

Abstract
Next-generation sequencing technologies, exponential increases in the availability of virus genomic data, and ongoing advances in phylogenomic methods have made genomic epidemiology an increasingly powerful tool for public health response to a range of mosquito-borne virus outbreaks. In this review, we offer a brief primer on the scope and methods of phylogenomic analyses that can answer key epidemiological questions during mosquito-borne virus public health emergencies. We then focus on case examples of outbreaks, including those caused by dengue, Zika, yellow fever, West Nile, and chikungunya viruses, to demonstrate the utility of genomic epidemiology to support the prevention and control of mosquito-borne virus threats. We extend these case studies with operational perspectives on how to best incorporate genomic epidemiology into structured surveillance and response programs for mosquito-borne virus control. Many tools for genomic epidemiology already exist, but so do technical and nontechnical challenges to advancing their use. Frameworks to support the rapid sharing of multidimensional data and increased cross-sector partnerships, networks, and collaborations can support advancement on all scales, from research and development to implementation by public health agencies.
Keywords: dengue, genomic epidemiology, yellow fever, arbovirus
In the last decade, we have seen many mosquito-borne virus threats of global concern (Figure 1). In the last 5 years alone, Zika virus (ZIKV) and chikungunya virus (CHIKV) have caused major pandemics, in addition to the growing burden of dengue virus (DENV) throughout the tropics and subtropics [1, 2]. Other regional mosquito-borne alphaviruses (eg, Mayaro virus [MAYV] and Ross River virus) and flaviviruses (eg, yellow fever virus [YFV], West Nile virus [WNV], Japanese encephalitis virus, and Saint Louis encephalitis virus) continue to cause substantial morbidity or mortality (Figure 1). Robust and timely epidemiological investigations are critical to respond to these pathogens and prevent further spread. Conventional epidemiological approaches, which leverage clinical data from laboratory diagnostic assays [3], case definitions, and contact tracing, are the necessary first step towards understanding outbreak dynamics. For example, these data can be used to estimate the basic reproduction number, epidemic peak, epidemic trajectory, cumulative case count, case-fatality ratio, latency period, and the predicted effect of interventions [4]. Yet, in many scenarios, these components may be difficult to measure and higher resolution may be required to support effective interventions. To help address these challenges, virus genomes are increasingly being used to complement traditional epidemiological data [5, 6]. These “genomic epidemiology” approaches can reveal explicit and otherwise hidden geographic, host and temporal histories of virus outbreaks, the association between virus strain and clinical outcome, strain dependency of vaccine effectiveness, the duration of asymptomatic transmissions, and mitigatable predictors of virus dispersal on spatial scales as fine as a household [7, 8]. Thus, in the era of “big data”, virus genomics has found a home in epidemiology.
Figure 1.
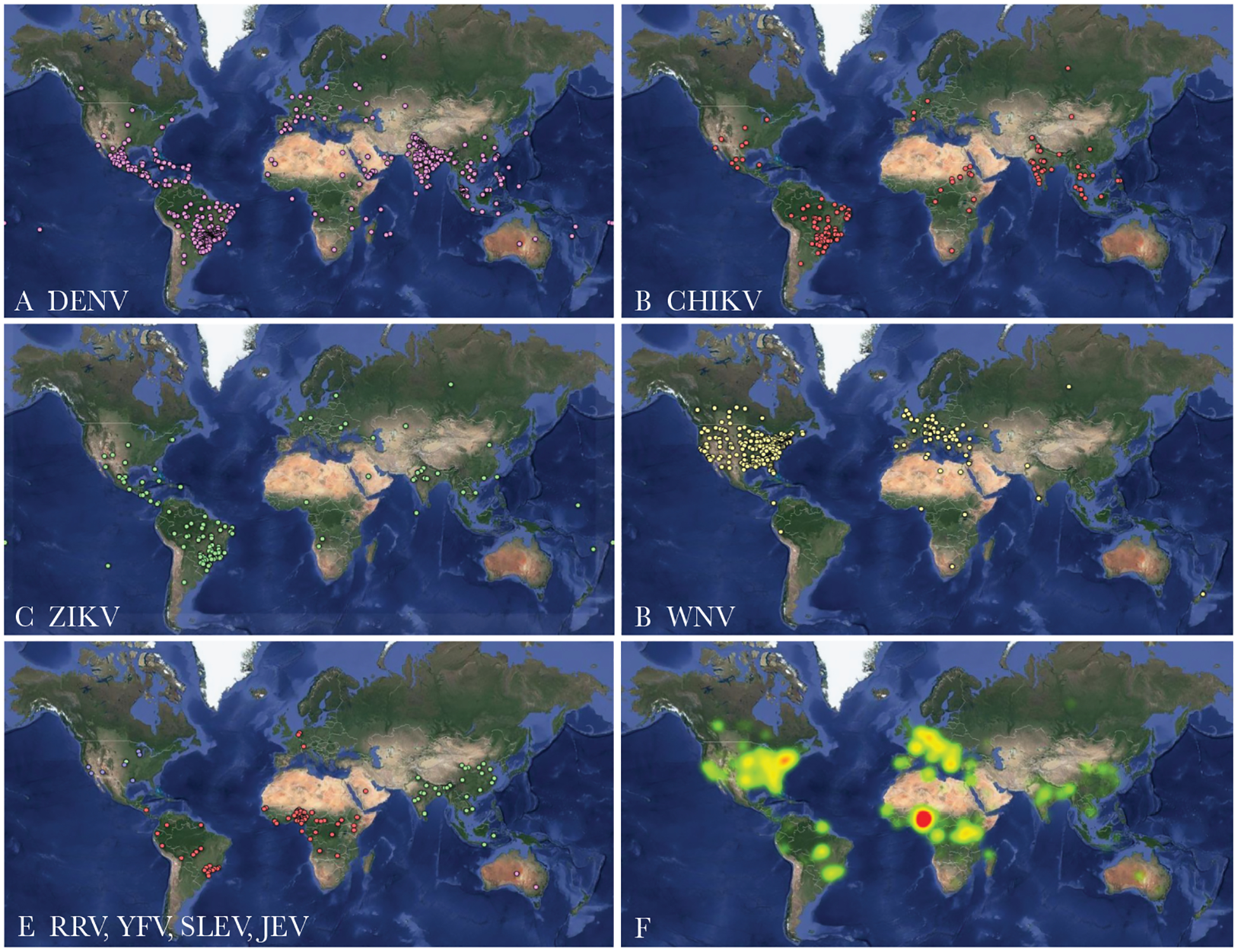
Healthmap-compiled reports of locally acquired or travel-associated cases of mosquito-borne viruses. (A) Dengue virus (DENV), (B) chikungunya virus (CHIKV), (C) ZIKA virus (ZIKV), (D) West Nile virus (WNV), (E) Ross River virus ([RRV] pink), yellow fever virus ([YFV] red), Saint Louis encephalitis virus ([SLEV] blue), and Japanese encephalitis virus ([JEV] green) case alerts from August 2018 to February 2019. (F) A heat-map of all the above reports combined. Data were accessed from (www.healthmap.org, with permission) and mapped using Google Fusion Tables (https://support.google.com/fusiontables/).
As virus genome sequence repositories continue to grow, enabled by advances in genomic sequencing technologies and bioinformatics [9], so too does the utility of genomic epidemiology. This deluge of genomic data, along with an increasing amount of open-access phylogenetic analysis software, offers a “world of possibilities” for enhancing mosquito-borne virus surveillance and outbreak response [10]. Nonetheless, the real-world implementation of such genomic technologies into operational public health remains challenging [7, 8] (Box 1). This review presents a variety of phylogenomic analyses that have been implemented to address key epidemiological questions and strengthen the public health response to mosquito-borne virus outbreaks, epidemics, and pandemics. We conclude with operational perspectives on incorporating genomic epidemiology into structured surveillance and response programs for these virus threats.
Box 1-. Challenges to incorporating genomic epidemiology into public health laboratories.
1. Aligning research and public health objectives:
Although public health needs during active mosquito-borne virus transmission are priority, meeting research goals can inform best practices for future outbreaks. Agreements regarding data ownership/usage through established collaborations can meet the needs of both public health practitioners and researchers.
2. Funding for reagents, salaries, sample collection, equipment and training:
Mosquito-borne virus epidemics strain public health resources and local economies [46]. The benefits of incorporating genomic epidemiology into mosquito-borne virus surveillance and control likely outweighs the cost, but cost-effectiveness analyses would be informative.
3. Generating tangible end results in a timely manner:
Public health agencies often work on limited budgets and personnel. Incorporating genomic epidemiology has the potential to increase individual workloads. Collaborations with academic institutions can ease the burden of extra work.
4. Linking clinical, epidemiological, and genomic data:
Merging complementary datasets, while removing any potential identifying information, will provide the more complete picture of virus transmission. The expertise required for these advanced analyses is considerable. The time necessary to obtain institutional review and approval to use clinical data and samples, however, can prolong the response time. This should be carefully planned before initiating the studies.
5. Standardized and reproducible bioinformatic pipelines
Accompanying software has expanded alongside NGS technology. An ever-changing computational environment will require standardization and documentation. Investment in NGS training and expertise is critical to accommodate these demands.
CASE STUDIES OF GENOMICS-INFORMED APPROACHES TO MOSQUITO-BORNE VIRUS OUTBREAKS
The trajectory of mosquito-borne virus outbreaks is influenced by a confluence of host, vector, and environmental factors, inevitably making the public health response largely context dependent. These factors include immunity in vertebrate host populations, vector abundance and competence, and their overlap in time and space [2]. Due to the lack of vaccines and effective antiviral treatments for most mosquito-borne viruses, mitigating outbreaks generally involves personal risk reduction and vector control. At a minimum, virus genomics can help enhance surveillance by identifying the etiological agents and specific genotypes to facilitate diagnostic assay development (eg, polymerase chain reaction primer design) [11]. At the other end of the spectrum, advanced genomic epidemiology, leveraging evolutionary and bioinformatics methods (Figure 2), can reconstruct local, granular transmission dynamics, pin-point emergence and transmission hotspots, identify drivers of epidemic spread and growth, and, thus, provide information for targeted control and prevention measures in near real time [8]. The bedrock of these advanced analyses is evolutionary biology, using phylogenomic inference to resolve many critical epidemiological features of mosquito-borne virus outbreaks because evolutionary time scales of ribonucleic acid viruses typically match epidemiological time scales (ie, many virus mutations arise during an epidemic) [5]. Software such as MEGA, R, RAxML, and PhyML can infer distance or character-based phylogenetic trees that are annotated with clinical, demographic, temporal, geographic, host species, and/or other critical phenotypic data [12–15]. This permits useful epidemiological inference through patterns of phylogenetic clustering and identification of genotype-phenotype associations. Bayesian packages like BEAST are frequently used because they can infer, for instance, the time-scale, geographic routes, and host of unsampled, ancestral viruses under a range of demographic and virological assumptions (discussed in detail in other reviews [5, 7, 8, 16]). Table 1 summarizes the epidemiological phenomena that can be elucidated using such evolutionary analytic methods, with specific examples. Below, we discuss examples from three pathogens in detail: DENV, ZIKV, and YFV.
Figure 2.
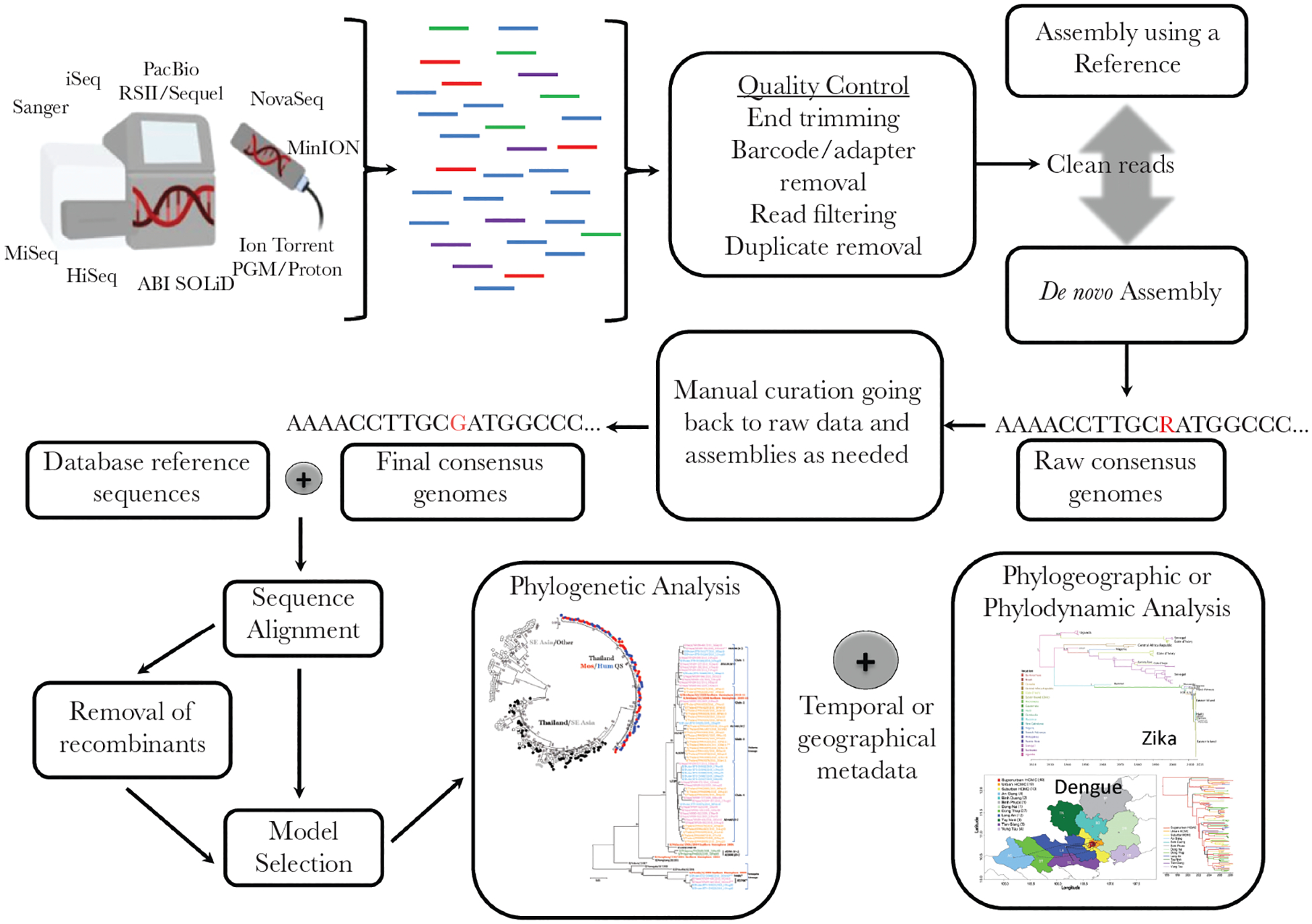
Wet-laboratory and dry-laboratory workflow for mosquito-borne virus genomic epidemiology. (Clockwise) Sequence reads are generated by a range of short or long read sequencing platforms (or older conventional methods) and undergo quality control before assembly using a reference sequence or de novo method (or both). Assembled genomes undergo manual curation by a trained bioinformatician. Consensus genomes are aligned with reference background data from public and other repositories. Advanced analyses require recombination detection (with consideration of removal), nucleotide model selection, and phylogenetic tree inference. Annotation of phylogenetic trees with epidemiological important metadata such as time, location, host, or clinical phenotype permits phylogeographic and/or phylodynamic analyses. Phylogeography images reproduced and adapted from [17, 89], under the creative commons.
Table 1.
Examples of Epidemiological Phenomenon That Can Be Elucidated Using Mosquito-Borne Virus Genome Sequence Data
| Epidemiological Phenomenon | Examples and References |
|---|---|
| Examining outbreak/case transmission linkage at a national or subnational scale | YFV, Uganda/Angola, 2016 [18] YFV, Brazil, 2016–2017 [20] CHIKV, Brazil, 2014 [19] DENV, Brazil, 2013 [21] |
| Confirming autochthonous spread in regions with first cases reported | ZIKV, Florida, 2016 [23] CHIKV, Florida, 2014–2015 [22] |
| Confirming geographic origin of imported or autochthonous arbovirus cases | RVF, China (ex-Angola), 2016 [24] YFV, China (ex-Africa), 2017 [25] JEV, Angola, 2016 [26] ZIKV, Florida, 2016 [23] CHIKV, Brazil, 2014 [19] DENV, Florida, 2013 [28] DENV, Puerto Rico, 1998–2007 [27] |
| Sentinel mosquito-borne virus case detection in otherwise undifferentiated neuroinvasive syndromes | SLEV, California, 2016 [29] Cache Valley Virus, Australia (ex-USA), 2013 [30] |
| Defining lineage host range | MAYV, South America, 2010–2015 [31] MVEV, Australia, 2015 [32] |
| Identifying lag between time of arbovirus emergence and first detection in a geographic area | ZIKV, Florida, 2016 [23] CHIKV, Brazil, 2014 [19] DENV, Peru, 2008–2015 [33] DENV, Brazil, 2006 [34] |
| Identifying patterns and predictors of virus spatial introduction and spread on various spatial scales | DENV, Thailand, 1994–2010 [35] DENV, Brazil, 1997–2008 [37] MAYV, Americas, 2010–2015 [31] WNV, USA, 1999–2004 [36] CHIKV, Nicaragua and USA, 2014–2015 [22] DENV, Vietnam, 2003–2008 [38] |
| Identifying the spatial distribution of strains of possible higher virulence or complications | ZIKV, French Polynesia and Americas, 2013–2016 [39] DENV, Puerto Rico, 1994 [40] |
| Identifying strains of higher vector competence and transmissibility | CHIKV, Indian Ocean, 2005 [41] CHIKV, Kenya, 2016 [42] DENV, Americas, 2001[43] |
| Examining role of virus recombination in virus emergence and spread | MAYV, Brazil, 2014–2015 [31] DENV, global, various years [44, 45] |
| Identifying virus strains of higher risk of vaccine failure | DENV, Asia and Americas, 2011–2014 [47] |
Abbreviations: CHIKV, chikungunya virus; DENV, dengue virus; JEV, Japanese encephalitis virus; MAYV, Mayaro virus; MVEV, Murray Valley encephalitis virus; RVF, Rift Valley Fever; SLEV, Saint Louis encephalitis virus; WNV, West Nile virus; YFV, yellow fever virus.
Dengue Viruses
As a result of the expanding range of Aedes aegypti (the primary DENV vector) and rapid urbanization, the burden of disease caused by DENV throughout the tropics is on the rise [1], resulting in the largest number of genomic epidemiology studies of any mosquito-borne virus. These studies have allowed fine-scale reconstructions of DENV spread between continents, countries, cities, suburbs, and households (Table 1 and [7]). Joint Bayesian analyses of human transport data with genomic data has resolved the role of airline traffic relative to other predictors such as distance and vector abundance, in the spread of DENV within South East Asia and Brazil, and has suggested that airline traffic contributes to international DENV dispersal [37, 48]. Although there have been many valuable mosquito-borne virus phylodynamic and phylogeographic studies that have leveraged envelope coding sequence data [28, 49] (Table 1), such epidemiological analyses are most informative with whole genome data, which offers phylogenetic resolution on very fine spatial and temporal scales [50]. However, even subgenomic sequence data is informative, particularly when combined with clinical data. For example, using envelope protein coding DENV sequences from breakthrough dengue infections in the CYD-14/15 (Dengvaxia) clinical trials, Rabaa et al [47] found that among DENV-4 genotypes, vaccine efficacy was lowest against genotype 1. Juraska et al [51] saw a similar sieve effect and noted that this association between strain and vaccine efficacy was modulated by host age, likely reflecting prior exposure risk to DENV before vaccination. Taken together, these molecular epidemiology studies have shown that Dengvaxia-induced protection is dependent on the genotypes of circulating DENV-4. Postlicensure, phylogenetic studies can offer an opportunity to identify future breakthrough infections as the circulation of specific DENV genotypes and immunity in the human population changes.
Zika Virus
Zika virus, another flavivirus primarily transmitted by A aegypti, caused a major pandemic in the Americas in 2015–2016, and it has been found in tropical regions of Asia and Africa [2, 52]. During the 2016 ZIKV outbreak in Florida, virus sequencing from clinical and entomological samples corroborated epidemiological evidence of local mosquito-borne transmission at suburban (subcounty level) granularity [23]. The phylogenomic resolution offered by near-complete ZIKV genome data also revealed that the outbreak was driven by multiple independent virus introductions, and that the local transmission had started months before the outbreak was recognized. These genomic analyses were further combined with regional country-level incidence and global transportation data to confirm that the Caribbean Islands contributed most to the seeding of the 2016 Florida outbreak, and they emphasized the role of human movement in the spread of ZIKV during the Americas pandemic [23].
The 2015–2017 ZIKV epidemic in Brazil was a valuable test case for the feasibility of deploying field-based, mobile sequencing capability during a public health emergency [53]. Despite the difficulty of whole-genome sequencing with low ZIKV viremia [11, 54], Faria et al [53] confirmed that ZIKV spread in the Americas originated from Brazil. Moreover, a genomic time-scaled reconstruction of the epidemic history of Brazilian ZIKV cases indicated cryptic (undetected) transmission that likely occurred for more than 1 year before the first case was detected [11, 53]. Such findings are important to understand the limitations of surveillance systems, especially in regions where mosquito-borne viruses have not previously been identified and may emerge [55]. Those findings have also been useful to inform subsequent seroepidemiological studies [56] and to clarify the population risk period of maternal ZIKV exposure. These studies emphasize the value of genomic epidemiology analyses for ZIKV, which has very similar symptoms and serological responses to DENV, thereby complicating conventional epidemiological investigations based on clinical case definitions and available serological assays [57].
Yellow Fever Virus
Yellow fever virus is another pathogenic flavivirus transmitted by Aedes and Haemagogus species mosquitoes in urban and sylvatic transmission cycles, respectively. Although a vaccine exists, coverage in endemic areas is limited and major epidemics, such as the 2015–2016 outbreak in Angola, continue to deplete the global vaccine supply [58]. The 2016–2017 YFV outbreak in Brazil is in part driven by the vaccine shortage and is a significant public health challenge with over 2000 cases and 676 deaths [20]. A genomic epidemiological study by Faria et al [20] in Minas Gerais (Southeastern Brazil) revealed many important components of this outbreak: (1) the sequence data confirmed that the outbreaks were not due to a reversion of the 17-DD YFV vaccine strain; (2) the virus was not the same strain that caused an outbreak in the region ~15 years prior, but rather a new introduction from elsewhere in Brazil; (3) molecular dating of the outbreak viruses showed that the surveillance system in Minas Gerais rapidly detected the first cases of human YFV; (4) the high estimated YFV dispersal velocity within Brazil suggests that intranational spread was aided by human activity; and, perhaps most importantly, (5) human cases were caused by continuous and direct sylvatic cycle (Haemagogus-primate) spillovers rather than urban cycle (A aegypti-human) spread [20]. This additional insight provided by high-resolution genomic analyses can be critical for assessing and controlling YFV outbreaks.
BEYOND RESEARCH: CHALLENGES AND APPROACHES TO INCORPORATING GENOMIC EPIDEMIOLOGY INTO STRUCTURED MOSQUITO-BORNE VIRUS SURVEILLANCE AND RESPONSE SYSTEMS
The rapid development of genomic tools to study virus outbreaks has facilitated the transition from genomic analysis solely as a research tool to the prospect of use in near-real-time public health investigations [59]. As next-generation sequencing (NGS) technology improves and costs decrease, public health departments may increasingly have the capability to identify and reconstruct outbreaks of mosquito-borne viruses closer to the source [8, 60]. The application of NGS technologies is already becoming increasingly routine for outbreaks of food-borne pathogens (eg, the US Centers for Disease Control and Prevention’s Pulsenet), nosocomial multidrug-resistant bacteria, influenza virus, hepatitis C virus, human immunodeficiency virus, and gonorrhoea [61]. Still, most genomic epidemiology studies of mosquito-borne virus outbreaks are retrospective in nature (Table 1) and are often communicated in publications many months after specimen collection is complete [62]. Next-generation sequencing can be implemented in real time within existing laboratory surveillance programs and serve direct needs of public health departments; however, a range of technical and nontechnical challenges currently restrict widespread adaptation (Box 1). These challenges are in addition to the methodological caveat of genomic sampling bias (ie, unsampled cases can strongly bias conclusions about the patterns of virus spread). Sampling bias remains a major issue for interpreting many genomic epidemiological studies [7], but it is also an issue that could be addressed by incorporating NGS in routine surveillance (eg, for a random sample of laboratory-confirmed cases). Perhaps the greatest challenge of using genomic epidemiology for routine public health practice is not the technology but its implementation.
Training and Resource Demands of Next-Generation Sequence-Enabled Epidemiology
Numerous NGS protocols developed for platforms such as the Oxford Nanopore MinION and Illumina’s MiSeq and iSeq 100 have drastically reduced upfront costs and shortened the time from sample preparation to raw data [63, 64]. Deployable hand-held sequencers like the MinION have received particularly enthusiastic coverage in the scientific literature [63]; however, the wet and dry laboratory training requirements of any NGS platform (regardless of size) are considerable [60]. This fact makes deployment of novel NGS technologies without appropriate training at best futile and at worst detrimental to outbreak response. Although there has been a recent push to prioritize NGS in public health laboratories and to train public health professionals in computational biology [61], a significant barrier to implementation appears to be insufficient staffing of individuals proficient in NGS sample preparation and data analysis. These trained professionals are necessary to implement standardized NGS and bioinformatic approaches to ensure that the results and analyses are easily repeatable, documented to include acceptable quality control steps and quality assurance processes, and therefore actionable for public health policy. Successful models of international and regional partnerships have contributed to improvements in critical pathogen wet laboratory and bioinformatic skills in a range of settings, including Africa, South America, and Asia (discussed in [7, 65]). However, the hiring and training of staff as well as purchasing the necessary equipment and reagents for genomic epidemiology ultimately requires dedicated and sustainable funding mechanisms. Acquiring these resources is often the major barrier for implementation because funding needs to be in place well before an outbreak begins, and funding needs to be allocated in the context of other potentially competing demands. For example, establishing a robust national field epidemiology program and basic reference molecular and serological diagnostic assay capability should occur before establishing NGS laboratory and bioinformatics capacity. However, spending on outbreak responses often outweighs spending on outbreak preparedness [65]. An illustrative example of reactive investment came after the 2013–2016 West African Ebola virus outbreak, where funds dedicated to capacity building in affected areas were transitioned to the ZIKV response in the Americas [66, 67].
The Need for Timely, Quality Sequence Data and Accompanying Metadata
Although mosquito-borne virus genomic data offers unique epidemiological insights (Table 1), it is necessary to integrate this data with routine and standardized public health data to get a complete picture of the outbreak (Box 2). For example, using genomics to identify transmission chains supplemented with ecological, entomological, and human mobility data will create higher resolution risk maps, as opposed to just one data source alone, and can identify mitigatable predictors of spread [8]. Thus, virus genomic data provides an extra layer of detail, but it is best applied in the context of “classical” epidemiological approaches and within a structured system. Contemporary bioinformatic and evolutionary biology methods now allow for joint analysis of genomic and nongenomic data to elucidate the relative roles of distance, vector abundance, climate, human movement, and human case counts in epidemic dispersal and epidemic size (Table 1) [68].
Box 2-. Other data sources that can complement genomic data in a “systems epidemiology” framework for mosquito-borne virus outbreak response and prevention.
1. Continuous and structured mosquito surveillance programs
Mosquito and virus surveillance provide information about vector abundance, distribution, infection rates, and time of infection (eg, recent introductions or overwintering of viruses into a mosquito population) to identify the patterns of transmission and to inform vector control.
2. Local clinical surveillance
The first reports of mosquito-borne autochthonous virus transmission are often from clinicians treating local patients. Confirmed diagnoses coupled with enhanced surveillance techniques (such as active surveillance or syndromic surveillance) help identify early transmission events and risk factors associated with arbovirus infections and their complications.
3. Traveler surveillance and situational awareness of arbovirus incidence in other countries
Clinical evaluation, including a travel history, and testing of specimens from febrile individuals returning to the country from abroad, provides a wealth of information about what, when, where, and how mosquito-borne viruses are being introduced [50]. In addition to conventional advisories from formal public health organizations, hybrid digital disease detection systems, such as HealthMap, can offer fine scale risk maps to tourist and VFR destinations abroad, and this can inform pre-test probability before microbiological testing [90].
4. Ecological data
Weather, land-use and human movement patterns are major predictors of mosquito-borne virus transmission. Temperature, precipitation, and humidity influence growth and reproductive rates in vector mosquito populations, as well as the rate at which mosquito-borne viruses will replicate. Air travel has been linked to arbovirus spread. These data can be used to inform models that elucidate risk of transmission.
5. Seroprevalence and vaccine coverage data
Population seroprevalence to arboviruses is a strong determinant of the risk of outbreaks for flaviviruses, alphaviruses and other arboviruses. Jointly analyzing seroprevalence data with genomic data can identify how population immunity shapes the dispersal patterns of mosquito-borne viruses.
Much attention focuses on improving the timely and open sharing of virus sequence data during outbreaks, acknowledging the multiple contributing technical, ethical, and political barriers to seamless genomic data sharing [69, 70]. Indeed, there have been marked improvements in outbreak virus sequence data sharing since the 2014 Ebola epidemic, such as the immediate publication of ZIKV whole-genome sequence data during the 2016 Florida outbreak on Github and Virological.org [23, 71]. Yet, timely access to well curated demographic, human movement, entomological, and other complementary metadata may be particularly challenging, especially in austere settings. Although encouraging progress has been made with initiatives such as WorldPop, FlowMinder, and VectorBase [72–74], these open-access datasets may often lack spatial granularity and do not cover all needs. Furthermore, the timely sharing of temporally and spatially explicit case data to accompany genomic data remains a pervasive issue. In a recent study, the World Health Organization drafted a pathogen sequence-sharing “code of conduct” that may facilitate the sharing of accompanying case metadata [75]. As whole-genome sequence and metadata repositories grow and offer epidemiological constructions on increasingly granular spatial and population scales, one important consideration is the potential for inadvertent breaches of case privacy [76]. Indeed, this has long been a risk for nongenomic spatial modeling and highlights the need for a robust ethical framework when undertaking public health and academic investigations into pathogen outbreaks [77, 78].
A final, yet critical, category of accompanying data that are vital for outbreak phylodynamic studies is population serological data that reflect age-specific exposure to epidemic mosquito-borne viruses. Such seroprevalence data are often the critical genotype-phenotype “missing link” in virus phylodynamic studies, yet they are often limited to costly and siloed natural infection cohort studies [79]. There are exciting efforts to establish “world serum banks”, and seroprevalence maps in part based on reported case incidence, but these initiatives have limitations or are still in their infancy [80, 81].
Translational Mosquito-Borne Virus Genomic Epidemiology Through Partnerships, Networks, and Collaborations
In general, mosquito-borne virus surveillance capabilities are split between clinical diagnostics (ie, patient-based) and environmental sampling (ie, mosquito-based). In the United States, both endemic and exotic mosquito-borne viruses are nationally notifiable diseases [82]. This passive clinical surveillance system used to report cases (eg, detection based on presentation of cases to clinical care) is often the first indication of an outbreak of an exotic virus, and it can trigger active investigations, environmental sampling, and emergency insecticide usage. On the other hand, some US districts screen for the now endemic WNV in mosquito collections from routine, active mosquito surveillance [83]. However, mosquito-based surveillance capabilities are heterogeneous throughout the United States, and many designated localities lack resources to implement routine surveillance, much less perform molecular tests [84]. Nevertheless, NGS does not need to be implemented everywhere. For example, smaller mosquito control and surveillance agencies can partner with larger public health departments for both routine genomic sampling and outbreak response.
Due to the nonuniform distribution of sophisticated laboratory (eg, NGS) and computational (eg, large data storage and phylogenetic software) resources primarily in academic settings and a few well funded public health institutions, genomic epidemiology studies can occur by three main mechanisms: (1) academic investigations using data shared by public health laboratories, (2) academic-public health laboratory partnerships, and (3) autonomous public health laboratory investigations.
Academic investigations from available or shared public health data with little or no direct input from the public health agencies (“Level 1”) is a common form of genomic epidemiology and is important for refining methods and revealing general transmission pathways and risk factors. However, because such investigations often rely on obtaining biospecimens well after the outbreak has occurred, they do not generally help to inform control measures in real time. In austere settings, such approaches may include so-called “parachute science” in which international academic researchers may reap high-impact journal publications (of high value for securing further research funding) but contribute little to the long-term public health or public health capacity of the study population [65].
Public health partnerships with academic institutions that have the tools and resources for genomic investigations (“Level 2”) are becoming more common. Because responses to rapidly evolving outbreaks need to be prompt and deliberate, successful partnerships require strong relationships and open communication among stakeholders, collaborators, and the community at large [65, 85]. This includes a clear understanding of what public health agencies need and what genomic epidemiology can provide. Such an understanding may be facilitated by implementation science studies, which measure end-user perceptions and expectations of genomic epidemiology tools (eg, Muscatello et al [86]). To facilitate the rapid flow of samples and information during an outbreak, the “roots” of these sorts of partnerships should be established well before an outbreak and should include approved protocols and plans for rapid sample processing, deidentifying clinical data, and data sharing [65]. However, the ethical landscape of performing any research during emerging epidemics, even on surveillance data, is complex [87]. Achieving pre-emptive, preapproved research protocols that are flexible enough to be rapidly adapted to a new, unexpected mosquito-borne virus outbreak remains a major challenge. Nevertheless, the collective benefit of strong collaborations with open data sharing is considerable and is exemplified by the WestNile 4K Project [88], where more than 50 academic and public health institutes are teaming up to assess the diversity of WNV strains currently circulating in the United States. Developing these types of long-term relationships between academic groups and public health laboratories has the potential to generate a positive feedback loop, where lessons learned from previous outbreak responses will inform future interventions.
CONCLUSIONS
Establishing training programs and building laboratory and computational capacity are the critical components for moving towards autonomous investigations by public health laboratories (“Level 3”). Putting all the tools and resources in the hands of those making the public health decisions may ultimately provide the most effective outbreak responses. Not only will this reduce turnaround times, but it also reduces the potential for miscommunication and unaligned priorities. However, multiple challenges remain to establishing autonomous public health genomic epidemiology capacity for mosquito-borne virus control (Box 1). During outbreaks of unexpected mosquito-borne viruses, such as ZIKV in the Americas, it is still valuable for public health laboratories to maintain collaborations within academic institutions to provide context-dependent support. More importantly, establishing networks of epidemiologists, entomologists, ecologists, microbiologists, molecular biologists, and bioinformaticians will remain an effective strategy to understand and control mosquito-borne virus outbreaks as they occur.
Financial support.
This work was funded by the US Department of Defense Global Emerging Infections Surveillance and Response System, a division of the Armed Forces Health Surveillance Branch (to R. G. J., S. P., and I. M. B).
Footnotes
Disclaimer. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, USUHS, the Centers for Disease Control and Prevention, nor the U.S. Government. Several of the authors are U.S. Government Employees. This work was prepared as part of their official duties. Title 17 U.S.C. § 105 provides that “Copyright protection under this title is not available for any work of the United States Government.” Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties. The funders had no role in the writing of this review article.
Supplement sponsorship. This supplement is sponsored by WRAIR, LANL, USAMRIID, PUCP (Pontificia Universidad Catolica del Peru), USAFSAM, NIH.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest.
References
- 1.Bhatt S, Gething PW, Brady OJ, et al. The global distribution and burden of dengue. Nature 2013; 496:504–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gould E, Pettersson J, Higgs S, Charrel R, de Lamballerie X. Emerging arboviruses: why today? One Health 2017; 4:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pang J, Chia PY, Lye DC, Leo YS. Progress and challenges towards point-of-care diagnostic development for dengue. J Clin Microbiol 2017; 55:3339–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vynnycky E, White RG. An introduction to infectious disease modeling. 1st ed. New York: Oxford University Press, 2010: pp 1–10. [Google Scholar]
- 5.Pybus OG, Tatem AJ, Lemey P. Virus evolution and transmission in an ever more connected world. Proc Biol Sci 2015; 282:20142878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ladner JT, Grubaugh ND, Pybus OG, Andersen KG. Precision epidemiology for infectious disease control. Nat Med 2019; 25:206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pollett S, Melendrez MC, Maljkovic Berry I, et al. Understanding dengue virus evolution to support epidemic surveillance and counter-measure development. Infect Genet Evol 2018; 62:279–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grubaugh ND, Ladner JT, Lemey P, et al. Tracking virus outbreaks in the twenty-first century. Nat Microbiol 2019; 4:10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pybus OG, Fraser C, Rambaut A. Evolutionary epidemiology: preparing for an age of genomic plenty. Philos Trans R Soc Lond B Biol Sci 2013; 368:20120193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes EC. RNA virus genomics: a world of possibilities. J Clin Invest 2009; 119:2488–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Metsky HC, Matranga CB, Wohl S, et al. Zika virus evolution and spread in the Americas. Nature 2017; 546:411–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014; 30:1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 2010; 59:307–21. [DOI] [PubMed] [Google Scholar]
- 14.Paradis E, Bolker B, Claude J, et al. Package ‘ape’. Available at: https://cran.r-project.org/web/packages/ape/ape.pdf. Accessed 5 June 2019.
- 15.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 2016; 33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol 2012; 29:1969–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rabaa MA, Ty Hang VT, Wills B, Farrar J, Simmons CP, Holmes EC. Phylogeography of recently emerged DENV-2 in southern Viet Nam. PLoS Negl Trop Dis 2010; 4:e766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hughes HR, Kayiwa J, Mossel EC, Lutwama J, Staples JE, Lambert AJ. Phylogeny of yellow fever virus, Uganda, 2016. Emerg Infect Dis 2018; 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nunes MR, Faria NR, de Vasconcelos JM, et al. Emergence and potential for spread of Chikungunya virus in Brazil. BMC Med 2015; 13:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faria NR, Kraemer MUG, Hill SC, et al. Genomic and epidemiological monitoring of yellow fever virus transmission potential. Science 2018; 361:894–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ortiz-Baez AS, Cunha MDP, Vedovello D, et al. Origin, tempo, and mode of the spread of DENV-4 genotype IIB across the state of Sao Paulo, Brazil during the 2012–2013 outbreak. Mem Inst Oswaldo Cruz 2019; 114:e180251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan Y, Pickett BE, Shrivastava S, et al. Differing epidemiological dynamics of Chikungunya virus in the Americas during the 2014–2015 epidemic. PLoS Negl Trop Dis 2018; 12:e0006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grubaugh ND, Ladner JT, Kraemer MUG, et al. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 2017; 546:401–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi Y, Zheng K, Li X, et al. Isolation and phylogenetic study of Rift Valley fever virus from the first imported case to China. Virol Sin 2017; 32:253–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cui S, Pan Y, Lyu Y, et al. Detection of yellow fever virus genomes from four imported cases in China. Int J Infect Dis 2017; 60:93–5. [DOI] [PubMed] [Google Scholar]
- 26.Simon-Loriere E, Faye O, Prot M, et al. Autochthonous Japanese encephalitis with yellow fever coinfection in Africa. N Engl J Med 2017; 376:1483–5. [DOI] [PubMed] [Google Scholar]
- 27.Santiago GA, McElroy-Horne K, Lennon NJ, et al. Reemergence and decline of dengue virus serotype 3 in Puerto Rico. J Infect Dis 2012; 206:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Munoz-Jordan JL, Santiago GA, Margolis H, Stark L. Genetic relatedness of dengue viruses in Key West, Florida, USA, 2009–2010. Emerg Infect Dis 2013; 19:652–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiu CY, Coffey LL, Murkey J, et al. Diagnosis of fatal human case of St. Louis encephalitis virus infection by metagenomic sequencing, California, 2016. Emerg Infect Dis 2017; 23:1964–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wilson MR, Suan D, Duggins A, et al. A novel cause of chronic viral meningoencephalitis: Cache Valley virus. Ann Neurol 2017; 82:105–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mavian C, Rife BD, Dollar JJ, et al. Emergence of recombinant Mayaro virus strains from the Amazon Basin. Sci Rep 2017; 7:8718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russell JS, Caly L, Kostecki R, et al. The first isolation and whole genome sequencing of murray valley encephalitis virus from cerebrospinal fluid of a patient with encephalitis. Viruses 2018; 10(6). pii: E319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pollett S, Cruz C, Torre A, et al. Transmission chains and a gravity dynamic of dengue 3 spread within a hyperendemic city in the Peruvian Amazon (poster). American Society of Tropical Medicine and Hygiene, 67th Annual Scientific Meeting, October 2018 (New Orleans, LA). [Google Scholar]
- 34.Mondini A, de Moraes Bronzoni RV, Nunes SH, et al. Spatio-temporal tracking and phylodynamics of an urban dengue 3 outbreak in São Paulo, Brazil. PLoS Negl Trop Dis 2009; 3:e448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Salje H, Lessler J, Maljkovic Berry I, et al. Dengue diversity across spatial and temporal scales: local structure and the effect of host population size. Science 2017; 355:1302–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pybus OG, Suchard MA, Lemey P, et al. Unifying the spatial epidemiology and molecular evolution of emerging epidemics. Proc Nat Acad Sci U S A 2012; 109:15066–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nunes MR, Palacios G, Faria NR, et al. Air travel is associated with intracontinental spread of dengue virus serotypes 1–3 in Brazil. PLoS Negl Trop Dis 2014; 8:e2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raghwani J, Rambaut A, Holmes EC, et al. Endemic dengue associated with the co-circulation of multiple viral lineages and localized density-dependent transmission. PLoS Pathog 2011; 7:e1002064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan L, Huang XY, Liu ZY, et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017; 358:933–6. [DOI] [PubMed] [Google Scholar]
- 40.Manokaran G, Finol E, Wang C, et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015; 350:217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weaver SC, Forrester NL. Chikungunya: evolutionary history and recent epidemic spread. Antiviral Res 2015; 120:32–9. [DOI] [PubMed] [Google Scholar]
- 42.Maljkovic Berry IE, Pollett S, Figueroa K, et al. Global outbreaks and origins of a chikungunya virus variant carrying mutations which may increase fitness for Aedes aegypti: revelations from the 2016 Mandera, Kenya outbreak. Am J Trop Med Hyg. 2019. May; 100(5):1249–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Armstrong PM, Rico-Hesse R. Differential susceptibility of Aedes aegypti to infection by the American and Southeast Asian genotypes of dengue type 2 virus. Vector Borne Zoonotic Dis 2001; 1:159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Worobey M, Rambaut A, Holmes EC. Widespread intra-serotype recombination in natural populations of dengue virus. Proc Nat Acad Sci U S A 1999; 96:7352–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waman VP, Kale MM, Kulkarni-Kale U. Genetic diversity and evolution of dengue virus serotype 3: a comparative genomics study. Infect Genet Evol 2017; 49:234–40. [DOI] [PubMed] [Google Scholar]
- 46.Lee BY, Alfaro-Murillo JA, Parpia AS, et al. The potential economic burden of Zika in the continental United States. PLoS Negl Trop Dis 2017; 11:e0005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rabaa MA, Girerd-Chambaz Y, Duong Thi Hue K, et al. Genetic epidemiology of dengue viruses in phase III trials of the CYD tetravalent dengue vaccine and implications for efficacy. Elife 2017; 6. pii: e24196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tian H, Sun Z, Faria NR, et al. Increasing airline travel may facilitate co-circulation of multiple dengue virus serotypes in Asia. PLoS Negl Trop Dis 2017; 11:e0005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thomas DL, Santiago GA R, et al. Reemergence of dengue in Southern Texas, 2013. Emerg Infect Dis 2016; 22:1002–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grubaugh ND, Saraf S, Gangavarapu K, et al. International travelers and genomics uncover a ‘hidden’ Zika outbreak. Available at: https://www.biorxiv.org/content/early/2018/12/14/496901.1. Accessed 31 January 2019. [DOI] [PMC free article] [PubMed]
- 51.Juraska M, Magaret CA, Shao J, et al. Viral genetic diversity and protective efficacy of a tetravalent dengue vaccine in two phase 3 trials. Proc Nat Acad Sci U S A 2018; 115:E8378–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siraj AS, Perkins TA. Assessing the population at risk of Zika virus in Asia - is the emergency really over? BMJ Glob Health 2017; 2:e000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Faria NR, Quick J, Claro IM, et al. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017; 546:406–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quick J, Grubaugh ND, Pullan ST, et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat Protoc 2017; 12:1261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardner LM, Bóta A, Gangavarapu K, Kraemer MU, Grubaugh ND. Inferring the risk factors behind the geographical spread and transmission of Zika in the Americas. PLoS Negl Trop Dis 2018; 12:e0006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Netto EM, Moreira-Soto A, Pedroso C, et al. High Zika virus seroprevalence in Salvador, Northeastern Brazil limits the potential for further outbreaks. mBio 2017; 8(6). pii: e01390–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Masel J, McCracken MK, Gleeson T, et al. Does prior dengue virus exposure worsen clinical outcomes of Zika virus infection? A systematic review, pooled analysis and lessons learned. PLoS Negl Trop Dis 2019; 13:e0007060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barrett AD. Yellow fever live attenuated vaccine: a very successful live attenuated vaccine but still we have problems controlling the disease. Vaccine 2017; 35:5951–5. [DOI] [PubMed] [Google Scholar]
- 59.Gardy JL, Loman NJ. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat Rev Genet 2018; 19:9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gargis AS, Kalman L, Lubin IM. Assuring the quality of next-generation sequencing in clinical microbiology and public health laboratories. J Clin Microbiol 2016; 54:2857–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Centers for Disease Control and Prevention. Advanced Molecular Detection. Available at: https://www.cdc.gov/amd/index.html. Accessed 29 January 2019.
- 62.Johansson MA, Reich NG, Meyers LA, Lipsitch M. Preprints: an underutilized mechanism to accelerate outbreak science. PLoS Med 2018; 15:e1002549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quick J, Loman NJ, Duraffour S, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016; 530:228–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kafetzopoulou LE, Pullan ST, Lemey P, et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 2019; 363:74–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yozwiak NL, Happi CT, Grant DS, et al. Roots, not parachutes: research collaborations combat outbreaks. Cell 2016; 166:5–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nature 2016. Available at: https://www.nature.com/news/zika-response-must-not-drain-research-funds-1.20511. Accessed 29 January 2019.
- 67.McNeil DG. Obama Administration to Transfer Ebola Funds to Zika Fight, New York Times, April 6, 2016. Available at: https://www.nytimes.com/2016/04/07/health/zika-virus-budget-ebola.html. Accessed 29 January 2019. [Google Scholar]
- 68.Lemey P, Rambaut A, Bedford T, et al. Unifying viral genetics and human transportation data to predict the global transmission dynamics of human influenza H3N2. PLoS Pathog 2014; 10:e1003932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Littler K, Boon WM, Carson G, et al. Progress in promoting data sharing in public health emergencies. Bull World Health Organ 2017; 95:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Modjarrad K, Moorthy VS, Millett P, Gsell PS, Roth C, Kieny MP. Developing global norms for sharing data and results during public health emergencies. PLoS Med 2016; 13:e1001935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Available at: https://github.com/. Accessed 5 June 2019
- 72.Available at: www.worldpop.org.uk/. Accessed 31 January 2019.
- 73.Available at: www.flowminder.org/. Accessed 29 January 2019.
- 74.Available at: https://www.vectorbase.org/. Accessed 29 January 2019.
- 75.World Health Organization. Public consultation - Pathogen genetic sequence data. Available at: https://www.who.int/blueprint/what/norms-standards/gsdsharing/en/. Accessed 29 January 2019.
- 76.Raza S, Luheshi L. Big data or bust: realizing the microbial genomics revolution. Microb Genom 2016; 2:e000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brownstein JS, Cassa CA, Mandl KD. No place to hide–reverse identification of patients from published maps. N Engl J Med 2006; 355:1741–2. [DOI] [PubMed] [Google Scholar]
- 78.Lee EC, Asher JM, Goldlust S, Kraemer JD, Lawson AB, Bansal S. Mind the scales: harnessing spatial big data for infectious disease surveillance and inference. J Infect Dis 2016; 214:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Katzelnick LC, Harris E. The use of longitudinal cohorts for studies of dengue viral pathogenesis and protection. Curr Opin Virol 2018; 29:51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Metcalf CJ, Farrar J, Cutts FT, et al. Use of serological surveys to generate key insights into the changing global landscape of infectious disease. Lancet 2016; 388:728–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Global Dengue Transmission Map. Available at: https://mrcdata.dide.ic.ac.uk/_dengue/dengue.php. Accessed 29 January 2019.
- 82.Centers for Disease Control & Prevention. National Notifiable Diseases Surveillance System. Available at: https://wwwn.cdc.gov/nndss/. Accessed 29 January 2019.
- 83.Centers for Disease Control & Prevention. West Nile Virus in the United States: guidelines for surveillance, prevention and control. Available at: https://www.cdc.gov/westnile/resources/pdfs/wnvGuidelines.pdf. Accessed 29 January 2019.
- 84.National Association of County and City Health Officials. Mosquito Control Capabilities in the US. Available at: https://www.naccho.org/uploads/downloadable-resources/Mosquito-control-in-the-U.S.-Report.pdf. Accessed 29 January 2019. [Google Scholar]
- 85.O’Connor DH, Osorio JE, Tanuri A, Kallas EG. Forging collaborative relationships in Brazil: from AIDS to ZIKV. Cell 2016; 166:2–4. [DOI] [PubMed] [Google Scholar]
- 86.Muscatello DJ, Chughtai AA, Heywood A, Gardner LM, Heslop DJ, MacIntyre CR. Translation of real-time infectious disease modeling into routine public health practice. Emerg Infect Dis 2017; 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Saenz C Zika virus: ethics preparedness for old and new challenges. Lancet Glob Health 2016; 4:e686. [DOI] [PubMed] [Google Scholar]
- 88.WestNile 4K. Available at: https://westnile4k.org/. Accessed 29 January 2019. [Google Scholar]
- 89.Lednicky J, Beau De Rochars VM, El Badry M, et al. Zika virus outbreak in Haiti in 2014: molecular and clinical data. PLoS Negl Trop Dis 2016; 10:e0004687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Available at: www.healthmap.org. Accessed 31 January 2019.