

Abstract
Heterochronic parabiosis (HPB) is known for its functional rejuvenation effects across several mouse tissues. However, its impact on biological age and long-term health remains unknown. Here, we performed extended (3-month) HPB, followed by a 2-month detachment period of anastomosed pairs. Old detached mice exhibited improved physiological parameters and lived longer than control isochronic mice. HPB drastically reduced the epigenetic age of blood and liver based on various clock models using two independent platforms. Remarkably, this rejuvenation effect persisted even after 2 months of detachment. Transcriptomic and epigenomic profiles of anastomosed mice showed an intermediate phenotype between old and young, suggesting a global multi-omic rejuvenation effect. In addition, old HPB mice showed gene expression changes opposite to aging, but akin to several lifespan-extending interventions. Altogether, we reveal that long-term HPB results in lasting epigenetic and transcriptome remodeling, culminating in the extension of lifespan and healthspan.
INTRODUCTION
Aging is the primary risk factor for chronic diseases 1–3. It brings accumulation of damage at many levels of biological organization and a pervasive and destructive decline of organ function, resulting in inevitable mortality. Although many attempts have been made to extend lifespan and ameliorate specific aging phenotypes through interventions, aging itself has generally been regarded as an irreversible process. However, the recent development and application of advanced aging biomarkers based on DNA methylation (i.e., methylation clocks) 4–7 have challenged this concept. With their assessment of the attenuated aging effect of various longevity interventions, including caloric restriction (CR), genetic models and cellular reprogramming, methylation clocks have been generally recognized as a robust predictor of organismal biological age, a measure of how old an organism is biologically rather than chronologically. Notably, DNA methylation clocks have successfully predicted the reversal of epigenetic age by several interventions, including reprogramming factor expression and treatment with drugs or blood components 8–12. Clocks have been also recently used to reveal and describe a natural rejuvenation event occurring during early embryonic development 13,14. However, in the case of interventions, it remains generally enigmatic whether the predicted reversal of epigenetic age is sustained, correlated with longer lifespan, and manifests in improved physiological function.
The heterochronic parabiosis (HPB) model has been used to study circulating factors that regulate the aging process since the 1950s 15–17. More recent work has established the model as a proof of concept that youthful circulation can restore old tissue functions 18,19. Indeed, the effects of HPB on the amelioration of aging phenotypes are evident across tissues including muscle 19, liver19, heart 20, brain 21,22 and bone 18,23. Remarkably, these effects are observed typically after only 4–5 weeks of parabiosis. Similar results are observed with acute heterochronic blood exchange (non-parabiosis), showing beneficial effects of “young blood” on muscle, liver and brain of old recipients 24. Heterochronic young blood plasma transfer also improves pathology of age-related diseases, such as in a model of Alzheimer’s disease in mice 25. Although HPB leads to diverse effects on old cells and tissues, our understanding of the molecular mechanisms involved remains limited. Likewise, whether the immediate effects of blood/plasma exchange are sustained after the procedure is still unknown. Lastly, due to the previous absence of a precise quantification method, it remains unclear whether HPB can slow or rewind epigenetic aging of an organism.
A previously reported 26,27, but seldom used HPB procedure is the detachment of mice following parabiosis. This technique has previously been used to verify cell engraftment using different tracer techniques 28. A recent investigation revealed the persistence of aged hematopoietic stem cells in the young bone marrow niche resulting from HPB months after surgical separation of parabionts 29, however, to our knowledge there have been no studies that investigate longevity or the long-term effects of HPB on healthspan. Although detachment involves performing a second surgery on the animals, it permits physiological and longevity measurements, which are difficult to obtain in the case of anastomosed mice.
Here, we report the results of a long-term HPB study followed by a detachment period. We found that old mice detached from young mice showed an extended lifespan and improvements across several dimensions of aging biology. By comprehensive epigenetic clock and RNA-seq analyses, we observed a robust reduction in epigenetic age of old mice following 3-month HPB, which was sustained even after a detachment period of 2 months. Notably, this rejuvenation effect was significantly stronger than that observed upon the more traditional, short-term HPB and is not accounted for by the effects of the surgery or changes in physical activity. We find that transcriptomic and epigenetic profiles of long-term HPB are intermediate between young and old isochronic pairs, and that HPB positively associates with the effects of common lifespan-extending interventions and counteracts aging-related gene expression changes. Our findings suggest the presence of profound and persisting molecular rejuvenation effects following exposure to young circulation, leading to extended lifespan and healthspan.
RESULTS
Long-term parabiosis followed by detachment extends lifespan and healthspan in mice
We used a long-term (3-month) parabiosis (either heterochronic or isochronic) period in mice, followed by detachment of the parabionts. Young mice started parabiosis at 3 months and old mice at 20 months of age (Fig. 1a). Transcriptomic and epigenetic profiling of the liver and blood was conducted to assess molecular changes resulting from HPB (Fig. 1b). For longevity and healthspan experiments, mice were detached and allowed 1 month of recovery after parabiosis prior to physiological data collection (Extended Data Fig. 1a). After separation from respective parabiosis pairs, a subset of mice were allowed to live freely until their natural death, in order to examine the effect of parabiosis on lifespan. We observed a 6-week extension in median lifespan and a 2-week extension in maximum lifespan in old mice detached from heterochronic pairs as compared to their isochronic controls (Fig. 1c). This was accompanied by an initial reduction in body weight coupled with better preservation of body weight in the final months of life (Fig. 1d). The initial drop in body weight appeared to be primarily caused by a reduction in fat mass, while the preservation of body weight was due to the maintenance of both lean and fat mass later in life (Fig. 1d). These changes in body composition were independent of changes in food consumption (Extended Data Fig. 1b). In addition to improvements in body composition, old heterochronic mice showed higher voluntary cage activity than isochronic controls (Fig. 1d and Extended Data Fig. 1c).
Fig. 1 |. Prolonged parabiosis followed by detachment leads to extended life span and health span.
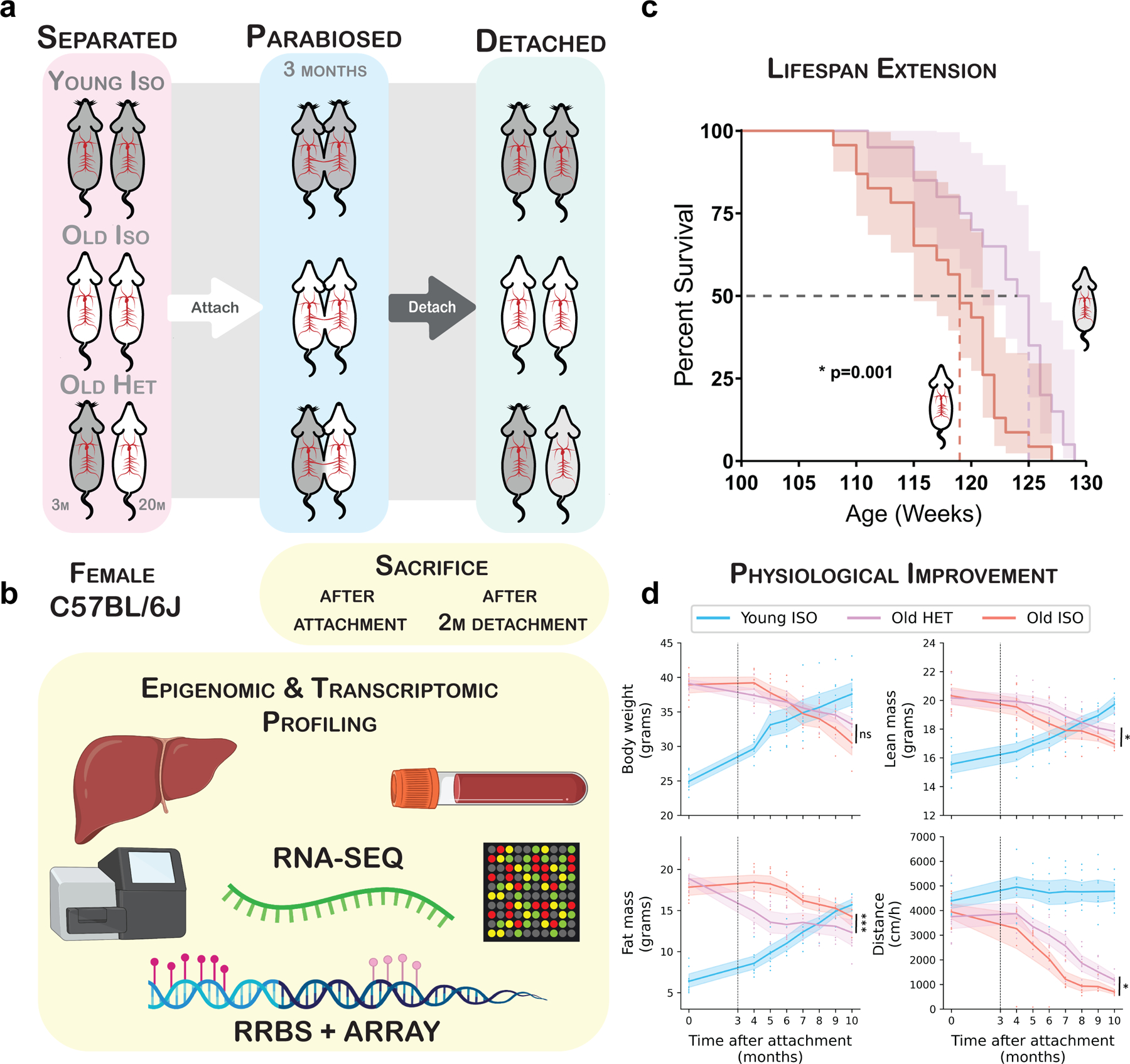
a, Overview of the parabiosis and detachment model. All pairs were anastomosed for 3 months, starting at 20 months of age for old mice and 3 months of age for young mice. At 23 months, old mice were detached and the remaining life span was assessed. b, Schematic of the molecular profiling methods applied to mouse tissues. c, Survival curves of detached old mice from isochronic (red, n = 23) and heterochronic (purple, n = 20) pairs. Kaplan–Meier curves with 95% confidence intervals are shown. The median life span is highlighted with dashed lines. A log-rank test comparing the two survival curves was used for statistical analysis (P = 0.001). d, Body weight, lean mass, fat mass and cage activity measurements before parabiosis and at monthly time points starting 1 month after detachment. Mean values with 95% confidence intervals are shown. A two-tailed Welch’s test was used to compare old isochronic and heterochronic groups: Pbody weight = 0.29; Plean mass = 0.049; Pfat mass = 0.00092; and Pcage activity = 0.015. ‘Young ISO’ denotes young mice from isochronic pairs (n = 10). ‘Old ISO’ denotes old mice from isochronic pairs (n = 7). ‘Old HET’ denotes old mice from heterochronic pairs (n = 8). The dashed lines show the time of detachment. *P < 0.05; **P < 0.01; ***P < 0.001.
Epigenetic age of old mice is reversed by parabiosis with a sustained effect after detachment
For epigenetic analyses, we subjected mice to the same prolonged attachment, and harvested tissues either immediately after the 3-month parabiosis procedure, or 2 months after detachment (Extended Data Fig. 1a). We first subjected the blood and liver of the animals to Reduced Representation Bisulfite Sequencing (RRBS) 30 (Table S1). Since blood samples taken immediately after detachment contained a mixture of old and young blood (Extended Data Fig. 2a–c), we initially chose to quantify methylation changes in blood after a 2-month detachment period. At this time point, we could investigate whether epigenomic remodeling upon exposure to young circulation persists without young blood contamination, as we did not observe blood crossover 2 months after detachment (Extended Data Fig. 2d). Additionally, we took liver as an example of a solid tissue to quantify the indirect effects on the methylome occurring immediately after parabiosis and following 2-month detachment.
To quantify the epigenetic age of the animals, we applied four RRBS-based epigenetic clocks to 6 different groups of mice: young isochronic, old isochronic, and old heterochronic mice, as well as detached variants of these three groups. Clocks used included two recently developed multi-tissue clocks 5,31, a blood-specific clock 6, and a maximum likelihood-based single-cell clock 14, which was modified to accommodate and profile epigenetic age in bulk data. Application of these clocks to blood RRBS data revealed a profound epigenetic age decrease (24 ± 3.8%) when comparing old isochronic and heterochronic pairs. Interestingly, this effect was sustained even after two months of detachment (reduction in age by 22.4 ± 6.4%) (Fig. 2a). Application of these same four clocks to liver data showed comparable immediate (16.8 ± 7.9%) and sustained (19.1 ± 5.4%) epigenetic age reduction as observed in blood (Fig. 2b). Together, these results indicate that long-term HPB rejuvenates the epigenome compared to isochronic pairs in both blood and liver. Moreover, these unbiased rejuvenation signals in solid organs (such as the liver) further suggest that long-term HPB acts in a systemic manner, leading to global epigenomic remodeling and organism-wide age reversal.
Fig. 2 |. Persistent epigenetic age reversal in blood and liver upon HPB assessed by RRBS-based aging clocks.
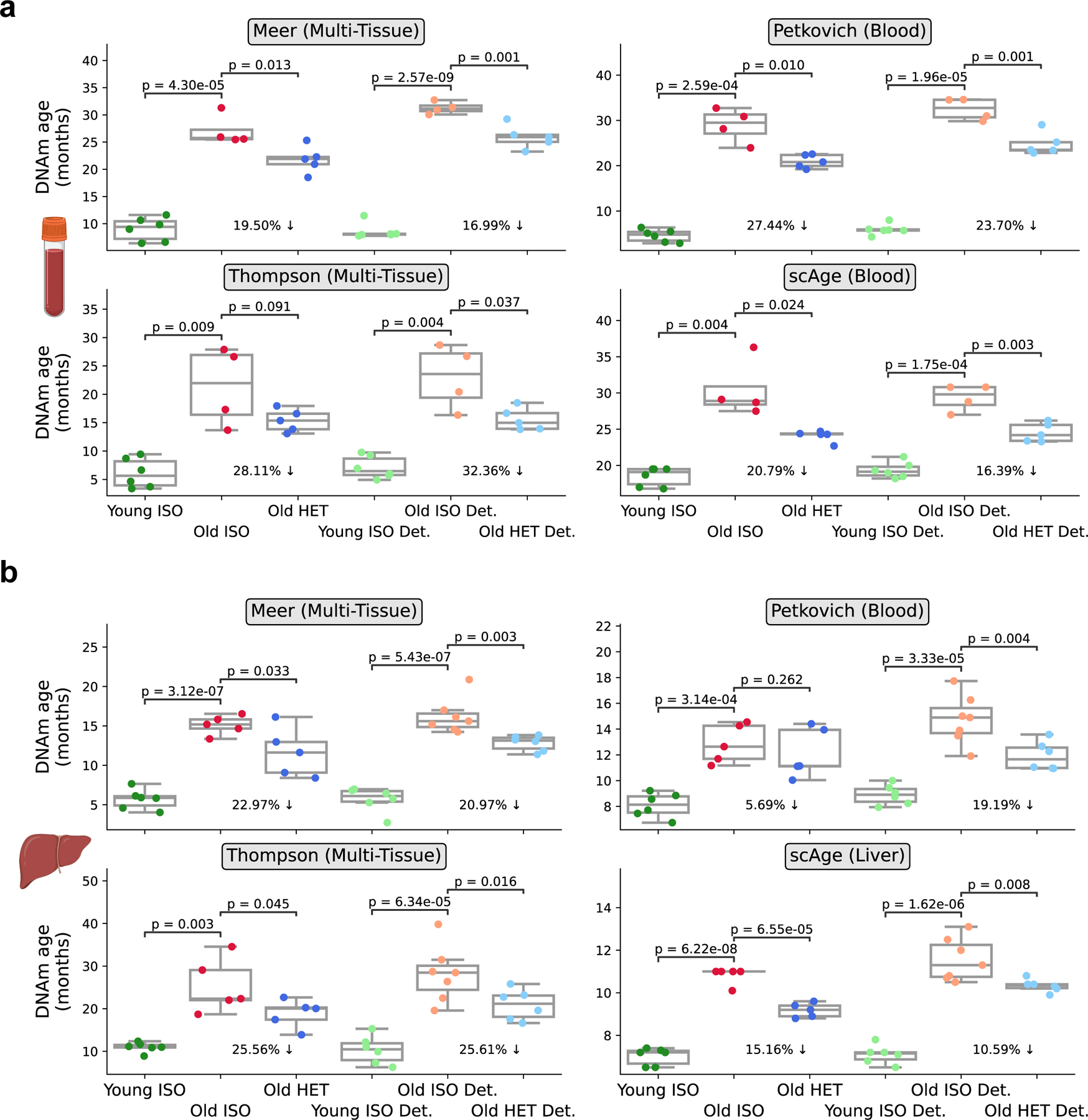
a, DNA methylation age of old heterochronic blood before and after detachment plotted with isochronic young and old controls assessed by the Meer multi-tissue clock, Petkovich blood clock, Thompson multi-tissue clock, and scAge blood clock. Young Iso (n = 6), Old iso (n = 4), Old HET (n = 5), Young ISO det. (n =6), Old ISO Det. (n = 4), Old HET Det. (n =5). B, DNA methylation age of old heterochronic liver before and after detachment plotted with isochronic young and old controls assessed by the Meer multi-tissue clock, Petkovich blood clock, Thompson multi-tissue clock, and scAge liver clock. Young Iso (n = 6), Old iso (n = 5), Old HET (n = 5), Young ISO det. (n =6), Old ISO Det. (n = 6), Old HET Det. (n =6) “Young ISO” denotes young mice from isochronic pairs, “Old ISO” denotes old mice from isochronic pairs, and “Old HET” denotes old mice from heterochronic pairs. “Det.” denotes detached old mice from isochronic or heterochronic pairs. Box plots represent median, 25–75 percentile and 1.5x IQR. One-tailed Welch’s t-tests assuming unequal variances were used for statistical analyses.
To confirm this epigenetic age decrease, we employed a completely independent analysis platform: methylation profiling of liver samples on the recently developed Mammalian Methylation Array 32,33 (Fig. 3, Table S2). We performed this profiling across 3 different long-term parabiosis groups (young isochronic, old isochronic, and old heterochronic) along with detached pairs of each of these groups. We applied four different array-specific epigenetic clocks. Two universal clocks were used: one based on relative age standardized to maximum lifespan and one based on log-linear transformed age standardized to the age of sexual maturity. Additionally, two mouse-specific clocks were used: a broad liver-specific clock and a clock tracking the epigenetic dynamics of the developmental process in the liver. Together, all four clocks were able to correctly predict the age of control isochronic samples with high precision and accuracy, and showed a highly significant decrease in epigenetic age when comparing old heterochronic and isochronic mice (22.3 ± 4.2% reduction in age) (Fig. 3a). Again, this effect was preserved after detachment, with a slight reduction in effect size (16.1 ± 3.4% reduction in age). Moreover, results from different platforms and organs were well correlated with each other (Extended Data Fig. 3a–b). These results from an independent platform further reinforce the notion that 3-month HPB leads to a profound, sustained, and systemic epigenetic rejuvenation.
Fig. 3 |. Sustained epigenetic age reversal in liver upon HPB assessed by microarray-based aging clocks.
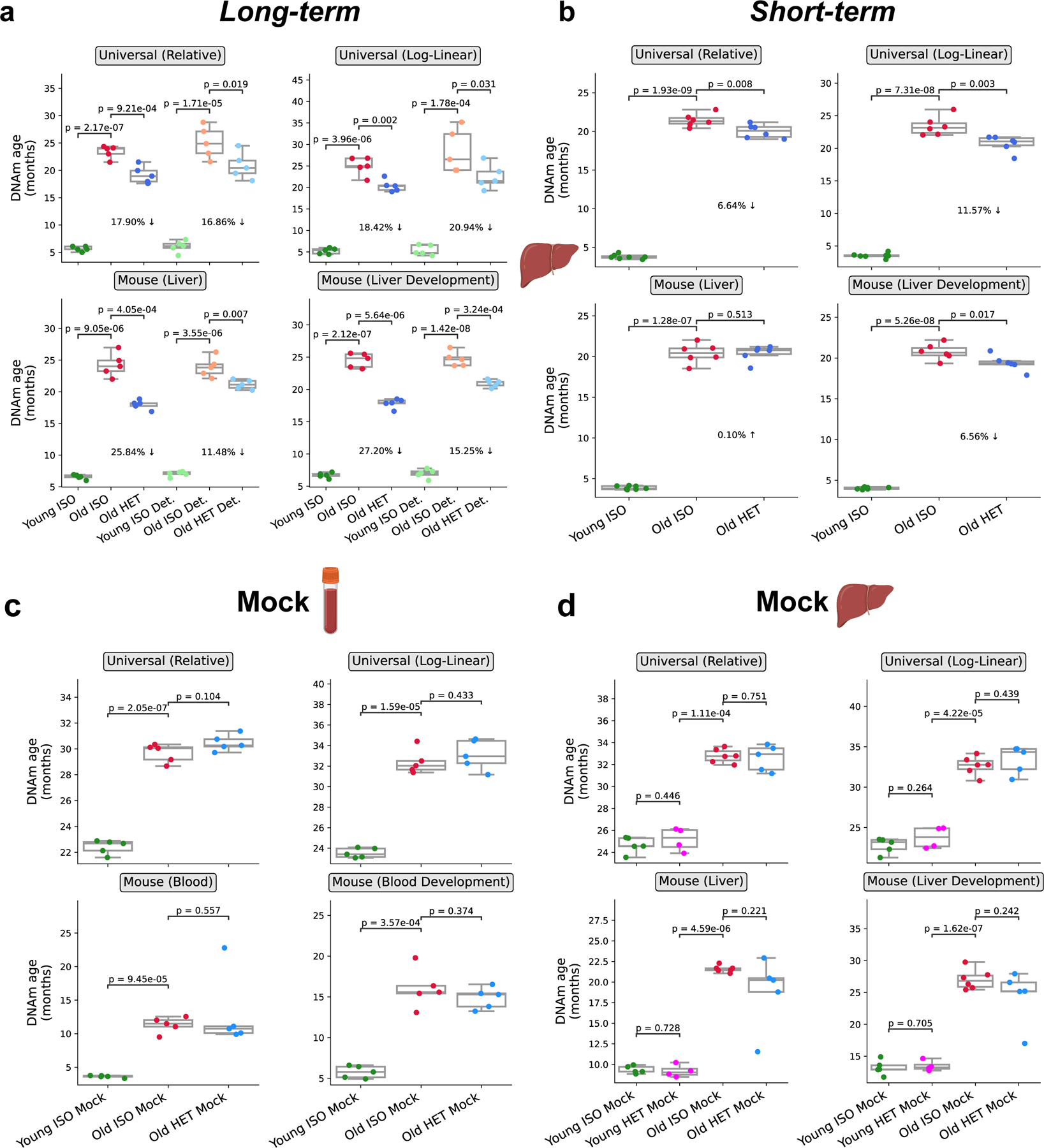
a, Epigenetic age of long-term (3-month) parabiosis liver samples from old heterochronic attached or detached mice, plotted with young and old isochronic controls, based on the universal relative age mammalian, universal log-linear transformed age mammalian, liver, and liver development clocks. (n = 5) per group. b, Epigenetic age of short-term (5-week) parabiosis liver samples from old heterochronic attached mice, plotted with young and old isochronic controls, based on the same clocks as (a). n = 6 per group. c-d, Epigenetic age of short-term (5-week) mock parabiosis blood (c) and liver (d) samples from old heterochronic attached mice, plotted with young and old isochronic controls, based on the same clocks as (a). (n = 5) per group for heterochronic mice and 6 per group for isochronic mice. Box plots represent 25–75 percentile and 1.5x IQR. One-tailed Welch’s t-tests were used for statistical analysis.
We then tested the difference between the commonly used 5-week, short-term protocol and our long-term (3-month) HPB protocol. Methylation array profiling of liver tissue was performed in three groups of animals (young isochronic, old isochronic, and old heterochronic) immediately after a 5-week attachment period. Only three of the four clocks applied detected a significant reduction in epigenetic age in heterochronic mice, and the effect size was dramatically smaller than in the long-term (3-month) parabiosis experiment (6.2 ± 4.1% reduction in age with short term HPB) (Fig. 3b). Notably, the clock trained specifically on mouse liver tissues did not detect this reduction. This suggests that while short-term parabiosis may induce some moderate epigenetic remodeling leading to mild rejuvenation readouts, a longer-term treatment clearly outperforms short-term attachment when it comes to immediate and particularly sustained rejuvenation of the liver epigenome.
To further determine whether a predicted epigenetic age-reversal (i.e., rejuvenation) occurs in the liver, an additional analysis of delta ages (the signed difference between epigenetic and chronological age) across all groups, including an untreated, nonsurgical control group, was performed (Extended Data Fig. 3c). This revealed that heterochronic pairs, both after 3-month attachment and after the 2-month detachment period, exhibited significantly lower epigenetic age compared to their chronological age. The same trend was observed with attached mice from short-term parabiosis experiments, but the effect size was dramatically stronger upon long-term treatment. In addition, the lack of difference between the old isochronic group and the old untreated control (Extended Data Fig. 3c) gives confidence that the parabiosis surgery itself does not alter the epigenome. We also tested this in young mice comparing the young isochronic group to a young nonsurgical control group (Extended Data Fig. 3d). Again, we found no significant difference with the age-matched non-surgical control mice. Additionally, we found that mean methylation was consistent among samples, suggesting that targeted epigenetic remodeling, manifesting itself in decreased epigenetic clock age, occurs as a result of long-term HPB (Extended Data Fig. 4).
A generally accepted mechanism for the health effects of HPB is youthful blood sharing. However, physical activity, especially in older populations can affect epigenetic age 34. The influence of physical activity on the epigenetic effects of HPB has been discussed but to our knowledge, not measured. Here, we developed a “mock” parabiosis model to attach mice without blood sharing for 5 weeks, matching our short-term HPB (Extended Data Fig. 5a–c). We observed a 32% decrease in voluntary cage activity in the old isochronic pairs compared to the young isochronic pair (Extended Data Fig. 5c–d). In contrast, activity in the heterochronic pair was similar to the young isochronic groups. Moreover, the heterochronic group had a trend of increased vertical activity when compared to the old isochronic group. Despite the changes in physical activity, the mock parabiosis did not alter epigenetic age in either blood or livers (Fig. 3c, d), suggesting blood sharing is the pre-requisite to obtain changes in the epigenetic age in the HPB model.
In addition to using clocks on the epigenetic data, we also explored the impact of HPB on the promoter methylation levels of individual genes. We identified several genes with significant differential mean promoter methylation across old isochronic and heterochronic mice, in both attached and detached samples (Extended Data Fig. 6d). Among the genes, we observed decreased promoter methylation in Ubl5, which has previously been linked to lifespan modulation in nematodes 35, as well as Mpped1, which was previously identified as an age-regulated gene in the brain across humans, macaques, and mice 36.
Taken together, we report that a systemic rejuvenation occurs in old animals upon exposure to youthful circulation, as assessed by 8 distinct epigenetic clocks across two independent DNA methylation analysis platforms. Furthermore, we show the slowing of epigenetic age is sustained even after a 2-month detachment period. These results present the first example of a systemic in vivo rejuvenation quantified by molecular biomarkers in mice.
Gene expression analyses reveal pathways responsive to long-term parabiosis
Given the notable epigenetic age reversal, we sought to concurrently elucidate the transcriptomic changes that occur as a result of HPB. We performed RNA-seq analyses of liver tissues in old isochronic and heterochronic mice, as well as in detached groups of both conditions, followed by differential gene expression and gene set enrichment analyses (GSEA) (Fig. 4a). Using the Hallmark, KEGG, REACTOME and GO BP gene sets obtained from MSigDB 37,38, we computed normalized enrichment scores comparing isochronic and heterochronic samples immediately after the 3-month parabiosis period, as well as after 2 months of detachment (Fig. 4a). Among samples taken immediately following the attachment period, we observed strong enrichment of the oxidative phosphorylation and mitochondrial biogenesis gene sets in heterochronic mice. Interestingly, oxidative phosphorylation is known to be disrupted during the aging process 39. Within this context, our results indicate that HPB may reverse some of the age-associated decline commonly associated with this critical energy production pathway. We also observed depletion of inflammatory and interferon gamma (IFN-gamma) response gene sets in heterochronic mice (i.e. enrichment of inflammation in old isochronic mice). IFN-gamma protein level is known to increase with age in some tissues (particularly in T-cells) 40, and inflammation is one of the crucial hallmarks of aging 2 associated with the development of various age-related pathologies, including cancer 41, Alzheimer’s disease 42 and chronic kidney disease 43. These results further support the rejuvenating effect of heterochronic parabiosis on murine transcriptomic profiles.
Fig. 4 |. Transcriptomic analyses of HPB reveal unique pathway enrichment and differential expression patterns.
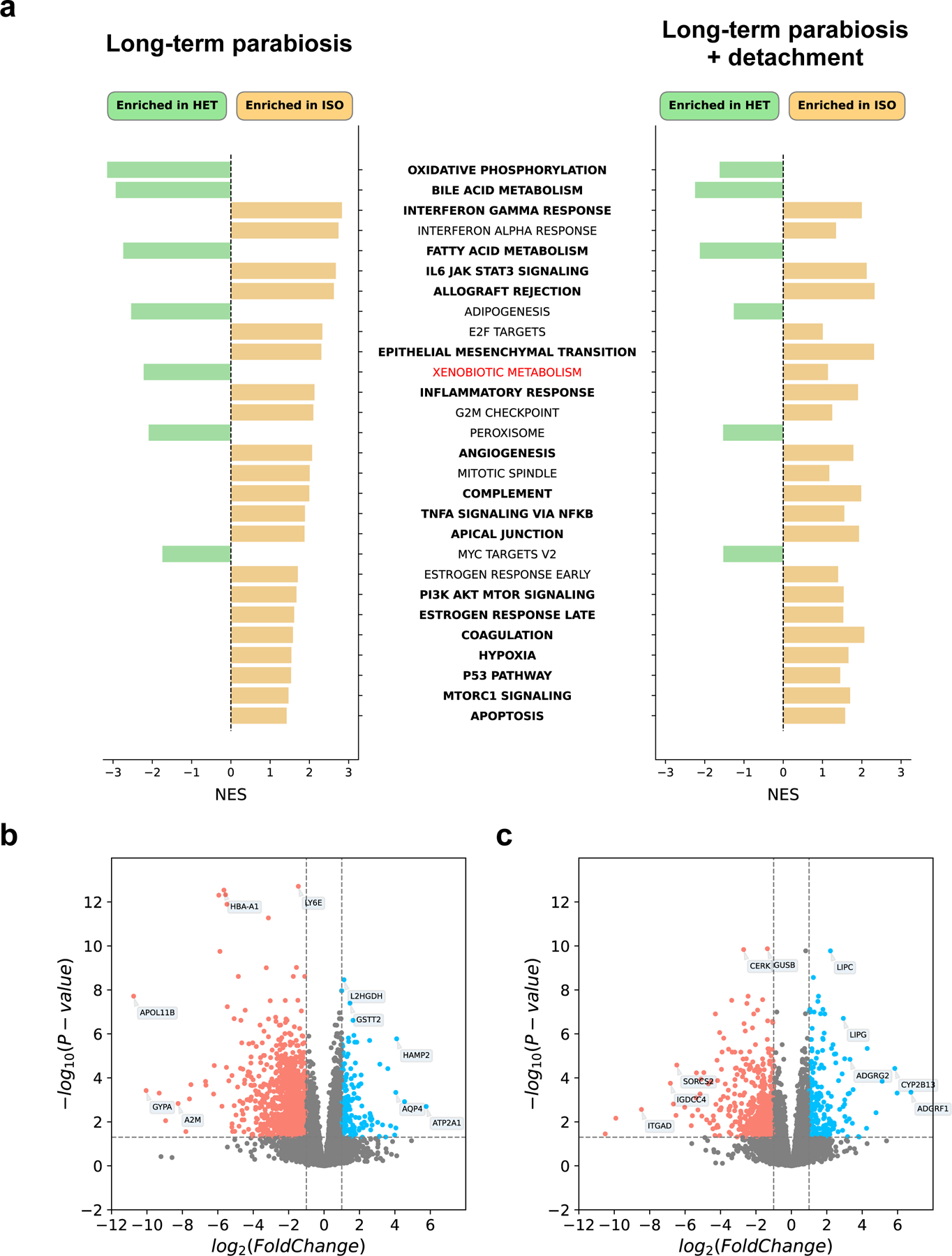
a, GSEA of old isochronic vs old. heterochronic mice (left), as well as of detached old isochronic vs. detached old heterochronic mice (right). Hallmark gene sets (n = 50) were used as input to GSEA. Only significant enrichments in attached groups (adjusted p-value < 0.05) are shown. Gene sets shown in green signify pathways enriched in heterochronic samples, and orange those enriched in isochronic samples. Terms in bold signify pathways that are significantly enriched in the same direction in both attached and detached groups. Terms in normal black font signify pathways that are enriched in the same direction in both attached and detached groups, but only significantly so in the attached comparison. The term in red signifies a pathway that is enriched in different directions in the attached and detached comparisons. Significant gene sets are ranked from top to bottom based on the absolute value of the normalized enrichment score in the attached group. b-c, Volcano plot of differentially expressed genes in attached (b) and detached (c) old heterochronic (n = 3) and old isochronic (n = 3) mice. Some highly significant genes are shown in each plot. Genes shown in red are significantly downregulated in heterochronic mice, while genes shown in blue are significantly upregulated in heterochronic mice, after multiple-testing correction.Two-tailed Benchamini-Hochberg corrected FDR was calculated to compare groups.
To investigate whether transcriptomic changes at the gene-set level are sustained, we compared the GSEA results from samples taken immediately after the attachment period to those after 2 months of detachment (Fig. 4a). We observed a strong positive correlation between the enrichment scores obtained for the attached and detached groups (Spearman ρ = 0.72, p-value < 10−16), and the top significantly enriched pathways in the detached analyses were common in identity and direction with those in the attached group, suggesting that transcriptomic changes in liver resulting directly from HPB are largely sustained even after prolonged detachment (Fig. 4a). Of note, the normalized enrichment scores and number of significantly enriched gene sets were generally decreased in the detached group, despite the same number of samples being analyzed (Fig. 4a). This may indicate the gradual reduction of the HPB effect following detachment.
We also conducted global differential gene expression analyses, comparing isochronic and heterochronic samples after the 3-month attachment period or after 2-month detachment. We identified 1044 significantly (adjusted p-value < 0.05) upregulated and 1855 downregulated genes in the post-attachment comparison (Fig. 4b), compared to 876 upregulated and 1034 downregulated genes in the detached comparison (Fig. 4c). Together, these data suggest that the liver transcriptome is rejuvenated upon HPB, and that this rejuvenation largely persists even after a 2-month detachment.
Dimensionality reduction reveals intermediate molecular phenotypes resulting from heterochronic parabiosis
Given the high dimensionality of epigenomic and transcriptomic data, we opted to use principal component analysis (PCA) to embed and visualize samples in two-dimensional space. We observed that heterochronic parabiosis samples clustered between young and old isochronic samples, closer to the young samples than the old (Fig. 5a). While clustering of short-term parabiosis also showed an intermediate transcriptomic profile for old heterochronic mice, these samples clustered much closer to the old isochronic group (Fig. 5b). Together with the epigenetic clock data, this further suggests that long-term parabiosis is a much more potent age-reversal intervention compared to short-term parabiosis.
Fig. 5 |. Dimensionality reduction highlights rejuvenated molecular profiles following HPB.
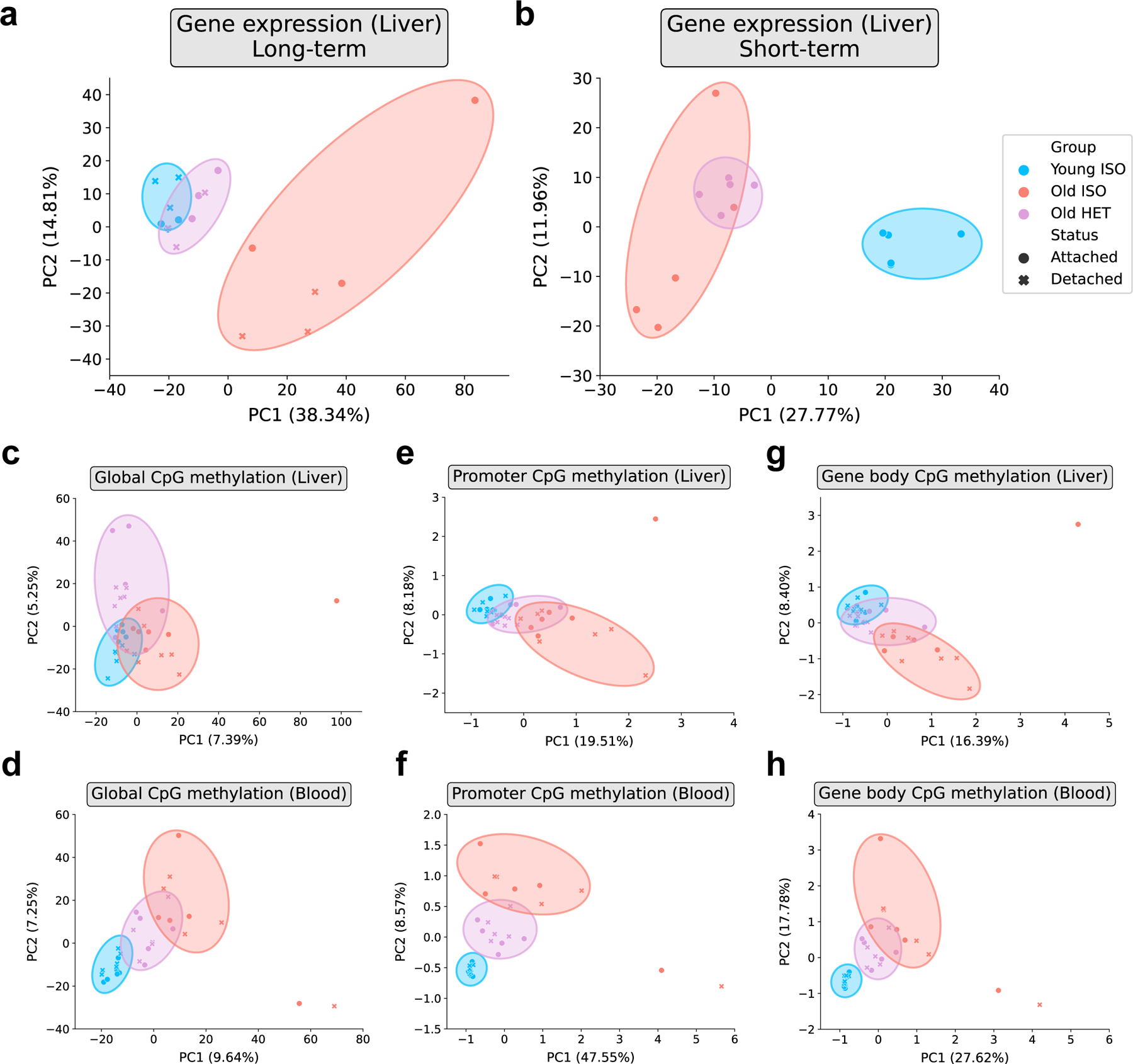
a, Principal Component Analysis (PCA) of liver RNA-seq data of mice following long-term parabiosis or 2 months after detachment (n = 3 per group). b, PCA of liver RNA-seq data of short-term parabiotic mice (n = 3 per group) c, PCA of liver RRBS data across all highly covered common CpGs (1,014,243 CpGs, n = 5–7 per group) d, PCA of blood RRBS data across all highly covered common CpGs (1,014,243 CpGs, n = 5–6 per group) e, PCA of liver RRBS data across gene promoters (11,842 promoters, n = 5–7 per group) f, PCA of blood RRBS data across gene promoters (11,842 promoters, n = 5–6 per group) g, PCA of liver RRBS data across gene bodies (13,811 gene bodies, n = 5–7 per group) h, PCA of blood RRBS data across gene bodies (13,811 gene bodies, n = 5–6 group). “Young ISO” denotes young isochronic mice, “Old ISO” denotes old isochronic mice, and “Old HET” denotes old heterochronic mice. Attached refers to samples taken immediately after the parabiosis period, while detached refers to samples taken after 2 months of detachment. Percentages of variance explained by the first two principal components are shown in parentheses on the axes.
We also performed PCA on the RRBS methylation data from liver and blood samples (Fig. 5c–h, Extended Data Fig. 6a–c). We analyzed methylation at individual CpG sites, as well as through concatenation into two types of genomic regions known to be functionally influenced by cytosine methylation: promoters (Table S3) and gene bodies (Table S4). Across both tissues, we observed that old heterochronic samples cluster in between old and young isochronic samples when analyzing over 1 million highly-covered CpGs (Fig. 5c–d). These results become especially pronounced when collapsing methylation to promoters and gene bodies, where heterochronic samples fall perfectly between the two isochronic groups (Fig. 5e–h).
Altogether, application of dimensionality reduction to our data suggests that old heterochronic mice have transcriptomic and epigenetic profiles intermediate between young isochronic and old isochronic mice. This effect is apparent across both the liver and blood, and we moreover observe that long-term parabiosis produces a more profound shift in transcriptomic profiles towards a young state compared to short-term parabiosis. This further indicates that global and sustained transcriptomic and epigenomic remodeling occurs as a result of long-term, but not short-term HPB.
Gene expression changes induced by heterochronic parabiosis mirror longevity intervention signatures and oppose aging
To further assess if HPB results in systemic rejuvenation of the transcriptome, we compared the identified gene expression changes with signatures associated with lifespan extension and aging, obtained through a previously published meta-analysis of multiple publicly available datasets. They include liver-specific gene signatures of individual interventions (such as caloric restriction and mutations leading to growth hormone deficiency), gene expression changes shared across different lifespan-extending interventions, and genes whose expression is associated with the quantitative effect of interventions on maximum and median lifespan. In contrast, aging signatures reflect age-related transcriptomic changes observed in the liver of mice and rats, as well as multi-tissue age-related alterations in each of these species.
We evaluated the association between parabiosis-induced changes and these signatures using a previously described GSEA-based approach 44. In heterochronic samples taken immediately after the 3-month parabiosis or after the 2-month detachment period, we observed a strong and significant negative association with 3 aging signatures, including the mouse liver, rat multi-tissue, and mouse multi-tissue aging signatures (adjusted permutation p-value < 5e-4) (Fig. 6a). On the contrary, there was a strong positive association between HPB in old mice and 4 out of 5 signatures from lifespan-extending interventions, including positive enrichment for median and maximum lifespan signatures and those for caloric restriction and growth hormone deficiency (adjusted permutation p-value < 5e-4). Of note, significant absolute normalized enrichment scores were consistently decreased in magnitude (20.4 ± 11.3% reduction) in detached samples compared to samples taken immediately after the long-term parabiosis period (Fig. 6a). Consistent with the functional enrichment analysis presented earlier, this suggests that the majority of transcriptomic changes are sustained after the detachment period, but the magnitude of the rejuvenating effect may be gradually diminished over time.
Fig. 6 |. The transcriptomic signatures of HPB align with longevity interventions and oppose aging.
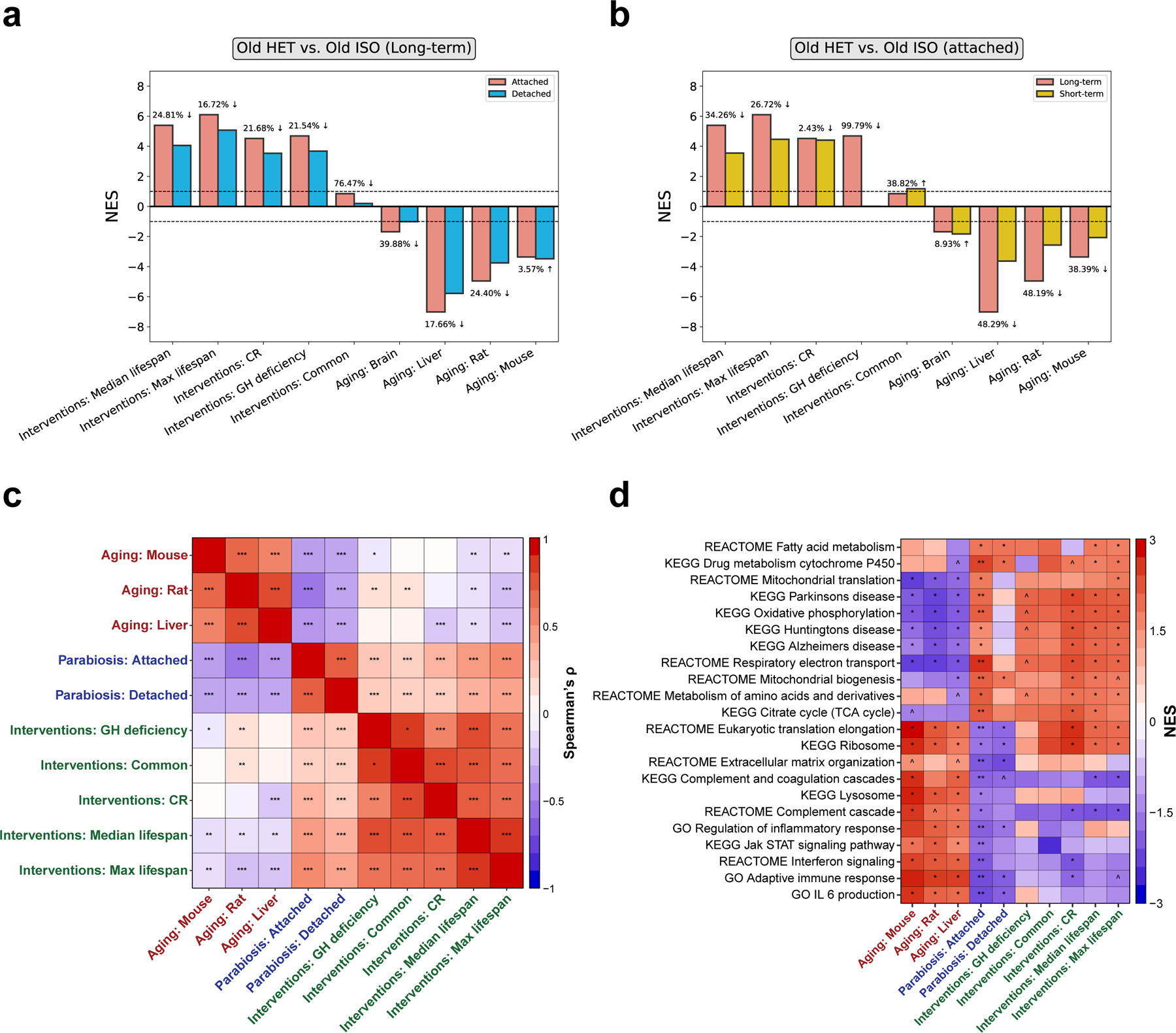
a, Association between gene expression changes induced by long-term parabiosis with (blue) and without (red) a 2-month detachment period, and signatures of aging and life span extension. The latter include gene signatures of individual interventions (CR and GH deficiency), common intervention signatures (interventions: common) and signatures associated with an effect on life span (maximum and median life span). The percentage decrease comparing the attached and detached samples is shown for each pair of bars. b, Association between gene expression changes induced by long-term (red) and short-term (yellow) parabiosis without detachment and signatures of aging and life span extension. The signatures analyzed are the same as in a. c, Spearman correlation matrix of gene expression signatures associated with aging (red labels), life span extension (green labels) and HPB (blue labels). d, Functional enrichment analyses of gene expression signatures. Only functions significantly associated with at least one signature are shown. Cells are colored based on NES. The entire list of enriched functions is provided in Supplementary Table 5. P values were generated using GSEA. ^Padj < 0.1; *Padj < 0.05; **Padj < 0.01; ***Padj < 0.001.
To refine the molecular resolution of longevity effects based on the duration of the parabiosis intervention, we also performed transcriptomic signature analysis for short-term (5-week) parabiosis samples. In accordance with the previous observations, we observed significant positive associations of HPB with several signatures of lifespan extension, while aging signatures demonstrated the opposite effect (Fig. 6b). This suggests that short-term parabiosis leads to important transcriptomic changes similar to those produced by established longevity interventions. However, the magnitude of association for short-term parabiosis was greatly reduced compared to long-term attachment. Therefore, long-term parabiosis seems to induce more profound lifespan-extending and rejuvenating effects at the level of gene expression and DNA methylation, highlighting the enhanced effect of the long-term HPB protocol.
To investigate mutual associations between the changes induced by heterochronic parabiosis, lifespan extension and aging, we calculated Spearman’s correlation coefficients of the log fold-change of genes for each pair of signatures. Consistent with the GSEA-based association analysis, the effects induced by HPB in old mice clustered together with the changes induced by lifespan-extending interventions and were positively correlated with them (Spearman’s ρ = 0.22; adjusted p-value < 2e-5), but negatively correlated with aging-related changes (Spearman’s ρ = −0.3; adjusted p-value < 3e-9) (Fig. 6c). Interestingly, based on our data, the negative association between aging and parabiosis in older animals was even more substantial than between aging and existing lifespan-extending interventions, such as caloric restriction (Spearman’s ρ = −0.22).
To understand which gene sets are responsible for the age-reversal and lifespan-extending effects of HPB, we performed functional GSEA for the above-mentioned aging and longevity signatures (Fig. 6d, Table S5). We observed that some functions that were upregulated in response to parabiosis, including the TCA cycle, respiratory electron transport and mitochondrial biogenesis, were also upregulated by lifespan-extending interventions, but downregulated during aging. At the same time, functions related to immune response, such as interferon signaling and complement and coagulation cascades, were significantly downregulated both in response to HPB and established lifespan-extending interventions, but upregulated with age. Therefore, HPB in old mice seems to counteract aging by activating genes related to metabolism and cellular respiration while inhibiting inflammatory response, similar to other longevity interventions.
In addition, to understand how the effects of parabiosis compare across the transcriptome and epigenome, we analyzed the correlation of the changes in promoter methylation and RNA expression comparing old isochronic and heterochronic mice, in both the attached and detached experimental groups (Extended Data Fig. 7a). Interestingly, we found that while the same readout modalities correlate well with each other across attached and detached groups (RRBS r = 0.67, RNA r = 0.72), there is much smaller correlation between parabiosis-induced changes when comparing the transcriptome and epigenome directly (r = 0.2). We also looked at the directional concordance of differentially expressed/methylated genes across readout types and attached/detached groups (Extended Data Fig. 7b). Again, we found very strong concordance when comparing the same assay types (RRBS or RNA) across attached and detached experiments (RRBS concordance = 88%, RNA concordance = 92%), but significantly less concordance when comparing RRBS with RNA in the same experiment (attached concordance = 65%, detached concordance = 60%, with 50% concordance signifying random effects). Taken together, we found that the effect of heterochronic parabiosis is largely conserved between attached and detached groups when accounting for each modality separately. However, there was smaller concordance between the magnitude and direction of changes induced by HPB when comparing promoter methylation and expression levels of individual genes. This suggests that parabiosis is not acting entirely through the epigenome to affect gene transcription, and that instead several layers of biological regulation are involved in the global rejuvenation effect we observe.
To summarize, we observed that HPB indeed rejuvenated old animals systemically, and this was supported by both epigenetic and transcriptomic remodeling induced by this intervention. Excitingly, signature analyses of HPB profiles appear to recapitulate gene expression effects of established lifespan-extending interventions, pointing to the potential utility of derivatives of this therapy in promoting healthy longevity.
Individual gene expression dynamics reveal putative longevity-associated mechanisms for HPB
To uncover the potential mechanisms underlying the rejuvenation effects we observed upon long-term parabiosis, we investigated the individual genes regulated in response to HPB and their association with longevity and aging based on the gene expression signatures (Fig. 7). We found that 27–45% of genes significantly changed in response to long-term and short-term HPB were also significantly associated with maximum and median lifespan extension in the same direction (Fisher’s exact test p-value < 0.03 for all models of HPB). Additionally, there was a significant overlap of genes downregulated in response to long-term heterochronic parabiosis and upregulated with age according to all tested aging signatures (Fisher’s exact test p-value < 0.004 for long-term attached and detached groups).
Fig. 7 |. Gene expression analyses hint at putative rejuvenation mechanisms of long-term HPB.
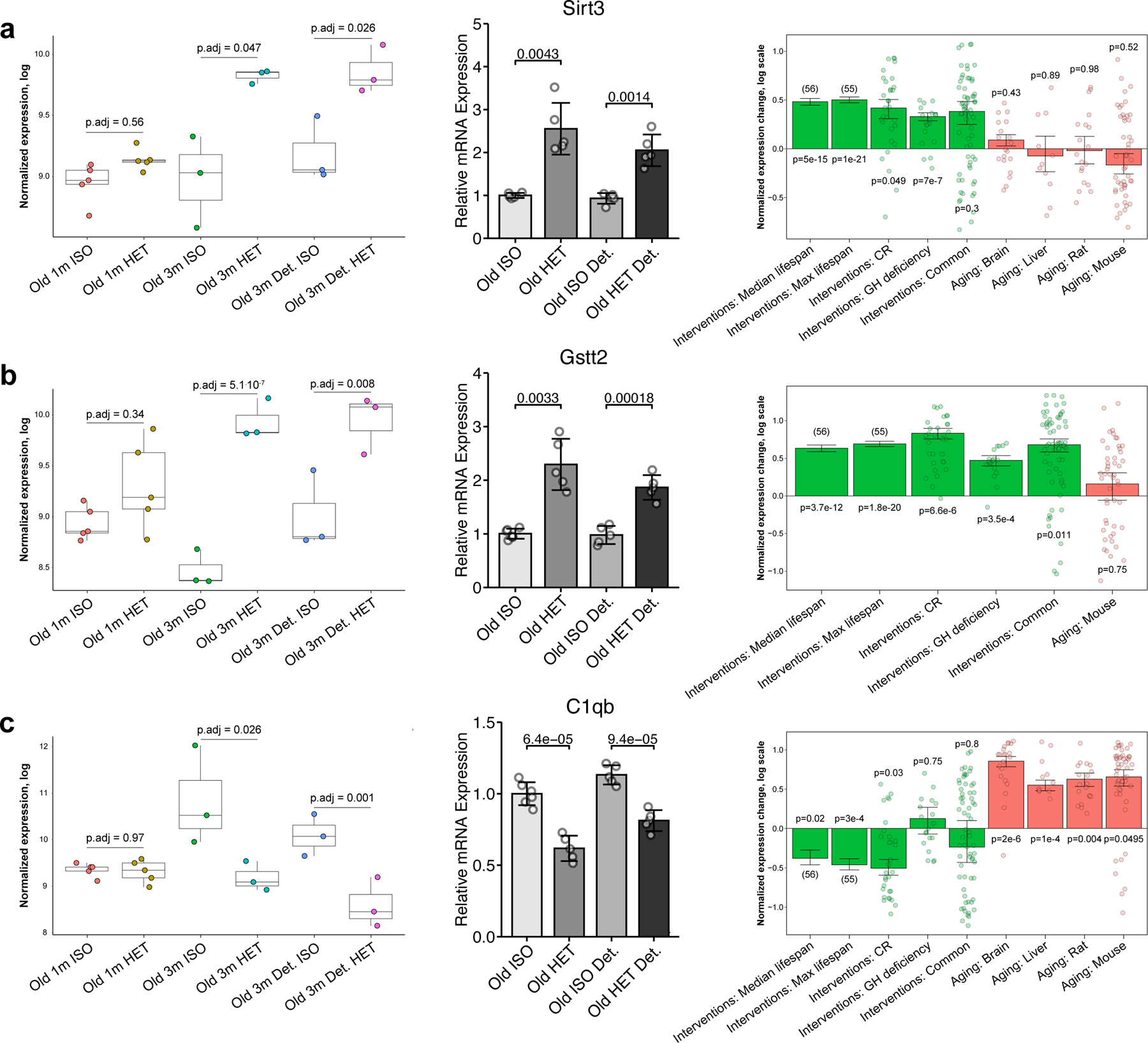
a–c, Expression of Sirt3 (a), Gstt2 (b) and C1qb (c) in mice subjected to parabiosis by RNA-seq analyses (left), RT–qPCR analyses (middle) and in response to established life span-extending interventions and aging (right). Left: for each gene, normalized expression in logarithmic scale is shown across different parabiosis groups: short-term isochronic (old 1 m ISO, n = 5) and heterochronic (old 1 m HET, n = 5) mice, long-term attached isochronic (old 3 m ISO, n = 3) and heterochronic mice (old 3 m HET, n = 3), long-term detached isochronic (old 3 m detached ISO, n = 3) and heterochronic mice (old 3 m detached HET, n = 3). Adjusted P values, indicating the difference in expression for each pair of isochronic and heterochronic mice, were calculated using a preplanned two-tailed Student’s t-test. The box plots represent the median, 25–75 percentiles and 1.5× IQR. Middle: for the RT–qPCR of each gene, each group has n = 5 biological replicates. The P value between connecting bars was calculated with a two-tailed Student’s t-test. Right: for every signature associated with life span-extending interventions (green) and aging (red), the means of the normalized log fold changes or slopes (for the signatures of the median and maximum life span) are presented. The error bars denote ± 1 s.e. Adjusted P values, indicating the difference of expression change from zero, were calculated with a mixed-effect linear model.
Among genes upregulated by long-term HPB in both attached and detached models, we identified Sirt3 (attached adjusted p-value < 0.047) (Fig. 7b). Sirt3 deficiency is known to promote cancer 45 and aging 46,47, while its overexpression has resulted in the decrease of reactive oxygen species levels and improvement of regenerative capacity in aged stem cells 47. Not surprisingly, its expression in liver is upregulated by many longevity interventions, including CR and growth hormone deficiency (adjusted p-value < 0.049), and is positively associated with the effect of interventions on median and maximum lifespan extension in mice (adjusted p-value < 10−14) (Fig. 7a).
Other important hits, connecting the effects of HPB with mechanisms of established longevity interventions, are genes involved in glutathione metabolism, such as Gstt2, encoding for glutathione S-transferase theta 2. Gstt2 was upregulated in response to long-term heterochronic parabiosis and remained increased after detachment (detached adjusted p-value < 0.008) (Fig. 7b). These findings point to some putative specific longevity mechanisms induced by long-term heterochronic parabiosis. Methionine metabolism was shown to play an important role in the regulation of longevity, and the increased blood level of glutathione is known to be an important biomarker of caloric 48 and methionine restriction 49. Consistent with this data, the expression of Gstt2 demonstrated significant positive association with the effect of various lifespan-extending interventions (adjusted p-value < 4×10−12 for signatures of median and maximum lifespan extension) (Fig. 7b).
Among genes downregulated by HPB and longevity interventions but upregulated with age, we identified several involved in the immune response such as C1qb, which encodes for one of the complement subcomponents (Fig. 7c). C1qb was downregulated in response to long-term heterochronic parabiosis both immediately after attachment and following 2 months of detachment (adjusted p-value < 0.03) (Fig. 7c), This gene appears to be positively associated with aging in liver and brain in both mice and rats (adjusted p-value < 0.05 for all aging signatures). At the same time, its expression in murine liver is downregulated by caloric restriction (adjusted p-value = 0.028) and demonstrates negative association with median and maximum lifespan extension induced by interventions (adjusted p-value < 0.023) (Fig. 7c). We confirmed the gene expression dynamics observed via RNA-seq for Sirt3, Gsst2, and C1qb by qRT-PCR, and observed concordant changes in protein expression as detected by western blotting (Fig. 7, Extended Data Fig. 6e and f).
We also observed a significant increase in Tert expression following long-term HPB (adjusted p-value = 4.3e-5), which appeared to be sustained in detached samples (adjusted p-value = 0.0014) (Extended Data Fig. 8a). Although Tert, encoding for telomerase reverse transcriptase, doesn’t demonstrate significant upregulation in response to various longevity interventions, the overexpression of this gene by itself has resulted in lifespan extension in healthy mice without induction of cancer 50.
Another interesting hit was Dnmt3b, which encodes one of the key enzymes involved in de novo methylation of the genome. We observed a significant decrease in Dnmt3b expression when comparing isochronic and heterochronic mice from long-term and short-term parabiosis (Extended Data Fig. 8a). This effect was also sustained in detached samples from long-term HPB. While de novo methylation occurs primarily during embryogenesis and appears to be responsible for a rejuvenation event around the time of gastrulation, this finding may additionally implicate Dnmt3b as a key driver in the modulation of epigenetic remodeling induced by HPB 13,14,51. We also observed significant downregulation of other genes induced by both short-term and long-term HPB, including Ly6e, Lmna and Pld1 (Extended Data Fig. 8a). Notably, the expression of these genes was at the same time negatively associated with the effect of longevity interventions on median and maximum lifespan, indicating that they may also mediate the beneficial effect of HPB. Consistent with our other data, the gene effect size was strongest in long-term attached samples, intermediate in long-term detached, and lowest in short-term attached samples, again reinforcing the utility of a long-term attachment period for transcriptomic rejuvenation.
To determine whether the observed gene expression changes were related to upstream epigenetic changes, we examined the mean promoter methylation levels of these genes (Extended Data Fig. 8b). Interestingly, we observed minimal differences in promoter methylation across samples for these genes, despite observing significant changes at the transcriptional level. This suggests that, at least for some genes, changes in promoter methylation are largely not responsible for changes in gene expression patterns in our data. Furthermore, for the genes that were found to have significant differential promoter methylation across isochronic and heterochronic mice in attached and detached samples (Extended Data Fig. 6d), none had significant changes in expression levels across any comparison. This finding further supports the notion that parabiosis acts through multiple layers of regulation, instead of only through methylome modifications.
Lastly, we also observed strong negative enrichment of senescence-associated secretory phenotype (SASP) genes (Table S6) in heterochronic mice immediately after long-term parabiosis (adjusted p-value = 4e-4) (Extended Data Fig. 9a). Expression of SASP-related genes is known to increase with age 52, suggesting that HPB ameliorates the pro-inflammatory phenotype of the aging liver. This negative enrichment was also observed in detached heterochronic samples (adjusted p-value = 0.01) and in short-term heterochronic samples (adjusted p-value = 0.008), but the effect in both was noticeably smaller (Extended Data Fig. 9b–c). Gene-specific analyses among several key SASP genes (Cdkn1a, Il1r1, Cxcl13, Igfbp7, Tgfb1) further revealed significant expression changes between old heterochronic and isochronic mice after 3-month attachment and after 2-month detachment, but not after short-term attachment (Extended Data Fig. 9d).
Together, these results suggest that long-term HPB causes profound transcriptomic change at the level of gene sets and individual genes associated with longevity and rejuvenation. STRING clustering of upregulated and downregulated genes further indicates commonalities in the regulatory gene ensembles that are modified as a result of HPB (Extended Data Fig. 10a–b). The observed shared genes and pathways point to the existence of crucial and interconnected molecular mechanisms driving the rejuvenation effect of long-term heterochronic parabiosis, some of which have been described in this work.
DISCUSSION
The history of HPB dates back to the mid-20th century, and it has re-emerged as an important model in aging research since 2005 19,26,53. In the past, most of these studies have focused primarily on phenotypes, such as tissue regeneration, brain function and stem cell characteristics. It has remained enigmatic whether this intervention could systematically reverse the epigenetic age of organisms and, moreover, whether the effects of HPB lasts after a prolonged detachment period. One challenge in this regard has been that the duration of circulatory system attachment in animals was too short to observe robust differences in epigenetic age. By adapting a long-term parabiosis protocol to include detachment and by using precise molecular biomarkers of aging, we demonstrate here for the first time that exposure to the young circulatory system leads to persistent and systemic slowing of epigenetic aging. Notably, this effect correlates with a longer lifespan, improved physiological parameters, and a globally rejuvenated epigenome and transcriptome.
We found that multiple epigenetic clocks point to slowing of epigenetic age, and the effect was observed independently using two different profiling platforms: high-throughput sequencing (i.e. RRBS) and microarrays. Importantly, we found that HPB decreases the epigenetic age acceleration (i.e. delta age) even compared to non-parabiosed control animals, suggesting that this effect is an actual reversal of epigenetic age rather than an amelioration of surgery-induced age acceleration. It is important to note that while almost all clocks showed significant epigenetic age reversal, some clocks showed only marginal differences. This most often happens when there exists incongruence between the tissues used for clock training and the actual tissues tested, i.e., the blood clock is not as precise in assessing epigenetic age in the liver.
The degree of rejuvenation we observed in our work is consistent with previous studies, which showed that HPB ameliorates aging phenotypes of tissues other than the blood, such as the muscle, liver and nervous system. While our study was under review, Yankova et al. published data using a similar HPB approach followed by detachment, where they showed a striking decrease in the life span of the young parabiont54. Although our life span analyses focused only on the old detached mice, our data and those of Yankova et al. were confirmatory. We both show a similar, modest increase in life span in old mice following detachment from HPB, despite differences in mouse strain, sex and exact timing of surgical interventions.
It is probable that cells and molecules in circulation contribute to the effect of parabiosis on solid organs and therefore affect cells resident to these tissues 22,55–57. Our finding further fortifies the view that aging is a systemic process 58. Additionally, we observed that gene expression changes induced by HPB in old mice (compared to isochronic controls) were negatively correlated with changes induced by aging, but positively correlated with changes induced by longevity interventions. Consistent with a recent study examining the effect of short-term parabiosis on single-cell gene expression 59, our findings indicate that what we observed is in fact multi-omic, systemic, and sustained biological rejuvenation. Although our methylation clock analyses showed a decrease in epigenetic age up to 30% from HPB, the lifespan extension effect on the detached mice, while still significant, was not as strong. This discrepancy may result from the numerous comorbidities occurring later in the mouse lifespan.
The complexity of the HPB model lends several caveats beyond sharing of “youthful” factors to understanding its biology. This includes, but is not limited to, contributions from cellular chimerism, surgical stress, and/or changes in physical activity. In regards to cellular chimerism, we did not believe this to be a factor in the maintenance of healthspan and reduced epigenetic age in our model. Although previous studies on congenic mouse strains reported that 3–5% of the blood from the parabiont remains in the body after 7 weeks of detachment 27, our model showed limited evidence of young cells in the old bone marrow or in circulation after the detachment period. The lack of bone marrow chimerism with HPB is in agreement with previous reports 29. Therefore, we believe any extension of healthspan and epigenetic alterations are based on the anti-aging effects during the anastomosis, with minimal contribution from the lingering young blood cells after detachment. Another effect of HPB, often discussed but seldom tested, is the effect on physical activity. We developed a non-surgical mock parabiosis model, enabling us to physically attach mice without blood sharing. We were surprised to see greater cage activity in the mock heterochronic pairs compared to the mock old isochronic group, which warranted further analysis to determine its effect on epigenetic remodeling. Despite the increased physical activity, we did not see any changes in epigenetic age, ruling out activity as a confounding variable to influence epigenetic age predictions in our HPB model. Lastly, we showed no effect of the parabiosis surgery itself on epigenetic age. While there are subtle differences in surgical methods across the parabiosis literature, it is important to know our model was not altering epigenetic age when compared to age-matched non-surgery controls. Together, these data help support the theory of blood factor transfer being the primary mechanism of rejuvenation.
There have been attempts to identify the components involved, and some factors from young blood like oxytocin were reported to recapitulate the physiological rejuvenation effect 60,61. It was also reported that following bone marrow transplantation, the blood of recipients maintains the epigenetic age of the donors, indicating that there may be factors intrinsic to blood cells and their precursors contributing to this effect 62. Another possibility could be that aged organisms accumulate damage, which can be effectively diluted by young blood during parabiosis experiments. This hypothesis is supported by a recent experiment that involved replacing the blood plasma with saline containing 5% albumin, leading to enhanced muscle repair and hippocampal neuro-regeneration 63; however, this has yet to be demonstrated using a longitudinal model. It is critical to note that there has been no evidence yet from aging clocks favoring either of these two hypotheses.
Overall, our findings reveal a robust decrease in epigenetic age upon long-term HPB, which remains strongly significant after a period of prolonged detachment. This is coupled with a strong positive correlation between parabiosis and longevity interventions and a negative correlation between parabiosis and aging based on several gene expression signatures. Gene set and pathway analyses revealed a positive enrichment of TCA cycle, oxidative phosphorylation, mitochondrial biogenesis and fatty-acid metabolism pathways as well as depletion (i.e. negative enrichment) of IFN-gamma and inflammatory response pathways in old heterochronic mice. Additionally, several longevity-mediated genes were identified, including Gstt2, Sirt3, C1qb and Tert, that showed sustained expression changes following long-term parabiosis. Importantly, we validate here that long-term (3-month) parabiosis is much more effective at rejuvenating the transcriptome and epigenome of mice compared to short-term (5-week) parabiosis. This long-lasting rejuvenation effect of HBP in old mice leads to the extension of healthspan and lifespan even after this traumatic surgery, and produces molecular profiles intermediate between young and old mice. It should also be noted that although we observed a strong conservation of effects using both modalities (RRBS and RNA-seq) across attached and detached groups, the concordance between the two omics readouts was smaller, likely indicating that parabiosis simultaneously affects different layers of biological regulation. At the moment, several interventions or biological events have been shown to reverse epigenetic age, including rejuvenation during early embryogenesis, rejuvenation by pharmaceutical treatments and rejuvenation via reprogramming factor expression. Given this, it would be interesting to investigate molecular commonalities resulting from these interventions, helping to design perturbations that recapitulate the rejuvenation effect without requiring a complex and impractical surgery such as parabiosis.
To conclude, our results indicate that epigenetic age can be systemically decelerated in a sustained manner following long-term exposure to young circulation. This opens exciting new avenues for research on parabiosis and its more translational derivatives for organismal rejuvenation.
METHODS
Mouse models
All animal care followed the guidelines and was approved by the Institutional Animal Care and Use Committees (IACUCs) at Duke Medical Center. Female C57BL/6J (Wild-type, Wt) mice were used at 3 months and 20 months of age. The selection of female mice was based on better temperament when conjoined and better survival rates in females compared to males after the surgeries. All mice were acquired from the NIA aging colony. Details about mouse number and survival across the cohorts and surgery protocols are shown in the table found in Extended Data Fig. 1a.
Parabiosis/detachment surgery
Parabiosis methods were performed as previously described 64, with modifications to the length of the protocol. Parabiosis surgery was carried out using 3 and/or 20-month-old female mice. Under isoflurane anesthesia, incisions are made through the skin and fascia on opposing lateral sides of the abdomen of each mouse followed by suturing of the fascia and suturing of the skin. The suturing of the underlying fascia allows anastomosis and angiogenesis through the natural wound healing process 64. Mouse pairs were prescreeneds to minimize differences in body size but were otherwise chosen indiscriminately. All pairs were maintained together for 3 months, then euthanized or detached for further analysis. The detachment surgery was performed under isoflurane anesthesia, separating the pairs by reversing the attachment site. Once both mice were separated, skin and fascia were sutured closed. For longevity analysis, all detached mice were allowed one month of recovery before entry into the study in an attempt to account for any premature deaths due to surgical complications. Experimental groups for the longevity study were isochronic old (Old ISO) and heterochronic old (Old HET). Mice used for longevity studies were not used for healthspan studies to prevent confounding effects, and allowed to live until natural death or recommendation of euthanasia by a veterinarian. For healthspan studies, all mice were detached after the 3-month parabiosis period and phenotypic and functional data collection started one month after detachment to allow for surgical recovery. Experimental groups included isochronic young (Young ISO), isochronic old (Old ISO) and heterochronic old (Old HET). The same groups were used for detachment cohorts. For epigenetic analyses, all mice were anastomosed for 3-months then analyzed while still attached or analyzed 2 months after detachment. Experimental groups included isochronic young (Young ISO), isochronic old (Old ISO) and heterochronic old (Old HET). The same groups were used for detachment cohorts and annotated with a “Det.” in our data sets and figures. We have previously shown a shared blood supply at 5 weeks 23 and here show some continued sharing at 3 months after surgery using flow cytometry as shown in Extended Data Fig. 2. To isolate the effects of blood sharing and physical attachment, we performed mock parabiosis (Extended Data Fig. 5). For mock parabiosis, we similarly selected mice as for regular parabiosis. Both mice were anesthetized by isoflurane and opposing lateral sides of the abdomen were sutured from elbow to knee (the same distance as standard parabiosis) to create a physical attachment between the two mice. Importantly, for the mock parabiosis, no incisions were made to the skin or fascia prior to attachment thus preventing the generation of shared vasculature. We confirmed separation of circulation by testing the blood glucose response in both parabionts to an IP glucose bolus delivered to one of the partners. All mock parabiosis pairs were attached for 5 weeks. Further details about mock parabiosis experiments are shown in Extended Data Fig. 5.
Genomic DNA and total RNA isolation
Five to seven samples were taken per group for RRBS, five DNA samples for microarray analyses, and three samples for RNA-seq analyses. The detailed number in each experimental group can be found in Table S1. DNA from samples was isolated by using the DNeasy Blood & Tissue Kit (Qiagen 69506), and then eluted from columns in 100 µl of 10 mM Tris-HCl buffer, pH 8.0. 2 µl of RNase A (Life Technologies) was added to each sample. Samples were incubated at room temperature for 2 min, and isolated genomic DNA was purified by using the Genomic DNA Clean & Concentrator™−10 kit (Zymo D4011). DNA was eluted in 25 µl of 10 mM Tris-HCl buffer, pH 8.0 and quantified using a Qubit 2.0 (Life Technologies AM2271). RNA from liver samples RNA was eluted by the Invitrogen Ambion RNAqueous Total RNA Isolation Kit (Invitrogen AM1912) in 60 µl of nuclease free water and quantified using Qubit 2.0 (Life Technologies).
RRBS library preparation and data processing
RRBS libraries were prepared following the protocol previously established 6, using 100 ng of DNA for each sample. Each library included 10 samples. To avoid an overlap of batch effect and age-related changes, we randomized samples into 9 separate libraries (Table S1). Liver and blood samples were analyzed separately. The libraries were sequenced using an Illumina HiSeq2500, with 150bp paired-end reads. 20% of mouse genomic DNA library was spiked in to compensate for low complexity of the libraries. RNA samples were sequenced separately. Adapter removal and quality trimming for the analysis of DNA methylation reads were performed using TrimGalore v0.4.1 following previously established protocols 5,6,65. For the genomic DNA methylation analyses, the trimmed reads were mapped to the mouse genome (GRCm38.p2/mm10) using Bismark v0.15.0. 65 Coverage files outputted by Bismark were then used for further analyses. To ensure high accuracy and precision in methylation levels, CpG sites that had less than 10-read-coverage were filtered out. When combining liver and blood samples into one matrix, a total of 1,014,243 CpGs were found to be covered at a depth greater than 10 across all samples.
For promoter methylation analyses, the promoter region of each gene was determined as [−1500, +500] bp from the transcription starting site following the direction of transcription by using the RefSeq annotation file MmRefSeqTSS.sga, found at https://ccg.epfl.ch/mga/mm10/refseq/refseq.html. Mean promoter methylation levels were calculated for each gene by taking the average of all CpGs covered in a particular promoter. To reduce noise from promoters with minimal CpG coverage, only promoters that had at least 5 CpG sites with valid data were retained for downstream analysis. This resulted in coverage of 11,842 promoters across all liver and blood RRBS samples. Gene body methylation analyses were conducted by concatenating all CpGs within a particular gene body (from transcription start site to transcription end site) and averaging methylation, just as in promoter methylation analyses (again with a ≥5 CpG filter). This resulted in coverage of 13,811 gene bodies across all blood and liver samples.
Application of mouse epigenetic clocks to RRBS datasets
Four clocks were applied to the RRBS dataset to characterize epigenetic age, including two whole-lifespan multi-tissue clocks (Meer et al. clock trained on 416 samples across 40 age groups, ranging from 1 week to 35-month-old & Thompson et al. clock trained on 1189 samples ranging from ~1 week to 35-month-old)5,31, a blood-based clock (trained on 141 mice across 16 age groups, ranging from 3- to 35-month-old) 6, and a recently developed maximum likelihood-based single-cell clock 14, modified to accommodate bulk data (Fig. 2). Epigenetic clock analyses for conventional elastic-net based clocks 5,6,31 were performed exactly following the protocols outlined in the respective original research articles.
In the case of the single-cell clock (scAge), several modifications were applied compared to its original application 13. Indeed, in the original scAge application to sparse single-cell data, single-cell profiles were predominantly binary, but this is not the case with bulk RRBS data. Bulk methylation levels are often fractional, given that many reads coming from different cells usually cover a single CpG. Therefore, to account for this biologically meaningful fractional methylation distribution, no forced binarization of RRBS profiles was performed prior to running the algorithm. This enabled epigenetic age predictions without modification to the input data. Given the targeted nature of RRBS, the top 25% age-associated CpGs per sample were used for predictions. As training data to the modified scAge framework, we utilized bulk C57BL/6J RRBS data from the previously discussed Thompson et al. study. Two CpG-specific reference tables were created: (1) bulk blood RRBS methylation profiles from 50 normally-fed C57BL/6J mice across 1,202,751 CpG sites and (2) bulk liver RRBS methylation profiles from 29 normally-fed C57BL/6J mice across 1,042,996 CpG sites on the positive strand. Based on dimensionality reduction analyses, two blood samples and one liver sample that were evident outliers were removed from downstream epigenetic aging analyses (Fig. 5, Supplementary Table 1). Welch’s one-tailed t-test assuming unequal variances was used for statistical testing.
Application of mouse epigenetic clocks to the DNA microarray dataset
We analyzed 5 samples in each group with the recently developed Infinium arrays HorvathMammalMethylChip40 and its expanded version HorvathMammalMethylChip320 32,33. Six clocks were applied in our analysis, including 2 clocks based on mouse liver (Mouse Liver and Mouse Liver Development), 2 clocks based on mouse blood (Mouse Blood and Mouse Blood Development), a universal mammalian clock based on age relative to maximum lifespan (Universal Relative), and a universal mammalian clock based on log-linear transformed age (Universal Log-Linear) (Fig. 3). The Mouse (Liver Development) clock was similar to the Mouse (Liver) clock, except that sites were sub-selected based on changes occurring during development in this organ. The epigenetic age data are in Table S2. Welch’s one-tailed t-tests assuming unequal variances were used for statistical testing.
RNA-seq analysis of heterochronic parabiosis
Paired-end RNA sequencing for mouse liver samples was performed on an Illumina NovaSeq 6000 S4 platform with 100 bp read length. Reads were mapped with STAR (version 2.5.2b) 66 and counted via featureCounts (version 1.5) 67. Data was passed through RLD transformation 68, and genes with |transformed normalized count| < 1 were filtered out. This left 22,772 genes, which were used for dimensionality reduction and further analyses (Fig. 4–7). For signature association analysis and identification of longevity-associated genes perturbed by heterochronic parabiosis, we further filtered out genes with low numbers of reads, keeping only the genes with at least 10 reads in at least 50% of the samples, which resulted in 12,374 detected genes according to Entrez annotations.
Differential gene expression analyses
Differential expression analyses (DEA) were conducted using DEseq2 on normalized count data. All samples were concatenated to a single data frame and normalized jointly prior to analysis, in order to minimize batch-effect bias. 15,481 valid genes were obtained when conducting DEA on long-term parabiosis samples comparing old isochronic and old heterochronic mice. Similarly, 14,522 valid genes were obtained when conducting DEA on long-term detached parabiosis samples comparing old detached isochronic and old detached heterochronic mice. These data were used to generate the volcano plots for attached and detached samples in Fig. 4. Volcano plots were generated using the package bioinfokit 2.0.6.
For signature association analyses (Fig. 6) and investigation of longevity-associated genes regulated by HPB (Fig. 7), differential expression of genes in response to heterochronic parabiosis compared to isochronic parabiosis was further analyzed using edgeR 69 separately for long-term attached, long-term detached and short-term attached models. Obtained p-values were adjusted for multiple comparison with the Benjamini-Hochberg method 70. The RLE method was used to obtain normalized expression of genes 71.
Dimensionality reduction
To identify trajectories of molecular profiles resulting from HPB, we conducted dimensionality reduction via Principal Component Analysis (PCA) on gene expression (RNA-seq) and methylation (RRBS) data (Fig. 5). For gene expression analyses, we used normalized, RLD-transformed gene count values from the liver across 22,272 genes in both short-term and long-term parabiosis samples (Fig. 5a–b). For sequencing-based methylation data, we used information at 1,014,243 highly-covered (≥10x) CpG sites in liver and blood samples. These data were then collapsed into 11,842 promoters and 13,811 gene bodies, each covered by at least 5 CpGs to ensure reliable mean methylation measurements. The percentage of the variance explained by the first two principal components was extracted and is shown across panels in Fig. 5.
Association of signature-based gene expression changes induced by parabiosis, aging and lifespan extending interventions
Gene expression signatures of lifespan-extending interventions, including: signatures of caloric restriction (CR), signatures of growth hormone deficiency, common gene expression changes across different interventions, and signatures associated with the effect of interventions on median and maximum lifespan, were obtained from Tyshkovskiy et al.44. Aging signatures, including the liver signature as well as the mouse and rat multi-tissue signatures, were obtained via a meta-analysis of age-related gene expression changes72. Association of gene expression log fold-changes associated with parabiosis, lifespan extension and aging were assessed using correlation and GSEA 37. For the correlation analysis, a Spearman correlation metric was calculated using the top 200 statistically significant genes for each pair of signatures. Clustering was subsequently performed with a complete hierarchical approach.
For the GSEA-based association analysis, we utilized an algorithm developed in Tyshkovskiy et al. 44. First, for every signature we specified 250 genes with the lowest p-values and separated them into up- and downregulated genes. These lists were subsequently considered as gene sets. Then, we ranked genes differentially expressed in response to parabiosis based on their p-values, calculated as described in the following methods section. Afterwards, we calculated normalized enrichment scores (NES) separately for up- and downregulated lists of gene sets as described in Tyshkovskiy et al. 44 and defined the final NES as a mean of the two. To calculate statistical significance of obtained NES, we performed permutation testing where we randomly assigned genes to the lists of gene sets, maintaining their size. To get the p-value of the association between parabiosis and a certain signature, we used 5,000 permutations and calculated the frequency of random final NES that are larger in magnitude than the observed final NES. To adjust for multiple testing, we performed a Benjamini-Hochberg correction 66. Final NES for association of HPB response with various longevity and aging signatures were used to generate barplots panels (Fig. 6a–b). Overlap between significant differentially expressed genes in response to HPB and lifespan-extending interventions or aging was assessed using Fisher’s exact test separately for sets of up- and downregulated genes.
Functional enrichment analysis
For the identification of enriched functions distinguishing isochronic and heterochronic mice, we performed functional GSEA 37 on a pre-ranked list of genes based on log10(p-value) corrected by the sign of regulation, calculated as:
where and are p-value and logFC of a certain gene, respectively, obtained from edgeR output, and is the signum function (equal to 1, −1 and 0 if value is positive, negative or equal to 0, respectively). REACTOME, KEGG and GO BP ontologies from the Molecular Signature Database (MSigDB) were used as gene sets for GSEA. The GSEA algorithm was performed separately for long-term attached and detached models via the fgsea package in R with 5000 permutations. A q-value cutoff of 0.1 was used to select statistically significant functions.
A similar analysis was performed for gene expression signatures associated with aging and lifespan extension. A heatmap colored by NES was built for manually chosen statistically significant functions (Fig. 6c–d). Clustering of functions was performed with complete hierarchical approach and Spearman correlation distance.
For parabiosis samples, we also conducted additional GSEA using the Hallmark gene sets, downloaded from MSigDB. We used a recent Python GSEA implementation, gseapy 1.0.0, to perform gene set enrichment. Only Hallmark gene sets with Benjamini-Hochberg adjusted p-values in below 0.05 in the attached comparison are shown in Fig. 4a. GSEA was conducted separately for long-term attached and long-term detached samples, with 5000 gene set permutations for statistical testing. Hallmark terms significantly enriched in the same direction across both attached and detached comparisons are shown in bold. Terms enriched in the same direction, but only significantly so in the attached comparison are shown in normal black font. The term enriched in opposite directions across attached and detached comparisons is shown in red font.
Cage activity
Voluntary activity was tested in the open field arena (Omnitech, Columbus, OH) illuminated at 340 lux as previously described by Huffman et al. 73. Mice were placed individually into the open field and baseline locomotion was monitored over 60 min.
Protein, western blotting and qRT-PCR mRNA expression
RNA isolation, complementary DNA synthesis and mRNA expression were performed as previously described 64. All primers used in this manuscript are:
Sirt3-Forward: GAGCGGCCTCTACAGCAAC
Sirt3-Reverse: GGAAGTAGTGAGTGACATTGGG
Gstt2-Forward: TTCCTGTACTCAAAGACGGAAGC
Gstt2-Reverse: CTGCCACCTGGTACTTGGAAC
C1qb-Forward: CGTCGGCCCTAAGGGTACT
C1qb-Reverse: GGGGCTGTTGATGGTCCTC
Protein isolation and western blotting was performed as described previously 74. Primary antibodies were as follows: Sirt3 (Cell Signaling Technologies, #5490), C1qb (Novus Biologicals, # NBP2–92455) and Gstt2 (ThermoFisher, # H00002953-D01P) Pan-tubulin (Cell Signaling #2194). Secondary antibody was an anti-rabbit IgG, HRP-linked antibody (Cell Signaling Technologies, #7074).
Statistics and reproducibility
No statistical method was used to predetermine sample size. No data were excluded from the analyses. Experimental mice were randomized after initial screening to minimize extreme body weight differences between pairings. The investigators were blinded to group allocations during experiments and outcome assessments following detachment.
Computational analyses were performed in Python 3.9.15, running with numpy 1.23.5 and pandas 1.5.2. Statistical analyses for epigenetic ages were performed with one-tailed Welch’s t-test, implemented in scipy 1.9.3. Statistical analyses for gene expression were performed with custom models from DEseq2 3.13 and edgeR 3.34.1 in R 4.0.3 69. Statistical analyses of gene expression signatures, gene set enrichment analyses, and epigenetic age profiling were performed as documented above.
For the statistical evaluation of body composition, activity, and food consumption data (Fig. 1d, Extended Data Fig. 1b–c) we used a custom test based on random simulations. We generated a randomly simulated point for each time point of old isochronic samples using the average and standard deviation of the old isochronic samples of each time point. In a similar way, we generated another randomly simulated point for each time point of old heterochronic samples. This process resulted in a simulated time series for old heterochronic samples and for old isochronic samples. We assessed these time series by summarizing differences for simulated old isochronic - old heterochronic pairs for all time points. Repeating the whole procedure 100,000 times, we calculated the p-value as the proportion of the cases when the difference between both time series was > 0, if the null hypothesis was ‘old isochronic > old heterochronic’; and < 0, if the null hypothesis was ‘old isochronic < old heterochronic’. If the p-value was less than 0.05, we rejected the null hypothesis and considered the difference significant. The null hypothesis was ‘old isochronic > old heterochronic’ in the case of body weight, lean mass, distance and vertical activity, and ‘old isochronic < old heterochronic’ in the case of fat mass and food consumption. Data distributions were assumed to be normal but this was not formally tested.
Extended Data
Extended Data Fig. 1: Timeline of molecular and physiological profiling.
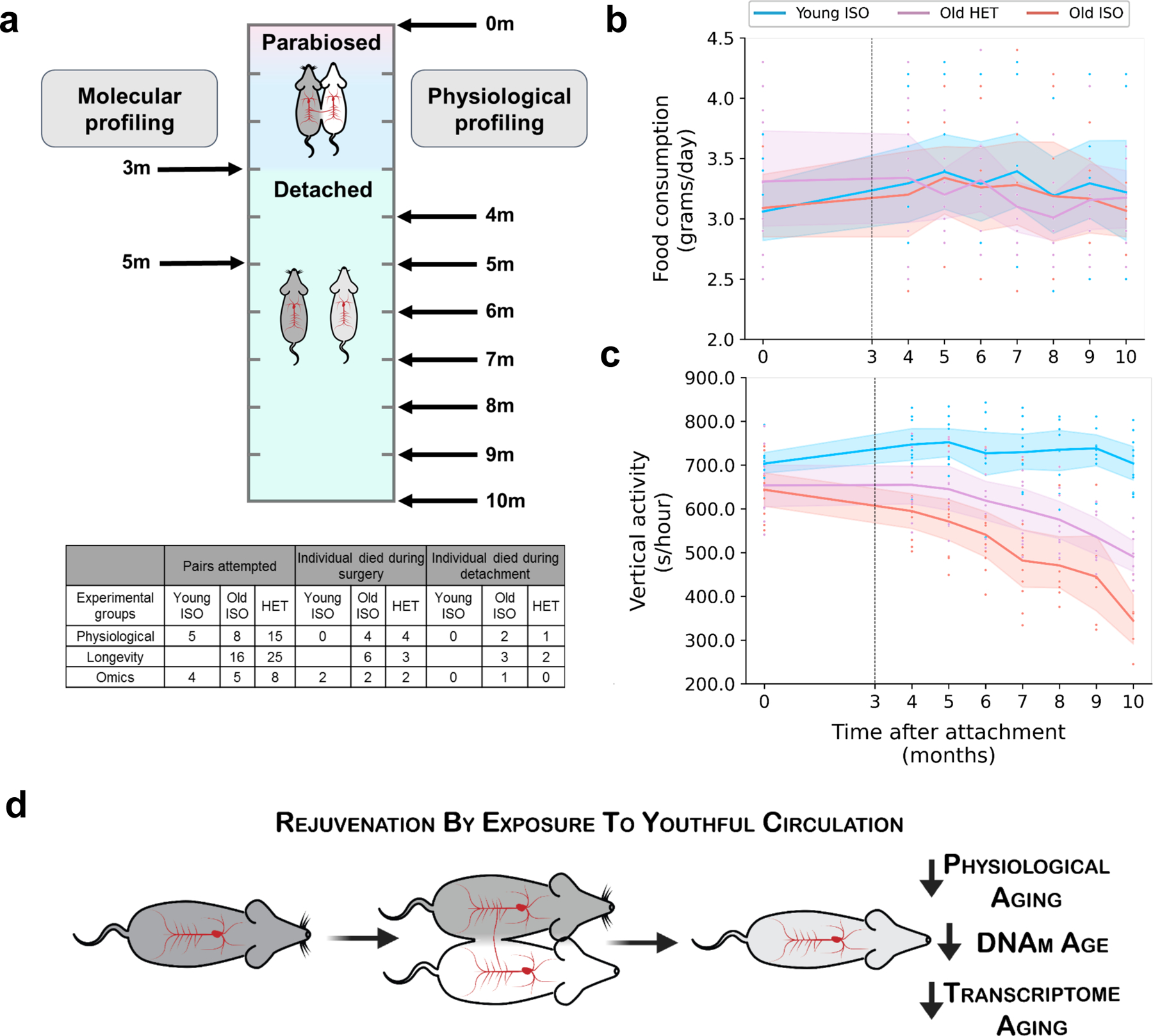
a, Schematic timeline of the molecular (transcriptomic & epigenetic) and physiological profiling presented in this study (top) as well as a table (bottom) highlighting mortality rates at various parts in the parabiosis protocol (numbers represent individual mice, not pairs). Samples were taken immediately after the parabiosis period (month 3), or after a 2-month detachment period (month 5). Physiological profiling was performed before the parabiosis experiment (month 0), and then every month from month 4 to month 10. Data in the table (bottom) is separated by experimental purposes and ages of the pairs. Mortality rate across experiments for each group: Young ISO: 11% mortality during PB, 0% during detach; Old ISO: 20% mortality during PB, 13% during detach. Old HET: 19% mortality during PB, 7% during detach. b, Changes in food consumption resulting from parabiosis. Lines depict mean changes with 95% confidence intervals. Individual observations are shown as points. Young isochronic (Young ISO) mice are shown in blue, old heterochronic (Old HET) mice are shown in purple, and old isochronic (Old ISO) mice are shown in red. Dashed line depicts time of detachment. c, Changes in vertical activity resulting from parabiosis. Lines depict mean changes with 95% confidence intervals. Individual observations are shown as points. Legend is the same as (b). d, Schematic of the experimental workflow and associated findings.
Extended Data Fig. 2: Blood mixture analyses after 3 months of parabiosis.
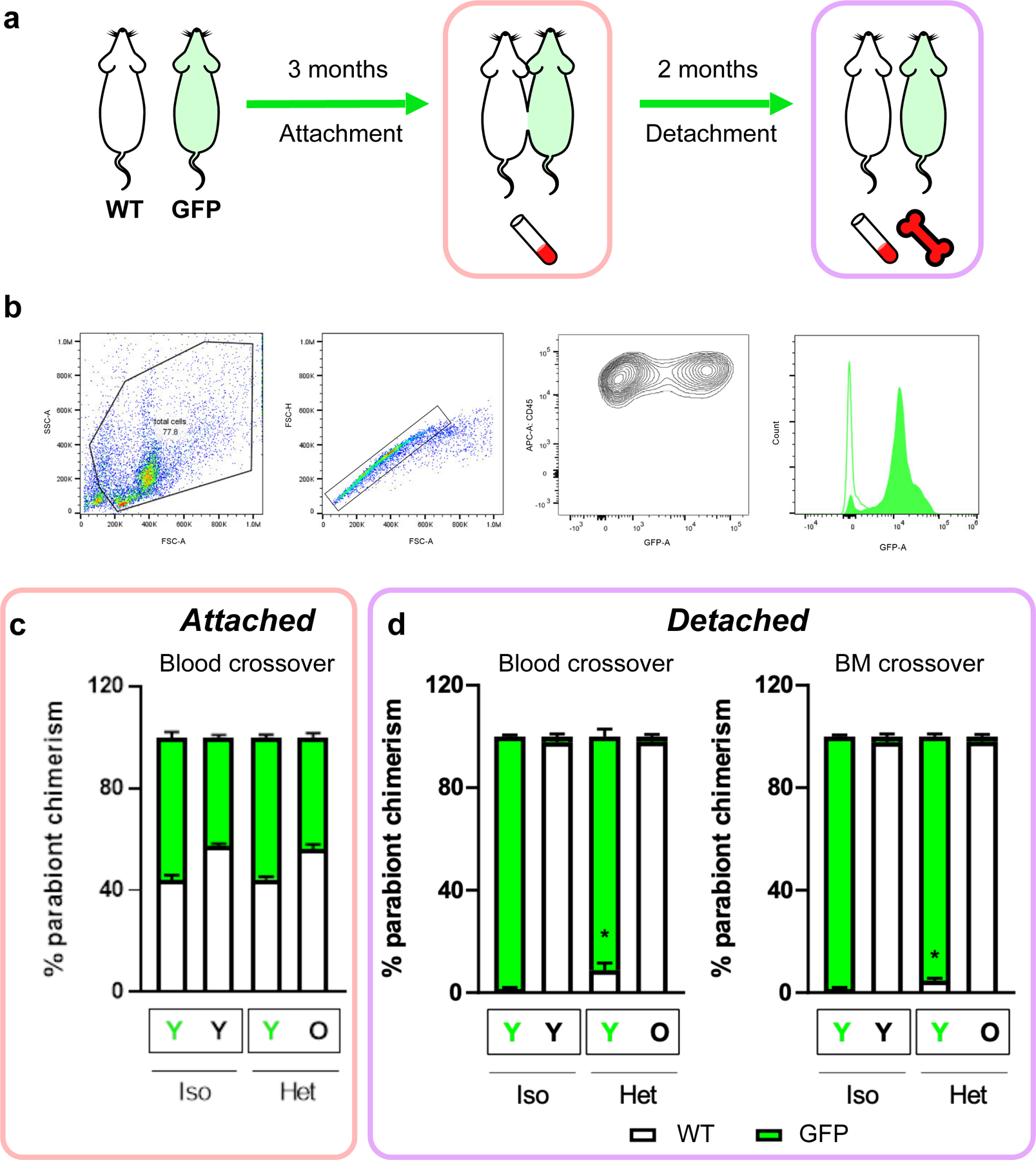
a, Schematic of experiments to analyze the blood chimerism after attachment and detachment. Green represents GFP (+) mice, white represents wild-type mice. b, Gating scheme applied to all downstream blood analysis between GFP and wild-type mice. Cells were ultimately gated on GFP (+) (x-axis) and CD45 (+) (y-axis) (right). c, Percentage of blood crossover during attachment. d, Percentage of blood (left) and bone marrow (BM, right) crossover after 2-month detachment, calculated as the % GFP (+) and (−) cells.
Extended Data Fig. 3. Relationship of DNAm age across tissues and platforms, delta age analysis, and effect of surgery.
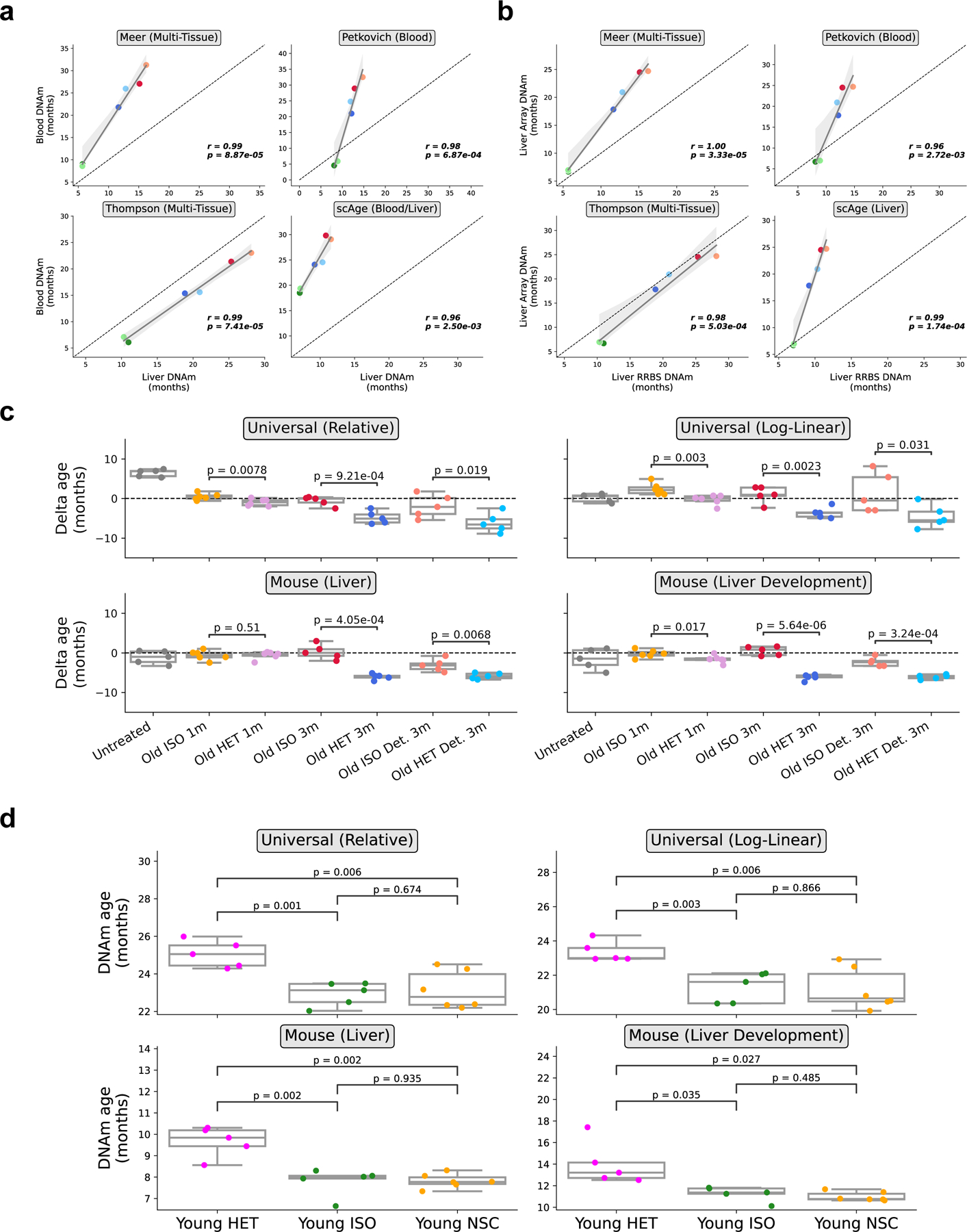
a, Scatterplots highlighting the association of liver and blood DNAm predictions based on RRBS sequencing across 6 different group: young isochronic (Young ISO), young isochronic detached (Young ISO Det.), old heterochronic (Old HET), old isochronic (Old ISO), old heterochronic detached (Old HET Det.), and old isochronic detached (Old ISO Det.). Each point depicts the mean DNAm prediction for a particular tissue for that group. The clock used for predictions is shown in the top part of each panel. The Pearson correlation (r) is shown in each panel, along with its associated two-tailed p-value. Linear regression lines (dark grey) with 95% confidence intervals (light grey) are shown. b, Scatterplots highlighting the association of liver DNAm predictions based on RRBS sequencing and methylation array profiling across 6 different groups (same legend as a). The clock used for RRBS predictions is shown in the top part of each panel. The mouse liver development clock was used for array predictions given that it showed the greatest changes in epigenetic age comparing long-term old heterochronic and isochronic mice. c, Delta age (epigenetic age minus chronological age) of liver samples from old heterochronic mice, old isochronic controls and untreated controls based on universal relative age mammalian, universal log-linear transformed age mammalian, liver, and development liver clocks. Dashed line denotes a delta age of 0 (epigenetic age = chronological age). Points above this line depict age acceleration (epigenetic age > chronological age), while points below this line depict age deceleration (epigenetic age < chronological age). n = 5 per group. d, Epigenetic age of liver samples from 8-month old non-surgical control mice (Young NSC), isochronic detached (Young ISO) and heterochronic detached mice (Young HET), based on the universal relative age mammalian, universal log-linear transformed age mammalian, liver, and liver development clocks. n = 5 per group for parabiosed mice and 6 per group for controls. Two-tailed Welch’s t-tests were used for statistical analyses.
Extended Data Fig. 4. Mean methylation in array and RRBS methylation profiles.
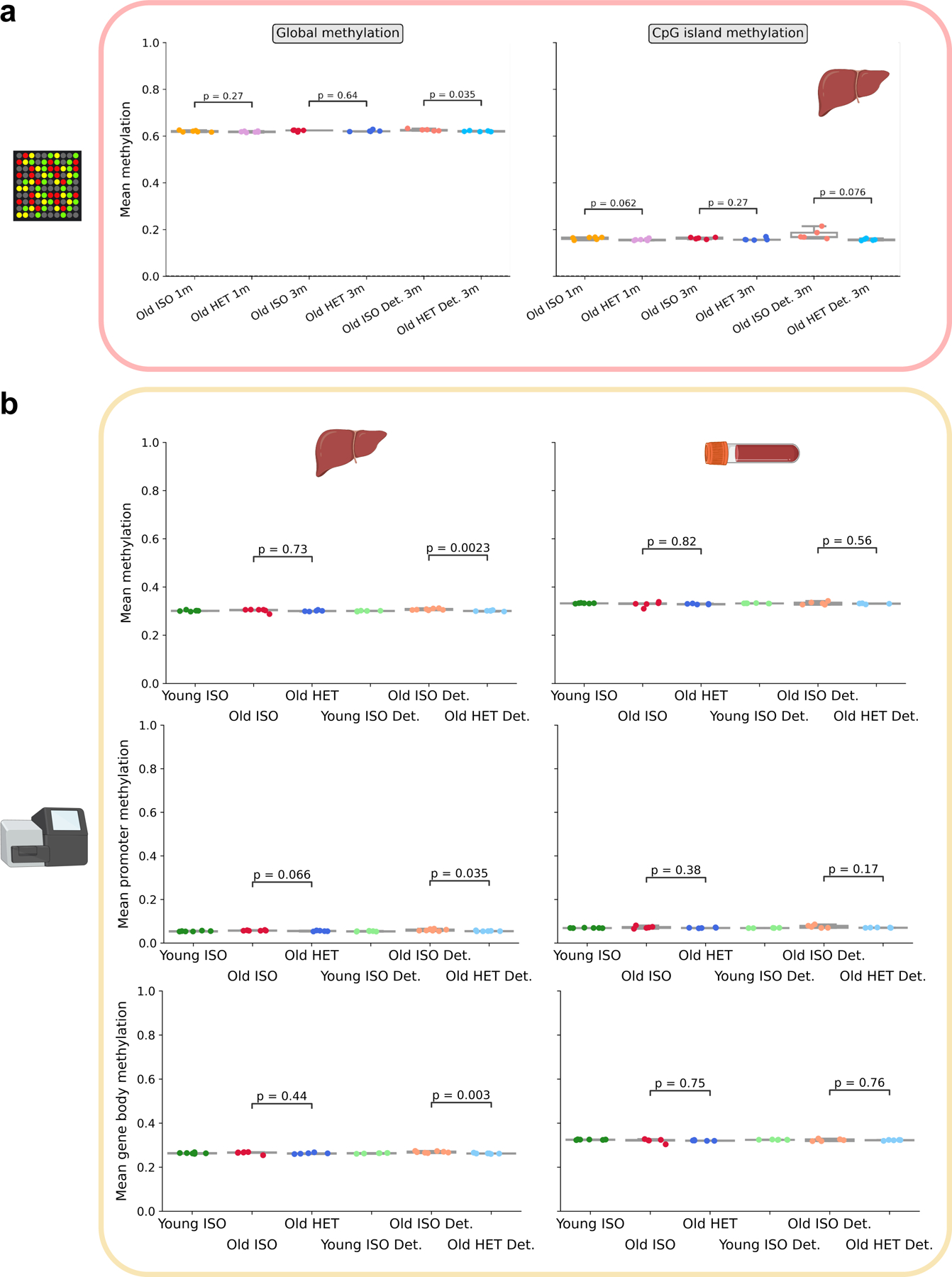
a, Mean liver methylation assayed by the Mammalian Methylation Array (HorvathMammalMethylChip40), both based on all sites in the array (left) and only sites in CpG islands (right). n = 5 samples per group. b, Mean global methylation (top), promoter methylation (middle), and gene body methylation (bottom) of liver (left) and blood (right) RRBS samples. Mean methylation was assayed across 1,014,243 CpGs (top), 11,842 promoters (middle), and 13,811 gene bodies (bottom). n = 4–7 samples per group. Two-tailed Welch’s t-test were used for statistical analysis. Schematic tissues in each panel indicate the source of the sample.
Extended Data Fig. 5. Physiological tests of a mock parabiosis procedure to assess the interaction of parabiosis and exercise.
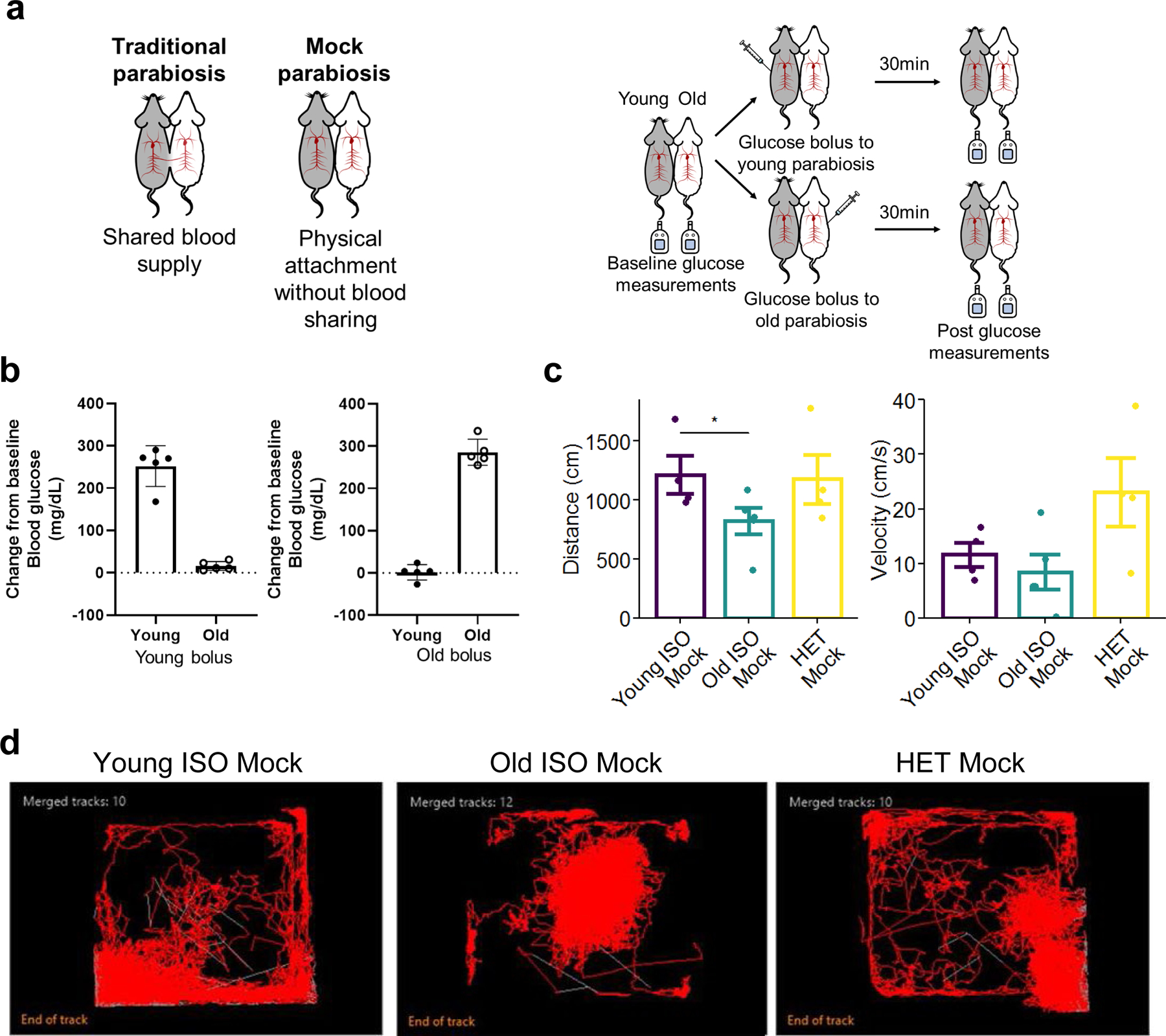
a, Schematic of the experiment to test blood sharing using glucose levels. Glucose is injected into one mouse and subsequently assessed in both mice in mock parabiosis pairs. b, Blood glucose level changes in pairs starting with a young (left) and old (right) glucose bolus. c, Moving distance in cm (left), and velocity (right) in the old isochronic, young isochronic and heterochronic mock parabiosis pairs. Standard error is shown as the error bar. pdistance(Young ISO—Old ISO) = 0.034. d, Movement tracking in the old isochronic, young isochronic and heterochronic mock parabiosis pairs.
Extended Data Fig. 6. Tissue specificity of RRBS epigenomic profiles, promoter methylation changes, and proteomic dynamics in HPB.
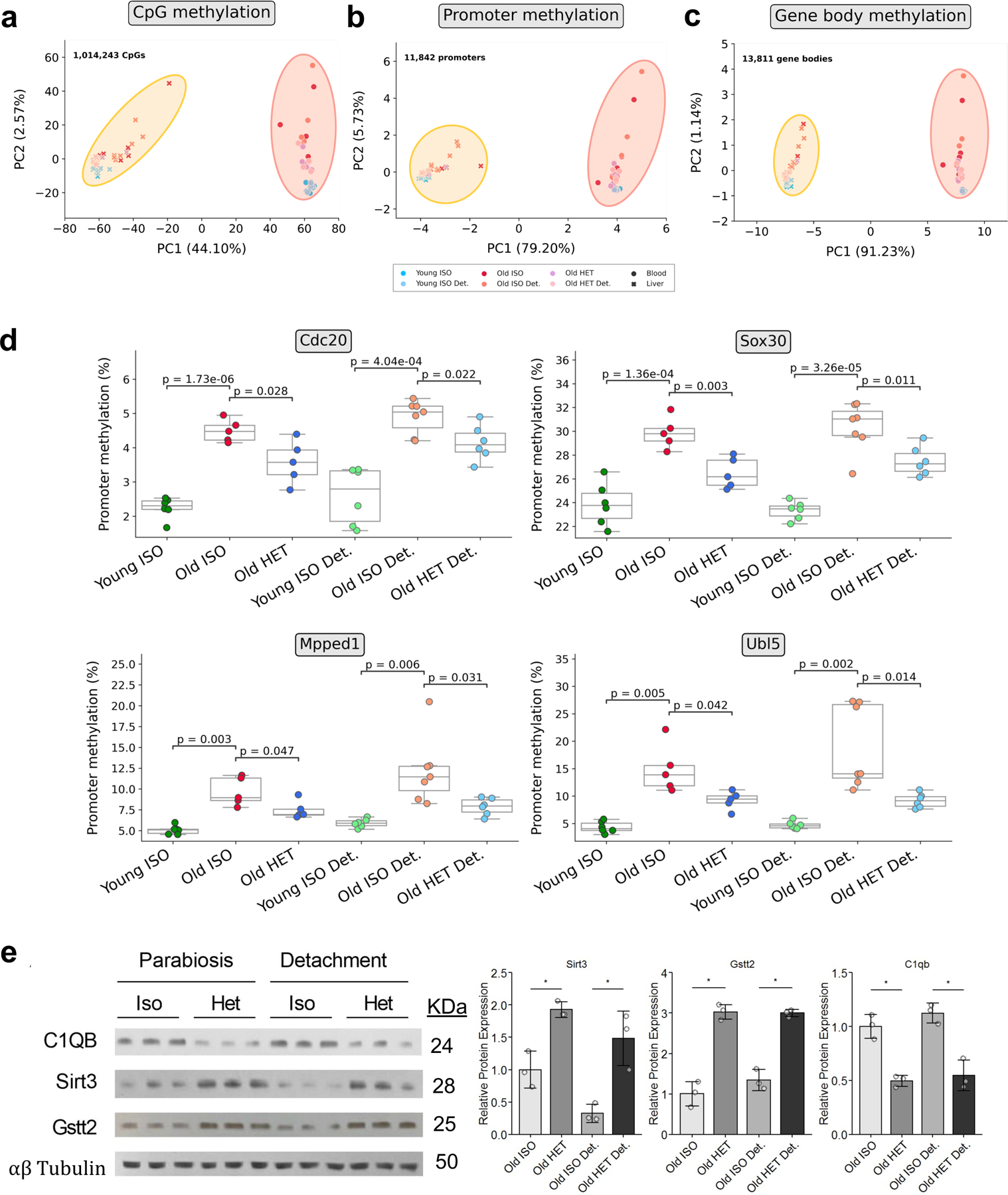
a-c, Principal component analysis (PCA) of CpG methylation across 1,014,243 CpG sites (a), 11,842 promoters (b), and 13,811 gene bodies (c) in n = 36 liver samples and n = 32 blood samples, with 6 different groups in both tissues: young isochronic (Young ISO), young isochronic detached (Young ISO Det.), old heterochronic (Old HET), old isochronic (Old ISO), old heterochronic detached (Old HET Det.), and old isochronic detached (Old ISO Det.). In all the PCA plots, tissue of origin is the largest source of variation (44–91%). d, Mean promoter methylation of Cdc20, Sox30, Mpped1, and Ubl5 across liver RRBS samples. Genes were identified after passing a significance threshold (p < 0.05) when comparing young and old mice, as well as old heterochronic and isochronic mice in both attached and detached groups. Two-tailed Welch’s t-tests were used for statistical analysis. e, Western blot validation (left) and relative protein expression quantification (right) of Sirt3, Gstt2 and C1qb in mice subjected to parabiosis.
Extended Data Fig. 7. Interactions between different omics modalities.
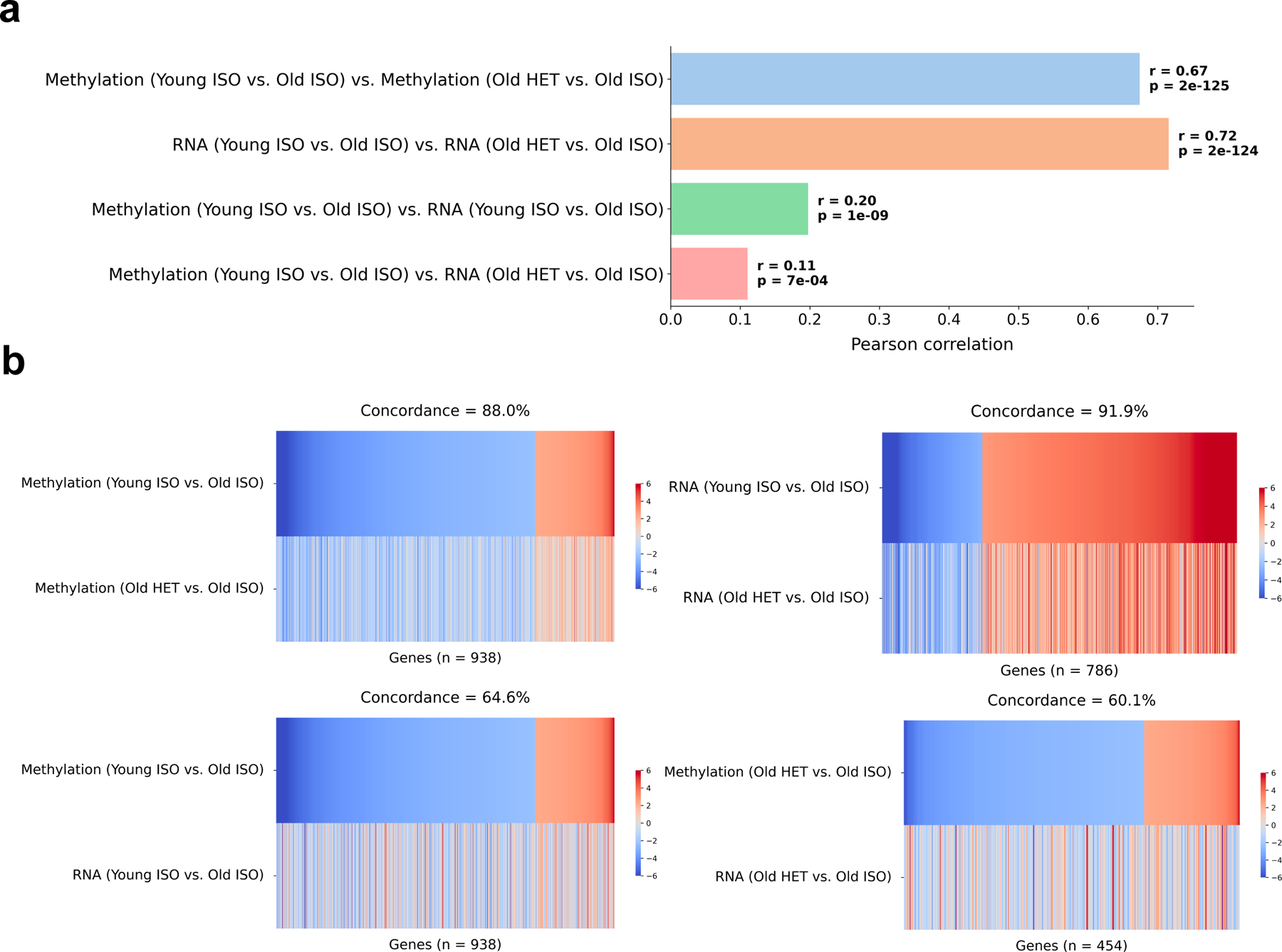
a, Correlation of changes across experimental groups and readout types. For each comparison in parentheses (i.e. Young ISO vs. Old ISO), a z-score was computed for either promoter methylation or gene expression. The Pearson correlation of these z-scores was then determined for genes passing a differential expression/methylation threshold of p < 0.05 for the first (left) group listed in each comparison. b, Heat maps highlighting the concordance of readouts and experimental groups. For each heat map, the z-score of genes that are significantly (p < 0.05) differentially expressed or methylated is shown. Genes are ordered by the z-score in the top group. Color bar on the right denotes z-score. The directional concordance, a measure of how directionally aligned the changes between the two readouts/groups are, is shown at the top of each heat map. 50% concordance represents random changes across groups.
Extended Data Fig. 8. Transcriptomic and epigenetic changes resulting from HPB.
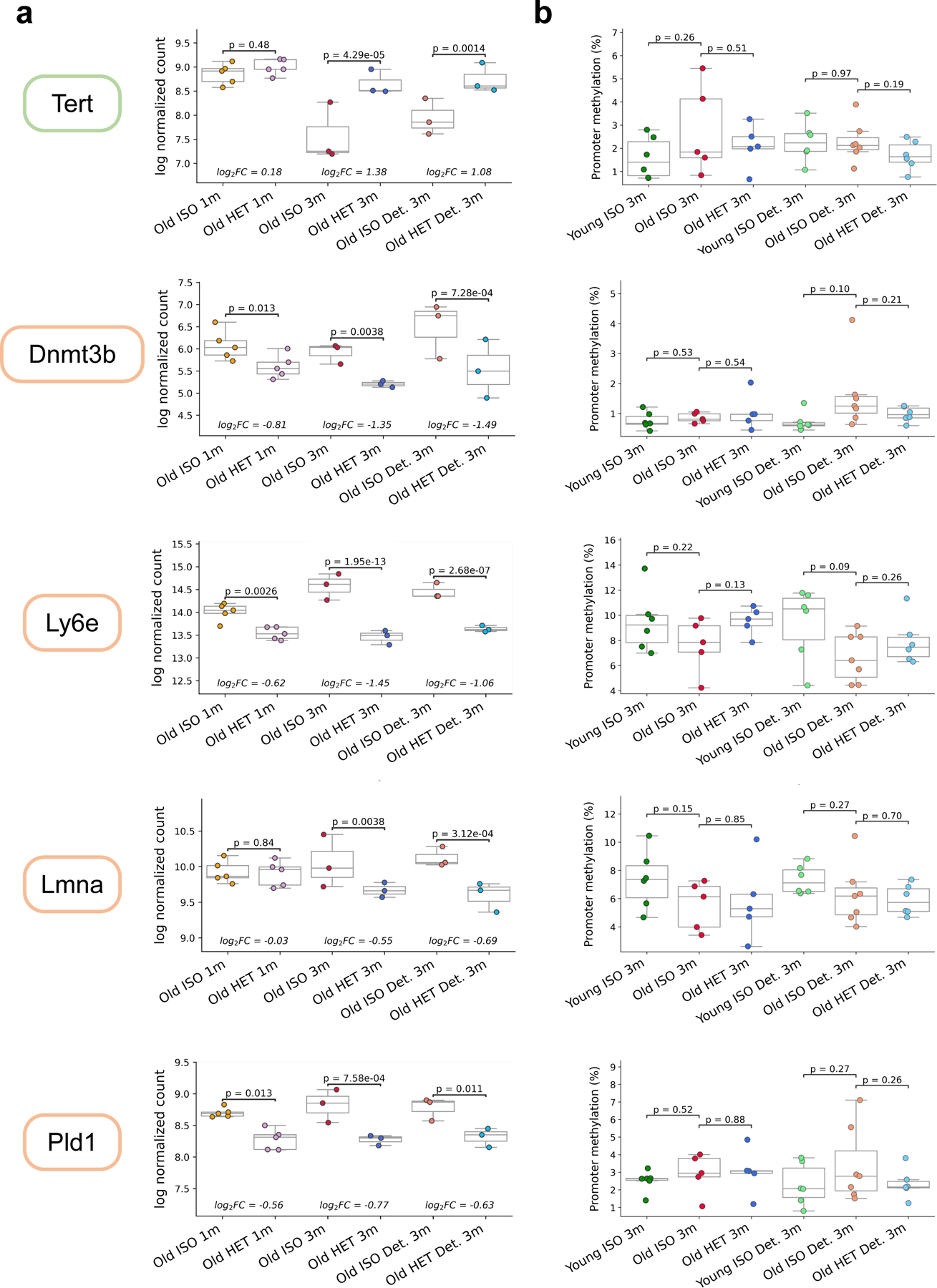
a, Boxplots of RLD-transformed, log-normalized counts of Tert, Dnmt3b, Ly6e, Lmna and Pld1 across 6 groups (from left to right: old short-term isochronic (n = 5), old short-term heterochronic (n = 5), old long-term isochronic (n = 3), old long-term heterochronic (n = 3), old long-term isochronic detached (n = 3), and old long-term heterochronic detached (n = 3). b, Mean promoter methylation of the genes mentioned in (a) across liver RRBS samples across 6 groups (from left to right: young long-term isochronic (n = 6), old long-term isochronic (n = 5), old long-term heterochronic (n = 5), young long-term isochronic detached (n = 6), old long-term isochronic detached (n = 7), and old long-term heterochronic detached (n = 6). Color of the box around each gene signifies directionality (green: gene expression increases in Old HET mice compared to ISO, orange: gene expression decreases in Old HET mice compared to ISO). Despite evident changes in expression patterns for these genes, no significant changes in promoter methylation are observed. N represents biological replicates for each group. P values determined by two-tailed Welch’s t-test. Box plots represent median, 25–75 percentile and 1.5x IQR.
Extended Data Fig. 9. SASP enrichment and differential expression across long-term and short-term HPB.
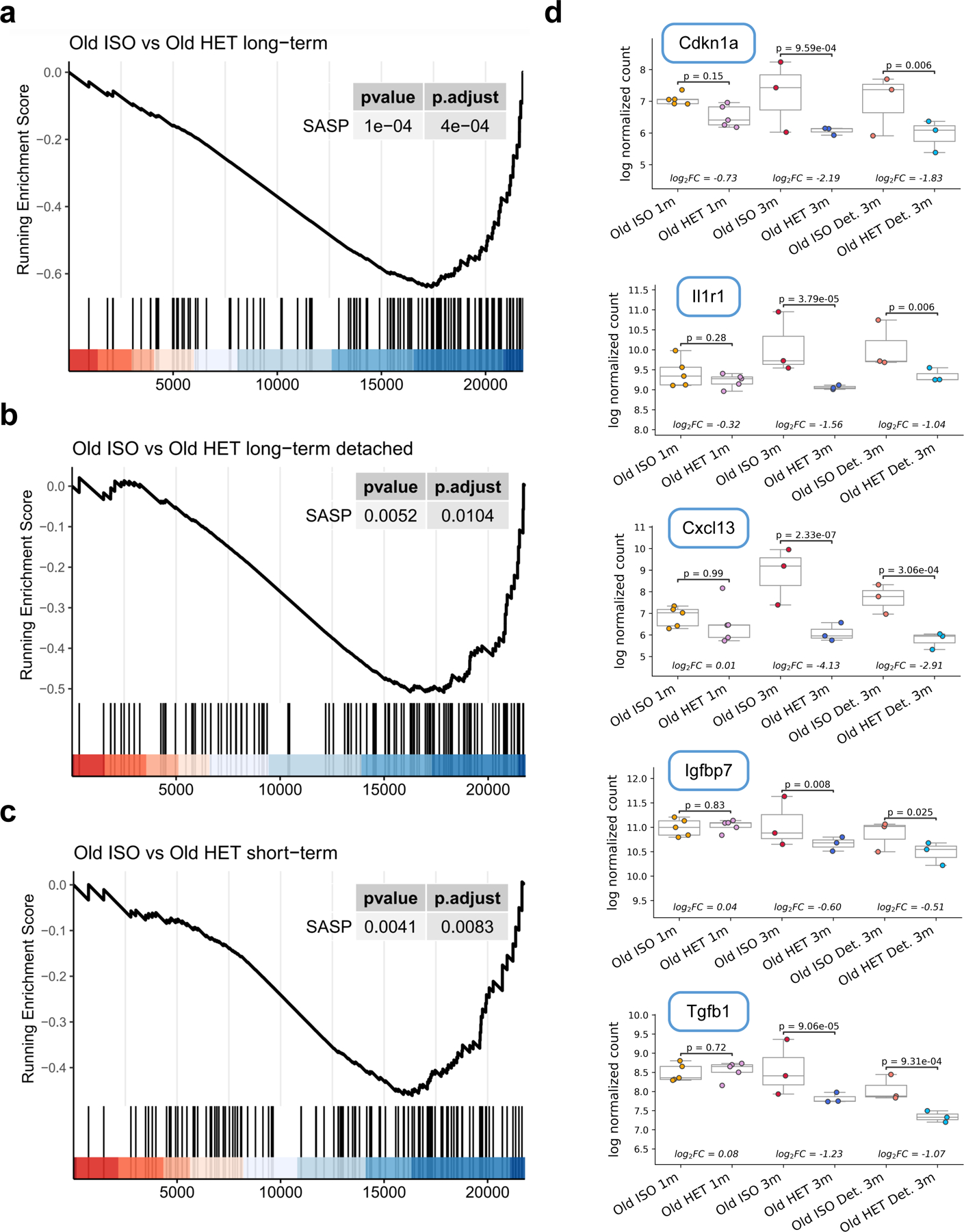
a-c, Running enrichment score for the senescence-associated secretory phenotype (SASP) gene set, comparing in (a) old isochronic and old heterochronic mice from the long-term HPB, in (b) old detached isochronic and old detached heterochronic mice from long-term HPB, and in (c) old detached isochronic and old detached heterochronic mice from short-term HPB. The P values for the gene set enrichment, along with the adjusted P value are shown in each panel. Positions of individual genes in the gene set are shown as black bars near the bottom of each panel. Long-term attached HPB shows the greatest negative enrichment for heterochronic samples, followed by detached long-term HPB and attached short-term HPB, respectively. d, Boxplots of RLD-transformed, log-normalized count of five SASP genes across 6 groups (from left to right: old short-term isochronic (n = 5), old short-term heterochronic (n = 5), old long-term isochronic (n = 3), old longterm heterochronic (n = 3), old long-term isochronic detached (n = 3), and old long-term heterochronic detached (n = 3). The log2 fold-change (log2FC) and associated P value are shown as calculated by two-tailed Welch’s test. Box plots represent median, 25–75 percentile and 1.5x IQR.
Extended Data Fig. 10. STRING network representation of significantly down- and upregulated genes.

a, STRING network representation of significantly downregulated genes in livers of old heterochronic mice (n = 421 genes) compared to isochronic mice immediately after detachment or after a 2-month detachment period (protein-protein interaction q-value < 1e-16). Genes were filtered based on directionality and absolute value of the log2FC. b, STRING network representation of significantly upregulated genes in livers of old heterochronic mice (n = 337 genes) compared to isochronic mice immediately after detachment or after a 2-month detachment period (protein-protein interaction q-value < 1e-16). Genes were filtered based on directionality and absolute value of the log2FC.
Supplementary Material
Acknowledgements
J.P.W. was supported by NIH grants K01AG056664 and R21AG065943. V.N.G. was supported by NIH grants R01AG067782, P01AG047200 and R01AG065403. D.E.L was supported by NIH training grant T32HL007057. S.H. acknowledges support from the Milky Way Research Foundation and the Epigenetic Clock Development Foundation, and A.Tyshkovskiy and S.E.D. acknowledges support from the Interdisciplinary Scientific and Educational School of Moscow University “Molecular Technologies of the Living Systems and Synthetic Biology”. We thank the Duke Behavioral Core for support on this project. We also thank Tiamat Fox for help with schematic figures. Figure icons were partially created with BioRender.com. The funders had no role in study design, data collection, data analysis, decision to publish, or preparation of the manuscript.
Footnotes
Competing interests
The authors do not report any conflicts of interest.
Data Availability
The sequencing data obtained in this study are deposited to GEO with the accession number GSE224447. Data used to generate graphs can be found in accompanying source data files.
REFERENCES
- 1.Brett JO & Rando TA Alive and well? Exploring disease by studying lifespan. Current opinion in genetics & development 26, 33–40, doi: 10.1016/j.gde.2014.05.004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G The hallmarks of aging. Cell 153, 1194–1217, doi: 10.1016/j.cell.2013.05.039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez-Otin C, Blasco MA, Partridge L, Serrano M & Kroemer G Hallmarks of aging: An expanding universe. Cell 186, 243–278, doi: 10.1016/j.cell.2022.11.001 (2023). [DOI] [PubMed] [Google Scholar]
- 4.Horvath S DNA methylation age of human tissues and cell types. Genome Biol 14, R115, doi: 10.1186/gb-2013-14-10-r115 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meer MV, Podolskiy DI, Tyshkovskiy A & Gladyshev VN A whole lifespan mouse multi-tissue DNA methylation clock. eLife 7, e40675, doi: 10.7554/eLife.40675 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petkovich DA et al. Using DNA Methylation Profiling to Evaluate Biological Age and Longevity Interventions. Cell Metab 25, 954–960 e956, doi: 10.1016/j.cmet.2017.03.016 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olova N, Simpson DJ, Marioni RE & Chandra T Partial reprogramming induces a steady decline in epigenetic age before loss of somatic identity. Aging Cell 18, e12877, doi: 10.1111/acel.12877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fahy GM et al. Reversal of epigenetic aging and immunosenescent trends in humans. Aging Cell 18, e13028, doi: 10.1111/acel.13028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horvath S et al. Reversing age: dual species measurement of epigenetic age with a single clock. bioRxiv, 2020.2005.2007.082917, doi: 10.1101/2020.05.07.082917 (2020). [DOI] [Google Scholar]
- 10.Lu Y et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 588, 124–129, doi: 10.1038/s41586-020-2975-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rando TA & Chang HY Aging, rejuvenation, and epigenetic reprogramming: resetting the aging clock. Cell 148, 46–57, doi: 10.1016/j.cell.2012.01.003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarkar TJ et al. Transient non-integrative expression of nuclear reprogramming factors promotes multifaceted amelioration of aging in human cells. Nature communications 11, 1545, doi: 10.1038/s41467-020-15174-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerepesi C, Zhang B, Lee SG, Trapp A & Gladyshev VN Epigenetic clocks reveal a rejuvenation event during embryogenesis followed by aging. Science Advances 7, eabg6082, doi: 10.1126/sciadv.abg6082 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Trapp A, Kerepesi C & Gladyshev VN Profiling epigenetic age in single cells. Nature Aging 1, 1189–1201, doi: 10.1038/s43587-021-00134-3 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lunsford WR, Mc CC, Lupien PJ, Pope FE & Sperling G Parabiosis as a method for studying factors which affect aging in rats. Gerontologia 7, 1–8 (1963). [DOI] [PubMed] [Google Scholar]
- 16.McCay CM, Pope F & Lunsford W Experimental prolongation of the life span. Bull N Y Acad Med 32, 91–101 (1956). [PMC free article] [PubMed] [Google Scholar]
- 17.Pope F, Lunsford W & McCay CM Experimental prolongation of the life span. J Chronic Dis 4, 153–158 (1956). [DOI] [PubMed] [Google Scholar]
- 18.Baht GS et al. Exposure to a youthful circulaton rejuvenates bone repair through modulation of beta-catenin. Nature communications 6, 7131, doi: 10.1038/ncomms8131 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conboy IM et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764, doi: 10.1038/nature03260 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Loffredo FS et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 153, 828–839, doi: 10.1016/j.cell.2013.04.015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ruckh JM et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell 10, 96–103, doi: 10.1016/j.stem.2011.11.019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villeda SA et al. Young blood reverses age-related impairments in cognitive function and synaptic plasticity in mice. Nat Med 20, 659–663, doi: 10.1038/nm.3569 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vi L et al. Macrophage cells secrete factors including LRP1 that orchestrate the rejuvenation of bone repair in mice. Nat Commun 9, 5191, doi: 10.1038/s41467-018-07666-0 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rebo J et al. A single heterochronic blood exchange reveals rapid inhibition of multiple tissues by old blood. Nature communications 7, 13363, doi: 10.1038/ncomms13363 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Middeldorp J et al. Preclinical Assessment of Young Blood Plasma for Alzheimer Disease. JAMA Neurol 73, 1325–1333, doi: 10.1001/jamaneurol.2016.3185 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conboy MJ, Conboy IM & Rando TA Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging Cell 12, 525–530, doi: 10.1111/acel.12065 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wright DE, Wagers AJ, Gulati AP, Johnson FL & Weissman IL Physiological migration of hematopoietic stem and progenitor cells. Science 294, 1933–1936, doi: 10.1126/science.1064081 (2001). [DOI] [PubMed] [Google Scholar]
- 28.Donskoy E & Goldschneider I Thymocytopoiesis is maintained by blood-borne precursors throughout postnatal life. A study in parabiotic mice. J Immunol 148, 1604–1612 (1992). [PubMed] [Google Scholar]
- 29.Ho TT et al. Aged hematopoietic stem cells are refractory to bloodborne systemic rejuvenation interventions. J Exp Med 218, doi: 10.1084/jem.20210223 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meissner A et al. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Research 33, 5868–5877, doi: 10.1093/nar/gki901 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thompson MJ et al. A multi-tissue full lifespan epigenetic clock for mice. Aging 10, 2832–2854, doi: 10.18632/aging.101590 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu AT et al. Universal DNA methylation age across mammalian tissues. bioRxiv, 2021.2001.2018.426733, doi: 10.1101/2021.01.18.426733 (2021). [DOI] [Google Scholar]
- 33.Arneson A et al. A mammalian methylation array for profiling methylation levels at conserved sequences. Nat Commun 13, 783, doi: 10.1038/s41467-022-28355-z (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fiorito G et al. DNA methylation-based biomarkers of aging were slowed down in a two-year diet and physical activity intervention trial: the DAMA study. Aging Cell 20, e13439, doi: 10.1111/acel.13439 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Durieux J, Wolff S & Dillin A The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell 144, 79–91, doi: 10.1016/j.cell.2010.12.016 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loerch PM et al. Evolution of the aging brain transcriptome and synaptic regulation. PloS one 3, e3329, doi: 10.1371/journal.pone.0003329 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Subramanian A et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liberzon A et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425, doi: 10.1016/j.cels.2015.12.004 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesnefsky EJ & Hoppel CL Oxidative phosphorylation and aging. Ageing Res Rev 5, 402–433, doi: 10.1016/j.arr.2006.04.001 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Bandres E et al. The increase of IFN-gamma production through aging correlates with the expanded CD8(+high)CD28(−)CD57(+) subpopulation. Clinical immunology (Orlando, Fla.) 96, 230–235, doi: 10.1006/clim.2000.4894 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Leonardi GC, Accardi G, Monastero R, Nicoletti F & Libra M Ageing: from inflammation to cancer. Immunity & ageing : I & A 15, 1, doi: 10.1186/s12979-017-0112-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai KSP et al. Peripheral inflammatory markers in Alzheimer’s disease: a systematic review and meta-analysis of 175 studies. J Neurol Neurosurg Psychiatry 88, 876–882, doi: 10.1136/jnnp-2017-316201 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Amdur RL et al. Inflammation and Progression of CKD: The CRIC Study. Clin J Am Soc Nephrol 11, 1546–1556, doi: 10.2215/CJN.13121215 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tyshkovskiy A et al. Identification and Application of Gene Expression Signatures Associated with Lifespan Extension. Cell metabolism 30, 573–593 e578, doi: 10.1016/j.cmet.2019.06.018 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez Herrera KN, Finley LW & Haigis MC The role of SIRT3 in regulating cancer cell metabolism. BMC Proceedings 6, P18, doi: 10.1186/1753-6561-6-s3-p18 (2012). [DOI] [Google Scholar]
- 46.Benigni A et al. Sirt3 Deficiency Shortens Life Span and Impairs Cardiac Mitochondrial Function Rescued by Opa1 Gene Transfer. Antioxidants & Redox Signaling 31, 1255–1271, doi: 10.1089/ars.2018.7703 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Brown K et al. SIRT3 reverses aging-associated degeneration. Cell reports 3, 319–327, doi: 10.1016/j.celrep.2013.01.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lang CA, Wu WK, Chen T & Mills BJ Blood glutathione: a biochemical index of life span enhancement in the diet restricted Lobund-Wistar rat. Progress in clinical and biological research 287, 241–246 (1989). [PubMed] [Google Scholar]
- 49.Richie JP Jr. et al. Methionine restriction increases blood glutathione and longevity in F344 rats. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 8, 1302–1307, doi: 10.1096/fasebj.8.15.8001743 (1994). [DOI] [PubMed] [Google Scholar]
- 50.Bernardes de Jesus B et al. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO molecular medicine 4, 691–704, doi: 10.1002/emmm.201200245 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Okano M, Bell DW, Haber DA & Li E DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99, 247–257, doi: 10.1016/s0092-8674(00)81656-6 (1999). [DOI] [PubMed] [Google Scholar]
- 52.Childs BG, Durik M, Baker DJ & van Deursen JM Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21, 1424–1435, doi: 10.1038/nm.4000 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conboy IM & Rando TA Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell cycle (Georgetown, Tex.) 11, 2260–2267, doi: 10.4161/cc.20437 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yankova T, Dubiley T, Shytikov D & Pishel I Three Month Heterochronic Parabiosis Has a Deleterious Effect on the Lifespan of Young Animals, Without a Positive Effect for Old Animals. Rejuvenation Res 25, 191–199, doi: 10.1089/rej.2022.0029 (2022). [DOI] [PubMed] [Google Scholar]
- 55.Castellano JM et al. Human umbilical cord plasma proteins revitalize hippocampal function in aged mice. Nature 544, 488–492, doi: 10.1038/nature22067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Salpeter SJ et al. Systemic regulation of the age-related decline of pancreatic beta-cell replication. Diabetes 62, 2843–2848, doi: 10.2337/db13-0160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Villeda SA et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477, 90–94, doi: 10.1038/nature10357 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rando TA & Wyss-Coray T Asynchronous, contagious and digital aging. Nat Aging 1, 29–35, doi: 10.1038/s43587-020-00015-1 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pálovics R et al. Molecular hallmarks of heterochronic parabiosis at single cell resolution. bioRxiv, 2020.2011.2006.367078, doi: 10.1101/2020.11.06.367078 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Conese M, Carbone A, Beccia E & Angiolillo A The Fountain of Youth: A Tale of Parabiosis, Stem Cells, and Rejuvenation. Open Med (Wars) 12, 376–383, doi: 10.1515/med-2017-0053 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elabd C et al. Oxytocin is an age-specific circulating hormone that is necessary for muscle maintenance and regeneration. Nature communications 5, 4082, doi: 10.1038/ncomms5082 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stolzel F et al. Dynamics of epigenetic age following hematopoietic stem cell transplantation. Haematologica 102, e321–e323, doi: 10.3324/haematol.2016.160481 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehdipour M et al. Rejuvenation of three germ layers tissues by exchanging old blood plasma with saline-albumin. Aging (Albany NY) 12, 8790–8819, doi: 10.18632/aging.103418 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baht GS et al. Meteorin-like facilitates skeletal muscle repair through a Stat3/IGF-1 mechanism. Nat Metab 2, 278–289, doi: 10.1038/s42255-020-0184-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krueger F & Andrews SR Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27, 1571–1572, doi: 10.1093/bioinformatics/btr167 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, doi: 10.1093/bioinformatics/bts635 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liao Y, Smyth GK & Shi W featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930, doi: 10.1093/bioinformatics/btt656 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, doi: 10.1186/s13059-014-0550-8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, doi: 10.1093/bioinformatics/btp616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300, doi: 10.1111/j.2517-6161.1995.tb02031.x (1995). [DOI] [Google Scholar]
- 71.Anders S & Huber W Differential expression analysis for sequence count data. Genome biology 11, R106, doi: 10.1186/gb-2010-11-10-r106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tyshkovskiy A et al. Distinct longevity mechanisms across and within species and their association with aging. Cell 186, 2929–2949 e2920, doi: 10.1016/j.cell.2023.05.002 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huffman KM et al. Exercise protects against cardiac and skeletal muscle dysfunction in a mouse model of inflammatory arthritis. Journal of applied physiology (Bethesda, Md. : 1985) 130, 853–864, doi: 10.1152/japplphysiol.00576.2020 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.White JP et al. The AMPK/p27(Kip1) Axis Regulates Autophagy/Apoptosis Decisions in Aged Skeletal Muscle Stem Cells. Stem Cell Reports 11, 425–439, doi: 10.1016/j.stemcr.2018.06.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data obtained in this study are deposited to GEO with the accession number GSE224447. Data used to generate graphs can be found in accompanying source data files.