

Abstract
INTRODUCTION
Although glymphatic function is involved in Alzheimer's disease (AD), its potential for predicting the pathological and clinical progression of AD and its sequential association with core AD biomarkers is poorly understood.
METHODS
Whole‐brain glymphatic activity was measured by diffusion tensor image analysis along the perivascular space (DTI‐ALPS) in participants with AD dementia (n = 47), mild cognitive impairment (MCI; n = 137), and normal controls (n = 235) from the Alzheimer's Disease Neuroimaging Initiative.
RESULTS
ALPS index was significantly lower in AD dementia than in MCI or controls. Lower ALPS index was significantly associated with faster changes in amyloid positron emission tomography (PET) burden and AD signature region of interest volume, higher risk of amyloid‐positive transition and clinical progression, and faster rates of amyloid‐ and neurodegeneration‐related cognitive decline. Furthermore, the associations of the ALPS index with cognitive decline were fully mediated by amyloid PET and brain atrophy.
DISCUSSION
Glymphatic failure may precede amyloid pathology, and predicts amyloid deposition, neurodegeneration, and clinical progression in AD.
Highlights
The analysis along the perivascular space (ALPS) index is reduced in patients with Alzheimer's disease (AD) dementia, prodromal AD, and preclinical AD.
Lower ALPS index predicted accelerated amyloid beta (Aβ) positron emission tomography (PET) burden and Aβ‐positive transition.
The decrease in the ALPS index occurs before cerebrospinal fluid Aβ42 reaches the positive threshold.
ALPS index predicted brain atrophy, clinical progression, and cognitive decline.
Aβ PET and brain atrophy mediated the link of ALPS index with cognitive decline.
Keywords: Alzheimer's disease, amyloid, analysis along the perivascular space, cognitive decline, glymphatic, neurodegeneration, progression
1. BACKGROUND
The glymphatic system is an essential fluid‐clearance system in the brain that has been identified in the rodent brain. 1 The highly organized cerebrospinal fluid (CSF) transport system subserves the influx of CSF into the brain parenchyma along the arterial perivascular spaces and subsequent transfer to the brain interstitial space. The system then directs the clearance of brain interstitial fluid along the venous perivascular into the meningeal and cervical lymphatic drainage vessels to remove waste. The accumulation of amyloid beta (Aβ) plaques and neurofibrillary tangles of hyperphosphorylated tau have been implicated in the cognitive dysfunction of Alzheimer's disease (AD). 2 Aβ is transported and removed from the central nervous system along the glymphatic pathways. 1 , 3 Although tau is an intracellular pathology characterized in AD, intracellular tau can release into extracellular space and be cleared from the brain along the glymphatic system. 4 , 5 , 6 , 7 Therefore, impaired brain clearance mechanisms may be an essential factor contributing to the deposition of pathological proteins in AD. 8 The novel fluid transport system provides a promising target for the prevention or treatment of AD.
Magnetic resonance imaging (MRI) tracer‐based studies have demonstrated the existence of a glymphatic system in the human brain 9 as previously discovered in rodents, through the intrathecal administration of gadolinium‐based contrast agent. This approach can detect glymphatic function directly. However, its invasive nature restricts the comprehensive human studies of the glymphatic system. Recently, a measure of perivascular clearance activity in the human brain using diffusion MRI called diffusion tensor image analysis along the perivascular space (DTI‐ALPS) has been proposed by Taoka et al., 10 which is calculated from the diffusivity along the deep medullary vein at the level of the lateral ventricular body. The reliability of the ALPS index as a measure of glymphatic activity was supported in a recent study that found a significant correlation between the ALPS index and glymphatic clearance function evaluated by the intrathecal angiography‐based method. 11 The ALPS index provides an opportunity for the non‐invasive investigation of the human glymphatic system. It has been studied in various neurological disorders. 11 , 12 , 13 , 14 In the field of AD, previous studies have observed a decreased ALPS index in AD patients compared to controls. 15 , 16 , 17 The ALPS index is also associated with cognitive performance in AD 10 , 15 and is negatively associated with amyloid and tau deposition on positron emission tomography (PET) images. 15 , 18 However, to our knowledge, no prior attempts have been made to investigate the potential of glymphatic activity in predicting the pathological and clinical progression of AD in longitudinal cohorts, or to determine whether it is a cause or a consequence of AD pathological and clinical progression.
In the present study, we used the ALPS index to investigate the cross‐sectional and longitudinal associations between glymphatic activity and clinical and pathological features of AD, including diagnosis, cognitive scores, and CSF and neuroimaging biomarkers. Taking advantage of the large‐scale and longitudinal measurements of the ALPS index and AD hallmarks in the Alzheimer's Disease Neuroimaging Initiative (ADNI) cohort, we further investigated the sequential relationship between glymphatic dysfunction, as measured by the ALPS index, and markers of AD pathology in the development of AD.
2. METHODS
2.1. Participants and study design
This study included two longitudinal follow‐up cohorts, the ADNI and the UK Biobank (UKB) study. The ADNI database was used for the main analyses, which included examining the relationship of the ALPS index with clinical and pathological features of AD, as well as its sequence association with core AD biomarkers. The UKB was used as a replication cohort for analyzing the role of the ALPS index in predicting clinical progression. A general scheme of the current study is depicted in Figure 1.
FIGURE 1.
Overview of the study. A, amyloid; Aβ, amyloid beta; AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; ALPS, analysis along the perivascular space; AV45, florbetapir; AV1451, flortaucipir; CN, cognitively normal; CSF, cerebrospinal fluid; FBB, florbetaben; FDG, fluorodeoxyglucose; MCI, mild cognitive impairment; MRI, magnetic resonance imaging; PACC, Preclinical Alzheimer's Cognitive Composite; PET, positron emission tomography; p‐tau181, phosphorylated tau 181; ROI, region of interest; SUVR, standard uptake value ratio; t‐tau, total tau.
2.1.1. ADNI cohort for main analysis
Data used in the main analysis were obtained from the ADNI database (http://adni.loni.usc.edu/). For the present study, we used data accessed from the ADNI database in March 2023. The ADNI was launched in 2003 as a public–private partnership and is led by principal investigator Michael W. Weiner, MD. The primary goal of ADNI is to test whether serial MRI, PET, and various clinical, biological, and neuropsychological markers can be combined to measure the progression of mild cognitive impairment (MCI) and early AD dementia. The ADNI study was approved by the local institutional review boards of all participating centers. All study participants provided written informed consent. Inclusion and exclusion criteria have been described previously. 19 ADNI participants were included in this study if they possessed available DTI and necessary covariate data (i.e., age, sex, education, and apolipoprotein E [APOE] ε4 genotype). Patients with AD dementia were diagnosed according to the National Institute of Neurological and Communication Disorders/Alzheimer's Disease and Related Disorders Association criteria, reported a Mini‐Mental State Examination (MMSE) score of between 20 and 26, a global Clinical Dementia Rating from 0.5 to 1.0, and a sum‐of‐boxes Clinical Dementia Rating (CDR‐SB) of 1.0 to 9.0. Patients diagnosed with MCI had an MMSE score between 24 and 30, objective memory loss as assessed by delayed recall of the Wechsler Memory Scale Logical Memory II, a CDR‐SB score of at least 0.5, preserved activities of daily living, and absence of dementia. Cognitively normal (CN) controls were defined as those with an MMSE score of at least 24 and a CDR‐SB score of 0 or 0.5. To sum up, a total of 47 AD dementia patients, 137 MCI patients, and 235 CN controls were included in the primary analysis.
RESEARCH IN CONTEXT
Systematic review: We reviewed literature in PubMed and found studies of glymphatic activity measured by analysis along the perivascular space (ALPS) index in Alzheimer's disease (AD) were all conducted in cross‐sectional cohorts. No prior attempts have been made to investigate the potential of glymphatic activity in tracking the pathological and clinical progression of AD and its sequential relationship with core AD biomarkers in longitudinal cohorts.
Interpretation: The ALPS index decreased in AD dementia, prodromal, and preclinical AD patients. We for the first time suggested the predictive effect of the ALPS index on longitudinal changes in amyloid positron emission tomography (PET) and AD signature region of interest volume, risk of amyloid‐positive transition and clinical progression, and cognitive decline. Amyloid PET and brain atrophy mediated the associations of ALPS index with cognitive decline.
Future directions: The ALPS index provides a rough measure of the global glymphatic activity and cannot reflect its regional heterogeneity. Developing and improving a precise and non‐invasive measure of regional glymphatic function is a promising future direction.
2.1.2. Replication cohort
The UKB study was approved by the National Information Governance Board for Health and Social Care and the National Health Service North West Multicenter Research Ethics Committee. 20 It is a population‐based cohort of > 500,000 participants aged 37 to 73 years recruited across the United Kingdom between 2006 and 2010. 20 All participants provided electronic informed consent at baseline assessment. To assess the relationship between baseline ALPS index and the risk of progression from non‐demented to AD, 36,050 non‐demented participants with available DTI data at the first imaging visit in UKB were screened. After excluding participants with missing follow‐up data, missing covariate data, and without incident AD within the first 3 years of follow‐up, 36,050 eligible participants were finally included in the replication analyses (Figure S1 in supporting information). Incident AD events were recorded using codes F00 and G30 of the International Classification of Diseases system. AD diagnosis data were ascertained by combining records from first occurrence data (Fields 131036‐131037 and 130836‐130837), algorithmically defined AD outcomes (Field 42020), hospital inpatient data summaries (Fields 41270 and 41280), and death registry data (Fields 40001 and 40002). Self‐reported AD records were excluded from the study. Detailed information about the data used for AD definitions in the UKB is provided in Table S1 in supporting information. Participants were considered at risk for AD from the first imaging visit (Field 53, instance 2) and were followed up until the date of first AD diagnosis, death, or the last recorded date (September 2023), whichever came first. Analyses were performed under UK Biobank application number 19542.
2.2. CSF biomarkers
CSF was collected by lumbar puncture from a subset of ADNI participants. Each sample was aliquoted to 500 µL in polypropylene tubes, shipped on dry ice to the ADNI Biomarker Core laboratory, and stored at −80°C. CSF Aβ42, phosphorylated tau 181 (p‐tau181), and total tau (t‐tau) concentrations were measured using Roche Elecsys and cobas e 601 automated immunoassay analyzer systems as previously described. 21 All CSF biomarker assays were performed in duplicate and averaged. Coefficients of variation for CSF analytes within and between lots were <15%. The cutoffs for CSF Aβ42, p‐tau181, and t‐tau were previously set at 1098 pg/mL, 22 , 23 26.64 pg/mL, 23 and 300 pg/ml, 24 , 25 respectively.
2.3. MRI imaging
In ADNI, T1‐weighted imaging, fluid‐attenuated inversion recovery (FLAIR) imaging, and DTI data were acquired for each participant using 3T scanners. Details of the imaging protocol can be found in the open‐source document (https://adni.loni.usc.edu/methods/documents/mri‐protocols/). Brain structural measures were derived from quality‐controlled T1‐weighted neuroimaging data. FreeSurfer was used to quantify the regional volumes according to the 2010 Desikan–Killiany atlas. We used volumetric data for the hippocampus and AD signature region of interest (ROI) composed of the entorhinal, inferior temporal, middle temporal, and fusiform areas. 26 We then calculated the residual hippocampal volume and the residual AD signature ROI volume from a linear regression of the imaging measure (y) against the intracranial volume (ICV; x) among CN subjects, as previously described. 27 The adjusted imaging measure is interpreted as the deviation from the value expected in a healthy individual with the observed ICV. Unless otherwise noted, “hippocampal volume” and “AD signature ROI volume” in the following refer to their residual metrics. White matter hyperintensities (WMH) and gray matter (GM) volume were automatically segmented by the ADNI core laboratory using the ADNI pipeline (https://files.alz.washington.edu/documentation/adni‐proto.pdf).
In UKB, all brain MRI were acquired on the standard 3T Siemens Skyra scanner with a 32‐channel head coil, according to a freely available protocol (https://www.fmrib.ox.ac.uk/ukbiobank/protocol/V4_23092014.pdf), document (https://biobank.ndph.ox.ac.uk/showcase/showcase/docs/brain_mri.pdf), and publication. 28 Further information on the image processing and quality control pipeline are available elsewhere. 28
2.4. ALPS index calculation
In both ADNI and UKB cohorts, the ALPS index was calculated from DTI using a semi‐automated and highly reliable pipeline developed and validated by Taoka et al. 10 Diffusivity maps along the x axis (right‐left), y axis (anterior‐posterior), z axis (inferior‐superior), and color‐coded fractional anisotropy (FA) maps were processed using the FMRIB Software Library (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki). To make the ROI templates, we randomly selected 100 ADNI participants and 150 UKB participants with age and sex matched. At the location where the direction of the deep medullary veins was perpendicular to the ventricular body, four spherical ROIs with a radius of 4 mm were placed on the bilateral projection (superior and posterior corona radiata) and association (superior longitudinal fasciculus) fibers on the FA map template. The ROI maps were non‐linearly warped to the Montreal Neurological Institute (MNI) space using FNIRT (https://fsl.fmrib.ox.ac.uk/fsl/fslwiki/FNIRT) and the probability maps were then generated and binarized as the templates. After that, the diffusivity maps along the x axis (right‐left), y axis (anterior‐posterior), z axis (inferior‐superior), and color‐coded FA maps for all participants were also warped to the MNI space. The diffusivity in the directions of the x axis (Dx), y axis (Dy), and z axis (Dz) of the ROIs on the projection and association fibers were calculated for each participant within the ROI templates and recorded as Dxproj, Dyproj, Dzproj, Dxassoc, Dyassoc, Dzassoc, respectively. The ALPS index was calculated as follows: ALPS index = . The ALPS indexes of the left and right hemispheres were calculated separately, and then we used the average ALPS index of the bilateral sides in the main analysis. A higher ALPS index represented better glymphatic activity. Two neurologists blinded to clinical data independently placed ROIs for inter‐observer reliability analysis. To exclude the potential influence of white matter integrity, we also calculated the whole‐brain white matter mean FA and mean diffusivity (MD) of ADNI individuals according to the prior work. 29
2.5. PET imaging
A subset of ADNI participants provided eligible PET data. The Aβ (florbetapir, or [18F]AV45; florbetaben, or FBB), tau (flortaucipir, or [18F]AV1451), and fluorodeoxyglucose (FDG) PET data were preprocessed using the ADNI pipeline (http://adni.loni.usc.edu/datasamples/pet/). Briefly, each participant's MRI image from the nearest available visit was segmented and parcellated using FreeSurfer (version 7.1.1) to define ROIs in native space. The mean standard uptake value ratio (SUVR) of each scan for targeted ROIs was calculated by dividing the tracer uptake in these regions by the value in a predefined reference region. For Aβ PET, the SUVRs were generated by averaging uptake ratios across AD summarized cortical regions (frontal, anterior/posterior cingulate, lateral parietal, and lateral temporal regions) and then normalized by a composite reference region (whole cerebellum, brainstem/pons, and eroded subcortical white matter). This composite reference region had more reliable longitudinal AV45 results in ADNI compared to using only the cerebellum as a reference region. 30 For note, Aβ PET in the following text refers only to AV45 PET, unless otherwise specified. For tau PET, a composite SUVR was calculated by referring AV1451 uptake in a weighted composite (MetaROI) of regions (bilateral entorhinal, amygdala, fusiform, inferior and middle temporal cortices) to the mean uptake of the inferior cerebellar GM. The tau PET SUVR values in Braak stage ROIs were extracted, including three Braak region groups (Braak I, Braak III–IV, and Braak V–VI) in the present study. The Braak II region (hippocampus) was not included because this region is known to be contaminated by off‐target binding in the choroid plexus. In addition, cortical uptake of AV45 and AV1451 in 68 ROIs defined by the Desikan–Killiany atlas 31 were also extracted from each PET scan that was coregistered with the corresponding individual structural MRI scan. For FDG PET images, a composite SUVR of each scan was calculated as the mean uptake of predefined MetaROIs (bilateral angular, posterior cingulate, and inferior temporal gyrus) relative to the mean of a pons/vermis reference region. Cutoffs for brain Aβ, tau, and FDG PET categories were as follows: 0.78 for AV45 SUVR, 0.74 for FBB SUVR, 1.37 for AV1451 MetaROI SUVR, 23 and 1.21 for FDG PET. 32 The cut points used in the present study are based on published articles or ADNI proposals.
2.6. Cognitive assessments
Previously validated Preclinical Alzheimer's Cognitive Composite (PACC) scores were used to represent global cognition in the present study. The PACC score has been associated with the detection of early cognitive decline in patients with AD. 33 In ADNI, the PACC score is the average of four z scored cognitive tests: Alzheimer's Disease Assessment Scale delayed recall word list test, MMSE, Logical Memory Delayed Recall test, and Digit Symbol coding test. Specific cognitive domains were also assessed using composite measures developed by ADNI that reflect memory and executive function (EF). 34 , 35 For each of the three cognitive measures, higher scores indicate more normal cognition and lower scores indicate more impaired cognition.
2.7. Biological status definition and AD profiles
Participants were classified into different amyloid (A) statuses according to the abnormal status of CSF Aβ42 and Aβ PET (AV45 and FBB). If any one of the three biomarkers indicates an abnormal level of amyloid pathology, the individual would be considered A+ status. Based on the CSF p‐tau181 levels and tau PET and the cutoffs described above, participants were classified into the aggregated AD‐tau positive (T+) group if either of these two biomarkers indicated the presence of tau pathology. Similarly, CSF t‐tau and FDG PET were used to determine neurodegeneration (N) status. Table S2 in supporting information lists detailed definitions of biological status. Unless otherwise noted, A/T/N statuses hereinafter refer to A/T/N statuses jointly defined by CSF or PET, respectively. To maximize the use of data, for participants who did not have a CSF assessment or PET scan at baseline (first ALPS index measurement), the corresponding measurements within the last 12 months before the baseline ALPS index measurement were used for imputation. For sensitivity analysis, we also used the biological status (A± , T± , and N±) defined separately by CSF or PET.
According to the clinical diagnosis and A status, participants were classified into five groups, including the A–CN, A+CN (preclinical AD 36 ), A–MCI, A+MCI (prodromal AD 37 ), and A+AD dementia. Then, aggregated tau and neurodegeneration groups were merged as a “TN” group according to the National Institute on Aging–Alzheimer's Association criteria for preclinical AD. 36 If any of the biomarkers indicate an abnormal level of tau or neurodegeneration pathology, the individual would be considered a TN+ status. Combining TN with A status, we subdivided CN participants into three biomarker subgroups, stage 0 (A–TN–), stage 1 (A+TN–), and stage 2 (A+TN+). Participants were accordingly classified into normal control (stage 0), preclinical AD (stage 1 and stage 2), prodromal AD (A+MCI), and A+AD dementia, following the AD continuum category. 36 , 37 , 38 Table S3 in supporting information provides detailed definitions of AD biological profiles.
2.8. Statistical analysis
All statistical analyses were performed using R software (version 4.2.3). Data were initially screened for outliers and those that fell outside three standard deviations (SDs) were removed. Raw data are presented in tables as mean (±SD) or number (%) unless otherwise noted. Normality tests were performed using the Shapiro–Wilk test and visual inspection of histograms. The ALPS index was normalized using the Box–Cox transformation. Z transformation was performed on the normally transformed ALPS index and AD variables when comparability of effect sizes was required. Non‐parametric Kruskal–Wallis tests, chi‐squared tests, and Spearman rank correlation were used to compare baseline characteristics between diagnostic groups, or to test associations between ALPS index and demographic factors. Group comparisons of the ALPS index were examined using analysis of covariance (ANCOVA) controlling for age, sex, education, and APOE ε4 carrier status (Model 1). Baseline diagnosis was also adjusted for comparing the ALPS index in different biological groups. A false discovery rate (FDR) of 0.05 was applied using the Benjamini–Hochberg approach for correction of multiple comparisons when appropriate.
Linear regression models were used to examine the cross‐sectional associations of the ALPS index with core AD biomarkers (including CSF, PET, and MRI indicators of amyloid pathology, tau pathology, and neurodegeneration) and cognition in Model 1. We then examined how the ALPS index correlated with these core AD biomarkers in participants stratified by CSF Aβ42 status (positive or negative), the established earliest biomarker of AD, 39 according to the predefined cutoffs. We also calculated the linear regression slopes for core AD biomarkers in CSF Aβ42 defined A± groups and tested whether the slopes were statistically significantly different. To investigate the longitudinal relationship among the ALPS index, core AD biomarkers, and cognition, we examined whether the baseline ALPS index could predict the longitudinal change rates of AD biomarkers and cognition. Additionally, we examined whether baseline levels of AD biomarkers and cognition could predict longitudinal changes in the ALPS index using linear regression in Model 1. We derived the slopes of each core AD biomarker (including CSF, PET, and MRI measures) and ALPS index using linear mixed‐effect (LME) models for all the participants with corresponding longitudinal data, adjusting for age and sex, and including a random slope and intercept. The slope of each cognitive score was calculated in a similar manner, but adjusted for age, sex, and education. As an additional analysis, we examined the interaction effect of baseline ALPS index with core AD variables on the longitudinal changes in PACC scores using linear regression in Model 1.
To further explore the sequential changes between the ALPS index and Aβ biomarkers, we first applied robust local weighted regression models to visualize the relationships among the ALPS index, CSF Aβ42, Aβ PET, and PACC score. The data above was scaled from 0% to 100%, with 0% representing minimal abnormalities and 100% representing maximal abnormalities, as performed elsewhere. 15 The CSF Aβ42 level and PACC score were modeled as the proxies for disease progression. Subsequently, we examined the relationship between ALPS index and the risk of A status progression from A– to A+ in baseline A–CN subjects using Kaplan–Meier curve and multivariable Cox proportional hazards regression adjusted for covariates in Model 1. In the Cox model, the baseline ALPS index was treated as a continuous variable and categorized as a dichotomous variable by the median level (< 1.25 and ≥1.25). Time variable in this Cox model was time to progression to A+ or the maximum follow‐up duration.
To assess whether baseline ALPS index could predict the risk of clinical conversion to AD dementia or MCI in ADNI participants and the risk of progression from non‐demented to incident AD in UKB baseline non‐demented participants, we constructed Kaplan–Meier curve and multivariable Cox proportional hazards regression in Model 1. In this section, baseline ALPS index was modeled as a continuous variable and categorized by tertiles (thresholds in ADNI: < 1.17, 1.17–1.34, ≥ 1.34; thresholds in UKB: < 1.30, 1.30–1.41, ≥ 1.41). The time variable was defined in the ADNI as the time to progression to MCI or AD dementia, or the maximum follow‐up duration. In the UKB, the time variable was measured as the time from the first imaging visit to the first AD diagnosis, death, or until the last recorded date (September 2023), whichever came first.
After the preliminary result and previous studies, 39 mediation analyses based on structural equation modeling were further used to investigate the directional dependencies among the ALPS index, amyloid (CSF Aβ42 and Aβ PET), neurodegeneration (AD signature ROI volume, hippocampal volume, or FDG PET), and cognitive function (PACC; implemented in the R package lavaan 0.6‐1.1). We first constructed simple mediation models to specify the mediation associations between ALPS index and AD biomarkers. Subsequently, three multiple mediation models including ALPS index, AD biomarkers, and cognition were constructed to examine the potential pathological pathways by which glymphatic function, assessed by the ALPS index, affects cognition (see eMethods in supporting information for more details). Covariates comprised age, sex, education, and APOE ε4 carrier status (Model 1).
In sensitivity analyses, we examined the influence of white matter pathology, the assessment location, and GM atrophy on the measurements of the glymphatic system by including WMH volume fraction (WMHVF; Model 2), mean FA and MD (Model 3), assessment site (Model 4), or GM volume fraction (GMVF; Model 5) as confounding factors in addition to the covariates in Model 1. For note, Model 5 was not applied in analyses involved in brain structure measures.
3. RESULTS
3.1. Cohort characteristics
Table 1 summarizes the clinical and demographic characteristics of the study population from ADNI at baseline. Among 419 participants, 229 (54.7%) were female, 204 (48.7%) were APOE ε4 carriers (ε4 +/‐ and ε4 +/+), and the mean (±SD) of age and education were 68.5 (±4.50) and 16.3 (±2.42), respectively. Longitudinal data of ALPS index and AD variables are also illustrated in Table 1. In the whole cohort, the ALPS index correlated with age (Spearman ρ = −0.253, P = 1.81×10−7), sex (median, 1.30 for females and 1.20 for males; P = 1.38×10−7), mean FA (ρ = 0.203, P = 3.63×10−5) and MD (ρ = −0.276, P = 1.11×10−8), WMHVF (ρ = −0.200, P = 8.20×10−5), and GMVF (ρ = 0.260, P = 1.94×10−7), but not with education (ρ = −0.089, P = 0.070) and APOE ε4 carrier status (P = 0.222). In addition, we tested whether the assessment location influence the ALPS index by using ANCOVA controlling for age, sex, education, and APOE ε4 genotype. The results showed a significant difference in the ALPS index across different assessment sites (F‐value = 1.477, P = 0.027). A general scheme of the current study is depicted in Figure 1.
TABLE 1.
Characteristics of the study population in ADNI.
| CN | MCI | AD dementia | Total | ||
|---|---|---|---|---|---|
| (N = 235) | (N = 137) | (N = 47) | (N = 419) | P value | |
| Age (years) | 68.7 (±3.85) | 68.2 (±5.17) | 68.0 (±5.36) | 68.5 (±4.50) | 0.980 |
| Female | 150 (63.8%) | 54 (39.4%) | 25 (53.2%) | 229 (54.7%) | 2.95×10−5 |
| Education (years) | 16.7 (±2.27) | 16.1 (±2.54) | 15.5 (±2.57) | 16.3 (±2.42) | 0.004 |
| APOE ε4 carrier status | |||||
| ε4−/− | 149 (63.4%) | 58 (42.3%) | 8 (17.0%) | 215 (51.3%) | 5.31×10−12 |
| ε4+/− | 82 (34.9%) | 55 (40.1%) | 30 (63.8%) | 167 (39.9%) | |
| ε4+/+ | 4 (1.7%) | 24 (17.5%) | 9 (19.1%) | 37 (8.8%) | |
| ALPS index | 1.28 (±0.187) | 1.24 (±0.198) | 1.17 (±0.143) | 1.26 (±0.189) | 3.16×10−4 |
| PACC score | 0.748 (±2.52) | −5.50 (±4.89) | −15.7 (±3.76) | −3.02 (±6.29) | <2.22×10−16 |
| ADNI‐MEM | 1.19 (±0.558) | 0.308 (±0.649) | −0.816 (±0.490) | 0.690 (±0.874) | <2.22×10−16 |
| ADNI‐EF | 1.21 (±0.799) | 0.504 (±0.869) | −0.859 (±1.07) | 0.758 (±1.07) | <2.22×10−16 |
| CSF Aβ42 (pg/ml) | 1270 (±563) | 1050 (±534) | 636 (±332) | 1110 (±568) | 2.85×10−12 |
| CSF p‐tau181 (pg/ml) | 20.2 (±8.32) | 25.3 (±13.1) | 34.3 (±14.6) | 23.7 (±11.9) | 1.34×10−8 |
| CSF t‐tau (pg/ml) | 226 (±80.8) | 266 (±117) | 344 (±130) | 254 (±108) | 4.19×10−7 |
| AV45 PET SUVR | 0.759 (±0.100) | 0.836 (±0.155) | 1.03 (±0.114) | 0.821 (±0.152) | 4.42×10−15 |
| FBB PET SUVR | 0.748 (±0.114) | 0.759 (±0.105) | 1.01 (±0.207) | 0.778 (±0.147) | 0.001 |
| AV1451 PET SUVR | 1.18 (±0.114) | 1.29 (±0.235) | 1.52 (±0.299) | 1.24 (±0.193) | 3.48×10−7 |
| FDG PET SUVR | 1.32 (±0.119) | 1.26 (±0.130) | 1.01 (±0.149) | 1.23 (±0.168) | <2.22×10−16 |
| Hippocampal volume (cm3) | 0.0235 (±0.705) | −0.494 (±0.993) | −1.76 (±0.757) | −0.321 (±0.972) | <2.22×10−16 |
| AD signature ROI volume (cm3) | 65.5 (±7.77) | 65.6 (±8.19) | 55.3 (±9.47) | 64.4 (±8.70) | 5.39×10−8 |
| WMHVF | 0.002 (±0.003) | 0.003 (±0.004) | 0.004 (±0.004) | 0.003 (±0.003) | 3.00×10−5 |
| GMVF | 0.431 (±0.021) | 0.420 (±0.026) | 0.402 (±0.025) | 0.424 (±0.025) | <2.22×10−16 |
| Mean FA | 0.243 (±0.017) | 0.237 (±0.016) | 0.228 (±0.017) | 0.239 (±0.017) | <2.22×10−16 |
| Mean MD | 0.00119 (±0.0001) | 0.00123 (±0.0001) | 0.00130 (±0.0001) | 0.00121 (±0.0001) | <2.22×10−16 |
| Longitudinal data (median, IQR, range) | |||||
| ALPS index | N = 133 | N = 89 | N = 29 | N = 251 | |
| Visits | 3 (1, 2−8) | 3 (2, 2−13) | 3 (1, 2−4) | 3 (2, 2−13) | 0.006 |
| Duration (years) | 2.1 (2, 0.45−9.5) | 2 (2, 0.21−11.11) | 1 (0.3, 0.18−2.26) | 2 (2, 0.2−11.1) | 1.68×10−8 |
| CSF Aβ42 | N = 77 | N = 31 | N = 6 | N = 114 | |
| Visits | 2 (1, 2−6) | 2 (1, 2−6) | 2 (0, 2−2) | 2 (1, 2−6) | 0.066 |
| Duration (years) | 2.3 (2, 1.95−9.92) | 3 (2, 1.77−11.11) | 2 (0.1, 1.95−2.26) | 2.2 (2, 1.8−11.1) | 0.096 |
| CSF p‐tau181 | N = 77 | N = 28 | N = 5 | N = 110 | |
| Visits | 2 (1, 2−6) | 3 (1, 2−6) | 2 (0, 2−2) | 2 (1, 2−6) | 0.042 |
| Duration (years) | 2.3 (2, 1.95−9.92) | 3.8 (2, 1.97−11.11) | 2 (0.1, 1.95−2.26) | 2.3 (2, 2−11.1) | 0.067 |
| CSF t‐tau | N = 78 | N = 28 | N = 5 | N = 111 | |
| Visits | 2 (1, 2−6) | 2.5 (1, 2−6) | 2 (0, 2−2) | 2 (1, 2−6) | 0.067 |
| Duration (years) | 2.3 (2, 1.95−9.92) | 3.3 (2, 1.97−11.11) | 2 (0.1, 1.95−2.26) | 2.3 (2, 2−11.1) | 0.087 |
| AV45 PET imaging | N = 118 | N = 64 | N = 7 | N = 189 | |
| Visits | 3 (2, 2−6) | 2.5 (1, 2−6) | 2 (0, 2−2) | 3 (2, 2−6) | 0.010 |
| Duration (years) | 4 (4.3, 1.54−9.92) | 3.8 (2, 1.72−9.98) | 2 (0, 1.95−2.26) | 4 (3.9, 1.5−10) | 5.65×10−4 |
| AV1451 PET imaging | N = 97 | N = 26 | N = 10 | N = 133 | |
| Visits | 2 (1, 2−4) | 2.5 (1, 2−6) | 2 (1, 2−3) | 2 (1, 2−6) | 0.605 |
| Duration (years) | 4 (3.1, 0.97−9.92) | 2.9 (5.7, 0.88−11.11) | 1.6 (1, 0.95−2.34) | 4 (3.2, 0.9−11.1) | 5.41×10−4 |
| FDG PET imaging | N = 22 | N = 28 | N = 4 | N = 54 | |
| Visits | 2 (0, 2−3) | 2 (1, 2−3) | 2 (0, 2−2) | 2 (0, 2−3) | 0.135 |
| Duration (years) | 2 (0.1, 1.76−6.58) | 2 (3.9, 1.77−6.98) | 2 (0, 1.95−2.03) | 2 (1.9, 1.8−7) | 0.222 |
| Structural MRI | N = 157 | N = 97 | N = 30 | N = 284 | |
| Visits | 3 (2, 2−8) | 3 (3, 2−11) | 3 (1, 2−4) | 3 (2, 2−11) | 4.32×10−4 |
| Duration (years) | 2.2 (2.3, 0.46−9.22) | 2.9 (2.2, 0.21−9.1) | 1 (1, 0.18−2.34) | 2.2 (2.1, 0.2−9.2) | 5.61×10−10 |
Notes: Raw data were presented as mean (± standard deviation [SD]) or number (percentage, %) in tables unless otherwise noted. Data were compared using two‐tailed Kruskal–Wallis tests or chi‐square tests.
Abbreviations: Aβ, amyloid beta; AD, Alzheimer's disease; ADNI, Alzheimer's Disease Neuroimaging Initiative; APOE, apolipoprotein E; ALPS, analysis along the perivascular space; AV45, florbetapir; AV1451, flortaucipir; CN, cognitively normal; CSF, cerebrospinal fluid; EF, executive function composite score; FA, fractional anisotropy; FBB, florbetaben; FDG, fluorodeoxyglucose; GMVF, gray matter volume fraction; IQR, interquartile range; MCI, mild cognitive impairment; MD, mean diffusivity; MEM, memory composite score; MRI, magnetic resonance imaging; p‐tau181, phosphorylated tau 181; PACC, Preclinical Alzheimer's Cognitive Composite; PET, positron emission tomography; ROI, region of interest; SUVR, standard uptake value ratio; t‐tau, total tau; WMLVF, white matter hyper hyperintensity volume fraction.
3.2. Comparisons of ALPS index among different diagnostic and biological groups
After correction for age, sex, education, and APOE ε4 carrier status (Model 1), patients with AD dementia had significantly lower baseline ALPS index compared to CN controls and MCI patients after FDR correction (P FDR = 1.39×10−4 and 0.010, respectively; Figure 2A). We then compared the ALPS index between different biological diagnostic groups (Figure 2B). Baseline characteristics of the study population stratified by diagnosis and A status are shown in Table S4 in supporting information. When the A status was defined by CSF Aβ42 and Aβ PET (AV45 and FBB), patients with A+CN, A+MCI, and A+AD dementia showed significantly decreased ALPS index compared to the A–CN controls (P FDR = 0.018, 0.001, and 2.02×10−4, respectively). Patients with A+AD dementia had a lower ALPS index compared to the A+CN individuals (P FDR = 0.031). The ALPS index was also decreased in patients with A+MCI compared to patients with A–MCI subjects at the very edge of significance after multiple correction (P FDR = 0.051). When the A status was defined by CSF Aβ42 or Aβ PET (AV45 and FBB) separately, the stepwise descending trend from A–CN, A+CN, through A+MCI to A+AD dementia remained (Figure S2A‐B in supporting information). In addition, when the CN individuals were divided into the preclinical AD stages 0 to 2, participants in the stage 1, A+MCI, and A+AD dementia groups had significantly more decreased ALPS index than those in the stage 0 group (P FDR = 0.033, 3.55×10−4, 3.04×10−5, respectively; Figure 2C). The A+MCI and A+AD dementia groups also had significantly lower ALPS index than the stage 2 group (P FDR = 0.033 and 0.002, respectively).
FIGURE 2.
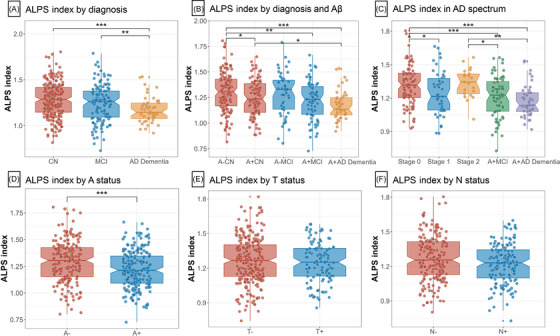
ALPS indexes by diagnosis and biological stages. The graphs display the 95% CIs around the median of raw values of each plasma biomarker. ALPS indexes were Box–Cox transformed for normalization prior to the analysis. Statistical analyses were conducted using analyses of covariance controlling for age, sex, education, and APOE ε4 carrier status (A–C). Comparing ALPS indexes in different biological groups, covariates include age, sex, education, APOE ε4 carrier status, and baseline diagnoses (D–F). A status was defined by CSF Aβ42 and amyloid PET (AV45 and FBB PET) (D). T status was defined by CSF p‐tau181 and AV1451 PET (E). N status was defined by CSF t‐tau and FDG PET (F). Significant P values after FDR corrected post hoc pairwise comparisons are marked with ***P < 0.001, *P<0.05, **P<0.01, ***P<0.001. “–” indicates negative; “+” indicates positive. A, amyloid; Aβ, amyloid beta; AD, Alzheimer's disease; ALPS, analysis along the perivascular space; APOE, apolipoprotein E; AV45, florbetapir; AV1451, flortaucipir; CI, confidence interval; CN, cognitively normal; CSF, cerebrospinal fluid; FBB, florbetaben; FDG, fluorodeoxyglucose; FDR, false discovery rate; MCI, mild cognitive impairment; N, neurodegeneration; PET, positron emission tomography; p‐tau181, phosphorylated tau 181; T, tau pathology; t‐tau, total tau.
We further examined the ALPS index in different biological groups. After correcting for age, sex, education, APOE ε4 carrier status, and baseline diagnosis, participants in the A+ group had a significantly lower ALPS index than the A– group (P = 2.92×10−4; Figure 2D), regardless of the definition of A status (Figure S2C‐D). No differences in ALPS index were observed between T± groups or N± groups (Figure 2E‐F and Figure S2E‐G). However, participants in the FDG PET defined N+ group had a significantly lower ALPS index than the corresponding N– group (P = 6.25×10−5; Figure S2H). When including WMHVF (Model 2), mean FA and MD (Model 3), assessment site (Model 4), or GMVF (Model 5) as covariates in addition to those of Model 1 in sensitivity analyses, the group difference results align with those of the primary analysis (Model 1; Table S5 in supporting information).
3.3. Association of ALPS index with core AD biomarkers
At baseline, the ALPS index was significantly correlated with Aβ biomarkers, including a positive correlation with CSF Aβ42 (β = 0.22, P = 1.85×10−5) and a negative correlation with Aβ PET burden (β = −0.20, P = 1.94×10−4; Table S6 in supporting information and Figure 3A). When participants were divided into two subgroups based on CSF Aβ42 status, a significant difference in the association of ALPS index with CSF Aβ42 was observed in the two subgroups (P for interaction = 0.003; Table S7 in supporting information), where the cross‐sectional association between ALPS index and CSF Aβ42 was maintained in the CSF A– subgroup (β = 0.32, P = 2.66×10−4) but not in the CSF A+ subgroup (β = 0.06, P = 0.434). Regarding tau biomarkers, ALPS index was negatively correlated with tau PET in the MetaROI regions (β = −0.14, P = 0.039), Braak III and IV (β = −0.14, P = 0.049), and Braak V and VI (β = −0.17, P = 0.017) regions, but not in Braak I region (β = −0.08, P = 0.241; Table S8 in supporting information). The ALPS index was also positively correlated with neuroimaging markers of neurodegeneration, that is, FDG PET, hippocampal volume, and AD signature ROI volume. There was no correlation of ALPS index with CSF p‐tau181 and CSF t‐tau levels. ,
FIGURE 3.
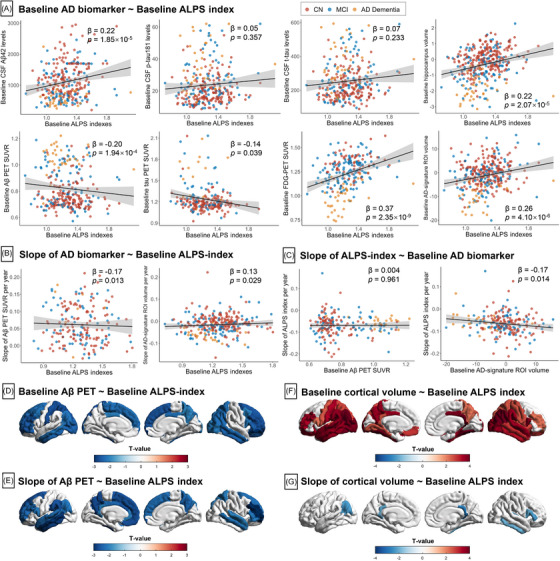
Association of ALPS indexes with core AD biomarkers. A, Cross‐sectional association between ALPS index and core AD variables. B, Prediction of longitudinal changes of core AD variables by baseline ALPS index. C, Association between baseline core AD variables and longitudinal slope of ALPS index. The points (red, CN; blue, MCI; yellow, AD dementia) and solid lines represent the individuals and regression lines, respectively. The regression coefficients (β) and P values were computed using a linear model across all participants, adjusting for age, sex, education, APOE ε4 carrier status. Association of baseline ALPS index with baseline and slope of regional Aβ PET SUVR (D–E) and cortical volume (F–G). The strength of associations with neuroimaging measures is shown in color scales representing the t value. Aβ, amyloid beta; AD, Alzheimer's disease; ALPS, analysis along the perivascular space; APOE, apolipoprotein E; CN, cognitively normal; FDG, fluorodeoxyglucose; MCI, mild cognitive impairment; p‐tau181, phosphorylated tau 181; PET, positron emission tomography; ROI, region of interest; SUVR, standard uptake value ratio; t‐tau, total tau.
Longitudinally, lower baseline ALPS indexes were related to faster rates of increase in Aβ PET burden (β = −0.17, P = 0.013) and faster rates of decrease in AD signature ROI volume (β = 0.13, P = 0.029; Figure 3B). In contrast, the baseline levels of Aβ PET burden did not predict longitudinal changes in ALPS index (Figure 3C), but higher baseline levels of AD signature ROI volume were associated with faster rates of decline in the ALPS index (β = −0.17, P = 0.014). As for tau PET, baseline ALPS index was not related to the slope of the tau PET SUVR either in the MetaROI regions (Table S6) or in different Braak regions (Table S8). In sensitivity analyses, the inclusion of WMHVF (Model 2), mean FA and MD (Model 3), assessment site (Model 4), or GMVF (Model 5) as confounding factors in addition to the covariates of Model 1 also resulted in significant longitudinal associations between ALPS index and slope of Aβ PET burden (Table S9 in supporting information).
Across 68 FreeSurfer‐defined brain regions, baseline ALPS index was negatively associated with baseline Aβ PET SUVR in 29 regions (Figure 3D). Baseline ALPS index was also negatively associated with rates of increase in Aβ PET SUVR in 26 regions (Figure 3E and Table S10 in supporting information). Regionally significant associations with both baseline and slope of Aβ PET burden included bilateral rostral middle frontal, right superior frontal, and bilateral frontal pole, and so on. As for tau PET, baseline ALPS index was negatively associated with baseline tau PET SUVR in 14 regions and with faster rates of increase in tau PET SUVR in 10 regions (Figure S3 and Table S11 in supporting information). The overlapping significant regions include left superior parietal, inferior parietal, paracentral, and so on. Regarding the regional MRI measures, baseline ALPS index was negatively correlated with cortical volume in 41 regions. The strongest associations were found in the inferior parietal region (Figure 3F and Table S12 in supporting information). Despite the significant correlation between the baseline ALPS‐index and the slope of the AD‐signature ROI volume, we observed no positive correlation between the baseline ALPS‐index and the slope of the cortical volume in any of the 68 ROI regions separately (Figure 3G).
3.4. ALPS index reduction may occur prior to Aβ and predict Aβ‐positive transition
To explore the sequential changes in glymphatic activity and Aβ biomarkers, the trajectories of ALPS index, Aβ PET deposition, and PACC score were modeled using CSF Aβ42 as a proxy for AD progression (Figure 4A). We observed that the abnormality of ALPS index prominently increased before the threshold of CSF Aβ42 positivity and then plateaued. The abnormality curves of Aβ PET SUVR and PACC score were initially flat and began to increase when the ALPS index approached the plateau (i.e., almost near the threshold for CSF Aβ42 positivity). When regarding PACC score as a surrogate for AD progression, the fitted curves for the abnormality of the ALPS index apparently increased before the fitted curves for the Aβ biomarkers (CSF Aβ42 and Aβ PET) increased markedly, then reached a plateau, and then gradually increased again (Figure 4B).
FIGURE 4.
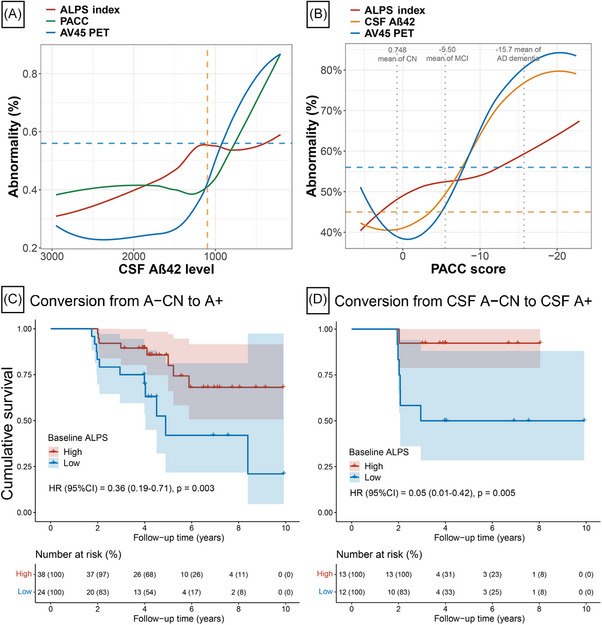
ALPS index reduction may occur prior to amyloid and predict amyloid‐positive transition. Trajectories of ALPS index, CSF Aβ42, Aβ PET (AV45 PET), and PACC score were modeled using CSF Aβ42 (A) or PACC score (B) as proxy of disease progression. The values of Aβ PET (AV45 PET) and PACC score were scaled to 0% to 100%, with 0% representing the minimal value and 100% representing the maximal value. The original ALPS index and CSF Aβ42 were reversed, with 100% representing the minimal value and 0% representing the maximal value. This normalization procedure made the higher values of abnormalities in ALPS index, CSF Aβ42, Aβ PET (AV45 PET), and PACC score represent the more severe disease state. C–D, The Kaplan–Meier curves showing cumulative probability of amyloid status progression. Results of multivariable Cox regression treating the ALPS index as a continuous variable after adjustment for age, sex, education, and APOE ε4 genotype are shown in the lower left. The table under the curve illustrates number at risk (%) at 2‐year intervals, to facilitate interpretation of the curves. A, amyloid; Aβ, amyloid beta; AD, Alzheimer's disease; APOE, apolipoprotein E; ALPS, analysis along the perivascular space; AV45, florbetapir; CN, cognitively normal; CSF, cerebrospinal fluid; CI, confidence interval; MCI, mild cognitive impairment; HR, hazard ratio; PACC, Preclinical Alzheimer's Cognitive Composite; PET, positron emission tomography.
We further investigated whether the baseline ALPS index could predict A+ conversion in baseline A–CN subjects. Among A–CN participants at baseline, when A status was defined by CSF Aβ42 or Aβ PET (AV45 and FBB PET), 19 (30.6%) participants converted to the corresponding A+ status within the median (interquartile range [IQR]) follow‐up period of 4.17 years (3.98–5.80), and 43 remained A– status after at least 3 years of follow‐up. A total of 39 CN participants in the A– group did not convert to A+ status during follow‐up, but were followed for < 3 years and were not included in the non‐converter group. Subjects who converted to A+ status had a significantly lower ALPS index at baseline compared to non‐converters (P = 0.016; Figure S4 in supporting information). In A–CN participants, those with higher ALPS index at baseline had a reduced risk of converting to A+ status (hazard ratio [HR; 95% confidence interval (CI)] = 0.36 [0.19–0.71], P = 0.003 when ALPS index was considered a continuous variable; HR [95% CI] = 0.27 [0.09–0.84], P = 0.024 when ALPS index was considered a binary variable; Figure 4C and Figure S5). Significant results remained when the A status was defined by either CSF Aβ42 levels or Aβ PET (AV45 and FBB PET) separately (Figure 4D and Figure S4‐5). Sensitivity analyses yielded similar results for the ALPS index in predicting Aβ‐positive transition as in the primary analysis (Table S13 in supporting information).
3.5. ALPS index predicts clinical progression in AD
In ADNI, among baseline non‐demented participants in the whole cohort, 45 (23.0%) participants experienced clinical progression (including 22 participants who progressed from CN to MCI, 20 participants who progressed from MCI to AD dementia, and three participants who progressed from CN to AD dementia) over a median (IQR) follow‐up of 4.12 years (3.92–6.23), and 151 participants remained clinically stable after at least 3 years of follow‐up. A total of 98 non‐demented participants did not show clinical progression during follow‐up, but were followed for < 3 years and were not included in the non‐converters group. Subjects who converted to MCI or AD dementia had a significantly lower ALPS index at baseline compared to non‐converters (P = 0.028; Figure S6 in supporting information). In the multivariable Cox regression, after the allowance for covariates in Model 1, we found that a higher baseline ALPS index was significantly associated with a lower risk of progression to MCI or AD dementia when the ALPS index was treated as a continuous variable (HR [95% CI] = 0.64 [0.47–0.88], P = 0.006; Figure 5A) or categorized by tertile (HR [95% CI] = 0.24 [0.11–0.56], P = 9.67×10−4 for high vs. low baseline ALPS group; Figure S7 in supporting information). These analyses were then repeated in baseline CN participants, and the results remained similar. A higher baseline ALPS index was significantly associated with a lower risk of progression from CN to MCI or AD dementia when treated as a continuous variable (HR [95% CI] = 0.54 [0.34–0.88], P = 0.013; Figure 5B) or categorized by tertile (HR [95% CI] = 0.32 [0.11–0.96], P = 0.043 for median vs. low baseline ALPS group, and (HR [95% CI] = 0.20 [0.06–0.62], P = 0.005 for high vs. low baseline ALPS group; Figure S7). In addition, the associations between ALPS index and risk of clinical progression remained significant in sensitivity analyses (Table S14 in supporting information).
FIGURE 5.
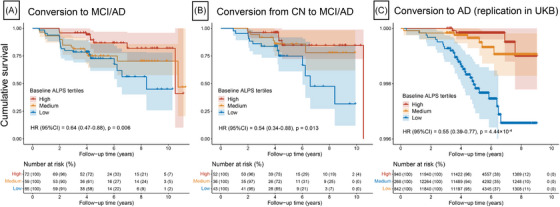
Associations of ALPS indexes with risk of clinical progression. The Kaplan–Meier curves of clinical progression to MCI/AD dementia in non‐demented participants (A), progression to MCI/AD dementia in CN participants (B), or progression to AD in UKB participants (C). Results of multivariable Cox regression treating the ALPS index as a continuous variable after adjustment for age, sex, education, and APOE ε4 carrier status are shown in the lower left. The table under the curve illustrates number at risk (%) at 2‐year intervals, to facilitate interpretation of the curves. AD, Alzheimer's disease; ALPS, analysis along the perivascular space; APOE, apolipoprotein E; CN, cognitively normal; CI, confidence interval; MCI, mild cognitive impairment; HR, hazard ratio; UKB, UK Biobank.
3.6. Replication for research of clinical progression
In the UKB, among the 36,050 non‐demented participants included in the first imaging visit (which was considered the baseline for our analysis), 40 incident AD events were recorded during a median (IQR) follow‐up period of 5.34 years (4.54–6.83; Table S15 in supporting information). The significant associations of the ALPS index with risk of clinical progression were successfully replicated in the UKB cohort. Participants with clinical progression had a significantly lower baseline ALPS index than non‐converters (P = 1.88×10−4; Figure S6). Participants with a higher baseline ALPS index had a lower risk of AD when the ALPS index was either treated as a continuous variable (HR [95% CI] = 0.55 [0.39–0.77], P = 4.44×10−4; Figure 5C) or categorized by tertile (HR [95% CI] = 0.26 [0.09–0.76], P = 0.013 for high vs. low baseline ALPS group; HR [95% CI] = 0.35 [0.15–0.81], P = 0.014 for median vs. low baseline ALPS group; Figure S7).
3.7. Association of ALPS index with cognitive function
Figure 6 shows the cross‐sectional and longitudinal associations of the ALPS index with PACC, memory, and EF composite scores. At baseline, all three of these cognitive tests were positively associated with baseline ALPS index (Figure 6A). Longitudinally, a higher baseline ALPS index was significantly associated with faster rates of decline in PACC (β = 0.19, P = 4.24×10−4), memory (β = 0.11, P = 0.038), and EF composite score (β = 0.16, P = 0.005; Figure 6B). In contrast, the baseline PACC, memory, and EF composite scores did not predict longitudinal changes in the ALPS index (Figure 6C). In sensitivity analyses, the longitudinal associations between baseline ALPS index and slope of decline in PACC were still statistically significant in all three sensitivity models (Table S16 in supporting information).
FIGURE 6.
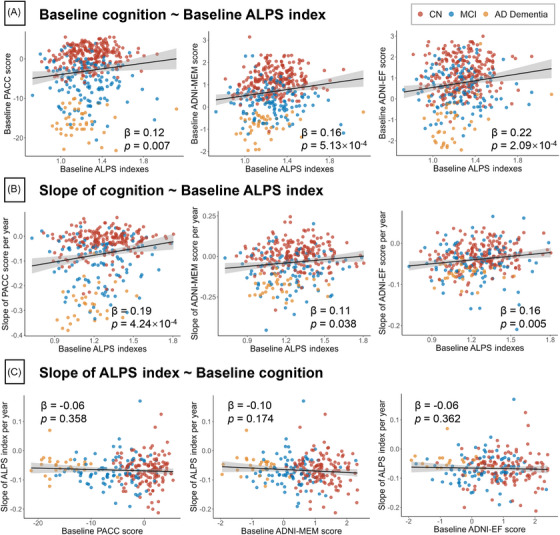
Associations of ALPS indexes with cognitive performance. A, Cross‐sectional association between ALPS and cognitive performance. B, Association between baseline ALPS and longitudinal slope of cognitive performance. C, Association between baseline cognitive performance and longitudinal slope of ALPS. The points (red, CN; blue, MCI; yellow, AD dementia) and solid lines represent the individuals and regression lines, respectively. The regression coefficients (β) and P values were computed using a linear model across all participants, adjusting for age, sex, education, APOE ε4 carrier status. AD, Alzheimer's disease; ALPS, analysis along the perivascular space; APOE, apolipoprotein E; CN, cognitively normal; EF, executive function composite score; PACC, Preclinical Alzheimer's Cognitive Composite; MCI, mild cognitive impairment; MEM, memory composite score.
3.8. Interaction of ALPS index with amyloid and neurodegeneration on cognitive decline
Based on the significant association of ALPS index with Aβ biomarkers (CSF Aβ42 and Aβ PET), neurodegeneration markers (AD signature ROI volume, hippocampal volume, and FDG PET), and cognitive function, we then tested whether the ALPS index had an interaction effect with these biomarkers in predicting longitudinal cognitive decline as measured by the PACC score. We found that baseline ALPS index significantly interacted with baseline CSF Aβ42 (β = −0.26, P = 0.027), AD signature ROI volume (β = −0.35, P = 0.001), hippocampal volume (β = −0.21, P = 0.026), and FDG PET SUVR (β = −0.30, P = 0.017) in predicting longitudinal cognitive decline, showing that individuals with low ALPS index had more pronounced positive associations of CSF Aβ42, AD signature ROI volume, hippocampal volume, and FDG PET with slopes of PACC scores than individuals with high ALPS index (Figure S8 in supporting information). Baseline ALPS index interacted with baseline Aβ PET SUVR with a strong trend toward statistical significance (β = 0.20, P = 0.051), indicating that negative associations of Aβ PET SUVR with slopes of PACC scores may be modified by ALPS index. However, baseline ALPS index did not show an interaction association with the slope of any of these biomarkers in predicting cognitive decline.
3.9. Mediation among ALPS index, amyloid, and neurodegeneration
The AD signature ROI volume is a robust neurodegeneration marker associated with the ALPS index and was therefore selected in the following simple mediation models, namely (1) ALPS → CSF Aβ42 → Aβ PET → AD signature ROI volume, (2) ALPS → CSF Aβ42 → Aβ PET, (3) ALPS → CSF Aβ42 → AD signature ROI volume, and (4) ALPS → Aβ PET → AD signature ROI volume (Figure S9A in supporting information). Results of the first model showed that ALPS index had a significant positive effect on the AD signature ROI volume (β = 0.284, P = 2.99×10−6), ALPS index was associated with CSF Aβ42 (β = 0.192, P = 0.009) and CSF Aβ42 was associated with Aβ PET (β = −0.563, P = 2.27×10−11), and in addition, Aβ PET was associated with brain volume (β = −0.273, P = 0.001). The indirect pathway of the effect of ALPS index on AD signature ROI volume via CSF Aβ42 and Aβ PET nearly reached a significant level (path β = 0.030, P = 0.071). CSF Aβ42 was a significant mediator for the association of ALPS index with Aβ PET (path β = 0.105, P = 0.027) or AD signature ROI volume (path β = 0.059, P = 0.033). Aβ PET significantly mediated the association between ALPS index and AD signature ROI volume (path β = 0.050, P = 0.018). When the hippocampal volume or FDG PET was used to represent neurodegeneration instead of AD signature ROI volume, the indirect pathway of the association of ALPS index on hippocampal volume (path β = 0.079, P = 0.003) or FDG PET (path β = 0.071, P = 0.025) via Aβ PET were both significant (Figure S10 in supporting information).
3.10. Potential pathological pathways by which ALPS index affects cognition
In the multiple mediation model comprised of baseline ALPS index, Aβ PET, AD signature ROI volume, and slope of PACC score (Figure 7), ALPS index was significantly associated with Aβ PET (β = −0.162, P = 0.014), and AD signature ROI volume (β = 0.268, P = 5.80×10−4). ALPS index showed no direct effect on the slope of PACC score (β = −0.028, P = 0.627), but its indirect effect was significant (β = 0.197, P = 3.85×10−4). The indirect pathway of the effect of ALPS index on the slope of PACC score via Aβ PET (path β = 0.079, P = 0.020) or AD signature ROI volume (path β = 0.100, P = 0.007) was significant. The proportion mediated by Aβ PET or AD signature ROI volume of the total mediation effect was 40.1% and 50.8%, respectively. There were no significant differences between the effects mediated by Aβ PET or AD trait ROI volume (P = 0.691). Two additional hypothetical structural equation models are shown in Figure S9B‐C, both indicating that the ALPS index indirectly affects cognitive performance via Aβ pathology or brain volume. Results of the corresponding models when hippocampal volume or FDG PET was used to represent neurodegeneration are shown in Figure S10 in supporting information. The sensitivity models yielded consistent results with the primary model, as shown in Figure S11 in supporting information.
FIGURE 7.
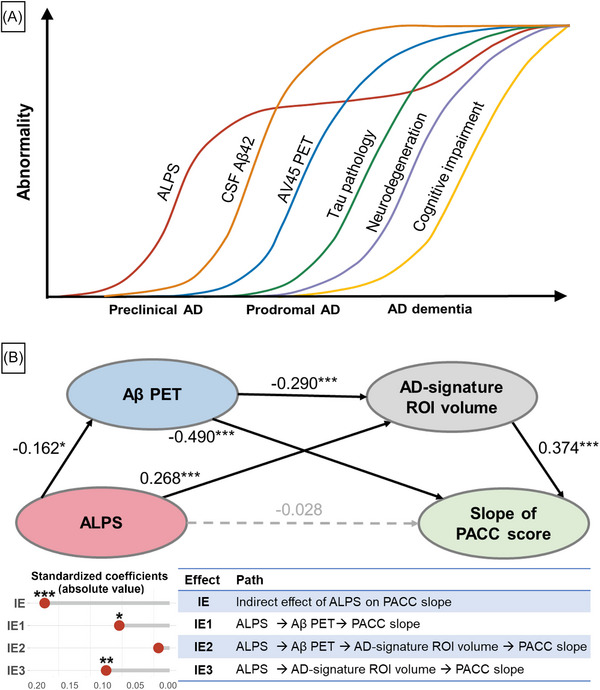
ALPS index predicts cognition change through amyloid and neurodegeneration. A, A model integrating the findings in the present study, together with previous studies, 39 depicts an approximative order of ALPS and AD core biomarkers in the AD continuum. B, Mediation analysis based on structural equation modeling. We computed the standardized coefficients (β) of each association. P values are visualized with *P<0.05, **P<0.01, ***P<0.001. The horizontal gray line in lollipop chart represents the absolute value of β, and the color of the circle represents the positive (orange) or negative (blue) direction of β. Aβ, amyloid beta; AD, Alzheimer's disease; ALPS, analysis along the perivascular space; AV45, florbetapir; CSF, cerebrospinal fluid; PACC, preclinical Alzheimer cognitive composite; IE, indirect effect; PET, positron emission tomography; ROI, region of interest.
4. DISCUSSION
In the current study, we used the ALPS index representing glymphatic activity and investigated its cross‐sectional and longitudinal relationships with clinical and pathological features of AD. We observed a significantly reduced ALPS index in AD dementia, and in the preclinical and prodromal stages of AD. Lower ALPS index predicts accelerated Aβ PET burden and AD signature ROI volume, higher risk of amyloid‐positive transition and clinical progression, and faster cognitive decline. The associations of ALPS index with cognitive decline were fully mediated by Aβ PET and brain atrophy. Synthesizing our and former findings, 39 one hypothetical cascade model of AD can be inferred (Figure 7A). Our findings suggest that glymphatic impairment indicated by the ALPS index may occur before significant Aβ deposits, and predict amyloid deposition, neurodegeneration, and clinical progression in AD.
The ALPS index is reduced in AD patients, 15 , 16 , 17 but few studies have examined it at different biological stages of AD. Our results filled the knowledge gap by showing that ALPS index decreased as early as the preclinical and prodromal stages of AD compared to A–CN controls, suggesting that glymphatic failure or impairment may occur in the early stages of AD. Moreover, glymphatic dysfunction may even occur prior to amyloid pathology. Using the PACC score as a proxy for disease progression, we observed that changes in glymphatic activity were present before the increase in Aβ PET burden and even before CSF Aβ42 reached the positive threshold (Figure 4B). Nevertheless, a recent study yielded inconsistent results. Using the Consortium to Establish a Registry for Alzheimer's Disease Neuropsychological Assessment Battery (CERAD‐NAB) total score as a surrogate for AD progression in a non‐linear curve‐fitting model, they suggested that the accelerating turning point of ALPS index abnormality might occur after the changes in Aβ deposition. 15 Different findings can result from various reasons. One possible explanation is that although both this previous work and ours used cognitive score as a proxy for disease progression to visualize the sequential changes of ALPS index and Aβ biomarkers in AD, 15 we used the PACC score, which is sensitive to cognitive decline in preclinical AD. 33 , 40 Our analysis can therefore more sensitively capture the sequential relationship between Aβ biomarkers and ALPS index in the early stages of AD, as the CERAD‐NAB total score is highly sensitive to cognitive decline in the late stages of AD. 41 , 42 From the perspective of late stages of AD, however, ours corroborates previous findings. 15 The abnormalities in the ALPS index lagging Aβ deposition observed by Hsu et al. may reflect the further impairment of the glymphatic system caused by Aβ deposition in late stages of AD. 15 Consistently, we found that the abnormality of the ALPS index gradually increased during the late stages of AD after the plateau, at which point Aβ pathology had accumulated to a certain level. Moreover, previous animal studies revealed that long‐term exposure to Aβ pathology can further compromise the glymphatic system. 43 Nonetheless, it cannot be ruled out that white matter damage unrelated to glymphatic function could be responsible for the alterations in the ALPS index after the plateau.
Another explanation for the discrepancy between our study and the previous one is that our study provides multiple longitudinal evidences supporting the determinant role of glymphatic activity in Aβ deposition. This strengthens the reliability of our conclusion compared to the previous cross‐sectional study. 15 We found that a lower baseline ALPS index predicted an accelerated rate of increase in Aβ PET burden with adjustment for age, sex, education, and APOE ε4 carrier status. Even after further correction for white matter pathology, this finding remained robust. In contrast, Aβ PET showed no apparent influence on the rate of ALPS index decline. When examined in 68 cortical regions, the atlas of cortical regions associated with ALPS index in cross‐sectional and longitudinal Aβ PET closely resembled those of brain regions considered to be particularly sensitive to early Aβ accumulation (i.e., posterior cingulate, superior frontal, and rostral middle frontal lobes). 44 , 45 This suggests that glymphatic activity may influence the progression of Aβ deposition in the early stages of AD. Additionally, lower baseline ALPS index predicted a higher risk of Aβ status progression, especially the CSF Aβ42 status progression. Together, one of the most critical findings of this study is that glymphatic failure may precede and contribute to the development of brain parenchymal Aβ deposits. This is concordant with animal studies demonstrating that glymphatic transport is significantly reduced before extensive Aβ deposition in brains of the AD mouse model. 43 Enhanced glymphatic activity in the early period can reduce Aβ burden and improve memory in an AD mice model. 46 While validation with pathophysiological studies is needed, our findings provide clues to the timing of interventions for the prevention and treatment of AD that target the glymphatic system.
In addition to Aβ biomarkers, the ALPS index predicted accelerated rates of brain atrophy in AD signature ROI regions, representing neurodegeneration downstream of Aβ in AD. 2 An interesting finding is that higher baseline levels of AD signature ROI volume were associated with faster decline in the ALPS index with adjustment for age, sex, education, and APOE ε4 carrier status. One possible explanation is that the decline of the ALPS index precedes the brain atrophy and exhibits early acceleration followed by deceleration. Therefore, in the advanced stages of AD with a certain degree of brain atrophy, glymphatic activity may have already undergone a period of rapid decline, while brain atrophy continues to change substantially. There is no proposed mechanism whereby impaired glymphatic function directly affects brain structure. Yet, our mediation analysis suggests that glymphatic activity may predict brain atrophy by affecting amyloid in AD. This is theoretically justifiable because the Aβ burden in early stages of AD is highly correlated with brain atrophy, and downstream pathologic events of Aβ accumulation may be responsible for the ongoing atrophy as disease progresses. 47
Consistent with previous study, we observed an association between the ALPS index and tau PET burden, 15 particularly pronounced in Braak III and VI regions. However, the longitudinal association was insignificant. The weaker ALPS associations with tau than amyloid may be attributed to the intracellular nature of AD tau pathology. Recent studies suggest that the extracellular space, which is cleared by the glymphatic system, serves as a conduit for the neuron‐to‐neuron propagation. 48 , 49 Therefore, a possible explanation of our findings is that the removal of extracellular tau may not be sufficient to affect tau deposition, but it may contribute somewhat to the spreading of tau throughout the brain.
Despite the mounting cross‐sectional evidence linking the glymphatic system to cognitive performance in AD, 10 , 15 , 16 , 50 we present the first preliminary evidence that lower ALPS index predicts faster cognitive decline and higher risk of clinical progression in AD adjusting for age, sex, education, and APOE ε4 carrier status. And the effects of white matter pathology or GM atrophy on the ALPS index were not sufficient to shake our main conclusions. Because our findings support that ALPS index alterations occur before apparent Aβ pathology and neurodegeneration, both of which contribute to cognitive dysfunction in AD, 2 , 39 we further examined the potential pathological pathways by which the ALPS index affects cognitive decline. Our study supports the previous finding that brain volume fully mediates the relationship between ALPS index and cognition. 50 Nonetheless, our novel contributions lie in the longitudinal design, the inclusion of Aβ biomarkers, and the use of the multiple mediation model. We found that the glymphatic function, assessed by the ALPS index, may protect against AD‐related cognitive decline mediated not only by brain volume but also by Aβ burden. And the mediation effects of the two were not significantly different. Different from the simple mediation model, we obtained the mediating effect of Aβ burden (or brain volume) in the association between the ALPS index and cognition controlling for brain volume (or Aβ burden) by constructing a multiple mediation model. This could explain why the beta of the indirect effect is not that high in our study.
This is a pioneering study investigating the sequential associations of the ALPS index with AD biomarkers and their influence on cognitive decline in large‐scaled and longitudinal cohorts, which provides novel insights into the underlying mechanism and role of glymphatic activity in AD. However, several caveats should be noted. First, the efficacy of the ALPS index in detecting human glymphatic function has not yet been substantially and rigorously validated by pathophysiological studies. While the ALPS index has been confirmed by glymphatic MRI 11 and has been extensively studied in various diseases, 14 , 15 , 51 it should be interpreted with caution. Second, the ALPS index was used to assess the global glymphatic activity and cannot reflect regional glymphatic dysfunction. Given the regional heterogeneity of glymphatic activity and the potentially heterogeneous effects of aggregated protein toxicity throughout the brain, studies investigating regional glymphatic function in AD‐susceptible networks are needed. Third, caution should be taken when interpreting the temporal sequence of glymphatic dysfunction and amyloid pathology. Although the trajectory plot with cognitive scores as proxy is an intuitive indication of that the ALPS index reduction may occur before Aβ deposition, this single evidence is cross‐sectional and may not accurately reflect the exact sequence of events. When interpreting the findings of this paper, it is important to note that our research conclusion is based not only on cross‐sectional findings but also on multiple longitudinal evidence. Besides, the predictive effect of the ALPS index on amyloid and brain atrophy is clearly demonstrated here, but we cannot definitively conclude their causal relationship from an observational cohort. Moreover, due to the lack of MCI diagnostic records and AD biomarker data in UKB, replication was not possible for some analyses.
5. CONCLUSIONS
In summary, we used the ALPS index to investigate the glymphatic system and suggested that the ALPS index reduction parallels CSF Aβ42 levels and may occur prior to amyloid pathology and neurodegeneration in the early stages of AD. This study demonstrates for the first time the predictive effect of the ALPS index on amyloid deposition, brain atrophy, clinical progression, and cognitive decline in AD, and the mediating effect of amyloid and neurodegeneration on the glymphatic–cognitive decline association. These findings extend the understanding of the relationships among glymphatic activity, AD pathology, and cognitive decline, and provide evidence for targeting the glymphatic system at an early stage to diminish glymphatic dysfunction and subsequent amyloid deposition, neurodegenerative process, and cognitive impairment in AD.
AUTHOR CONTRIBUTIONS
Jin‐Tai Yu had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Conceptualization: Jin‐Tai Yu; methodology: Shu‐Yi Huang, Ya‐Ru Zhang, Yu Guo; acquisition, analysis, or interpretation of data: all authors; writing—original draft: Shu‐Yi Huang, Jing Du; critical revision of the manuscript for important intellectual content: Shu‐Yi Huang, Ya‐Ru Zhang, Yu Guo, Jin‐Tai Yu; data curation and formal analysis: Shu‐Yi Huang, Jing Du, Peng Ren, Bang‐Sheng Wu; obtained funding: Jin‐Tai Yu; administrative, technical, or material support: Jian‐Feng Feng, Wei Cheng, Jin‐Tai Yu; all authors read and approved the final manuscript.
CONFLICTS OF INTEREST STATEMENT
The authors declare no conflict of interest related to this work. Author disclosures are available in the supporting information.
CONSENT STATEMENT
ADNI was approved by the institutional review boards of all participating institutions. All ADNI participants provided written informed consent according to the Declaration of Helsinki before study enrollment. The UK Biobank has research tissue bank approval from the North West Multi‐centre Research Ethics Committee (Ref 21/NW/0157) (https://www.ukbiobank.ac.uk/learn‐more‐about‐uk‐biobank/about‐us/ethics) and provided oversight for this study. All UK Biobank participants provided electronic informed consent at baseline assessment.
Supporting information
Supporting information
Supporting information
ACKNOWLEDGMENTS
The authors want to acknowledge the study participants and all the investigators involved in the Alzheimer's Disease Neuroimaging Initiative (ADNI) and UK Biobank for their helpful contributions to data collection. Data used in preparation for this paper were obtained from the ADNI database (http://adni.loni.usc.edu/). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf. Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie; Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC; Johnson & Johnson Pharmaceutical Research & Development LLC; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California. This study used the UK Biobank Resource under application number 19542. This study was supported by grants from the Science and Technology Innovation 2030 Major Projects (2022ZD0211600), the National Natural Science Foundation of China (82071201, 82071997), Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01), Research Start‐up Fund of Huashan Hospital (2022QD002), Excellence 2025 Talent Cultivation Program at Fudan University (3030277001), Shanghai Talent Development Funding for The Project (2019074), Shanghai Rising‐Star Program (21QA1408700), and ZHANGJIANG LAB, Tianqiao and Chrissy Chen Institute, and the State Key Laboratory of Neurobiology and Frontiers Center for Brain Science of Ministry of Education, Fudan University.
Huang S‐Y, Zhang Y‐R, Guo Y, et al. Glymphatic system dysfunction predicts amyloid deposition, neurodegeneration, and clinical progression in Alzheimer's disease. Alzheimer's Dement. 2024;20:3251–3269. 10.1002/alz.13789
Shu‐Yi Huang and Ya‐Ru Zhang contributed equally to the present work.
Data used in preparation for this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp‐ontent/uploads/how_to_apply/16ADNI_Acknowledgement_List.pdf.
REFERENCES
- 1. Iliff JJ, Wang M, Liao Y, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med. 2012;4:147ra11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA research framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xu Z, Xiao N, Chen Y, et al. Deletion of aquaporin‐4 in APP/PS1 mice exacerbates brain Abeta accumulation and memory deficits. Mol Neurodegener. 2015;10:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamada K, Cirrito JR, Stewart FR, et al. In vivo microdialysis reveals age‐dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci. 2011;31:13110‐13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iliff JJ, Chen MJ, Plog BA, et al. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 2014;34:16180‐16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ishida K, Yamada K, Nishiyama R, et al. Glymphatic system clears extracellular tau and protects from tau aggregation and neurodegeneration. J Exp Med. 2022;219:e20211275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Harrison IF, Ismail O, Machhada A, et al. Impaired glymphatic function and clearance of tau in an Alzheimer's disease model. Brain. 2020;143:2576‐2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nedergaard M, Goldman SA. Glymphatic failure as a final common pathway to dementia. Science. 2020;370:50‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Eide PK, Ringstad G. MRI with intrathecal MRI gadolinium contrast medium administration: a possible method to assess glymphatic function in human brain. Acta Radiol Open. 2015;4:2058460115609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taoka T, Masutani Y, Kawai H, et al. Evaluation of glymphatic system activity with the diffusion MR technique: diffusion tensor image analysis along the perivascular space (DTI‐ALPS) in Alzheimer's disease cases. Jpn J Radiol. 2017;35:172‐178. [DOI] [PubMed] [Google Scholar]
- 11. Zhang W, Zhou Y, Wang J, et al. Glymphatic clearance function in patients with cerebral small vessel disease. Neuroimage. 2021;238:118257. [DOI] [PubMed] [Google Scholar]
- 12. Cacciaguerra L, Carotenuto A, Pagani E, et al. Magnetic resonance imaging evaluation of perivascular space abnormalities in neuromyelitis optica. Ann Neurol. 2022;92:173‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Carotenuto A, Cacciaguerra L, Pagani E, Preziosa P, Filippi M, Rocca MA. Glymphatic system impairment in multiple sclerosis: relation with brain damage and disability. Brain. 2022;145:2785‐2795. [DOI] [PubMed] [Google Scholar]
- 14. Si X, Guo T, Wang Z, et al. Neuroimaging evidence of glymphatic system dysfunction in possible REM sleep behavior disorder and Parkinson's disease. NPJ Parkinsons Dis. 2022;8:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hsu JL, Wei YC, Toh CH, et al. Magnetic resonance images implicate that glymphatic alterations mediate cognitive dysfunction in Alzheimer disease. Ann Neurol. 2023;93:164‐174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamagata K, Andica C, Takabayashi K, et al. Association of MRI indices of glymphatic system with amyloid deposition and cognition in mild cognitive impairment and Alzheimer disease. Neurology. 2022;99:e2648‐2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Steward CE, Venkatraman VK, Lui E, et al. Assessment of the DTI‐ALPS parameter along the perivascular space in older adults at risk of dementia. J Neuroimaging. 2021;31:569‐578. [DOI] [PubMed] [Google Scholar]
- 18. Ota M, Sato N, Nakaya M, et al. Relationships between the deposition of amyloid‐β and tau protein and glymphatic system activity in Alzheimer's disease: diffusion tensor image study. J Alzheimers Dis. 2022;90:295‐303. [DOI] [PubMed] [Google Scholar]
- 19. Petersen RC, Aisen PS, Beckett LA, et al. Alzheimer's disease neuroimaging initiative (ADNI): clinical characterization. Neurology. 2010;74:201‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sudlow C, Gallacher J, Allen N, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bittner T, Zetterberg H, Teunissen CE, et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of beta‐amyloid (1‐42) in human cerebrospinal fluid. Alzheimers Dement. 2016;12:517‐526. [DOI] [PubMed] [Google Scholar]
- 22. Schindler SE, Gray JD, Gordon BA, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimers Dement. 2018;14:1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Meyer PF, Pichet Binette A, Gonneaud J, Breitner JCS, Villeneuve S. Characterization of Alzheimer disease biomarker discrepancies using cerebrospinal fluid phosphorylated tau and AV1451 positron emission tomography. JAMA Neurol. 2020;77:508‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen SD, Huang YY, Shen XN, et al. Longitudinal plasma phosphorylated tau 181 tracks disease progression in Alzheimer's disease. Transl Psychiatry. 2021;11:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mattsson N, Cullen NC, Andreasson U, Zetterberg H, Blennow K. Association between longitudinal plasma neurofilament light and neurodegeneration in patients with Alzheimer disease. JAMA Neurol. 2019;76:791‐799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moscoso A, Grothe MJ, Ashton NJ, et al. Longitudinal associations of blood phosphorylated tau181 and neurofilament light chain with neurodegeneration in Alzheimer disease. JAMA Neurol. 2021;78:396‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jack CR Jr, Wiste HJ, Weigand SD, et al. Different definitions of neurodegeneration produce similar amyloid/neurodegeneration biomarker group findings. Brain. 2015;138:3747‐3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Alfaro‐Almagro F, Jenkinson M, Bangerter NK, et al. Image processing and quality control for the first 10,000 brain imaging datasets from UK Biobank. Neuroimage. 2018;166:400‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He P, Shi L, Li Y, et al. The association of the glymphatic function with Parkinson's disease symptoms: neuroimaging evidence from longitudinal and cross‐sectional studies. Ann Neurol. 2023;94:672‐683. [DOI] [PubMed] [Google Scholar]
- 30. Palmqvist S, Mattsson N, Hansson O. Alzheimer's Disease Neuroimaging I. Cerebrospinal fluid analysis detects cerebral amyloid‐beta accumulation earlier than positron emission tomography. Brain. 2016;139:1226‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Desikan RS, Segonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968‐980. [DOI] [PubMed] [Google Scholar]
- 32. Landau SM, Harvey D, Madison CM, et al. Comparing predictors of conversion and decline in mild cognitive impairment. Neurology. 2010;75:230‐238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Donohue MC, Sperling RA, Salmon DP, et al. The preclinical Alzheimer cognitive composite: measuring amyloid‐related decline. JAMA Neurol. 2014;71:961‐970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gibbons LE, Carle AC, Mackin RS, et al. A composite score for executive functioning, validated in Alzheimer's Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 2012;6:517‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Crane PK, Carle A, Gibbons LE, et al. Development and assessment of a composite score for memory in the Alzheimer's Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav. 2012;6:502‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:280‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dubois B, Feldman HH, Jacova C, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG‐2 criteria. Lancet Neurol. 2014;13:614‐629. [DOI] [PubMed] [Google Scholar]
- 38. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7:263‐269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Jack CR Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mormino EC, Papp KV, Rentz DM, et al. Early and late change on the preclinical Alzheimer's cognitive composite in clinically normal older individuals with elevated amyloid beta. Alzheimers Dement. 2017;13:1004‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seo EH, Lee DY, Lee JH, et al. Total scores of the CERAD neuropsychological assessment battery: validation for mild cognitive impairment and dementia patients with diverse etiologies. Am J Geriatr Psychiatry . 2010;18:801‐809. [DOI] [PubMed] [Google Scholar]
- 42. Rossetti HC, Munro Cullum C, Hynan LS, Lacritz LH. The CERAD neuropsychologic battery total score and the progression of Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24:138‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Peng W, Achariyar TM, Li B, et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer's disease. Neurobiol Dis. 2016;93:215‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Villeneuve S, Rabinovici GD, Cohn‐Sheehy BI, et al. Existing Pittsburgh Compound‐B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain. 2015;138:2020‐2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jagust WJ, Landau SM, Alzheimer's Disease Neuroimaging Initiative . Temporal dynamics of beta‐amyloid accumulation in aging and Alzheimer disease. Neurology. 2021;96:e1347‐e1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee Y, Choi Y, Park EJ, et al. Improvement of glymphatic‐lymphatic drainage of beta‐amyloid by focused ultrasound in Alzheimer's disease model. Sci Rep. 2020;10:16144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chételat G, Villemagne VL, Bourgeat P, et al. Relationship between atrophy and β‐amyloid deposition in Alzheimer disease. Ann Neurol . 2010;67:317‐324. [DOI] [PubMed] [Google Scholar]
- 48. Lewis J, Dickson DW. Propagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016;131:27‐48. [DOI] [PubMed] [Google Scholar]
- 49. Medina M, Avila J. The role of extracellular Tau in the spreading of neurofibrillary pathology. Front Cell Neurosci. 2014;8:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chang HI, Huang CW, Hsu SW, et al. Gray matter reserve determines glymphatic system function in young‐onset Alzheimer's disease: evidenced by DTI‐ALPS and compared with age‐matched controls. Psychiat Clin Neurosci. 2023;77:401‐409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jiang D, Liu L, Kong Y, et al. Regional glymphatic abnormality in behavioral variant frontotemporal dementia. Ann Neurol. 2023;94:442‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information
Supporting information