

Abstract
PURPOSE
Although immune checkpoint inhibitors (ICI) have extended survival in patients with non–small-cell lung cancer (NSCLC), acquired resistance (AR) to ICI frequently develops after an initial benefit. However, the mechanisms of AR to ICI in NSCLC are largely unknown.
METHODS
Comprehensive tumor genomic profiling, machine learning–based assessment of tumor-infiltrating lymphocytes, multiplexed immunofluorescence, and/or HLA-I immunohistochemistry (IHC) were performed on matched pre- and post-ICI tumor biopsies from patients with NSCLC treated with ICI at the Dana-Farber Cancer Institute who developed AR to ICI. Two additional cohorts of patients with intervening chemotherapy or targeted therapies between biopsies were included as controls.
RESULTS
We performed comprehensive genomic profiling and immunophenotypic characterization on samples from 82 patients with NSCLC and matched pre- and post-ICI biopsies and compared findings with a control cohort of patients with non-ICI intervening therapies between biopsies (chemotherapy, N = 32; targeted therapies, N = 89; both, N = 17). Putative resistance mutations were identified in 27.8% of immunotherapy-treated cases and included acquired loss-of-function mutations in STK11, B2M, APC, MTOR, KEAP1, and JAK1/2; these acquired alterations were not observed in the control groups. Immunophenotyping of matched pre- and post-ICI samples demonstrated significant decreases in intratumoral lymphocytes, CD3e+ and CD8a+ T cells, and PD-L1–PD1 engagement, as well as increased distance between tumor cells and CD8+PD-1+ T cells. There was a significant decrease in HLA class I expression in the immunotherapy cohort at the time of AR compared with the chemotherapy (P = .005) and the targeted therapy (P = .01) cohorts.
CONCLUSION
These findings highlight the genomic and immunophenotypic heterogeneity of ICI resistance in NSCLC, which will need to be considered when developing novel therapeutic strategies aimed at overcoming resistance.
Genomic and immunophenotypic changes shape acquired resistance to #ICI in #NSCLC à Link to the paper @BiagioRicciutMD @GLambertiMD @DanaFarber #LCSM
INTRODUCTION
Immune checkpoint inhibitors (ICI) produce durable responses and extend survival in patients with advanced non–small-cell lung cancer (NSCLC).1 Unfortunately, most responders to ICI will eventually develop disease progression. Although serial tumor genomic profiling has led to the identification of resistance mechanisms to targeted therapies in subsets of lung cancer such as those with EGFR mutations or ALK rearrangements,2 the landscape of acquired resistance (AR) to ICI in NSCLC remains unknown. Recent reports have identified defects in interferon gamma (INFγ) signaling and impaired HLA class I antigen presentation as potential mechanisms of AR to ICI in solid tumors.3,4 However, these studies were limited by the retrospective design, small sample sizes, and the lack of a control group. Although rational strategies have been developed to overcome resistance to targeted therapies, there are no approved immunotherapies to overcome resistance to ICI in NSCLC. Thus, understanding the mechanisms of AR to immunotherapy is essential for the development of novel strategies tailored to overcoming specific resistance mechanisms. Here, we analyzed a large cohort of patients with NSCLC and matched pre- and post-ICI samples to characterize the genomic and immunophenotypic landscape of AR to PD-(L)1 inhibition.
METHODS
Detailed methods, including statistical analysis and methods used for genomic, immunophenotypic, and machine learning (ML) analysis, are reported in the Supplementary Methods.
Patient Population
Patients with NSCLC who had consented to correlative research protocols at the Dana-Farber Cancer Institute, received treatment with ICI, and developed AR between two repeat biopsies were included. Patients who received other systemic therapies including targeted therapies or chemotherapy were included as controls. AR to systemic treatments was defined as the development of disease progression after an objective response (either partial response [PR] or complete response [CR]) or stable disease (SD) for ≥3 months.
Immunohistochemistry
The PD-L1 tumor proportion score, beta-2-microglobulin (B2M), and HLA class I expression were assessed with immunohistochemistry (IHC) using validated monoclonal antibodies as summarized in the Supplementary Methods.
Tumor Genomic Profiling
Targeted exome next-generation sequencing was performed using the validated OncoPanel assay, as previously described.5 Tumor mutational burden (TMB) was determined using OncoPanel.3
For each gene, the absolute copy number was estimated based on the tumor purity (p) and the weighted average of segmented log2 ratios across the gene (l) using the formula
To quantify aneuploidy levels and aneuploidy score, targeted sequencing data were analyzed using ASCETS (Arm-level Somatic Copy-number Events in Targeted Sequencing), as previously described (Supplementary Methods).6,7
Tumor Similarity Score
To assess similarity between tumor pairs and determine which pairs arose from shared lineage, a score was computed using an internal pipeline on the basis of the frequency (fevent) of each single-nucleotide variant, small insertion/deletion, gene-level copy-number alteration (CNA), and structural variant in our NSCLC cohort and pevent, computed as the square of fevent, representing the likelihood of observing the event independently in two samples. The formula for similarity score computation for a tumor pair is outlined below, where s = sample and fi = frequency of event i in the cohort.
Samples deemed to arise from shared lineage were included in the analysis.
Multiplexed immunofluorescence
Multiplexed immunofluorescence was performed on histologic tissue samples by staining 5-micron formalin-fixed, paraffin-embedded whole-tissue sections with standard, primary antibodies, as previously described (Supplementary Methods).8-10 To examine the interactions between PD-L1 and PD-1 in single-cell data, a k-nearest searching algorithm was used to identify cell neighbors within a 20-micron radius. PD-L1+ cells located in proximity to PD-1+ cells were labeled as PD-1+ interactors. To quantify the PD-1–PD-L1 interaction, all potential PD-1–PD-L1 interaction pairs were normalized by total PD-L1+ cells in each sample.
ML Assessment of Tumor-Associated Immune Cells
Whole-slide images were converted into tiles of size 2,048 × 2,048 pixels using PathML, as previously described.11 Model inference was carried out to identify the following cell types: lymphocytes, epithelial cells, macrophages, and neutrophils. K-nearest neighbor minimum spanning tree graphs, on the basis of spatial proximity, were generated for each cell type using the centroid locations (Supplementary Methods).
Statistical Analysis
Categorical and continuous variables were summarized using descriptive statistics. The Wilcoxon signed-rank test was used to test for a difference in median of paired observations, and Fisher's exact test or χ2 was used to test for associations between categorical variables (Supplementary Methods). All statistical analyses were performed using R version 3.6.1.
RESULTS
Patient Characteristics
The schema for this study is shown in the Data Supplement (Fig S1, online only). Among 1,757 patients with NSCLC who received ICI at Dana-Farber Cancer Institute, 1,498 had progressed at the data lock, with a median follow-up of 37.4 months (95% CI, 35.0 to 39.9). Of these, 742 (49.5%) patients developed AR to immunotherapy, defined as an objective response or SD for at least 3 months followed by disease progression, of whom 461 (62.1%) had AR after 6 months of treatment. The cumulative risk of AR over time among all-comers with an objective response or SD ≥3 months is shown in the Data Supplement (Fig S2). Among patients with AR, 82 had matched pre- and post-ICI tissue available for correlative analysis (Data Supplement, Table S1). At the time of AR, 25 (30.5%) experienced systemic progression, while 57 (69.5%) had oligoprogression, defined as disease progression in ≤3 metastatic sites. In this cohort, the median age of patients was 65 years (range, 24-81), 57.3% were women, 79.3% had a history of tobacco use, and 86.6% had adenocarcinoma histology. Driver mutations in KRAS were identified in 34.1% of cases. Immunotherapies received were PD-(L)1 monotherapy (56.1%), combined PD-1 and cytotoxic T-cell lymphocyte-4 (CTLA-4) blockade (6.1%), and chemoimmunotherapy (37.8%). Importantly, among these patients, 74.4% received a PD-(L)1–containing regimen as the only intervening therapy between the tumor biopsies. Immunotherapy was given as first line in 59.8% patients and as ≥second line in 40.2% of cases.
In this cohort, 2.4% of patients had a CR, 41.5% had a PR, and 56.1% had SD ≥3 months as the best response to treatment (Data Supplement, Fig 3A). The median progression-free survival (PFS) and overall survival (OS) were 7.9 months (95% CI, 6.8 to 10.4) and 35.0 months (95% CI, 24.7 to 45.1), respectively (Data Supplement, Figs 3B-3C). The median OS from the time of AR and from the post-ICI biopsy was 17.3 months (95% CI, 12.9 to 33.3) and 10.9 months (95% CI, 6.9 to 14.8), respectively (Data Supplement, Fig S4), and the median time between pre- and post-ICI tumor biopsies was 18.9 months. Among the 82 patients with AR to ICI, pre- and post-ICI correlative analyses were available as detailed in the Data Supplement (Table S2 and Fig S5).
Two additional cohorts including a total of 138 patients with NSCLC who had matched tumor biopsies pre- and post-chemotherapy (N = 32), pre- and post-targeted therapies (N = 89), or both (N = 17) were included as controls (Data Supplement, Table S2). The median time between pre- and post-treatment tumor biopsies was 19.8 months in the chemotherapy cohort and 17.7 months in the targeted therapy cohort. Baseline clinicopathologic characteristics of these patients are summarized in the Data Supplement (Table S3). The biopsy site and the correlative analyses performed on each tumor biopsy in patients who received immunotherapy, targeted therapies, and chemotherapy are summarized in the Data Supplement (Table S4). In each of these three cohorts, the tumor purity was similar between pre- and post-treatment tumor samples (Data Supplement, Fig S6).
Genomic Correlates of AR to PD-(L)1 Blockade in NSCLC
Genomic changes at the time of AR to ICI were identified in 62.0% (49/79) of patients with pre- and post-ICI tumor genomic profiling (Fig 1). Acquired mutations were identified in 27.8% of cases, while acquired CNAs were found in 49.4% of cases. The frequency of genomic changes identified at the time of resistance by the type of immunotherapy received is shown in the Data Supplement (Fig S7). Among patients with acquired mutations, the most common included loss-of-function alterations in STK11 (8.9%), B2M (6.3%), SMARCA4 (6.3%), NF1/2 (5.1%), APC (3.8%), CDKN2A/B (3.8%), MAP3K1 (2.5%), MAP2K4 (2.5%), KEAP1 (2.5%), MTOR (1.3%), JAK1 (1.3%), and JAK2 (1.3%; Fig 2A). Acquired activating mutations at the time of AR to immunotherapies included PIK3CA (3.8%), SOS1 (1.3%), PDGFRA (1.3%), ERBB2 (1.3), and BRAF (1.3%). Importantly, no acquired mutations in the selected list of genes were identified in the control cohort of patients with pre- and post-chemotherapy tumor genomic profiling (Fig 2B). Similarly, acquired mutations in STK11, KEAP1, B2M, APC, CDKN2A/B, JAK1, NF1/2, MAP3K1, MAP2K4, PDGFRA, and ERBB2 were not detected among patients with AR to EGFR tyrosine kinase inhibitor therapy (N = 72), as well as among patients with AR to ALK (N = 8), MET (N = 4), RET (N = 2), ROS1 (N = 2), and BRAF (N = 1) inhibitor therapy, further suggesting that acquired mutations in these genes are more likely to develop as specific mechanisms of resistance to ICI. As expected, among patients with intervening targeted therapies, we found acquired mutations in EGFR (19.1%), PIK3CA (7.8%), KRAS (3.3%), and BRAF (2.2%), which are known resistance mechanisms to these therapies in NSCLC12 (Fig 2C). The distribution of acquired genomic changes in patients receiving intervening chemotherapy or targeted therapy is shown in the Data Supplement (Fig S8).
FIG 1.

Summary of the genomic and immunophenotypic changes identified at the time of acquired resistance to PD-(L)1–based therapies in patients with NSCLC whose tumor underwent comprehensive genomic profiling at the DFCI. Only acquired genomic alterations in the post-ICI biopsy that were not present in the pre-ICI biopsy are displayed. Samples without acquired genomic changes at the time of resistance are not shown. Driver mutations shown in the oncoprint represent mutations identified at baseline, before the start of immunotherapy. Variants predicted to be benign or originating from clonal hematopoiesis of indeterminate potential are not shown. CR, complete response; CTLA-4, cytotoxic T-cell lymphocyte-4; DFCI, Dana-Farber Cancer Institute; ICI, immune checkpoint inhibitor; NSCLC, non–small-cell lung cancer; PFS, progression-free survival; PR, partial response; SD, stable disease.
FIG 2.
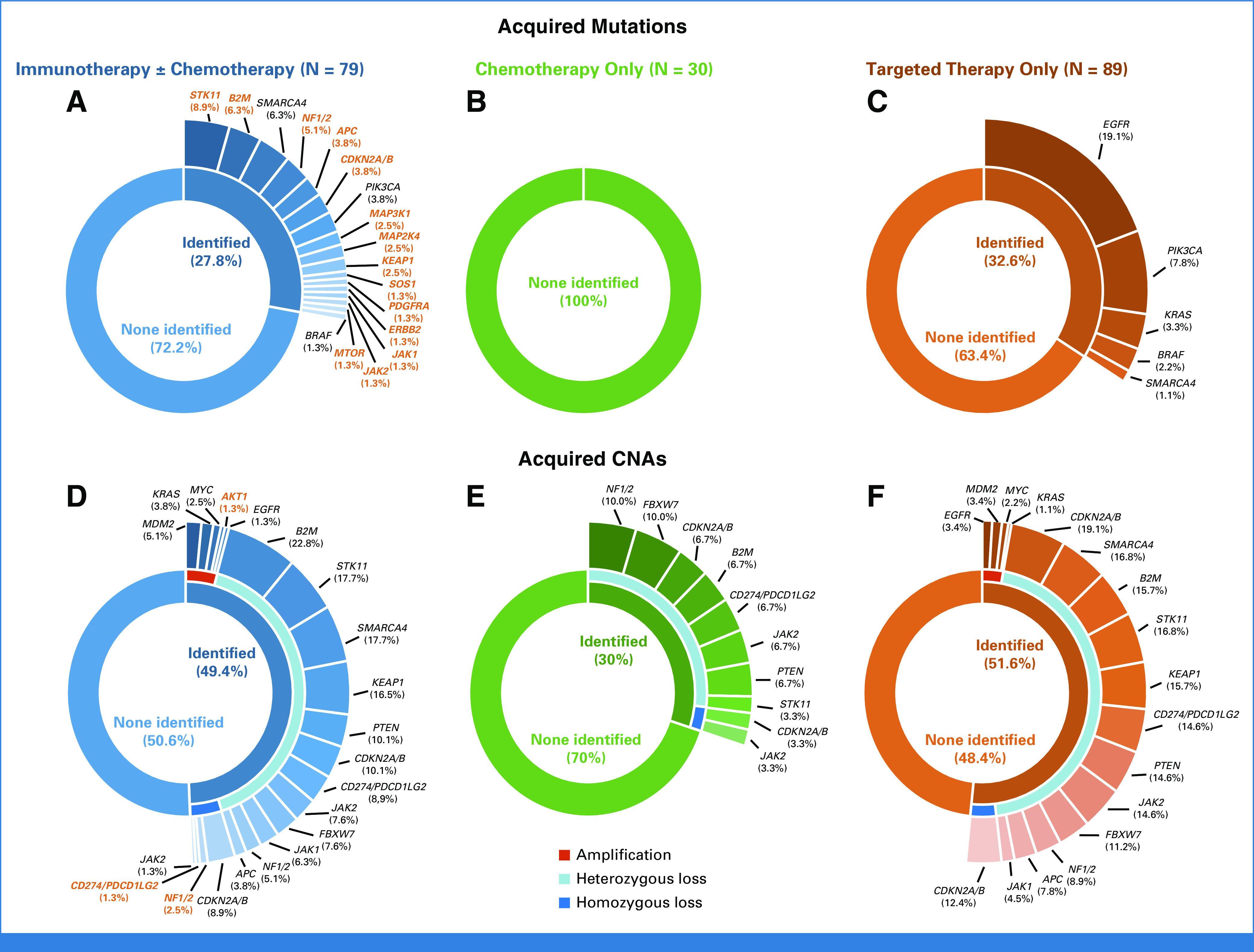
Pie-donut plots depicting the rate of acquired mutation at the time of resistance to (A) PD-(L)1–based therapies, (B) chemotherapy, and (C) targeted therapy in patients with NSCLC and matched pre- and post-treatment tumor genomic profiling. Pie-donut plots depicting the rate of acquired CNAs at the time of resistance to (D) PD-(L)1–based therapies, (E) chemotherapy, and (F) targeted therapy. Labels of genomic alterations uniquely identified in the PD-(L)1–based therapies cohort are in orange. CNA, copy-number alteration; NSCLC, non–small-cell lung cancer.
We next examined acquired CNAs in each of these three cohorts. Among patients treated with ICI, the most common acquired heterozygous losses included B2M (22.8%), STK11 (17.7%), SMARCA4 (17.7%), and KEAP1 (16.5%). Additional acquired heterozygous deletions included PTEN (10.1%), CDKN2A/B (10.1%), CD274/PDCD1LG2 (8.9%), JAK2 (7.6%), FBXW7 (7.6%), and JAK1 (6.3%). We also identified homozygous deletion in CDKN2A/B (8.9%), NF1/2 (2.5%), CD274/PDCD1LG2 (1.3%), and JAK2 (1.3%; Fig 2D). Of note, acquired heterozygous deletions with concurrent acquired mutations were found in STK11 (5.1%), SMARCA4 (2.5%), KEAP1 (1.3%), B2M (1.3%), and JAK1 (1.3%), indicating biallelic inactivation of these genes (Fig 1). Among gene-level copy gains, acquired high-level amplification was found in MDM2 (5.1%), KRAS (3.8%), MYC (2.5%), AKT1 (1.3%), and EGFR (1.3%) in ICI-resistant samples (Fig 2D). Among patients with AR to chemotherapy or targeted therapies, we also noted similar acquired CNA patterns (Figs 2E and 2F). However, no biallelic losses in STK11, KEAP1, SMARCA4, B2M, JAK1, and CD274/PDCD1LG2 were noted in these control cohorts.
Because 25% of patients who developed AR to ICI in our cohort also received at least one additional line of therapy between the pre- and post-ICI tumor biopsy, we also examined acquired genomic changes in a third control cohort of patients (N = 17) who received multiple lines of therapy including both chemotherapy and targeted therapy (but not immunotherapy) between tumor biopsies. Similarly, in this cohort, we only identified acquired EGFR and ERBB2 mutations, which were developed as resistance mechanisms to targeted therapy (Data Supplement, Figs S9A-S9B). No acquired mutations in STK11, KEAP1, B2M, JAK1, and APC were noted. In examining CNAs in this cohort, we again noted similar patterns of heterozygous deletions as was observed in the immunotherapy cohort and in the control groups of patients with tumor genomic profiling performed before and after chemotherapy or targeted therapy (Data Supplement, Fig S9C). A list of the baseline and acquired genomic changes identified at the time of AR to ICI is shown in the Data Supplement (Tables S5 and S6).
We next examined whether there was a difference in the frequency of acquired genomic alterations according to the time to developing resistance to ICI (3-6 months v ≥6 months13). In all-comers treated with ICI, there was no difference in the frequency of acquired genomic alterations between patients who developed AR within 6 months of ICI initiation and those who developed AR ≥6 months of treatment (Data Supplement, Figs S10A-S10C). However, among patients who received PD-(L)1 monotherapy, the frequency of acquired mutations at the time of AR was significantly higher in cases who developed resistance ≥6 months after ICI initiation versus those who developed AR within the first 6 months of treatment (58.3% v 10.5%, P < .01; Data Supplement, Figs S11A-S11C), possibly reflecting the emergence of resistant clones after prolonged exposure to PD-1 monotherapy. By contrast, there was no difference in the frequency of acquired mutations and CNAs according to the time to AR among patients who developed resistance to chemoimmunotherapy (Data Supplement, Figs S11D-S11F). Subgroup analyses showing the frequency of acquired genomic changes by best response to ICI ± chemotherapy, line of therapy, and PD-L1 expression levels (<1%, 1%-49%, and ≥50%) are shown in the Data Supplement (Fig S12).
In this cohort, there was no difference in the median TMB or in the median aneuploidy score (Data Supplement, Figs S13A-S13B) between pre- and post-ICI samples, suggesting that acquired mutations and CNAs were not simply the result of increased mutational load and tumor aneuploidy. Similarly, no differences in TMB or aneuploidy were noted in the two control cohorts (Data Supplement, Fig S13). A swimmer's plot summarizing time to AR to ICIs, length of time on therapy for individual patients, and key acquired mutations at AR is shown in the Data Supplement (Fig S14).
Immunophenotypic Changes in NSCLC With AR to PD-(L)1 Blockade
We next sought to determine whether there were changes in the tumor immunophenotype after development of AR to ICI in NSCLC. Of the 82 patients who developed AR, 16 had matched pre- and post-ICI digitalized hematoxylin and eosin–stained slides that passed quality metrics. We used our validated ML approach to quantify immune cell types (PathML11; Data Supplement, Fig S15). We noted that the density of tumor-infiltrating lymphocytes (TILs) significantly decreased after treatment with ICI (median 88 v 36 cells/mm2, P = .02, Fig 3A). By contrast, there was no difference in TIL density among patients with pre- and post-chemotherapy (P = .62) and pre- and post-targeted therapy (P = .51) tumor samples (Fig 3A). There was no difference in tumor-infiltrating macrophages or neutrophils before versus after immunotherapy, chemotherapy, or targeted therapy (Figs 3B and 3C). A representative case of pre- and post-ICI tumor biopsy showing a marked decrease in TILs as assessed by PathML at the time of AR to immunotherapy is shown in Figures 3D-3E.
FIG 3.
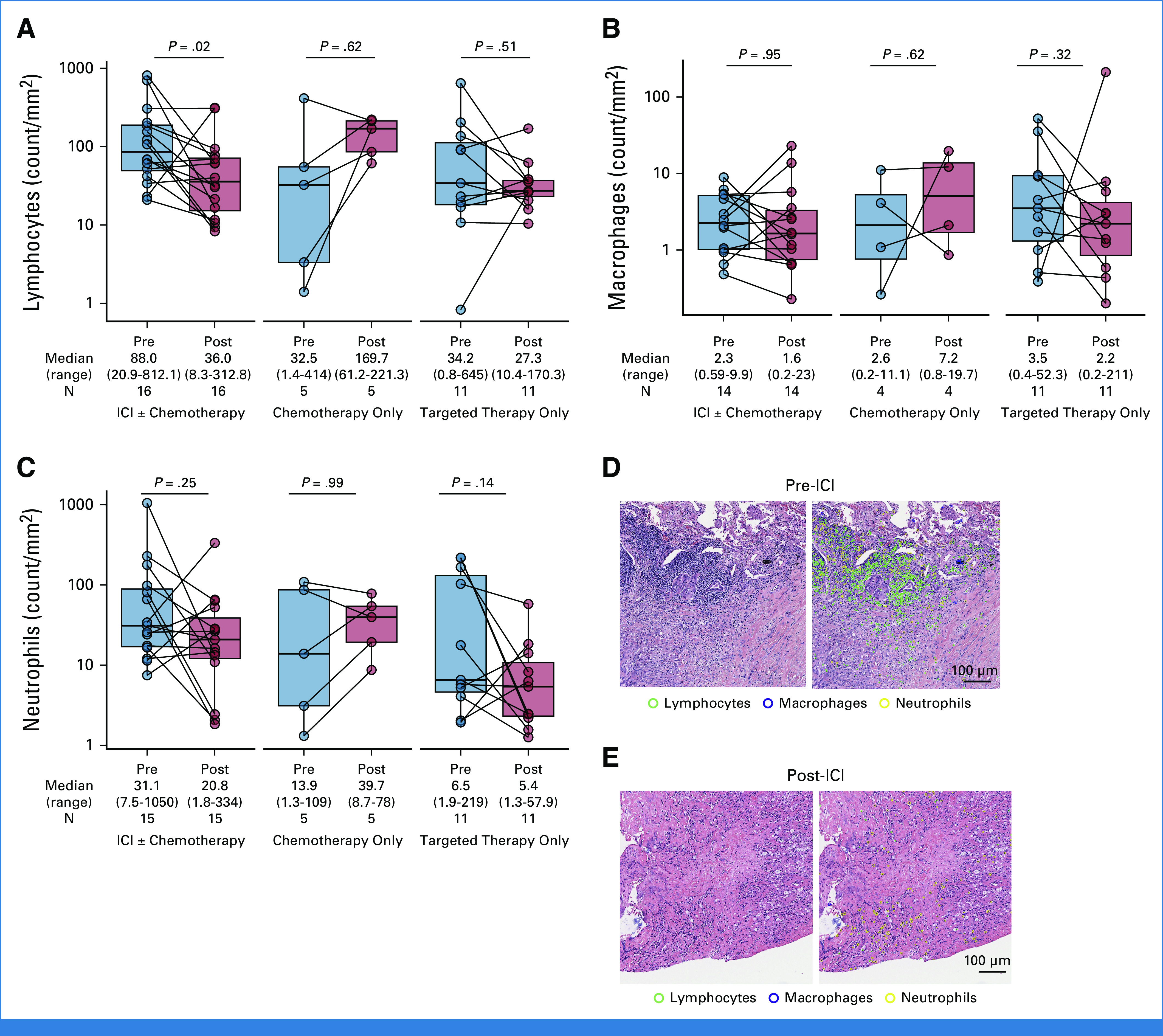
Paired box plots showing the change in the density of tumor-infiltrating (A) lymphocytes, (B) macrophages, and (C) neutrophils among patients with matched H&E-stained slides before versus after intervening immunotherapy, chemotherapy, or targeted therapy. Immune cell density was quantified on digital pathology imaging using the validated machine learning algorithm PathML. Representative images of (D) a pre-ICI sample and (E) a matched postimmunotherapy tumor sample that underwent immune cell subset deconvolution using PathML are shown, reflecting a significant decrease in intratumoral lymphocytes in the immunotherapy resistant sample. Groups were compared using the paired Wilcoxon signed-rank test. H&E, hematoxylin and eosin; ICI, immune checkpoint inhibitor.
To further characterize the immunophenotypic changes in NSCLC with AR to ICI, we next performed mIF to assess 21 different markers (Supplementary Methods) on tumors from six patients with tissue available from pre- and post-ICI samples. We noted a significant decrease in tumor-infiltrating CD3e+ T cells (P = .03), CD8a+ T cells (P = .03), and PD-1+ cells (P = .03) in post-ICI samples, compared with pre-ICI samples (Fig 4). There was also a significant decrease in CD3e+PD-1+, CD8a+PD-1+, and CD4+PD-1+ T cells, as well as CD45+ cells in post- versus pre-ICI tumor samples (P < .05, Data Supplement, Fig S16). There was no difference in the density of T regulatory cells, M1/M2 macrophages (CD68+, CD163+, CD206+), CD11+, TCF1+, and SMA+ cells (Data Supplement, Fig S16), as well as in PD-L1 expression on tumor cells and nontumor cells, and in total between pre- and post-ICI samples as assessed by mIF (Data Supplement, Fig S17), or in PD-L1 expression as assessed by IHC (Data Supplement, Fig S18). Spatial immunophenotyping showed a significant decrease in PD-1–PD-L1 engagement by mIF at the time of AR (P = .03, Fig 4E, Data Supplement, Fig S19). Representative images of pre- and post-ICI samples that underwent mIF are shown in Figures 4F and 4G. Finally, we noted a significant increase in the neighbor distance between tumor cells and CD8+PD-1+ T cells (P = .03, Fig 4H). Ray plots showing increased tumor cells to CD8+PD-1+ T cells distance at AR are shown in Figures 4I and 4L. A detailed summary of immunophenotypic changes identified at the time of AR is reported in the Data Supplement (Table S7).
FIG 4.
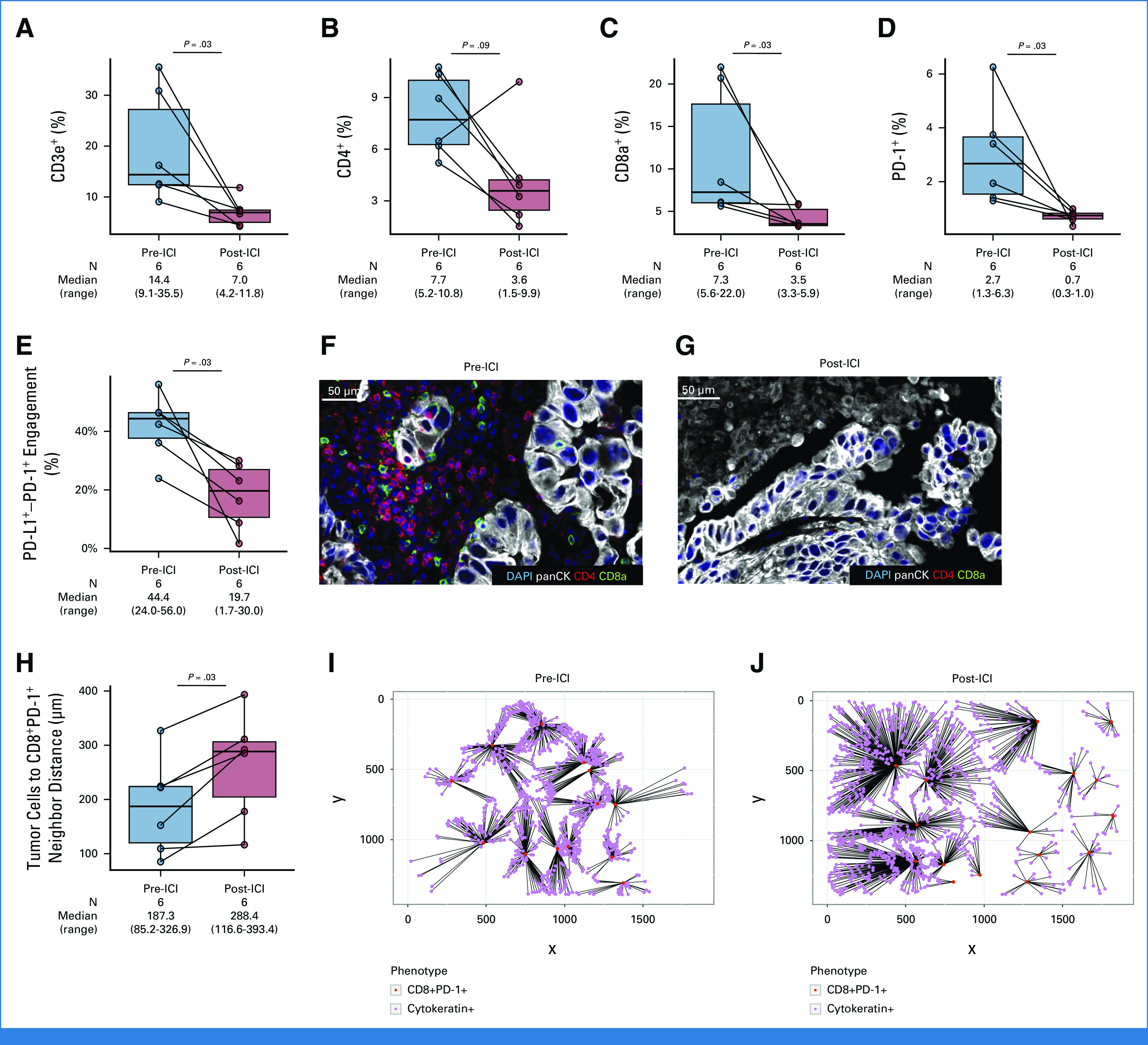
Paired box plots showing changes in (A) CD3e+, (B) CD4+, (C) CD8a+, and (D) PD-1+ T cells in patients with pre- and post-ICI tumor samples that underwent multiplexed immunofluorescence. (E) Paired box plot showing a significant decrease in PD-1–PD-L1 engagement at the time of AR to ICI. Representative case of a patient with (F) pre- and (G) post-immunotherapy multiplexed immunofluorescence showing significant decrease in tumor-infiltrating CD4 and CD8a T cells. (H) Paired box plots showing significant increase in the neighbor distance between tumor cells (cytokeratin+) and CD8+PD-1+ T cells at the time of AR to immunotherapy. Representative ray plots showing increase in the neighbor distance between tumor cells (cytokeratin+) and CD8+PD-1+ T cells between (I) pre- and (J) post-ICI tumor samples. AR, acquired resistance; ICI, immune checkpoint inhibitor.
HLA Class I Expression at the Time of AR to PD-(L)1–Based Therapies
Increased HLA class I expression correlates with improved outcomes to ICI across cancer types.14 However, whether changes in HLA class I expression mediate the development of AR to ICI in lung cancer is unknown. To determine whether changes in HLA class I expression contribute to the development of AR to ICI, we performed pan-HLA class I IHC on NSCLC samples from patients with available pre- and post-ICI tumor tissue (N = 8), as well as in the two control cohorts of patients with pre- and post-chemotherapy and targeted therapy with available tissue. We found that NSCLC samples collected at the time of AR had a marked decrease in HLA class I expression compared with pre-ICI samples (median H score: 230 v 300, P = .06). By contrast, there was a slight increase in HLA class I expression between pre- and post-chemotherapy (N = 7, P = .06) and pre- and post-targeted therapy (N = 9, P = .19) NSCLC samples (Fig 5A). When comparing the difference in HLA class I H scores, there was a significant drop in HLA class I expression in the immunotherapy cohort at the time of AR compared with the chemotherapy (P = .005) and the targeted therapy (P = .01) cohorts (Fig 5B). Figure 5C illustrates a representative case with a marked decrease HLA class I expression in an ICI-resistant sample compared with baseline.
FIG 5.
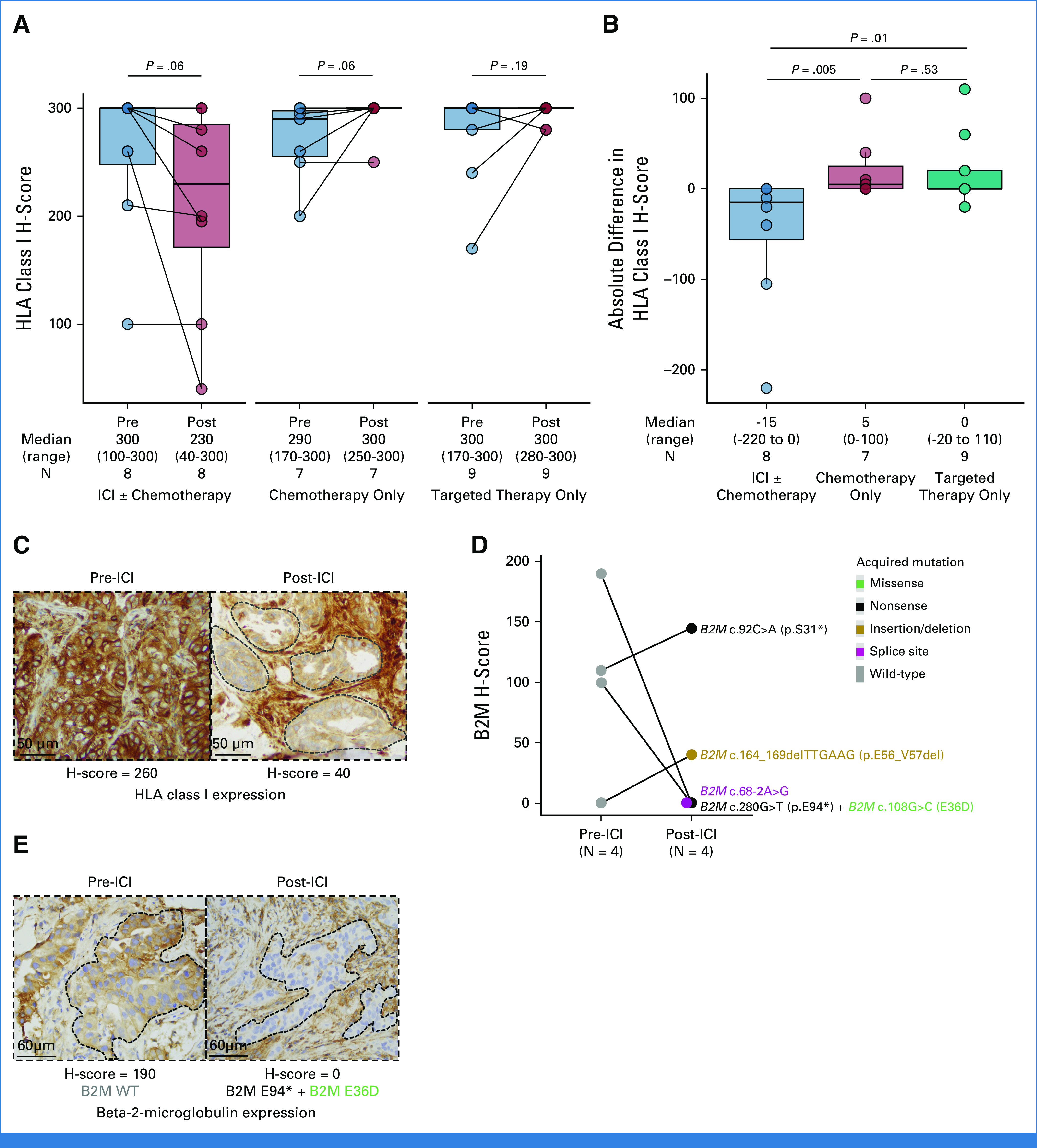
(A) Paired box plot showing absolute changes in HLA class I H-score among patients with pre- and post-ICI, chemotherapy, and targeted therapy tumor samples that underwent HLA class I IHC. (B) Percent change in HLA class I H-score among patients with pre- and post-ICI, chemotherapy, and targeted therapy tumor samples that underwent HLA class I IHC. (C) Representative case of pre- and post-ICI tumor biopsy showing significant decrease in HLA class I expression at the time of acquired resistance. (D) Change in B2M expression by H-score between pre- and post-ICI tumor samples. Individual acquired mutations are also reported. (E) Representative case of decreased B2M expression by IHC in a patient with acquired B2M nonsense mutation (c.280G>T; p.E94*) with concurrent B2M missense mutation (c.108G>C; E36D). Groups were compared using the paired Wilcoxon signed-rank test. B2M, beta-2-microglobulin; ICI, immune checkpoint inhibitor; IHC, immunohistochemistry.
We finalty tested whether the acquired mutations in the B2M gene found at the time of AR to ICI were associated with changes in B2M expression. Among the five patients with acquired B2M mutations, four had matched pre- and post-ICI tissue available for IHC. Two patients (50%) with acquired B2M mutations including a nonsense + missense mutations (p.E94* + p.E36D) and splice site mutation (c.68-2A>G) had abolished B2M expression by H-score (190 to 0 and 100 to 0, respectively; Figs 5D and 5E). In the remaining two cases (B2M in-frame deletion [p.E56_V57del], and B2M nonsense mutation [p.S31*]), there was mild increase in B2M expression by H-score (110 to 145 and 0 to 40, respectively; Fig 5D).
DISCUSSION
In this study, we examined matched pre- and post-ICI samples from patients who developed acquired resistance to ICI. We identified recurrent acquired genomic changes, decreased TILs and HLA class I expression in tumor biopsies at disease progression on ICI, not seen in patients treated with chemotherapy or targeted therapies.
Previous studies have shown that loss-of-function mutations in STK11, KEAP1, and SMARCA4 drive primary resistance to ICI in lung cancer.15,16 Here, we demonstrate that these alterations can also mediate AR to ICI. This is significant as these mutations create vulnerabilities exploitable by novel therapies to restore ICI sensitivity.17-20
Additionally, 6.3% of patients acquired B2M mutations, which resulted in abolished B2M expression by IHC in 50% of evaluable cases. B2M alterations have previously been reported in nonresponders compared with responders to PD-1 and CTLA-4 inhibitors among patients with melanoma, and to associate with reduced B2M expression.21 In a previous analysis of 14 ICI-resistant NSCLC samples, an acquired homozygous deletion of B2M that caused lack of cell-surface HLA class I expression in the tumor and a matched patient-derived xenograft was identified, and CRISPR-mediated knockout of B2M in an immunocompetent lung cancer mouse model conferred resistance to PD-1 blockade in vivo, confirming the role of B2M loss in resistance to ICIs.3 Although our results are also supportive of mechanistic role of B2M loss in mediating AR to ICI, these findings should be interpreted with caution, given the small sample size.
Additionally, we found an acquired biallelic loss of JAK1, as well as an acquired homozygous deletion in JAK2 in two different patients. JAK1 and JAK2 are essential signal transducers of the INFγ pathway, therefore, in the setting of AR to ICI, a tumor may become insensitive to INFγ because of loss of JAK1/2.22 Consistently, previous evidence from whole-exome sequencing of four ICI-resistant melanomas revealed acquired loss in JAK1 and JAK2, and preclinical modeling of JAK1 and JAK2 truncating mutations also resulted in cancer cells insensitivity to INFγ, supporting a mechanistic role of these mutations in mediating resistance to ICI.4
Other acquired genomic alterations previously associated with an impaired antitumor immune response through the activation of the MAPK, PI3K/Akt/mTOR, and wingless type/β-catenin pathways23-26 were observed among patients with AR to ICI but not among those who were treated with chemotherapy. These included activating mutations in PIK3CA, SOS1, ERBB2, and BRAF, and loss-of-function mutations in NF1/NF2 and APC. Importantly, drugs targeting these pathways are either FDA approved or under investigation, and preclinical evidence has shown that PI3K/Akt/mTOR and MAPK inhibition may synergize with immunotherapies to resensitize resistant tumors to ICI. Nonetheless, these results remain exploratory and preclinical validation is necessary.
Finally, we demonstrated decreased TILs and HLA class I expression at the time of AR to ICI. Reduced HLA expression has been recognized as a mechanism of escape from antitumor immunity,27 and the most common mechanisms of HLA-I losses are reversible defects that can be pharmacologically exploited to restore HLA expression.27
Limitations of this study include the heterogeneity of our cohort (ICI ± chemotherapy) and the small sample size of some subgroups (HLA IHC, and mIF). Although small (25%), a fraction of patients received another line of therapy in addition to ICI between tumor biopsies. Nonetheless, we included a third control cohort of patients who received multiple lines of therapy including both chemotherapy and targeted therapy between tumor biopsies, which addresses this limitation. Another limitation is the inclusion of patients who developed AR after ≥3 months of ICI therapy. We used this threshold to reflect the enrollment of patients treated with ≥second-line ICI in this historical cohort, in which the median PFS from ICI is <6 months.28-30
In conclusion, mechanisms of AR to ICI are heterogeneous and require personalized post-ICI strategies.
Biagio Ricciuti
Honoraria: Targeted Oncology
Consulting or Advisory Role: Regeneron, AstraZeneca, Amgen, Guidepoint Inc
Navin R. Mahadevan
Stock and Other Ownership Interests: Vertex, Roche, AstraZeneca, Johnson & Johnson
Research Funding: Bristol Myers Squibb (Inst), Daiichi Sankyo (Inst)
Yvonne Y. Li
Stock and Other Ownership Interests: g.Root Biomedical Services
Liam Spurr
Employment: National Resilience, Inc
Andy J. Pangilinan
Employment: Dana Farber Cancer Hospital
Kathleen L. Pfaff
Employment: Dana-Farber Cancer Institute
Patents, Royalties, Other Intellectual Property: receive royalties for patent on a high content screening assay for iPSC-derived motor neuron survival (work done prior to joining DFCI)
Mizuki Nishino
Consulting or Advisory Role: AstraZeneca
Research Funding: Daiichi Sankyo (Inst), Canon Medical System (Inst), Konica Minolta (Inst)
Andrew D. Cherniack
Employment: LabCorp
Stock and Other Ownership Interests: Merck
Consulting or Advisory Role: BirdsEye Bio
Research Funding: Bayer
Bruce E. Johnson
Consulting or Advisory Role: Novartis, Daiichi Sankyo, Checkpoint Therapeutics, G1 Therapeutics, Jazz Pharmaceuticals, GlaxoSmithKline, Genentech, AstraZeneca, Hummingbird Diagnostics, BlueDot, Merus NV, Abdera Thereapeutics, Simcere
Patents, Royalties, Other Intellectual Property: Dana-Farber Cancer Institute
Scott J. Rodig
Leadership: Immunitas
Stock and Other Ownership Interests: Immunitas
Honoraria: Perkin Elmer, Bristol Myers Squibb
Consulting or Advisory Role: Bristol Myers Squibb
Research Funding: Bristol Myers Squibb, Merck, Affimed Therapeutics, Kite, a Gilead company
Patents, Royalties, Other Intellectual Property: Patent pending for use of Anti-galectin1 antibodies for diagnostic use
Travel, Accommodations, Expenses: Roche, Bristol Myers Squibb
Lynette M. Sholl
Stock and Other Ownership Interests: Moderna Therapeutics
Consulting or Advisory Role: Genentech (Inst), Lilly (Inst), AstraZeneca
Research Funding: Roche/Genentech (Inst), Bristol Myers Squibb (Inst)
Peter Sorger
Leadership: RareCyte, Applied BioMath, NanoString Technologies, Flagship Pioneering, Glencoe Software
Stock and Other Ownership Interests: RareCyte, Applied BioMath, Glencoe Software
Honoraria: Novartis
Consulting or Advisory Role: Merck
Research Funding: Merck (Inst)
Sandro Santagata
Honoraria: Novartis
Consulting or Advisory Role: BioReference Laboratories
Patents, Royalties, Other Intellectual Property: US9696313B2 HSF1 as a marker in tumor prognosis and treatment Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products. In some aspects, the invention provides methods of tumor diagnosis, prognosis, treatment-specific prediction, or treatment selection, the methods comprising assessing the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation correlate with poor outcome in cancer, eg, breast cancer, US20150241436A1 Hsf1 and hsf1 cancer signature set genes and uses relating thereto Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products. In some aspects, the invention provides methods of tumor diagnosis, prognosis, treatment-specific prediction, or treatment selection, the methods comprising assessing the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation correlate with poor outcome in cancer, eg, breast cancer. In some aspects, the invention relates to the HSF1 cancer program genes, HSF1 cancer signature set genes, subsets thereof, and uses in tumor diagnosis, prognosis, treatment-specific prediction, treatment selection, or drug discovery, among others, US20170037480A1 Hsf1 in tumor stroma Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products in tumor stroma. In some aspects, the invention provides methods of tumor prognosis, treatment-specific prediction, or treatment selection, the methods comprising measuring the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor that comprises tumor-associated stromal cells. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation in tumor-associated stromal cells correlate with poor outcome in cancer. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells and specifically in cancer cells. In some embodiments HSF1 expression or activation is measured using an antibody that specifically binds to HSF1. In some embodiments HSF1 expression or activation is measured by measuring expression of genes that are regulated by HSF1 in tumor-associated stromal cells. In some aspects, the invention relates to inhibiting HSF1 in tumor-associated stromal cells as an approach to cancer therapy, US20170037480A1 Hsf1 in tumor stroma Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products in tumor stroma. In some aspects, the invention provides methods of tumor prognosis, treatment-specific prediction, or treatment selection, the methods comprising measuring the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor that comprises tumor-associated stromal cells. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation in tumor-associated stromal cells correlate with poor outcome in cancer. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells and specifically in cancer cells. In some embodiments HSF1 expression or activation is measured using an antibody that specifically binds to HSF1. In some embodiments HSF1 expression or activation is measured by measuring expression of genes that are regulated by HSF1 in tumor-associated stromal cells. In some aspects, the invention relates to inhibiting HSF1 in tumor-associated stromal cells as an approach to cancer therapy, US20180306796A1 Methods and compositions relating to proteasome inhibitor resistance Abstract In some aspects, the disclosure provides methods of modulating the level of proteasome inhibitor resistance of a cell, the methods comprising manipulating the level of expression or activity of a subunit of the 19S proteasome in the cell. In some aspects, cells in which the level of a 19S subunit is modulated, eg, reduced, are provided. In some aspects, methods of identifying agents that reduce proteasome inhibitor resistance are provided. In some aspects, methods of classifying cancers according to predicted proteasome inhibitor resistance are provided. In some aspects, methods of killing or inhibiting proliferation of cancer cells, eg, proteasome inhibitor resistant cancer cells, are provided. In some aspects, methods of treating cancer, eg, proteasome inhibitor resistant cancer, are provided, “Combination Treatments of Hsp90 Inhibitors for Enhancing Tumor Immunogenicity and Methods of Use Thereof” Application pending, “Targeted Manipulation of the Proteasome Subunit Expression Levels as a Method for Curing Cancer” Application pending
Renato Umeton
Patents, Royalties, Other Intellectual Property: Patent: Portable medical device and method for quantitative retinal image analysis through a smartphone, Patent: Epstein barr virus genotipic variants and uses thereof as risk predictors, biomarkers and therapeutic targets of multiple sclerosis
Mark M. Awad
Consulting or Advisory Role: Merck, Pfizer, Bristol Myers Squibb, Foundation Medicine, Novartis, Gritstone Bio, Mirati Therapeutics, EMD Serono, AstraZeneca, Instil Bio, AstraZeneca, Regeneron, Janssen, Affini-T Therapeutics
Research Funding: Genentech/Roche (Inst), Lilly (Inst), AstraZeneca (Inst), Bristol Myers Squibb (Inst), Amgen (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb Foundation
Open Payments Link: https://openpaymentsdata.cms.gov/physician/1127368
No other potential conflicts of interest were reported.
PRIOR PRESENTATION
Presented in part at ASCO 2022 annual meeting, Chicago, IL, June 3-7; and at the Society for Immunotherapy of Cancer 2022, Boston, MA, November 8-12, 2022.
SUPPORT
Supported by the Elva J. and Clayton L. McLaughlin Fund for Lung Cancer Research and LUNGSTRONG. B.R. was supported by the Barbara Wilson Gomez Endowed Fellowship in Thoracic Oncology, and the Society for Immunotherapy of Cancer Astra-Zeneca Lung Cancer Clinical Fellowship Award.
See accompanying Editorial, p. 1211
B.R. and G.L. are co-first authors.
AUTHOR CONTRIBUTIONS
Conception and design: Biagio Ricciuti, Giuseppe Lamberti, Scott J. Rodig, Peter Sorger, Renato Umeton, Mark M. Awad
Financial support: Bruce E. Johnson, Peter Sorger, Sandro Santagata, Mark M. Awad
Administrative support: Andy J. Pangilinan, Elinton Lee, Bruce E. Johnson, Mark M. Awad
Provision of study materials or patients: Danielle Haradon, Mark M. Awad
Collection and assembly of data: Biagio Ricciuti, Giuseppe Lamberti, Jia-Ren Lin, Joao V. Alessi, Yvonne Y. Li, Federica Pecci, Alessandro Di Federico, Deepti Venkatraman, Adriana P. Barrichello, Malini Gandhi, Andy J. Pangilinan, Danielle Haradon, Elinton Lee, Emma L. Welsh, Mizuki Nishino, Lynette M. Sholl, Peter Sorger, Mark M. Awad
Data analysis and interpretation: Biagio Ricciuti, Giuseppe Lamberti, Sreekar R. Puchala, Navin R. Mahadevan, Jia-Ren Lin, Joao V. Alessi, Alexander Chowdhury, Yvonne Y. Li, Xinan Wang, Liam Spurr, Victor R. Vaz, Hersh Gupta, Kathleen L. Pfaff, Mizuki Nishino, Andrew D. Cherniack, Bruce E. Johnson, Jason L Weirather, Ian D Dryg, Scott J. Rodig, Lynette M. Sholl, Sandro Santagata, Renato Umeton, Mark M. Awad
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Genomic and Immunophenotypic Landscape of Acquired Resistance to PD-(L)1 Blockade in Non–Small-Cell Lung Cancer
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Biagio Ricciuti
Honoraria: Targeted Oncology
Consulting or Advisory Role: Regeneron, AstraZeneca, Amgen, Guidepoint Inc
Navin R. Mahadevan
Stock and Other Ownership Interests: Vertex, Roche, AstraZeneca, Johnson & Johnson
Research Funding: Bristol Myers Squibb (Inst), Daiichi Sankyo (Inst)
Yvonne Y. Li
Stock and Other Ownership Interests: g.Root Biomedical Services
Liam Spurr
Employment: National Resilience, Inc
Andy J. Pangilinan
Employment: Dana Farber Cancer Hospital
Kathleen L. Pfaff
Employment: Dana-Farber Cancer Institute
Patents, Royalties, Other Intellectual Property: receive royalties for patent on a high content screening assay for iPSC-derived motor neuron survival (work done prior to joining DFCI)
Mizuki Nishino
Consulting or Advisory Role: AstraZeneca
Research Funding: Daiichi Sankyo (Inst), Canon Medical System (Inst), Konica Minolta (Inst)
Andrew D. Cherniack
Employment: LabCorp
Stock and Other Ownership Interests: Merck
Consulting or Advisory Role: BirdsEye Bio
Research Funding: Bayer
Bruce E. Johnson
Consulting or Advisory Role: Novartis, Daiichi Sankyo, Checkpoint Therapeutics, G1 Therapeutics, Jazz Pharmaceuticals, GlaxoSmithKline, Genentech, AstraZeneca, Hummingbird Diagnostics, BlueDot, Merus NV, Abdera Thereapeutics, Simcere
Patents, Royalties, Other Intellectual Property: Dana-Farber Cancer Institute
Scott J. Rodig
Leadership: Immunitas
Stock and Other Ownership Interests: Immunitas
Honoraria: Perkin Elmer, Bristol Myers Squibb
Consulting or Advisory Role: Bristol Myers Squibb
Research Funding: Bristol Myers Squibb, Merck, Affimed Therapeutics, Kite, a Gilead company
Patents, Royalties, Other Intellectual Property: Patent pending for use of Anti-galectin1 antibodies for diagnostic use
Travel, Accommodations, Expenses: Roche, Bristol Myers Squibb
Lynette M. Sholl
Stock and Other Ownership Interests: Moderna Therapeutics
Consulting or Advisory Role: Genentech (Inst), Lilly (Inst), AstraZeneca
Research Funding: Roche/Genentech (Inst), Bristol Myers Squibb (Inst)
Peter Sorger
Leadership: RareCyte, Applied BioMath, NanoString Technologies, Flagship Pioneering, Glencoe Software
Stock and Other Ownership Interests: RareCyte, Applied BioMath, Glencoe Software
Honoraria: Novartis
Consulting or Advisory Role: Merck
Research Funding: Merck (Inst)
Sandro Santagata
Honoraria: Novartis
Consulting or Advisory Role: BioReference Laboratories
Patents, Royalties, Other Intellectual Property: US9696313B2 HSF1 as a marker in tumor prognosis and treatment Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products. In some aspects, the invention provides methods of tumor diagnosis, prognosis, treatment-specific prediction, or treatment selection, the methods comprising assessing the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation correlate with poor outcome in cancer, eg, breast cancer, US20150241436A1 Hsf1 and hsf1 cancer signature set genes and uses relating thereto Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products. In some aspects, the invention provides methods of tumor diagnosis, prognosis, treatment-specific prediction, or treatment selection, the methods comprising assessing the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation correlate with poor outcome in cancer, eg, breast cancer. In some aspects, the invention relates to the HSF1 cancer program genes, HSF1 cancer signature set genes, subsets thereof, and uses in tumor diagnosis, prognosis, treatment-specific prediction, treatment selection, or drug discovery, among others, US20170037480A1 Hsf1 in tumor stroma Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products in tumor stroma. In some aspects, the invention provides methods of tumor prognosis, treatment-specific prediction, or treatment selection, the methods comprising measuring the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor that comprises tumor-associated stromal cells. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation in tumor-associated stromal cells correlate with poor outcome in cancer. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells and specifically in cancer cells. In some embodiments HSF1 expression or activation is measured using an antibody that specifically binds to HSF1. In some embodiments HSF1 expression or activation is measured by measuring expression of genes that are regulated by HSF1 in tumor-associated stromal cells. In some aspects, the invention relates to inhibiting HSF1 in tumor-associated stromal cells as an approach to cancer therapy, US20170037480A1 Hsf1 in tumor stroma Abstract In some aspects, the invention relates to Heat Shock Protein-1 (HSF1) gene and HSF1 gene products in tumor stroma. In some aspects, the invention provides methods of tumor prognosis, treatment-specific prediction, or treatment selection, the methods comprising measuring the level of HSF1 expression or HSF1 activation in a sample obtained from the tumor that comprises tumor-associated stromal cells. In some aspects, the invention relates to the discovery that increased HSF1 expression and increased HSF1 activation in tumor-associated stromal cells correlate with poor outcome in cancer. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells. In some embodiments, the methods comprise measuring HSF1 expression or activation specifically in tumor-associated stromal cells and specifically in cancer cells. In some embodiments HSF1 expression or activation is measured using an antibody that specifically binds to HSF1. In some embodiments HSF1 expression or activation is measured by measuring expression of genes that are regulated by HSF1 in tumor-associated stromal cells. In some aspects, the invention relates to inhibiting HSF1 in tumor-associated stromal cells as an approach to cancer therapy, US20180306796A1 Methods and compositions relating to proteasome inhibitor resistance Abstract In some aspects, the disclosure provides methods of modulating the level of proteasome inhibitor resistance of a cell, the methods comprising manipulating the level of expression or activity of a subunit of the 19S proteasome in the cell. In some aspects, cells in which the level of a 19S subunit is modulated, eg, reduced, are provided. In some aspects, methods of identifying agents that reduce proteasome inhibitor resistance are provided. In some aspects, methods of classifying cancers according to predicted proteasome inhibitor resistance are provided. In some aspects, methods of killing or inhibiting proliferation of cancer cells, eg, proteasome inhibitor resistant cancer cells, are provided. In some aspects, methods of treating cancer, eg, proteasome inhibitor resistant cancer, are provided, “Combination Treatments of Hsp90 Inhibitors for Enhancing Tumor Immunogenicity and Methods of Use Thereof” Application pending, “Targeted Manipulation of the Proteasome Subunit Expression Levels as a Method for Curing Cancer” Application pending
Renato Umeton
Patents, Royalties, Other Intellectual Property: Patent: Portable medical device and method for quantitative retinal image analysis through a smartphone, Patent: Epstein barr virus genotipic variants and uses thereof as risk predictors, biomarkers and therapeutic targets of multiple sclerosis
Mark M. Awad
Consulting or Advisory Role: Merck, Pfizer, Bristol Myers Squibb, Foundation Medicine, Novartis, Gritstone Bio, Mirati Therapeutics, EMD Serono, AstraZeneca, Instil Bio, AstraZeneca, Regeneron, Janssen, Affini-T Therapeutics
Research Funding: Genentech/Roche (Inst), Lilly (Inst), AstraZeneca (Inst), Bristol Myers Squibb (Inst), Amgen (Inst)
Travel, Accommodations, Expenses: Bristol Myers Squibb Foundation
Open Payments Link: https://openpaymentsdata.cms.gov/physician/1127368
No other potential conflicts of interest were reported.
REFERENCES
- 1. Shields MD, Marin-Acevedo JA, Pellini B. Immunotherapy for advanced non–small cell lung cancer: A decade of progress. Am Soc Clin Oncol Educ Book. 2021;41:1–23. doi: 10.1200/EDBK_321483. [DOI] [PubMed] [Google Scholar]
- 2. Recondo G, Facchinetti F, Olaussen KA, et al. Making the first move in EGFR-driven or ALK-driven NSCLC: First-generation or next-generation TKI? Nat Rev Clin Oncol. 2018;15:694–708. doi: 10.1038/s41571-018-0081-4. [DOI] [PubMed] [Google Scholar]
- 3. Gettinger S, Choi J, Hastings K, et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 2017;7:1420–1435. doi: 10.1158/2159-8290.CD-17-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zaretsky JM, Garcia-Diaz A, Shin DS, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med. 2016;375:819–829. doi: 10.1056/NEJMoa1604958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Garcia EP, Minkovsky A, Jia Y, et al. Validation of OncoPanel: A targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch Pathol Lab Med. 2017;141:751–758. doi: 10.5858/arpa.2016-0527-OA. [DOI] [PubMed] [Google Scholar]
- 6. Spurr LF, Touat M, Taylor AM, et al. Quantification of aneuploidy in targeted sequencing data using ASCETS. Bioinformatics. 2021;37:2461–2463. doi: 10.1093/bioinformatics/btaa980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Taylor AM, Shih J, Ha G, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33:676–689.e3. doi: 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bogusz AM, Baxter RHG, Currie T, et al. Quantitative immunofluorescence reveals the signature of active B-cell receptor signaling in diffuse large B-cell lymphoma. Clin Cancer Res. 2012;18:6122–6135. doi: 10.1158/1078-0432.CCR-12-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin J-R, Chen Y-A, Campton D, et al. High-plex immunofluorescence imaging and traditional histology of the same tissue section for discovering image-based biomarkers. Nat Cancer. 2023;4:1036–1052. doi: 10.1038/s43018-023-00576-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ricciuti B, Wang X, Alessi JV, et al. Association of high tumor mutation burden in non–small cell lung cancers with increased immune infiltration and improved clinical outcomes of PD-L1 blockade across PD-L1 expression levels. JAMA Oncol. 2022;8:1160–1168. doi: 10.1001/jamaoncol.2022.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rosenthal J, Carelli R, Omar M, et al. Building tools for machine learning and artificial Intelligence in cancer research: Best practices and a case study with the PathML toolkit for computational pathology. Mol Cancer Res. 2022;20:202–206. doi: 10.1158/1541-7786.MCR-21-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cooper AJ, Sequist LV, Lin JJ. Third-generation EGFR and ALK inhibitors: Mechanisms of resistance and management. Nat Rev Clin Oncol. 2022;19:499–514. doi: 10.1038/s41571-022-00639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schoenfeld AJ, Antonia SJ, Awad MM, et al. Clinical definition of acquired resistance to immunotherapy in patients with metastatic non-small-cell lung cancer. Ann Oncol. 2021;32:1597–1607. doi: 10.1016/j.annonc.2021.08.2151. [DOI] [PubMed] [Google Scholar]
- 14. Sabbatino F, Liguori L, Polcaro G, et al. Role of human leukocyte antigen System as A predictive biomarker for checkpoint-based immunotherapy in cancer patients. Int J Mol Sci. 2020;21:7295. doi: 10.3390/ijms21197295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ricciuti B, Arbour KC, Lin JJ, et al. Diminished efficacy of programmed death-(ligand)1 inhibition in STK11- and KEAP1-mutant lung adenocarcinoma is affected by KRAS mutation status. J Thorac Oncol. 2022;17:399–410. doi: 10.1016/j.jtho.2021.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alessi JV, Ricciuti B, Spurr LF, et al. SMARCA4 and other SWItch/Sucrose Nonfermentable family genomic alterations in NSCLC: Clinicopathologic characteristics and outcomes to immune checkpoint inhibition. J Thorac Oncol. 2021;16:1176–1187. doi: 10.1016/j.jtho.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 17. Xue Y, Meehan B, Fu Z, et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat Commun. 2019;10:557. doi: 10.1038/s41467-019-08380-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Papillon JPN, Nakajima K, Adair CD, et al. Discovery of orally active inhibitors of Brahma homolog (BRM)/SMARCA2 ATPase activity for the treatment of Brahma related gene 1 (BRG1)/SMARCA4-Mutant cancers. J Med Chem. 2018;61:10155–10172. doi: 10.1021/acs.jmedchem.8b01318. [DOI] [PubMed] [Google Scholar]
- 19. Koppula P, Olszewski K, Zhang Y, et al. KEAP1 deficiency drives glucose dependency and sensitizes lung cancer cells and tumors to GLUT inhibition. iScience. 2021;24:102649. doi: 10.1016/j.isci.2021.102649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kitajima S, Tani T, Springer BF, et al. MPS1 inhibition primes immunogenicity of KRAS-LKB1 mutant lung cancer. Cancer Cell. 2022;40:1128–1144.e8. doi: 10.1016/j.ccell.2022.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sade-Feldman M, Jiao YJ, Chen JH, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun. 2017;8:1136. doi: 10.1038/s41467-017-01062-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shin DS, Zaretsky JM, Escuin-Ordinas H, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7:188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 24. Yang B, Li X, Fu Y, et al. MEK inhibition remodels the immune landscape of mutant KRAS tumors to overcome resistance to PARP and immune checkpoint inhibitors. Cancer Res. 2021;81:2714–2729. doi: 10.1158/0008-5472.CAN-20-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Collins NB, Al Abosy R, Miller BC, et al. PI3K activation allows immune evasion by promoting an inhibitory myeloid tumor microenvironment. J Immunother Cancer. 2022;10:e003402. doi: 10.1136/jitc-2021-003402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Prasad M, Zorea J, Jagadeeshan S, et al. MEK1/2 inhibition transiently alters the tumor immune microenvironment to enhance immunotherapy efficacy against head and neck cancer. J Immunother Cancer. 2022;10:e003917. doi: 10.1136/jitc-2021-003917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hazini A, Fisher K, Seymour L. Deregulation of HLA-I in cancer and its central importance for immunotherapy. J Immunother Cancer. 2021;9:e002899. doi: 10.1136/jitc-2021-002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373:1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–265. doi: 10.1016/S0140-6736(16)32517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]