

Abstract
INTRODUCTION
Degradation of fractal patterns in actigraphy independently predicts dementia risk. Such observations motivated the study to understand the role of fractal regulation in the context of neuropathologies.
METHODS
We examined associations of fractal regulation with neuropathologies and longitudinal cognitive changes in 533 older participants who were followed annually with actigraphy and cognitive assessments until death with brain autopsy performed. Two measures for fractal patterns were extracted from actigraphy, namely, α 1 (representing the fractal regulation at time scales of <90 min) and α 2 (for time scales 2 to 10 h).
RESULTS
We found that larger α 1 was associated with lower burdens of Lewy body disease or cerebrovascular disease pathologies; both α 1 and α 2 were associated with cognitive decline. They explained an additional significant portion of the variance in the rate of cognitive decline above and beyond neuropathologies.
DISCUSSION
Fractal patterns may be used as a biomarker for cognitive resilience against dementia‐related neuropathologies.
Keywords: actigraphy, Alzheimer's disease, cognition, reserve, resilience, wearable
1. BACKGROUND
Many people do not develop symptoms of cognitive impairment despite significant pathological changes in the brain such as the accumulation of Alzheimer's disease (AD) pathologies, 1 , 2 implying cognitive resilience. 3 , 4 Understanding the mechanisms and contributing factors to cognitive resilience may guide intervenable targets that prevent or slow cognitive impairment in older adults, especially those with or at risk for neurodegenerative diseases such as AD. 5 , 6 , 7
Using multi‐modal and functional imaging technology, studies have shown that connectivity and segregation of functional networks in the brain contribute to or underlie cognitive resilience. 8 , 9 This network concept is appealing because cognitive processes involve the coordination of multiple brain regions, and the spatiotemporal pattern of neural interactions, instead of activation of isolated brain regions, is believed to be the key for normal cognitive function. 10 In parallel, the network control property has also been proposed as a key concept in fractal physiology—a rapidly growing interdisciplinary field that is focused on understanding the dynamics and complexity of physiological outputs or signals in health and diseases. 11 , 12 , 13 A striking finding in fractal physiology is that a wide range of physiological outputs such as neural activity, heart rate, and motor activity display intrinsic fractal fluctuations (ie, similar temporal structures at different time scales). 14 , 15 , 16 , 17 , 18 , 19 Alterations in fractal patterns are associated with aging and pathological conditions, and predict adverse health outcomes including disability and mortality. 12 , 13 , 20 Fractal physiological fluctuations are believed to represent the adaptability and integrity of coupled regulatory networks. 19
RESEARCH IN CONTEXT
Systematic review: We searched PubMed using the search terms “fractal”, “cognitive resilience”, “cognitive reserve”, “cognitive function”, “dementia”, and “neuropathology” in relevant combinations published between 2010 and up to September 10, 2023, with no language restrictions. Published studies show that degraded fractal regulation predicts future risk of Alzheimer's dementia and faster cognitive decline. Additionally, studies have discovered several proxies of cognitive resilience/reserve including functional connectivity and segregation based on multi‐model brain imaging techniques. However, no studies assessed whether fractal regulation is directly linked to brain neuropathologies or offset cognitive decline against neuropathologies.
Interpretation: New findings show that fractal regulation explains an additional significant portion of the variance in global cognitive decline independent of known brain pathologies. Altogether, fractal regulation may be a biomarker for cognitive resilience. The actigraphy‐based approach used in the current study is unobtrusive, noninvasive, and has long‐term monitoring capabilities that are scalable to large populations.
Future directions: Future studies are warranted to investigate whether fractal regulation provides an additional pillar in cognitive resilience defined previously based on imaging modalities.
Our recent studies showed that altered fractal patterns in ambulatory actigraphy predicted cognitive decline and future risk of Alzheimer's dementia independent of many other known risk factors. 21 Besides, changes in fractal patterns over time sped up with the clinical progression of AD. 22 , 23 Nevertheless, many risk factors for AD may not be mediated via the pathologies of AD and related dementias. Therefore, in this current work we ought to further examine (1) the associations of fractal regulation with dementia‐related neuropathologies, and (2) whether better maintained fractal regulation provides cognitive resilience and offsets the negative effects of pathologies on cognitive decline in older adults. To achieve these goals, we analyzed longitudinal cognition and actigraphy data as well as postmortem brain pathological data collected from deceased participants in the Rush Memory and Aging Project (MAP). 24
2. METHODS
2.1. Participants
The MAP is a community‐based epidemiological clinical‐pathologic cohort study conducted by the Rush Alzheimer's Disease Center beginning in 1997. 24 In 2005, the project began to monitor participants using daily actigraphy every 1 to 2 years (see Data collection and pre‐processing). 24 The MAP was approved by an Institutional Review Board (IRB) of Rush University Medical Center. Written informed and repository consents, and an Anatomical Gift Act for brain donation were obtained from all participants. The dataset used here was frozen on December 2, 2022. This current study included deceased participants who had at least one actigraphy assessment, had cognition assessed, and an autopsy with a completed neuropathologic assessment. The Partners Healthcare Inc. (now the Mass General Brigham) IRB approved the current study. The study was performed in accordance with the ethical standards as laid down in the 1964 Declaration of Helsinki and its later amendments or comparable ethical standards.
2.2. Data collection and pre‐processing
The Actical device (Philips Respironics, Bend, OR) was used to collect actigraphy signals. Participants wore it on their non‐dominant wrists for up to 14 days during the annual assessment. The device sensed three‐dimensional acceleration with a sampling rate of 32 Hz. The acceleration data were integrated into activity counts of every 15‐s epoch. The activity count recordings were subject to quality screenings to identify (1) isolated large spikes with amplitudes more than 10 standard deviations from the individual global mean levels, and (2) >240 consecutive epochs (60 min) with zero activity counts during daytime (eg, potentially occurring when subjects took the device off). The identified data points or segments were marked as gaps and were excluded from the fractal analysis. 20 , 21 , 23
2.3. Fractal analysis of actigraphy (α 1 and α 2)
To assess fractal regulation, detrended fluctuation analysis (DFA) was performed to examine the temporal correlations of actigraphy across a range of timescales. DFA calculates the fluctuation amplitude, F(n), as a function of timescale n. A power‐law relationship between F(n) and n, that is, F(n)∼nα , indicates a fractal structure. Detailed procedures for performing the DFA and related quality control have been published elsewhere 20 , 21 , 23 , 25 , and a customized program is openly available at Zenodo. 26 , 27
The exponent α quantifies the temporal correlation. If α = 0.5, there is no correlation in the signal (ie, similar to white noise); if α > 0.5, there are positive correlations, where large values are more likely to be followed by large values (and vice versa); if α < 0.5, there are negative correlations, where large values are more likely to be followed by small values (and vice versa). For many physiological signals of healthy young adults, α values are close to 1.0, indicating the most complex regulatory mode. In humans, aging and dementia lead to degraded fractal patterns in motor activity fluctuations that can be characterized by different changes in the temporal correlations over two distinct timescale regions with the boundary at ≈1.5 to 2 h. 22 As reported in previous studies, 20 , 21 , 23 two scaling exponents of F(n) were calculated: α 1 at < 90 min, and α 2 from 2 h up to 10 h. The transitional region of timescales between 1.5 and 2 h was omitted.
2.4. Assessment of cognition
A battery of 21 neuropsychological tests was performed annually to assess cognitive function; 19 tests were used to assess performance in five domains of cognition, specifically episodic memory, semantic memory, working memory, perceptual speed, and visuospatial ability. Individual tests within each domain were first converted to z scores using the mean and standard deviation (SD) from the baseline evaluation of all MAP participants; they were then averaged to yield a summary measure of overall cognitive function. This process minimizes floor and ceiling effects and other sources of random variability. 2 For all z scores including the global cognition score, 0 represented the mean, and 1 represented 1 SD of the baseline score of all MAP participants. Larger scores indicate better cognitive performance.
2.5. Postmortem autopsy for brain pathologies
Brain autopsy was performed following a standard protocol at death. Staff performing autopsy and examinations were blinded to clinical data. In total, 10 neuropathological indices were obtained, including amyloid β, hyperphosphorylated tau tangles (paired helical filaments, or PHFtau tangles), Lewy bodies, limbic‐predominant age‐related TDP‐43 encephalopathy neuropathological change (LATE‐NC), hippocampal sclerosis, chronic gross infarcts, chronic microscopic infarcts, atherosclerosis, arteriolosclerosis, and cerebral amyloid angiopathy (CAA). Additional information can be found in prior MAP works. 28 , 29
Amyloid β and PHFtau tangles represented the estimated burden of amyloid (% area occupied) and density of tangles (per mm2) across eight brain regions. They were right‐skewed and square root transformation was used for analyses.
Lewy bodies were initially classified as none, nigral‐predominant, limbic‐type, and neocortical‐type, and were recategorized to “neocortical disease (1)” if initially staged as neocortical‐type and “no (0)” if otherwise. A four‐stage classification was also initially applied to LATE‐NC (ie, none, amygdala, amygdala + limbic, and amygdala + limbic + neocortical) and was then recategorized to “no (0)” if initially classified as none or amygdala and “yes (1)” if otherwise. 28
Hippocampal sclerosis was graded as absent (0) or present (1) based on severe neuronal loss and gliosis in hippocampal section CA1 and/or subiculum. Gross infarcts and microscopic infarcts were both graded as no (0; if no infarcts were detected) or yes (1; if one or more infarcts were detected). Atherosclerosis, arteriolosclerosis, and CAA were all initially staged as none, mild, moderate, and severe, and were dichotomized to “no (0)” if initially classified as none or mild and to “yes (1)” if otherwise.
2.6. Statistical analysis
To examine the associations of fractal regulation with dementia‐related neuropathologies, we first fitted linear or logistic regression models with the fractal regulation α 1 as a continuous predictor. Linear regressions were used for amyloid β and PHFtau that were considered separately as a continuous outcome variable; logistic regressions were used for the other eight pathological outcomes that were considered separately as a dichotomous variable. The same set of models were then repeated to test the associations of the fractal regulation α 2 with these neuropathologies. All models were adjusted for age at death, sex, and education. We used the actigraphy proximate to death for calculating α 1 and α 2. As sensitivity analyses, we restricted the dataset to participants with a time interval between the actigraphy proximate to death and death ≤2 years and repeated the above‐mentioned models.
A series of mixed‐effects models were used to test whether better maintained fractal regulation provides cognitive resilience and offsets the negative effects of pathologies on cognitive decline. Specifically, we first performed a linear mixed‐effects model (ie, model A) to determine the longitudinal changes in global cognition after considering demographics (ie, age at death, sex, and education) 30 , 31 and the 10 neuropathologies. The model included a term for time in years to death and included the corresponding interactions of this time variable with demographics and neuropathologies. We then augmented this model by further including α 1 (or α 2 separately) proximate to death (as well as the interaction between α 1 or α 2 with the time variable, and we separately denoted the models as model B1 and model B2, respectively. Evidence of cognitive resilience was rendered when α 1 in model B1 (or α 2 in model B2) was still associated with the rate of change in global cognition (ie, the interaction of the time lag with α 1 or α 2). 32 Additionally, we also performed a reference model (model C) without considering any covariates to determine the variance of the rate of cognitive decline and separately, an augmented model (model D) with adjustment of demographics and apolipoprotein E gene (APOE) ε4 carrier status (as well as their interactions with the time variable). With the two sets of nested models (ie, models C, D, A, B1, and separately, models C, D, A, B2), we would be able to specifically examine how much variance of the rate of cognitive decline can be respectively explained by demographics and APOE ε4 carrier status, neuropathologies, and α 1 or α 2. Similarly, we also performed sensitivity analyses to restrict the analysis based on participants who had their last actigraphy within 2 years before death. Additionally, we have previously reported that total daily activity derived from actigraphy was related to cognitive resilience. 29 We thus separately augmented the models B1 and B2 by further including total daily activity (and its interaction with the time variable) to examine whether any observed results were independent from the effect of total daily activity.
TABLE 1.
Participant demographics, clinical characteristics, and pathological disease burden.
| Measure | Mean (SD) or N (%) | Range |
|---|---|---|
| Demographics | ||
| Age | ||
| At baseline (years) | 82.8 (5.8) | 59.0–99.8 |
| At death (years) | 91.1 (6.0) | 65.9–108.3 |
| Sex | ||
| Female | 384 (72.0%) | – |
| Male | 149 (28.0%) | – |
| Education (years) | 14.7 (2.8) | 5–25 |
| APOE ε4 carrier status | ||
| Carriers | 114 (21.4) | |
| Non‐carriers | 412 (77.3%) | |
| Cognition | ||
| Mini‐Mental State Examination | ||
| At baseline | 27.4 (3.0) | 1–30 |
| At last actigraphy visit | 23.8 (6.3) | 0–30 |
| Pathological markers | ||
| Amyloid β, square root transformed | 1.82 (1.18) | 0–4.79 |
| PHFtau tangles, square root transformed | 2.36 (1.36) | 0–7.32 |
| Lewy bodies | ||
| 0 (no) | 453 (85.0%) | – |
| 1 (neocortical disease) | 80 (15.0%) | – |
| LATE‐NC | ||
| 0 (none or amygdala) | 339 (63.6%) | – |
| 1 (limbic or neocortical) | 194 (36.4%) | – |
| Hippocampal sclerosis | ||
| 0 (no) | 485 (91.0%) | – |
| 1 (yes) | 48 (9.0%) | – |
| Chronic gross infarcts | ||
| 0 (no) | 327 (61.4%) | – |
| 1 (yes) | 206 (38.6%) | – |
| Chronic microinfarcts | ||
| 0 (no) | 360 (67.5%) | – |
| 1 (yes) | 173 (32.5%) | – |
| Atherosclerosis | ||
| 0 (none‐mild) | 400 (75.0%) | – |
| 1 (moderate‐severe) | 133 (25.0%) | – |
| Arteriolosclerosis | ||
| 0 (none‐mild) | 380 (71.3%) | – |
| 1 (moderate‐severe) | 153 (28.7%) | – |
| Cerebral amyloid angiopathy | ||
| 0 (none‐mild) | 351 (65.9%) | – |
| 1 (moderate‐severe) | 182 (34.1%) | – |
| Fractal regulation | ||
| α 1 | 0.88 (0.07) | 0.60–1.11 |
| α 2 | 0.77 (0.11) | 0.39–1.30 |
| Others | ||
| Interval between last actigraphy visit and death (years) | 2.28 (2.19) | 0.02–12.91 |
APOE, apolipoprotein E; LATE‐NC, limbic‐predominant age‐related TDP‐43 encephalopathy neuropathological change; PHF, paired helical filament.
All statistical analyses were performed using MATLAB (Ver. R2022a, The MathWorks Inc., Natick, MA, USA). Statistical significance was determined a priori at a nominal level of alpha = 0.05.
3. RESULTS
There were 533 decedents who had assessment of actigraphy and had all 10 neuropathological indices. Table 1 summarizes their demographics, clinical characteristics, and pathological disease burden.
Among the 10 neuropathologies, α 1 was associated with Lewy bodies, gross chronic infarcts, chronic microinfarcts, and arteriolosclerosis. Specifically, for each 1 SD increase in α 1, the odds of having neocortical Lewy body disease decreased by 21.0% (95% confidence interval [CI]: 0.4% to 37.3%). Similarly, the odds of having gross chronic infarcts, chronic microinfarcts, and arteriolosclerosis decreased by 22.7% (95% CI: 7.7% to 35.2%), 18.7% (95% CI: 2.6% to 32.2%), and 20.5% (95% CI: 4.0% to 34.2%), respectively (Table 2; Table S1). Consistent results with comparable or relatively greater effect sizes were observed from sensitivity analyses using 324 decedents who had their last actigraphy (ie, proximate to death) within 2 years before the time of death (Table 2; Table S2). Nevertheless, α 1 was not associated statistically with the other six neuropathologies; α 2 was not associated statistically with any of the ten neuropathologies (Table 2; Tables S1 and S2).
TABLE 2.
Linear regression models examining the cross‐sectional associations of fractal regulation with neuropathologies.
| Neuropathology | α 1 | α 2 | ||
|---|---|---|---|---|
| Estimate (95% CI) | p value | Estimate (95% CI) | p value | |
| Amyloid β | −0.008 (−0.106, 0.090) | 0.871 | 0.047 (−0.052, 0.146) | 0.349 |
| PHFtau tangles | 0.003 (−0.108, 0.115) | 0.953 | −0.063 (−0.176, 0.049) | 0.270 |
| Odds ratio (95% CI) | p value | Odds ratio (95% CI) | p value | |
|---|---|---|---|---|
| Lewy body disease | 0.790 (0.627, 0.996) | 0.047 | 0.996 (0.780, 1.270) | 0.972 |
| LATE‐NC | 0.994 (0.831, 1.188) | 0.945 | 0.992 (0.829, 1.187) | 0.929 |
| Hippocampal sclerosis | 0.804 (0.600, 1.078) | 0.144 | 0.980 (0.730, 1.318) | 0.896 |
| Gross chronic infarcts | 0.773 (0.648, 0.923) | 0.005 | 0.893 (0.748, 1.066) | 0.210 |
| Chronic microinfarcts | 0.813 (0.678, 0.974) | 0.025 | 1.021 (0.851, 1.224) | 0.822 |
| Atherosclerosis | 0.862 (0.710, 1.047) | 0.134 | 0.989 (0.811, 1.206) | 0.916 |
| Arteriolosclerosis | 0.795 (0.658, 0.960) | 0.017 | 1.028 (0.852, 1.241) | 0.769 |
| Cerebral amyloid angiopathy | 1.111 (0.927, 1.331) | 0.255 | 1.043 (0.871, 1.250) | 0.643 |
| Sensitivity analysis | Estimate (95% CI) | p value | Estimate (95% CI) | p value |
|---|---|---|---|---|
| Amyloid β | −0.090 (−0.217, 0.037) | 0.164 | 0.002 (−0.124, 0.127) | 0.978 |
| PHFtau tangles | −0.072 (−0.192, 0.047) | 0.233 | −0.118 (−0.235, −0.001) | 0.049 |
| Odds ratio (95% CI) | p value | Odds ratio (95% CI) | p value | |
|---|---|---|---|---|
| Lewy body disease | 0.694 (0.504, 0.955) | 0.025 | 0.975 (0.700, 1.359) | 0.881 |
| LATE‐NC | 1.076 (0.854, 1.355) | 0.533 | 0.995 (0.796, 1.244) | 0.965 |
| Hippocampal sclerosis | 1.109 (0.721, 1.705) | 0.637 | 0.850 (0.562, 1.286) | 0.439 |
| Gross chronic infarcts | 0.774 (0.618, 0.970) | 0.026 | 0.910 (0.732, 1.131) | 0.392 |
| Chronic microinfarcts | 0.743 (0.587, 0.941) | 0.014 | 1.007 (0.803, 1.262) | 0.954 |
| Atherosclerosis | 0.833 (0.654, 1.062) | 0.140 | 1.014 (0.797, 1.289) | 0.909 |
| Arteriolosclerosis | 0.802 (0.632, 1.017) | 0.069 | 1.064 (0.847, 1.338) | 0.592 |
| Cerebral amyloid angiopathy | 1.056 (0.836, 1.335) | 0.646 | 1.105 (0.880, 1.389) | 0.390 |
Note: Models were adjusted for age at death, sex, and education. Results represent changes in the level (for continuous outcomes) or the odds (for dichotomized outcomes) for each 1 standard deviation increase in α 1 or α 2. Sensitivity analysis was performed in participants with the time interval between last actigraphy and death of ≤2 years. Detailed results are summarized in Tables S1 and S2. LATE‐NC, limbic‐predominant age‐related TDP‐43 encephalopathy neuropathological change; PHF, paired helical filament.
The longitudinal analysis included 519 decedents who had finished at least two cognitive assessments. The reference model (ie, model C) demonstrated that global cognition progressively declined over time with a rate of −0.1069 (standard error [SE]: 0.0046; p < 0.0001; Table S3) per year. The decline rate had a variance of 0.0084 (Table 3). After further adjusting for demographics and APOE ε4 carrier status (model D), the variance reduced to 0.0076, meaning that demographics together with APOE ε4 carrier status explained about 9.31% of the variance of cognitive decline (Table 3; note that only APOE ε4 carrier status was significantly associated with cognitive decline in this model; Table S3). The 10 neuropathologies together explained 33.51% of the variance (ie, in model A; Table 3) on top of demographics (note that in this model, APOE ε4 carrier status was no longer significantly associated with cognitive decline, meaning that its effect was explained by certain neuropathologies; Table S3). Finally, in model B1, α 1 was still associated with cognitive decline after considering the effects of demographics, APOE ε4 carrier status, and all 10 neuropathologies. For each 1 SD increase in α 1, the rate of cognitive decline was offset by 0.0170 (SE: 0.0037; p < 0.0001; Figure 1; Table S3), an effect that is equivalent to that of a more than a half‐unit (close to a half SD) decrease in PHFtau tangles or a more than a 2‐unit (close to 1.8 SD) decrease in amyloid β (note, both amyloid β and PHFtau tangles were square root transformed; Table S3). Besides, α 1 explained an additional 3.03% of the variance in cognitive decline on top of demographics and neuropathologies (Table 3). The observation remained statistically significant after further adjusting for total daily activity, with a slight decrease in the effect size (Table S3). In model B2, α 2 was also associated with cognitive decline after considering the effects of demographics, APOE ε4 carrier status, and all 10 neuropathologies. For each 1 SD increase in α 2, the rate of cognitive decline was offset by 0.0110 (SE: 0.0038; p = 0.0035; Figure 1; Table S3), an effect that is equivalent to that of an about 0.4‐unit decrease in PHFtau tangles or a 1.3‐unit decrease in amyloid β (Table S3). Besides, α 2 explained an additional 1.59% of the variance in cognitive decline on top of demographics and neuropathologies (Table 3). The observation became not statistically significant after further adjusting for total daily activity (Table S3). In sensitivity analysis based on 311 participants who had their last actigraphy within 2 years before the time of death, all above observations were retained with relatively greater effect sizes (Table 3 and Table S4).
TABLE 3.
Linear mixed‐effects models examining the contribution of fractal regulation to longitudinal cognitive decline.
| Primary analysis | Sensitivity analysis | |||
|---|---|---|---|---|
| Nested models | Remaining variance | % variance explained | Remaining variance | % variance explained |
| C (reference) | 0.0084 | – | 0.0072 | – |
| D (C + demographics + APOE) | 0.0076 | 9.31% | 0.0067 | 6.56% |
| A (D + pathologies) | 0.0048 | 33.51% | 0.0042 | 34.48% |
| B1 (A + α 1) | 0.0045 | 3.03% | 0.0038 | 5.39% |
| B2 (A + α 2) | 0.0046 | 1.59% | 0.0040 | 2.80% |
Note: Results were from two sets of nested models (ie, set 1: C, D, A, B1; set 2: C, D, A, B2). The % of variance explained represents variance of the rate of cognitive decline explained by the added variables. Specifically, in model D, it is the variance explained by demographics (age, sex, education, and apolipoprotein E [APOE] ε4 carrier status); in model A, it is the variance explained by all 10 neuropathologies on top of demographics and APOE ε4 carrier status; in model B1, it is the variance explained by α 1 on top of demographics, APOE ε4 carrier status, and neuropathologies; in model B2, it is the variance explained by α 2 on top of demographics, APOE ε4 carrier status, and neuropathologies.
FIGURE 1.
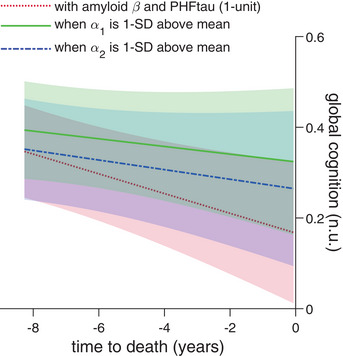
Better maintained fractal regulation buffers cognitive decline associated with neuropathologies. The red dotted line shows a representative apolipoprotein E (APOE) ε4 non‐carrier woman with age at death and years of education both at the corresponding average levels of all participants and with amyloid β and PHFtau burdens (both 1‐unit in terms of the square root transformed version of the two variables). The representative participant has faster global cognitive decline with aging. The green line shows a similar representative participant, but with the corresponding α 1 at 1 SD above the average levels. Similarly, the blue dash‐dotted line shows a similar representative participant, but with the corresponding α 2 at 1 SD above the average levels.
4. DISCUSSION
The clinical expression of neuropathologies demonstrates great heterogeneity, 1 suggesting that resilience exists accounting for the inconsistencies between pathology and cognitive performance. Such cognitive resilience has attracted much attention especially in AD. 4 , 30 , 31 , 33 In this large sample of community‐dwelling older adults, we found that better maintained fractal regulation, especially at smaller time scales, was able to offset the longitudinal cognitive decline caused by neuropathologies, with a considerable large effect size equivalent to that of an around half‐unit change in PHFtau tangles or an around 2‐unit change in amyloid β. The results suggest that better maintained fractal regulation offers resilience to cognitive decline due to neuropathologies. Besides, both fractal regulation measures explained a significant portion of the variance of global cognitive decline above and beyond demographics, APOE ε4 carrier status, and the 10 neuropathologies that are known to impair cognition, suggesting additional pathways independent of pathological changes in the brain that support cognitive intactness.
We operationalized resilience as residual decline after regressing out the effects of brain pathologies. 32 A few prior studies have used a similar approach based on brain imaging. For example, the brain functional networks have been hypothesized to maintain an efficient topological organization to support cognitive health in pre‐symptomatic genetic frontotemporal dementia patients. 8 In another study of AD patients, the segregation of the brain's connectome that forms distinct functional networks has been proposed to support cognitive reserve in AD. 9 Nevertheless, brain imaging is expensive and reliant significantly on patient cooperation. To enhance the study of cognitive resilience, especially in large population cohorts and for scalable applications, there is a need for non‐invasive tools that can assess and monitor resilience. 5
Wearable devices offer a distinct advantage in this regard, as they can continuously monitor physiological data in natural settings without disrupting people's daily routines. This provides a significant edge over imaging modalities. 20 , 34 Similar to the concept of a network control property supporting these imaging findings, fractal organization in physiological outputs is believed to represent the adaptability and plasticity of the underlying regulatory networks. 12 Fractal regulation in physiology is believed to reflect the integrative property of the complex networks in which biological processes that function at different time scales are coupled or orchestrated to ensure optimal performance of the body. Our findings also support the network theory of cognition resilience.
In our previous study of longitudinal antemortem motor activity data of MAP participants, we observed that both the two α’s progressively decreased with aging within the individual, with a greater rate of decline in α 2. 23 It is thus plausible that the better maintained fractal regulation at the end of life reflects a slower declining profile of fractal regulation. To explore this possibility, we examined whether the longitudinal changes in the fractal metrics played a similar role of resilience as the fractal metrics proximate to death by replacing the annual change in α 1 or α 2 (ie, the random‐slope of a separate mixed‐effects model for the change of α over time 23 ) with α 1 or α 2 itself in the linear mixed‐effects model. We observed similar results, that is, a slower decline in α during late life counteracted the longitudinal decline in global cognition caused by neuropathologies. These additional results support the finding that better‐maintained fractal regulation delineates cognitive resilience to brain pathologies. However, the late‐life changing profile of fractal regulation can be difficult to model due to unforeseeable events, for example, the development of multiple comorbidities or hospitalizations. The use of the fractal metrics derived from actigraphy proximate to death was thus preferred in this current work as they may indicate a residue state after many years’ declines, where a higher value represents better maintained function in terms of fractal regulation.
Future studies are warranted to enhance the understanding of the generation and maintenance of fractal regulation. In this current study, we observed significant associations of α 1 with neocortical Lewy body disease and markers for cerebrovascular diseases (ie, gross chronic infarcts, chronic microinfarcts, and arteriolosclerosis). In a prior study, we reported that α 1 (ie, representing the fractal regulation at smaller time scales) predicted incident Alzheimer's dementia and longitudinal cognitive changes. 21 The prediction ability of α 1 for Alzheimer's dementia and cognitive decline thus may be partially driven by Lewy bodies and cerebrovascular diseases. The observation that α 1 was still associated with cognitive decline after considering all neuropathologies further suggests that such a predictive ability of α 1 is independent of and beyond the known neuropathologies. While it is limited for infering a causal directionality, future effort should focus on elucidating the neural correlates and mechanisms underlying the change in α 1. Additionally, based on prior animal studies, the intrinsic circadian function plays a key role in generating and maintaining a fractal structure at larger timescales (ie, the α 2). 35 , 36 The current observation that α 2 was not associated with any of the 10 studied neuropathologies whereas it was able to offset cognitive decline independently from the neuropathologies therefore further highlights the urgency in identifying the pathophysiological pathways underlying the link between circadian function and AD and related dementias.
Our findings also encourage additional analyses toward differentiating specific resilience capacities against different neurodegenerative diseases. For example, whether fractal regulation overlaps with cognitive resilience defined by the imaging modalities or offers a new dimension to measure this important modulating factor of cognition is unclear. Additionally, novel genetic analyses may help identify molecular mechanisms mediating the resilience offered by fractal regulation and encourage further research to examine the genetic components of fractal regulation, and what genetic activities may interact with fractal regulation to affect cognition in neurodegenerative diseases.
The current study has notable strengths, including its longitudinal assessment of cognitive profiles in a large community‐based cohort of older adults, extending until death, and thorough postmortem histopathological examinations for dementia‐related neuropathologies. However, it is important to acknowledge certain limitations. First, cognitive decline has multifactorial causes. While our findings suggest an association between fractal metrics and cognitive decline beyond the influence of the 10 examined neuropathologies, the study was not specifically designed to identify whether this resilience factor is neuropathology specific. Second, chronic comorbidities, linked to accelerated cognitive decline, were not directly integrated into the statistical models. Although some comorbidities may be indicated by neuropathological variables, incorporating their clinical manifestations directly into models could provide a more comprehensive understanding. Lastly, the accumulation of brain pathology may commence decades before death. For a more robust examination of cognitive resilience, future studies should incorporate in vivo data on neuropathologies.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest. Author disclosures are available in the supporting information.
CONSENT STATEMENT
Written informed and repository consents, and an Anatomical Gift Act for brain donation were obtained from all participants.
Supporting information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information
ACKNOWLEDGMENTS
This study was supported by the National Institute on Aging (NIA) grant (number RF1AG064312) and the BrightFocus Foundation Alzheimer's Research Program (number A2020886S). P.L. is also supported by the Brigham Research Institute under the Fund to Sustain Research Excellence Program and a start‐up fund from the Department of Anesthesia, Critical Care and Pain Medicine at Massachusetts General Hospital. L.G. is also supported by the Alzheimer's Association grant (number AACSF‐23‐1148490). The MAP is supported by NIA grant R01AG17917. K.H. is also supported by a start‐up fund from the Department of Anesthesia, Critical Care and Pain Medicine at Massachusetts General Hospital and an NIA grant (number R01AG083799).
Li P, Gao C, Yu L, et al. Delineating cognitive resilience using fractal regulation: Cross‐sectional and longitudinal evidence from the Rush Memory and Aging Project. Alzheimer's Dement. 2024;20:3203–3210. 10.1002/alz.13747
Contributor Information
Peng Li, Email: pli9@mgh.harvard.edu.
Kun Hu, Email: khu1@mgh.harvard.edu.
REFERENCES
- 1. Price JL, Buckles VD, Roe CM, et al. Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026‐1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bennett DA, Wilson RS, Boyle PA, Buchman AS, Schneider JA. Relation of neuropathology to cognition in persons without cognitive impairment. Ann Neurol. 2012;72:599‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stern Y. What is cognitive reserve? Theory and research application of the reserve concept. J Int Neuropsychol Soc JINS. 2002;8:448‐460. [PubMed] [Google Scholar]
- 4. Stern Y. Cognitive reserve in ageing and Alzheimer's disease. Lancet Neurol. 2012;11:1006‐1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stern Y, Barnes CA, Grady C, Jones RN, Raz N. Brain reserve, cognitive reserve, compensation, and maintenance: operationalization, validity, and mechanisms of cognitive resilience. Neurobiol Aging. 2019;83:124‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Negash S, Wilson RS, Leurgans SE, et al. Resilient brain aging: characterization of discordance between Alzheimer's disease pathology and cognition. Curr Alzheimer Res. 2013;10:844‐851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dobyns L, Zhuang K, Baker SL, et al. An empirical measure of resilience explains individual differences in the effect of tau pathology on memory change in aging. Nat Aging. 2023;3:229‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rittman T, Borchert R, Jones S, et al. Functional network resilience to pathology in presymptomatic genetic frontotemporal dementia. Neurobiol Aging. 2019;77:169‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ewers M, Luan Y, Frontzkowski L, et al. Segregation of functional networks is associated with cognitive resilience in Alzheimer's disease. Brain J Neurol. 2021;144:2176‐2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McIntosh AR. Towards a network theory of cognition. Neural Netw Off J Int Neural Netw Soc. 2000;13:861‐870. [DOI] [PubMed] [Google Scholar]
- 11. Goldberger AL. Non‐linear dynamics for clinicians: chaos theory, fractals, and complexity at the bedside. Lancet Lond Engl. 1996;347:1312‐1314. [DOI] [PubMed] [Google Scholar]
- 12. Goldberger AL. Fractal dynamics in physiology: alterations with disease and aging. Proc Natl Acad Sci U S A. 2002;99(Suppl 1):2466‐2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. West BJ. Fractal physiology and the fractional calculus: a perspective. Front Physiol. 2010;1:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ivanov PC, Amaral L A, Goldberger A L, Havlin S, Rosenblum M G, Struzik Z R, Stanley H E. Multifractality in human heartbeat dynamics. Nature. 1999;399:461‐465. [DOI] [PubMed] [Google Scholar]
- 15. Mäkikallio TH, Huikuri HV, Mäkikallio A, et al. Prediction of sudden cardiac death by fractal analysis of heart rate variability in elderly subjects. J Am Coll Cardiol. 2001;37:1395‐1402. [DOI] [PubMed] [Google Scholar]
- 16. Nunes Amaral LA, Ivanov PC, Aoyagi N, et al. Behavioral‐independent features of complex heartbeat dynamics. Phys Rev Lett. 2001;86:6026‐6029. [DOI] [PubMed] [Google Scholar]
- 17. Hu K, Chen Z, Hilton MF, et al. Non‐random fluctuations and multi‐scale dynamics regulation of human activity. Phys ‐Stat Mech Its Appl. 2004;337:307‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hu K, Shea SA, vanderLeest HT, et al. Fractal patterns of neural activity exist within the suprachiasmatic nucleus and require extrinsic network interactions. Plos One. 2012;7:e48927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pittman‐Polletta BR, Scheer FAJL, Butler MP, Shea SA, Hu K. The role of the circadian system in fractal neurophysiological control. Biol Rev Camb Philos Soc. 2013;88:873‐894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li P, Gao L, Hu C, et al. More random motor activity fluctuations predict incident frailty, disability, and mortality. Sci Transl Med. 2019;11:eaax1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li P, Yu L, Lim ASP, et al. Fractal regulation and incident Alzheimer's disease in elderly individuals. Alzheimers Dement J Alzheimers Assoc. 2018;14:1114‐1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu K, Van Someren EJW, Shea SA, Scheer FAJL. Reduction of scale invariance of activity fluctuations with aging and Alzheimer's disease: involvement of the circadian pacemaker. Proc Natl Acad Sci U S A. 2009;106:2490‐2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li P, Yu L, Yang J, et al. Interaction between the progression of Alzheimer's disease and fractal degradation. Neurobiol Aging. 2019;83:21‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bennett DA, Schneider JA, Buchman AS, et al. Overview and findings from the Rush Memory and Aging Project. Curr Alzheimer Res. 2012;9:646‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao C, et al. Approaches for assessing circadian rest‐activity patterns using actigraphy in cohort and population‐based studies. Curr Sleep Med Rep. 2023. doi: 10.1007/s40675-023-00267-4 [DOI] [Google Scholar]
- 26. Li P, pliphd/MATLAB‐detrended‐fluctuation‐analysis: an MATLAB application for detrended fluctuation analysis. 2019;doi: 10.5281/zenodo.3407188 [DOI]
- 27. Li, P. pliphd/Actigraphy: ezActi2. 2023;doi: 10.5281/ZENODO.8411607 [DOI]
- 28. Boyle PA, Wang T, Yu L, et al. To what degree is late life cognitive decline driven by age‐related neuropathologies? Brain. 2021;144:2166‐2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Memel M, Buchman AS, Bennett DA, Casaletto K. Relationship between objectively measured physical activity on neuropathology and cognitive outcomes in older adults: resistance versus resilience? Alzheimers Dement Amst Neth. 2021;13:e12245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Xu H, Yang R, Qi X, et al. Association of lifespan cognitive reserve indicator with dementia risk in the presence of brain pathologies. JAMA Neurol. 2019;76:1184‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ossenkoppele R, Lyoo CH, Jester‐Broms J, et al. Assessment of demographic, genetic, and imaging variables associated with brain resilience and cognitive resilience to pathological tau in patients with Alzheimer disease. JAMA Neurol. 2020;77:632‐642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stern Y, Albert M, Barnes CA, Cabeza R, Pascual‐Leone A, Rapp PR. A framework for concepts of reserve and resilience in aging. Neurobiol Aging. 2022(22). doi: 10.1016/j.neurobiolaging.2022.10.015. S0197‐4580 00254‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Negash S, Xie S, Davatzikos C, et al. Cognitive and functional resilience despite molecular evidence of Alzheimer's disease pathology. Alzheimers Dement J Alzheimers Assoc. 2013;9:e89‐e95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steinhubl SR, Muse ED, Topol EJ. The emerging field of mobile health. Sci Transl Med. 2015;7:283rv3‐283rv3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu K, Scheer FaJL, Ivanov PC, Buijs RM, Shea SA. The suprachiasmatic nucleus functions beyond circadian rhythm generation. Neuroscience. 2007;149:508‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li P, Chiang WY, Escobar C, et al. Fractal regulation in temporal activity fluctuations: a biomarker for circadian control and beyond. JSM Biomark. 2017;3:1008. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information
Supporting Information
Supporting Information
Supporting Information