

Abstract
Ischemia-reperfusion injury (IRI) during orthotopic liver transplantation (OLT) contributes to graft rejection and poor clinical outcomes. The disulfide form of high mobility group box 1 (diS-HMGB1), an intracellular protein released during OLT-IRI, induces pro-inflammatory macrophages. How diS-HMGB1 differentiates human monocytes into macrophages capable of activating adaptive immunity remains unknown. We investigated if diS-HMGB1 binds toll-like receptor (TLR) 4 and TLR9 to differentiate monocytes into pro-inflammatory macrophages that activate adaptive immunity and promote graft injury and dysfunction. Assessment of 106 clinical liver tissue and longitudinal blood samples revealed that OLT recipients were more likely to experience IRI and graft dysfunction with increased diS-HMGB1 released during reperfusion. Increased diS-HMGB1 concentration also correlated with TLR4/TLR9 activation, polarization of monocytes into pro-inflammatory macrophages, and production of anti-donor antibodies. In vitro, healthy volunteer monocytes stimulated with purified diS-HMGB1 had increased inflammatory cytokine secretion, antigen presentation machinery, and reactive oxygen species production. TLR4 inhibition primarily impeded cytokine/chemokine and costimulatory molecule programs, whereas TLR9 inhibition decreased HLA-DR and reactive oxygen species production. diS-HMGB1–polarized macrophages also showed increased capacity to present antigens and activate T memory cells. In murine OLT, diS-HMGB1 treatment potentiated ischemia-reperfusion–mediated hepatocellular injury, accompanied by increased serum alanine transaminase levels. This translational study identifies the diS-HMGB1/TLR4/TLR9 axis as potential therapeutic targets in OLT-IRI recipients.
Keywords: ischemia-reperfusion injury, disulfide-HMGB1, TLR activation, pro-inflammatory macrophage, alloimmunity
1. Introduction
Orthotopic liver transplantation (OLT) is a life-saving treatment for end-stage liver diseases.1,2 During the transplant process, the donor liver is at risk of ischemia-reperfusion injury (IRI), which is associated with increased incidence of rejection episodes and lower graft and patient survival.3,4 IRI involves a complex sterile inflammatory response, accompanied by the release of damage-associated molecular patterns (DAMPs) into the extracellular environment.5–7 Liver resident macrophages expressing pattern recognition receptors (PRRs) bind DAMPs, become activated, and produce cytokine/chemokine programs that recruit recipient-derived inflammatory cells. Recruited host monocytes are present in the graft within 6 hours post-reperfusion and undergo distinct alterations in response to IRI stress that drive their activation and differentiation into monocyte-derived macrophages (MDMs).8,9 PRR activation also links innate and adaptive immunity, as their signals improve the antigen-presenting capacity of macrophages to host T cells.10 The mechanism by which OLT-IRI immune cascades regulate human MDM recruitment/enhancement of antigen presentation and subsequent adaptive alloimmunity remains to be elucidated.
The function of high mobility group box (HMGB) 1, an ubiquitously-expressed DNA binding protein classified as a DAMP, is determined by the oxidation of 3 cysteine residues. Fully-reduced all-thiol HMGB1 (aT-HMGB1) acts as a chemo-attractant, partially-oxidized disulfide HMGB1 (diS-HMGB1) induces pro-inflammatory cytokine secretion in monocytes/macrophages, and terminally-oxidized all-sulfonyl HMGB1 (aS-HMGB1) is thought to be non-immunogenic.11–13 We reported that post-reperfusion portal vein blood (liver flush [LF]) from IRI+ OLT patients contains increased concentrations of diS-HMGB1 compared to IRI– patients.14 IRI+ pre-reperfusion biopsies comprise macrophages with hyperacetylated, lysosomal diS-HMGB1 that increase by 2 hours post-reperfusion.14 This suggests that diS-HMGB1 secreted from macrophages induces a self-perpetuating cycle of inflammation in OLT-IRI by activating infiltrating macrophages. Although our data support the idea that diS-HMGB1 has the potential to function as a main driver of OLT injury,14 definitive evidence showing diS-HMGB1’s ability to worsen liver IRI and hepatocellular damage is lacking.
HMGB1 has multiple receptors15 with diS-HMGB1 reported to bind toll-like receptor (TLR) 4.13 HMGB1 can also bind unme-thylated DNA fragments and be endocytosed by the receptor for advanced glycation end products to trigger activation of intracellular TLR9, but the oxidative state of HMGB1 activating TLR9 is unknown.16 Both TLR4 and TLR9 serve as HMGB1 sensors that exacerbate the innate immune response in murine hepatic IRI.17,18 Post-reperfusion portal LF from IRI+ OLT patients containing increased concentrations of diS-HMGB1 induces higher activation of both TLR4-transfected and TLR9-transfected reporter cells,14,19 suggesting a dual role for these PRRs in diS-HMGB1 pro-inflammatory signaling. However, the definitive mechanism(s) underpinning how diS-HMGB1 differentiates monocytes into pro-inflammatory macrophages that promote graft inflammation and prime alloreactive T cells in human OLT are unknown. We hypothesized that diS-HMGB1 binds TLR4 and/or TLR9, causing monocyte differentiation into pro-inflammatory sentinel, which stimulate alloimmunity through cytokine secretion and alloantigen presentation in OLT-IRI.
2. Materials and methods
2.1. Study design, and sample and data collection
106 adult primary OLT recipients were recruited into a study approved by the UCLA Institutional Research Board (IRB #13–000143) (Table 1). All participants provided informed consent in writing, and all research was conducted in accordance with the Declaration of Helsinki. Routine standard-of-care and immune-suppressive therapy were administered as previously described.20 Donor and recipient clinical information, sample collection, data set collection, and analysis details were described previously.4,14,19
Table 1.
Recipient, donor, and transplant characteristics (n = 106).
| Recipient | Donor | ||
|---|---|---|---|
| diS-HMGB1 | 413 ± 333 | Age (y) | 39 ± 16 |
| Age (y) | 57 ± 11 | Sex | |
| Sex | Female | 48 (45) | |
| Female | 45 (42) | Male | 58 (55) |
| Male | 61 (58) | Race | |
| Race | Asian | 7 (7) | |
| Asian | 8 (7) | Black/African American | 8 (8) |
| Black/African American | 5 (5) | White/Caucasian | 55 (52) |
| White/Caucasian | 42 (40) | Hispanic/Latino | 33 (31) |
| Hispanic/Latino | 48 (45) | Other | 3 (3) |
| Other | 3 (3) | Status | |
| Liver disease etiology | DBD | 102 (96) | |
| AIH | 5 (5) | DCD | 4 (4) |
| ALF | 3 (3) | Warm ischemia (min) | 52 ± 11 |
| EtOH | 29 (27) | Cold ischemia (h) | 7.6 ± 3.3 |
| HBV | 4 (4) | Cause of death | |
| HCV | 34 (32) | Trauma | 37 (35) |
| NAFLD/NASH | 19 (18) | CVS | 32 (30) |
| PBC | 2 (2) | Anoxia | 35 (33) |
| PSC | 4 (4) | Other | 2 (2) |
| Other/unknown | 6 (6) | DM | 20 (12) |
| LFTs, opening | HTN | 35 (22) | |
| AST | 1091 ±1007 | CAD | 6 (4) |
| ALT | 545 ± 492 | LFTs at procurement | |
| Bilirubin | 8 ± 6.2 | ALT | 58 ± 89 |
| INR | 1.5 ± 0.3 | Bilirubin | .95 ± 0.69 |
| Transplant liver type | |||
| Liver/kidney | 11 (10) | Transplant, Recipient + Donor | |
| Split right tri-segment | 1 (1) | LIRI | |
| Whole liver | 94 (89) | LIRI+ | 47 (44) |
| MELD at list | 26 ± 11 | LIRI − | 59 (56) |
| MELD at transplant | 33 ± 9 | ABO | |
| Identical | 95 (90) | ||
| Compatible | 11 (10) | ||
| Donor risk index | 1.49 ± 0.36 | ||
| Sharing | |||
| Local | 63 (59) | ||
| Regional | 41 (39) | ||
| National | 2 (2) |
Values are mean ± SD or n (%).
AIH, autoimmune hepatitis; ALF, acute liver failure; ALT, alanine transaminase; AST, aspartate aminotransferase; CAD, coronary artery disease; CVS, cerebral vasospasm; DBD, donation after brain death; DCD, donation after cardiac death; DM, diabetes mellitus; EtOH, alcohol-associated; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; HCV, hepatitis C virus; HTN, hypertension; INR, international normalized ratio; LFT, liver function test; LIRI, liver ischemia-reperfusion injury; MELD, model for end-stage liver disease; NAFLD, non-alcoholic fatty liver disease; NASH, non-alcoholic steatohepatitis; PBC, primary biliary cirrhosis; PSC, primary sclerosing cholangitis.
2.2. Animals
C57BL/6 male mice at 8–10 weeks of age were used (Jackson Laboratory). Animals were housed in a UCLA animal facility under specific pathogen-free conditions and received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals.21 All studies were reviewed and approved by the UCLA Animal Research Committee (ARC #1999–094).
2.3. Mouse liver transplantation
We used an established liver IRI mouse model of ex vivo hepatic cold storage (18 hours), followed by syngeneic OLT.22 When required, diS-HMGB1 (100 ng/g) was directly injected into the portal vein immediately following liver reperfusion (diS-OLT). Animals were sacrificed at 6 hours post-reperfusion, the peak of hepatocellular damage in this model, and liver and serum samples were collected. The sham group was subjected to the same procedures except for OLT. Severity of IRI was graded using Suzuki histologic criteria, hepatocellular death, and graft function, as described previously.22
2.4. Disulfide-HMGB1 quantification
diS-HMGB1 was quantified by combining ELISA quantification and Western blot densitometry, as described previously.14
Additional materials and methods information may be found online in the Supplementary Method section.
3. Results
3.1. Disulfide-HMGB1 released during liver reperfusion correlates with hepatocellular injury and graft dysfunction and polarizes pro-inflammatory macrophages in human OLT recipients
To determine if diS-HMGB1 levels correlated with post-OLT clinical outcomes, we retrospectively analyzed the correlation between diS-HMGB1 levels in human OLT LF samples and clinical outcomes in a cohort of 106 OLT patients (Fig. 1A). The recipient, donor, and transplant characteristics are summarized in Table 1. IRI was scored by degree of neutrophil infiltration and hepatocyte necrosis in 2-hour post-reperfusion biopsies, as described previously.4 IRI+ (n = 47) patients showed higher diS-HMGB1 compared with IRI– patients (n = 59) (P = .0001) (Table 2, Supplementary Fig. S1A). OLT recipients were 2.4-times more likely to experience IRI for every 333 ng/mL of diS-HMGB1 released during reperfusion (P = .001). Longer donor warm ischemia time positively correlated with diS-HMGB1 concentration (P = .04) (Table 2, Supplementary Fig. S1B). No other recipient or donor factors correlated with LF diS-HMGB1 concentration (Supplementary Tables S1 and S2).
Figure 1.
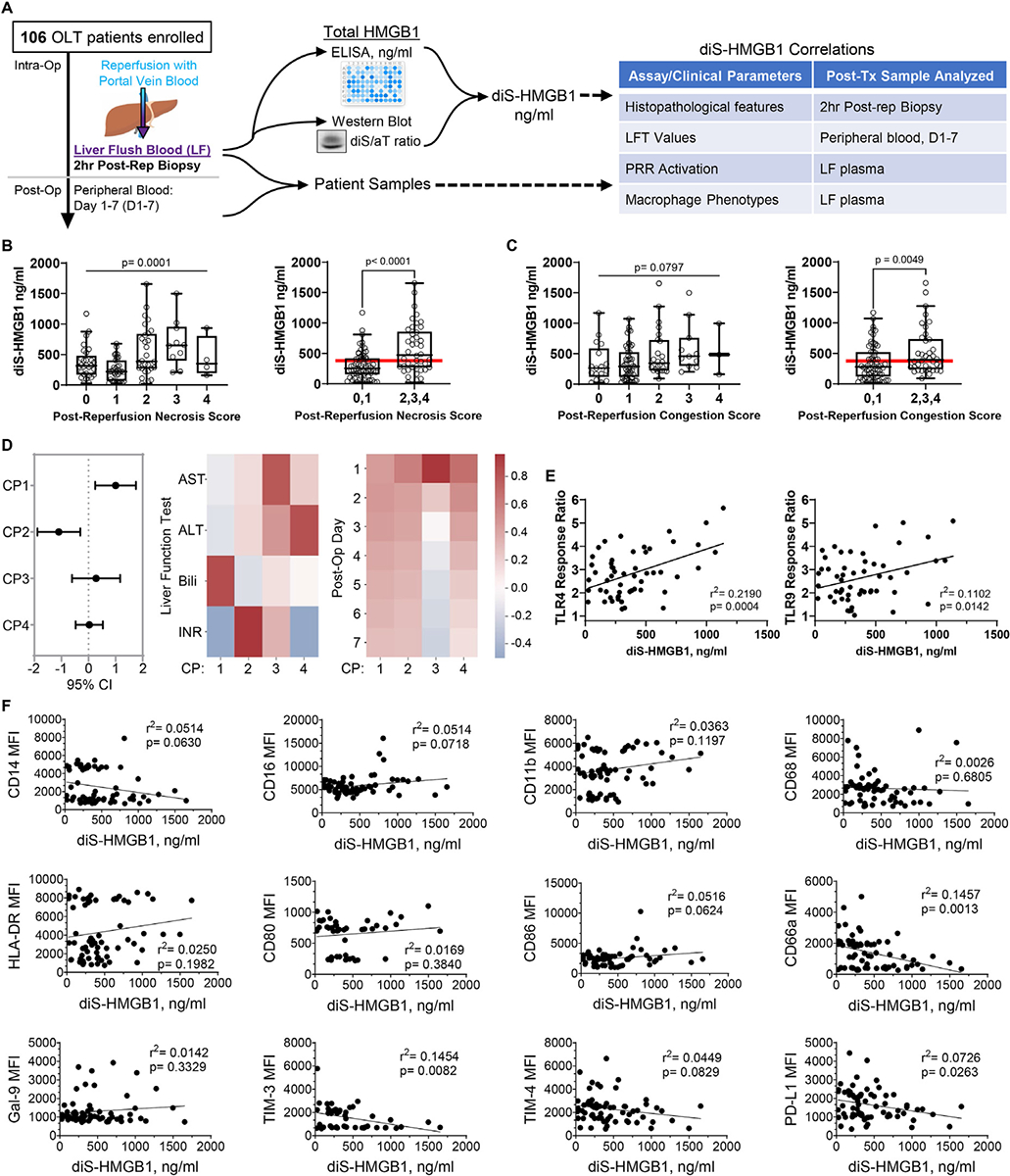
Disulfide-HMGB1 released during liver reperfusion correlates with a pro-inflammatory profile in OLT patients. diS-HMGB1 concentration in LF samples from 106 OLT patients were correlated with histopathologic features on 2-hour post-reperfusion biopsies, liver function test (LFT) data for the first 7 days post-transplant, ability of LF to activate TLR4 and TLR9 reporter cell lines, and surface marker expression of healthy volunteer monocytes cultured with recipient LF for 3 days in vitro. (A) Patient sample collection and analysis overview. (B-F) diS-HMGB1 concentration was assessed in LF patient samples regardless of IRI status and correlated with: severity (0, none; 1, minimal; 2, mild; 3, moderate; and 4, severe) of 2-hour post-reperfusion biopsies and by grouping of negative (0 and 1) vs positive (2, 3, and 4) for (B) necrosis and (C) sinusoidal congestion with red line indicating overall population mean diS-HMGB1 concentration (n = 106); (D) LFT values for the first week post-transplant summarized by canonical polyadic (CP) decomposition (n = 106); (E) LF activation of human TLR4-transfected and TLR9-transfected HEK-Blue reporter cell lines (n = 54); (F) surface marker expression of healthy volunteer monocytes after 3-day culture with LF samples (n = 69). Data are (B, C) Tukey plots: whiskers notate either 1.5 times the interquartile range or minimum and maximum values, whichever is closer to the median; boxes notate interquartile ranges; and lines indicate median values with all points shown; (D) left to right: regression coefficients and 95% CIs of component regression against LF diS-HMGB1 concentration, heatmap of component association with each of the 4 LFTs, and heatmap of component association with each of the 7 days post-transplant; warmer colors denote positive associations, cooler colors denote negative associations; (E, F) dot plots and lines of best fit for individual patient TLR activation scores or surface marker mean fluorescence intensities (MFIs) and corresponding LF diS-HMGB1 concentration.
Table 2.
Characteristics correlated with diS-HMGB1 concentration in LF samples.
| Characteristic, continuous | r | P |
| Donor warm ischemia (min) | 0.204 | .04* |
| Characteristic, categorical | diS-HMGB1 (ng/ml), mean ± SD | P |
| LIRI | .0001* |
Two cohort characteristics correlated with diS-HMGB1 in patient LF samples using simple linear regression and ANOVA models for continuous and categorical variables, respectively; r values represent the correlation coefficients of continuous factors with diS-HMGB1;
P <.05. Correlations of all other characteristics with diS-HMGB1 concentration were not significant and shown in Supplementary Tables S1 and S2.
LIRI, liver ischemia-reperfusion injury.
To determine if diS-HMGB1 levels associate with hepatocellular injury in human OLT, we correlated diS-HMGB1 concentration in LF samples from 106 OLT recipients with the following: (1) histologic features on 2-hour post-reperfusion liver biopsies, and (2) liver function tests (LFTs) from peripheral blood draws for the first 7 days post-transplant. Indeed, higher concentrations of diS-HMGB1 in recipient LF correlated with increased hepatocellular necrosis (0-to-4 severity score: P = .0001; negative [0 and 1] vs positive [2, 3, and 4] for feature: P < .0001) (Fig. 1B) and sinusoidal congestion (0-to-4 severity score: P = .0797; negative [0 and 1] vs positive [2, 3, and 4] for feature: P = .0049) (Fig. 1C). To identify common patterns across LFTs in the first week post-transplant and their relationship with diS-HMGB1, we applied a dimensionality reduction technique called canonical polyadic decomposition (Fig. 1D; Supplementary Methods). The first 4 components contributed to ~85% of dataset variance and were therefore analyzed further (Supplementary Fig. S2). Component 1 positively correlated with diS-HMGB1 (β = 1.00; 95% CI: 0.25–1.75) and was associated with high bilirubin and low international normalized ratio (INR), whereas component 2 negatively correlated with diS-HMGB1 (β = −1.01; 95% CI: −1.89, −0.30) and was associated with high INR. Components 1 and 2 demonstrated sustained association to each post-transplant day, indicating the pattern did not have a temporal relationship. However, components 3 (β = 0.28; 95% CI: −0.61, 1.17) and 4 (β = 0.03; 95% CI: −0.47, 0.53) did not correlate with diS-HMGB1 and contained higher LFT values at Day 1 that tapered over the first 7 days post-transplant, suggesting transient and resolving high LFT values did not correlate with diS-HMGB1. Therefore, histology and LFT results across time suggest that diS-HMGB1 correlates with hepatocellular damage and inflammation that does not resolve within the first week post-OLT.
Next, we investigated whether diS-HMGB1 concentration correlates with the ability of LF samples to activate TLR-transfected HEK-293 reporter cell lines. diS-HMGB1 concentration in LF positively correlated with TLR4 (r2 = 0.22, P = .0004) and TLR9 (r2 = 0.11, P = .01) activation (Fig. 1E). Based on these findings, we questioned if diS-HMGB1 present in LF could alter the phenotype of monocytes in 3-day cultures (n = 69) (Fig. 1F). Lower diS-HMGB1 concentrations correlated with the expression of proteins associated with dampening innate and adaptive immune responses, including CD66a (r2 = 0.15, P = .001), TIM-3 (r2 = 0.15, P = .008), and PD-L1 (r2 = 0.07, P = .03). These results support the hypothesis that generation of diS-HMGB1 during OLT-IRI can drive macrophage polarization to a pro-inflammatory profile in recipients via TLR4 and TLR9 signaling.
3.2. Disulfide-HMGB1 increases human monocyte cytokine production through TLR4 and TLR9 signaling
To elucidate the role of diS-HMGB1 in pro-inflammatory MDM polarization, we developed an in vitro culture system to model the stimulation of OLTrecipient infiltrating monocytes by diS-HMGB1 released from the donor allograft upon reperfusion. Monocytes were cultured with purified diS-HMGB1 or the non-immunogenic, all-sulfonyl form of HMGB1 (aS-HMGB1) at 600 ng/mL, the mean value of diS-HMGB1 previously detected in IRI+ patient LF samples.14 After 24-hour stimulation with diS-HMGB1 or aS-HMGB1, we analyzed culture supernatant cytokines and chemokines by multiplex Luminex. diS-HMGB1–stimulated monocytes showed an increase in 9 pro-inflammatory cytokines, 2 anti-inflammatory cytokines, 7 chemokines, and 4 growth factors (Fig. 2A). Increases that reached statistical significance included pro-inflammatory cytokines interleukin (IL)-1β (P = .05), IL-6 (P = .05), and interferon (IFN)-α2 (P = .004); anti-inflammatory cytokines IL-10 (P = .02) and IL-1RA (P = .02); chemokines MDC (P = .03) and MCP-1 (P = .05); and growth factors EGF (P = .03), GM-CSF (P = .05), and IL-7 (P = .05). These data showed that diS-HMGB1 triggers monocyte-derived inflammatory soluble mediators that can induce a local pro-inflammatory environment, recruit myeloid cells into the liver, and support myeloid/lymphoid cell differentiation and activation.
Figure 2.

Disulfide-HMGB1 increases human monocyte pro-inflammatory phenotype through TLR4 and TLR9. Healthy volunteer monocytes were stimulated with 600 ng/mL aS-HMGB1 or diS-HMGB1 and either 10 μg/ml TAK-242 (stored in dimethyl sulfoxide [DMSO]; TLR4 inhibitor, TLRi), 1:1000 dilution of DMSO (to match dilution used for TAK-242; TLR4 control, TLR4c), the synthetic oligonucleotide TLR9 antagonist ODN2088 (TLR9 inhibitor, TLR9i) at 5 μM, inactive oligonucleotide control ODN2088c (TLR9 control, TLR9c) at 5 μM, or both inhibitors or controls in combination (TLR4+9i/TLR4+9c) for 24 hours, at which time supernatants were collected and analyzed for cytokine secretion. (A) Log2 fold-change of cytokine concentrations in diS-HMGB1-treated compared with that of cytokine concentrations in aS-HMGB1-treated supernatants (n = 4). Data are Tukey plots: whiskers notate either 1.5 times the interquartile range or minimum and maximum values, whichever is closer to the median; boxes notate interquartile ranges; and lines indicate median values with all points shown. One-sample t tests, *P < .05, ** P < .01. (B) Log2 fold-change of cytokine concentrations in diS-HMGB1+inhibitor or diS-HMGB1+control supernatants compared with that of cytokine concentrations in supernatants of diS-HMGB1 alone (n = 4). Vertical axis notates inhibitor or control treatments for each row while the horizontal axis notates cytokines. Point size represents the P value of cytokine concentration under inhibitor or control treatments compared with that of concentration under diS-HMGB1 treatment alone, with larger points having lower, more significant P values. Cooler colors denote a decrease compared with diS-HMGB1 alone and warmer colors denote an increase compared with diS-HMGB1 alone. Filled points indicate inhibitors, while corresponding outlined points immediately below each inhibitor indicate respective controls. All cytokines were tested with each inhibitor and control. The dot plot shows inhibitor/control combinations where the inhibitor had a P value of .05 compared with diS-HMGB1 alone using a one-sample t test.
As TLR4 and TLR9 are diS-HMGB1 receptors and elevated concentrations of diS-HMGB1 elaborated from the allograft into LF positively correlated with TLR4 and TLR9 activation, we examined the requirement for 1 or both PRRs in the diS-HMGB1–induced cytokine/chemokine profile. Monocytes were stimulated for 24 hours with diS-HMGB1 with/without the following TLR4 or TLR9 pharmacologic inhibitors: TAK-242 (a TLR4 small-molecule inhibitor stored in dimethyl sulfoxide [DMSO] [TLR4i]), ODN-2088 (a TLR9 antagonist oligonucleotide [TLR9i]), both inhibitors in combination (TLR4+9i), or appropriate controls (DMSO, TLR4c; inactive oligonucleotide ODN-2088c, TLR9c; TLR4+9c); all 24 cytokines were tested under each condition (Fig. 2B). Production of 19 of the 24 cytokines were altered by TLR4 and/or TLR9 inhibition. TLR4i decreased IL-1β (P = .05), IL-6 (P = .04), IFN-γ (P = .006), TNF-α (P = .05), IL-1RA (P = .05), IL-8 (P = .05), MDC (P = .02), MIP-1a (P = .03), EGF (P = .03), Flt-3 (P = .03), and IL-7 (P = .01). TLR9i decreased MDC (P = .002) and increased MIP-1a (P = .01), MCP-3 (P = .04), and IP-10 (P = .04). Combined TLR4i+9i treatment increased MIP-1a (P = .02), decreased IL-1α (P = .02), IFN-α2 (P = .03), and IL-10 (P = .01) and decreased MDC (P = .0002). These data confirm that TLR4 and TLR9 are the principal PRRs regulating the diS-HMGB1–induced pro-inflammatory macrophage secretome.
3.3. Disulfide-HMGB1 triggers macrophage polarization to a pro-inflammatory phenotype and function through TLR4 and TLR9 signaling
Having established that TLR4 and TLR9 control the cytokine profile of diS-HMGB1–stimulated monocytes, we next asked if diS-HMGB1 regulates MDM polarization through TLR4, TLR9, or both. We first cultured monocytes with diS-HMGB1 or aS-HMGB1 for 5 days and assessed their T cell activation molecules (HLA-DR and CD86), T cell inhibitory molecules (PD-L1 and TIM-3) and Fc receptors (CD16 and CD64) by flow cytometry. diS-HMGB1 increased expression of HLA-DR (P = .06), CD86 (P = .02), and CD64 (P = .02) and decreased PD-L1 (P = .04) (Fig. 3A). TIM-3 and CD16 expression did not differ between aS-HMGB1 and diS-HMGB1 stimulation. Further, diS-HMGB1’s ability to upregulate HLA-DR, CD86, and CD64 while sup-pressing PD-L1 was blocked by neutralizing HMGB1 antibodies (αHMGB1) (Fig. 3B) (HLA-DR: P < 0.0001; CD86: P = .0007; CD64: P = .0002; PD-L1: P = .001). These data reveal diS-HMGB1 can enhance MDM-mediated recipient innate-adaptive crosstalk through upregulated antigen presentation machinery and Fc receptors and downregulation of inhibitory molecules.
Figure 3.
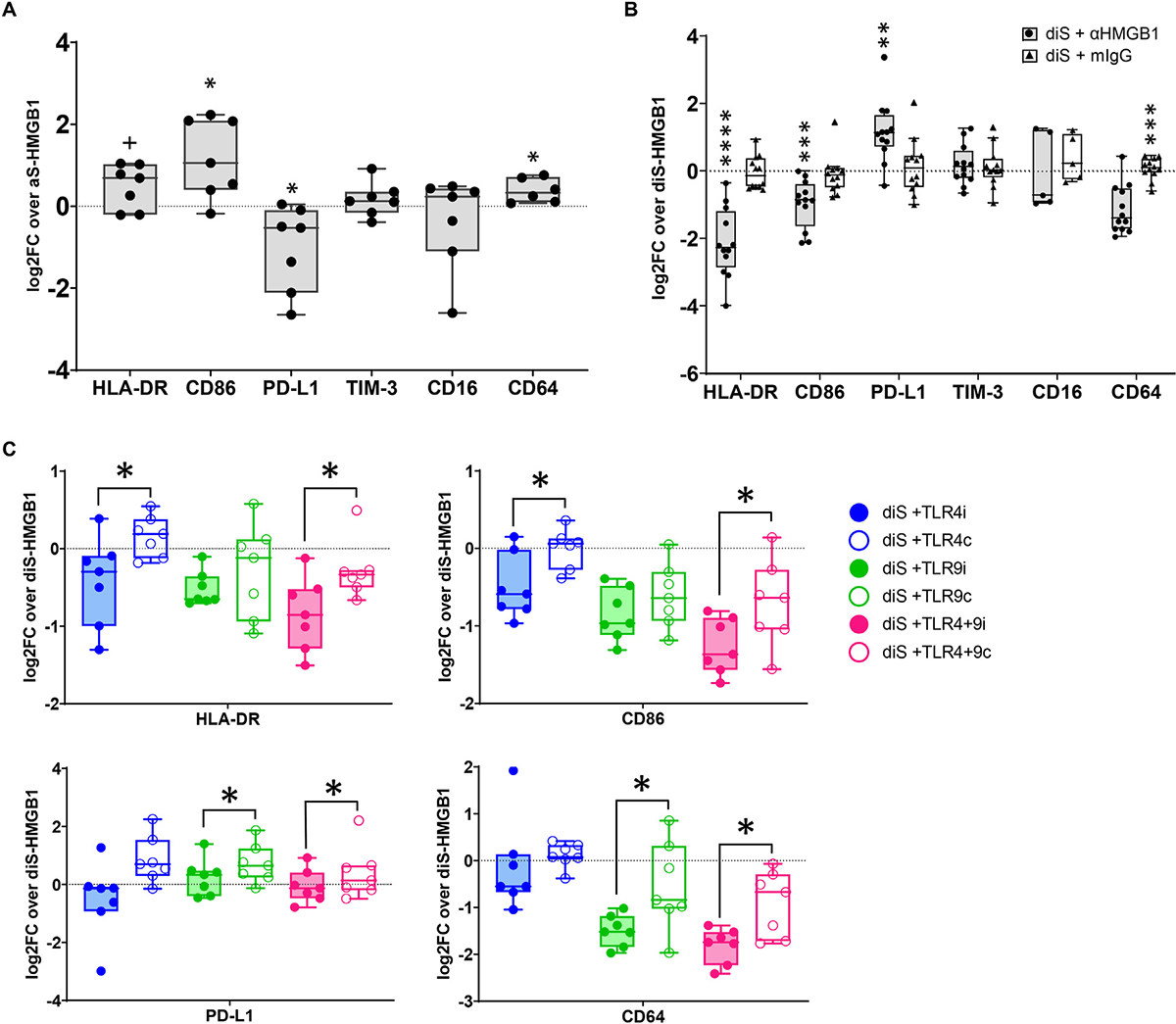
Disulfide-HMGB1 increases human macrophages pro-inflammatory phenotype through TLR4 and TLR9. Healthy volunteer monocytes were stimulated with aS-HMGB1 or diS-HMGB1, and either 2.25 ng/mL HMGB1 neutralizing antibody (αHMGB1) or isotype control (mIgG) or 10 μg/ml TAK-242 (stored in dimethyl sulfoxide [DMSO]; TLR4 inhibitor, TLRi), 1:1000 dilution of DMSO (to match dilution used for TAK-242; TLR4 control, TLR4c), the synthetic oligonucleotide TLR9 antagonist ODN2088 (TLR9 inhibitor, TLR9i) at 5 μM, inactive oligonucleotide control ODN2088c (TLR9 control, TLR9c) at 5 μM, or both inhibitors or controls in combination (TLR4+9i/TLR4+9c) on Day 0 and cultured for 5 days. Surface marker expression was assessed by flow cytometry on Day 5. (A) Log2 fold-change of each marker on diS-HMGB1-stimulated vs aS-HMGB1-stimulated macrophages from the same healthy volunteer donor (CD64 n = 6, all others n = 7). (B) Log2 fold-change of each marker on diS-HMGB1-stimulated macrophages during αHMGB1 treatment vs diS-HMGB1 stimulation alone (CD16 n = 5, all others n = 12). (C) Log2 fold-change of each marker on diS-HMGB1–stimulated macrophages during TLR inhibition or control treatment vs diS-HMGB1 stimulation alone (n = 7). Data are Tukey plots: whiskers notate either 1.5 times the interquartile range or minimumandmaximumvalues, whichever is closer to themedian; boxes notate interquartile ranges; and lines indicatemedian values with all points shown. (A, B) One-sample t tests, +P <.06, * P <.05, ** P <.01, *** P <.001, **** P <.0001 (C) Paired t tests between inhibitor and control conditions, * P <.05
We next investigated the specific roles of TLR4 and TLR9 as essential regulators of diS-HMGB1 MDM polarization. We stimulated donor monocytes with diS-HMGB1 together with TLR4i, TLR9i, TLR4+9i, or respective controls. Surface marker expression on MDMs treated with inhibitors was compared with MDMs from the same donor treated with controls (Fig. 3C). TLR4i decreased HLA-DR (P = .05) and CD86 (P = .04), whereas TLR9i decreased CD64 (P = .03) and PD-L1 (P = .01). Combined TLR4+9i decreased HLA-DR (P = .02), CD86 (P = .04), CD64 (P = .01), and PD-L1 (P = .05), confirming the regulatory role of TLR4 and TLR9 on their expression. These data reveal TLR4 and TLR9 each have unique, yet cooperative, impacts on diS-HMGB1–induced expression of CD86, HLA-DR, CD64, and PD-L1, and therefore both contribute to diS-HMGB1–mediated innate-adaptive interactions.
3.4. Disulfide-HMGB1 increases human macrophage reactive oxygen species production through TLR9 signaling
Macrophages produce reactive oxygen species (ROS) that can damage parenchymal liver cells, promoting alloreactivity through the release of donor HLA antigens.23,24 To examine if diS-HMGB1–polarized MDMs produce ROS via TLR4 and/or TLR9, we stimulated monocytes with diS-HMGB1, lipopolysaccharide (LPS) (TLR4 ligand), or ODN2216 (TLR9 agonist) on Day 0 and cultured cells for 5 days (Fig. 4A). diS-HMGB1 stimulated higher ROS production compared with both LPS (P = .002) and ODN2216 (P = .0096). We then assessed diS-HMGB1–mediated ROS production in the presence of TLR4i, TLR9i, or respective controls (Fig. 4B, C). TLR9i decreased ROS production (P = .003) but TLR4i had no effect (P = .29). Neutralizing diS-HMGB1 decreased ROS production (P = .0003) (Fig. 4D) to levels equivalent to TLR9i treatment (P = .5) (Fig. 4E). These data suggest that diS-HMGB1 induction of ROS production by MDMs is mediated through TLR9.
Figure 4.

Disulfide-HMGB1 increases reactive oxygen species (ROS) production in human macrophages via TLR9. Healthy volunteer monocytes were stimulated with 10 ng/mL LPS (TLR4 ligand), the synthetic oligonucleotide ODN2216 (TLR9 agonist), or diS-HMGB1 ± 2.25 ng/mL HMGB1 neutralizing antibody (αHMGB1) or isotype control (mIgG) or 10 μg/mL TAK-242 (stored in dimethyl sulfoxide [DMSO]; TLR4 inhibitor [TLRi]), 1:1000 dilution of DMSO (to match dilution used for TAK-242; TLR4 control [TLR4c]), the synthetic oligonucleotide TLR9 antagonist ODN2088 (TLR9 inhibitor [TLR9i]) at 5 μM, or inactive oligonucleotide control ODN2088c (TLR9 control [TLR9c]) at 5 μM on Day 0. ROS production was assessed on Day 5 by flow cytometry. (A) Log2 fold-change of ROS levels in diS-HMGB1-stimulated macrophages vs macrophages stimulated by LPS (n = 10) or ODN2216 (n = 6). (B-D) Log2 fold-change of ROS levels in diS-HMGB1–stimulated macrophages under (B) TLR4i/TLR4c, (C) TLR9i/TLR9c, or (D) αHMGB1/mIgG treatment (n = 6/group) vs diS-HMGB1 alone. (E) Comparison of ROS levels under TLR9i and αHMGB1 treatments (n = 6). Data are Tukey plots: whiskers notate either 1.5 times the interquartile range or minimum and maximum values, whichever is closer to the median; boxes notate interquartile ranges; and lines indicate median values. All points are shown. (A-D) One-sample t tests, (E) paired t test. ns, not significant, **P <.01, *** P < .001, **** P < .0001.
3.5. Disulfide-HMGB1–polarized human macrophages have increased capacity to present antigens and activate T cells
To address if diS-HMGB1–polarized macrophages are poised to stimulate an adaptive immune response, we cultured monocytes with patient LF samples and analyzed their PD-L1:CD86 ratio, a measurement that reflects macrophage capacity to suppress or activate T cells.25 LF from IRI+ patients induced a lower PD-L1:CD86 ratio than the corresponding IRI– LF samples (P = .001) (Fig. 5A). Notably, regardless of patient IRI status, higher diS-HMGB1 concentrations in LF correlated with a lower PD-L1:CD86 ratio (r2 = 0.0841, P = .02) (Fig. 5B), suggesting that increased diS-HMGB1 leads to greater capacity for MDMs to activate T cells. To determine if this lower ratio is linked to increased post-OLTalloimmunity, we compared the LF-induced PD-L1:CD86 ratio to the presence of post-transplant donor-specific HLA antibodies (DSA) in OLT patients. LF from post-transplant DSA producers (n = 15) stimulated a lower PD-L1:CD86 ratio on macrophages compared with the LF from patients who did not have post-OLT DSA (n = 26) (P = .04) (Fig. 5C). To confirm diS-HMGB1’s capacity to upregulate co-stimulation/co-inhibitory molecules, we compared the PD-L1:CD86 ratio of macrophages stimulated with purified diS-HMGB1 and aS-HMGB1. Consistent with patient LF, diS-HMGB1 stimulated a lower PD-L1:CD86 ratio compared with aS-HMGB1-stimulated macrophages from the same donor (P = .02) (Fig. 5D).
Figure 5.
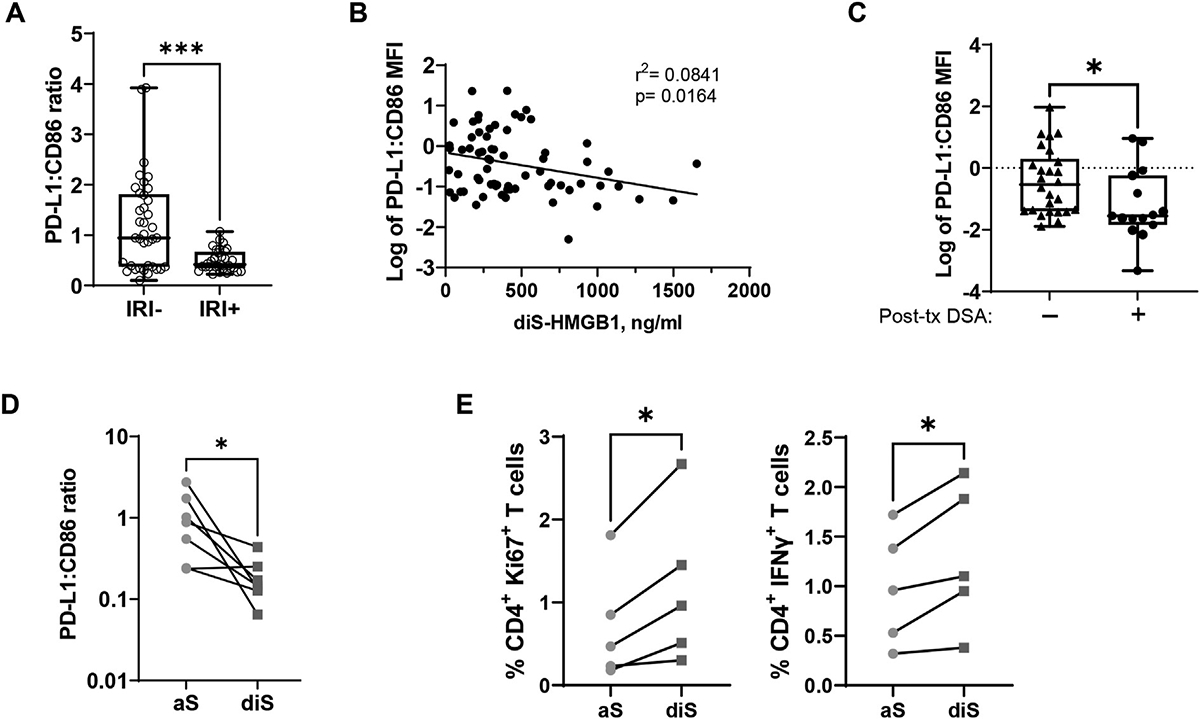
Disulfide-HMGB1 decreases macrophage PD-L1:CD86 ratio and increases T cell activation. (A) Healthy donor monocytes were cultured for 3 days with OLT recipient LF. Surface marker expression of PD-L1 and CD86 were assessed by flow cytometry on Day 3. The ratio of PD-L1 mean fluorescence intensity (MFI) to CD86 MFI was calculated and stratified by patient IRI status (IRI–, n = 37; IRI+, n = 32). Unpaired Mann-Whitney. (B) The log of the PD-L1:CD86 ratio as described in (A) was correlated with the corresponding diS-HMGB1 concentration in each recipient’s LF sample regardless of IRI status (n = 69). (C) Longitudinal post-transplant blood samples from 41 OLT recipients were tested for the presence of anti-donor HLA antibodies (donor-specific antibodies [DSAs]) through HLA-coated single-antigen bead testing. Then, the log of the PD-L1:CD86 ratio induced by recipient LF as described in (A) was stratified by the presence or absence of DSA in each recipient (detectable or not; Post-tx DSA–, n = 26; Post-tx DSA+, n = 15). Unpaired t test. (D) Healthy volunteer monocytes were stimulated with 600 ng/mL aS-HMGB1 or diS-HMGB1 on Day 0 and cultured for 5 days, at which time PD-L1 and CD86 expression was assessed by flow cytometry. The ratio of PD-L1 MFI vs CD86 MFI was compared between aS-HMGB1–stimulated and diS-HMGB1–stimulated macrophages from the same monocyte donor (n = 7). Paired t test. (E) Healthy volunteer monocytes were stimulated with 600 ng/mL aS-HMGB1 or diS-HMGB1 on Day 0 and cultured for 5 days. CD4+ T cells from the same monocyte donor were then co-cultured with Day 5 macrophages and SARS-CoV-2 peptides, a model antigen standing in for donor peptides, for 24 hours. CD4+ T cells were analyzed for proliferation by Ki67 staining and pro-inflammatory cytokine production by IFN-γ staining at the 24-hour time point. Percentages of CD4+ T cells positive for Ki67 or IFN-γ are shown. CD4+ T cells co-cultured with diS-HMGB1–stimulated macrophages were compared with CD4+ T cell co-cultured with aS-HMGB1–stimulated macrophages from the same healthy donor (n = 5). Paired t test. Data are (A, C) Tukey plots: whiskers notate either 1.5 times the interquartile range or minimum and maximum values, whichever is closer to the median; boxes notate interquartile ranges; and lines indicate median values, all points are shown. *P < .05, ***P < .001.
Collectively, our data support the notion that diS-HMGB1–polarized MDMs have increased capacity to present antigens and activate T cells that can then influence antibody production and class-switching in B cells. To investigate this possibility, we developed a co-culture assay modeling indirect allorecognition, where recipient MDMs present donor peptides to recipient alloreactive memory T cells. Human CD4+ T cells were co-cultured with diS-HMGB1–derived or aS-HMGB1–derived MDMs and SARS-CoV-2 spike protein peptide pools for 24 hours. diS-HMGB1–stimulated MDMs induced higher Ki67 (P = .02) and IFN-γ (P = .02) expression in CD4+T cells compared with their aS-HMGB1-stimulated counterparts (Fig. 5E). These data indicate that diS-HMGB1 can directly influence adaptive immune activation.
3.6. Disulfide-HMGB1 increases hepatocellular injury in a murine OLT-IRI model
To determine if diS-HMGB1 mediates hepatocellular injury, we used a syngeneic OLT murine model of IRI with extended 18-hour cold storage that mimics the marginal human OLT setting.22 We collected the PBS used to flush the cold-stored liver immediately before the transplant surgery (PBS LF) and blood 6 hours post-reperfusion (post-OLT) (Fig. 6A). diS-HMGB1 was the principal form of HMGB1 present at both pre-reperfusion and post-reperfusion phases compared with aT-HMGB1 (PBS, P = .01; post-OLT, P = .05) (Fig. 6B, C). The diS-HMGB1 to aT-HMGB1 ratio also increased between the end of cold storage (2.9 ± 0.4) and 6 hours post-reperfusion (5.5 ± 1.0) (P = .01). To determine if diS-HMGB1 potentiates the severity of hepatic IRI, recipient mice (n = 6/group) were conditioned with diS-HMGB1 (100 ng/g; portal vein) immediately following reperfusion (Fig. 6D) and compared with OLT without diS-HMGB1 adjunct (diS-OLT vs OLT). At 6 hours post-reperfusion, diS-HMGB1–treated IR-stressed OLT displayed augmented hepatocellular injury compared with controls (Fig. 6E). These correlated with increased Suzuki grading of liver IRI (OLT = 5 ± 1.3 vs diS-OLT = 7 ± 1.8; P = .05) (Fig. 6F); frequency of TUNEL+ hepatocytes (determined by nuclear morphology) (OLT = 49.17 ± 12.27 vs diS-OLT = 85.83 ± 18.15; P = .002) (Fig. 6G, H) and hepatocellular damage (serum alanine transaminase (ALT): OLT = 4436 ± 1349 vs diS-OLT = 10 161 ± 5950 IU/L; P = .04) (Fig. 6I). These data demonstrated that diS-HMGB1 augments hepatocellular injury and liver dysfunction post-transplant.
Figure 6.
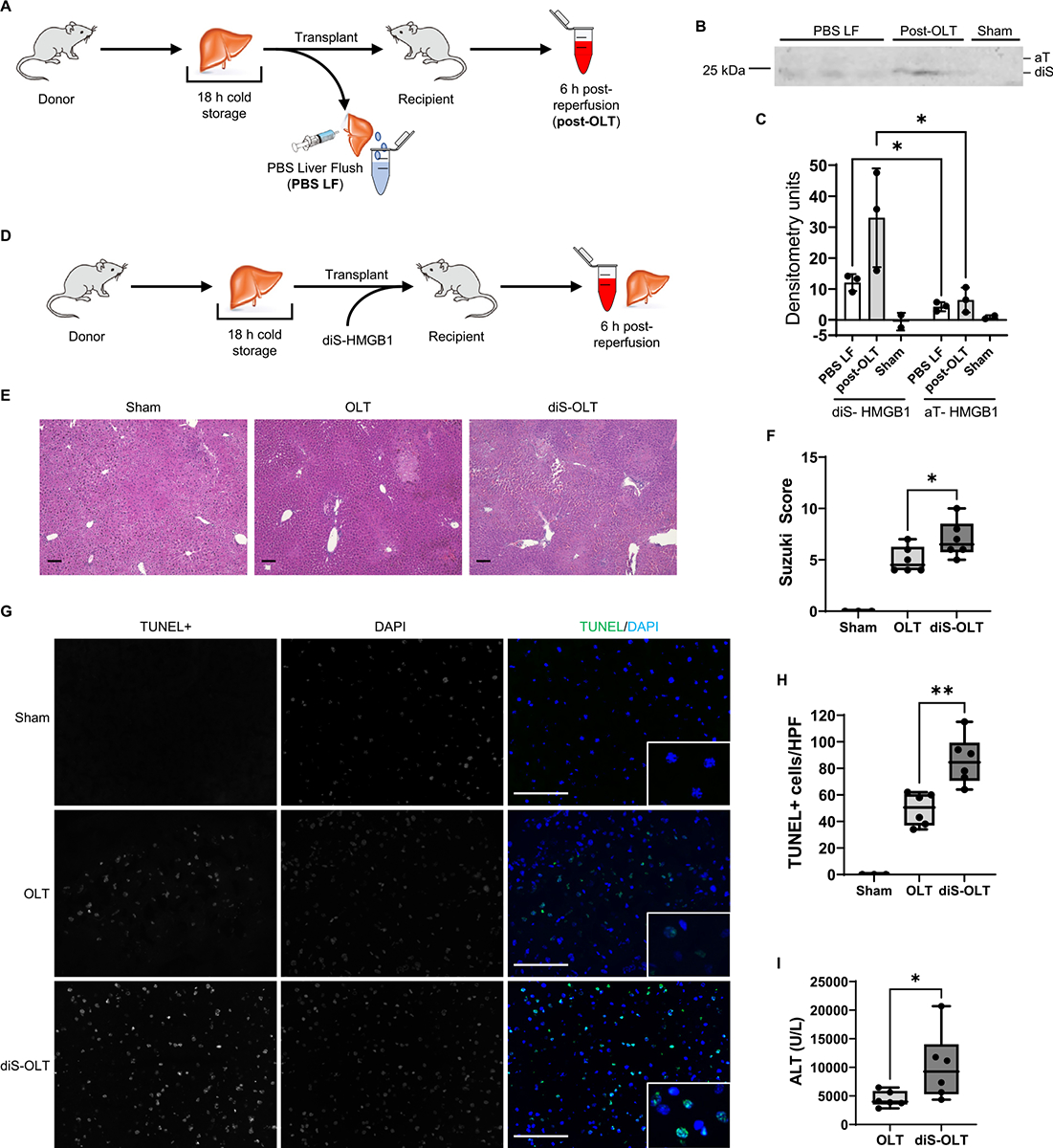
Disulfide-HMGB1 potentiates mouse OLT-IRI. C57BL/6 mice subjected to syngeneic OLT-IRI were analyzed for presence of diS-HMGB1 after ischemia only and after ischemia/reperfusion, and for the effect of exogenous diS-HMGB1 on histologic Suzuki scoring, DNA damage by TUNEL staining, and alanine transaminase (ALT) levels in serum. (A) Experimental design and sample collection workflow to determine serum diS-HMGB1 concentrations in a syngeneic OLT mouse model with extended 18-hour cold storage. Sham control animals were subjected to abdominal opening but no transplantation; serum from sham animals was collected 6 hours after closing. (B) Non-reducing Western blot of PBS LF (n = 3), post-OLT (n = 3), and sham (n = 2) serum samples collected as noted in (A). (C) Densitometry quantification of diS-HMGB1 (bottom band) and aT-HMGB1 (top band) levels in Western blot from (B). (D) Experimental design for mice subjected to syngeneic OLT with 18-hour extended cold storage and administration of 100 ng/g exogenous diS-HMGB1 (diS-OLT). This dose was determined to be equivalent to the highest diS-HMGB1 concentration found in the LF of IRI+ recipients. OLT control animals underwent the same transplant procedure without diS-HMGB1 administration. (E) Representative hematoxylin-and-eosin–stained images of sham (n = 3), OLT (n = 6), and diS-OLT (n = 6) liver tissue collected 6 hours post-reperfusion, 100, scale bar 100 μm. (F) Suzuki histologic grading of liver IRI severity in tissues shown in (E). (G) Representative images of terminal deoxy-nucleotidyl transferase dUTP nick end labeling (TUNEL) staining in sham (n = 3), OLT (n = 6), and diS-OLT (n = 6) liver tissue collected 6 hours after reperfusion; main panel: 400×, scale bar 100 μm, inlay: 1000×; HPF, high-power field. (H) Quantification of TUNEL+ hepatocytes in tissues shown in (G). (H) ALT levels in OLT and diS-OLT mouse serum collected 6 hours after reperfusion (n = 6/group). Data are (C) mean ± SD or (F,H,I) Tukey plots: whiskers notate either 1.5 times the interquartile range or minimum and maximum values, whichever is closer to the median; boxes notate interquartile ranges; and lines indicate median values with all points shown. *P < .05, **P < .01, paired t test for Western blots, unpaired t tests for mouse data.
4. Discussion
This translational study aimed to test the hypothesis that diS-HMGB1 is a key mediator of myeloid cell inflammation and alloimmunity in OLT-IRI through TLR4 and/or TLR9. diS-HMGB1 levels corresponded with increased hepatocellular necrosis, congestion, and graft dysfunction that failed to resolve during the first week post-transplant, and heightened immune activation in human OLT-IRI. TLR4 and TLR9 were key innate immune receptors driving diS-HMGB1–mediated human MDM polarization to a pro-inflammatory phenotype capable of promoting graft inflammation and T cell priming. TLR4 and TLR9 inhibition impaired macrophage commitment toward the pro-inflammatory phenotype induced by diS-HMGB1 alone—TLR4 ablation abrogated expression of HLA class II and co-stimulatory CD86 and cytokine production, whereas TLR9 silencing downregulated CD64 and PD-L1 expression and chemokine/ROS production. Given the increase of co-stimulatory molecules and MHC class II and downregulation of co-inhibitory molecules, diS-HMGB1–polarized MDMs were more efficient in driving antigen-specific memory T cell activation. These in vitro data highlight the novel function of the diS-HMGB1/TLR4/TLR9 axis in myeloid cell regulation, capacity for antigen presentation and capability to drive T cell–mediated alloimmunity in the mechanism of liver IRI. We also provide what we believe is the first direct evidence that administration of diS-HMGB1 exacerbated IR–mediated hepatocellular injury in a clinically-relevant murine OLT model. As such, diS-HMGB1 and its TLR4/TLR9 cognate receptors may represent putative therapeutic targets for preventing IRI in solid organ transplants and, potentially, more broadly in other disease states involving ischemic injury/sterile inflammation, such as stroke and myocardial infarction.16,26–28
diS-HMGB1 was present in murine circulation at 6 hours post-reperfusion in an in vivo model of hepatic cold ischemia that mimics marginal human OLT-IRI, consistent with the presence of diS-HMGB1 in human OLT-IRI.14 Further, injection of exogenous diS-HMGB1 into the liver at the time of reperfusion augmented IR-induced hepatocyte necrosis, sinusoidal congestion, and vacuolization, and DNA damage, resulting in increased ALT levels. Although prior studies reported that extracellular HMGB1 causes injury in a murine model of liver IRI via TLR4 signaling,18,29 we believe this is the first report showing the importance of HMGB1 oxidation state in OLT damage. These results are consistent with the concept that diS-HMGB1 is a key driver of OLT inflammation/injury and support pre-clinical assessment for therapeutics dampening diS-HMGB1, such as HMGB1 neutralizing antibodies or TLR4/9 inhibitors.30 Future work in murine models of IRI can explore the role of diS-HMGB1–skewed MDMs and utilize TLR4/TLR9 knockout mice to understand their role in diS-HMGB1–mediated injury.
The importance of diS-HMGB1 to clinical outcomes was corroborated by our findings in OLT patients. diS-HMGB1 concentration in reperfusion effluent correlated with severity of hepatocellular injury and early allograft dysfunction (EAD). We did not find a sex-dependent effect of diS-HMGB1 concentration on hepatocyte necrosis or sinusoidal congestion. Furthermore, OLT patient plasma containing diS-HMGB1 had greater potential to activate TLR4/TLR9 signaling in reporter cells and induce a pro-inflammatory macrophage phenotype. Similar to the L-GrAFT scoring system that uses the rate of change of aspartate transaminase, bilirubin, and platelet counts and maximum INR value to assess EAD,31 diS-HMGB1 release in patient LF correlated with a low rate of change for bilirubin and INR, suggesting that measuring its level could identify OLT recipients at risk for EAD.
Purified diS-HMGB1 was a potent driver of MDM polarization in macrophage phenotype/function, comparable with MDMs polarized with OLT patient plasma containing diS-HMGB1. These MDMs were characteristic of M1 macrophages expressing increased antigen presentation machinery and secreting greater amounts of inflammatory cytokines, chemokines, and growth factors. Oxidative stress from ROS production also contributes to graft injury because it is detrimental to liver parenchymal cell survival.24,32,33 These results expand on previous findings of M1 macrophages initiating and sustaining IRI pathology.18,34
Inhibition of TLR4 and TLR9 signaling impaired multiple facets of the diS-HMGB1–induced MDM phenotype. Consistent with previous studies, TLR4 regulated a broader cytokine/chemokine pro-inflammatory response and TLR9 mainly produced chemokines involved in leukocyte recruitment/ROS generation, while both TLRs controlled surface marker expression.14,35,36 A number of transcriptional regulators downstream of TLR4 and TLR9 modulate the surface markers explored in this study. LPS (TLR4 ligand) and bacterial CpG-DNA (TLR9 agonist) are capable of inducing a functional interaction of NF-κB with the promoter element of HLA-DRA to induce MHC-II gene expression and of increasing CD86 expression in both a TLR4-dependent or TLR9-dependent manner.37–39 Interestingly, neutralization of diS-HMGB1 increased expression of the immune checkpoint PD-L1 but TLR inhibition decreased its expression. TLR9 is reported to induce PD-L1 in dendritic cells (DCs), suggesting that TLR2, CXCR4, or another HMGB1 receptor not examined in this study competitively regulates myeloid PD-L1 to ultimately decrease its expression.15,38 Additionally, TLR9 controlled diS-HMGB1–induced ROS production, although evidence exists supporting both TLR4-mediated and TLR9-mediated ROS production. CpG-DNA increases ROS production during intracellular infection of DCs with Salmonella, a model for ROS production, while also increasing presentation of Salmonella peptides.10 Salmonella infection in TLR9−/− mice confirmed this pathway, supporting our TLR9-dependent ROS mechanism.10,40 A separate study concluded that TLR4, not TLR9, contributes to macrophage Salmonella–induced ROS production in vitro.41 Collectively, these data document the complex and divergent effects of diS-HMGB1–mediated TLR4 vs TLR9 signaling on MDM polarization and function.
An increasing body of data emphasizes the importance of donor and recipient TLR-mediated signals in promoting alloreactive T/B cell responses and graft rejection.42 Simultaneous deletion of TLR adaptor molecules MyD88 and Trif in donor skin prolonged allograft survival, diminished donor cell migration to draining lymph nodes, and delayed graft infiltration by host T cells.43 T cells stimulated by MyD88-silenced DCs increased the production of TH2-type cytokines, accompanied by decreased donor-specific alloreactivity.44 Correspondingly, DCs treated with HMGB1 promoted T cell proliferation/differentiation, whereas HMGB1 blockade ameliorated chronic cardiac transplant vasculopathy and decreased T cell infiltration/pro-inflammatory cytokine production.45,46 Our data support TLR4 and TLR9 signaling in enhancing antigen-presenting activity of MDMs and their capacity to stimulate alloreactivity. MDMs exposed to diS-HMGB1 stimulated increased CD4+ T cell proliferation and IFN-γ production, implying that IRI+ patients have increased ability to promote T cell–mediated and B cell–mediated alloimmunity and that diS-HMGB1 promotes this activated state. In agreement with this concept, we found that higher diS-HMGB1 concentrations correlated with a lower PD-L1:CD86 ratio when monocytes were stimulated with either IRI+ LF or purified diS-HMGB1. Additionally, a lower PD-L1:CD86 ratio in LF correlated with post-transplant production of DSA. Therefore, it is reasonable to consider that diS-HMGB1 blockade would reduce alloreactivity and improve OLT outcomes.
A limitation of this study is the inherent property of diS-HMGB1 as a transient oxidative form of HMGB1.14 To account for this, we confirmed that a single dose of diS-HMGB1 on Day 0 and repeated doses of diS-HMGB1 through Day 5 showed comparable MDM phenotypes (Supplementary Fig. S3). A second limitation is the receptor repertoire of diS-HMGB1 extending beyond TLR4 and TLR9.15 As seen in our cytokine/phenotyping data, IL-12p40, IL-12p70, G-CSF, and GM-CSF secretion were modified by diS-HMGB1 adjunct but not TLR4 or TLR9 inhibition. diS-HMGB1 neutralization had an opposite effect of TLR inhibition on PD-L1 expression, suggesting perhaps an alternate receptor regulates PD-L1 expression. Future studies should explore the effect of other receptors, such as TLR2 or CXCR4, individually or in concert with TLR4 and TLR9 pathways on MDM phenotypes.15 We focused on MDMs because they can promote alloimmunity. However, HMGB1 induces pyroptosis in hepatocytes and activates neutrophils, 2 cell types present during OLT-IRI.17,47,48 CD4+ T cells also augment IRI in an antigen-independent manner and express the diS-HMGB1 receptor TLR4.49,50 Future studies can focus on diS-HMGB1–mediated activation of other cell types to increase understanding of post-OLT inflammation.
In summary, our data are consistent with a model whereby interactions between diS-HMGB1, released from IRI-stressed donor allografts, and TLR4/TLR9 molecules induce MDM polarization to an M1-like phenotype with increased expression of both pro-inflammatory cytokines and antigen presentation/co-stimulation molecules. Macrophage-derived ROS contributes to hepatocyte death, likely generating donor HLA antigens processed and presented via indirect allorecognition by diS-HMGB1–polarized macrophages. These macrophages then go on to present donor-derived peptides to activate alloreactive T cells and DSA production. The current findings also provide a basis for using therapeutics to alter macrophage polarization and improve OLT outcomes, including antioxidants and TLR4/TLR9 neutralizing antibodies and/or small molecule antagonists.29,51
Supplementary Material
Acknowledgments
The authors thank Dr A. Sette for the kind gift of the SARS-CoV-2 spike protein peptide pools used in this study. The authors hereby express their gratitude toward all organ donors and their families, for giving the gift of life and the gift of knowledge, by their generous donation.
Funding
This work was supported by NIH grants PO1 AI120944 (to J.W.K.W. and E.F.R.), Ruth L. Kirschstein National Research Service Award T32CA009120 and L60 MD011903 (to R.A.S.), and U01AI148119 (to A.S.M.).
Abbreviations:
- ALT
alantine transaminase
- aS-HMGB1
terminally-oxidized all-sulfonyl HMGB1
- aT-HMGB1
fully-reduced all-thiol HMGB1
- DAMP
damage-associated molecular pattern
- DC
dendritic cell
- diS-HMGB1
partially-oxidized disulfide HMGB1
- diS-OLT
mice that were subjected to cold ischemia transplants with 100 ng diS-HMGB1 per 1 g mouse weight injected into the portal vein during reperfusion
- DSA
donor-specific HLA antibody
- EAD
early allograft dysfunction
- HMGB
high mobility group box
- IFN
interferon
- INR
international normalized ratio
- IRI
ischemia-reperfusion injury
- LF
portal vein blood collected immediately following graft reperfusion from human OLT recipients (liver flush)
- LFT
liver function test
- LPS
lipopolysaccharide
- MDM
monocyte-derived macrophage
- OLT
orthotopic liver transplantation
- PRR
pattern recognition receptor
- ROS
reactive oxygen species
- TLR
toll-like receptor
- TLR4i
TLR4 inhibitor
- TLR4+9i
TLR4 inhibitor and TLR9 antagonist in combination
- TLR9i
TLR9 antagonist
Footnotes
Disclosure
The authors of this manuscript have no conflicts of interest to disclose as described by the American Journal of Transplantation.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ajt.2023.08.002.
Data availability
No data are available for this publication.
References
- 1.Zhai Y, Petrowsky H, Hong JC, Busuttil RW, Kupiec-Weglinski JW. Ischaemia-reperfusion injury in liver transplantation–from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10(2):79–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim WR, Lake JR, Smith JM, et al. OPTN/SRTR 2017 annual data report: liver. 2019;19(S2):184–283. [DOI] [PubMed] [Google Scholar]
- 3.Ali JM, Davies SE, Brais RJ, et al. Analysis of ischemia/reperfusion injury in time-zero biopsies predicts liver allograft outcomes. Liver Transpl. 2015;21(4):487–499. [DOI] [PubMed] [Google Scholar]
- 4.Sosa RA, Zarrinpar A, Rossetti M, et al. Early cytokine signatures of ischemia/reperfusion injury in human orthotopic liver transplantation. JCI Insight. 2016;1(20):e89679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Giwa S, Lewis JK, Alvarez L, et al. The promise of organ and tissue preservation to transform medicine. Nat Biotechnol. 2017;35(6):530–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yue S, Zhou H, Wang X, Busuttil RW, Kupiec-Weglinski JW, Zhai Y. Prolonged ischemia triggers necrotic depletion of tissue-resident macrophages to facilitate inflammatory immune activation in liver ischemia reperfusion injury. J Immunol. 2017;198(9):3588–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni M, Zhang J, Sosa R, et al. T-cell immunoglobulin and mucin domain-containing protein-4 is critical for kupffer cell homeostatic function in the activation and resolution of liver ischemia reperfusion injury. Hepatology. 2021;74(4):2118–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lahiri A, Lahiri A, Das P, Vani J, Shaila MS, Chakravortty D. TLR 9 activation in dendritic cells enhances salmonella killing and antigen presentation via involvement of the reactive oxygen species. PLoS One. 2010;5(10):e13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Venereau E, Casalgrandi M, Schiraldi M, et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or pro-inflammatory cytokine release. J Exp Med. 2012;209(9):1519–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang H, Lundback P, Ottosson L, et al. Redox modification of cysteine residues regulates the cytokine activity of high mobility group box-1 (HMGB1). Mol Med. 2012;18:250–259. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Yang H, Hreggvidsdottir HS, Palmblad K, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A. 2010;107(26): 11942–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sosa RA, Terry AQ, Kaldas FM, et al. Disulfide-HMGB1 drives ischemia-reperfusion injury in human liver transplantation. Hepatology. 2021;73(3):1158–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bertheloot D, Latz E. HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol. 2017;14(1):43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tian Y, Charles EJ, Yan Z, et al. The myocardial infarct-exacerbating effect of cell-free DNA is mediated by the high-mobility group box 1-receptor for advanced glycation end products-Toll-like receptor 9 pathway. J Thorac Cardiovasc Surg. 2019;157(6):2256–2269.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll-like receptor 9 inhibition confers protection from liver ischemia-reperfusion injury. Hepatology. 2010;51(2):621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsung A, Sahai R, Tanaka H, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med. 2005; 201(7):1135–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sosa RA, Rossetti M, Naini BV, et al. Pattern recognition receptor-reactivity screening of liver transplant patients: potential for personalized and precise organ matching to reduce risks of ischemia-reperfusion injury. Ann Surg. 2020;271(5):922–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito T, Naini BV, Markovic D, et al. Ischemia-reperfusion injury and its relationship with early allograft dysfunction in liver transplant patients. Am J Transplant. 2020;21(2):614–625. [DOI] [PubMed] [Google Scholar]
- 21.National Research Council. Guide for the Care and Use of Laboratory Animals. National Research Council; 2011. [Google Scholar]
- 22.Nakamura K, Kageyama S, Kaldas FM, et al. Hepatic CEACAM1 expression indicates donor liver quality and prevents early transplantation injury. J Clin Invest. 2020;130(5):2689–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paardekooper LM, Dingjan I, Linders PTA, et al. Human monocyte-derived dendritic cells produce millimolar concentrations of ROS in phagosomes per second. Front Immunol. 2019;10:1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Andrade KQ, Moura FA, dos Santos JM, de Araujo OR, de Farias Santos JC, Goulart MO. Oxidative stress and inflammation in hepatic diseases: therapeutic possibilities of N-acetylcysteine. Int J Mol Sci. 2015;16(12):30269–30308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shen T, Chen X, Chen Y, Xu Q, Lu F, Liu S. Increased PD-L1 expression and PD-L1/CD86 ratio on dendritic cells were associated with impaired dendritic cells function in HCV infection. J Med Virol. 2010; 82(7):1152–1159. [DOI] [PubMed] [Google Scholar]
- 26.Tian J, Avalos AM, Mao SY, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–496. [DOI] [PubMed] [Google Scholar]
- 27.Muhammad S, Barakat W, Stoyanov S, et al. The HMGB1 receptor RAGE mediates ischemic brain damage. J Neurosci. 2008;28(46):12023–12031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wu H, Ma J, Wang P, et al. HMGB1 contributes to kidney ischemia reperfusion injury. J Am Soc Nephrol. 2010;21(11):1878–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsung A, Hoffman RA, Izuishi K, et al. Hepatic ischemia/reperfusion injury involves functional TLR4 signaling in nonparenchymal cells. J Immunol. 2005;175(11):7661–7668. [DOI] [PubMed] [Google Scholar]
- 30.Yang H, Wang H, Andersson U. Targeting Inflammation Driven by HMGB1. Front Immunol. 2020;11:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agopian VG, Harlander-Locke MP, Markovic D, et al. Evaluation of early allograft function using the liver graft assessment following transplantation risk score model. JAMA Surg. 2018;153(5): 436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conde de la Rosa L, Schoemaker MH, Vrenken TE, et al. Superoxide anions and hydrogen peroxide induce hepatocyte death by different mechanisms: involvement of JNK and ERK MAP kinases. J Hepatol. 2006;44(5):918–929. [DOI] [PubMed] [Google Scholar]
- 33.Ma X, Han S, Zhang W, et al. Protection of cultured human hepatocytes from hydrogen peroxideinduced apoptosis by relaxin3. Mol Med Rep. 2015;11(2):1228–1234. [DOI] [PubMed] [Google Scholar]
- 34.Dal-Secco D, Wang J, Zeng Z, et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J Exp Med. 2015;212(4):447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.El-Zayat SR, Sibaii H, Mannaa FA. Toll-like receptors activation, signaling, and targeting: an overview. Bull Natl Res Centre. 2019;43(1):187. [Google Scholar]
- 36.Blackwell SE, Krieg AM. CpG-A-induced monocyte IFN-gamma-inducible protein-10 production is regulated by plasmacytoid dendritic cell-derived IFN-alpha. J Immunol. 2003;170(8):4061–4068. [DOI] [PubMed] [Google Scholar]
- 37.Lee KW, Lee Y, Kim DS, Kwon HJ. Direct role of NF-kappaB activation in Toll-like receptor-triggered HLA-DRA expression. Eur J Immunol. 2006;36(5):1254–1266. [DOI] [PubMed] [Google Scholar]
- 38.Tel J, Hato SV, Torensma R, et al. The chemotherapeutic drug oxaliplatin differentially affects blood DC function dependent on environmental cues. Cancer Immunol Immunother. 2012;61(7):1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W, An EK, Hwang J, Jin JO. Mice plasmacytoid dendritic cells were activated by lipopolysaccharides through toll-like receptor 4/myeloid differentiation factor 2. Front Immunol. 2021;12:727161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhan R, Han Q, Zhang C, Tian Z, Zhang J. Toll-Like receptor 2 (TLR2) and TLR9 play opposing roles in host innate immunity against Salmonella enterica serovar Typhimurium infection. Infect Immun. 2015; 83(4):1641–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.West AP, Brodsky IE, Rahner C, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alegre ML, Chong A. Toll-like receptors (TLRs) in transplantation. Front Biosci (Elite Ed). 2009;1(1):36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKay D, Shigeoka A, Rubinstein M, Surh C, Sprent J. Simultaneous deletion of MyD88 and Trif delays major histocompatibility and minor antigen mismatch allograft rejection. Eur J Immunol. 2006;36(8): 1994–2002. [DOI] [PubMed] [Google Scholar]
- 44.Bao W, Qin X, Guan N, et al. MyD88-silenced dendritic cells induce T-cell hyporesponsiveness and promote Th2 polarization in vivo. Cytotherapy. 2015;17(9):1240–1250. [DOI] [PubMed] [Google Scholar]
- 45.Chen Y, Zhang W, Bao H, He W, Chen L. High mobility group box 1 contributes to the acute rejection of liver allografts by activating dendritic cells. Front Immunol. 2021;12:679398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zou H, Yang Y, Gao M, et al. HMGB1 is involved in chronic rejection of cardiac allograft via promoting inflammatory-like mDCs. Am J Transplant. 2014;14(8):1765–1777. [DOI] [PubMed] [Google Scholar]
- 47.Hou W, Wei X, Liang J, et al. HMGB1-induced hepatocyte pyroptosis expanding inflammatory responses contributes to the pathogenesis of acute-on-chronic liver failure (ACLF). J Inflamm Res. 2021;14:7295–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Xia Y, Sun B, et al. Neutrophil extracellular traps induced by the hypoxic microenvironment in gastric cancer augment tumour growth. Cell Commun Signal. 2023;21(1):86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen X, Wang Y, Gao F, et al. CD4 T cells promote tissue inflammation via CD40 signaling without de novo activation in a murine model of liver ischemia/reperfusion injury. Hepatology. 2009;50(5): 1537–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gonzalez-Navajas JM, Fine S, Law J, et al. TLR4 signaling in effector CD4+ T cells regulates TCR activation and experimental colitis in mice. J Clin Invest. 2010;120(2):570–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Farooq M, Batool M, Kim MS, Choi S. Toll-like receptors as a therapeutic target in the era of immunotherapies. Front Cell Dev Biol. 2021;9:756315. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No data are available for this publication.