

Abstract
Purpose:
Plexiform neurofibromas (PNF) are benign peripheral nerve sheath tumors (PNST) associated with neurofibromatosis type 1 (NF1). Despite similar histological appearance, these neoplasms exhibit diverse evolutionary trajectories, with a subset progressing to malignant peripheral nerve sheath tumor (MPNST), the leading cause of premature death in individuals with NF1. Malignant transformation of PNF often occurs through development of atypical neurofibroma (ANF) precursor lesions characterized by distinct histopathological features and CDKN2A copy number loss. While genomic studies have uncovered key driver events promoting tumor progression, the transcriptional changes preceding malignant transformation remain poorly defined.
Experimental Design:
Here we resolve gene expression profiles in PNSTs across the neurofibroma-to-MPNST continuum in NF1 patients and mouse models, revealing early molecular features associated with neurofibroma evolution and transformation.
Results:
Our findings demonstrate that ANF exhibit enhanced signatures of antigen presentation and immune response, which are suppressed as malignant transformation ensues. MPNSTs further displayed deregulated survival and mitotic fidelity pathways, and targeting key mediators of these pathways, CENPF and BIRC5, disrupted growth and viability of human MPNST cells lines and primary murine Nf1-Cdkn2a mutant Schwann cell precursors. Finally, neurofibromas contiguous with MPNST manifested distinct alterations in core oncogenic and immune surveillance programs, suggesting that early molecular events driving disease progression may precede histopathological evidence of malignancy.
Conclusions:
If validated prospectively in future studies, these signatures may serve as molecular diagnostic tools to augment conventional histopathological diagnosis by identifying neurofibromas at high risk of undergoing malignant transformation, facilitating risk-adapted care.
INTRODUCTION
Neurofibromatosis type 1 (NF1) is a cancer predisposition syndrome that affects approximately 1 in 3000 individuals and results from mutations in the NF1 tumor suppressor gene, which encodes neurofibromin, a GTPase activating protein for p21Ras (1,2). Plexiform neurofibromas (PNF) are benign peripheral nerve sheath tumors (PNST) and are a hallmark of NF1, occurring in 50% of persons with NF1 (3,4). PNF are heterogenous neoplasms that despite similar histological appearances, can exhibit disparate biological behavior and growth kinetics across the lifespan of an individual (4,5).
The lifetime risk of developing malignant peripheral nerve sheath tumor (MPNST) is approximately 8–16% in individuals with NF1 (6,7). MPNST is a devastating form of sarcoma that is recalcitrant to chemotherapy and radiation and represents the leading cause of premature death in persons with NF1. Cure is only possible through wide marginal excision, which is often not feasible due to entanglement with vital structures. For patients presenting with unresectable disease, median overall survival is approximately five months (8). Progression of PNF to MPNST often occurs with little to no early warning, and clinical indicators such as pain, change in consistency on palpation, nodularity, increased FDG-PET avidity and rapid growth may only manifest once malignant transformation has already taken place. Thus, identifying key molecular alterations and biomarkers that precede malignant transformation is of critical importance for early detection and to inform risk-adapted care.
Malignant transformation of PNF often occurs through the development of atypical neurofibroma (ANF), which typically appear as distinct nodular lesions (DNLs) (9) and exhibit characteristic histopathological features including atypia, increased cellularity, loss of neurofibroma architecture, and/or mitotic index >1/50 high-power fields (HPFs) and <3/10 HPFs (10). Recent consensus criteria have categorized these lesions as atypical neurofibromatous neoplasms of uncertain biological potential (ANNUBP) when two or more of these criteria are fulfilled, whereas neurofibromas with isolated atypia in the absence of other features are termed neurofibroma with atypia (NF with atypia) (10). It remains unknown whether ANNUBP may exhibit other distinguishing characteristics from NF with atypia on a molecular level. Loss of the 9p21 locus harboring the CDKN2A tumor suppressor gene has been identified as a key “second hit” in development of ANF/ANNUBP and is present in the majority of ANF/ANNUBP and MPNST (11–16). Pathogenic variants in PRC2 complex genes and other diverse genetic events have been identified in MPNST (12,13,16,17), yet are characteristically absent in PNF and ANF/ANNUBP (15). Loss of Cdkn2a and/or its alternate reading frame (Arf) in genetically engineered mice with conditional knockout of Nf1 is sufficient to engender the development of both ANF/ANNUBP and MPNST (18,19), but the penetrance of malignant transformation is only 50% with loss of a single Arf allele (18). Similarly, not all patients with histopathologically defined ANF or DNLs on imaging will develop MPNST, and the timing of malignant transformation remains unpredictable. Therefore, we reasoned that additional factors intrinsic to neoplastic Schwann cell precursors or within the tumor microenvironment may critically govern the malignant potential of ANF/ANNUBP precursor lesions.
While genomic studies have advanced our knowledge of key driver events promoting neurofibroma progression, our understanding of transcriptional and microenvironmental dynamics orchestrating the progression and malignant transformation of PNF and ANF/ANNUBP precursor lesions remains limited. PNST are highly heterogenous neoplasms and evolution across multiple stages of the neurofibroma-to-MPNST continuum can be observed even within a single specimen. To overcome this challenge, we utilized a multiplexed, hybridization-based approach to quantify gene expression in spatially defined tissue regions. Through multi-regional and longitudinal profiling of human PNST samples (n=35), we identify alterations in signal transduction, immune response, and mitotic fidelity associated with disease progression and trace their dynamics across multiple stages of the neurofibroma to MPNST continuum within individual patients, providing insight into the nascent events associated with the evolution of MPNST precursor lesions. We further demonstrate that these core molecular signatures are retained in genetically engineered mice harboring conditional loss of Nf1 and Cdkn2a/Arf in neural crest derived Schwann cell precursors that recapitulate the development of MPNST from existing PNF and ANNUBP. Finally, gene signature and pathway enrichment analyses comparing isolated lesions at discrete stages of PNST development as well as neurofibromas contiguous with adjacent MPNST provides preliminary insight into potential biomarkers of disease progression, that if validated prospectively in future studies, may help guide risk adapted care.
METHODS
Ethical considerations
Archived samples, associated clinical data and imaging were collected under approval by the Indiana University Institutional Review Board (IRB Protocol #17332). Animal studies were conducted according to the guidelines established by the Indiana University Institutional Animal Care and Use Committee (IACUC Protocol #21009).
Sample selection
Samples were selected retrospectively by querying the Indiana University pathology archives with the search terms “neurofibromatosis type 1”, “ANNUBP”, “atypical neurofibroma”, “neurofibroma with atypia,” “plexiform neurofibroma,” and “MPNST.” Priority was given to cases of NF with atypia or ANNUBP that had additional samples from the same patient classified as PNF and/or MPNST. Several isolated PNST cases were also included in the analysis.
Slide review, microdissection and RNA extraction from FFPE tissues
Hematoxylin and eosin (H&E)-stained tissue sections from each tumor were scanned using an Aperio ScanScope CS digital slide scanner at 20x magnification. A board-certified pathologist, with expertise in diagnosing NF1-associated nerve sheath tumors digitally annotated tissue areas meeting consensus diagnostic criteria (10). Areas of hemorrhage and necrosis were excluded from the analysis.
Formalin fixed paraffin embedded (FFPE) tissues were microdissected from 5 μm thick sections on freshly cut slides. Total RNA was extracted using a RNeasy FFPE Kit (Qiagen). RNA concentration and fragmentation index were quantified on an Agilent bioanalyzer. Gene expression analysis was conducted on the nCounter® Sprint Profiler (NanoString Technologies, Inc.) using the nCounter® human PanCancer IO360™ probe set (NanoString Technologies, Inc.). Fragmentation index (percent greater than 200 bp) was used to normalize the loading of 50 ng total input RNA in a final volume of 5 μL with the capture probe set. Probes and target transcripts were hybridized for 20 hours at 65°C per the manufacturer’s recommendations. Hybridized samples were loaded into a nCounter® cartridge and quantified on the nCounter® Sprint Profiler. Quality control, normalization, differential expression and pathway analysis were performed using nSolver Advanced Analysis Software (version 4.0, Nanostring Technologies, Inc.) as further detailed below.
Quality Control
Adequate signal was observed across all samples in the data set as indicated by the heatmap of raw counts in (Supplemental Figure 1, Supplemental Table 1). 40 probes below threshold with raw counts less than 25 (below the level of the background) were excluded from further analysis (Supplemental Table 2).
Normalization
Data were normalized using both positive control probes and housekeeping genes. Prior to hybridization, non-natural RNA sequences (positive control) probes were added to the samples at known concentrations to control for variations in pipetting and hybridization across samples. The read counts of each sample were then normalized by a correction factor computed by the geometric mean level of the control probes of across all samples divided by the levels of the control probes in individual samples.
To account for differences in RNA loading and quality, a set of housekeeping genes exhibiting low variance across all samples profiled were selected using the geNorm algorithm to form a normalization constant for each sample (Supplemental Table 3 and 4). Housekeeping probes with significant sample-to sample-variability and were excluded from the housekeeping normalization. Normalized and log2 transformed normalized counts for each probe and sample used in the downstream differential gene expression analysis are presented in Supplemental Table 5 and 6, respectively.
Differential gene expression and pathway analysis
Statistical analysis of differentially expressed genes and signature pathway scoring was performed using the Differential Expression (DE) and PathView modules of the nSolver Advanced Analysis Platform (version 4.0, NanoString Technologies, Inc.). Multiplicity adjusted p-values <0.05 were considered statistically significant. Results of differential gene expression (Supplemental Table 7 and 8) and PathView analysis (Supplemental Table 9) used in downstream plotting and data visualization are presented in the supplemental data. Sample metadata used for analysis are provided in Supplemental Table 10.
RNAseq analysis
RNAseq data from murine peripheral nerve sheath tumors arising in DhhCre mice harboring Nf1-floxed alleles and Ink4a/Arf heterozygosity, as described by Chaney et al. (19), were obtained from the Gene Expression Omnibus (GEO) database (GSE148249). Raw count matrices with sample metadata deposited on GEO were reanalyzed using the Omics Playground v2.7.18 Platform. RNAseq data from Nf1flox/flox;PostnCre+ and Nf1flox/flox;Arf flox/flox;PostnCre+ mice, available under GEO accession GSE213786 and GSE232707. Publicly available RNA sequencing data was also obtained from the TARGET TCGA GTEx database using the UCSC Xena browser (RRID:SCR_018938). Differential gene expression analysis was performed using the UCSC Xena browser to compare gene expression profiles between TCGA MPNST (n=10) and GTEx Normal Nerve samples (n=335). A volcano plot of the resulting data was generated using GraphPad Prism 9.5.1 software.
Principal Component Analysis (PCA)
PCA was conducted in IDEP.92 (20) using log2 transformed, normalized counts as input under default settings.
Heatmaps
The pathway heatmap in Figure 2C was generated using the nSolver Advanced Analysis Platform (version 4.0, Nanostring Technologies, Inc). The gene expression heatmap in Supplemental Figure 3 was generated using iDEP.92 (20). Log2 transformed, z-score normalized counts from the top 100 most variable genes were clustered by correlation coefficient with average linkage. Heatmaps in Figure 5B and C were generated using Morpheus with log2 transformed, z-score normalized counts from all genes clustered by correlation coefficient with average linkage. Heatmaps in Supplemental Figure 8B and Figure 6C were generated using the Omics Playground v2.7.18 platform (21) and were generated using the ComplexHeatmap R/Bioconductor package (RRID:SCR_017270) on scaled log-expression values (z-score) using Euclidean distance and Ward linkage. The standard deviation was used to rank the genes for the reduced heatmaps. Rows represent individual genes, and their respective expression log2 normalized, z-score transformed expression values across each sample in the data. Columns represent individual samples, with annotation tracks below indicating sample IDs and above indicating status of contiguity with MPNST.
Figure 2. Pathway analysis identifies three molecular NF1-PNST subclusters traversing conventional histopathological classifiers.
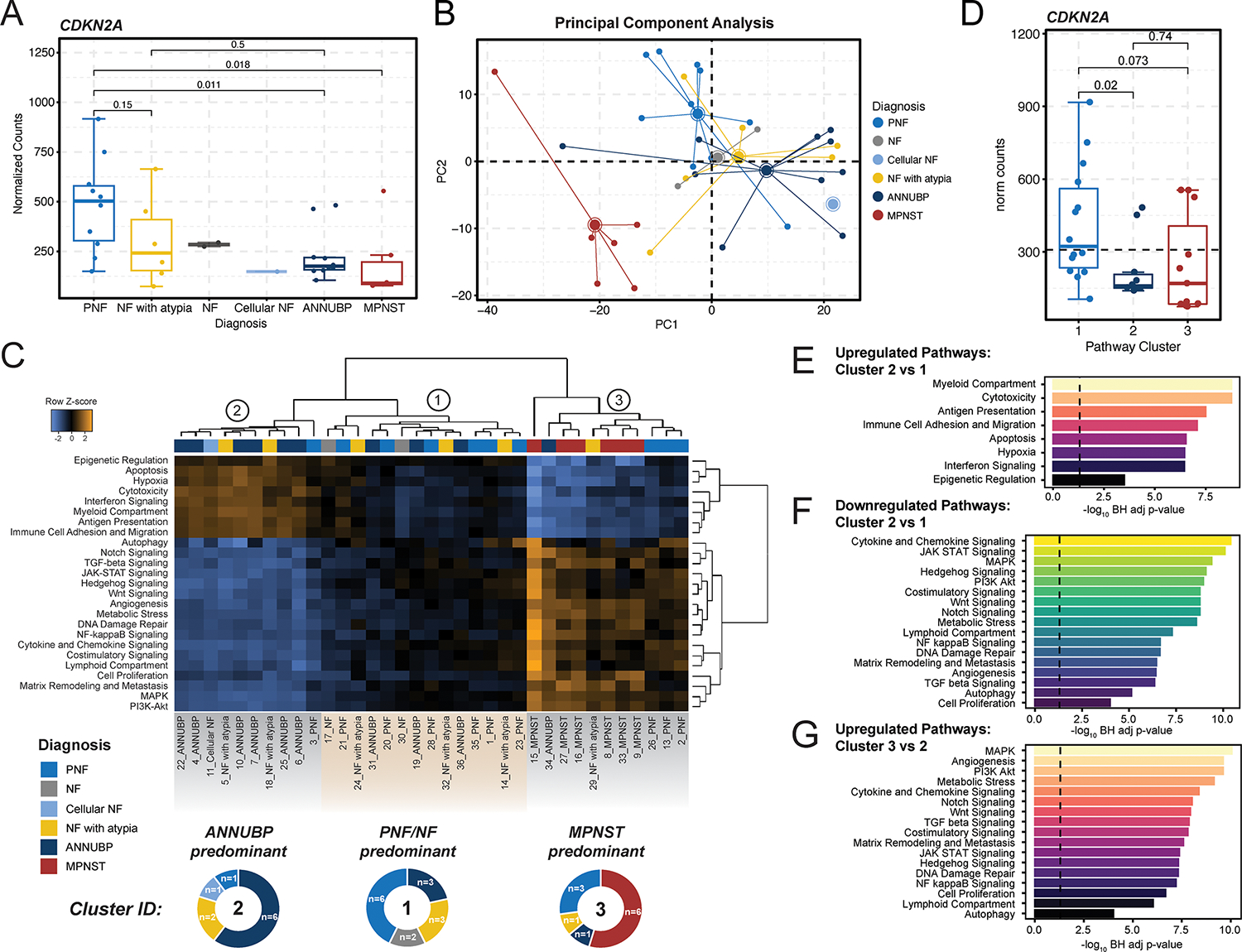
(A) Box and whisker plot depicting CDKN2A mRNA expression (normalized counts) by tumor subtype. Dots represent individual samples. Whiskers extend from the minima to maxima that are no further than 1.5x the inter-quartile range spanning the first to third quartiles. The center line represents the median. The box spans the 25th to 75th percentiles. Data beyond the whiskers are outliers and are plotted as individual points. P-values represent unpaired, two-tailed t-tests between groups. (B) Principal component analysis demonstrating global variation in gene expression between PNF (n=10), NF (n=2), cellular NF (n=1), NF with atypia (n=6), ANNUBP (n=10), and MPNST (n=6) based on principal components 1 and 2 (PC1 and PC2). (C) Heatmap of differentially expressed pathways. Rows represent individual pathways and their z-score normalized signature scores across each sample in the data. Columns represent individual samples, with annotation tracks above indicating tissue diagnosis as noted on the figure legend. Hierarchical clustering revealed three predominating clusters: Cluster 1 (n=14 samples), Cluster 2 (n=10 samples), and Cluster 3 (n=11 samples). Cluster 1 is predominately comprised of NF and PNF lesions, Cluster 2 of ANNUBP, and Cluster 3 of MPNST. The proportion of each tissue diagnosis within each cluster is illustrated in the donut plots below each cluster in the heatmap. (D) Box and whisker plot depicting CDKN2A mRNA expression (normalized counts) stratified by molecular cluster. Dots represent individual samples. Whiskers extend from the minima to maxima that are no further than 1.5x the inter-quartile range spanning the first to third quartiles. The center line represents the median. The box spans the 25th to 75th percentiles. Data beyond the whiskers are outliers and are plotted as individual points. P-values represent unpaired, two-tailed t-tests between groups. Barplots depicting differential enrichment of pan-cancer pathways relevant to tumor progression, immune response and microenvironment. Signatures are plotted in rank order based on −log10 Bejamini Hochberg adjusted p-values for pathways upregulated in Cluster 2 vs Cluster 1 (E), downregulated in Cluster 2 vs Cluster 1 (F) and upregulated in Cluster 3 vs Cluster 2 (G). The dashed line represents a false discovery rate of 0.05.
Figure 5. Clinical vignettes.
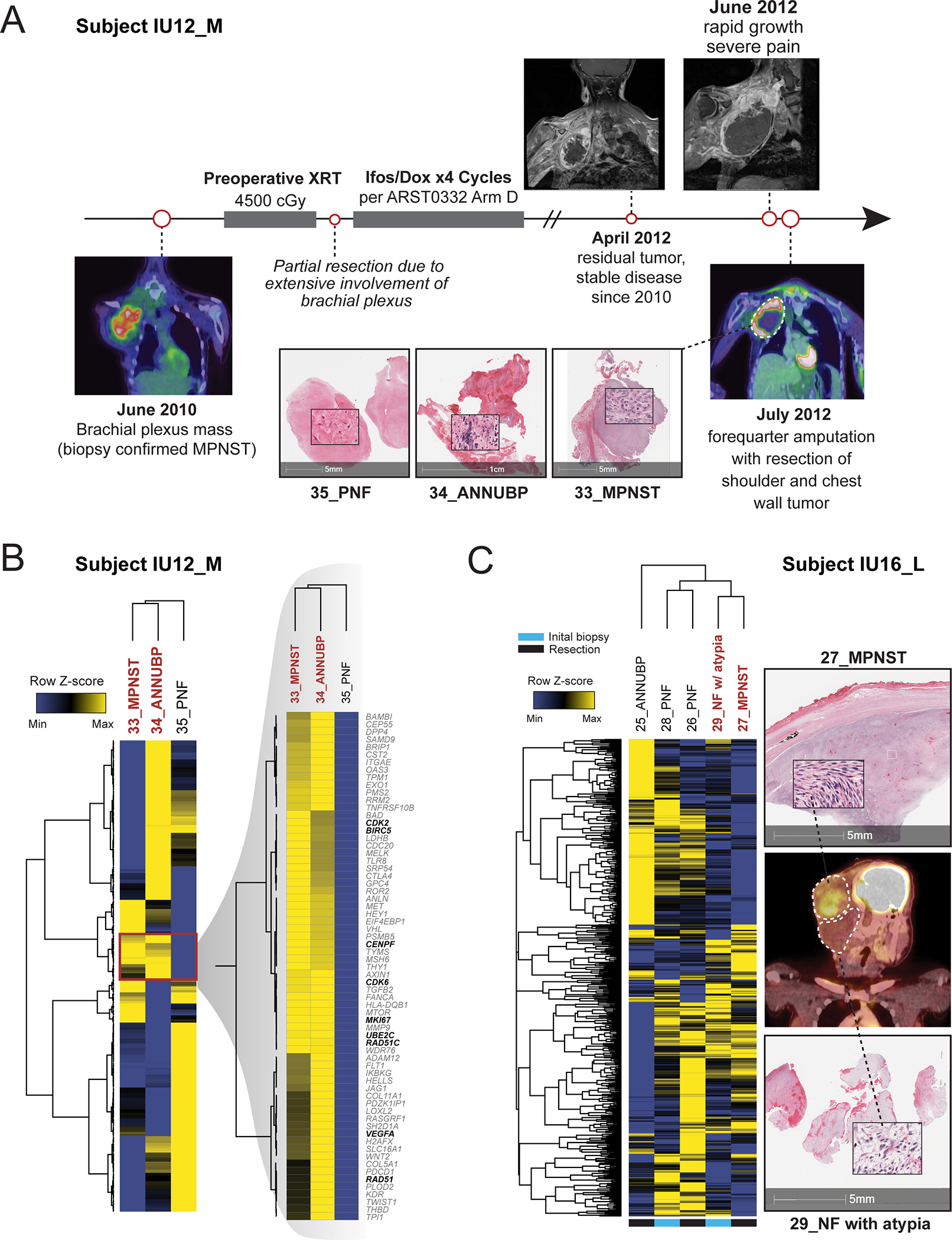
(A) Subject IU12_M, a 17-year-old female with NF1 presented with a large, FDG-PET avid, right upper extremity mass confirmed to be high-grade MPNST on biopsy. Despite aggressive treatment with radiation, chemotherapy and attempted debulking, the patient had recurrence of disease two years after initial presentation. A forequarter amputation with resection of the shoulder and chest wall mass was performed. Multi-regional tissue sampling revealed regions of tumor consistent with PNF (35_PNF), ANNUBP (34_ANNUBP) and MPNST (33_MPNST). (B) Hierarchically clustered heatmap of all genes with inset magnification demonstrating overlapping molecular signatures shared by 34_ANNUBP and 35_MPNST. Select genes involved in cell cycle and mitotic fidelity, DNA repair and immune evasion are bolded. Rows represent individual genes and their respective expression log2 normalized, z-score transformed expression values across the three samples. Columns represent individual samples. Rows and columns were clustered by correlation coefficient with average linkage. (C) Multiregional profiling of subject IU16_L, a 13-year-old female with NF1 who presented with a right sided, FDG avid occipital mass. Multiple samples from an initial core needle biopsy (28-PNF, 29_NF with atypia) and from a subsequent surgical resection (26_PNF) sharing overlapping molecular signatures with the adjacent MPNST (27_MPNST). A hierarchically clustered heatmap of all genes is shown. Rows represent individual genes and their respective expression log2 normalized, z-score transformed expression values across each sample. Columns represent individual samples. Rows and columns were clustered by correlation coefficient with average linkage. Annotations below indicate the sample origin, whether from the initial biopsy or the subsequent tumor resection.
Figure 6. Neurofibromas contiguous with MPNST are transcriptionally distinct from discrete PNF/ANNUBPs not associated with malignant transformation.
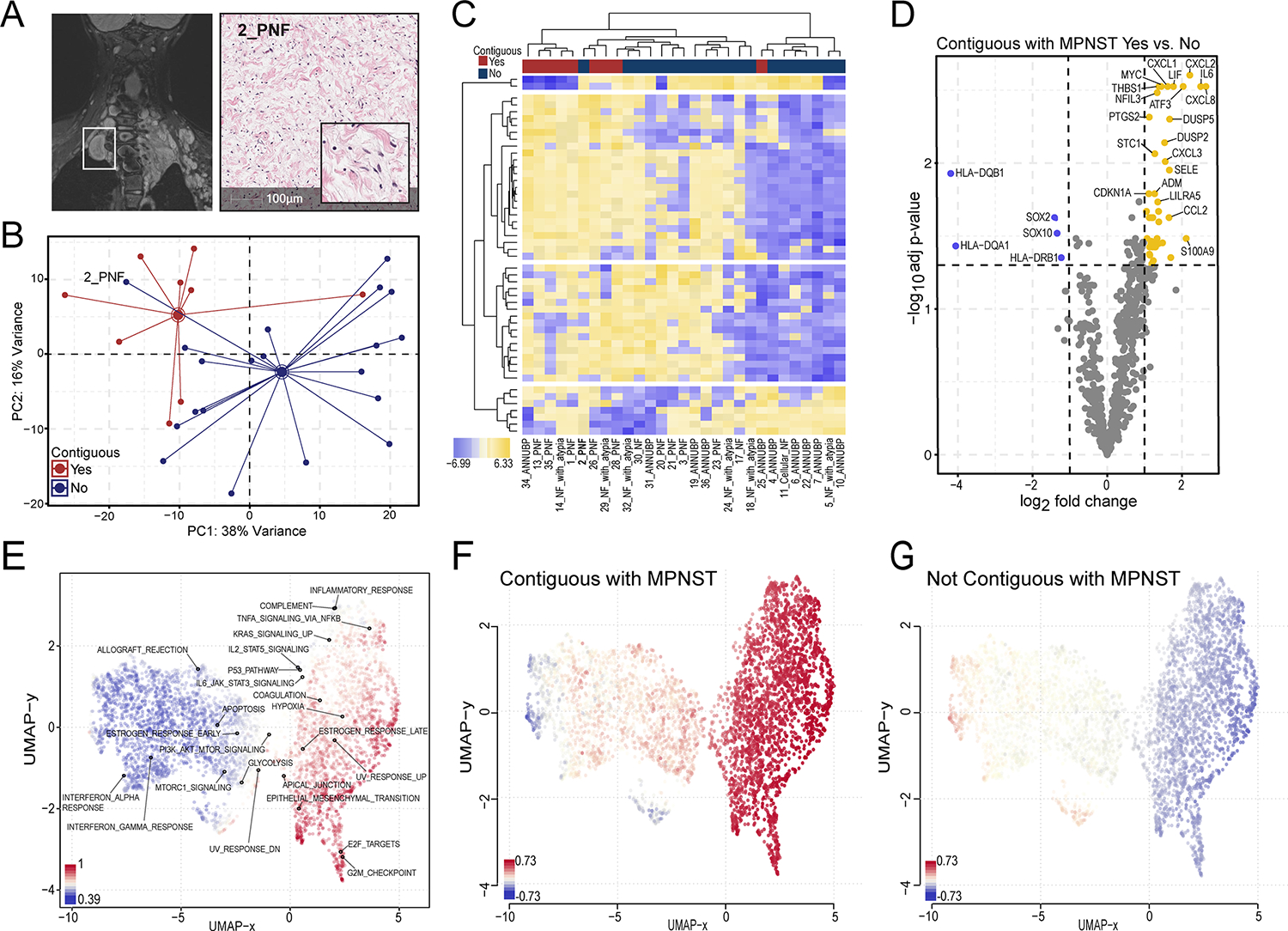
(A) MRI and corresponding H&E-stained section of 2_PNF resected from subject IU13_B, an 8-year-old female with innumerable neurofibromas involving the mediastinal and cervical-thoracic paraspinal region. The lesion, enclosed in the white rectangle, appeared as a distinct nodular lesion on MRI. (B) Principal component analysis demonstrating distinct clustering of neurofibromas contiguous with MPNST (red) from discrete lesions not contiguous with MPNST (blue) across PC1 and PC2. (C) Heatmap depicting the expression pattern of the top 50 variable genes sorted by 2-way, unsupervised hierarchical clustering. Rows represent individual genes, and their respective log2 transformed, z-score normalized expression values across each sample in the data, with yellow corresponding to increased expression and blue to decreased expression according to the figure legend. Columns represent individual samples. Annotation tracks above indicate contiguity with MPNST. Sample IDs appear below. (D) Volcano plot showing the top differentially expressed genes in neurofibromas that were and were not contiguous with MPNST. The x-axis represents the log2 fold change in gene expression, whereas the y-axis represents the −log10 Benjamini Hochberg adjusted p-value. An adjusted p-value of 0.05 was set as the false discovery threshold denoted by the dotted line. Differentially expressed genes with log2 fold changes ≥1 are colored yellow, whereas those with log2 fold changes ≤ −1 are colored blue. (E) UMAP clustering of gene sets colored by standard-deviation (sd.X) using a covariance distance metric with superimposed annotations corresponding to Hallmark collection gene sets. UMAP clustering of gensets colored by relative expression comparing lesions contiguous with MPNST (F) versus neurofibromas not contiguous or associated with malignant transformation (G). Blue represents downregulation and red represents upregulation of gene sets as shown in the figure legend.
Gene Set Signature Maps
Uniform manifold approximation and projection (UMAP) plots were generated by Omics Playground v2.7.18 (21). Clustering of features was computed using a normalized log-expression matrix (logCPM), with covariance as the distance metric. UMAP clustered gene sets were colored by standard-deviation (sd.X). Labels reflect Hallmark gene set enrichment.
Single cell RNAsequencing
scRNA-seq data from four human MPNST samples with NF1 were downloaded from GEO (GSE179043) (22). Data preprocessing and visualization was performed using Scanpy (RRID:SCR_018139) in Python. Low-quality cells with less than 200 genes, genes expressed in less than 3 cells, and outlier cells with less than the 2%tile or greater than 98%tile of the number of genes detected, or greater than 20% mitochondrial counts were discarded. Filtered data from the four samples were concatenated. The top 4,000 highly variable genes were selected with the Seurat v3 method. An scVI model (23) was trained on the raw counts with sample and quality metrics (percentage of mitochondrial counts and total counts) as covariates. The latent representation and normalized expression values from the scVI model were extracted for downstream analysis to compute the neighborhood graph, UMAP coordinates, and Leiden clusters at a resolution of 0.5. After identifying cellular communities with similar transcriptional profiles, the decoupler package (24) was used to perform over-representation analysis (ORA) of cell type markers against canonical human cell type markers obtained from the PanglaoDB database (25). ORA estimates were converted to activation scores and ranked to identify the top five predicted cell types for each cluster. Final annotations were assigned manually following assessment of the enrichment results. To further distinguish malignant from non-malignant cells, the infercnvpy package (https://github.com/icbi-lab/infercnvpy) was used to detect copy number variations (CNVs) from single cell transcriptomic data. The average gene expression over genomic regions was compared between immune cell types (as references cells) and query cells, using a window size of 25. CNV scores were computed for each cell based on the number and magnitude of CNV events detected and projected onto the UMAP coordinates for visual confirmation.
Immunohistochemistry
5 μm thick tissue sections were deparaffinized, hydrated and transferred to 0.1M EDTA (pH 8.0) for antigen retrieval in a pressure cooker. Sections were then treated with 3% hydrogen peroxide for 10 min, rinsed and blocked with 5% normal goat serum in TBST (TBS buffer with 0.1% Tween-20), and incubated overnight at 4 °C or 45 minutes at room temp (CD31) with primary antibodies diluted in blocking buffer: CD31 (3528S, 1:100, Cell Signaling), Survivin (MA5–15077, 1:200, Invitrogen), and CENPF (PA5–84637, 1:200, Invitrogen) and H3K27me3 (9733S, 1:100, Cell Signaling). Sections were then incubated with secondary antibodies for 1 hour at room temperature (goat anti-rabbit, ab205718, 1:1000, Abcam or goat anti-mouse, ab205719 1:1000, Abcam). VECTASTAIN DAB was applied for 10 minutes and the reaction was terminated by rinsing in distilled water. Counterstaining was performed with modified Mayer’s hematoxylin (Vector), and the sections were dehydrated, cleared and coverslipped. Slide images were acquired on an Aperio ScanScope CS at 20x magnification. CD31+ staining blood vessels were counted manually on five randomly selected high-power fields per slide. Quantitative immunohistochemical analysis for BIRC5 and CENPF was conducted using the Cytonuclear IHC module of HALO Image Analysis software (version 2.0.5, Indica Labs). Cytonuclear analysis settings were optimized for each stain and the intensity of nuclear staining was scored as negative (0, blue), weakly positive (1+, yellow), moderately positive (2+, orange), or strongly positive (3+, red). All positive cells (1–3+) were used for statistical analysis in GraphPad Prism software as described below.
Culture of Human MPNST cell lines
Human MPNST cell lines JH-2–002 (26) and JH-2–103 (27) were obtained from the Johns Hopkins NF1 Biospecimen Repository. ST-8814 and S462 cells (28) were obtained from Dr. Andrew Tee (Cardiff University). Immortalized human normal (hTERT ipn02.3 2λ) and NF1 deficient neurofibroma Schwann cells (hTERT NF1 ipNF95.6) (29) were obtained from Dr. Peggy Wallace. Cells were authenticated by STR analysis. Cells were cultured in either DMEM (NF90.8, ST88–14, ipn02.3 2 2λ and ipNF95.6) or DMEM/F12 (JH-2–002 and JH-2–103) media supplemented with 10% FBS (Harvest Midsci), 1% glutamine (Gibco), 1% penicillin/streptomycin (Lonza), and prophylactic 5 μg/mL Plasmocin (Invivogen). Trypsin-EDTA 0.05% (Gibco) was used to dissociate cells for passaging upon reaching confluence. Cultures were tested for mycoplasma and confirmed to be negative prior to experimentation.
Isolation and culture of primary murine Nf1−/−Arf −/− and Nf1−/−Cdkn2a−/− DNSCs
Embryonic day 13.5 (E13.5) mice were extracted by cesarean section from freshly euthanized pregnant females. Dorsal root ganglia (DRGs) and spinal nerve roots were isolated under a stereomicroscope and treated with 20 mg/mL collagenase. After centrifugation and washing in PBS (#20012027, Gibco), the cell pellet was resuspended in DNSC complete media consisting of serum-free DMEM/F12 media (Gibco) supplemented with 0.2% heparin (StemCell), 30% glucose (Gibco), 7.5% sodium bicarbonate (Gibco), 1M HEPES (Gibco), 1% N2 supplement (Gibco), 1% glutamine (Gibco), 1% sodium pyruvate (Gibco), 1% penicillin/streptomycin (Lonza), 20 ng/mL epidermal growth factor (Sigma-Aldrich), 40 ng/mL basic fibroblast growth factor (PeproTech), 2% B27 (without vitamin A) (Gibco), 40 μg/mL amphotericin B/Fungizone (Gibco), and prophylactic 5 μg/mL Plasmocin (Invivogen). Cells were cultured in low adhesion plates for 7–10 days to form neurospheres, which were then transferred to fibronectin-coated plates for subsequent passaging.
Genotyping of Nf1flox/flox; Arf flox/flox and Nf1flox/flox;Cdkn2aflox/flox DNSCs was confirmed by PCR using the following primers:
Nf1
P1: 5’-AATGTGAAATTGGTGTCGA GTAAGGTAACCAC-3’,
P2: 5’-TTAAGAGCATCTGCTGCTCTTAGAGGGAA-3’,
P3: 5’-TCAGACTGATTGTTGTACCTGAT GGTTGTACC-3’
Arf:
P1: Forward (19243): 5’-ACT GCA GCC AGA CCA CTA GG-3’
Reverse (19244): 5’-AGC TCG GAG ATT GAG AAA GC-3’
Cdkn2a:
P1: Forward (INK20): 5’-GTTTCCATTGCGAGGCTGCTCCGTAAGC-3’
Reverse (INK21): 5’-CTTTAGGGCGTTCCTTTCCCACTTCTGC-3’
P2: Forward (INK10): 5’-CCAAGTGTGCAAACCCAGGCTCC-3’
Reverse (INK11): 5’-TTGTTGGCCCAGGATGCCGACATC-3’
Nf1flox/flox; Arf flox/flox and Nf1flox/flox;Cdkn2aflox/flox DNSCs were incubated with Cre recombinase expressing adenovirus (Ad5CMVCre-GFP High Titer, University of Iowa Viral Vector Core) at 1:1000 dilution for 24 hours. The cells were then replenished with fresh DNSC complete media. Cre-mediated recombination of the floxed Nf1, Arf, and Cdkn2a alleles in the resulting Nf1−/−Arf −/− and Nf1−/−Cdkn2a−/− DNSCs was confirmed by PCR and western blot. Dissociation of DNSCs for passaging was performed using 1X TrypLE express enzyme (Thermo Fisher Scientific). Prior to experimentation, cultures were tested for mycoplasma and confirmed to be negative.
Drug Treatment
Human MPNST cell lines and murine DNSCs were plated at a density of 5,000 cells per well in 96-well plates. Cells were allowed to adhere overnight and then treated with increasing concentrations of YM155 (MedChemExpress, HY-10194) or LQZ-7I (MedChemExpress, HY-136538). Cell viability was assessed using the CellTiter-Glo® Assay (Promega) 48–72 hours post-treatment. Endpoint luminescence was measured using a SynergyH4 plate reader with filters and settings as follows: 528/20 and hole filter sets, top read, 4 mm read height, gain 135, 0.5 second integration time.
siRNA transfection
Human MPNST cell lines were reverse transfected with 10 nM siRNA against CENPF (Santa Cruz Biotechnology, sc-37563), BIRC5 (Santa Cruz Biotechnology, sc-29499) or scrambled control (Santa Cruz Biotechnolgy, sc-37007) using Lipofectamine 3000 transfection reagent (Invitrogen) according to the manufacturer’s instructions. Cells were plated in the transfection complex at a density of 5,000 cells per well in 96-well plates. After 24 hours of transfection, cell lysates were collected to confirm protein knockdown by Western blot. Viability was assessed via CellTiter-Glo® Assay 48–72 hours post-transfection as described above. Luminescent values were normalized to the average of the siControl for each cell line.
Western Blot Analysis
Lysis buffer was prepared using cOmplete Mini Protease Inhibitor Cocktail (#11836153001, Roche), PhosSTOP Phosphotase Inhibitor Cocktail (#4906837001, Roche) and xTractor buffer (#635671, Takara Bio). Cell lysates were collected, and protein concentrations were determined using Pierce BCA Protein Assay Kit (#23227, Thermo Fisher Scientific). Isolated proteins were fractionated using NuPAGE 4–12% Bis-Tris Gels (Invitrogen Cat#NP0322BOX) and electro-transferred to PVDF membranes. Immunoblots were carried out using antibodies specific to CENPF (ab223847, 1:1000 dilution, Abcam), Survivin (MA5–15077, 1:1000, Invitrogen) and Vinculin (ab219649, 1:1000 dilution, Abcam). After incubation with primary antibody, appropriate HRP conjugated secondary antibody was used (anti-rabbit, #NA934V, 1:2000 dilution, GE Healthcare).
Statistical analysis
Statistical analyses were performed in R or using GraphPad Prism 9.5.1 software (GraphPad, La Jolla, CA). One way analysis of variance (ANOVA) with post-hoc correction for multiple comparisons or unpaired t-tests were used to evaluate statistically significant differences between groups as detailed in the accompanying figure legends. Adjusted p-values ≤ 0.05 were considered statistically significant. For dose response curves, the IC50s were calculated for each line using nonlinear regression in GraphPad Prism.
Data availability
Raw RCC files, raw counts and sample metadata pertaining to spatial gene expression profiling of human PNST specimens described here are available from Gene Expression Omnibus under accession GSE239561. Bulk RNAseq for comparing TCGA MPNST vs GTEx normal nerve are in Supplemental Table 11, Supplementary Table 12 and publicly available through the UCSC RNA-seq Toil Recompute Compendium (30) hosted through the UCSC Xena Browser. Bulk RNAseq from Nf1flox/flox;DhhCre and Ink4a/Arf +/−;Nf1flox/flox;DhhCre murine PNST generated by Chaney et al. (19) are available from GEO under accession number GSE148249. Bulk RNAseq data from Nf1flox/flox;PostnCre and Nf1flox/flox;Arf flox/flox;PostnCre + mice are available from GEO under accession numbers GSE213786 and GSE232707.
RESULTS
PNF, ANNUBP, and MPNST exhibit distinct global gene expression programs.
A cohort of 35 PNST samples was assembled by retrospective review of the Indiana University Pathology archives. 32 samples were obtained from subjects who met clinical diagnostic criteria for NF1, two additional samples were from a subject where the diagnosis of NF1 was strongly suspected pending genetic confirmation, and one case consisted of a sporadic neurofibroma in which the subject did not have NF1 (Figure 1A). Some samples contained multiple PNST subtypes within a single lesion, and in select cases, multiple specimens were collected longitudinally from the same individual during clinically indicated procedures (Supplemental Table 13, Figure 1B). NF with atypia were distinguished from ANNUBP based on published consensus diagnostic criteria (10). Two samples with a histopathological diagnosis of neurofibroma (NF) and one with cellular NF were included in the hierarchical clustering and pathway analysis but excluded from statistical comparisons based on tumor histology due to their underrepresentation in the data set. 4 out of 6 MPNSTs were negative for H3K27Me3 staining by immunohistochemistry (Supplemental Figure 2). Tissues were microdissected from selected regions of interest conforming to consensus diagnostic criteria (Figure 1C) and subjected to RNA expression profiling across a high-content panel of 770 genes involved in tumor progression, microenvironment, and immune response (PanCancer IO360™ Panel, NanoString Technologies, Inc.), thus allowing for spatially restricted, multiplexed quantification of gene expression from tumors across the neurofibroma to MPNST continuum (Figure 1D).
Figure 1. Spatial gene expression profiling of NF1-associated PNST.
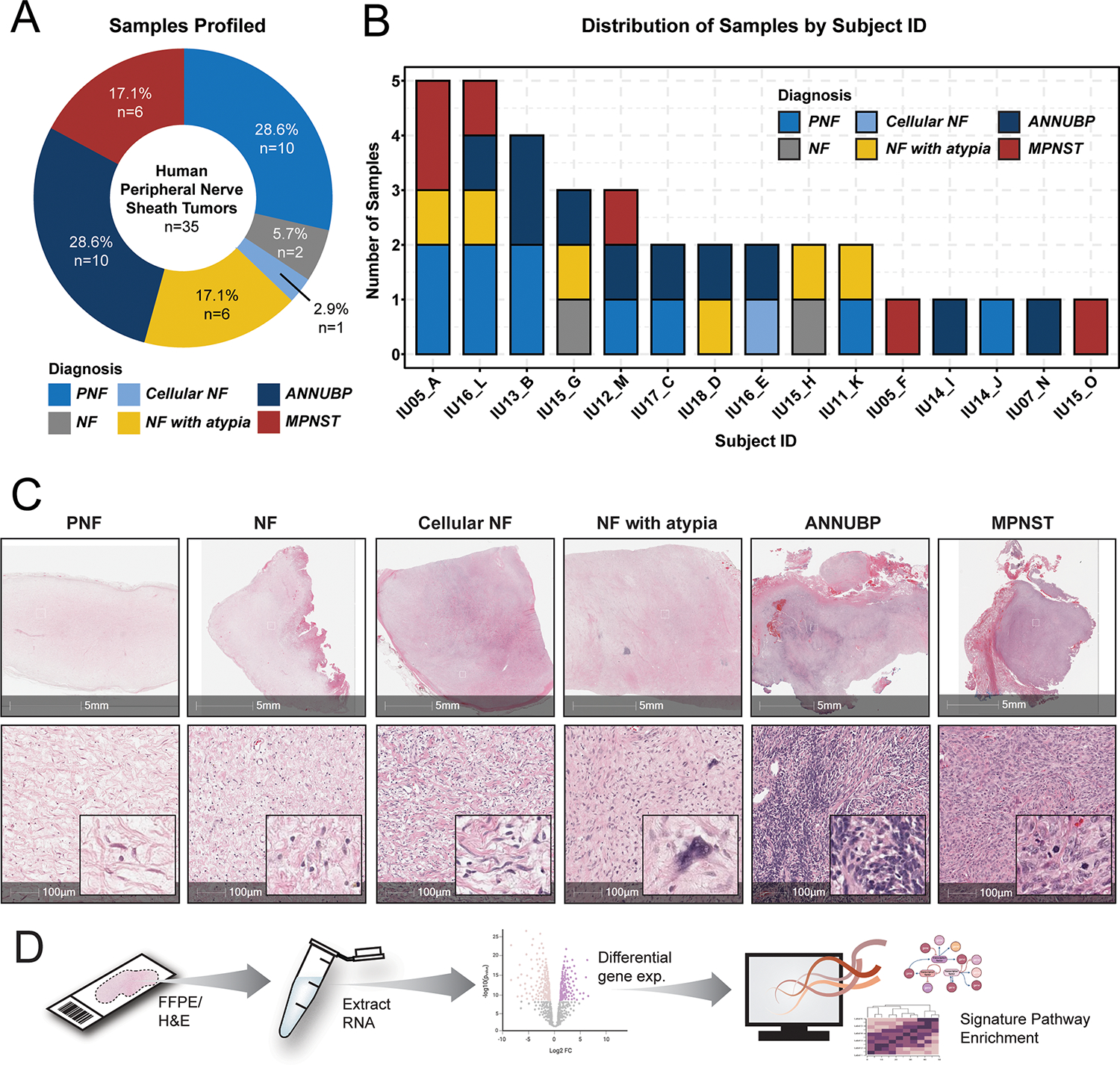
(A) Donut plot depicting the distribution of samples by tissue diagnosis. (B) Stacked barplot illustrating the distribution of tumor subtypes by subject ID. (C) Representative photomicrographs of H&E-stained sections of each tumor subtype represented in the analysis including neurofibroma (NF), plexiform neurofibroma (PNF), cellular neurofibroma (cellular NF), neurofibroma with atypia (NF with atypia), atypical neurofibromatous neoplasm of uncertain biological potential (ANNUBP), and malignant peripheral nerve sheath tumor (MPNST). 5 mm (top panel) and 100 μm scale bars (bottom panel) denote the magnification, with insets at high power. (D) Schematic depicting experimental workflow and analysis. Tissue was microdissected from annotated regions and RNA was extracted for gene expression profiling using the using the Nanostring nCounter platform (IO360 panel).
Copy number loss of the 9p21 chromosome cytoband encoding the CDKN2A tumor suppressor locus has been identified in the majority of ANF/ANNUBP and MPNST (11–15). Seeking confirmation of these findings, we compared CDKN2A mRNA expression across the various PNST subtypes in our dataset. CDKN2A exhibited variable expression in PNF, NF with atypia, and ANNUBP. Nonetheless, we observed reduced CDKN2A expression both in ANNUBP and MPNST relative to PNF (Figure 2A). These findings are concordant with previously published data suggesting that increased Cdkn2a expression in PNF impedes the genesis of ANNUBP and malignant transformation by inducing senescence-mediated growth arrest in Schwann cells with biallelic Nf1 loss (18).
PNF, ANNUBP, and MPNST were largely distinguishable by principal component analysis (PCA) and unsupervised hierarchical clustering of the top variable features within the dataset (Figure 2B and Supplemental Figure 3), although considerable transcriptional heterogeneity was observed. Notably, NF with atypia were highly heterogeneous and did not cluster discretely by unsupervised methods. Non-malignant lesions from the same subject clustered more closely irrespective of tissue diagnosis, whereas MPNSTs segregated distinctly. Taken together, these findings indicate that PNF, ANNUBP, and MPNST broadly possess distinctive global gene expression signatures, but substantial transcriptional heterogeneity exists even within tumors of the same histopathological subtype.
Pathway analysis identifies three molecular NF1-PNST subclusters traversing conventional histopathological classifiers.
To further dissect the molecular heterogeneity across the PNST continuum, we implemented an unsupervised approach to computationally stratify samples agnostic of tumor histology based on pathway enrichment analysis of gene signatures related to cancer signal transduction, immune response, and the tumor microenvironment. Our analysis revealed three distinct sub-clusters predominated by PNF (Cluster 1), ANNUBP (Cluster 2), and MPNST (Cluster 3), each characterized by unique transcriptional programs (Figure 2C). As above, NF with atypia exhibited heterogeneous dispersion across all subclusters. CDKN2A mRNA expression was highest in Cluster 1 (PNF predominant) and significantly reduced in both Cluster 2 (ANNUBP predominant) and Cluster 3 (MPNST predominant) (Figure 2D). Cluster 2 exhibited robust enrichment of pathways associated with immune system processes, antigen presentation, cytotoxicity, interferon signaling, hypoxia, and epigenetic regulation (Figure 2E), while Cluster 3 showed enhanced signatures of MAPK, PI3K/AKT, Notch, WNT, TGF-beta, JAK/STAT, Hedgehog and NF kappa B pathway transcriptional output as well as enrichment of gene signatures associated with autophagy, angiogenesis, metabolic stress, matrix remodeling and metastasis, DNA damage repair, and cellular proliferation (Figure 2F and G). Overall, these findings suggest a coordinated deregulation of both microenvironmental and tumor cell autonomous processes as neurofibromas progress across the PNST continuum.
ANNUBP are typified by signatures of enhanced immune surveillance and T cell infiltration.
To further examine the transcriptional alterations in signatures of antigen presentation and immune response across the neurofibroma to MPNST continuum, we performed differential gene expression analysis, which revealed genes involved in antigen presentation, including HLA-DPA1 and HLA-DPB1, to be among the top upregulated genes in Cluster 2 lesions (Figure 3A–C). Conversely, multiple HLA associated genes including HLA-DQA1 and HLA-DQB1 were found to be markedly downregulated in MPNST and Cluster 3 lesions (Figure 3D–F). Concordantly, we observed that ANNUBP are heavily infiltrated with CD4+FOXP3− and CD8+FOXP3− T cells, which were significantly diminished in MPNST and replaced by an increase in regulatory, FOXP3+ T cells (Figure 3G and H). Signatures of cytotoxicity (Figure 3I) in MPNST and Cluster 3 lesions were also reduced and accompanied by increased expression of genes associated with immune exhaustion including CTLA4 (Figure 3J).
Figure 3. ANNUBP and Cluster 2 lesions are characterized by enhanced signatures of antigen presentation, immune surveillance and T cell infiltration.
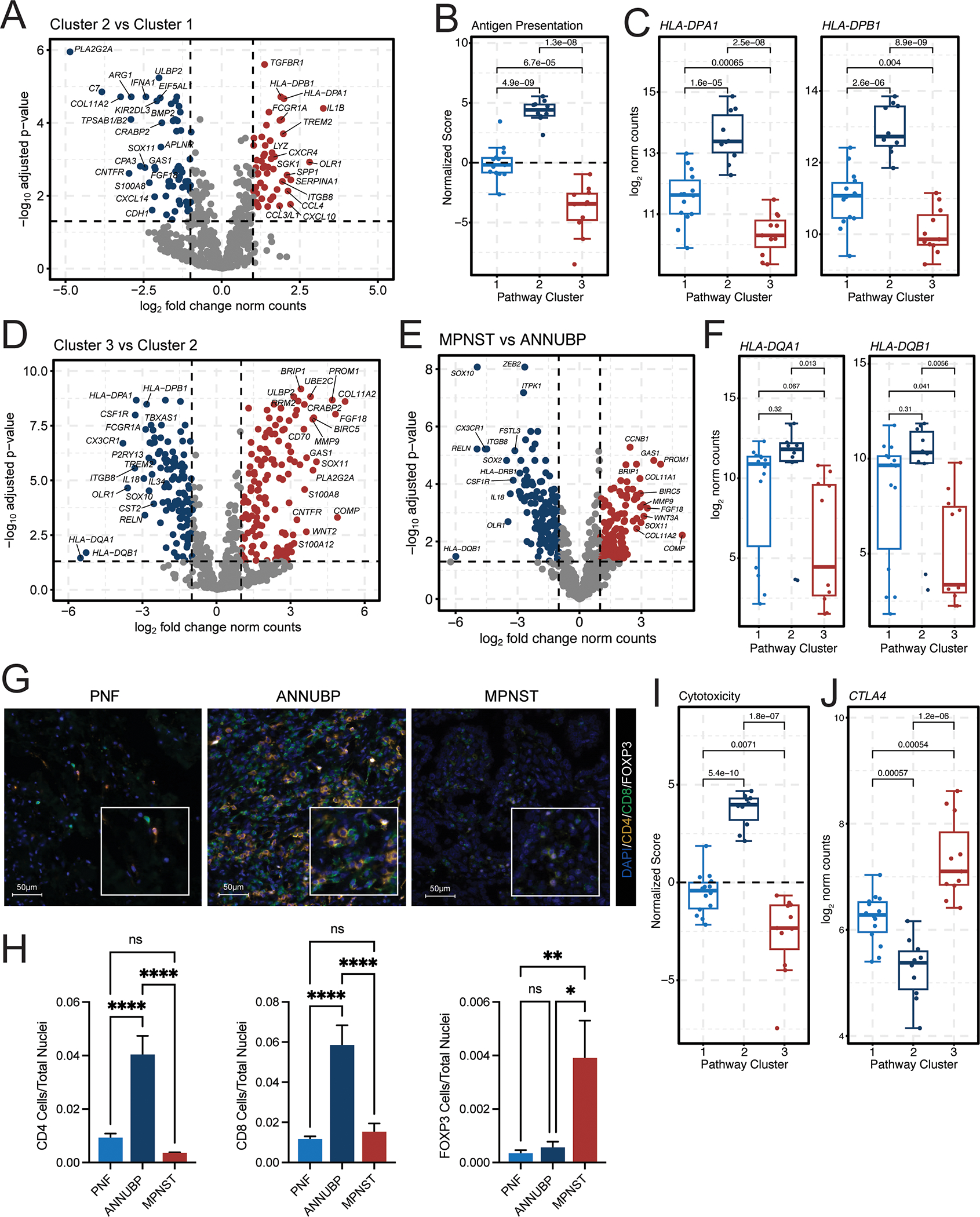
(A) Volcano plots illustrating the top differentially expressed genes in Cluster 2 (n=10) versus Cluster 1 (n=14) lesions. The x-axis represents the log2 fold change in gene expression, whereas the y-axis represents the −log10 Benjamini Hochberg adjusted p-value. An adjusted p-value of 0.05 was set as the false discovery threshold as denoted by the dotted line. Genes with log2 fold changes ≥1 are colored red, whereas those with log2 fold changes ≤ −1 are colored blue. (B) Box and whisker plot of normalized antigen presentation signature scores in Cluster 1 (n=14), Cluster 2 (n=10), and Cluster 3 (n=11) tumors. Dots represent individual samples. Whiskers extend from the minima to maxima that are no further than 1.5x the inter-quartile range spanning the first to third quartiles. The center line represents the median. The box spans the 25th to 75th percentiles. Data beyond the whiskers are outliers and are plotted as individual points. P-values represent unpaired, two-tailed t-tests between groups as shown. (C) Box and whisker plots depicting HLA-DPA1 and HLA-DPB1 mRNA expression (normalized counts) stratified by molecular cluster. P-values represent unpaired, two-tailed t-tests between groups. (D) Volcano plot illustrating the top differentially expressed genes in Cluster 3 (n=11) vs Cluster 2 (n=10) lesions. (E) Volcano plot showing top differentially expressed genes in MPNST (n=6) vs ANNUBP (n=10). (F) Box and whisker plots depicting HLA-DQA1 and HLA-DQB1 mRNA expression (normalized counts) stratified by molecular cluster. (G) Representative photomicrographs of T cell subsets identified by immunofluorescence staining of CD4 (orange), CD8 (green) and FOXP3 (white) in PNF, ANNUBP and MPNST tissue sections. DAPI (blue) is shown as a nuclear counterstain. Magnification denoted by 100 μm scale bars with inset high-power magnification as shown. (H) Barplots reflecting quantitative analysis of T cell subsets normalized to the total number of nuclei per high power field (HPF). Error bars reflect standard error of the mean (SEM). PNF (n=6, ROI=30), ANNUBP (n=4, ROI=20) and MPNST (n=5, ROI=25) were analyzed by one-way ANOVA using Tukey’s multiple comparisons test. (I) Box and whisker plot of normalized cytotoxicity signature scores in Cluster 1 (n=14), Cluster 2 (n=10), and Cluster 3 (n=11) tumors. P-values represent unpaired, two-tailed t-tests between groups as shown. (J) Box and whisker plot depicting CTLA4 mRNA expression (normalized counts) stratified by molecular cluster. P-values represent unpaired, two-tailed t-tests between groups.
Deregulation of genes involved angiogenesis and mitotic fidelity typifies malignant transformation of PNF and ANF/ANNUBP precursors.
Angiogenesis has been implicated in the malignant transformation of PNF in children with NF1 (31). In comparing differentially expressed genes in Cluster 3 tumors and MPNSTs relative to Cluster 1 lesions and PNFs respectively (Supplemental Figure 4A and B), we identified genes associated with angiogenesis including MMP9 (32), FGF18, and VEGFA (33) to be among the top-upregulated genes associated with malignant transformation (Supplemental Figure 4C). Immunohistochemical staining for CD31 positive blood vessels across PNF, ANNUBP, and MPNST in our dataset was consistent with these findings, demonstrating a significant increase in tumor vascularity in MPNST (Supplemental Figure 4D and E). Reanalysis of integrated, publicly available scRNA datasets (22) demonstrated that proangiogenic marker genes are highly expressed by a number of cell lineages in MPNST including both tumor cells, which highly express VEGFA, SERPINH1, HEY1, and FGFR1 as well as constituents of the tumor microenvironment including macrophages and dendritic cells, expressing MMP9 and fibroblasts, which abundantly express FGF18 (Supplemental Figure 4F and G, Supplemental Figure 5). Collectively, these data provide additional insight regarding the angiogenic signatures characterizing MPNSTs identified in our dataset.
Aberrant cellular proliferation and pro-survival signaling are hallmarks of malignant transformation (34). Genes associated with cellular proliferation and survival/apoptosis were also highly dysregulated in MPNST and Cluster 3 tumors (Supplemental Figure 6A–D). CENPF and BIRC5 were among the top differentially expressed genes comprising these signatures (Supplemental Figure 6E, Figure 4A and B). CENPF encodes centromere protein-F, a kinetochore-associated protein that safeguards chromosome segregation during mitosis (35). Immunohistochemical staining confirmed elevated CENPF protein expression in ANNUBP and a marked increase in CENPF in MPNST as compared to both PNF and ANNUBP (Figure 4C and D). BIRC5 encodes the Baculoviral Inhibitor of Apoptosis (IAP) Repeat Containing 5 protein, also known as Survivin, which plays an essential role in preventing apoptosis via caspase inactivation. Survivin also participates in chromosomal alignment and segregation during mitosis as part of the chromosomal alignment and segregation during mitosis as part of the chromosome passenger complex (CPC) (36). IHC analysis independently confirmed an increase in the percentage of BIRC5 positive cells in MPNST relative to PNF (Figure 4E and F). We further validated these findings by comparing gene expression in normal nerve and MPNST from the Genotype-Tissue Expression (GTEx) and The Cancer Genome Atlas (TCGA) databases, respectively, which revealed BIRC5 and CENPF to be among the most highly upregulated genes in MPNST (Supplemental Figure 7).
Figure 4. Genetic and pharmacologic disruption of BIRC5 and CENPF abrogates viability of human MPNST cell lines and primary murine Schwann cell precursors.
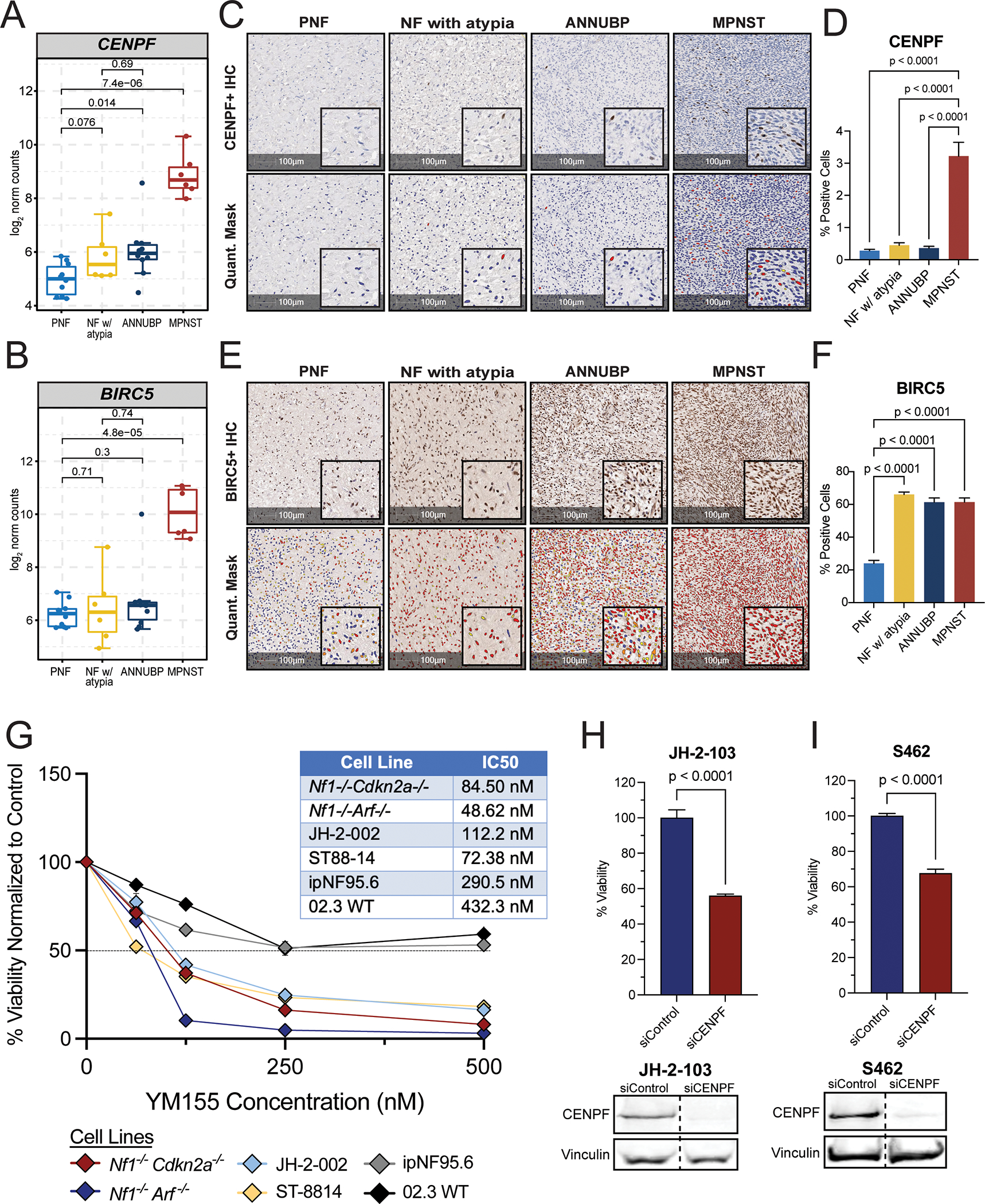
(A) Box and whisker plot depicting CENPF mRNA expression (log2 normalized counts) by tumor subtype: PNF (n=10), NF with atypia (n=6), ANNUBP (n=10), and MPNST (n=6). Dots represent individual samples. Whiskers extend from the minima to maxima that are no further than 1.5x the inter-quartile range spanning the first to third quartiles. The center line represents the median. The box spans that 25th to 75th percentiles. Data beyond the whiskers are outliers and are plotted as individual points. P-values represent unpaired t-tests between groups. (B) Box and whisker plot of BIRC5 mRNA expression (log2 normalized counts) by tumor subtype: PNF (n=10), NF with atypia (n=6), ANNUBP (n=10), and MPNST (n=6). P-values represent unpaired, two-tailed t-tests between groups. (C) Representative photomicrographs of tumor sections across the PNST continuum immunohistochemically stained for CENPF. Magnification is denoted by 100 μm scale bars with inset high-power magnification as shown. The HALO cytonuclear mask used to quantify CENPF staining is shown in the bottom panel. Blue cells indicate negative staining for CENPF, yellow indicate weak positive (1+), orange indicate moderate positive (2+) and red indicate strong positive (3+) CENPF staining. (D) Barplot depicting CENPF positive cells as a percentage of total cells per field. Error bars reflect standard error of the mean (SEM). PNF (n=5, ROI=42), NF with atypia (n=5, ROI=35), ANNUBP (n=6, ROI=49) and MPNST (n=4, ROI=49) were analyzed by one-way ANOVA using Tukey’s multiple comparisons test. (E) Representative photomicrographs of tumor sections across the PNST continuum immunohistochemically stained for BIRC5. Magnification denoted by 100 μm scale bars with inset high-power magnification as shown. The HALO cytonuclear mask used to quantify BIRC5 staining is shown in the bottom panel. Blue cells indicate negative staining for BIRC5, yellow indicate weak positive (1+), orange indicate moderate positive (2+) and red indicate strong positive (3+) BIRC5 staining. (F) Barplot depicting BIRC5+ cells as a percentage of total cells per field. Error bars reflect standard error of the mean (SEM). PNF (n=9, ROI=144), NF with atypia (n=4, ROI=38), ANNUBP (n=10, ROI=95) and MPNST (n=5, ROI=101) were analyzed by one-way ANOVA using Tukey’s multiple comparisons test. P-values represent unpaired, two-tailed t-tests between groups. (G) Mean viability of primary murine Schwann cell precursors (Nf1−/−;Cdkn2a−/− and Nf1−/−;Arf−/−), human MPNST cell lines (JH-2–002 and ST8814), and human wild-type (ipn02.3 2λ) and NF1 mutant Schwann cell lines (ipNF95.6) as a function of increasing concentrations of YM115 (Survivin inhibitor). Error bars represent SEM of n=6 technical replicates per condition. The experiment was repeated twice per cell line with similar results. The IC50 of each line was determined by nonlinear regression in GraphPad Prism and reported in the adjacent table. (H and I) Barplots depicting percent viability of human MPNST cells lines (JH-2–103 and S462) following siRNA-mediated depletion of CENPF (n=8 replicates per line) vs control (n=8 replicates per line). Error bars represent the SEM. P-values reflect unpaired, two-tailed t-tests between groups. The experiment was repeated three times and the graph reflects pooled results from all three experiments. CENPF protein expression was detected by western blot in human MPNST cell lines (JH-2–103 and S462) following transfection with siRNA against CENPF vs scrambled, non-targeting control. Vinculin is shown as the loading control.
Murine ANF/ANNUBP and MPNST driven by Cdkn2a/Arf loss reflect core molecular features of their human counterparts.
ANF/ANNUBP development and malignant transformation occurs only in a small subset of individuals with NF1. Due to the limited availability of clinical samples, preclinical disease models that recapitulate key genetic driver events in specific cells of origin have played a critical role in understanding the biology and evolution of tumors along the neurofibroma to MPNST continuum. Conditional genetic inactivation of Nf1 and Arf in Schwann cell precursors driven by Periostin Cre (PostnCre) gives rise to tumors that recapitulate human ANNUBP and progress to MPNST with high penetrance, dependent on gene dosage (18). Similar results were observed in mice with conditional biallelic inactivation of Nf1 driven by Desert hedgehog Cre (DhhCre) and germline loss of Cdkn2a (Ink4a/Arf) (19).
We reanalyzed publicly available RNAseq data from tumors arising in Ink4a/Arf +/−;Nf1flox/flox;DhhCre mice, as well as RNAseq data generated from Nf1flox/flox;Arf flox/flox;PostnCre mice developed in our laboratory. We evaluated the coherence of gene signature enrichments within PNSTs derived from these genetically engineered mouse models (GEMMs) (18,19) and our patient derived neurofibromas and MPNSTs described above. T-distributed stochastic neighbor embedding (t-SNE) (Supplemental Figure 8A) and unsupervised hierarchical clustering of top variable features (Supplemental Figure 8B) demonstrated distinct transcriptional profiles of plexiform neurofibromas, paraspinal tumors (PST) reminiscent of human ANF, and GEM-PNST akin to human MPNST, arising in Ink4a/Arf +/−;Nf1flox/flox;DhhCre mice, as reported by Chaney and colleagues (19). Enriched genes were queried against publicly available databases including Gene Ontology, KEGG, and Reactome. DNA repair emerged as the top enrichment in Cluster S4 and was strongly upregulated in GEM-PNST and to a lesser extent in PST. This finding is consistent with results from clinical samples in our dataset, which also identified enrichment of DNA repair signatures in MPNST compared to PNF and ANF/ANNUBP (Supplemental Figure 9A and B). Gene Set Enrichment Analysis (GSEA) also demonstrated enhancement of cell cycle/proliferation and apoptosis-related signatures in GEM-PNST (Supplemental Figure 8C). We observed robust expression of Birc5 in GEM-PNST/MPNST arising in both models (Supplemental Figure 8D and E), consistent with our analysis of patient derived samples. Cenpf expression demonstrated upregulation in MPNST compared to both PNF (p=0.0647) and ANNUBP (p=0.0783) in the PostnCre model, though not achieving significance. In the DhhCre model,Cenpf expression was significantly increased in GEM-PNST compared to PST (p=0.0035), but was not significant compared to NF (p=0.1126) (Supplemental Figure 8F and G).
Disruption of BIRC5 and CENPF impairs MPNST cell viability
Having observed concordant molecular features between human NF1-associated PNST samples and pre-clinical murine models, we aimed to investigate the biological significance of BIRC5 and CENPF as potential regulators of pro-survival and mitotic fidelity pathways in MPNST. We treated human MPNST cell lines (ST88–14, JH-2–002), primary murine MPNST precursors lacking Nf1 and either Arf (Nf1−/−Arf −/−) or Cdkn2a (Nf1−/−Cdkn2a−/−), human wild-type (ipn02.3 2λ) and NF1 mutant Schwann cells (ipNF95.6)with pharmacologic inhibitors of BIRC5 (Survivin), YM155 and LQZ-7I, and observed a significant reduction in cell viability with both drugs in human MPNST and murine MPNST precursor ecell lines, with minimal effect on the viability of human wild-type and NF1 mutant Schwann cells (Figure 4G, Supplemental Figure 10), suggesting that BIRC5 inhibitors preferentially target MPNST cells. Likewise, siRNA-mediated depletion of CENPF (Figure 4H and I) or BIRC5 (Supplemental Figure 10B–E) impaired viability of human MPNST cell lines. Together, these results demonstrate the utility of integrated, cross-species analysis to delineate putative targets with functional relevance to MPNST biology.
Identification of MPNST-like gene expression programs in precursor lesions associated with malignant transformation
NF1-associated PNST exhibit substantial intratumoral heterogeneity. Lesions at multiple stages of the neurofibroma to MPNST continuum can co-exist discretely within an individual or even within a single tumor itself. This heterogeneity poses unique challenges for diagnosis and treatment, as early identification of malignant transformation is crucial for preventing morbidity and mortality in persons with NF1. Needle biopsy specimens can be confounded by sampling bias, and furthermore, even neurofibromas that conform to uniform histopathological criteria can exhibit diverse molecular features as shown above.
To further illustrate this concept, subject IU12-M, a 17-year-old female with NF1, presented with a large, FDG-avid, right upper extremity mass, which was confirmed to be high-grade MPNST on biopsy (Figure 5A). Due to extensive involvement of the brachial plexus, complete surgical resection was not feasible. She received pre-operative radiation followed by a partial resection and debulking procedure. Her tumor remained stable for two years following initial radiation and four cycles of chemotherapy with ifosfamide and doxorubicin. The patient subsequently returned to clinic with severe pain and MRI revealed significant tumor growth. A forequarter amputation was performed with resection of the shoulder and chest wall tumor. Multi-regional tissue sampling revealed regions of tumor consistent with PNF, ANNUBP, and MPNST. The PNF component of the lesion (35_PNF) retained a molecular signature associated with predominantly benign lesions (Cluster 1). However, the ANNUBP portion of the mass (34_ANNUBP) stratified molecularly in Cluster 3 exhibiting enhanced expression of BIRC5, CENPF, MET, CDK6, and MKI67, which overlapped closely with the adjacent malignancy (35_MPNST) (Figure 5B).
Similarly, another individual, subject IU16-L, a 13-year-old female with NF1 presented to clinic at age 13 with a large, FDG-PET avid, right, cervical and occipital neck mass (Figure 5C). Molecular profiling of multiple tumor regions from an initial needle biopsy and a subsequent surgical resection revealed components of the tumor spanning the neurofibroma-to-MPNST continuum. Notably, several histopathologically benign-appearing regions of tumor obtained from the initial needle biopsy (29_NF with atypia) and subsequent surgical resection (26_PNF) were characterized by a Cluster 3-like molecular signature (Figure 2A) and were transcriptionally similar to the adjacent malignancy (27_MPNST).
We conducted a detailed review of the clinical records from subjects in our dataset to identify PNF and ANF/ANNUBP precursor lesions that were contiguous with adjacent MPNST. All microdissected samples were scrutinized by an expert clinical pathologist to confirm a lack of morphological evidence of cross-contamination of malignant cells, thus allowing for the transcriptomic analysis of histopathologically benign lesions contiguous with MPNST. Interestingly, we found that none of the tumors in Cluster 2 (ANNUBP predominant) were contiguous with MPNST. This may reflect a clinical bias to preemptively resect lesions histologically classified as ANNUBP due their recognized potential for malignant transformation. Conversely, four out of the five (80%) histopathologically benign lesions computationally stratified in Cluster 3 (Figure 2A, 34_ANNUBP, 29_NF with atypia, 26_PNF, 13_PNF), were associated with the development of MPNST. The fifth lesion, 2_PNF, was obtained from subject IU13-B, an 8-year-old female who had innumerable NF1-associated neurofibromas involving her mediastinum and cervical-thoracic paraspinal region. Over a four-year period, she underwent multiple biopsies and tumor resections, which identified lesions histopathologically consistent with PNF (2_PNF, 21_PNF) and ANNUBP (4_ANNUBP, 22_ANNUBP). 2_PNF corresponded to a DNL that was resected due to interval growth and clustered within the MPNST predominant Cluster 3 (Figure 6A). Extensive internal and external histopathological review revealed no evidence of malignant transformation within the lesion. Notably, after 5 years of follow up, the patient developed a large posterior chest mass consistent with high-grade MPNST and ultimately succumbed to widespread metastatic disease following aggressive surgery, chemotherapy and radiation.
Based on these findings, we conducted principal component analysis and differential gene expression profiling to compare precursor lesions contiguous with MPNST to discrete PNF and ANF/ANNUBPs not associated with malignant transformation. Lesions contiguous with MPNST were broadly distinguishable from isolated PNF, NF with atypia, and ANNUBPs by principal component analysis (PCA) and unsupervised hierarchical clustering (Figure 6B and C), indicating that they harbor distinct global gene expression programs. Notably, the sole outlier, 2_PNF, was a nodular neurofibroma resected from subject IU13-B that clustered with MPNST-contiguous lesions (Figure 6B and C, labeled) and this patient subsequently developed high grade MPNST as described above.
Further differential gene expression profiling (Figure 6D) and GSEA (Figure 6E–G) revealed upregulation of genes involved TNF-alpha signaling, inflammatory responses, hypoxia, epithelial mesenchymal transition and angiogenesis in neurofibromas contiguous with MPNST. Conversely, genes involved in progenitor cell differentiation, interferon-gamma signaling and antigen presentation (HLA-DQA1, HLA-DQB1, HLA-DRB1) were downregulated (GO Biological Processes). IL6, CXCL8, and HLA-DQB1 emerged as the top three most significantly differentially expressed genes in neurofibromas contiguous with MPNST. To determine the ability of IL6, CXCL8, and HLA-DQB1 (Supplemental Figure 11A–C) to distinguish neurofibromas contiguous with MPNST from discrete lesions not associated with malignant transformation, multiple logistic regression analysis was performed (Supplemental Figure 11D) that revealed an area under the receiver operating characteristic (ROC) curve of 0.9778 (SE=0.02246, 95% CI: 0.9338 to 1.000, p<0.0001), suggesting that IL6, CXCL8, and HLA-DQB1 exhibited excellent discriminative power in distinguishing neurofibromas contiguous with MPNST in our dataset (Tjur’s R Squared= 0.72, NPP= 95.00%, PPP= 88.89%, Cutoff= 0.5) (Supplemental Figure 11E). Together, our findings suggest that neurofibromas contiguous with MPNST exhibit distinct alterations in core oncogenic and immune surveillance programs. Further investigation is needed to evaluate the prospective validity of these signatures as molecular diagnostic tools to complement conventional histopathological diagnosis and guide risk-adapted care.
DISCUSSION
This study provides insights into the nascent transcriptional programs present in PNF and ANF/ANNUBP precursor lesions, both in isolation and concurrent with malignant transformation. While PNF, ANNUBP, and MPNST exhibit distinct global gene expression profiles, our findings demonstrate significant molecular heterogeneity even among tumors of a given histological subtype. NF with atypia, in particular, were transcriptionally diverse and did not segregate discretely by PCA or unsupervised clustering methods. These observations suggest a potential role for molecular profiling to more accurately ascribe biological phenotypes within the context of consensus histopathological classifications schemes (10).
Differential gene expression and pathway level enrichment analyses revealed deregulation of tumor cell autonomous factors and the tumor microenvironment at discrete stages of PNST development. ANNUBP exhibited gene signature alterations associated with antigen presentation and immune response, including dendritic cell function, immune cell chemotaxis and T cell activation. The ANNUBP predominant cluster, Cluster 2, also exhibited robust enrichment of pathways associated with hypoxia, and epigenetic regulation. Interestingly, previously published studies suggest that hypoxic tumor microenvironments drive epigenetic dysregulation and subsequent immunosuppression (37). Specifically, hypoxia impairs immune cell cytotoxicity and reduces interferon-gamma signaling, both of which are critical to the anti-tumor immune response and demonstrated enrichment in ANNUBP in this dataset (37,38). Conversely, compared to ANNUBPs, MPNSTs were characterized by further disruptions in apoptotic and angiogenic signature genes, as well as genes involved in mitotic processes and cell cycle integrity.
Unsupervised, computationally-based stratification identified alterations in core molecular signatures across the PNST continuum agnostic of tumor histology. These findings were cross validated in two independent genetically engineered mouse models that spontaneously recapitulate the progression of MPNST from existing PNF and ANF/ANNUBP lesions driven by combined inactivation of Nf1 and Cdkn2a/Arf in Schwann cell precursors. Targeting key regulators of survival and mitotic fidelity pathways, including CENPF and BIRC5 (Survivin), potently disrupted the viability of human MPNST cell lines and primary murine Nf1-Ink4a/Arf mutant Schwann cell precursors in vitro. These results are congruent with other published work implicating Survivin as a potential therapeutic target in MPNST (39) and demonstrate the utility of integrated, cross-species omic analysis to identify functionally relevant therapeutic candidates in NF1-associated PNST and other orphan cancers where patient samples are limited.
Early detection of malignant transformation in patients with NF1 is crucial to prevent adverse outcomes. Intratumoral heterogeneity and sampling bias in needle biopsy specimens pose unique diagnostic challenges. Moreover, even neurofibromas that conform to uniform histopathological criteria can exhibit diverse molecular phenotypes, growth kinetics, and clinical behavior. Neurofibromas contiguous with MPNST exhibited enrichment of genes involved in epithelial mesenchymal transition, angiogenesis, proliferation, and hypoxic responses. Conversely, genes involved in antigen presentation were downregulated, suggesting a potential role of impaired anti-tumor immune responses in driving PNST evolution and transformation, which warrants further investigation. These results suggest that some of the molecular events driving progression of neurofibroma to MPNST may be present before such changes manifest histopathologically.
While this study represents a key step toward establishing molecular signatures that define PNST behavior, there are several key limitations. Firstly, our study cohort was intentionally selected, focusing on subjects with ANF (NF with atypia and ANNUBP lesions) who also developed other tumors along the PNST continuum, including PNF and MPNST. Given the inherent selection bias, the incidence of MPNST in our study cohort is significantly higher than the 8–13% rate of malignant conversion in the general NF1 patient population. Furthermore, samples in this study were obtained retrospectively from subjects who were not followed as part of a formal natural history study. Therefore, it is possible that the prevalence of malignant transformation could be underestimated if subjects subsequently developed MPNST and sought care outside of our healthcare system. Although previous treatments including radiation, chemotherapy, and targeted therapies were documented based on review of the electronic medical record (Supplemental Table 13), the sample size of this cohort is insufficiently powered to investigate the impact of these interventions on the tumor transcriptome. Genomic characterization of the tumor samples in our study was also limited primarily to a subset of MPNSTs that underwent sequencing for clinical indications and information regarding germline pathogenetic variants and/or microdeletions of NF1 were not available. Finally, while the probe set employed here included a high content panel of genes with established relevance to cancer biology, tumor progression, and microenvironment, it does not provide comprehensive whole transcriptome coverage. Thus, other key transcriptional alterations may not have been captured in these data.
In addition to transcriptomics, combining multiple technologies including but not limited to ATAC-seq, methylation profiling and cell-free DNA (cfDNA) as predictive biomarkers may likely enhance our ability to accurately forecast the biological behavior and stratify risk of malignant transformation of neurofibroma precursor lesions. For instance, cutaneous neurofibroma exhibit site-specific methylation changes in genes associated with RAS dependent signaling that may contribute to their distinct growth kinetics in comparison to PNF (40). Recently, global methylation profiling revealed that most ANF exhibit an epigenetic profile similar to benign PNST (41). However, similar to our study, a subset of NF with atypia and ANNUBP clustered distinctly with MPNST, suggesting that histopathological diagnosis alone may be insufficient to ascribe the potential risk of malignant transformation associated with these lesions. Ultra-low pass whole gene sequencing of cfDNA has shown promise in distinguishing PNF from MPNST based on copy number alterations and has demonstrated potential for monitoring treatment responses and early detection of relapsed disease (42). Recently, multi-omic analysis conducted by the international Genomics of MPNST (GeM) consortium has further identified specific somatic copy number aberrations in both MPNST samples and cfDNA that function as a surrogate for H3K27 trimethylation loss and predict prognosis (16).
Prospective validation of biomarkers predictive of malignant transformation is limited by the practice of preemptively resecting DNLs/ANNUBP at many NF centers including our own. DNLs/ANNUBP that are inoperable, however, offer a valuable opportunity to elucidate molecular features that prognosticate the biological behavior of these lesions. Our findings suggest that suppression of immune surveillance and antigen presentation, along with upregulation of transcriptional programs associated with proliferation, survival, and developmental regression may indicate the emergence of malignant transformation. If validated prospectively, targeted gene expression analysis could augment conventional histopathological criteria to improve risk stratification and clinical decision-making for surveillance and early intervention in the treatment and prevention of MPNST.
Supplementary Material
TRANSLATIONAL RELEVANCE.
Transformation of benign plexiform neurofibromas (PNF) to malignant peripheral nerve sheath tumors (MPNST) is the leading cause of premature death in individuals with neurofibromatosis type 1 (NF1), one of the most common cancer predisposition syndromes affecting 1 in 3000 individuals. Transcriptional changes preceding malignant transformation remain poorly defined and no biomarkers capable of reliably predicting risk of transformation have been identified. Notably, NF1-associated PNST exhibit substantial heterogeneity and lesions at multiple stages across the PNST continuum can co-exist discretely within an individual or single tumor. Biopsies can be confounded by sampling bias and even neurofibroma conforming to uniform histopathological criteria can exhibit diverse features. This study identifies early molecular events preceding histopathological evidence of malignancy, which if validated prospectively in future studies, may serve to augment conventional histopathological diagnosis by identifying neurofibromas at high risk of undergoing malignant transformation, thus facilitating risk-adapted care.
ACKNOWLEDGEMENTS
This work was supported by a K08 Mentored Clinical Scientist Research Career Development Award to SDR by the National Institute of Neurological Disorders and Stroke (NIH/NINDS, K08-NS128266–02), R01-NS128025–01 (to DWC) and a Developmental and Hyperactive RAS Tumor SPORE funded by the National Cancer Institute (NIH/NCI, U54-CA196519–07) led by DWC. SDR is also supported by the Francis S. Collins Scholars Program in Neurofibromatosis Clinical and Translational Research funded by the Neurofibromatosis Therapeutic Acceleration Program (2004757180) and startup funding from the Department of Pediatrics at the Indiana University School of Medicine, the Indiana University Simon Comprehensive Cancer Center, and the Indiana University Precision Health Initiative. We thank Katie Jackson and Heather Daniel for administrative support.
Footnotes
Conflict of Interest Statement
The authors have no financial conflicts to disclose.
REFERENCES
- 1.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 1990;63(4):843–9 doi 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 2.Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, et al. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell 1990;62(1):187–92 doi 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- 3.Gross AM, Singh G, Akshintala S, Baldwin A, Dombi E, Ukwuani S, et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–51 doi 10.1093/neuonc/noy067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nguyen R, Dombi E, Widemann BC, Solomon J, Fuensterer C, Kluwe L, et al. Growth dynamics of plexiform neurofibromas: a retrospective cohort study of 201 patients with neurofibromatosis 1. Orphanet J Rare Dis 2012;7:75 doi 10.1186/1750-1172-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dombi E, Solomon J, Gillespie AJ, Fox E, Balis FM, Patronas N, et al. NF1 plexiform neurofibroma growth rate by volumetric MRI: relationship to age and body weight. Neurology 2007;68(9):643–7 doi 10.1212/01.wnl.0000250332.89420.e6. [DOI] [PubMed] [Google Scholar]
- 6.Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet 2002;39(5):311–4 doi 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uusitalo E, Rantanen M, Kallionpaa RA, Poyhonen M, Leppavirta J, Yla-Outinen H, et al. Distinctive Cancer Associations in Patients With Neurofibromatosis Type 1. J Clin Oncol 2016;34(17):1978–86 doi 10.1200/JCO.2015.65.3576. [DOI] [PubMed] [Google Scholar]
- 8.Kim A, Stewart DR, Reilly KM, Viskochil D, Miettinen MM, Widemann BC. Malignant Peripheral Nerve Sheath Tumors State of the Science: Leveraging Clinical and Biological Insights into Effective Therapies. Sarcoma 2017;2017:7429697 doi 10.1155/2017/7429697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higham CS, Dombi E, Rogiers A, Bhaumik S, Pans S, Connor SEJ, et al. The characteristics of 76 atypical neurofibromas as precursors to neurofibromatosis 1 associated malignant peripheral nerve sheath tumors. Neuro Oncol 2018;20(6):818–25 doi 10.1093/neuonc/noy013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miettinen MM, Antonescu CR, Fletcher CDM, Kim A, Lazar AJ, Quezado MM, et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum Pathol 2017;67:1–10 doi 10.1016/j.humpath.2017.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beert E, Brems H, Daniels B, De Wever I, Van Calenbergh F, Schoenaers J, et al. Atypical neurofibromas in neurofibromatosis type 1 are premalignant tumors. Genes Chromosomes Cancer 2011;50(12):1021–32 doi 10.1002/gcc.20921. [DOI] [PubMed] [Google Scholar]
- 12.Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 2014;46(11):1227–32 doi 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brohl AS, Kahen E, Yoder SJ, Teer JK, Reed DR. The genomic landscape of malignant peripheral nerve sheath tumors: diverse drivers of Ras pathway activation. Sci Rep 2017;7(1):14992 doi 10.1038/s41598-017-15183-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carrio M, Gel B, Terribas E, Zucchiatti AC, Moline T, Rosas I, et al. Analysis of intratumor heterogeneity in Neurofibromatosis type 1 plexiform neurofibromas and neurofibromas with atypical features: Correlating histological and genomic findings. Hum Mutat 2018;39(8):1112–25 doi 10.1002/humu.23552. [DOI] [PubMed] [Google Scholar]
- 15.Pemov A, Hansen NF, Sindiri S, Patidar R, Higham CS, Dombi E, et al. Low mutation burden and frequent loss of CDKN2A/B and SMARCA2, but not PRC2, define premalignant neurofibromatosis type 1-associated atypical neurofibromas. Neuro Oncol 2019;21(8):981–92 doi 10.1093/neuonc/noz028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cortes-Ciriano I, Steele CD, Piculell K, Al-Ibraheemi A, Eulo V, Bui MM, et al. Genomic Patterns of Malignant Peripheral Nerve Sheath Tumor (MPNST) Evolution Correlate with Clinical Outcome and Are Detectable in Cell-Free DNA. Cancer Discov 2023;13(3):654–71 doi 10.1158/2159-8290.CD-22-0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014;514(7521):247–51 doi 10.1038/nature13561. [DOI] [PubMed] [Google Scholar]
- 18.Rhodes SD, He Y, Smith A, Jiang L, Lu Q, Mund J, et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum Mol Genet 2019;28(16):2752–62 doi 10.1093/hmg/ddz095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chaney KE, Perrino MR, Kershner LJ, Patel AV, Wu J, Choi K, et al. Cdkn2a Loss in a Model of Neurofibroma Demonstrates Stepwise Tumor Progression to Atypical Neurofibroma and MPNST. Cancer Res 2020;80(21):4720–30 doi 10.1158/0008-5472.CAN-19-1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ge SX, Son EW, Yao R. iDEP: an integrated web application for differential expression and pathway analysis of RNA-Seq data. BMC Bioinformatics 2018;19(1):534 doi 10.1186/s12859-018-2486-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akhmedov M, Martinelli A, Geiger R, Kwee I. Omics Playground: a comprehensive self-service platform for visualization, analytics and exploration of Big Omics Data. NAR Genom Bioinform 2020;2(1):lqz019 doi 10.1093/nargab/lqz019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu LMN, Zhang F, Rao R, Adam M, Pollard K, Szabo S, et al. Single-cell multiomics identifies clinically relevant mesenchymal stem-like cells and key regulators for MPNST malignancy. Sci Adv 2022;8(44):eabo5442 doi 10.1126/sciadv.abo5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lopez R, Regier J, Cole MB, Jordan MI, Yosef N. Deep generative modeling for single-cell transcriptomics. Nat Methods 2018;15(12):1053–8 doi 10.1038/s41592-018-0229-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Badia IMP, Velez Santiago J, Braunger J, Geiss C, Dimitrov D, Muller-Dott S, et al. decoupleR: ensemble of computational methods to infer biological activities from omics data. Bioinform Adv 2022;2(1):vbac016 doi 10.1093/bioadv/vbac016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franzen O, Gan LM, Bjorkegren JLM. PanglaoDB: a web server for exploration of mouse and human single-cell RNA sequencing data. Database (Oxford) 2019;2019 doi 10.1093/database/baz046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pollard K, Banerjee J, Doan X, Wang J, Guo X, Allaway R, et al. A clinically and genomically annotated nerve sheath tumor biospecimen repository. Sci Data 2020;7(1):184 doi 10.1038/s41597-020-0508-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Calizo A, Zhang L, Pino JC, Lyu Y, Pollard K, et al. CDK4/6 inhibition enhances SHP2 inhibitor efficacy and is dependent upon RB function in malignant peripheral nerve sheath tumors. Sci Adv 2023;9(47):eadg8876 doi 10.1126/sciadv.adg8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gampala S, Shah F, Zhang C, Rhodes SD, Babb O, Grimard M, et al. Exploring transcriptional regulators Ref-1 and STAT3 as therapeutic targets in malignant peripheral nerve sheath tumours. Br J Cancer 2021;124(9):1566–80 doi 10.1038/s41416-021-01270-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li H, Chang LJ, Neubauer DR, Muir DF, Wallace MR. Immortalization of human normal and NF1 neurofibroma Schwann cells. Lab Invest 2016;96(10):1105–15 doi 10.1038/labinvest.2016.88. [DOI] [PubMed] [Google Scholar]
- 30.Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 2017;35(4):314–6 doi 10.1038/nbt.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gesundheit B, Parkin P, Greenberg M, Baruchel S, Senger C, Kapelushnik J, et al. The role of angiogenesis in the transformation of plexiform neurofibroma into malignant peripheral nerve sheath tumors in children with neurofibromatosis type 1. J Pediatr Hematol Oncol 2010;32(7):548–53 doi 10.1097/MPH.0b013e3181e887c7. [DOI] [PubMed] [Google Scholar]
- 32.Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol 2000;2(10):737–44 doi 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci 2001;22(4):201–7 doi 10.1016/s0165-6147(00)01676-x. [DOI] [PubMed] [Google Scholar]
- 34.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000;100(1):57–70 doi 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 35.Yang ZY, Guo J, Li N, Qian M, Wang SN, Zhu XL. Mitosin/CENP-F is a conserved kinetochore protein subjected to cytoplasmic dynein-mediated poleward transport. Cell Res 2003;13(4):275–83 doi 10.1038/sj.cr.7290172. [DOI] [PubMed] [Google Scholar]
- 36.Garg H, Suri P, Gupta JC, Talwar GP, Dubey S. Survivin: a unique target for tumor therapy. Cancer Cell Int 2016;16:49 doi 10.1186/s12935-016-0326-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ma S, Zhao Y, Lee WC, Ong LT, Lee PL, Jiang Z, et al. Hypoxia induces HIF1alpha-dependent epigenetic vulnerability in triple negative breast cancer to confer immune effector dysfunction and resistance to anti-PD-1 immunotherapy. Nat Commun 2022;13(1):4118 doi 10.1038/s41467-022-31764-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miar A, Arnaiz E, Bridges E, Beedie S, Cribbs AP, Downes DJ, et al. Hypoxia Induces Transcriptional and Translational Downregulation of the Type I IFN Pathway in Multiple Cancer Cell Types. Cancer Res 2020;80(23):5245–56 doi 10.1158/0008-5472.CAN-19-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghadimi MP, Young ED, Belousov R, Zhang Y, Lopez G, Lusby K, et al. Survivin is a viable target for the treatment of malignant peripheral nerve sheath tumors. Clin Cancer Res 2012;18(9):2545–57 doi 10.1158/1078-0432.CCR-11-2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grit JL, Johnson BK, Dischinger PS, C JE, Adams M, Campbell S, et al. Distinctive epigenomic alterations in NF1-deficient cutaneous and plexiform neurofibromas drive differential MKK/p38 signaling. Epigenetics Chromatin 2021;14(1):7 doi 10.1186/s13072-020-00380-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kresbach C, Dottermusch M, Eckhardt A, Ristow I, Paplomatas P, Altendorf L, et al. Atypical Neurofibromas reveal distinct epigenetic features with proximity to benign peripheral nerve sheath tumor entities. Neuro Oncol 2023. doi 10.1093/neuonc/noad053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szymanski JJ, Sundby RT, Jones PA, Srihari D, Earland N, Harris PK, et al. Cell-free DNA ultra-low-pass whole genome sequencing to distinguish malignant peripheral nerve sheath tumor (MPNST) from its benign precursor lesion: A cross-sectional study. PLoS Med 2021;18(8):e1003734 doi 10.1371/journal.pmed.1003734. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw RCC files, raw counts and sample metadata pertaining to spatial gene expression profiling of human PNST specimens described here are available from Gene Expression Omnibus under accession GSE239561. Bulk RNAseq for comparing TCGA MPNST vs GTEx normal nerve are in Supplemental Table 11, Supplementary Table 12 and publicly available through the UCSC RNA-seq Toil Recompute Compendium (30) hosted through the UCSC Xena Browser. Bulk RNAseq from Nf1flox/flox;DhhCre and Ink4a/Arf +/−;Nf1flox/flox;DhhCre murine PNST generated by Chaney et al. (19) are available from GEO under accession number GSE148249. Bulk RNAseq data from Nf1flox/flox;PostnCre and Nf1flox/flox;Arf flox/flox;PostnCre + mice are available from GEO under accession numbers GSE213786 and GSE232707.