification assay for misfolded alpha‐synuclein in cerebrospinal fluid and brain tissue in relation to Lewy body disease stage and pathology burden. Acta Neuropathol. 2024;147(1):18. doi: 10.1007/s00401-023-02663-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fischer DL, Seeley WW. A precision medicine approach to Dementia Care: syndrome, Etiology, and Copathology. Pract Neurol (Fort Wash Pa). 2023;2023:17‐22. [PMC free article] [PubMed] [Google Scholar]
- 29. Yamada M, Komatsu J, Nakamura K, et al. Diagnostic Criteria for Dementia with Lewy Bodies: updates and Future Directions. J Mov Disord. 2020;13(1):1‐10. doi: 10.14802/jmd.19052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14(4):535‐562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Attems J, Toledo JB, Walker L, et al. Neuropathological consensus criteria for the evaluation of Lewy pathology in post‐mortem brains: a multi‐centre study. Acta Neuropathol. 2021;141(2):159‐172. doi: 10.1007/s00401-020-02255-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information